

Abstract
Intracellular cytokine staining (ICS) is a powerful method for identifying functionally distinct lymphocyte subsets, and for isolating these by fluorescence activated cell sorting (FACS). Although transcriptomic analysis of cells sorted on the basis of ICS has many potential applications, this is rarely performed because of the difficulty in isolating intact RNA from cells processed using standard fixation and permeabilization buffers for ICS. To address this issue, we compared three buffers shown previously to preserve RNA in nonhematopoietic cells subjected to intracellular staining for their effects on RNA isolated from T lymphocytes processed for ICS. Our results showed that buffers containing the recombinant ribonuclease inhibitor RNasin or high molar concentrations of salt yielded intact RNA from fixed and permeabilized T cells. As proof of principle, we successfully used the buffer containing RNasin to isolate intact RNA from CD4+ T cells that were sorted by FACS on the basis of specific cytokine production, thus demonstrating the potential of this approach for coupling ICS with transcriptomic analysis.
Keywords: intracellular cytokine staining, FACS, RNA isolation, T cells, transcriptomics
1. Introduction
Intracellular cytokine staining (ICS) is a common method for analysis of immune cells, especially T cells. This typically requires fixation of previously stimulated cells with paraformaldehyde and membrane permeabilization using a detergent, followed by staining with appropriate anti-cytokine antibodies. Cells are then analyzed by flow cytometry to identify functional subsets. Although cells stained in this manner can be isolated by fluorescence-activated cell sorting (FACS), this is seldom done since they are no longer viable and their constituents undergo chemical modifications due to the fixation step that complicate subsequent extraction and analysis. These modifications include cross-linkage between nucleic acids and proteins and covalent modification of RNA by addition of monomethylol groups to the bases (Farragher, Tanney et al. 2008). Downstream analysis of these cells including transcriptomics depends upon the development of methods that allow isolation of high quality nucleic acids from these cells. This is partly because cell surface cytokine capture assay, an alternative method that allows purification of cytokine-secreting cells, requires bifunctional antibodies and is laborious, while suffering from limited sensitivity (Kunnath-Velayudhan, Goldberg et al. 2017). In addition, currently available commercial cell surface cytokine capture assays are limited to a few cytokines (http://www.miltenyibiotec.com).
Research over recent decades resulted in protocols that allow successful extraction of nucleic acids from fixed cells, as shown in the context of formalin-fixed paraffin embedded (FFPE) tissue samples (Farragher, Tanney et al. 2008). The most successful of these methods uses proteinase K digestion prior to acid-phenol chloroform extraction and carrier precipitation. Proteinase K readily destroys proteins despite their highly cross-linked nature. Use of proteinase K was adopted by many commercial kits that enable successful RNA isolation from FFPE samples. Transcriptomic studies performed using RNA isolated by these methods showed that while fragmentation and modifications of isolated RNA is a concern, there is high correlation of transcriptome profiles between fresh frozen and FFPE samples (Hedegaard, Thorsen et al. 2014). However, when one of those commercial methods was applied to perform transcriptomic studies of primary human CD4+ T cells infected with HIV-1, the RNA isolated from these cells showed degradation that likely occurred during the ICS procedure (Iglesias-Ussel, Marchionni et al. 2013).
More recent reports have proposed modifications in the buffers used for intracellular staining in addition to the use of modified RNA isolation protocols to isolate intact RNA from fixed and permeabilized cells. One of these buffers called RNA preserving hybridization buffer, was used in fluorescent in-situ hybridization experiments of mouse induced pluripotent stem cells (Klemm, Semrau et al. 2014). Another buffer called high salt buffer contained 2.0 M NaCl and was used for intracellular staining for cytokeratin in a human renal cell line (Nilsson, Krawczyk et al. 2014). A third buffer condition included a commercially available recombinant protein inhibitor to mammalian RNases (RNasin) and was used to isolate intact RNA from primary human radial glial (Thomsen, Mich et al. 2016) and pancreatic (Hrvatin, Deng et al. 2014) cells after intracellular protein staining and FACS sorting. All these studies showed that RNA isolated by these methods was suitable for downstream analysis including transcriptome profiling. In addition, a high correlation was observed between transcriptome profiles of live cells and cells which underwent fixation and permeabilization (Hrvatin, Deng et al. 2014, Klemm, Semrau et al. 2014, Thomsen, Mich et al. 2016). However, these protocols were neither validated for cytokine staining nor used with hematopoietic cell types. In the current study, we assessed these buffers in the context of ICS of murine T cells and subsequent isolation of intact RNA.
2. Materials and Methods
2.1. Mice
Six- to 8-wk-old female wild-type C57BL/6 mice were obtained from The Jackson Laboratory. All mice were maintained in specific pathogen-free conditions. All procedures involving the use of animals were in compliance with protocols approved by the Einstein Institutional Animal Use and Biosafety Committees.
2.2. Preparation of murine splenocytes and stimulation
Splenocyte suspensions were prepared by gently forcing spleens through a 70 μm cell strainer. RBC lysis was performed using RBC lysing buffer Hybri-Max (Sigma). The cells were washed with media [RPMI medium (Gibco) supplemented with FBS (10%; Atlanta Biologicals), penicillin-streptomycin (1%; Gibco), HEPES (1%; Gibco), beta-mercaptoethanol (0.1%; Gibco), essential amino acids (0.5%; Gibco) and non-essential amino acids (0.5%; Gibco)] and plated in 96-well round-bottom plates with 2 million splenocytes per well. For stimulation, cells were incubated in the presence of Phorbol 12-Myristate 13-Acetate (PMA,1 μg/ml, Sigma), ionomycin (1 μg/ml, Sigma), brefeldin A (5 μg/ml; Sigma) and monensin (5 μM, Sigma) for 4 hours at 37°C. In some experiments, T cells or CD4+ T cells were isolated from splenocytes using Pan T Cell Isolation Kit II or CD4+ T Cell Isolation Kit (Miltenyi Biotec) respectively.
2.3. Intracellular cytokine staining and FACS analysis and sorting
After stimulation, splenocytes were washed with PBS and incubated with viability dye (LIVE/DEAD Fixable Blue (or Violet for sorting experiments) Dead Cell Stain, Molecular Probes) diluted in PBS for 30 minutes at 4°C. All incubation steps were performed in tubes shielded from light. Subsequently, the cells were washed with FACS buffer (PBS containing FBS [2%; Atlanta Biologicals] and sodium azide [0.05%; Sigma]) and blocked for 30 minutes at 4°C with FACS buffer containing 10% normal rat serum and 10% normal mouse serum. Antibodies specific for cell surface markers diluted in FACS buffer were added directly to this mixture and incubated for 30 minutes at 4°C. The cells were then washed with PBS and fixed by incubation with paraformaldehyde (4% in PBS; Electron Microscopy Sciences) for 10 minutes at room temperature. The cells were washed twice with permeabilization buffer (PBS with FBS [2%], sodium azide [0.05%], and saponin [0.1%; Sigma]) and blocked for 30 minutes at room temperature with permeabilization buffer containing 10% normal rat serum. Antibodies diluted in permeabilization buffer were added directly to this mixture. After overnight (16 hours) staining at 4°C, cells were washed with permeabilization buffer and PBS and resuspended in FACS buffer. The cells were kept on ice until the analysis using an LSR II flow cytometer (BD Biosciences).
When alternate ICS conditions were used, the above protocol was followed until the end of the fixation step and the buffers used for subsequent steps varied. For experiments involving the RNA preserving hybridization buffer (Klemm, Semrau et al. 2014), the cells were permeabilized, blocked and stained in RNA preserving hybridization buffer (Table 1). The cells were then washed with RNA preserving hybridization buffer and PBS before resuspending in FACS buffer. For experiments involving the high salt buffer (Nilsson, Krawczyk et al. 2014), the cells were permeabilized, blocked and stained in high salt buffer (Table 1). The cells were then washed with high salt buffer and resuspended in high salt buffer lacking saponin. For experiments involving buffer containing RNasin (RNasin® Plus RNase Inhibitor, Promega) (Thomsen, Mich et al. 2016), the cells were permeabilized, blocked and stained in RNasin containing buffer (Table 1). The cells were then washed with RNasin-containing buffer and resuspended in RNasin-containing buffer lacking saponin. In all experiments, antibodies were diluted in corresponding staining buffers.
Table 1.
Summary of the ICS staining buffers used and the quality of RNA obtained
| Buffer | Composition | Yield (ng)*,# | 280/260 ratio* | RIN* |
|---|---|---|---|---|
| standard permeabilization buffer | PBS containing FBS (2%), sodium azide (0.05%) saponin (0.1%) and normal rat serum (10%); pH 7.4 | 575.7 (477.0 – 615.0) | 2.01 (1.81 – 2.13) | 3.8 (2.3 – 5.2) |
| RNA preserving hybridization buffer | 2× SSC containing ammonium sulfate (2.1 M), EDTA (10 mM), ultrapure BSA (1 mg/ml), formamide (25% [40% v/v]) and saponin (0.1%); pH 5.2 | NP | NP | NP |
| high salt buffer | PBS containing sodium azide (0.05%) saponin (0.1%), purified rat IgG (0.02 mg/ml), ultrapure BSA (1 mg/ml) and sodium chloride (2.0 M); pH 7.4 | 585.3 (416.4 – 747.0) | 2.03 (1.72 – 2.15) | 9.1 (7.8 – 10.0) |
| buffer containing RNase inhibitor | PBS containing sodium azide (0.05%), saponin (0.1%), purified rat IgG (0.02 mg/ml), ultrapure BSA (1 mg/ml) and RNase inhibitor (RNasin® Plus RNase Inhibitor, 0.5 U/ml); pH 7.4 | 574.9 (492.0 – 645.0) | 1.98 (1.70 – 2.13) | 9.0 (8.1 – 10.0) |
average value with range in parenthesis
total yield per million cells
NP not performed
Results represent two independent experiments (n = 3)
For experiments involving FACS sorting of IFNγ-producing CD4+ T cells, the cells were permeabilized, blocked and stained in RNasin-containing buffer (Table 1). The cells were then washed with RNasin-containing buffer and resuspended in RNasin-containing buffer lacking saponin. Cell sorting was performed with a BD FACS Aria (BD Biosciences) and IFNγ-producing CD4+ T cells were collected into a buffer which contained RNasin (0.5 U/ml), ultrapure BSA (1 mg/ml), sodium azide (0.05%), and sodium chloride (2.0 M) in PBS.
The following antibodies were used for staining: CD4-APC-Cy7 (Clone RM4-5, Tonbo Biosciences), CD8α-PE-Cy5 (Clone 53-6.7, Tonbo Biosciences), B220-PE-Cy5 (Clone RA3-6B2, BD Biosciences), MHC II-PE-Cy5 (Clone M5/114.15.2, eBioscience), IFNγ-Alexa Fluor 700 (Clone XMG1.2, BD Biosciences), TNF-Alexa Fluor 488 (Clone MP6-XT22, BD Biosciences), IL-2-PE-Cy7 (Clone JES6-5H4, eBioscience) and GM-CSF-APC (Clone MP1-22E9, BD Biosciences). For sorting experiments, the following antibodies were used: CD4-APC-Cy7 (Clone RM4-5, Tonbo Biosciences), CD8α-PE-Cy5 (Clone 53-6.7, Tonbo Biosciences), B220-PE-Cy5 (Clone RA3-6B2, BD Biosciences), MHC II-PE-Cy5 (Clone M5/114.15.2, eBioscience), and IFNγ-APC (Clone XMG1.2, BD Biosciences).
2.4. RNA isolation and assessment of quality
RNA isolation from pelleted cells was performed using RecoverAll™ Total Nucleic Acid Isolation Kit for FFPE (Ambion) as per manufacturer’s instructions (see the main text for details). Isolated RNA was quantified using a Nanodrop 2000 instrument (Thermo Fisher Scientific Inc.) and RNA quality was assessed using Bioanalyzer Nano or Pico Chips (Agilent Technologies). Pico chips were used to analyze RNA obtained from sorting experiments since these chips allow analysis of samples with minute amounts of RNA (0.05 – 5 ng/μl).
3. Results and Discussion
3.1. Optimization of RNA isolation from fixed cells
For the RNA isolations described in this report, we used a commercial kit called RecoverAll™ Total Nucleic Acid Isolation Kit for FFPE (Ambion). We omitted early steps of deparaffinization from the manufacturer’s recommended protocol and began with the step of protease digestion. We assessed the quality of isolated RNA by measuring the 260:280 nm absorbance ratio using a nanodrop spectrophotometer, and by estimating RNA Integrity Number (RIN) (Schroeder, Mueller et al. 2006)) using a Bioanalyzer (Agilent Technologies). A lower 260:280 ratio (ideal is 2) indicates contamination of the RNA preparation by proteins while a lower RIN (ideal is 10) indicates RNA degradation. Based on the results of preliminary experiments, we modified conditions for protease digestion to 30 min at 50°C (instead of suggested 15 min at 50°C followed by 15 min at 80°C) since the suggested conditions resulted in degraded RNA (data not shown), consistent with previous reports (Iglesias-Ussel, Marchionni et al. 2013, Thomsen, Mich et al. 2016).
3.2. Assessment of RNA preserving hybridization buffer
The buffer called RNA preserving hybridization buffer was previously described in fluorescent in-situ hybridization experiments of mouse induced pluripotent stem cells (Table 1) (Klemm, Semrau et al. 2014). Given the harsh buffer conditions (high molarity, 25% formamide, and acidic pH), we first tested the suitability of this buffer for ICS. We stimulated naive CD4+ T cells in a polyclonal fashion and performed ICS for IFNγ, IL-2 and TNF. While cells stained under standard buffer condition showed production of these cytokines upon stimulation, we were unable to detect any positive cytokine staining for cells stained using RNA preserving hybridization buffer (data not shown). We did not perform additional experiments with this buffer.
3.3. Assessment of high salt buffer
The buffer called high salt buffer was originally described for intracellular staining for cytokeratin in a human renal cell line and contained 2.0 M NaCl (Table 1) (Nilsson, Krawczyk et al. 2014). In that study, this buffer resulted in considerably reduced fluorescence intensity for cytokeratin 7/8 when compared to samples processed with a standard control buffer (Nilsson, Krawczyk et al. 2014). When we assessed this buffer for ICS of polyclonally stimulated murine CD4+ T cells, we observed levels of IFNγ, IL-2, and TNF comparable to the standard buffer condition (Fig. 1A). However, extension of our antibody panel to include additional targets such as IL-3 and GM-CSF (Kunnath-Velayudhan, Goldberg et al. 2017) showed relatively high background with anti-GM-SCF antibody with this buffer compared to standard buffer, which precluded accurate assessment of GM-CSF-producing cells (Fig. 1A). Since binding characteristics may vary among different antibody clones targeting the same cytokine, it is possible that staining using a different antibody clone to GM-CSF would have eliminated the high background obtained with high salt buffer. Since we were able to isolate intact RNA from purified T cells incubated under this buffer condition (Fig. 1B, Table 1), we believe that this buffer has applicability in conditions where cytokine staining is unaffected.
Figure 1.
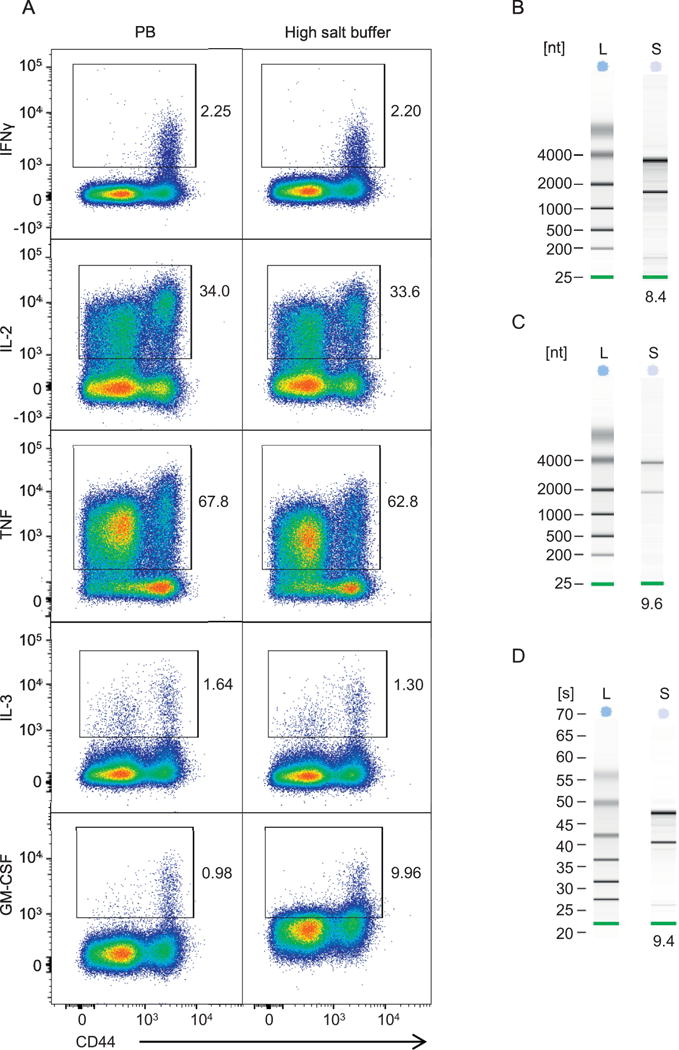
Assessment of modified intracellular staining buffers. (A) Comparison of high salt buffer to standard permeabilization buffer (PB). Polyclonally stimulated splenocytes from C57BL/6 mice underwent ICS using two buffer conditions as indicated. Cells were then analyzed by flow cytometry and the expression of various cytokines by CD4+ T cells are shown. Refer to Supplementary Figure 1 for gating strategy. (B) Quality of RNA isolated from cells permeabilized with high salt buffer. CD4+ T cells were purified from naïve splenocytes, fixed with paraformaldehyde and permeabilized with high salt buffer. The cells were incubated for 16 hours in the same buffer before RNA isolation. Quality of isolated RNA was assessed by Agilent Bioanalyzer nano chips. nt, nucleotide, L, ladder; S, test sample. The number below the lane indicates corresponding RIN. (C) The experiments were performed as in Panel B except cells were permeabilized and incubated in permeabilization buffer containing RNase inhibitor. (D) CD4+ T cells isolated from naïve murine spleens underwent polyclonal stimulation and ICS staining for IFNγ using a permeabilization buffer containing RNase inhibitor. The cells were then analyzed with FACS Aria and IFNγ-producing CD4+ T cells were sorted into collection buffer containing 0.5 U/ml of RNase inhibitor. Quality of isolated RNA was assessed by Agilent Bioanalyzer pico chips which allows analysis of samples with low RNA concentration (0.05 – 5 ng/μl. s, seconds; L, ladder; S, test sample. The number below the lane indicates corresponding RIN. Results shown represent two independent experiments with n = 3 except the sorting experiments described in panel D which represents two independent experiments with n = 2.
3.4. Assessment of buffer containing RNase inhibitor
The third buffer tested included a commercially available recombinant protein inhibitor to mammalian RNases (RNasin® Plus RNase Inhibitor, Promega), and used purified bovine serum albumin (BSA) instead of fetal bovine serum (FBS) to eliminate RNases present in the FBS (Huang, Zhao et al. 2014). This buffer was successfully used in previous studies to isolate intact RNA from primary human radial glial (Thomsen, Mich et al. 2016) and pancreatic (Hrvatin, Deng et al. 2014) cells after intracellular protein staining and FACS sorting. We were able to isolate intact RNA from purified T cells after fixation and permeabilization using this buffer (Fig. 1C, Table 1). We tested whether this buffer can be used to isolate RNA from FACS-sorted CD4+ T cells that secrete a specific cytokine. We performed ICS of polyclonally stimulated naive CD4+ T cells using this buffer and sorted IFNγ-producing cells into a collection buffer containing RNase inhibitor. Analysis of RNA isolated from these cells showed high RIN indicating intact RNA (Fig. 1D).
4. Conclusions
Our results showed that the previously described RNA preserving hybridization buffer is not compatible with ICS, and buffer containing high salt, although able to preserve RNA integrity, may not be suitable for intracellular staining with certain antibodies. However, the use of a buffer containing the RNase inhibitor RNasin and reagents containing minimal amounts of RNases preserves RNA integrity during ICS and is compatible with FACS-sorting. Thus, with optimization of ICS buffer conditions for desired cytokines, it is possible to isolate intact RNA from lymphocyte subsets that secrete specific cytokines. In contrast to the limited choice of commercial cell surface cytokine capture assays, this approach should be applicable to the analysis of cells secreting a much broader range of cytokines.
Supplementary Material
Acknowledgments
We thank Lydia Tesfa from flow cytometry facility for help with sorting experiments.
Funding
This work was supported by NIH/NIAD grant AI45889 awarded to SAP. Core resources for flow cytometry and genomics (Agilent Bioanalyzer) were supported by the Einstein Cancer Center (CA13330).
Abbreviations
- FACS
fluorescence-activated cell sorting
- ICS
intracellular cytokine staining
- FFPE
formalin-fixed, paraffin-embedded
- RIN
RNA integrity number
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest:
None
References
- Farragher SM, Tanney A, Kennedy RD, Paul Harkin D. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochem Cell Biol. 2008;130(3):435–445. doi: 10.1007/s00418-008-0479-7. [DOI] [PubMed] [Google Scholar]
- Hedegaard J, Thorsen K, Lund MK, Hein AM, Hamilton-Dutoit SJ, Vang S, Nordentoft I, Birkenkamp-Demtroder K, Kruhoffer M, Hager H, Knudsen B, Andersen CL, Sorensen KD, Pedersen JS, Orntoft TF, Dyrskjot L. Next-generation sequencing of RNA and DNA isolated from paired fresh-frozen and formalin-fixed paraffin-embedded samples of human cancer and normal tissue. PLoS One. 2014;9(5):e98187. doi: 10.1371/journal.pone.0098187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, Deng F, O’Donnell CW, Gifford DK, Melton DA. MARIS: method for analyzing RNA following intracellular sorting. PLoS One. 2014;9(3):e89459. doi: 10.1371/journal.pone.0089459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Zhao M, Wei N, Wang X, Cao H, Du Q, Liang Z. Site-specific RNase A activity was dramatically reduced in serum from multiple types of cancer patients. PLoS One. 2014;9(5):e96490. doi: 10.1371/journal.pone.0096490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias-Ussel M, Marchionni L, Romerio F. Isolation of microarray-quality RNA from primary human cells after intracellular immunostaining and fluorescence-activated cell sorting. J Immunol Methods. 2013;391(1–2):22–30. doi: 10.1016/j.jim.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm S, Semrau S, Wiebrands K, Mooijman D, Faddah DA, Jaenisch R, van Oudenaarden A. Transcriptional profiling of cells sorted by RNA abundance. Nat Methods. 2014;11(5):549–551. doi: 10.1038/nmeth.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunnath-Velayudhan S, Goldberg MF, Saini NK, Johndrow CT, Ng TW, Johnson AJ, Xu J, Chan J, Jacobs WR, Jr, Porcelli SA. Transcriptome Analysis of Mycobacteria-Specific CD4(+) T Cells Identified by Activation-Induced Expression of CD154. J Immunol. 2017;199(7):2596–2606. doi: 10.4049/jimmunol.1700654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson H, Krawczyk KM, Johansson ME. High salt buffer improves integrity of RNA after fluorescence-activated cell sorting of intracellular labeled cells. J Biotechnol. 2014;192(Pt A):62–65. doi: 10.1016/j.jbiotec.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ER, Mich JK, Yao Z, Hodge RD, Doyle AM, Jang S, Shehata SI, Nelson AM, Shapovalova NV, Levi BP, Ramanathan S. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nat Methods. 2016;13(1):87–93. doi: 10.1038/nmeth.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.