

Abstract
Background
Systemic inflammation is associated with increased cognitive decline and risk for Alzheimer’s disease. Microglia (MG) activated during systemic inflammation can cause exaggerated neuroinflammatory responses and trigger progressive neurodegeneration. Dimethyl fumarate (DMF) is a FDA-approved therapy for multiple sclerosis. The immunomodulatory and anti-oxidant properties of DMF prompted us to investigate whether DMF has translational potential for the treatment of cognitive impairment associated with systemic inflammation.
Methods
Primary murine MG cultures were stimulated with lipopolysaccharide (LPS) in the absence or presence of DMF. MG cultured from nuclear factor (erythroid-derived 2)-like 2-deficient (Nrf2−/−) mice were used to examine mechanisms of DMF actions. Conditioned media generated from LPS-primed MG were used to treat hippocampal neuron cultures. Adult C57BL/6 and Nrf2−/− mice were subjected to peripheral LPS challenge. Acute neuroinflammation, long-term memory function, and reactive astrogliosis were examined to assess therapeutic effects of DMF.
Results
DMF suppressed inflammatory activation of MG induced by LPS. DMF suppressed NF-κB activity through Nrf2-depedent and Nrf2-independent mechanisms in MG. DMF treatment reduced MG-mediated toxicity towards neurons. DMF suppressed brain-derived inflammatory cytokines in mice following peripheral LPS challenge. The suppressive effect of DMF on neuroinflammation was blunted in Nrf2−/− mice. Importantly, DMF treatment alleviated long-term memory deficits and sustained reactive astrogliosis induced by peripheral LPS challenge. DMF might mitigate neurotoxic astrocytes associated with neuroinflammation.
Conclusions
DMF treatment might protect neurons against toxic microenvironments produced by reactive MG and astrocytes associated with systemic inflammation.
Electronic supplementary material
The online version of this article (10.1186/s12974-018-1125-5) contains supplementary material, which is available to authorized users.
Keywords: Microglia, Neuroinflammation, Dimethyl fumarate, Cognitive impairment, Astrocytes
Background
Dementia causes severe memory loss and destroys functional independence in affected individuals. Mounting evidence indicates that systemic inflammation acts as a significant driver of neurodegeneration and development of dementia [1]. Acute systemic inflammatory traumas not only cause profound cognitive dysfunction, such as delirium, but also lead to long-term cognitive decline and increase risk of developing dementia [2–4]. Low-grade peripheral inflammation chronically affects the volume of the hippocampus and increases risk of cognitive deficits in late life [5, 6]. During the progression of aging, the human body accumulates inflammatory attacks through repeated microbial infections, tissue injuries, or chronic disorders, such as obesity, diabetes, or atherosclerosis. Systemic inflammation caused by these inflammatory comorbidities can robustly alter brain inflammatory status through inducing phenotypic changes of the brain resident immune cells, microglia (MG).
MG play critical roles in maintaining tissue homeostasis of the brain. Upon stimulation, MG undergo a dynamic spectrum of activation patterns and change their functional properties [7–9]. Classically activated MG produce robust pro-inflammatory cytokines, including tumor necrosis α (TNFα), interleukin-1β (IL-1β), IL-12, and IL-6. The inflammatory activation of reactive MG contributes to the pathogenesis of neurodegeneration in various human neurological disorders [10]. Dysregulation in microglial activation leads to accumulations of “primed” MG that tend to react strongly to the stimuli from the periphery and get activated towards inflammatory phenotypes. Activated inflammatory MG trigger and orchestrate the process leading to chronic neuroinflammation and neurodegeneration [10].
Dimethyl fumarate (DMF) is a fumaric acid ester that is commonly used to treat psoriasis [11, 12]. The delayed-released DMF is approved as a therapy for the treatment of multiple sclerosis (MS) after showing its efficacy on reducing relapse rate and the number of new lesions in phase III clinical trials [13, 14]. DMF and its metabolites, monomethyl fumarate (MMF), exhibit immunomodulatory and anti-oxidant properties [15]. DMF stimulates anti-inflammatory responses and reduces oxidative stress by activating the nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a transcription factor regulating anti-oxidant response pathways [16, 17]. Under normal conditions, Nrf2 is sequestered in the cytoplasm by Kelch ECH-associated protein 1 (Keap1) binding and is degraded through the ubiquitin-proteasome pathway. Application of DMF stabilizes Nrf2 and promotes its transcriptional activities. Nrf2 binds the antioxidant response elements (ARE) located within gene promoters and induces expressions of detoxification and anti-oxidant enzymes such as hemoxygenase-1 (HO-1), NADPH quinone oxidoreductase-1 (NQO1), glutamate-cysteine ligase, catalytic subunit (Gclc), and glutamate-cysteine ligase regulatory subunit (Gclm). However, the DMF mechanism of action is not fully understood. Nrf2-independent mechanisms may be involved in the therapeutic effects of DMF in MS [18, 19].
In chronic autoimmune disorders, such as MS, MG are thought to sustain the inflammatory responses that are initially triggered by infiltrating T cells from the periphery. Activated MG propagate neuroinflammation and drive disease evolution into the progressive phase, leading to irreversible demyelination and axonal loss [9, 20]. Reactive MG induced by systemic inflammation might result in progressive inflammation inside the brain, leading to neurodegeneration and subsequently cognitive impairment. In this study, we investigated the potential effects of DMF on modulating reactive MG associated with systemic inflammation. Our results demonstrate that DMF significantly suppresses acute neuroinflammation following peripheral lipopolysaccharide (LPS) challenge in mice. DMF treatment alleviates long-term memory deficits and mitigates reactive A1-like astrocytes associated with systemic inflammation.
Methods
Animals
C57BL/6, Cx3cr1gfp/+, and Nrf2−/− mice were purchased from the Jackson Laboratory and bred at the animal facility of Indiana University School of Medicine, Fort Wayne. Mice were housed and maintained at 25 °C under a 12-h light/12-h dark cycle with ad libitum access to food and water. Adult female mice aged at 12–16 weeks were used for studies. All animal procedures in this study were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by Purdue Animal Care and Use Committee.
Reagents
Dimethyl fumarate (DMF) was purchased from Tocris Bioscience (Minneapolis, MN). Lipopolysaccharide (LPS; Escherichia coli O55:B5) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). JC-1 dye (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) was purchased from Thermo Fisher Scientific. Antibodies used for fluorescence-activated cell sorting (FACS) analysis were purchased from BioLegend (San Diego, CA): PE-conjugated anti-mouse CD11b (clone: M1/70) and CD86 (clone: PO3), PE/Cy7-conjugated anti-mouse CD80 (clone: 16-10A1), and APC-conjugated anti-mouse CD40 (clone: 3/23). For the western blot analysis, rabbit anti-Nrf2 and anti-HO-1 antibodies were purchased from Proteintech (Chicago, IL). Rabbit anti-phospho-IKK-α/β and rabbit anti-inducible nitric oxide synthase (iNOS) antibodies were purchased from Cell Signaling (Danvers, MA). For immunostaining of astrocytes, mouse anti-glial fibrillary acidic protein (GFAP) antibody was purchased from Cell Signaling (Danvers, MA); rabbit anti-aquaporin-4 (Aqp4) antibody was purchased from Sigma-Aldrich. Recombinant mouse TNFα and IL-1α were purchased from BioLegend (San Diego, CA).
Cell culture and drug treatment
Primary MG cells were obtained as previously described with some modifications [21]. Briefly, cells were cultured in medium DMEM/F12 containing 10% heated-inactivated fetal bovine serum, 2 mM l-glutamine, and 1X antibiotic/antimycotic. The medium was supplemented with 5 ng/ml granulocyte macrophage-colony stimulating factor (GM-CSF) on days in vitro (DIV) 5 and 10 after plating. On DIV 15, MG were harvested by shaking the flasks. The purity of MG cells was examined using FACS analysis. More than 95% of harvested cells were positive for CD11b expression (Additional file 1: Figure S1A). MG cells were seeded at desired densities and pre-treated with DMF (0, 10, and 100 μM in culture medium containing 0.001% DMSO final concentration) for 1 h and then stimulated with 100 ng/ml LPS for various times as indicated in the text and/or figure legends. The working concentration of LPS was prepared from the stock using culture medium as the diluent. Enriched astrocyte cultures were prepared as previously described [22]. Briefly, cells were harvested and maintained in the same medium for primary MG in the absence of GM-CSF. On DIV 8, MG and oligodendrocyte precursor cells growing on the top of confluent astrocyte monolayer were detached by shaking at 250 rpm for 6 h. The astrocyte layer was detached using trypsin EDTA and re-plated to cell culture flasks. On DIV 20, enriched astrocytes were prepared and seeded at desire density for experiments. The purity of enriched astrocyte cultures was examined using the GFAP and Aqp4 immunostaining analysis, showing 70–75% of cells are GFAP+ and Aqp4+ astrocytes (Additional file 1: Figure S1C). The astrocyte cultures were pre-treated with DMF (0 and 100 μM) for 1 h followed by TNFα (30 ng/ml) and IL-1α (3 ng/ml) stimulation for 24 h as indicated.
RNA isolation and real-time PCR
Total RNAs were isolated from cells or tissues using RNeasy RNA isolation kit (Qiagen). cDNAs were prepared by reverse transcription using High-Capacity cDNA Reverse Transcription Kit with random primers (Life Technologies, CA). Quantitative real-time PCR (qPCR) was performed using TaqMan gene expression probes and PCR Master Mix (Applied Biosystems, Life Technologies, CA).
Immunocytochemistry and confocal microscopy
Primary MG were treated as described. After treatment, cells were fixed in 2% paraformaldehyde following with phosphate-buffered saline (PBS) wash. Cells were permeabilized and blocked with 3% normal goat serum for 30 min at room temperature (RT). Cells were incubated with primary antibodies, rabbit anti-Iba1 (WAKO, Richmond, VA), rabbit anti-NF-κB p65 or anti-cleaved Caspase-3 (Cell Signaling, Danvers, MA) for 1 h at RT. After washing, cells were incubated with Alexa Fluor 594-conjugated goat anti-rabbit (Life Technologies, CA) for 1 h at RT. After washing, cell nuclei were counterstained with Hoechst 33342 (Life Technologies, CA). Images were acquired on Fluoview FV10i confocal microscope with an × 60 objective using the FV10iO software (Olympus) and analyzed using ImageJ, NIH.
Western blotting
Cell lysates were prepared with cold RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.15% SDS, 1 mM phenylmethylsulfonyl fluoride, phosphatase inhibitor, and protease inhibitor cocktail). Proteins were resolved in 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). The membrane was blocked and incubated with specific primary antibodies. After incubating with secondary antibodies, the blots were detected using ImmobilonTM Western Chemiluminescent HRP Substrate (EMD Millipore, MA).
Analysis of LPS-induced neuroinflammation after DMF treatment
Adult Cx3cr1gfp/+ mice were randomly assigned to different treatment groups and intraperitoneally (ip) injected with 0.9% saline or LPS (1 mg/Kg). For the DMF-treated group, mice were administered DMF (45 mg/Kg, ip) at 1 h before and 8 h after LPS injection [23, 24]. The ip administration route was performed to achieve higher bioavailability of DMF during shorter period of time after dosing and to decrease the potential stress to the sick LPS-treated mice. The brains were harvested 24 h post-injection. For imaging analysis, the harvested brains were fixed with 4% paraformaldehyde and cryoprotected with 30% sucrose. Serial 15-μm frozen sections were cut in a cryostat, mounted on superfrost plus slides, and stored at − 20 °C until use. Brain slides were prepared with ProLong Gold anti-fade mountant (Invitrogen). The fluorescence microscope BX53 (Olympus, PA), Nuance FX multispectral imaging system, and Nuance software (PerkinElmer) were used for image acquisition and analysis. For FACS analysis, brain mononuclear cells were isolated as previously described [21, 25]. The isolated cells were incubated with an antibody cocktail containing anti-CD86, CD80, and CD40. After staining, cells were subjected to FACS analysis. Mononuclear cell populations, including MG and infiltrating immune cells, were gated as GFP+ cells. For analysis of pro-inflammatory cytokines, the harvested brains were homogenized and subjected to RNA isolation. The expression levels of inflammatory mediators were measured using qPCR with interested TaqMan gene expression probes.
Cell viability assay
Primary MG were treated with DMF (0, 10, and 100 μM) for 1 h followed by LPS (1 μg/ml) stimulation. To produce LPS/DMF-free conditioned-medium (CM), the culture medium was changed to neurobasal medium (Life Technologies, CA) containing 2 mM l-glutamine and 1 × N2 supplement (Life Technologies, CA) at 6 h after LPS stimulation to wash out LPS and DMF. The microglial CM was collected 24 h after LPS. The microglial CM was applied to differentiate HT22 hippocampal neurons. Mouse HT22 hippocampal neurons were differentiated as previously described before exposed to microglial CM [21]. At 24 h after incubating with microglial CM, the viability of neurons was examined using a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay.
Mitochondrial membrane potential measurement
The mitochondrial membrane potential was detected using fluorescent probe JC-1. Cells were incubated with 10 μg/ml of JC-1 dye for 10 min at 37 °C followed by PBS wash. Healthy cells with polarized mitochondria predominantly exhibited orange-red fluorescence due to an aggregated form of JC-1 (Ex 535 nm, Em 590 nm). When cells contained depolarized mitochondria, they displayed green fluorescence reflected by a monomeric form of JC-1 (Ex 485 nm, Em 530 nm). Six to eight fields from each treatment group in an experiment were acquired using inverted fluorescence microscope DMI6000B with an attached digital camera (Leica Microsystems, IL).
Immunohistochemistry
The mouse brains were harvested and processed to prepare brain sections as previously described [21, 25]. Brain sections were blocked and incubated with primary antibodies, rabbit anti-Iba1 (1:2500, WAKO, VA), or rabbit anti-GFAP (1:3000, Cell Signaling, Danvers, MA) for 2 h at RT. After the secondary antibody incubation, slides were developed with 3,3′-diaminobenzidine (DAB) liquid substrate (DAKO, CA). Then, slides were counterstained with Mayer’s hematoxylin (Sigma-Aldrich) and mounted with Permount (FisherSci). Two brain sections from each mouse were stained and analyzed. Stained slides were imaged (microscope: BX53, camera: DP72, Olympus) and analyzed using CellSens software (Olympus) by an experimenter blinded to experimental conditions.
Novel object recognition test
The test was performed using a square arena (50 cm × 50 cm × 34.5 cm). Adult C57BL/6J mice were randomly assigned to different treatment groups. Mice were handled for 3 days for habituation of handling. Mice were then acclimated to the arena over 3 days, one 10-min trial per day. Mice were subjected to a single-dose LPS (1 mg/Kg, ip) injection. DMF (30 mg/Kg, ip) was administered every other day for 3 days as indicated. The changes of body weight were measured daily to monitor mice recovering from sickness following LPS injection. Four days after LPS injection, mice were exposed to the same arena containing two identical objects (henceforth “familiar” object) for 10 min, as the acquisition session. Twenty-four hours after the acquisition, the recognition session was performed by replacing one of the familiar objects with a novel object. Any-maze software (Stoelting, IL) was used to track and analyze the movements of the mice. The behavioral experiments were performed by researchers blinded to treatment groups. Results were expressed as percentage of time exploring each object.
Statistical analysis
Data are presented as mean ± SEM. For in vivo studies with the groups containing ≥ 7 mice, a normal distribution of the data was confirmed by Shapiro-Wilk normality test. Comparisons between two groups were done using the unpaired t test, whereas comparisons between multiple groups were done by one-way ANOVA followed by Bonferroni post hoc test using GraphPad Prism software. Statistical significance was determined with p values < 0.05.
Results
DMF suppresses inflammatory activation of MG
We first asked whether DMF is capable of modulating phenotypes of MG in response to LPS stimulation. We examined expressions of maturation markers, CD86, CD80, and CD40, on MG cells stimulated with LPS in the absence or presence of DMF. The LPS stimulation induced microglial activation resulting in the increase of these surface markers, whereas DMF treatment significantly suppressed these markers in a dose-dependent manner (Fig. 1a and Additional file 2: Figure S2A). In addition, DMF was able to inhibit morphological changes and Iba1 upregulation in MG stimulated with LPS (Fig. 1b). The concentrations of DMF used in these studies did not affect the cell viability of MG, suggesting that the suppressive effect of DMF on MG is not mediated through cytotoxicity (Additional file 1: Figure S1B).
Fig. 1.
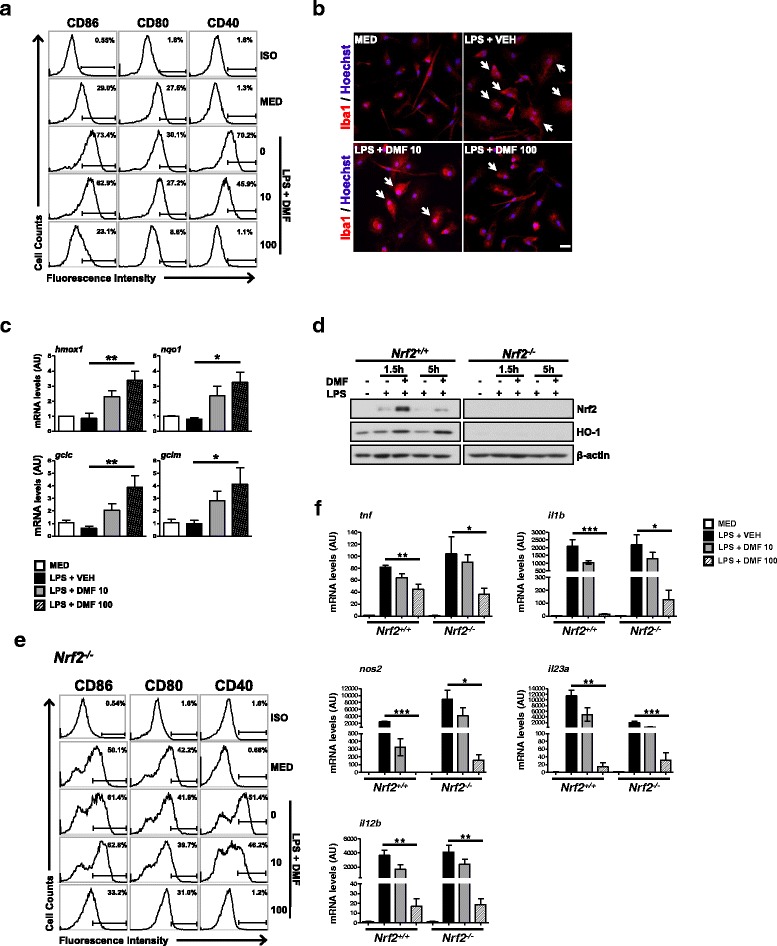
DMF suppresses LPS-induced reactive MG. Primary MG were stimulated by LPS (100 ng/ml) in the absence or presence of DMF for various time periods. a Microglial surface markers, CD86, CD80, and CD40, were examined at 18 h after stimulation using FACS analysis. Representative flow plots from five independent experiments are shown. ISO, the isotype control antibody. MED, cell culture medium. b Morphological changes of MG at 18 h after stimulation were examined using Iba1 immunostaining. Arrows indicate activated MG with increased expression of Iba1 and enlarged cell bodies. Cell nuclei were stained blue with Hoechst 33342. Scale bar, 20 μm. c The expressions of DMF-induced Nrf2 target genes were examined at 3 h after stimulation. The mRNA levels of hmox1, nqo1, gclc, and gclm were quantified using qPCR. Data presented are from 3 to 5 independent experiments. *p < 0.05, **p < 0.01, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test. d The Nrf2+/+ and Nrf2−/− microglial cells were harvested for western blot analysis to examine Nrf2 and HO-1 proteins at 1.5 and 5 h after stimulation. Representative results are shown from three independent experiments. e The CD86, CD80, and CD40 expressed on the surface of Nrf2−/− MG were examined at 18 h after stimulation using FACS analysis. Representative flow plots from three independent experiments are shown. f Expressions of inflammatory mediators were examined in Nrf2+/+ and Nrf2−/− MG at 3 h after stimulation. The mRNA levels of tnf, il1b, nos2, il23a, and il12b in treated MG were quantified using qPCR. Data presented are from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test.
DMF is shown as a strong inducer of Nrf2 in animal models of MS [16, 17]. We also found that the expressions of Nrf2 target genes were significantly upregulated in MG treated with DMF (Fig. 1c). To investigate whether Nrf2 mediates actions of DMF in MG, we compared effects of DMF on Nrf2+/+ and Nrf2−/− MG stimulated with LPS. In Nrf2+/+ MG, LPS stimulation slightly increased the level of Nrf2 protein, but not its downstream target, HO-1 (Fig. 1d). The DMF treatment significantly upregulated the protein levels of Nrf2 and HO-1. In contrast, Nrf2−/− MG did not exhibit induction of Nrf2 nor HO-1, after being treated with DMF, demonstrating the deficiency of Nrf2. We next analyzed the effects of DMF on Nrf2−/− MG stimulated with LPS. The DMF suppressed the induction of surface markers in Nrf2−/− MG after LPS stimulation, similar to its action on Nrf2+/+ MG (Fig. 1e and Additional file 2: Figure S2B). These results indicate that DMF might act on multiple pathways to modulate microglial phenotype in response to LPS stimulation.
The release of pro-inflammatory cytokines indicates activated MG moving towards an inflammatory phenotype. Further analyzing expressions of cytokines, we found that DMF suppressed LPS-induced mRNA expressions of TNF-α, IL-1β, iNOS, IL-23p19, and IL-12p40 in Nrf2+/+ MG (Fig. 1f). DMF was capable of suppressing the induction of inflammatory mediators in Nrf2−/− MG stimulated with LPS. We noticed that the suppressive effect of DMF on some of the inflammatory mediators in Nrf2−/− MG was not as powerful as its suppressive action in Nrf2+/+ MG. Taken together, these results suggest that DMF suppresses inflammatory activation of reactive MG induced by LPS potentially through both Nrf2-dependent and Nrf2-independent mechanisms.
DMF inhibits LPS-induced NF-κB activation in MG
NF-κB regulates the transcription of a variety of pro-inflammatory genes in different immune cells. To test whether DMF acts on NF-κB signaling pathway to regulate microglial phenotypes, we examined the nuclear translocation of NF-κB p65 induced by LPS in the absence or presence of DMF. When stimulated with LPS, about 80% of MG cells exhibited nuclear p65 immunoreactivity (Fig. 2a, b). DMF treatment significantly suppressed nuclear translocation of p65 in both Nrf2+/+ and Nrf2−/− MG stimulated with LPS. Notably, Nrf2−/− MG exhibited a higher level of p65 nuclear translocation at the resting state and upon LPS stimulation, suggesting that Nrf2 deficiency might promote inflammatory phenotypes of MG. In addition, DMF abolished LPS-induced phosphorylation of IKK-α/β in both Nrf2+/+ and Nrf2−/− MG (Fig. 2c). Taken together, these results demonstrate the Nrf2-independent mechanisms mediating actions of DMF to suppress LPS-induced NF-κB activity in MG.
Fig. 2.
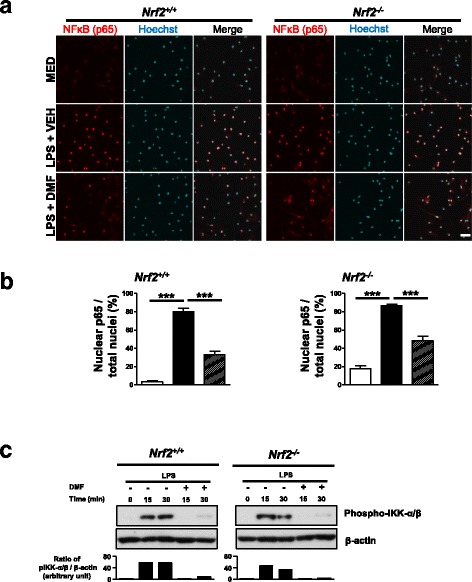
DMF acts on Nrf2-independent mechanisms to suppress NF-κB activation induced by LPS. Primary Nrf2+/+ and Nrf2−/− MG were stimulated with LPS (100 ng/ml) in the absence or presence of DMF (100 μM) for various time periods. a Nuclear translocation of NF-κB (p65) was examined at 15 min after stimulation. Representative images of p65 immunostaining from three independent experiments are shown. Hoechst 33342-stained cell nuclei are depicted in blue. MED, cell culture medium. VEH, vehicle control. Scale bar, 20 μm. b The percentage of p65-positive nuclei/total cell nuclei in each treatment group was quantified. Data presented are from three independent experiments. ***p < 0.001, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test. c The western blot analysis of phospho-IKKα/β at 15 or 30 min after stimulation. Data presented are representative blots from three independent experiments
DMF reduces MG-mediated neurotoxicity
We next asked whether manipulating microglial phenotypes and functions by DMF can foster neuroprotection. We exposed differentiated HT22 hippocampal neurons to CM derived from MG stimulated with LPS in the absence or presence of DMF. After 24 h of exposure, there was a substantial reduction in cell viability of neurons incubated with CM generated from LPS-primed MG (Fig. 3a). The induction of inflammatory phenotype in MG was examined after collecting CM, showing increased iNOS protein in MG stimulated with LPS (Additional file 2: Figure S2C). The DMF treatment was able to suppress the LPS-induced iNOS in MG. When MG were stimulated with LPS in the presence of DMF, they generated less toxic CM, resulting in more survived neurons after incubation. In consistent with neuronal cell viability, neurons exposed to CM derived from LPS-primed MG showed increased Caspase-3 activation, indicating increased neuronal cell death induced by microglial CM (Fig. 3b, c). In contrast, the CM generated from DMF-treated MG did not induce significant Caspase-3 activation in neurons compared to the vehicle-treated controls.
Fig. 3.
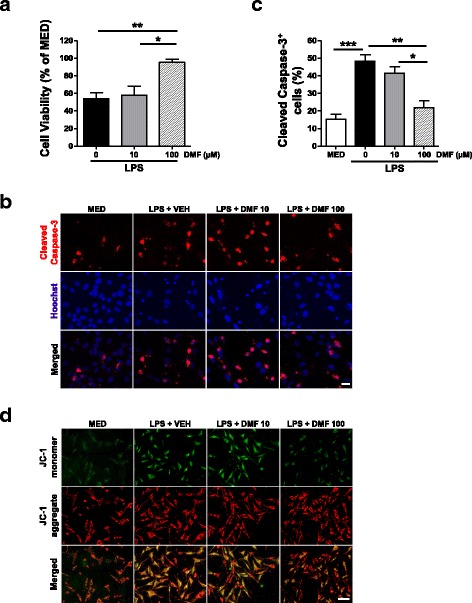
DMF suppresses MG-mediated neurotoxicity. MG were stimulated with LPS (1 μg/ml) in the absence or presence of DMF. Microglial CM was collected and applied to cultured hippocampal neurons. Neurons were exposed to microglial CM for 24 h. a The cell viability was assessed by MTT assay. Data presented are from four independent experiments. *p < 0.05, **p < 0.01, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test. b The Caspase-3 activation was examined by immunostaining to detect cell death in neurons. Cell nuclei were stained blue with Hoechst 33342. Representative images are from three independent experiments. MED, cell culture medium. VEH, vehicle control. Scale bar, 20 μm. c The cleaved Caspas-3+ cells were quantified and compared with total cells (Hoechst-stained cells). Data presented are percentage of cleaved Caspas-3+ cells in each treatment group. Results presented are from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test. d Representative images show a shift of JC-1 fluorescence from orange red (mitochondrial membrane polarized) to green (mitochondrial membrane depolarized) in neurons incubated with CM from LPS-primed MG for 6 h. Neurons incubated with the CM generated from DMF-treated MG (100 μM) show decreased JC-1 green fluorescence. MED, cell culture medium. VEH, vehicle control. Scale bar, 40 μm. Results are from three independent experiments
Mitochondrial dysfunction plays a critical role in the degeneration and death of neurons [26]. We monitored mitochondrial health status using molecular probe JC-1 to distinguish energized (high membrane potential) and de-energized mitochondria (membrane depolarization) in neurons after short-term (6 h) exposure to microglial CM (Fig. 3d). Incubating neurons with CM generated from LPS-primed MG resulted in a shift of JC-1 fluorescence emission from orange red (JC-1 aggregates) to green (JC-1 monomer). These data indicate that CM from LPS-primed MG leads to disrupted membrane potential of mitochondria in neurons. In contrast, the CM generated from DMF-treated MG did not significantly cause mitochondrial abnormalities in neurons. Taken together, our results demonstrate that DMF might protect neurons from inflammatory insults by modulating toxic factors released from reactive MG associated with inflammation.
DMF suppresses LPS-induced acute neuroinflammation
To investigate whether DMF modulates microglial activation in vivo, we treated Cx3cr1gfp/+ mice with a single dose of LPS injection (1 mg/Kg, ip) and examined the brain 24 h post-injection. The LPS injection led to increased GFP+ cells in the hippocampus of the brain, indicating acute neuroinflammation in response to LPS challenge (Fig. 4a). The administration of DMF was able to reduce numbers and morphological changes of GFP+ cells in the hippocampus. We further isolated brain mononuclear cells and examined maturation markers of GFP+ cells, which include activated MG and potentially infiltrating peripheral immune cells, such as monocytes/macrophages, following LPS challenge. The LPS challenge significantly upregulated CD80 expression on GFP+ cells, whereas DMF treatment reduced CD80 upregulation induced by LPS challenge (Fig. 4b, c). The expressions of CD86 and CD40 on the surface of GFP+ cells were not increased, while examined at 24 h after LPS challenge (data not shown). In addition, DMF treatment significantly suppressed de novo cytokine production within the brain following peripheral LPS challenge (Fig. 4d). Intriguingly, the suppressive effect of DMF on brain-derived inflammatory cytokine was blunted in LPS-challenged Nrf2−/− mice. The suppression of IL-1β and TNFα mRNAs by DMF treatment was partially retained in Nrf2−/− mice. However, the suppression of Ccl2 mRNA by DMF treatment was not observed in Nrf2−/− mice. These data indicate that the Nrf2-dependent mechanism mediates the actions of DMF to suppress acute neuroinflammation induced by LPS. Taken together, these results suggest that DMF treatment alleviates acute inflammatory responses in the brain induced by peripheral LPS challenge.
Fig. 4.
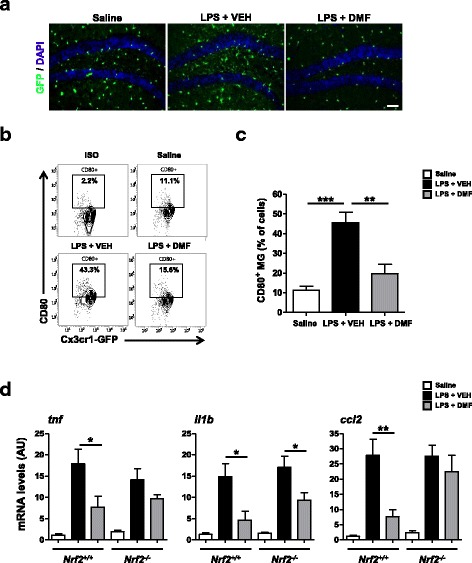
DMF reduces LPS-induced acute neuroinflammation. Cx3cr1gfp/+ mice were treated with a single-dose LPS injection (1 mg/Kg, ip). DMF (45 mg/Kg) was administered through ip injection. At 24 h after LPS injection, the mouse brains were harvested and subjected to analyses. a Representative images show GFP+ cells in the hippocampus of Cx3cr1gfp/+ mice that received saline, LPS + vehicle (VEH), or LPS + DMF treatment. N = 7 mice/group. Cell nuclei were stained with DAPI (blue). Scale bar, 40 μm. b Mononuclear cells were isolated from the brain homogenates and subjected to FACS analysis. The levels of CD80 marker on gated GFP+ cell populations were examined. Representative flow plots from three treatment groups, saline, LPS + VEH, LPS + DMF are shown. N = 7 mice/group. ISO, the isotype control antibody. c The proportion of CD80+ cells was determined. N = 7 mice/group. **p < 0.01, ***p < 0.001, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test. d Effects of DMF treatment on inflammatory mediators induced by LPS injection in Nrf2+/+ (Cx3cr1gfp/+) or Nrf2−/− mice. The mRNA levels of pro-inflammatory cytokines or chemokine (tnf, il1b, or ccl2) in the brain were measured using qPCR. N = 7 mice/group for Nrf2+/+ mice. N = 5–6 mice/group for Nrf2−/− mice. *p < 0.05, **p < 0.01, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test.
DMF alleviates cognitive deficits induced by systemic immune challenge
We further investigated whether DMF treatment can alleviate the impairment of cognition driven by systemic inflammation. We treated mice with a single dose of LPS (1 mg/Kg, ip) and administered three doses of DMF (30 mg/Kg, ip) as shown in Additional file 3: Figure S3A. The changes of body weight were monitored daily to ensure that mice recovered from transient sickness after LPS injection (Additional file 3: Figure S3B). At 5 days after LPS challenge, mice were subjected to the novel object recognition task (Fig. 5a). The 24-h delayed paradigm was used to access the long-term memory function [27]. Mice that received saline injection showed a clear preference for the novel object, but mice that received LPS injection did not (Fig. 5b). The total exploratory or locomotor activity in mice was not affected at the time when tasks were performed (Additional file 3: Figure S3C). These results show that LPS-treated mice were not able to distinguish between the novel and familiar objects, indicating an impairment in recognition memory. In contrast, mice that received DMF treatment following LPS challenge exhibited a preference towards the novel object, suggesting that DMF treatment rescues the long-term memory deficits caused by systemic immune challenge.
Fig. 5.
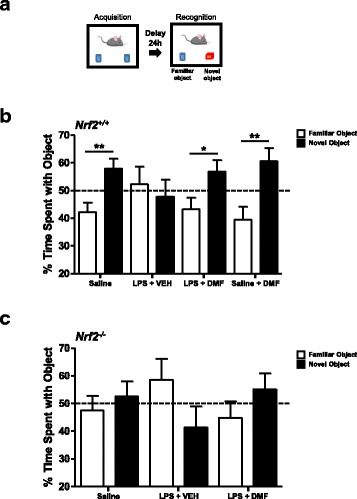
DMF treatment alleviates cognitive deficits following systemic immune challenge. a This figure illustrates the novel object recognition task using a 24-h retention interval. b Adult C57BL/6 mice were subjected to saline or LPS injection. DMF or vehicle (VEH) treatment was administered every other day for 3 days following LPS injection. At 5 days after LPS injection, the mice were subjected to the novel object recognition task. The time that mice spent with familiar or novel object was measured. Percentage of exploration time on the object was calculated. N = 10–16 mice/group. *p < 0.05, **p < 0.01, compared to familiar object by unpaired Student’s t test. Data of saline, LPS + VEH, and LPS + DMF treatment are from three independent experiments; data of saline + DMF treatment are from two independent experiments. c Adult Nrf2−/− mice were subjected to saline, LPS + VEH, or LPS + DMF injection as described. At 5 days after LPS injection, the mice were subjected to the novel object recognition task. The time that mice spent with familiar or novel object was measured. Percentage of exploration time on the object was calculated. N = 8–12 mice/group. There was no statistical significance between the familial and the novel object when data were analyzed by unpaired Student’s t test. Data presented are from two independent experiments
Moreover, we examined Nrf2−/− mice that were systemically challenged with LPS and subjected to novel object recognition task. We observed exacerbation of sickness with significant weight loss and increased mortality rate (about 20–30%) following LPS challenge in Nrf2−/− mice (Additional file 3: Figure S3D), suggesting that Nrf2 plays an important role in alleviating systemic inflammation. The Nrf2−/− mice exhibited the impairment in recognition memory regardless of LPS challenge (Fig. 5c and Additional file 3: Figure S3E). The administration of DMF, although was not able to fully rescue the memory deficits, tended to mitigate effects of LPS challenge. These results suggest that Nrf2 dysfunction might alter the cognitive function of the brain.
DMF suppresses chronic neuroinflammation associated with systemic immune challenge
The systemic immune responses, although being resolved, still led to a long-lasting impact to the brain. There was a slight increase of Iba1-expressing MG in the hippocampus of LPS-treated mice, compared to saline- or DMF-treated mice, even 2 weeks after LPS injection (Fig. 6a, c). It is noteworthy that we observed profound GFAP+ astrocytes in the hippocampus of LPS-treated mice compared to saline-injected mice (Fig. 6b, d). The GFAP+ astrocytes were significantly reduced in the hippocampus of DMF-treated mice. These results indicate that systemic immune challenge triggers robust but sustained responses in astrocytes and that DMF administration alleviates reactive astrocytes in response to inflammation. A recent study shows that TNFα, IL-1α, and C1q released from inflammatory MG transform astrocytes towards neurotoxic A1 astrocytes [28]. We analyzed whether DMF modulates expressions of IL-1α and C1q in the brain following peripheral LPS challenge. In addition to TNFα, DMF treatment significantly reduced the expressions of IL-1α and C1q in the brain induced by LPS injection (Fig. 6e). We further tested whether DMF mitigates A1 phenotype in primary astrocytes. In vitro, TNFα and IL-1α were able to induce A1 astrocyte specific genes, including ggta1, h2-d1, and serping1. The expression of these genes was suppressed in DMF-treated astrocytes (Fig. 6f). Taken altogether, these results suggest that the protective effect of DMF on rescuing neuronal dysfunction in LPS-challenged mice might potentially be mediated through its suppressive effect on the generation of neurotoxic A1 astrocytes.
Fig. 6.
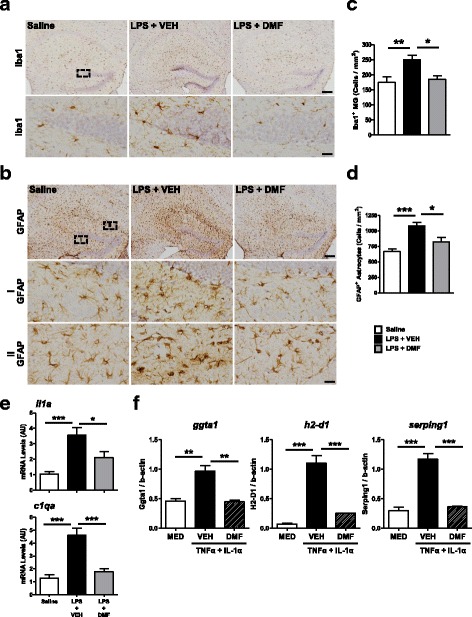
DMF alleviates reactive astrogliosis following systemic immune challenge. a Iba1+ MG and b GFAP+ astrocytes were examined by immunohistochemistry at 2 weeks after a single-dose LPS injection. DMF was administered every other day for 3 days following LPS injection. Representative images show the hippocampus of mice from three treatment groups, saline, LPS + VEH, LPS + DMF. N = 4–5 mice/group. High magnification images of circled areas are shown at the lower panel. Quantification results of c Iba1+ MG and d GFAP+ astrocytes are shown. *p < 0.05, **p < 0.01, and ***p < 0.001 by one-way ANOVA with Bonferroni’s post hoc multiple comparison test. Scale bar, 200 μm (low magnification) or 20 μm (high magnification). e The mRNA levels of inflammatory molecules, il1a and c1qa, in the brain were quantified using qPCR. N = 7 mice/group. *p < 0.05, ***p < 0.001, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test. f Enriched astrocyte cultures were stimulated with TNFα (30 ng/ml) and IL-1α (3 ng/ml) in the absence or presence of DMF. The expressions of A1 signature genes were measured at 24 h after stimulation. The mRNA levels of ggta1, h2-d1, and serping1 were quantified using qPCR. Data presented are from three independent experiments. **p < 0.01, ***p < 0.001, by one-way ANOVA with Bonferroni’s post hoc multiple comparison test.
Discussion
In the present study, we investigated the translational potential of DMF for the treatment of cognitive impairment associated with systemic inflammation. DMF exerts distinct neuroprotective effects and clinical efficacy for the treatment of relapsing-remitting MS [13, 14]. Here, we report a direct modulatory effect of DMF on inflammatory MG induced by LPS. DMF effectively suppresses maturation markers, morphological changes, and production of inflammatory mediators in cultured MG. In line with our results, several studies also report that DMF shows effective suppression on LPS-induced production of inflammatory cytokines in human fetal and adult MG cultures and in murine MG cultures [29, 30].
As a consequence of inflammatory activation, the toxic factor released from reactive MG is able to induce neurotoxicity [31]. We show that the CM from LPS-primed MG induces Caspase-3 activation in neurons after 24 h of exposure, while shorter time of exposure (6 h) results in disrupted mitochondrial membrane potential in neurons. DMF reduces inflammatory phenotype of MG and ameliorates MG-mediated neurotoxicity. Similar to our results, DMF is reported to reduce neuronal dysfunction through mitigating inflammatory activation of MG induced by LPS [32]. MMF is shown to preserve neuronal viability through modulating microglial phenotype stimulated with virus-infected monocytes [33].
In addition to in vitro studies, we identify that DMF reduces acute neuroinflammation following peripheral LPS challenge in mice. DMF administration reduces morphological changes and maturation marker induction in brain-associated immune cells, including resident and infiltrating immune cells, following LPS challenge. Given that DMF is reported to suppress M1 polarization of monocytes/macrophages in different disease models, we speculate that the reduced neuroinflammation in LPS/DMF-treated mice could be contributed by the actions of DMF on peripheral immune cells [19, 34]. LPS is a strong activator of Toll-like receptor 4 signaling, which induces inflammatory mediators robustly. Excessive pro-inflammatory cytokines in the brain can impair neuronal viability and lead to cognitive dysfunction [35–39]. DMF administration significantly suppresses the expression of pro-inflammatory cytokines in the brain following peripheral LPS challenge, supporting its potential to ameliorate cognitive dysfunction associated with systemic inflammation [40, 41]. Our results provide promising evidence that DMF treatment rescues deficits in long-term recognition memory affected by systemic inflammation.
Several studies have reported that Nrf2-independent mechanisms mediate the action of DMF [19, 32, 42, 43]. In this study, we also observe that Nrf2-independent actions of DMF modulate inflammatory MG and neuroinflammation in mice following LPS challenge. However, we acknowledge that the suppressive effect of DMF on LPS-induced neuroinflammation is blunted in Nrf2−/− mice. Nrf2 is reported exhibiting immunomodulatory and anti-inflammatory activities in T cells [44]. The nuclear factor NF-κB is a key transcription factor regulating immune responses. It is known that there is a cross-talk between Nrf2 and NF-κB signaling pathways [45]. Activation of Nrf2 attenuates NF-κB activity, which is possibly mediated by HO-1-derived bilirubin in endothelial cells [46]. Nrf2 knockdown is associated with increased MafK activity, which positively promotes p65 acetylation and its subsequent transactivation activity in hepatocytes [47]. Our findings provide the evidence of cross-talk between Nrf2 and NF-κB signaling pathways in MG. The deficiency of Nrf2 in MG results in a higher level of p65 nuclear translocation at the resting state. This result suggests potential role of Nrf2 in regulating the intrinsic homeostasis of MG. Moreover, Nrf2−/− mice show exaggerated sickness response to LPS challenge, demonstrating the important role of Nrf2-mediated anti-inflammatory responses.
When examining long-term effects of peripheral LPS challenge to the brain, we observed profound reactive astrocytes in the hippocampus of LPS-treated mice. It is noteworthy that astrocytes are able to produce chemokine Ccl2 in response to cytokine stimulation in animal models. The astrocyte-derived chemokine Ccl2 is found to recruit circulating Ccr2+ monocytes infiltrating into the brain, which exacerbates the degeneration of neurons [48, 49]. DMF administration significantly suppresses transcription of Ccl2 mRNA in the brain induced by LPS challenge. Our results suggest potential modulatory effects of DMF on astrocytes. In line with our findings, DMF, but not MMF, is shown to reduce the secretion of Ccl2 from cultured astrocytes stimulated with pro-inflammatory cytokines [50]. Moreover, we observed that DMF inhibits expressions of essential molecules required for the generation of neurotoxic A1 astrocytes, including TNFα, IL-1α, and C1q, in the mouse brain following LPS challenge. Our subsequent examinations showed that the induction of A1 signature genes is suppressed by DMF treatment in cultured astrocytes, suggesting the action of DMF-regulating astrocyte phenotypes. Genetic over-expression of Nrf2 in astrocytes is reported to rescue neurodegeneration in the amyotrophic lateral sclerosis (ALS) animal model [51]. It would require further investigation to examine the potential of DMF regulating phenotypic or functional polarization of reactive astrocytes associated with neuroinflammation.
Systemic inflammation is reported to exacerbate pre-existing conditions and accelerate cognitive decline in several models of neurodegeneration including traumatic brain injury and prion disease [52, 53]. We acknowledge that the mice used in this study are young healthy adults. Future studies to investigate effects of DMF on systemic inflammation superimposed upon neurodegenerative conditions are warranted. Recent studies identify that Nrf2 deficiency exacerbates cognitive deficits and pathological features in mouse models of Alzheimer’s disease [54–56]. Loss of Nrf2 in the mouse brain is found to recapitulate the dysregulated pathways in human aging and Alzheimer’s disease using the transcriptomic analysis. In line with our findings of cognitive impairment in Nrf2−/− mice, the long-term potentiation is found decreased in the hippocampus of Nrf2−/− mice. Although the dosing regimen we selected to treat LPS-challenged Nrf2−/− mice is not able to fully rescue the memory deficits, our results reveal a critical role of Nrf2 in the cognitive function and a potential of DMF as a therapeutic strategy to fight Alzheimer’s disease.
Conclusions
The present study demonstrates that DMF attenuates microglial responses towards the inflammatory phenotype induced by LPS stimulation. The DMF treatment alleviates the acute microglial activation, neuroinflammation, long-term memory deficits, and responses of reactive astrocytes in mice following systemic immune challenge. The DMF treatment has the translational potential to protect neurons against toxic microenvironments in the brain associated with systemic inflammation.
Additional files
Figure S1. Characterization of microglial cultures, cell toxicity of DMF, and enriched astrocyte cultures. (PDF 134 kb)
Figure S2. Inflammatory activation of MG induced by LPS. (PDF 146 kb)
Figure S3. Cognitive assessment following systemic immune challenge and DMF treatment. (PDF 154 kb)
Acknowledgements
The authors thank Barbara A. Scofield for the skilled technical assistance.
Funding
This work was financially supported by IU Foundation, Lutheran Foundation, and Steel Dynamics Foundation. J-H. Yen was supported by Anna Yoder MS Foundation.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on a reasonable request.
Authors’ contributions
HCP contributed to the study design, performed the experiments, and wrote the manuscript. P-CK contributed to the research data, analyzed the data, and reviewed and edited the manuscript. ETC contributed to the research data of histology/immunohistochemistry staining and analyzed the data. HJM contribute to the research data of western blotting. RDS and F-LC contributed to the discussion and reviewed and edited the manuscript. J-HY contributed to the study design and discussion and reviewed and edited the manuscript. I-CY conceived the study, designed the experiments, and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Consent for publication
Not applicable. This article does not contain any individual person’s data.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12974-018-1125-5) contains supplementary material, which is available to authorized users.
Contributor Information
Hallel C. Paraiso, Email: paraisoh@ipfw.edu
Ping-Chang Kuo, Email: kuop@iu.edu.
Eric T. Curfman, Email: mindstroller@gmail.com
Haley J. Moon, Email: moonhj@iu.edu
Robert D. Sweazey, Email: rsweazey@iu.edu
Jui-Hung Yen, Email: jimyen@iu.edu.
Fen-Lei Chang, Phone: 2060-481-0578, Email: fchang@iu.edu.
I-Chen Yu, Phone: 260-481-0572, Email: ivoyu@iu.edu.
References
- 1.Teeling JL, Perry VH. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience. 2009;158:1062–1073. doi: 10.1016/j.neuroscience.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 2.Widmann CN, Heneka MT. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014;13:630–636. doi: 10.1016/S1474-4422(14)70017-1. [DOI] [PubMed] [Google Scholar]
- 3.Witlox J, Eurelings LS, de Jonghe JF, Kalisvaart KJ, Eikelenboom P, van Gool WA. Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta-analysis. JAMA. 2010;304:443–451. doi: 10.1001/jama.2010.1013. [DOI] [PubMed] [Google Scholar]
- 4.Inouye SK, Westendorp RG, Saczynski JS. Delirium in elderly people. Lancet. 2014;383:911–922. doi: 10.1016/S0140-6736(13)60688-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, Tylavsky FA, Newman AB. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA. 2004;292:2237–2242. doi: 10.1001/jama.292.18.2237. [DOI] [PubMed] [Google Scholar]
- 6.Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR. Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry. 2008;64:484–490. doi: 10.1016/j.biopsych.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shemer A, Erny D, Jung S, Prinz M. Microglia plasticity during health and disease: an immunological perspective. Trends Immunol. 2015;36:614–624. doi: 10.1016/j.it.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. Clin Dev Immunol. 2013;2013:208093. doi: 10.1155/2013/208093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- 11.Altmeyer PJ, Matthes U, Pawlak F, Hoffmann K, Frosch PJ, Ruppert P, Wassilew SW, Horn T, Kreysel HW, Lutz G, et al. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J Am Acad Dermatol. 1994;30:977–981. doi: 10.1016/S0190-9622(94)70121-0. [DOI] [PubMed] [Google Scholar]
- 12.Mrowietz U, Asadullah K. Dimethylfumarate for psoriasis: more than a dietary curiosity. Trends Mol Med. 2005;11:43–48. doi: 10.1016/j.molmed.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, Yang M, Raghupathi K, Novas M, Sweetser MT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 14.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 15.Linker RA, Gold R. Dimethyl fumarate for treatment of multiple sclerosis: mechanism of action, effectiveness, and side effects. Curr Neurol Neurosci Rep. 2013;13:394. doi: 10.1007/s11910-013-0394-8. [DOI] [PubMed] [Google Scholar]
- 16.Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 17.Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, Rhodes KJ. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Assmann JC, Krenz A, Rahman M, Grimm M, Karsten CM, Kohl J, Offermanns S, Wettschureck N, Schwaninger M. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J Clin Invest. 2014;124:2188–2192. doi: 10.1172/JCI72151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, Cree BA, Sobel RA, Wipke BT, Steinman L, et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci U S A. 2016;113:4777–4782. doi: 10.1073/pnas.1603907113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giannetti P, Politis M, Su P, Turkheimer F, Malik O, Keihaninejad S, Wu K, Reynolds R, Nicholas R, Piccini P. Microglia activation in multiple sclerosis black holes predicts outcome in progressive patients: an in vivo [(11)C](R)-PK11195-PET pilot study. Neurobiol Dis. 2014;65:203–210. doi: 10.1016/j.nbd.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Yu IC, Kuo PC, Yen JH, Paraiso HC, Curfman ET, Hong-Goka BC, Sweazey RD, Chang FL. A combination of three repurposed drugs administered at reperfusion as a promising therapy for postischemic brain injury. Transl Stroke Res. 2017;8:560–77. doi: 10.1007/s12975-017-0543-5. [DOI] [PubMed] [Google Scholar]
- 22.Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013;(71). 10.3791/50079. [DOI] [PMC free article] [PubMed]
- 23.Lin R, Cai J, Kostuk EW, Rosenwasser R, Iacovitti L. Fumarate modulates the immune/inflammatory response and rescues nerve cells and neurological function after stroke in rats. J Neuroinflammation. 2016;13:269. doi: 10.1186/s12974-016-0733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao Y, Miao W, Liu Z, Han W, Shi K, Shen Y, Li H, Liu Q, Fu Y, Huang D, Shi FD. Dimethyl fumarate and monomethyl fumarate promote post-ischemic recovery in mice. Transl Stroke Res. 2016;7:535–547. doi: 10.1007/s12975-016-0496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo PC, Yu IC, Scofield BA, Brown DA, Curfman ET, Paraiso HC, Chang FL, Yen JH. 3H-1,2-Dithiole-3-thione as a novel therapeutic agent for the treatment of ischemic stroke through Nrf2 defense pathway. Brain Behav Immun. 2017;62:180–192. doi: 10.1016/j.bbi.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 26.Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med. 2014;20:179–187. doi: 10.1016/j.molmed.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Moore SJ, Deshpande K, Stinnett GS, Seasholtz AF, Murphy GG. Conversion of short-term to long-term memory in the novel object recognition paradigm. Neurobiol Learn Mem. 2013;105:174–185. doi: 10.1016/j.nlm.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michell-Robinson MA, Moore CS, Healy LM, Osso LA, Zorko N, Grouza V, Touil H, Poliquin-Lasnier L, Trudelle AM, Giacomini PS, et al. Effects of fumarates on circulating and CNS myeloid cells in multiple sclerosis. Ann Clin Transl Neurol. 2016;3:27–41. doi: 10.1002/acn3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. 2010;7:30. doi: 10.1186/1742-2094-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 32.Peng H, Li H, Sheehy A, Cullen P, Allaire N, Scannevin RH. Dimethyl fumarate alters microglia phenotype and protects neurons against proinflammatory toxic microenvironments. J Neuroimmunol. 2016;299:35–44. doi: 10.1016/j.jneuroim.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Ambrosius B, Faissner S, Guse K, von Lehe M, Grunwald T, Gold R, Grewe B, Chan A. Teriflunomide and monomethylfumarate target HIV-induced neuroinflammation and neurotoxicity. J Neuroinflammation. 2017;14:51. doi: 10.1186/s12974-017-0829-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han R, Xiao J, Zhai H, Hao J. Dimethyl fumarate attenuates experimental autoimmune neuritis through the nuclear factor erythroid-derived 2-related factor 2/hemoxygenase-1 pathway by altering the balance of M1/M2 macrophages. J Neuroinflammation. 2016;13:97. doi: 10.1186/s12974-016-0559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, Manalastas A, Hilfiker M, Pfister S, Schwerdel C, et al. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. 2012;9:151. doi: 10.1186/1742-2094-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pott Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain. 2008;131:1880–1894. doi: 10.1093/brain/awn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T. IL-1 receptor/toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun. 2011;25:1281–1289. doi: 10.1016/j.bbi.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 38.Moraes CA, Santos G, de Sampaio e Spohr TC, D'A vila JC, Lima FR, Benjamim CF, Bozza FA, Gomes FC. Activated microglia-induced deficits in excitatory synapses through IL-1beta: implications for cognitive impairment in sepsis. Mol Neurobiol. 2015;52:653–663. doi: 10.1007/s12035-014-8868-5. [DOI] [PubMed] [Google Scholar]
- 39.Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa AC, O'Banion MK. Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24:243–253. doi: 10.1016/j.bbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Czerniawski J, Guzowski JF. Acute neuroinflammation impairs context discrimination memory and disrupts pattern separation processes in hippocampus. J Neurosci. 2014;34:12470–12480. doi: 10.1523/JNEUROSCI.0542-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czerniawski J, Miyashita T, Lewandowski G, Guzowski JF. Systemic lipopolysaccharide administration impairs retrieval of context-object discrimination, but not spatial, memory: evidence for selective disruption of specific hippocampus-dependent memory functions during acute neuroinflammation. Brain Behav Immun. 2015;44:159–166. doi: 10.1016/j.bbi.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brennan MS, Patel H, Allaire N, Thai A, Cullen P, Ryan S, Lukashev M, Bista P, Huang R, Rhodes KJ, Scannevin RH. Pharmacodynamics of dimethyl fumarate are tissue specific and involve NRF2-dependent and -independent mechanisms. Antioxid Redox Signal. 2016;24:1058–1071. doi: 10.1089/ars.2015.6622. [DOI] [PubMed] [Google Scholar]
- 43.Gillard GO, Collette B, Anderson J, Chao J, Scannevin RH, Huss DJ, Fontenot JD. DMF, but not other fumarates, inhibits NF-kappaB activity in vitro in an Nrf2-independent manner. J Neuroimmunol. 2015;283:74–85. doi: 10.1016/j.jneuroim.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Rockwell CE, Zhang M, Fields PE, Klaassen CD. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J Immunol. 2012;188:1630–1637. doi: 10.4049/jimmunol.1101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, Kong AN. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. 2008;76:1485–1489. doi: 10.1016/j.bcp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, Tsui TY, Bach FH. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. 2004;172:3553–3563. doi: 10.4049/jimmunol.172.6.3553. [DOI] [PubMed] [Google Scholar]
- 47.Hwang YJ, Lee EW, Song J, Kim HR, Jun YC, Hwang KA. MafK positively regulates NF-kappaB activity by enhancing CBP-mediated p65 acetylation. Sci Rep. 2013;3:3242. doi: 10.1038/srep03242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hennessy E, Griffin EW, Cunningham C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1beta and TNF-alpha. J Neurosci. 2015;35:8411–8422. doi: 10.1523/JNEUROSCI.2745-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parillaud VR, Lornet G, Monnet Y, Privat AL, Haddad AT, Brochard V, Bekaert A, de Chanville CB, Hirsch EC, Combadiere C, et al. Analysis of monocyte infiltration in MPTP mice reveals that microglial CX3CR1 protects against neurotoxic over-induction of monocyte-attracting CCL2 by astrocytes. J Neuroinflammation. 2017;14:60. doi: 10.1186/s12974-017-0830-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galloway DA, Williams JB, Moore CS. Effects of fumarates on inflammatory human astrocyte responses and oligodendrocyte differentiation. Ann Clin Transl Neurol. 2017;4:381–391. doi: 10.1002/acn3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hennessy E, Gormley S, Lopez-Rodriguez AB, Murray C, Murray C, Cunningham C. Systemic TNF-alpha produces acute cognitive dysfunction and exaggerated sickness behavior when superimposed upon progressive neurodegeneration. Brain Behav Immun. 2017;59:233–244. doi: 10.1016/j.bbi.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muccigrosso MM, Ford J, Benner B, Moussa D, Burnsides C, Fenn AM, Popovich PG, Lifshitz J, Walker FR, Eiferman DS, Godbout JP. Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun. 2016;54:95–109. doi: 10.1016/j.bbi.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rojo AI, Pajares M, Rada P, Nunez A, Nevado-Holgado AJ, Killik R, Van Leuven F, Ribe E, Lovestone S, Yamamoto M, Cuadrado A. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–451. doi: 10.1016/j.redox.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Branca C, Ferreira E, Nguyen TV, Doyle K, Caccamo A, Oddo S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2017;26:4823–4835. doi: 10.1093/hmg/ddx361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer’s disease-like pathology in the APP/ PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging. 2015;36:664–679. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Characterization of microglial cultures, cell toxicity of DMF, and enriched astrocyte cultures. (PDF 134 kb)
Figure S2. Inflammatory activation of MG induced by LPS. (PDF 146 kb)
Figure S3. Cognitive assessment following systemic immune challenge and DMF treatment. (PDF 154 kb)
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on a reasonable request.