

Abstract
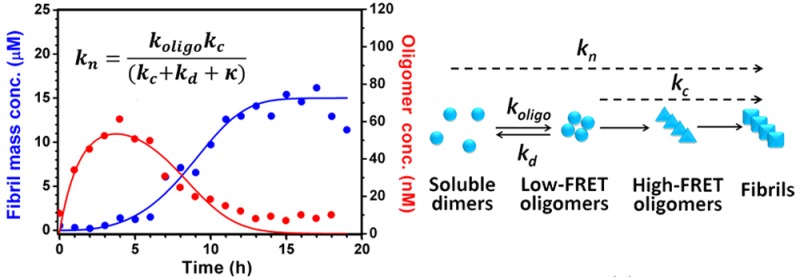
The self-assembly of polypeptides into amyloid structures is associated with a range of increasingly prevalent neurodegenerative diseases as well as with a select set of functional processes in biology. The phenomenon of self-assembly results in species with dramatically different sizes, from small oligomers to large fibrils; however, the kinetic relationship between these species is challenging to characterize. In the case of prion aggregates, these structures can self-replicate and act as infectious agents. Here we use single molecule spectroscopy to obtain quantitative information on the oligomer populations formed during aggregation of the yeast prion protein Ure2. Global analysis of the aggregation kinetics reveals the molecular mechanism underlying oligomer formation and depletion. Quantitative characterization indicates that the majority of Ure2 oligomers are relatively short-lived, and their rate of dissociation is much higher than their rate of conversion into growing fibrils. We identify an initial metastable oligomer, which can subsequently convert into a structurally distinct oligomer, which in turn converts into growing fibrils. We also show that fragmentation is responsible for the autocatalytic self-replication of Ure2 fibrils, but that preformed fibrils do not promote oligomer formation, indicating that secondary nucleation of the type observed for peptides and proteins associated with neurodegenerative disease does not occur at a significant rate for Ure2. These results establish a framework for elucidating the temporal and causal relationship between oligomers and larger fibrillar species in amyloid forming systems, and provide insights into why functional amyloid systems are not toxic to their host organisms.
Introduction
The self-assembly of soluble proteins into insoluble and highly structured amyloid fibrils rich in β-sheet structure is associated with a variety of human disorders, including Alzheimer’s and Parkinson’s diseases, type II diabetes, and the prion diseases.1,2 In addition, the formation of amyloid fibrils has been found to be a common or generic property of polypeptide molecules, and also to be associated with a number of diverse biological functions in living organisms.1,3 Over the past decade, oligomeric intermediates that form during the early stages of amyloid fibril formation or dissociate from mature fibrils have become of increasing interest4 because such species, rather than the fibrils, have been shown to be toxic to cells and are now thought to be the major pathogenic agent in neurodegenerative disease.1,2,5−7 Understanding the nature and dynamics of the oligomers is not only of intrinsic interest, but has the potential to provide a catalyst for the development of therapeutic strategies for protein misfolding diseases.2,8,9
Conventional biochemical and biophysical methods, such as the thioflavin T (ThT) fluorescence assay,10 circular dichroism spectroscopy (CD),11 electron microscopy (EM)12 and atomic force microscopy (AFM)13 measurements, are able to provide ensemble information about the aggregation kinetics of amyloidogenic proteins as well as the conformation and morphology of amyloid fibrils and the intermediate prefibrillar species populated during their formation. The metastable and heterogeneous nature of protein oligomers, however, limits the detailed characterization of such prefibrillar species. Recently, a variety of novel approaches have been developed and applied for this purpose. Fluorescence correlation spectroscopy, for example, provides information about the nature and distribution of species formed in the early stages of fibril formation.14,15 Additionally, mass spectrometry and NMR spectroscopy have been applied to detect and analyze the properties of oligomers with a range of different structures.16−18 Single molecule fluorescence spectroscopy, however, offers a particularly powerful approach for exploring the formation and properties of oligomers, as it has the ability to investigate individual molecular species and to reveal conformational dynamics that may be averaged in the ensemble experiment.19,20 These techniques have been shown to be able to identify and characterize the low-populated, heterogeneous and transient species formed during fibril assembly of several amyloidogenic proteins.21−25
Ure2 from Saccharomyces cerevisiae is the protein determinant of the yeast prion state [URE3]26 and provides an important system for probing amyloid formation and prion propagation. The Ure2 protein is a dimer in solution; each monomer contains 354 amino acids and consists of two domains.27,28 The unstructured and flexible N-terminal domain (residues 1–93) is primarily responsible for prion conversion and propagation in vivo and for formation of amyloid fibrils in vitro.29−31 The globular C-terminal domain (residues 94–354) is similar to glutathione transferases in structure32,33 and has both glutathione-dependent peroxidase activity34 and glutaredoxin activity.35 Ure2 is also a negative regulatory factor of nitrogen metabolism, as in its native state the protein interacts with the transcription factor Gln3 and represses the uptake of poor nitrogen sources.36 Therefore, when Ure2 converts into the aggregated prion state, Gln3 is released and activates the expression of the genes related to the metabolism of less favorable nitrogen sources.
While the aggregated prion form of Ure2 is tolerated by yeast cells, precursor aggregates of Ure2 are toxic to mammalian cells,37,38 as are amyloid aggregates of other proteins.39 It is therefore interesting to study the oligomers formed and the structural changes occurring during Ure2 fibril formation, and to compare them to disease-related models. In a previous study we identified by AFM prefibrillar intermediates of Ure2 with a range of different sizes formed during the aggregation process.31 A soluble oligomeric species formed during the early stages of Ure2 aggregation was separated and characterized by biochemical and spectroscopic methods.40 Taken together, these results suggest a connection between the population of oligomeric species and the course of Ure2 amyloid assembly into mature fibrils. Theoretical modeling has previously enabled the kinetic parameters that describe the growth and breakage of Ure2 fibrils to be defined, allowing the contribution of individual molecular steps to be correlated with prion propensity;41 however, data on the oligomeric populations of Ure2, which would allow this type of mechanistic analysis to be carried out, have not previously been available. In the present study, we have applied single molecule fluorescence resonance energy transfer (smFRET) to investigate in detail the intermolecular assembly and aggregation process of Ure2. This approach has enabled oligomerization during the initial lag phase to be observed, and two types of Ure2 oligomers with different assembly modes have been identified. Furthermore, using theoretical analysis combined with single molecule and ensemble kinetic data, we describe the formation and depletion pathway of oligomers, and propose a multistep mechanism for Ure2 fibril formation, in which initial oligomerization is followed by conformational conversion to β-sheet-containing oligomers that are then able to grow to form mature amyloid fibrils.
Methods
Mutant Construction, Protein Expression, Purification and Labeling
The single point cysteine variants (V9C, S53C and S68C) of Ure2 were obtained by overlapping PCR using a synthetic wild-type URE2 gene as template42 and ligated into the mini-pRSETa vector. All the mutants constructed in this study were confirmed by DNA sequencing. Full-length Ure2 was expressed in E. coli C41(DE3) cells with a His6-tag and purified by nickel-affinity chromatography as described previously.42,43 Purified Ure2 was dialyzed into 50 mM Tris–HCl (pH 8.4) buffer containing 200 mM NaCl and 500 μM TCEP at 4 °C. Protein purity was checked by SDS-PAGE, and the protein concentration was determined by the absorbance at 280 nm for full-length Ure2 using a molar extinction coefficient of 48,220 M–1 cm–1.42 The details of fluorescence labeling of Ure2 are described in the Supporting Information (SI) Methods.
Single-Molecule FRET Measurement of Ure2 Oligomers
SmFRET experiments were carried out using a home-built confocal microscope or total internal reflection fluorescence (TIRF) microscope based on a Nikon Ti-E inverted microscope similar to that described previously.44 The details of the instrumentation, experimental procedures and data analysis used for confocal smFRET and TIRF smFRET are described in the SI Methods.
Developing a Model for Kinetic Data Fitting
We set out here to develop a quantitative model that describes the experimental observables: the total fibril mass concentration M(t) and the total oligomer concentration O(t). In addition to M(t) and O(t), the model explicitly considers the concentration of native state dimeric Ure2 m(t), and of fibrils P(t). In particular, the model describes explicitly the formation of oligomers through dimer association with rate constant koligo, their conversion to fibrils with rate constant kc, their destruction (rate constant kd), and fibril growth and fragmentation (rate constants k+ and k–, respectively). Addition of further complexity to this coarse-grained model is in principle readily possible within the master equation formalism; such additional details, including differentiating between multiple structural classes within the oligomer subpopulations, would, however, require further experimental constraints than are currently available, in order to avoid overfitting.45 We used the Ure2 dimer concentration for m(t) rather than the monomer concentration, as evidence suggests Ure2 remains in its dimeric form throughout the aggregation reaction (see Results).
The rate equations for the model can be written as a master equation:
| 1 |
| 2 |
| 3 |
| 4 |
where we have left out terms with negligible contributions to the overall kinetics, such as the effects of nonelongation steps on monomer depletion.46 Any larger oligomers are expected to form from growth of smaller oligomers; all oligomers ultimately grow from the initial interaction of a pair of dimeric Ure2 molecules. The physically reasonable choice of overall reaction order for oligomer formation is therefore 2.0. (For further explanation, see SI Methods.)
These equations were solved for early times in the aggregation process, and the solutions used to derive a first-order self-consistent expression for M(t) (see also SI Methods):
| 5 |
with A = αk+kc/κ2(kl + κ), kl = kc + kd, 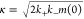 and
α = koligom(0)2.
and
α = koligom(0)2.
The time-dependent evolution of the oligomer population depends only on koligo, kl and m(t). In turn, m(t) depends only on κ and A, or κ, kl, and αk+kc. Overall therefore, the dynamics of the dimer and oligomer populations depend on the following four combinations of rate parameters: k+k–, k+kc, kl, and koligo. Moreover the fitted values of koligo and kl are approximately independent of the values chosen for k+k– and k+kc, provided that these two parameter combinations give a reasonable fit to the fibril mass concentration.
Relating Model Rate Constants to Fundamental Reaction Steps
Experiments indicate that we can resolve the observed oligomers into two structurally distinct populations, with a low-FRET oligomer formed initially and subsequently converting into a high-FRET oligomer, which in turn converts to fibrils. The data are not, however, sufficiently detailed to allow a full kinetic analysis to be carried out on both populations individually. The avoidance of overfitting necessitates, therefore, that we consider together the different structural classes of oligomers, and examine the overall fluxes that lead to their generation or depletion. We can, however, incorporate elements of our knowledge of the oligomer subpopulations in the overall interpretation of the results. We did so by determining how each species contributes to the total oligomer formation, depletion and conversion rate constants in our coarse-grained model. The concentration of the later high-FRET oligomer species changes very little over the time course of the aggregation reaction compared to that of the earlier low-FRET species, and is present at significantly lower concentrations than the earlier low-FRET species over the times most relevant to the fitting procedure. Moreover, we show below that high-FRET oligomers are likely to be formed from conversion of low-FRET oligomers. Therefore, the rate constants for total oligomer formation and dissociation obtained from the fitting process can be interpreted as approximately the rate constants for low-FRET oligomer formation and dissociation. The rate constant kc gives the approximate proportionality between the overall oligomer concentration and the rate of formation of fibrils from oligomers, and so contains information on both the conversion of low-FRET to high-FRET oligomers, as well as the conversion of high-FRET oligomers to fibrils. The concentration data on high-FRET oligomers indicated that the steady-state approximation is likely to be valid here, in which case we can explicitly write kc in terms of the rate constants of a more detailed kinetic model featuring two separate oligomeric species (see SI Methods). The “conversion” rate constant would then be proportional to the rate constant for transformation of low-FRET oligomers to high-FRET oligomers, as well as to the rate constant for conversion of high-FRET oligomers to fibrillar species.
Fitting the Combined smFRET/ThT Data to the Model
The ThT component of the data was fitted globally to the analytical expression for the full time-course fibril concentration to obtain values for A and κ using the online fitting platform Amylofit.47 Then, the smFRET component of the data was fitted to our early time expression for O(t) using Mathematica to give approximate values for koligo and kl. Having established that kd ≫ kc, we can set kd = kl. Combining these conclusions with A and κ yields approximate values for k+k– and k+kc. The availability of these approximate rate constants as trial parameters enabled a numerical fit of our combined smFRET/ThT data to eqs 1–4 to be carried out, yielding robust rate constants and verifying the consistency of our model with the experimental data. For the fitting of the combined smFRET/ThT data, a ratio of 1.5:1 was chosen for k+k–(S68C):k+k–(V9C) (see Results). For a full description of the kinetic model and fitting methods, see SI Methods.
Results
Single Molecule FRET Measurements Can Monitor the Formation of Ure2 Oligomers
Since Ure2 has no intrinsic cysteine residues, single cysteine mutations were introduced to specific sites within the Ure2 N-terminal region to allow covalent linkages to maleimide-functionalized dyes. In order to avoid perturbing the process of Ure2 fibril formation, we chose mutation sites that are located near the turn of the β-strand in the structural models of the Ure2 amyloid fibril core.48 The residues selected were V9C, S53C and S68C, of which V9C and S53C are near the N-terminal and C-terminal ends of the fibril core (Figure 1A), respectively, and should be sensitive reporters of the conformational changes associated with aggregation. The formation of fibrils by these derivatives was then monitored using ThT fluorescence. The results showed that both Ure2 S53C and S68C formed fibrils at essentially the same rate as that of wild-type Ure2, while V9C formed fibrils more slowly, as reflected in a longer lag time than the wild type (Figure S1A). A possible reason for this change is that V9 is located in one of the regions of the prion domain that have been shown to be important for formation of Ure2 amyloid31 and may indeed participate in the formation of the first β-strand in Ure2 fibrils;49 the source of this difference in rate is addressed further in the kinetic analysis below. For the case of the dye-labeled proteins, ThT assays could not be used because of potential FRET effects between ThT and the labeled fluorophores; the kinetics of fibril formation by the labeled proteins were therefore monitored using turbidity measured at OD400, which indicates that fluorescence labeling does not significantly perturb the fibril formation rate of the mutants (Figure S1B). Furthermore, the aggregates formed by these unlabeled and labeled Ure2 proteins were imaged using transmission electron microscopy (TEM) and each showed a similar fibrillar morphology to that of wild-type Ure2 fibrils (Figure S1C).
Figure 1.
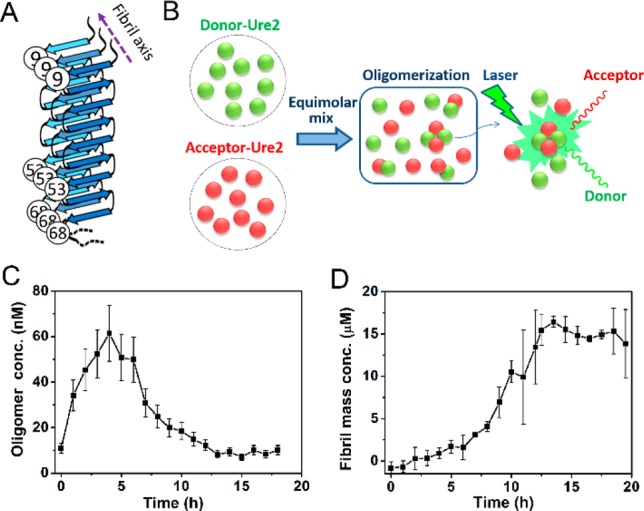
Oligomerization of Ure2 monitored by confocal single molecule FRET. (A) Schematic figure to indicate the cysteine mutations and fluorescence labeling sites that were used in this study, based on a previously suggested structural model of Ure2 fibrils.48 (B) Scheme for smFRET detection of Ure2 oligomers. (C) The concentration of AF555/AF647 labeled Ure2-S68C oligomers throughout the aggregation reaction. (D) Ensemble kinetics of the aggregation of 15 μM (dimeric concentration) unlabeled Ure2-S68C monitored by ThT fluorescence. All the aggregation reactions were carried out at 18 °C in an Innova 4230 incubator with shaking at 150 rpm in 50 mM Tris–HCl (pH 8.4) buffer containing 200 mM NaCl.
To check the efficiency of fibril formation, we also measured the fraction of labeled protein in the supernatant and the pellet by SDS-PAGE after the aggregation reaction reached a plateau, and most of the soluble protein was found to have converted into the insoluble fibrillar form, with less than 10% of the protein remaining in the supernatant (Figure S1D). Fluorophore attachment on the sites selected here does not, therefore, alter the ability of Ure2 to form fibrils or the nature of the fibrillar products. The self-assembly process of the fluorophore-labeled Ure2, as monitored by single molecule fluorescence experiments, can therefore be considered to be the same as that of unlabeled Ure2. Note that since Ure2 is a highly stable dimer in solution,42,50 and the dimeric unit of Ure2 seldom dissociates during fibril formation (Figure S2); we therefore refer to the dimer concentration throughout this study, unless otherwise stated.
Next we used smFRET, which has been used in studies of other amyloidogenic proteins22−24 to detect the oligomeric species formed during the Ure2 fibril formation process. The soluble oligomer formed between AF555- and AF647-labeled Ure2 gave well-defined FRET signals and could be detected at the single molecule level by monitoring coincident bursts in both donor and acceptor channels when the oligomers diffused across the diffraction-limited focus (Figure 1B). The numbers of selected FRET events were converted to oligomer concentrations (see SI Methods) and plotted against time (Figure 1C). Meanwhile, the progress of fibril formation of the unlabeled Ure2 in bulk solution was monitored by the ThT fluorescence assay under the same incubation conditions (Figure 1D). As shown in the smFRET data (Figure 1C), the number of oligomers rises to its highest level within 5 h, which occurs during the lag phase of the ensemble ThT curve (Figure 1D), reflecting the assembly and increase in the number of oligomers. After 5 h, the soluble oligomeric species were observed to decrease in concentration as the Ure2 becomes sequestered into mature fibrils that are not detected by the confocal smFRET technique. The decrease in the number of oligomers corresponds well with the onset of fibril formation reflected in the bulk ThT assay under the same conditions (Figure 1D).
We assessed the stability of the oligomers formed at different time points by taking an aliquot from the aggregation reaction and diluting it into buffer; we then monitored whether or not there was any decrease in the smFRET burst rate corresponding to the detection of individual oligomers during a 1 h window immediately after dilution. Coverslips were pretreated with unlabeled protein to avoid adsorption of the labeled sample onto the surface. We observed by TIRF imaging that pretreatment of the surface with either BSA or unlabeled Ure2 was equally effective at suppressing the adsorption of the fluorescently labeled sample (Figure S3A). However, when the diluted sample was loaded onto BSA-coated coverslips, oligomer dissociation was evident by confocal smFRET within 1 h (Figure S3B). In contrast, when we used coverslips pretreated with unlabeled Ure2, the oligomers remained at a constant level during the measurement time (Figure S3B–F), a finding that can be rationalized by the desorption of unlabeled protein from the surface into solution, thus stabilizing the oligomers. Therefore, in order to avoid underestimating oligomer concentrations, we used coverslips coated with unlabeled Ure2 when performing smFRET experiments.
These results demonstrate the power of single molecule techniques for the observation of low populations of oligomers. It is evident that aggregates of Ure2 form during the lag phase observed by ThT fluorescence, in agreement with theoretical modeling.51
SmFRET Measurements Reveal the Absence of Significant Oligomer Formation via Secondary Nucleation During Ure2 Fibril Formation
The two generic mechanisms that lead to the formation of fibrils are the primary nucleation pathway, during which new oligomers are generated by the direct association of soluble protein or peptide molecules, and secondary pathways, where existing fibrils have the propensity to generate the formation of new fibrils, either through fragmentation or through surface catalyzed secondary nucleation. In the latter case, nucleation of new fibrils takes place on, and is catalyzed by, the surface of existing fibrils,46 the rate of which therefore depends on the mass concentration of existing fibrils. In the case of Aβ42 aggregation,52 whose kinetics are dominated by the surface-catalyzed secondary nucleation pathway, it has been shown explicitly that the majority of small oligomers present during the reaction are produced during secondary nucleation.52 In earlier studies of Ure2, we have shown that fragmentation events are important determinants of the rate of fibril formation under both quiescent and shaking conditions41,53,54 although the production of oligomers through surface-catalyzed secondary nucleation in the case of Ure2 has not previously been discounted.
We measured the generation of Ure2 oligomers directly using smFRET in the presence of 1% preformed mature fibrils to provide a surface for oligomer formation if secondary nucleation were to occur at a significant rate for Ure2. The time course of oligomerization observed by smFRET in the presence of the added fibrils shows a similar initial rate to that of the unseeded system, and the quantity of oligomers detected at each time point is not increased, indicating that the rate of oligomer production during secondary nucleation is insignificant compared to the rate of direct association of dimers to form oligomers during primary nucleation. This was confirmed by explicit fitting to kinetic models featuring oligomer formation during primary nucleation (Figure 2A) and during secondary nucleation (Figure 2B). The former yields a good fit; the latter a poor fit, a result that is very different from the findings for Aβ4252 (Figure 2C). The quantity of Ure2 oligomers under seeded conditions appears to be lower than in the absence of preformed fibrils, which is likely to be a result of the rapid depletion of native Ure2 by association with, and elongation of, the pre-existing fibril ends. Together with the kinetic analysis of the ensemble fibril formation of Ure2 (SI Methods and Figure S4), this observation confirms that the proliferation of Ure2 fibrils results from fragmentation and not from secondary nucleation.
Figure 2.

Absence of a fibril-catalyzed secondary nucleation process for Ure2. (A, B) Ensemble aggregation kinetics of 15 μM unlabeled Ure2-S68C monitored by ThT fluorescence under unseeded (blue) or seeded (red) conditions (upper panels). Ure2 oligomers were then detected under unseeded (blue) or seeded (red) conditions by confocal single molecule FRET (lower panels). The incubation conditions were the same as in Figure 1. (A) The data fit well to a model that generates oligomers during primary nucleation. (B) A model that generates oligomers during secondary nucleation cannot fit the data. (C) The presence of seeds (right-hand columns) drastically increases the concentration of Aβ42 oligomers measured at a single time point in the lag phase of an Aβ42 aggregation experiment52 (right panel), but do not increase the production of Ure2 oligomers in this study (left panel), indicating fundamentally different mechanisms of oligomer formation for these two systems.
Analysis of Oligomer Populations Reveals the Existence of an Oligomer Conformational Conversion Step
The results so far have established that Ure2 oligomers are formed predominantly from the free association of dimers during primary nucleation. We can demonstrate using the following simple argument that only a minority of these oligomers ultimately become fibrils, and that both an oligomer dissociation pathway and a conformational conversion step are needed. Taking a conservative estimate of the initial oligomer formation rate of 20 nM/h (Figure 1C), and noting that minimal native Ure2 depletion occurs over the first 4 h of aggregation, a concentration of oligomers of at least 80 nM will be formed in the first 4 h. The rate of oligomer formation declines subsequently, but does not cease until all Ure2 is depleted from solution at ∼10–12 h; we therefore estimate the lower bound on the total concentration of oligomers that form during primary nucleation to be 100 nM. Mature Ure2 fibrils are observed to have lengths typically greater than 100 nm, and mostly on the micrometer scale. Given that the interchain distance within an amyloid fibril is ca. 0.5 nm, fibrils must typically contain at least 100 Ure2 dimers. In the smFRET experiments the concentration of Ure2 dimers incorporated into fibrils is approximately 15 μM, so the final concentration of fibrils is at most 150 nM. Ure2 has also been shown to follow fragmentation-dominant kinetics, so fibril formation through primary nucleation is insignificant compared to the total formation of fibrils through fragmentation. The total concentration of fibrils formed through primary nucleation must therefore be at least an order of magnitude less than the total concentration of fibrils formed, and so is significantly less than 15 nM. This value is far lower than the concentration of oligomers formed during primary nucleation observed in our smFRET experiments, and leads us to conclude that most oligomers must dissociate rather than elongate. Given that oligomers undergo faster dissociation than the fibrils, the oligomers must be structurally distinct from fibrils. In the following two sections we demonstrate that new fibrils are likely to originate from structural conversion of these oligomers, although this must occur much more slowly than dissociation. This result was confirmed by comparing fitted values for the oligomer conversion and dissociation rate constants (see kinetic analysis below).
Two Types of Oligomeric Species with Different Structures Can Be Observed
To investigate directly the possibility of structurally distinct subpopulations of Ure2 oligomers, potentially related by additional conformational conversion steps, we performed an smFRET efficiency distribution analysis. Here we used S53C rather than S68C Ure2, because both these variants show similar fibril formation kinetics to the wild-type protein, but the S53 residue is closer to the fibril core than is S68 and is thus expected to be more sensitive to structural changes within the oligomers. The experiment was carried out in a similar manner to that described above except that AF488 instead of AF555 was used to label Ure2 in order to reduce cross talk between the donor and acceptor fluorophore in the smFRET experiments, allowing detection of population distributions not apparent when using the AF555 dye as donor. We calculated the FRET efficiency of selected oligomers at different time points during fibril formation to obtain a FRET efficiency distribution histogram (Figure 3). At the early stages within the lag time, for example after 1 h of aggregation in the case of S53C, only one broad, low FRET efficiency distribution with a maximum of 0.40 was observed. As the aggregation reaction progressed, but before significant fibril mass had formed, an additional higher FRET efficiency distribution with a maximum of 0.68 appeared, suggesting these later oligomers contain a more compact assembly of Ure2 molecules than the initial low-FRET oligomers (Figure 3A). Despite the slow kinetics of fibril formation, the existence of two different types of oligomers was also observed for the V9C mutant (Figure 3B), where the low and high FRET distributions peaked at 0.46 and 0.66, respectively. High-FRET oligomers appear after low-FRET oligomers but before fibrils, suggesting that they are formed by conversion of the low-FRET oligomers. Additionally, the lack of an increase in the later high-FRET oligomer population as the fibrils start to form indicates that there is no significant production of surface-catalyzed secondary nuclei (Figure 3C,D). It should be noted that FRET efficiency is not only related to the distance between the donor and acceptor but also to their dipole orientation. The similar change in the FRET efficiency when dyes were introduced at different sites (i.e., S53C and V9C) indicates that the signal reflects principally the change in the average distances between dyes, and not the change of orientation of dyes, as there is little probability of the latter occurring simultaneously at two different residue sites.
Figure 3.
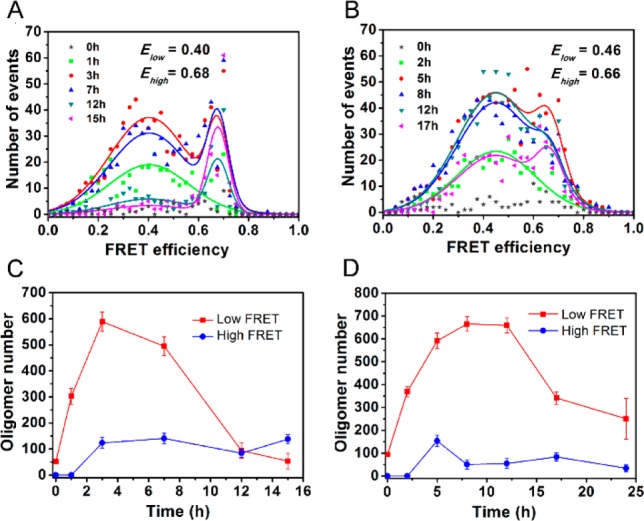
Different types of Ure2 oligomers revealed by confocal single molecule FRET. (A,B) SmFRET efficiency distribution of selected oligomers of AF488/AF647-labeled Ure2 at different incubation times. The FRET distributions at different time points were fitted globally to double Gaussian functions giving the average peak positions indicated. (C,D) Population of low- and high-FRET oligomers of Ure2 at different incubation times. The incubation conditions were the same as in Figure 1. (A,C) Ure2-S53C. (B,D) Ure2-V9C.
An interesting finding in our smFRET study is the observation of a lower FRET efficiency for the initially formed oligomers for S53C than for V9C but a similar FRET efficiency of the late phase oligomers for the two variants. This observation provides clues as to the structures of the two types of oligomers. Since the V9 residue is located in the hydrophobic region (residues 9–21) of the N-terminal domain and the S53 residue is in the Q/N rich region (residues 44 to 80), the closer intermolecular distances within the early stage oligomers of V9C than S53C strongly suggests that the initial intermolecular oligomerization involves hydrophobic interactions. After the reorganization of the initial oligomers to β-sheet-containing oligomers, both V9 and S53 sites would be included in the amyloid structure and should have similar interchain distances between the same residues aligned along the fibril axis, thus explaining the similarity of the intermolecular FRET efficiency of the late phase oligomers (the high-FRET species) of the two mutants.
To obtain further evidence to support the coexistence and interconversion of two types of oligomers, we carried out a dot blot assay using the conformation-specific antibodies A11 and OC that have been used previously for identification of specific types of oligomers.55,56 We first probed the two types of oligomers formed during aggregation of full-length Ure2, but the signals obtained in the experiments were very weak, probably caused by the blocking effect of the globular C-terminal domain surrounding the N-terminal prion domain of Ure2. Therefore, we used the N-terminal prion domain fragment (residues 1–93) to perform the dot blot assay (see SI Methods). The results (Figure S5) show that two types of oligomers exist during aggregation of the Ure2 prion domain and that the concentration of relatively disordered oligomers (A11 reactive species) reaches a maximum earlier than the β-sheet rich oligomers (OC reactive species), indicating a possible conversion from the former to the latter, consistent with our conclusions based on the smFRET results. Taken together, these results indicate that the two oligomeric intermediates have distinct structural properties, consistent with the existence of a conformational conversion step between the earlier and later appearing oligomers.
Ure2 Oligomers That Disaggregate from Mature Fibrils Have Structures Similar to Those of the Oligomers Appearing Later in the Aggregation Reaction
We next probed by smFRET the structures of the Ure2 species dissociated from fibrils. The mature fibrils formed from an equimolar mixture of AF488-Ure2 and AF647-Ure2 were incubated in fresh buffer for at least 1 h before detection by confocal smFRET, under the same conditions as for the aggregation reaction. Upon incubation, the oligomers dissociating from fibrils reached a concentration of 0.1–0.2% of the total Ure2 fibril mass concentration, the same proportion as observed in the combined smFRET/ThT data set at the end of the aggregation reaction. These oligomers showed a broad FRET distribution, mainly between 0.2 and 0.8, and could not be precisely fitted by a Gaussian function due to the extremely low occurrence of such species. In order to detect large or insoluble species, which may not be observed during the confocal smFRET experiment, we applied TIRF with smFRET to probe the dissociated oligomers in order to increase the detection efficiency for larger species (see SI Methods). In addition, in order to obtain a higher number of disaggregated oligomers, we sonicated the AF488/AF647 fibrils before carrying out the smFRET measurements. The resulting mixture of oligomers and small fibrils in TIRF images showed coincident signals in both AF488 and AF647 channels, and the FRET distribution of the samples of Ure2 S53C and V9C could be fitted by double Gaussian functions (Figure 4). Two major populations centered at around 0.64 and around 0.8 were observed, the lower of which was similar to the high-FRET distribution observed during the aggregation reaction (Figure 3), and can be attributed to disaggregated oligomers. The species showing higher FRET values, with a maximum at around 0.8, can be attributed to small fibrils, indicating the more compact structure within amyloid fibrils. This observation is remarkably similar to that in previous studies of α-synuclein.22 The results of TIRF experiments, therefore, demonstrate that the oligomers that disaggregate from Ure2 fibrils have the same structural properties as the oligomeric species formed in the later stages of the aggregation reaction. This finding strongly suggests that the high-FRET oligomers contain β-sheet structure similar to that found in mature amyloid fibrils, and that this type of oligomeric species is able to convert to elongation-competent fibril-type species.
Figure 4.
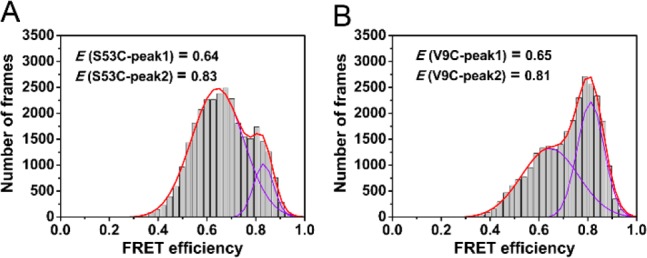
Single molecule TIRF measurements of disaggregated fibrils. (A,B) SmFRET distribution histogram of Ure2 oligomers disaggregated from AF488/AF647-labeled fibrils. Data were fitted to a double Gaussian function (continuous line) to obtain the FRET values of the two species. (A) Ure2-S53C. (B) Ure2-V9C.
Kinetic Analysis of Combined smFRET and ThT Data Yields a Quantitative Understanding of Oligomer Formation, Dissociation and Conversion
Analysis of the kinetics of the aggregation reaction is a crucial step in understanding the microscopic mechanism of amyloid formation. Previous theoretical work has provided an analytical solution to the kinetics of fibril formation involving fragmentation,57 and by globally fitting the ThT curves over a range of concentrations58 to this expression, two combined kinetic parameters, knk+ and k+k– can be obtained, where kn, k+ and k– represent the amyloid nucleation, elongation and fragmentation rates, respectively. To study how the mutations affect these rates, and thus to gain further structural and mechanistic insight into the nucleation process, we performed smFRET experiments to compare the oligomer formation of the two Ure2 mutants, V9C and S68C.
Previous kinetic modeling of amyloid aggregation used a single coarse-grained reaction step to represent the “primary nucleation” pathway by which new fibrils are generated via an initial association step. Here, the availability of accurate kinetic data on the total concentration of oligomeric intermediates allows us to devise a less coarse-grained kinetic model that explicitly includes intermediates in the nucleation step. The model remains partly coarse-grained, however, as it makes no distinction between different oligomer types; nevertheless, it provides additional insights into the nature of the nucleation process. In this model, oligomers are formed through an initial assembly process, occurring with rate constant koligo, and subsequently convert into growth-competent fibril-type species with a rate constant kc. These species can then elongate by dimer addition with rate constant k+, and fragment with rate constant k–. The oligomers can also dissociate with rate constant kd (that we have shown above is much larger than kc). Note that we can approximately interpret oligomerization and dissociation as fundamental reaction steps; however, the conversion step is in fact a coarse-grained step that contains information on the transformation of low-FRET to high-FRET oligomers, as well as on the subsequent conversion of the latter species to fibrils (see Methods and SI for full details). The accurate determination of reaction orders with respect to dimers requires kinetic data for a range of initial dimer concentrations. Given just one initial dimer concentration, however, we can make the reasonable assumption of a reaction order of 0 for conversion and 2 for oligomer formation. Any inaccuracy in these reaction orders is effectively incorporated into our definitions of kc and koligo, and does not significantly affect the quality of the fitting (see SI Methods).
An accurate analytical solution for the time dependence of the fibril mass concentration in our model can be derived by extension of previous approaches.57,59 The solution is identical to the analytical solution for the kinetics of a fragmenting system, upon which the kinetic analysis of the bulk ThT experiments in the SI is based, except that the fibril nucleation rate knm(0)nc is resolved in terms of the microscopic processes introduced in our oligomer model. Specifically, we obtain
| 6 |
where 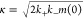 and α = koligom(0)nc. This
result, combined with an analysis of the equation governing oligomer
kinetics, reveals that the kinetics of this system are controlled
by the parameter combinations koligo, kd, k+kc, and k+k–. The combined rate parameters k+kc and k+k– can be determined with order-of-magnitude accuracy.
and α = koligom(0)nc. This
result, combined with an analysis of the equation governing oligomer
kinetics, reveals that the kinetics of this system are controlled
by the parameter combinations koligo, kd, k+kc, and k+k–. The combined rate parameters k+kc and k+k– can be determined with order-of-magnitude accuracy.
A numerical procedure was used to fit the combined smFRET and bulk ThT data to this model (see Methods for further details). The numerical fits are reasonable given the accuracy of the data, and show that our coarse-grained model provides a good description of the system (Figure 5). The minor divergence between the fitted curve and the data for S68C oligomers at the latest times is consistent with the fact that disaggregation from fibrils yields a small population of oligomers at equilibrium, yet to avoid overfitting there is no explicit fibril disaggregation step in the model. Fitting gives a value for koligo of 1.6 × 10–3 μM–1 h–1 for both variants (Figure 5A,B), and a value of kd of 0.60 h–1 for S68C, and 0.45 h–1 for V9C. This result indicates that the kinetics of oligomer formation by S68C and V9C are broadly similar, but that the S68C oligomers dissociate more readily by a factor of approximately 1.3. Given that the rates of oligomer formation are very similar, it is likely that the reaction order of oligomerization is approximately the same in the two mutants at this concentration range.
Figure 5.
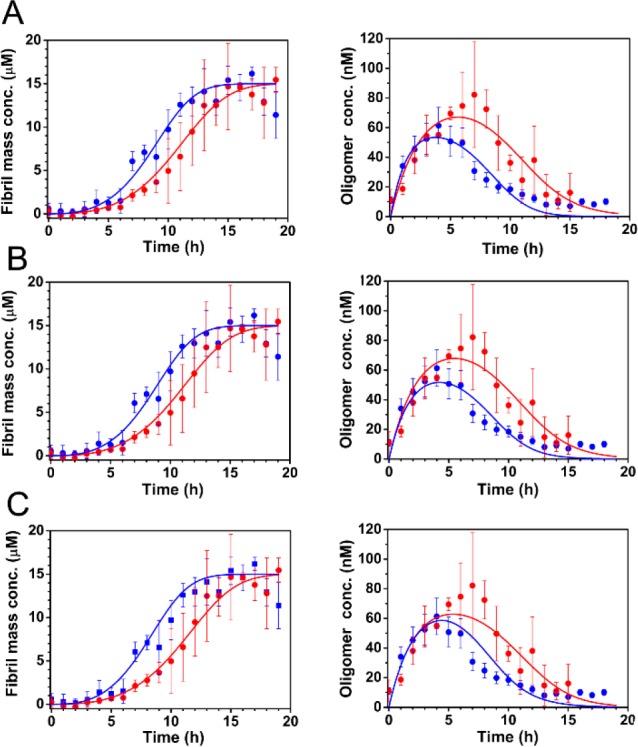
Fitting of combined smFRET/ThT data to models indicates a probable effect of mutations on the dissociation of oligomers, but not on their formation. (A–C) The bulk aggregation kinetics of 15 μM unlabeled Ure2-V9C (red) and Ure2-S68C (blue) monitored by ThT fluorescence (left panels) and the concentration of AF555/AF647 labeled Ure2-V9C (red) and Ure2-S68C (blue) oligomers throughout the aggregation reaction monitored by confocal smFRET (right panels) were globally fitted to a theoretical model (see Methods) including the formation, dissociation, and conversion of oligomers, and the elongation and fragmentation of fibrils. The incubation conditions were the same as in Figure 1. (A) Allowing both koligo and kd to differ for each mutant gives good fits, with a mean squared error of 1.54. (B) If koligo is constrained to be the same for both mutants, the model fits the data equally well, with a mean squared error of 1.58. (C) If neither koligo nor kd is allowed to differ, the fit is less good, especially around the time when the oligomer concentration is at a maximum, with a mean squared error of 1.87. This result therefore implies that koligo is the same for the two variants, while the values of kd may differ slightly.
The differences in k+kc and k+k– between the V9C and S68C variants are less than an order of magnitude, indicating that these parameters are similar for both variants. An order-of-magnitude estimate for k+ of 40 μM–1 h–1 was obtained from analysis of the seeded aggregation experiment monitored by ThT fluorescence (see SI Methods and Figure S7 for details). This value allows us to calculate order-of-magnitude estimates for k– and kc, of 1 × 10–4 h–1 and 2 × 10–3 h–1 respectively. We note that kc is 2 orders of magnitude smaller than kd, as expected from our analysis of oligomer dissociation. To estimate the differences in these parameters between each variant, we used a ratio k+(S68C):k+(V9C) of 1.5:1, as indicated by seeded bulk experiments (see SI Methods and Figure S7), and a ratio k–(S68C):k–(V9C) of 1:1 as indicated by fragmentation rate measurements (see SI Methods and Figure S8). This series of steps then allows us to calculate a ratio kc(S68C):kc(V9C) of 1.4:1. Although this value is rather sensitive both to experimental error and to errors in the parameter ratios that are used, it can be interpreted as demonstrating that the conversion rates are similar, although possibly somewhat larger in S68C. Taken together, these results indicate that the V9C mutation decreases the fibril elongation rate and oligomer dissociation rate, leaving the fibril fragmentation rate and oligomer formation rate unaffected. Furthermore, they suggest that the oligomer conversion rate may also be slightly decreased by the V9C mutation.
Discussion
We have applied single molecule FRET measurements to investigate the aggregation behavior of the yeast prion protein Ure2 and to observe the low populations of transient oligomers formed during the aggregation reaction that are challenging to detect by ensemble methods. The single molecule FRET observations indicate that the majority of the oligomers formed during the initial step of the nucleation process dissociate back to the native dimeric state, but a small population of oligomers undergoes a conformational conversion step leading to formation of elongation-competent species. Quantitative analysis of a combination of bulk and single molecule data has provided detailed information about the rates of the microscopic kinetic steps in the formation of amyloid fibrils, and how these rates are altered by point mutations in the prion domain. Based on our observations, a mechanism has been proposed for the formation of Ure2 amyloid fibrils in which native Ure2 forms relatively disordered oligomers, probably driven by hydrophobic intermolecular interactions, only a small proportion of which then rearrange to form structurally more compact β-sheet containing oligomers that are able to convert further to elongation-competent fibrillar species and grow by addition of native dimers to form mature amyloid fibrils (Figure 6).
Figure 6.
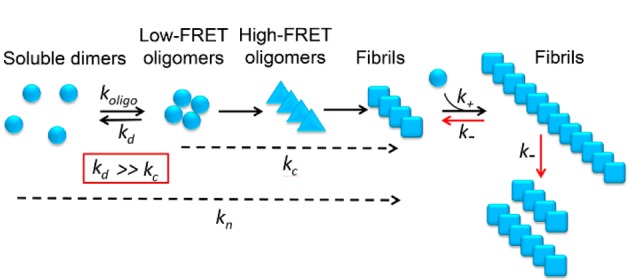
Proposed model for the aggregation pathway of Ure2. Native dimeric Ure2 forms relatively disordered oligomers driven by hydrophobic interactions and either dissociates back to the native state or undergoes conformational conversion to form more compact oligomers containing β-sheet structure, which can in turn convert into growth-competent fibrillar species. Fragmentation of fibrils then contributes to their proliferation.
Primary nucleation of amyloidogenic proteins, in which a native protein converts into an elongation-competent species, is a crucial step in fibril formation. Molecular simulations of the aggregation of Aβ42 indicate that primary nucleation occurs via intermediate disordered non-β oligomers, which not only facilitates encounters between the monomeric proteins but also provides an environment that facilitates their conversion to fibrillar β-structure.60,61 Nonspecific intermolecular interactions, such as hydrophobic interactions, play a crucial role in the formation of the initial disordered oligomers,60 while the intra- and intermolecular hydrogen bonding within β-sheets is considered to be the driving force for the subsequent conformational conversion.62 This formation of hydrogen bonds compensates for the disruption of the hydrophobic interactions in the initial disordered oligomers, thus favoring conformational reorganization to β-sheet structure in order to reach the lowest energy state. The conformational reorganization between early relatively disordered oligomers and β-rich elongation-competent species during amyloid formation has also been suggested by experimental studies of other amyloidogenic proteins such as α-synuclein,22 Aβ40,63 and the yeast prion protein Sup35.64 In agreement with theoretical and experimental results for other amyloid proteins, both theoretical analysis of oligomer concentrations and direct observation of two types of Ure2 oligomers possessing different assembly and emergence times (Figure 3) provide additional evidence for the oligomerization/conversion model as a generic feature of amyloid nucleation.
In order to obtain a quantitative understanding of the fundamental reaction steps that contribute to the amyloid formation process, analytical methods have been used to describe fibril growth and to obtain the microscopic kinetic parameters.46,57 Since the advent of single molecule techniques, it has become feasible to probe the nucleation process directly in real time throughout the aggregation reaction.22−25 However, the kinetic analysis in previous studies has either focused on the early stage of oligomer formation22,25 or made use of a highly coarse-grained nucleated polymerization model in which secondary processes were not considered and in which the fitted rate constants were difficult to relate to specific reaction steps.24 In this study, we provide a kinetic analysis of the combined single molecule FRET and ensemble ThT data (Figure 5 and Figure 2) that considers not only the oligomer formation process but also the elongation and fragmentation processes, providing quantitative information about the complete aggregation pathway of an amyloid protein. Another advance in the present study is the determination of an expression for the bulk primary nucleation rate kn, which is found to depend not only on the rate constants of oligomer formation, dissociation and conversion, but also on the rate constants for fibril elongation and fragmentation. Global analysis of the oligomer formation kinetics, measured by smFRET, and the fibril formation kinetics, monitored by the ensemble ThT assay, results in two independent parameters koligo and kd and two combined parameters kck+ and k+k−, where the latter two can be decomposed by direct measurement of k+ or of k–. Thus, the theoretical analysis of the smFRET data provides detailed quantitative information about the fundamental steps in the process of amyloid nucleation.
Our study also provides new structural insights into the aggregation mechanism of Ure2. The disordered N-terminal domain consists of a hydrophobic region (residues 15–42) and two Q/N repeat regions (residues 1–14 and 43–89).31 In a previous study, we have demonstrated that deletion of residues 1–42 eliminates the ability of Ure2 to form fibrils at measurable rates, while deletion of either residues 1–14 or residues 15–42 increases the lag time significantly,31 indicating the importance of this region in the aggregation reaction. We observed that the V9C mutation causes an increase in the length of the lag phase for fibril formation compared with that of wild-type Ure2. Kinetic analysis of the bulk ThT variable-concentration data under both unseeded and cross-seeded conditions reveals that this effect is predominantly due to a decrease in the elongation rate caused by the V9C mutation. We then investigated the effect of the mutation on the nucleation process in detail by kinetic analysis of the combined smFRET and bulk ThT data. The rates of both oligomer dissociation and oligomer conversion are slightly decreased in the V9C mutant relative to WT Ure2, while the rate of oligomer formation is unaffected, indicating that V9C forms a more stable oligomer. However, the V9C mutation has a negligible effect on the overall rate of formation of elongation-competent oligomers (kn). Taken together, these results show that the V9 residue plays an important role in the elongation of Ure2 fibrils, consistent with a recent structural study using site-directed spin labeling and electron paramagnetic resonance (EPR), which demonstrated that the residues 8–12 form the first β-strand in the fibrils.49
The toxicity of amyloid oligomers has been demonstrated to be correlated with the degree of exposure of hydrophobic surface.65,66 Since oligomers assembled by functional amyloidogenic proteins, such as Ure2 in this study, and Sup35 in a previous study,64 have similar conformational features (for example, reactivity with A11 and OC antibodies) and a similar formation and conversion pathway to that of toxic amyloidogenic proteins such as α-synuclein22 and Aβ,60,63 the difference between functional and disease-related amyloid remains to be elucidated. In this study, we observed a gradual decrease in the concentration of oligomers accompanying the onset of ThT fluorescence. Calculations and theoretical analysis of the smFRET data has led to the conclusion that the majority of Ure2 oligomers formed do not ultimately become fibrils but are depleted by dissociation. In contrast, the populations of Aβ40 and α-synuclein oligomers have been observed to follow a single exponential process and to remain at a significant equilibrium concentration even after all the monomers are depleted.22,23,25 A possible explanation for the accumulation of Aβ40 and α-synuclein oligomers is that they convert very slowly relative to their formation, and become kinetically trapped once monomers are depleted, as the residual population of oligomers can no longer grow through monomer addition into fibrils. Another possible explanation for the difference is that the fibrils of Aβ40 and α-synuclein may not be as stable as the Ure2 fibrils, such that the equilibrium between oligomers and fibrils favors oligomers to a much greater extent than is the case with Ure2. In either of the above cases, Aβ40 and α-synuclein oligomers should have higher stability relative to the native state than Ure2 oligomers, and therefore would not be expected to dissociate significantly. The accumulation of relatively high concentrations of oligomers formed by disease-related amyloidogenic proteins could therefore be the cause of their toxicity.
A further potential reason for the higher toxicity of disease-related amyloidogenic proteins is that oligomers are generated not only during primary nucleation but also during secondary nucleation. For the case of Ure2, we have been able to demonstrate directly the lack of formation of surface-catalyzed secondary nuclei using smFRET measurements (Figure 2). Several structural studies indicate that the N-terminal domain of Ure2 forms an in-register parallel β-sheet structure with its C-terminal domain decorating the fibril core.67,68 Compared with other amyloidogenic proteins which show monomer-dependent secondary nucleation, such as Aβ42,52 α-synuclein69 and IAPP,70 the surface of Ure2 fibrils is likely to be blocked by the presence of its C-terminal globular domain, thus restricting the access of native Ure2 to the fibrillar structure and inhibiting the ability of the pre-existing aggregates to catalyze the nucleation of the native protein. The absence of surface-catalyzed secondary nucleation will greatly reduce the generation of oligomers,52 which is another possible reason for the lower toxicity of functional amyloids. Taken together, the low stability of Ure2 oligomers and the absence of secondary nucleation suggests that the functional yeast prions may replicate and propagate by fragmentation rather than secondary nucleation and hence avoid significant populations of potentially toxic oligomers, as occurs in the aggregation of neurodegenerative disease-related proteins. This study paves the way for the detailed study and comparison of further examples of both disease-related and functional amyloid systems, particularly the variety of amyloidogenic proteins that play structural and functional roles in bacteria.71,72 The understanding gained from such studies will not only shed light on the mechanisms by which amyloid structures are harnessed for functional roles, but may also provide clues as to possible new therapeutic strategies to combat amyloid disease.
Acknowledgments
We thank Dr. Xun Li (Nikon Instruments Co. Ltd. Beijing Branch) and Dr. Zeyong Zhi (Beijing Coolight Technology) for helpful discussions about single molecule instrumentation. We thank David Klenerman and George Meisl (University of Cambridge) for helpful discussions regarding data analysis. Our electron microscopy work was performed at the Center for Biological Imaging (Institute of Biophysics, Chinese Academy of Sciences).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10439.
Methods, Figures, Tables (PDF)
Author Contributions
¶ These authors contributed equally.
The Perrett group acknowledges support from the Chinese Ministry of Science and Technology [2017YFA0504000], the National Natural Science Foundation of China [31110103914, 21673278, 31300631], the National Laboratory of Biomacromolecules, and the CAS Center of Excellence in Biomacromolecules. T.P.J.K. acknowledges support from the Engineering and Physical Sciences Research Council, the Biotechnology and Biological Sciences Research Council, the European Research Council and the Frances and Augustus Newman Foundation. A.J.D. was supported by the Schiff Foundation. T.C.T.M. was supported by Peterhouse and St John’s College, Cambridge; and the Swiss National Science Foundation. C.M.D. and T.P.J.K. acknowledge support from the Cambridge Centre for Misfolding Diseases.
The authors declare no competing financial interest.
Supplementary Material
References
- Chiti F.; Dobson C. M. Annu. Rev. Biochem. 2017, 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Knowles T. P.; Vendruscolo M.; Dobson C. M. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Fowler D. M.; Koulov A. V.; Balch W. E.; Kelly J. W. Trends Biochem. Sci. 2007, 32, 217–224. 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Bemporad F.; Chiti F. Chem. Biol. 2012, 19, 315–327. 10.1016/j.chembiol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Cleary J. P.; Walsh D. M.; Hofmeister J. J.; Shankar G. M.; Kuskowski M. A.; Selkoe D. J.; Ashe K. H. Nat. Neurosci. 2005, 8, 79–84. 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Winner B.; Jappelli R.; Maji S. K.; Desplats P. A.; Boyer L.; Aigner S.; Hetzer C.; Loher T.; Vilar M.; Campioni S.; Tzitzilonis C.; Soragni A.; Jessberger S.; Mira H.; Consiglio A.; Pham E.; Masliah E.; Gage F. H.; Riek R. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 4194–4199. 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Munoz M. J.; Castillo-Carranza D. L.; Krishnamurthy S.; Paulucci-Holthauzen A. A.; Sengupta U.; Lasagna-Reeves C. A.; Ahmad Y.; Jackson G. R.; Kayed R. Neurobiol. Dis. 2014, 71, 14–23. 10.1016/j.nbd.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Cheng B.; Gong H.; Xiao H.; Petersen R. B.; Zheng L.; Huang K. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 4860–4871. 10.1016/j.bbagen.2013.06.029. [DOI] [PubMed] [Google Scholar]
- Guerrero-Munoz M. J.; Castillo-Carranza D. L.; Kayed R. Biochem. Pharmacol. 2014, 88, 468–478. 10.1016/j.bcp.2013.12.023. [DOI] [PubMed] [Google Scholar]
- LeVine H. 3rd Methods Enzymol. 1999, 309, 274–284. 10.1016/S0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Bertucci C.; Bolognesi M. L.; Cavalli A.; Melchiorre C.; Andrisano V. ChemBioChem 2007, 8, 2152–2161. 10.1002/cbic.200700427. [DOI] [PubMed] [Google Scholar]
- Newcomb C. J.; Moyer T. J.; Lee S. S.; Stupp S. I. Curr. Opin. Colloid Interface Sci. 2012, 17, 350–359. 10.1016/j.cocis.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. F.; Knowles T. P.; Dobson C. M.; Macphee C. E.; Welland M. E. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15806–15811. 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath S.; Meuvis J.; Hendrix J.; Carl S. A.; Engelborghs Y. Biophys. J. 2010, 98, 1302–1311. 10.1016/j.bpj.2009.12.4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes J. M.; Casares S.; Ruedas-Rama M. J.; Fernandez E.; Castello F.; Varela L.; Orte A. Int. J. Mol. Sci. 2012, 13, 9400–9418. 10.3390/ijms13089400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulla N.; Zhou M.; Arimon M.; Gairi M.; Giralt E.; Robinson C. V.; Dobson C. M. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7828–7833. 10.1073/pnas.0812227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paslawski W.; Mysling S.; Thomsen K.; Jorgensen T. J.; Otzen D. E. Angew. Chem., Int. Ed. 2014, 53, 7560–7563. 10.1002/anie.201400491. [DOI] [PubMed] [Google Scholar]
- Fusco G.; Chen S. W.; Williamson P. T. F.; Cascella R.; Perni M.; Jarvis J. A.; Cecchi C.; Vendruscolo M.; Chiti F.; Cremades N.; Ying L.; Dobson C. M.; De Simone A. Science 2017, 358, 1440–1443. 10.1126/science.aan6160. [DOI] [PubMed] [Google Scholar]
- Banerjee P. R.; Deniz A. A. Chem. Soc. Rev. 2014, 43, 1172–1188. 10.1039/C3CS60311C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler B.; Hofmann H. Curr. Opin. Struct. Biol. 2013, 23, 36–47. 10.1016/j.sbi.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Orte A.; Birkett N. R.; Clarke R. W.; Devlin G. L.; Dobson C. M.; Klenerman D. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 14424–14429. 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremades N.; Cohen S. I.; Deas E.; Abramov A. Y.; Chen A. Y.; Orte A.; Sandal M.; Clarke R. W.; Dunne P.; Aprile F. A.; Bertoncini C. W.; Wood N. W.; Knowles T. P.; Dobson C. M.; Klenerman D. Cell 2012, 149, 1048–1059. 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P.; Orte A.; Clarke R. W.; Bolognesi B.; Hook S.; Ganzinger K. A.; Meehan S.; Wilson M. R.; Dobson C. M.; Klenerman D. Nat. Struct. Mol. Biol. 2012, 19, 79–83. 10.1038/nsmb.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas S. L.; Garcia G. A.; Kumar S.; Kjaergaard M.; Horrocks M. H.; Shivji N.; Mandelkow E.; Knowles T. P.; Mandelkow E.; Klenerman D. Nat. Commun. 2015, 6, 7025. 10.1038/ncomms8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iljina M.; Garcia G. A.; Horrocks M. H.; Tosatto L.; Choi M. L.; Ganzinger K. A.; Abramov A. Y.; Gandhi S.; Wood N. W.; Cremades N.; Dobson C. M.; Knowles T. P.; Klenerman D. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E1206–1215. 10.1073/pnas.1524128113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner R. B. Science 1994, 264, 566–569. 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- Lian H. Y.; Jiang Y.; Zhang H.; Jones G. W.; Perrett S. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 535–545. 10.1016/j.bbapap.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Thual C.; Komar A. A.; Bousset L.; Fernandez-Bellot E.; Cullin C.; Melki R. J. Biol. Chem. 1999, 274, 13666–13674. 10.1074/jbc.274.19.13666. [DOI] [PubMed] [Google Scholar]
- Taylor K. L.; Cheng N.; Williams R. W.; Steven A. C.; Wickner R. B. Science 1999, 283, 1339–1343. 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- Masison D. C.; Wickner R. B. Science 1995, 270, 93–95. 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Li H.; Zhu L.; Zhou J. M.; Perrett S. J. Biol. Chem. 2004, 279, 3361–3369. 10.1074/jbc.M310494200. [DOI] [PubMed] [Google Scholar]
- Bousset L.; Belrhali H.; Janin J.; Melki R.; Morera S. Structure 2001, 9, 39–46. 10.1016/S0969-2126(00)00553-0. [DOI] [PubMed] [Google Scholar]
- Umland T. C.; Taylor K. L.; Rhee S.; Wickner R. B.; Davies D. R. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 1459–1464. 10.1073/pnas.98.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai M.; Zhou J. M.; Perrett S. J. Biol. Chem. 2004, 279, 50025–50030. 10.1074/jbc.M406612200. [DOI] [PubMed] [Google Scholar]
- Zhang Z. R.; Perrett S. J. Biol. Chem. 2009, 284, 14058–14067. 10.1074/jbc.M901189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinder D.; Coschigano P. W.; Magasanik B. J. Bacteriol. 1996, 178, 4734–4736. 10.1128/jb.178.15.4734-4736.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieri L.; Bucciantini M.; Nosi D.; Formigli L.; Savistchenko J.; Melki R.; Stefani M. J. Biol. Chem. 2006, 281, 15337–15344. 10.1074/jbc.M511647200. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Jackson A. P.; Zhang Z. R.; Han Y.; Yu S.; He R. Q.; Perrett S. PLoS One 2010, 5, e12529 10.1371/journal.pone.0012529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciantini M.; Giannoni E.; Chiti F.; Baroni F.; Formigli L.; Zurdo J.; Taddei N.; Ramponi G.; Dobson C. M.; Stefani M. Nature 2002, 416, 507–511. 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- Catharino S.; Buchner J.; Walter S. Biol. Chem. 2005, 386, 633–641. 10.1515/BC.2005.074. [DOI] [PubMed] [Google Scholar]
- Wang Y. Q.; Buell A. K.; Wang X. Y.; Welland M. E.; Dobson C. M.; Knowles T. P.; Perrett S. J. Biol. Chem. 2011, 286, 12101–12107. 10.1074/jbc.M110.208934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrett S.; Freeman S. J.; Butler P. J.; Fersht A. R. J. Mol. Biol. 1999, 290, 331–345. 10.1006/jmbi.1999.2872. [DOI] [PubMed] [Google Scholar]
- Fei L.; Perrett S. J. Biol. Chem. 2009, 284, 11134–11141. 10.1074/jbc.M809673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Ge X.; Lv Z.; Zhi Z.; Chang Z.; Zhao X. S. Biochem. J. 2011, 438, 505–511. 10.1042/BJ20110264. [DOI] [PubMed] [Google Scholar]
- Burnham K. P.; Anderson D. R.. Model selection and multimodel inference: a practical information-theoretic approach; Springer: New York, 2003. [Google Scholar]
- Cohen S. I.; Vendruscolo M.; Welland M. E.; Dobson C. M.; Terentjev E. M.; Knowles T. P. J. Chem. Phys. 2011, 135, 065105. 10.1063/1.3608916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisl G.; Kirkegaard J. B.; Arosio P.; Michaels T. C.; Vendruscolo M.; Dobson C. M.; Linse S.; Knowles T. P. Nat. Protoc. 2016, 11, 252–272. 10.1038/nprot.2016.010. [DOI] [PubMed] [Google Scholar]
- Kajava A. V.; Baxa U.; Wickner R. B.; Steven A. C. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7885–7890. 10.1073/pnas.0402427101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo S.; Chiang V.; Guo Z. J. Struct. Biol. 2012, 180, 374–381. 10.1016/j.jsb.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Galani D.; Fersht A. R.; Perrett S. J. Mol. Biol. 2002, 315, 213–227. 10.1006/jmbi.2001.5234. [DOI] [PubMed] [Google Scholar]
- Arosio P.; Knowles T. P.; Linse S. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. 10.1039/C4CP05563B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9758–9763. 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Chen L. J.; Wang H. Y.; Wang Y. Q.; Perrett S. Protein Eng., Des. Sel. 2011, 24, 69–78. 10.1093/protein/gzq100. [DOI] [PubMed] [Google Scholar]
- Xu L. Q.; Wu S.; Buell A. K.; Cohen S. I.; Chen L. J.; Hu W. H.; Cusack S. A.; Itzhaki L. S.; Zhang H.; Knowles T. P.; Dobson C. M.; Welland M. E.; Jones G. W.; Perrett S. Philos. Trans. R. Soc., B 2013, 368, 20110410. 10.1098/rstb.2011.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe C. G. J. Biol. Chem. 2008, 283, 29639–29643. 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. Science 2003, 300, 486–489. 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Knowles T. P.; Waudby C. A.; Devlin G. L.; Cohen S. I.; Aguzzi A.; Vendruscolo M.; Terentjev E. M.; Welland M. E.; Dobson C. M. Science 2009, 326, 1533–1537. 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- Cohen S. I.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. Mol. Biol. 2012, 421, 160–171. 10.1016/j.jmb.2012.02.031. [DOI] [PubMed] [Google Scholar]
- Garcia G. A.; Cohen S. I.; Dobson C. M.; Knowles T. P. Phys. Rev. E Stat Nonlin Soft Matter Phys. 2014, 89, 032712. 10.1103/PhysRevE.89.032712. [DOI] [PubMed] [Google Scholar]
- Saric A.; Chebaro Y. C.; Knowles T. P.; Frenkel D. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 17869–17874. 10.1073/pnas.1410159111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saric A.; Michaels T. C. T.; Zaccone A.; Knowles T. P. J.; Frenkel D. J. Chem. Phys. 2016, 145, 211926. 10.1063/1.4965040. [DOI] [PubMed] [Google Scholar]
- Auer S.; Meersman F.; Dobson C. M.; Vendruscolo M. PLoS Comput. Biol. 2008, 4, e1000222. 10.1371/journal.pcbi.1000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Culyba E. K.; Powers E. T.; Kelly J. W. Nat. Chem. Biol. 2011, 7, 602–609. 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan R.; Goodman J. L.; Mukhopadhyay S.; Pacheco C. D.; Lemke E. A.; Deniz A. A.; Lindquist S. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11172–11177. 10.1073/pnas.1209527109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B.; Kumita J. R.; Barros T. P.; Esbjorner E. K.; Luheshi L. M.; Crowther D. C.; Wilson M. R.; Dobson C. M.; Favrin G.; Yerbury J. J. ACS Chem. Biol. 2010, 5, 735–740. 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- Campioni S.; Mannini B.; Zampagni M.; Pensalfini A.; Parrini C.; Evangelisti E.; Relini A.; Stefani M.; Dobson C. M.; Cecchi C.; Chiti F. Nat. Chem. Biol. 2010, 6, 140–147. 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- Baxa U.; Wickner R. B.; Steven A. C.; Anderson D. E.; Marekov L. N.; Yau W. M.; Tycko R. Biochemistry 2007, 46, 13149–13162. 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- Kryndushkin D. S.; Wickner R. B.; Tycko R. J. Mol. Biol. 2011, 409, 263–277. 10.1016/j.jmb.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell A. K.; Galvagnion C.; Gaspar R.; Sparr E.; Vendruscolo M.; Knowles T. P.; Linse S.; Dobson C. M. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 7671–7676. 10.1073/pnas.1315346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschak A. M.; Miranker A. D. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12341–12346. 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer E. B.; Claessen D.; Gras S. L.; Perrett S. Biochem. Soc. Trans. 2012, 40, 728–734. 10.1042/BST20120013. [DOI] [PubMed] [Google Scholar]
- Evans M. L.; Chapman M. R. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843, 1551–1558. 10.1016/j.bbamcr.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.