

Abstract
Membrane phosphoinositides control organization and dynamics of the actin cytoskeleton by regulating the activities of several key actin-binding proteins. Twinfilin is an evolutionarily conserved protein that contributes to cytoskeletal dynamics by interacting with actin monomers, filaments, and the heterodimeric capping protein. Twinfilin also binds phosphoinositides, which inhibit its interactions with actin, but the underlying mechanism has remained unknown. Here, we show that the high-affinity binding site of twinfilin for phosphoinositides is located at the C-terminal tail region, whereas the two actin-depolymerizing factor (ADF)/cofilin–like ADF homology domains of twinfilin bind phosphoinositides only with low affinity. Mutagenesis and biochemical experiments combined with atomistic molecular dynamics simulations reveal that the C-terminal tail of twinfilin interacts with membranes through a multivalent electrostatic interaction with a preference toward phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2), PI(4,5)P2, and PI(3,4,5)P3. This initial interaction places the actin-binding ADF homology domains of twinfilin in close proximity to the membrane and subsequently promotes their association with the membrane, thus leading to inhibition of the actin interactions. In support of this model, a twinfilin mutant lacking the C-terminal tail inhibits actin filament assembly in a phosphoinositide-insensitive manner. Our mutagenesis data also reveal that the phosphoinositide- and capping protein–binding sites overlap in the C-terminal tail of twinfilin, suggesting that phosphoinositide binding additionally inhibits the interactions of twinfilin with the heterodimeric capping protein. The results demonstrate that the conserved C-terminal tail of twinfilin is a multifunctional binding motif, which is crucial for interaction with the heterodimeric capping protein and for tethering twinfilin to phosphoinositide-rich membranes.
Keywords: actin, membrane, protein-lipid interaction, protein-protein interaction, molecular dynamics, atomistic simulation
Introduction
The dynamic interplay between the actin cytoskeleton and plasma membrane is critical for several cellular processes, such as migration, morphogenesis, endocytosis, and phagocytosis. Coordinated polymerization of actin filaments provides a force for generation of membrane invaginations in endocytic processes (1, 2). In migrating cells, polymerization of actin filaments against the plasma membrane at the leading edge pushes the membrane forward to generate plasma membrane protrusions, such as lamellipodia and filopodia (3–6).
Although actin polymerization controls the geometry of cellular membranes, membrane phospholipids, especially PI(4,5)P2,2 reciprocally regulate the organization and dynamics of the actin cytoskeleton. Typically, an increase in the plasma membrane PI(4,5)P2 density leads to actin filament assembly beneath the membrane, whereas a decrease in PI(4,5)P2 concentration results in diminished actin filament assembly (7–10). Phosphoinositides regulate actin filament assembly and disassembly by directly interacting with several actin-binding proteins (11). The activities and the plasma membrane targeting of actin-binding proteins promoting actin filament assembly in cells, such as N-WASP, Dia1, and Dia2, are often positively regulated by phosphoinositides. In contrast, proteins promoting actin filament disassembly (e.g. ADF/cofilins and gelsolin) and preventing actin filament assembly (e.g. heterodimeric capping protein) are inhibited by interactions with phosphoinositides (12–17). Binding sites for phosphoinositides and actin overlap on surfaces of ADF/cofilins and heterodimeric capping protein, providing a molecular explanation as to why these proteins are inhibited through interactions with PI(4,5)P2 (18–21).
Twinfilin is an evolutionarily conserved actin-binding protein that regulates cytoskeletal dynamics in organisms from yeasts to mammals (22). Lower eukaryotes, such as yeasts and Drosophila, have one twinfilin gene (23, 24), whereas mammals have two genes, namely twinfilin-1 and twinfilin-2 (25). All twinfilins consist of two actin-depolymerizing factor homology (ADF-H) domains separated by a short linker and followed by a short C-terminal tail region. Twinfilins are involved in several actin-dependent cellular and developmental processes, such as cell migration (26, 27), endocytosis (28, 29), epithelial-to-mesenchymal transition (30), morphology of inner ear stereocilia (31), axonal growth of neurons (27), and platelet activation (32). Moreover, human twinfilin-1 has been shown to facilitate resistance to chemotherapy agents in breast cancer and lymphoma (26, 30).
Twinfilins contribute to cytoskeletal dynamics through a complex mechanism that involves interactions with actin monomers, actin filaments, and heterodimeric capping protein. Both yeast and mammalian twinfilins bind ADP-actin monomers with high affinity and inhibit their nucleotide exchange and assembly into filament ends (23, 33, 34). Mammalian twinfilins can additionally cap actin filament barbed ends with a preference toward ADP-actin–containing filament ends (29, 35, 36). Moreover, the budding yeast twinfilin, together with the cyclase-associated protein, can accelerate actin filament depolymerization from the barbed and pointed ends (37). The high-affinity ADP-actin monomer–binding site is located in the C-terminal ADF-H domain of twinfilin, whereas the presence of both actin-binding ADF-H domains is required for twinfilin's filament barbed end capping and filament depolymerization activities (34, 35, 37). In addition to actin, yeast and mammalian twinfilins interact with the heterodimeric capping protein. Mutagenesis studies demonstrated that the C-terminal tail of twinfilin is critical for interaction with capping protein and for proper subcellular localization of twinfilin in cells (38, 39). However, this interaction does not appear to affect the actin-related activities of twinfilin or capping protein, at least not in vitro, and thus the biological role of the twinfilin-capping protein interaction has remained largely unknown.
Budding yeast twinfilin and mammalian twinfilin-1 and twinfilin-2 also interact with phosphoinositides. Interaction with PI(4,5)P2 inhibits the ability of twinfilin to bind actin monomers and decelerate actin filament assembly (25, 38). Hence, similarly to structurally related ADF/cofilins, the actin interactions of twinfilin are inhibited by phosphoinositides (38, 39). However, the mechanism of twinfilin-PI(4,5)P2 interaction and the location of the phosphoinositide-binding site(s) in twinfilin are unknown. Also, the cellular roles and possible effects of the phosphoinositide interaction on other biochemical functions of twinfilin remain to be elucidated.
Here, we utilized a combination of mutagenesis and biochemical experiments together with atomistic molecular dynamics simulations to expose how mouse twinfilin-1 (hereafter twinfilin) interacts with phosphoinositide-rich membranes. Our results revealed that twinfilin is initially tethered to the phosphoinositide-rich membrane through its conserved C-terminal tail. Further on, we found that this interaction forces the actin-binding ADF-H domains in close proximity to the membrane and subsequently leads to inactivation of actin binding by twinfilin. Thus, twinfilin utilizes a novel two-step mechanism for interactions with phosphoinositide-rich membranes.
Results
Twinfilin binds phosphoinositide-rich membranes via electrostatic interactions
Previous studies on twinfilin-phosphoinositide interactions were performed with the non-quantitative native gel electrophoresis approach using micelles comprising only phosphoinositides (25, 38). Here, we applied vesicle cosedimentation, coflotation, and fluorometric assays with vesicles based on more physiological lipid compositions to study the mechanism by which twinfilin interacts with lipids. Cosedimentation assays performed with vesicles containing 5% phosphoinositides (PI(3)P, PI(4)P, PI(3,4)P2, PI(3,5)P2, PI(4,5)P2, or PI(3,4,5)P3) mixed with other abundant lipid species found at the inner leaflet of the plasma membrane (see “Experimental procedures”) revealed that twinfilin has highest affinity toward vesicles containing PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3 (Fig. 1A and Fig. S1). Twinfilin therefore displays the strongest binding to lipids with a high net negative charge and may additionally prefer PIPs having a phosphate group in the C-5 position of the inositol ring. We also tested whether the twinfilin-lipid interaction is sensitive to phosphoinositide density by carrying out a vesicle cosedimentation assay with vesicles of different PI(4,5)P2 densities (0–20%). These experiments revealed a relatively linear correlation between the amount of cosedimenting twinfilin and the PI(4,5)P2 density of the vesicles (Fig. 1B). Thus, compared with the sharp phosphoinositide-density thresholds reported for N-WASP and cofilin binding to membranes (19, 40), twinfilin does not respond in a switchlike manner to a small increase in phosphoinositide density, although it nevertheless preferentially binds vesicles with a high PI(4,5)P2 density.
Figure 1.

Twinfilin-1 binds lipids via electrostatic interactions. A, a vesicle cosedimentation assay. Lipid composition of vesicles was POPC:POPE:POPS:PIP 55:20:20:5. In the “no PIP” samples, the amount of POPC was 60%. The final protein and lipid concentrations were 2 and 500 μm, respectively. Results are shown as individual data points and as the mean from four experiments. Error bars represent standard deviations. B, a vesicle cosedimentation assay with increasing PI(4,5)P2 density. Vesicle composition was POPC:POPE:POPS 60:20:20, and PI(4,5)P2 concentration was 0, 2, 4, 6, 8, 10, or 20%. The concentration of POPC was decreased correspondingly. The final protein and lipid concentrations were 2 and 250 μm, respectively. C, a vesicle cosedimentation assay with increasing NaCl concentration. The assay was done in 20 mm HEPES, pH 7.4, and varying concentrations of NaCl (0, 100, 250, or 500 mm). The final protein and lipid concentrations were 2 and 500 μm, respectively. Results are shown as individual data points and as the mean of four experiments. Error bars represent standard deviations. D, DPH anisotropy of 40 μm vesicles with lipid composition of POPC:POPS:POPE:PI(4,5)P2:DPH 50:20:20:10:1/500 with increasing twinfilin-1 or MIM I-BAR concentrations.
To examine whether twinfilin interacts with membranes via electrostatic interactions, we performed vesicle cosedimentation assays at different sodium chloride concentrations (0, 100, 250, and 500 mm). The results revealed a clear negative correlation between twinfilin-vesicle interactions and an increasing salt concentration, suggesting that twinfilin associates with membranes through electrostatic interactions (Fig. 1C). Next, we utilized a 1,6-diphenyl-1,3,5-hexatriene (DPH) anisotropy assay to examine whether twinfilin inserts into a lipid bilayer. In this assay, the insertion of a protein motif into the bilayer changes rotational diffusion and hence the anisotropy of the hydrophobic DPH probe (41). Whereas the I-BAR domain of missing-in-metastasis (MIM) protein, containing a membrane-inserting amphipathic helix (42), resulted in an increase in the DPH anisotropy, twinfilin did not induce a detectable effect on DPH anisotropy even at high protein concentrations (Fig. 1D). Together, these data show that twinfilin interacts with negatively charged phosphoinositides via electrostatic interactions and does not penetrate the hydrophobic acyl chain region of the lipid bilayer.
The positively charged C-terminal tail facilitates interaction of twinfilin with phosphoinositides
Because PI(4,5)P2 inhibits the actin-binding function of twinfilin (25, 38), we hypothesized that the binding site for PI(4,5)P2 overlaps with the actin-binding sites in the ADF-H domains (43, 44). In support of this hypothesis, ADF/cofilins, which are entirely composed of a structurally similar ADF-H domain, bind PI(4,5)P2 (12, 19, 45) although with relatively low affinity (21). We thus expressed and purified different domains of twinfilin (Fig. 2A) and applied a vesicle cosedimentation assay to study their interactions with lipid vesicles. Surprisingly, the isolated N-terminal ADF-H domain (TWF1(1–174)) did not exhibit detectable affinity toward lipids, and the C-terminal ADF-H domain (TWF1(169–316)) bound PI(4,5)P2-rich vesicles only with very low affinity. Moreover, a construct comprising the two ADF-H domains, thus lacking only the C-terminal tail (TWF1(1–316)), bound phosphoinositide-rich vesicles with low affinity. In contrast, full-length twinfilin (TWF1(1–350)) and the protein containing the C-terminal ADF-H domain and the C-terminal tail (TWF1(169–350)) bound lipid vesicles with a relatively high affinity with apparent dissociation constants of 247 and 414 μm, respectively (Fig. 2B). We therefore reasoned that the C-terminal tail region of twinfilin is essential for high-affinity interactions with lipids. This result was confirmed by a coflotation assay where the twinfilin construct containing the C-terminal ADF-H domain and the tail region (TWF1(169–350)) was enriched with lipids in fraction 2, whereas the isolated ADF-H domain without the tail (TWF1(169–316)) failed to accumulate in the lipid-containing fraction (Fig. 2C).
Figure 2.
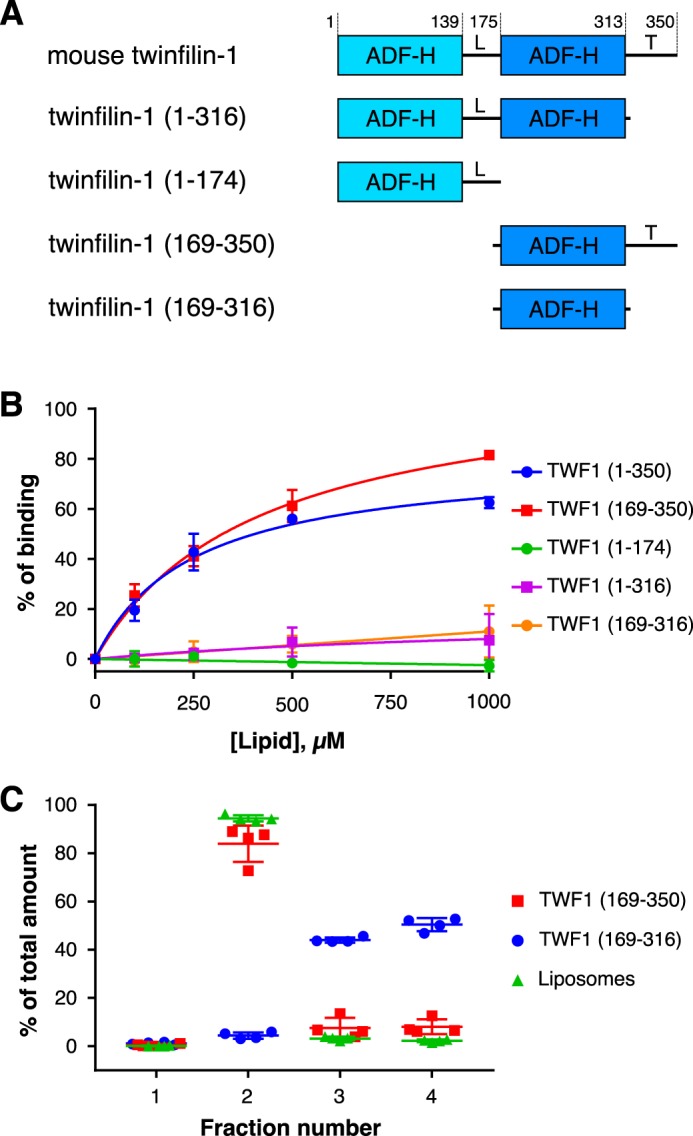
The tail region of twinfilin is crucial for its interactions with lipid vesicles. A, the domain organization of mouse twinfilin-1 and its truncated versions used in this study. The protein is composed of two actin-binding ADF-H domains, a linker region (L), and a C-terminal tail (T). B, a cosedimentation assay performed at different lipid concentrations. The final protein concentration was 2 μm, and the lipid concentration varied from 0 to 1000 μm. The lipid composition was POPC:POPE:POPS:PI(4,5)P2 50:20:20:10 in all experiments. Data were fitted to the equation y = Vmax × [x]/(KD + [x]). Results are shown as individual data points and as the mean of four experiments. Error bars represent standard deviations. C, a vesicle coflotation assay with final protein and lipid concentrations of 1 μm and 1 mm, respectively. The lipid composition was as in B.
We also tested whether the isolated C-terminal tail of twinfilin could interact with lipids. For this purpose, we prepared a construct in which the C-terminal tail of twinfilin is fused to glutathione S-transferase (GST). Importantly, in a cosedimentation assay, this fusion protein displayed much stronger binding toward phosphoinositide-rich vesicles compared with GST alone, which also exhibited some interaction with lipids (Fig. S2A). Moreover, in a coflotation assay, the GST-twinfilin tail fusion protein was enriched with lipids in fraction 2, whereas GST alone was mainly enriched in non-lipid–containing fractions 3 and 4 (Fig. S2B). The isolated C-terminal tail of twinfilin hence binds phosphoinositide-rich membranes with high affinity.
The C-terminal tail of twinfilin contains several positively charged residues and one aromatic residue (Phe-323), which are likely to contribute to phosphoinositide binding. Many of these are conserved in evolution from yeast and Drosophila to mammals (Fig. 3A). To map the phosphoinositide-binding site in the twinfilin tail, we performed vesicle cosedimentation assays with truncated proteins (Fig. 3B). As shown above, a protein lacking the entire tail region (TWF1(1–316)) did not interact with vesicles. Addition of nine residues (TWF1(1–325)), including four positively charged residues and one aromatic residue, did not detectably increase the binding. However, including five additional residues (TWF1(1–330)) increased lipid binding significantly, and the construct lacking the seven C-terminal residues of the tail (TWF1(1–343)) bound phosphoinositide-rich vesicles with affinity similar to that of the full-length protein (Fig. 3B).
Figure 3.

Clusters of positively charged residues in the tail of twinfilin facilitate its lipid interactions. A, a sequence alignment of tail regions from mouse and human twinfilin-1 as well as fruit fly and budding yeast twinfilins. Clusters of positively charged residues mutated to alanines are highlighted with *, +, and ×. The conserved phenylalanine mutated for biochemical experiments is highlighted with #. B, a vesicle cosedimentation assay preformed on full-length twinfilin-1 and C-terminally truncated proteins. C, vesicle cosedimentation assays carried out on wild-type twinfilin-1 and mutant proteins. The final protein and lipid concentrations were 2 and 500 μm, respectively, and the lipid composition was POPC:POPS:POPE:PI(4,5)P2 50:20:20:10. Results are shown as individual data points and as the mean of four experiments. Error bars represent standard deviations. Statistical significance was calculated with Student's t test. ***, p < 0.001; *, p < 0.05. n.d., not detectable.
To further map the PI(4,5)P2-binding site, we generated three mutant twinfilins in which clusters of positively charged residues of the tail were replaced by alanines (TWF1(H317A,H319A,K320A), TWF1(K325A,K327A), and TWF1(K332A,K333A,R335A,R336A)) and a mutant twinfilin in which the conserved phenylalanine was replaced by alanine (TWF1(F323A)) (Fig. 3A). All mutant proteins displayed reduced binding to phosphoinositide-rich vesicles in a cosedimentation assay (Fig. 3C). However, the phenylalanine-to-alanine mutant (TWF1(F323A)) displayed a less pronounced defect in lipid binding compared with the charge-neutralized mutants (Fig. 3C). Together, these results revealed that the clusters of positively charged residues in the C-terminal tail comprise the high-affinity phosphoinositide-binding site of twinfilin. In contrast, the ADF-H domains of twinfilin bind lipids only with low affinity.
Molecular mechanism underlying the binding of twinfilin to phosphoinositide-rich membranes
To reveal the mechanism by which twinfilin binds to PI(4,5)P2-rich membranes, we utilized atomistic molecular dynamics (MD) simulations. Experimental results discussed above suggest that the C-terminal tail plays an important role in this process. Crystal and solution structures of the N-terminal and C-terminal ADF-H domains have been determined (35, 43, 44, 46), but the structure of the 34-residue-long C-terminal tail region (residues 317–350) has remained unknown. To explore the secondary structure of the twinfilin tail, we first utilized unbiased MD simulations of the C-terminal tail region detached from the rest of the protein. The MD simulations indicated the C-terminal tail region to be mainly unfolded with transient structures consistent with α-helical and partially β-turn conformations. Then we constructed a more complete description for a twinfilin fragment containing the C-terminal ADF-H domain and the 34-residue C-terminal tail, whose structure was consistent with the above described simulations. Details of these simulations are discussed in the supporting information.
To elucidate the structural mechanism of the twinfilin interaction with a PI(4,5)P2-rich membrane, we performed a series of simulations with different twinfilin fragments using asymmetric membranes (see “Experimental procedures”). We first compared the membrane binding of the C-terminal ADF-H domain with the tail in an α-helical conformation (system 5; see Table S1) and the ADF-H domain alone without the tail (system 10). The ADF-H domain linked to the α-helical tail interacted more strongly with lipid bilayer compared with the ADF-H domain without the tail (Fig. S4, Movie S3, and Movie S4). We next studied the interaction of the α-helical tail attached to the ADF-H domain (system 5) more closely. In four independent simulations, this protein bound to the PI(4,5)P2-rich leaflet with the C-terminal tail leading the binding process (Fig. 4A and Movie S3). This could be visualized by following the distance between any lipid in a membrane and residues Arg-267 and Arg-269 in the actin-binding region of the ADF-H domain and residues Lys-323 and Lys-325 in the C-terminal tail region (Fig. 4B). In all four simulations, the residues in the tail region bound lipids first followed by the residues in the ADF-H domain (Fig. 4C). The C-terminal tail remained on the membrane surface instead of being inserted into the membrane. Interestingly, of the key residues in the tail that interacted with PI(4,5)P2 or other lipids (Lys-320, Ser-322, Lys-325, Lys-327, Gly-328, Gly-331, Lys-332, Arg-333, Arg-336, Arg-337, and Arg-340), eight are charged (lysines and arginines), and of the residues in the ADF-H domain that interacted with lipids (Ser-265, Arg-267, Arg-269, Arg-285, and Asp-298), four of the five are also charged (arginines and aspartic acid) (Fig. S5, left panel). This strongly suggests that the binding process is driven by electrostatics. We note that two of six repeats of system 5 hovered early on onto a negatively charged PI(4,5)P2-free leaflet due to sensitivity to initial conditions. These simulation repeats were interrupted because it would require much larger timescales to reproduce the binding process on the other leaflet.
Figure 4.

Atomistic simulations to explore twinfilin-membrane interaction. A, snapshots from a simulation with the C-terminal ADF-H domain and the tail (TWF1(169–350)) together with a phosphoinositide-rich membrane (system 5 in Table S1). The lipid composition in the upper membrane leaflet was POPC:POPS:POPE 60:20:20, and that in the lower leaflet was POPC:POPS:POPE:PI(4,5)P2 50:20:20:10. The tail of twinfilin is shown in yellow, and PI(4,5)P2 in the membrane is depicted in light red. B, the final conformation of TWF1(169–350) on a membrane. PI(4,5)P2 in the membrane is depicted in light red. Two residues at the actin-binding region of the ADF-H domain (Arg-267 and Arg-269) and two residues in the twinfilin tail (Lys-325 and Lys-327) are highlighted. C, minimum distance between lipids of the PI(4,5)P2-rich leaflet and residues Arg-267, Arg-269, Lys-325, and Lys-327 during four independent simulations. Residues in the C-terminal tail (Lys-325 and Lys-327) equilibrated much faster onto the plasma membrane compared with the residues in the ADF-H domain (Arg-267 and Arg-269).
To test the role of electrostatic interactions between the membrane and the protein as the driving force for membrane binding, we neutralized all the positively charged residues in the C-terminal tail of the protein starting from the α-helical conformation (system 11). In all six repeats of the membrane-binding simulations (Fig. S5, right panel), we observed unstable and unspecific association with the negatively charged leaflet of the membrane, although the secondary structure of the C-terminal tail remained helical throughout simulations. The unspecific adsorption is evident from the contact frequency plots shown in Fig. S5, which not only shows the divergence in the set of residues that contact the membrane but also the diminished membrane-interacting role of the C-terminal tail in all six repeats (Fig. S5, right panel). These simulations further support the importance of the charged residues in the C-terminal tail for proper initial association with the membrane and convergence to a well-defined membrane-binding mode.
To confirm that the α-helical conformation is important for membrane binding of the C-terminal tail, we constructed two additional twinfilin structures: the C-terminal ADF-H domain combined with the tail either in a random coil conformation (system 6) or in a β-sheet conformation (system 7). In five independent simulations, we observed only weak binding coupled to short residence times spent on the membrane or no binding at all as shown by the atom number densities estimated from the simulations for each tail conformation (Movie S5, Movie S6, and Fig. S6). The overlap of the densities clearly shows the convergence of the membrane-binding interactions of the C-terminal tail and C-terminal ADF-H domain regions of twinfilin in four of six simulations when the tail is in an α-helical conformation (Fig. S6, upper panel). However, other conformations of the tail fail to bind the membrane, interact unspecifically with it, or associate with the PI(4,5)P2-free leaflet (Fig. S6, middle and lower panels). The weaker lipid binding of the β-sheet conformation compared with that of the α-helical conformation is supported by the presence of a more pronounced positive electrostatic potential in the tail region when it is in the α-helical conformation (Fig. S3C) compared with the β-sheet conformation (Fig. S3F). We also performed simulations of the isolated C-terminal tail in the α-helical conformation (system 9). These simulations revealed that the α-helical tail binds the membrane also in the absence of the ADF-H domain (Movie S7). All charged amino acid residues in the tail interact strongly with phosphoinositides, again highlighting the importance of electrostatic interactions in the membrane-binding process.
Finally, we performed a Define Secondary Structure of Protein (DSSP) analysis for the initial simulation structures and the last 50 ns of simulations of the C-terminal tail to reveal possible conformation changes (Fig. S7). The secondary structure of the protein remained almost entirely unchanged within the timescales of the simulations in all repeats regardless of the initial secondary structure of the C terminus and interactions with the membrane. In the case of the random coil simulations, however, the initially present short stretch of the helix at the very C terminus appeared to be less stable, adopting some structure or showing signs of becoming unstructured (Fig. S7).
Collectively, our atomistic MD simulations provided compelling evidence that twinfilin binds to phosphoinositide-rich membranes by its C-terminal tail, which does not penetrate the bilayer but instead lies along the membrane plane, interacting simultaneously with several lipid headgroups (Fig. S4). The driving force for membrane binding is the electrostatic interactions. The binding is the strongest when the tail has a transient α-helical structure that renders strong electrostatic interactions between the charged residues and PI(4,5)P2 possible. Moreover, independent simulations of this structure provided evidence for a two-step mechanism where the strong association of the C-terminal tail with lipids places the ADF-H domain in close proximity to the membrane. The ADF-H domain subsequently interacts with phosphoinositides through charged residues that are also critical for its actin binding, hence providing a structural explanation for the PI(4,5)P2-induced inactivation of twinfilin-actin interactions.
The C-terminal tail is crucial for regulation of twinfilin by phosphoinositides
To test the model where the C-terminal tail tethers twinfilin to the phosphoinositide-rich membrane, we applied an actin filament sedimentation assay. Here, we compared the abilities of full-length twinfilin (TWF1(1–350)) and a protein lacking the C-terminal tail (TWF1(1–316)) to inhibit actin filament assembly (i.e. to shift actin from the pellet (F-actin) to the supernatant (G-actin) fraction) in the absence and presence of PI(4,5)P2-rich vesicles. Both full-length twinfilin and the protein lacking the C-terminal tail efficiently inhibited actin filament assembly in the absence of lipids (Fig. 5, A and B). However, the inhibition effect of full-length twinfilin compared with mutant TWF1(1–316) was slightly stronger due to its higher affinity toward filament barbed ends (35). When these proteins were first incubated with PI(4,5)P2-rich vesicles, the ability of full-length twinfilin to inhibit actin polymerization was severely diminished, although lipids could not completely inhibit full-length twinfilin possibly due to saturation of exposed PI(4,5)P2 lipids at high protein concentrations (Fig. 5A). In contrast, the ability of the protein lacking the C-terminal tail to inhibit actin filament assembly was only mildly affected by PI(4,5)P2-rich vesicles (Fig. 5B). Together with the mutagenesis and MD simulations presented above, these biochemical data show that the C-terminal tail is crucial for anchoring twinfilin to the membrane and subsequently inhibiting the actin-binding activity of the nearby ADF-H domain by forcing it into close contact with the phosphoinositide-rich membrane.
Figure 5.

The C-terminal tail is critical for regulation of twinfilin by PI(4,5)P2. An actin filament cosedimentation assay was performed in the absence and presence of phosphoinositide-rich vesicles for the full-length twinfilin (TWF1(1–350)) (A) and for a protein where the C-terminal tail was deleted (B). Actin and lipid concentrations were 3 μm and 1 mm, respectively, and the lipid composition was POPC:POPS:POPE:PI(4,5)P2 40:20:20:20. Each data point is the mean of four independent experiments. Error bars represent standard deviations. Please note that the full-length and the C-terminally deleted twinfilins efficiently increased the amount of monomeric actin in the absence of lipids. However, only the full-length twinfilin could be efficiently inhibited by addition of phosphoinositide-rich vesicles. The sigmoidal behavior in TWF1(1–350) + 1 mm lipid samples in A most likely result from saturation of PI(4,5)P2 with high concentrations of TWF1.
The binding sites for phosphoinositides and capping protein overlap in the C-terminal tail of twinfilin
Twinfilins also interact with the heterodimeric capping protein, and the C-terminal tail is important in this interaction (39). To examine whether the phosphoinositide- and capping protein–binding sites overlap the C-terminal tail of twinfilin, we utilized the microscale thermophoresis (MST) method (47–49) to measure the affinities of wildtype and mutant twinfilins to EGFP-tagged capping protein. Consistent with an earlier study (39), twinfilin lacking the C-terminal tail (TWF1(169–316)) exhibited only low affinity toward capping protein (Fig. 6D), whereas the full-length twinfilin (TWF1(1–350)) and the protein lacking the N-terminal ADF-H domain (TWF1(169–350)) bound capping protein with submicromolar affinities (Fig. 6, A and B). Importantly, also the isolated tail fused to GST (TWF1(316–350)) bound capping protein, although the affinity was ∼5-fold lower compared with that of the full-length protein (Fig. 6C). Please note that the decrease in the thermophoretic signal at high TWF1(169–316) concentrations may also result from nonspecific interactions of this protein and EGFP at high concentrations, leading to quenching of EGFP fluorescence.
Figure 6.

Microscale thermophoresis (MST) assay demonstrating that the C-terminal tail of twinfilin-1 interacts with capping protein. A–H, the normalized changes in thermophoretic signals with 50 nm EGFP-capping protein in the presence of different concentrations of full-length twinfilin and twinfilin fragments (A–D) or proteins with point mutations in the C-terminal tail (E–H). Each curve represents the fitting of an individual experiment. KD represents a mean of three to four independent experiments, and a standard deviation is shown.
Next, we measured the binding affinities of the mutant twinfilins that displayed defects in PI(4,5)P2 interactions with the EGFP-tagged capping protein. Three mutants, TWF1(F323A), TWF1(K325A,K327A), and TWF1(K332A,K333A,R335A,R336A), exhibited only very low (∼20-fold decreased) affinity to capping protein (Fig. 6, F–H), whereas one mutant, TWF1(H317A,H319A,K320A), displayed about 5-fold lower affinity to capping protein compared with the wildtype protein (Fig. 6E). Together, these results demonstrate that the isolated C-terminal tail of twinfilin is sufficient for interactions with capping protein and that the binding sites for PI(4,5)P2-rich membranes and capping protein overlap in the C-terminal tail of twinfilin.
Discussion
Twinfilin is an evolutionarily conserved actin-binding protein, whose activity is inhibited by phosphoinositides. In this work, we utilized a combination of mutagenesis and biochemical studies as well as atomistic molecular dynamics simulations to elucidate the mechanism underlying the interaction between twinfilin and phosphoinositide-rich membranes. Strikingly, we found that the high-affinity lipid-binding site is located at the C-terminal tail of twinfilin and that the actin-binding ADF-H domains bind phosphoinositide-rich membranes only with low affinity. We also demonstrated that the binding sites for capping protein and phosphoinositides overlap in the C-terminal tail of twinfilin. The atomistic simulation data provided evidence that twinfilin is initially tethered to the membrane through its C-terminal tail, which has a transient α-helical structure, and that this interaction subsequently forces the ADF-H domains to associate with the membrane, hence resulting in inhibition of their actin-binding activities. This paradigm is also supported by biochemical data demonstrating that a twinfilin mutant lacking the C-terminal tail regulates actin dynamics in a phosphoinositide-insensitive manner.
Twinfilin is composed of two ADF/cofilin–like ADF-H domains. Our cosedimentation and DPH anisotropy experiments as well as atomistic simulations demonstrated that, similarly to ADF/cofilins (19, 21), twinfilin interacts with negatively charged phospholipids through electrostatic interactions without penetrating the hydrophobic acyl chain region of the bilayer. However, the isolated N-terminal ADF-H domain of twinfilin does not bind lipids with detectable affinity, and the C-terminal ADF-H domain displays only relatively modest binding to lipids in vitro. These results are consistent with a recent study demonstrating that ADF/cofilins display only transient, low-affinity interactions with phosphoinositide-rich membranes (21). Importantly, our experiments revealed that full-length twinfilin binds membranes more strongly compared with ADF/cofilins and that the C-terminal tail of twinfilin is critical for this high-affinity lipid binding. Thus, the ADF-H domains appear to interact with membranes only with low affinity and seem to require additional protein motifs (e.g. the C-terminal tail in the case of twinfilin) for strong association with membranes.
Our data revealed that the isolated C-terminal tail of twinfilin is sufficient for interactions with both phosphoinositides and capping protein. Moreover, our mutagenesis experiments demonstrated that the positively charged residues (Lys-325, Lys-327, Lys-332, Lys-333, Arg-335, and Arg-336) and the aromatic residue (Phe-323) at the central region of the tail are critical for interactions with both phosphoinositides and capping protein. Residues near the beginning of the tail (His-317, Lys-319, and Lys-320) also contribute to these interactions but are not as crucial as the residues in the central region. The overlapping binding sites suggest that, in addition to actin binding (25, 38), phosphoinositides inhibit interactions between twinfilin and capping protein. However, because both twinfilin and capping protein bind lipids with high affinity (20, 50), it is technically difficult to provide direct experimental evidence (e.g. by using pulldown, vesicle cosedimentation, or vesicle coflotation assays) that twinfilin does not bind capping protein in the presence of phosphoinositide-rich membranes.
Our atomistic MD simulations and biochemical data provide evidence for a novel two-step mechanism for twinfilin inhibition by phosphoinositides (Fig. 7). Whereas the other actin-binding proteins examined so far, including ADF/cofilins and capping protein (19, 21, 50), directly associate with membranes through their actin-binding sites, twinfilin does not utilize its actin-binding site for initial interactions with the membrane. The high-affinity actin-binding site is located in the C-terminal ADF-H domain of twinfilin (34). Our simulation data suggest that the adjacent C-terminal tail is a relatively flexible, extended structure with some α-helical content. The tail also contains the high-affinity lipid-binding site, and when encountering a phosphoinositide-rich membrane it can tether the protein on the surface of the lipid bilayer. Interestingly, a cluster of positively charged arginines in the tail has a spacing of (n, n +3) or (n, n + 4), creating a surface of positive charge on one side of the helical tail that interacts with lipids (Fig. S8). This further supports the proposed partially helical structure as the lipid-binding interface. Because the C-terminal ADF-H domain binds actin and phosphoinositides through an overlapping interface, tethering this domain to the membrane through the C-terminal tail results in its association with phosphoinositides and consequent inhibition of the actin-binding activity (see Movie S3). The membrane-bound, inactive twinfilin molecule can then be released from the membrane either spontaneously (assuming that its off-rate from the membrane is rather rapid) or following PI(4,5)P2 hydrolysis.
Figure 7.
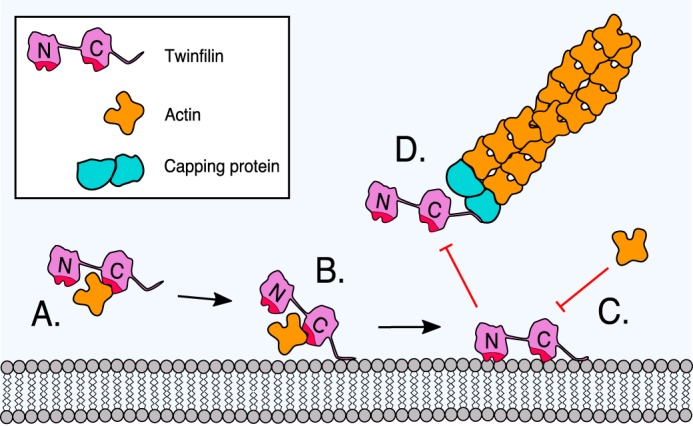
A working model for the inhibition of twinfilin-1 by a phosphoinositide-rich membrane. A, twinfilin-1 interacts, through its C-terminal ADF-H domain, with an ADP-actin monomer and prevents its association with filament ends. B, the C-terminal tail of twinfilin-1 can anchor the protein onto a phosphoinositide-rich membrane through electrostatic interactions between the positively charged residues of the protein and negatively charged lipid headgroups. C, membrane-tethering (driven by the C-terminal tail) places the actin-binding ADF-H domains of twinfilin in contact with the membrane. Because the ADF-H domains associate with phosphoinositide through an interface that overlaps with the actin-binding sites, this leads to inhibition of the actin-binding activities of twinfilin-1. D, because the binding sites for phosphoinositides and capping protein overlap in the tail of twinfilin-1, phosphoinositide binding is also expected to inhibit the interaction of twinfilin with capping protein.
In cells, twinfilin localizes to regions of high actin turnover, such as lamellipodia, filopodia, cell–cell junctions, and sites of endocytosis (25, 29). Interestingly, these cellular regions are also enriched in specific phosphoinositides and are under dynamic regulation of several phosphoinositide kinases and phosphatases (7, 51–53). Moreover, epidermal growth factor (EGF)-induced hydrolysis of PI(4,5)P2 has been suggested to release cofilin from the plasma membrane to activate the protein in carcinoma cells (54). We thus hypothesize that phosphoinositides can control the subcellular localization of twinfilin and regulate its activities in cells during lamellipodial protrusions and retractions. Because twinfilin promotes actin filament disassembly and inhibits actin filament assembly (23, 25, 29, 34, 37), a local decrease in the plasma membrane PI(4,5)P2 density may lead to a displacement of twinfilin from the membrane and subsequently unleash it to promote actin filament disassembly. To study the in vivo role of twinfilin-phosphoinositide interaction in the future, it will be important to identify twinfilin mutants that specifically inhibit its lipid-binding activity. Because twinfilin is linked to several diseases, such as lymphoma (26) and breast cancer (30) progression and chemoresistance, it will also be important to elucidate the possible role of twinfilin-phosphoinositide interactions, as well as other regulatory mechanisms of twinfilin, in these disorders.
Experimental procedures
Plasmids
Mouse twinfilin-1 cDNA was amplified with Phusion polymerase (Thermo Fisher) and cloned to pGAT2 bacterial expression vector using NcoI and HindIII restriction (Thermo Fisher). Point mutations were introduced to twinfilin-1 using the quick-change mutagenesis method with the KAPA HiFi HotStart ReadyMix PCR kit (Kapa Biosystems). Truncated cDNAs were generated by introducing a premature stop codon to twinfilin-1 gene with the KAPA HiFi HotStart ReadyMix PCR kit. A tail fragment of twinfilin-1 was cloned to the pGEX-6P-1 bacterial expression vector with SalI and EcoRI restriction enzymes (Thermo Fisher). Mouse capping protein subunits α1 and β2 were cloned to pRSFDuet1 bacterial expression vector containing N-terminal EGFP protein with EcoRI/HindIII and NdeI/XhoI, respectively.
Proteins
All proteins were expressed in either Escherichia coli BL21 (DE3) cells in Luria broth with isopropyl 1-thio-β-d-galactopyranoside induction or in E. coli BL21 (DE3) pLysS cells in autoinduction Luria broth (Formedium). Cells were lysed with an EmulsiFlex-C3 homogenizer (Avestin). GST-tagged twinfilins were immobilized with glutathione-agarose beads (Thermo Fisher and Sigma-Aldrich) and washed several times with 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2.5 mm CaCl2 buffer, and GST tag was cleaved with 10 units/ml thrombin (Sigma-Aldrich). Proteins were further purified with a Superdex-75 gel filtration column (GE Healthcare) in 20 mm Tris-HCl, pH 7.5, 50 mm NaCl buffer. GST-tagged tail fragment of twinfilin-1 was eluted from beads with 20 mm glutathione without thrombin cleavage. His-tagged EGFP-capping protein was immobilized with nickel-nitrilotriacetic acid-agarose beads and washed with 20 mm Tris-HCl, pH 7.5, 250 mm NaCl, 25 mm imidazole buffer. Protein was eluted with 20 mm Tris-HCl, pH 7.5, 250 mm NaCl, 250 mm imidazole buffer and further purified with a Superdex-200 gel filtration column (GE Healthcare) in 20 mm HEPES, pH 7.4, 100 mm NaCl buffer.
Lipids and membrane probes
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; Avanti catalog number 850457), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE; Avanti catalog number 850757), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS; Avanti catalog number 840034), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′-phosphate) (PI(3)P; Avanti catalog number 850150), l-α-phosphatidylinositol-4-phosphate (PI(4)P; Avanti catalog number 840045), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,4′-bisphosphate) (PI(3,4)P2; Avanti catalog number 850153), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,5′-bisphosphate) (PI(3,5)P2; Avanti catalog number 850154), l-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2; Avanti catalog number 840046), and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,4′,5′-trisphosphate) (PI(3,4,5)P3, Avanti catalog number 850156) were purchased from Avanti Polar Lipids. Rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (rhodamine-DHPE; Thermo Fisher catalog number L1392) was purchased from Thermo Fisher. DPH (catalog number D208000 Aldrich) was purchased from Merck.
Lipid vesicle cosedimentation assay
Multilamellar vesicles were prepared as described earlier (19). In most experiments (differences are specified in figure legends), 2 and 500 μm proteins and lipid vesicles, respectively, were mixed in 20 mm HEPES, pH 7.4, 100 mm NaCl reaction buffer and incubated for 30 min at room temperature. Lipid composition in vesicles was commonly POPC:POPE:POPS:PI(4,5)P2:rhodamine-POPE 50:18:20:10:2, thus mimicking the inner leaflet of the plasma membrane. However, in lipid-specificity assays, we used a phosphoinositide concentration of 5% to avoid saturated binding and hence to detect differences in binding with various phosphoinositide species more clearly. The reaction mixture was centrifuged at 100,000 rpm with a Beckman TLA-100 rotor for 30 min at room temperature. Equal amounts of supernatants and pellets were loaded for SDS-PAGE and after electrophoresis were stained with Brilliant Blue G (Sigma-Aldrich). Intensities of twinfilin-1 bands were quantified with ImageJ. In each assay, the amount of nonspecific aggregation of protein was taken into account through a control sample consisting of only a protein and a reaction buffer. This background was always subtracted from other samples.
Lipid coflotation assay
Interactions between proteins and multilamellar lipid vesicles were examined with a lipid coflotation assay (55). The lipid composition was POPC:POPS:POPE:PI(4,5)P2:rhodamine-POPE 50:20:20:10:2. Protein and lipids with final concentrations of 1 μm and 1 mm, respectively, were incubated for 30 min in 20 mm HEPES, pH 7.4, containing 0.3 m sucrose and then brought to 30% sucrose concentration. The reaction mixture was overlaid with 25 and 0% sucrose in 20 mm HEPES, pH 7.4, buffer and centrifuged at 240 000 × g for 30 min at 4 °C. Four fractions were collected as described elsewhere (56) and analyzed by SDS-PAGE. Protein amounts were quantified with ImageJ, and liposome amounts were measured with an Agilent Cary Eclipse fluorescence spectrometer at 550- and 580-nm excitation and emission wavelengths, respectively.
DPH anisotropy assay
The fluorescence anisotropy of DPH was measured in a 3-mm-path-length quartz cuvette with PerkinElmer Life Sciences LS-55 spectrometer. The final concentration of lipid vesicles was 40 μm in 100 μl of reaction buffer (20 mm HEPES, pH 7.4, 100 mm NaCl). Lipid composition was POPC:POPE:POPS:PI(4,5)P2:DPH 50:20:20:10:1/500, and lipids were extruded through 100-nm filters before measurements.
Actin filament cosedimentation assay
The final concentration of 3 μm muscle actin was polymerized for 30 min in a modified F-buffer (5 mm Tris-HCl, pH 8, 0.05 mm CaCl2, 0.5 mm 1,4-dithiothreitol (DTT), 0.2 mm ATP, 100 mm KCl, 1.25 mm MgCl2, 1 mm EGTA) with or without 0–10 μm full-length or C-terminally deleted twinfilin and 1 mm multilamellar vesicles. Spectrin-actin seeds were used to initialize the polymerization as described earlier (57). Lipid composition of vesicles was POPC:POPE:POPS:PI(4,5)P2 40:20:20:20. Actin filaments were sedimented by centrifugation at 75,000 rpm for 30 min with a TLA-100 rotor. Equal amounts of supernatant and pellet fractions were analyzed by SDS-PAGE, and intensities of actin bands were determined with ImageLab (Bio-Rad).
Microscale thermophoresis
MST measurements were performed in 20 mm sodium phosphate buffer, pH 7.4, 150 mm NaCl, 0.05% Tween 20, 0.1 mg/ml BSA, 5 mm DTT with Monolith standard treated capillaries (NanoTemper Technologies) and Monolith NT.115 (NanoTemper Technologies). The light-emitting diode (LED) power and the MST power were 40 and 60%, respectively. The concentration of EGFP-capping protein was 50 nm, and the concentration of twinfilins ranged from 0.7629 nm to 25 μm. Changes in thermophoretic signals were analyzed with MO.Affinity Analysis software (NanoTemper Technologies). Data were fitted with Prism (GraphPad Software) as described earlier (47) with the following equation,
| (1) |
where [BL] is the concentration of formed complexes, [B0] is the total concentration of binding sites, KD is the dissociation constant, [L] is the concentration of twinfilin, and h represents the Hill coefficient.
MD simulations and analysis
The atomistic CHARMM36 force field was used for proteins and lipids as described earlier (58, 59), and the TIP3P-CHARMM model was used for water (60). The protein structure for twinfilin was taken from the Protein Data Bank (code 2HD7) (35). Twinfilin was explored in 11 different systems with or without a membrane (see Table S1). For simulations in water solution without a membrane (systems 1–4), the C-terminal tail was placed in a box of 11 × 11 × 11 nm3 to ensure no interaction with its image even in the coil-starting simulations and to confirm that the peptide was always very well-solvated. For the bilayer simulations (systems 5–10), lipid bilayers of a size of about 11 × 11 nm2 were used (61). The upper leaflet had a lipid composition of POPC:POPE:POPS:PI(4,5)P2 50:20:20:10, matching the lipid content of membranes considered in the experiments. The lower leaflet had a lipid composition of POPC:POPE:POPS 60:20:20 to confine PI(4,5)P2 and its specificity to the upper leaflet. The concentration of NaCl was 100 mm in every simulation system. To build systems 5 and 7 (Table S1), i.e. the C-terminal domain attached to the 34-residue tail in either a β-sheet or an α-helical structure, we performed clustering analysis as explained in the supporting information. In every system studied, the protein was placed at a distance of more than 3 nm from the upper leaflet and the periodic image of the lower image. This yielded a very large simulation box, which subsequently guaranteed well-solvated lipids with more than 120 water molecules per lipid in all cases.
All simulations were performed in the NPT ensemble using GROMACS 5.0.4 (62). The temperature in all the systems was maintained at 303 K using the Nosé–Hoover coupling method (63, 64) with a time constant of 1 ps. The temperature of the protein, lipids, and solvent molecules was controlled independently. The pressure coupling was done with the Parrinello–Rahman semi-isotropic barostat (65) with a coupling constant of 1 ps and a reference pressure of 1 bar. The equations of motion were integrated with a time step of 2 fs. The LINCS algorithm (66) constrained all bonds involving hydrogens. A cutoff radius of 1.2 nm was used to switch off van der Waals interactions, and the smooth particle mesh Ewald technique (67) was used to calculate long-range Coulomb interactions. Electrostatic representations in Fig. S2 were drawn with the Adaptive Poisson-Boltzmann Solver (APBS) electrostatics (68). Each system was simulated over times (see Table S1) that were sufficiently long for observing and analyzing protein conformational behavior and/or the binding process. To improve sampling, the simulations for the key systems were repeated several times (Table S1).
Author contributions
M. H., M. K., and G. E. formal analysis; M. H., M. K., and G. E. investigation; M. H., M. K., and G. E. visualization; M. H., M. K., and G. E. methodology; M. H., M. K., and G. E. writing-original draft; M. H., I. V., and P. L. writing-review and editing; I. V. and P. L. supervision; I. V. and P. L. funding acquisition; P. L. conceptualization.
Supplementary Material
Acknowledgments
We thank Mitro Miihkinen, Konstantin Kogan, Tommi Kotila, Anna-Liisa Nyfors, and Danielle Bansfield for excellent technical assistance. Hongxia Zhao and Yosuke Senju are acknowledged for technical advice. CSC-IT Center for Science is thanked for computational resources.
This work was supported by ProLipids Centre of Excellence Grants 272130 and 307415 from the Academy of Finland (to P. L. and I. V.), a fellowship from the Doctoral School in Health Sciences (to M. H.), and European Research Council Advanced Grant CROWDED-PRO-LIPIDS (Grant 290974; to I. V.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8, Movies S1–S7, Table S1, and supporting molecular dynamics simulation results.
- PI
- phosphatidylinositol
- PIP
- phosphatidylinositol phosphate
- ADF
- actin-depolymerizing factor
- ADF-H
- actin-depolymerizing factor homology
- N-WASP
- neural Wiskott–Aldrich syndrome protein
- I-BAR
- inverse Bin-Amphiphysis-Rvs
- DPH
- 1,6-diphenyl-1,3,5-hexatriene
- TWF1
- twinfilin-1
- MD
- molecular dynamics
- MST
- microscale thermophoresis
- EGFP
- enhanced GFP
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- MIM
- missing-in-metastasis.
References
- 1. Mooren O. L., Galletta B. J., and Cooper J. A. (2012) Roles for actin assembly in endocytosis. Annu. Rev. Biochem. 81, 661–686 10.1146/annurev-biochem-060910-094416 [DOI] [PubMed] [Google Scholar]
- 2. Weinberg J., and Drubin D. G. (2012) Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol. 22, 1–13 10.1016/j.tcb.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krause M., and Gautreau A. (2014) Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 15, 577–590 10.1038/nrm3861 [DOI] [PubMed] [Google Scholar]
- 4. Ridley A. J. (2011) Life at the leading edge. Cell 145, 1012–1022 10.1016/j.cell.2011.06.010 [DOI] [PubMed] [Google Scholar]
- 5. Pollard T. D., and Cooper J. A. (2009) Actin, a central player in cell shape and movement. Science 326, 1208–1212 10.1126/science.1175862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lehtimäki J., Hakala M., and Lappalainen P. (2017) Actin filament structures in migrating cells. Handb. Exp. Pharmacol. 235, 123–152 10.1007/164_2016_28 [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto M., Hilgemann D. H., Feng S., Bito H., Ishihara H., Shibasaki Y., and Yin H. L. (2001) Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J. Cell Biol. 152, 867–876 10.1083/jcb.152.5.867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raucher D., and Sheetz M. P. (2000) Cell spreading and lamellipodial extension rate is regulated by membrane tension. J. Cell Biol. 148, 127–136 10.1083/jcb.148.1.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rozelle A. L., Machesky L. M., Yamamoto M., Driessens M. H., Insall R. H., Roth M. G., Luby-Phelps K., Marriott G., Hall A., and Yin H. L. (2000) Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr. Biol. 10, 311–320 10.1016/S0960-9822(00)00384-5 [DOI] [PubMed] [Google Scholar]
- 10. Cunningham C. C., Vegners R., Bucki R., Funaki M., Korde N., Hartwig J. H., Stossel T. P., and Janmey P. A. (2001) Cell permeant polyphosphoinositide-binding peptides that block cell motility and actin assembly. J. Biol. Chem. 276, 43390–43399 10.1074/jbc.M105289200 [DOI] [PubMed] [Google Scholar]
- 11. Saarikangas J., Zhao H., and Lappalainen P. (2010) Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 90, 259–289 10.1152/physrev.00036.2009 [DOI] [PubMed] [Google Scholar]
- 12. Yonezawa N., Nishida E., Iida K., Yahara I., and Sakai H. (1990) Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J. Biol. Chem. 265, 8382–8386 [PubMed] [Google Scholar]
- 13. Janmey P. A., and Stossel T. P. (1987) Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 325, 362–364 10.1038/325362a0 [DOI] [PubMed] [Google Scholar]
- 14. Ramalingam N., Zhao H., Breitsprecher D., Lappalainen P., Faix J., and Schleicher M. (2010) Phospholipids regulate localization and activity of mDia1 formin. Eur. J. Cell Biol. 89, 723–732 10.1016/j.ejcb.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 15. Gorelik R., Yang C., Kameswaran V., Dominguez R., and Svitkina T. (2011) Mechanisms of plasma membrane targeting of formin mDia2 through its amino terminal domains. Mol. Biol. Cell. 22, 189–201 10.1091/mbc.E10-03-0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prehoda K. E., Scott J. A., Mullins R. D., and Lim W. A. (2000) Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science 290, 801–806 10.1126/science.290.5492.801 [DOI] [PubMed] [Google Scholar]
- 17. Rohatgi R., Ho H. Y., and Kirschner M. W. (2000) Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 150, 1299–1310 10.1083/jcb.150.6.1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yonezawa N., Homma Y., Yahara I., Sakai H., and Nishida E. (1991) A short sequence responsible for both phosphoinositide binding and actin binding activities of cofilin. J. Biol. Chem. 266, 17218–17221 [PubMed] [Google Scholar]
- 19. Zhao H., Hakala M., and Lappalainen P. (2010) ADF/cofilin binds phosphoinositides in a multivalent manner to act as a PIP2-density sensor. Biophys. J. 98, 2327–2336 10.1016/j.bpj.2010.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim K., McCully M. E., Bhattacharya N., Butler B., Sept D., and Cooper J. A. (2007) Structure/function analysis of the interaction of phosphatidylinositol 4,5-bisphosphate with actin-capping protein: implications for how capping protein binds the actin filament. J. Biol. Chem. 282, 5871–5879 10.1074/jbc.M609850200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Senju Y., Kalimeri M., Koskela E. V., Somerharju P., Zhao H., Vattulainen I., and Lappalainen P. (2017) Mechanistic principles underlying regulation of the actin cytoskeleton by phosphoinositides. Proc. Natl. Acad. Sci. U.S.A. 114, E8977–E8986 10.1073/pnas.1705032114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poukkula M., Kremneva E., Serlachius M., and Lappalainen P. (2011) Actin-depolymerizing factor homology domain: a conserved fold performing diverse roles in cytoskeletal dynamics. Cytoskeleton 68, 471–490 10.1002/cm.20530 [DOI] [PubMed] [Google Scholar]
- 23. Goode B. L., Drubin D. G., and Lappalainen P. (1998) Regulation of the cortical actin cytoskeleton in budding yeast by twinfilin, a ubiquitous actin monomer-sequestering protein. J. Cell Biol. 142, 723–733 10.1083/jcb.142.3.723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wahlström G., Vartiainen M., Yamamoto L., Mattila P. K., Lappalainen P., and Heino T. I. (2001) Twinfilin is required for actin-dependent developmental processes in Drosophila. J. Cell Biol. 155, 787–796 10.1083/jcb.200108022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vartiainen M. K., Sarkkinen E. M., Matilainen T., Salminen M., and Lappalainen P. (2003) Mammals have two twinfilin isoforms whose subcellular localizations and tissue distributions are differentially regulated. J. Biol. Chem. 278, 34347–34355 10.1074/jbc.M303642200 [DOI] [PubMed] [Google Scholar]
- 26. Meacham C. E., Ho E. E., Dubrovsky E., Gertler F. B., and Hemann M. T. (2009) In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nat. Genet. 41, 1133–1137 10.1038/ng.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang D., Zhang L., Zhao G., Wahlström G., Heino T. I., Chen J., and Zhang Y. Q. (2010) Drosophila twinfilin is required for cell migration and synaptic endocytosis. J. Cell Sci. 123, 1546–1556 10.1242/jcs.060251 [DOI] [PubMed] [Google Scholar]
- 28. Pelkmans L., Fava E., Grabner H., Hannus M., Habermann B., Krausz E., and Zerial M. (2005) Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436, 78–86 10.1038/nature03571 [DOI] [PubMed] [Google Scholar]
- 29. Helfer E., Nevalainen E. M., Naumanen P., Romero S., Didry D., Pantaloni D., Lappalainen P., and Carlier M.-F. (2006) Mammalian twinfilin sequesters ADP-G-actin and caps filament barbed ends: implications in motility. EMBO J. 25, 1184–1195 10.1038/sj.emboj.7601019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bockhorn J., Dalton R., Nwachukwu C., Huang S., Prat A., Yee K., Chang Y.-F., Huo D., Wen Y., Swanson K. E., Qiu T., Lu J., Park S. Y., Dolan M. E., Perou C. M., et al. (2013) MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat. Commun. 4, 1393 10.1038/ncomms2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peng A. W., Belyantseva I. A., Hsu P. D., Friedman T. B., and Heller S. (2009) Twinfilin-2 regulates actin filament lengths in cochlear stereocilia. J. Neurosci. 29, 15083–15088 10.1523/JNEUROSCI.2782-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stritt S., Beck S., Becker I. C., Vögtle T., Hakala M., Heinze K. G., Du X., Bender M., Braun A., Lappalainen P., and Nieswandt B. (2017) Twinfilin 2a is a regulator of platelet reactivity and turnover in mice. Blood 130, 1746–1756 10.1182/blood-2017-02-770768 [DOI] [PubMed] [Google Scholar]
- 33. Vartiainen M., Ojala P. J., Auvinen P., Peränen J., and Lappalainen P. (2000) Mouse A6/twinfilin is an actin monomer-binding protein that localizes to the regions of rapid actin dynamics. Mol. Cell. Biol. 20, 1772–1783 10.1128/MCB.20.5.1772-1783.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ojala P. J., Paavilainen V. O., Vartiainen M. K., Tuma R., Weeds A. G., and Lappalainen P. (2002) The two ADF-H domains of twinfilin play functionally distinct roles in interactions with actin monomers. Mol. Biol. Cell. 13, 3811–3821 10.1091/mbc.E02-03-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paavilainen V. O., Hellman M., Helfer E., Bovellan M., Annila A., Carlier M.-F., Permi P., and Lappalainen P. (2007) Structural basis and evolutionary origin of actin filament capping by twinfilin. Proc. Natl. Acad. Sci. U.S.A. 104, 3113–3118 10.1073/pnas.0608725104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nevalainen E. M., Skwarek-Maruszewska A., Braun A., Moser M., and Lappalainen P. (2009) Two biochemically distinct and tissue-specific twinfilin isoforms are generated from the mouse Twf2 gene by alternative promoter usage. Biochem. J 417, 593–600 10.1042/BJ20080608 [DOI] [PubMed] [Google Scholar]
- 37. Johnston A. B., Collins A., and Goode B. L. (2015) High-speed depolymerization at actin filament ends jointly catalysed by twinfilin and Srv2/CAP. Nat. Cell Biol. 17, 1504–1511 10.1038/ncb3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Palmgren S., Ojala P. J., Wear M. A., Cooper J. A., and Lappalainen P. (2001) Interactions with PIP2, ADP-actin monomers, and capping protein regulate the activity and localization of yeast twinfilin. J. Cell Biol. 155, 251–260 10.1083/jcb.200106157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Falck S., Paavilainen V. O., Wear M. A., Grossmann J. G., Cooper J. A., and Lappalainen P. (2004) Biological role and structural mechanism of twinfilin-capping protein interaction. EMBO J. 23, 3010–3019 10.1038/sj.emboj.7600310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Papayannopoulos V., Co C., Prehoda K. E., Snapper S., Taunton J., and Lim W. A. (2005) A polybasic motif allows N-WASP to act as a sensor of PIP2 density. Mol. Cell 17, 181–191 10.1016/j.molcel.2004.11.054 [DOI] [PubMed] [Google Scholar]
- 41. Zhao H., and Lappalainen P. (2012) A simple guide to biochemical approaches for analyzing protein-lipid interactions. Mol. Biol. Cell. 23, 2823–2830 10.1091/mbc.E11-07-0645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saarikangas J., Zhao H., Pykäläinen A., Laurinmäki P., Mattila P. K., Kinnunen P. K., Butcher S. J., and Lappalainen P. (2009) Molecular mechanisms of membrane deformation by I-BAR domain proteins. Curr. Biol. 19, 95–107 10.1016/j.cub.2008.12.029 [DOI] [PubMed] [Google Scholar]
- 43. Paavilainen V. O., Merckel M. C., Falck S., Ojala P. J., Pohl E., Wilmanns M., and Lappalainen P. (2002) Structural conservation between the actin monomer-binding sites of twinfilin and actin-depolymerizing factor (ADF)/cofilin. J. Biol. Chem. 277, 43089–43095 10.1074/jbc.M208225200 [DOI] [PubMed] [Google Scholar]
- 44. Paavilainen V. O., Oksanen E., Goldman A., and Lappalainen P. (2008) Structure of the actin-depolymerizing factor homology domain in complex with actin. J. Cell Biol. 182, 51–59 10.1083/jcb.200803100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gorbatyuk V. Y., Nosworthy N. J., Robson S. A., Bains N. P., Maciejewski M. W., dos Remedios C. G., and King G. F. (2006) Mapping the phosphoinositide-binding site on chick cofilin explains how PIP2 regulates the cofilin-actin interaction. Mol. Cell 24, 511–522 10.1016/j.molcel.2006.10.007 [DOI] [PubMed] [Google Scholar]
- 46. Goroncy A. K., Koshiba S., Tochio N., Tomizawa T., Sato M., Inoue M., Watanabe S., Hayashizaki Y., Tanaka A., Kigawa T., and Yokoyama S. (2009) NMR solution structures of actin depolymerizing factor homology domains. Protein Sci. 18, 2384–2392 10.1002/pro.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wienken C. J., Baaske P., Rothbauer U., Braun D., and Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100 10.1038/ncomms1093 [DOI] [PubMed] [Google Scholar]
- 48. Jerabek-Willemsen M., André T., Wanner R., Roth H. M., Duhr S., Baaske P., and Breitsprecher D. (2014) Microscale thermophoresis: interaction analysis and beyond. J. Mol. Struct. 1077, 101–113 10.1016/j.molstruc.2014.03.009 [DOI] [Google Scholar]
- 49. Scheuermann T. H., Padrick S. B., Gardner K. H., and Brautigam C. A. (2016) On the acquisition and analysis of microscale thermophoresis data. Anal. Biochem. 496, 79–93 10.1016/j.ab.2015.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pleskot R., Pejchar P., Žárský V., Staiger C. J., and Potocký M. (2012) Structural insights into the inhibition of actin-capping protein by interactions with phosphatidic acid and phosphatidylinositol (4,5)-bisphosphate. PLoS Comput. Biol. 8, e1002765 10.1371/journal.pcbi.1002765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Balla T. (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137 10.1152/physrev.00028.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schink K. O., Tan K.-W., and Stenmark H. (2016) Phosphoinositides in control of membrane dynamics. Annu. Rev. Cell Dev. Biol. 32, 143–171 10.1146/annurev-cellbio-111315-125349 [DOI] [PubMed] [Google Scholar]
- 53. Di Paolo G., and De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- 54. van Rheenen J., Song X., van Roosmalen W., Cammer M., Chen X., Desmarais V., Yip S.-C., Backer J. M., Eddy R. J., and Condeelis J. S. (2007) EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J. Cell Biol. 179, 1247–1259 10.1083/jcb.200706206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dharmalingam E., Haeckel A., Pinyol R., Schwintzer L., Koch D., Kessels M. M., and Qualmann B. (2009) F-BAR proteins of the syndapin family shape the plasma membrane and are crucial for neuromorphogenesis. J. Neurosci. 29, 13315–13327 10.1523/JNEUROSCI.3973-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pykäläinen A., Boczkowska M., Zhao H., Saarikangas J., Rebowski G., Jansen M., Hakanen J., Koskela E. V., Peränen J., Vihinen H., Jokitalo E., Salminen M., Ikonen E., Dominguez R., and Lappalainen P. (2011) Pinkbar is an epithelial-specific BAR domain protein that generates planar membrane structures. Nat. Struct. Mol. Biol. 18, 902–907 10.1038/nsmb.2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Casella J. F., Maack D. J., and Lin S. (1986) Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. J. Biol. Chem. 261, 10915–10921 [PubMed] [Google Scholar]
- 58. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 10.1021/ct300400x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 10.1021/jp101759q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. B 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 61. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 10.1002/jcc.20945 [DOI] [PubMed] [Google Scholar]
- 62. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindah E. (2015) Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- 63. Nosé S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 10.1063/1.447334 [DOI] [Google Scholar]
- 64. Melchionna S., Ciccotti G., and Holian B. L. (1993) Hoover NPT dynamics for systems varying in shape and size. Mol. Phys. 78, 533–544 10.1080/00268979300100371 [DOI] [Google Scholar]
- 65. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 10.1063/1.328693 [DOI] [Google Scholar]
- 66. Hess B., Bekker H., Berendsen H. J., and Fraaije J. G. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO%3B2-H [DOI] [Google Scholar]
- 67. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 68. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.