

ABSTRACT
Various microorganisms play key roles in the nitrogen (N) cycle. Quantitative PCR (qPCR) and PCR amplicon sequencing of N cycle functional genes allow us to analyze the abundance and diversity of microbes responsible for N-transforming reactions in various environmental samples. However, analysis of multiple target genes can be cumbersome and expensive. PCR-independent analysis, such as metagenomics and metatranscriptomics, is useful but expensive, especially when we analyze multiple samples and try to detect N cycle functional genes present at a relatively low abundance. Here, we present the application of microfluidic qPCR chip technology to simultaneously quantify and prepare amplicon sequence libraries for multiple N cycle functional genes as well as taxon-specific 16S rRNA gene markers for many samples. This approach, named the nitrogen cycle evaluation (NiCE) chip, was evaluated by using DNA from pure and artificially mixed bacterial cultures and by comparing the results with those obtained by conventional qPCR and amplicon sequencing methods. Quantitative results obtained by the NiCE chip were comparable to those obtained by conventional qPCR. In addition, the NiCE chip was successfully applied to examine the abundance and diversity of N cycle functional genes in wastewater samples. Although nonspecific amplification was detected on the NiCE chip, this can be overcome by optimizing the primer sequences in the future. As the NiCE chip can provide a high-throughput format to quantify and prepare sequence libraries for multiple N cycle functional genes, this tool should advance our ability to explore N cycling in various samples.
IMPORTANCE We report a novel approach, namely, the nitrogen cycle evaluation (NiCE) chip, by using microfluidic qPCR chip technology. By sequencing the amplicons recovered from the NiCE chip, we can assess the diversities of N cycle functional genes. The NiCE chip technology is applicable to analysis of the temporal dynamics of N cycle gene transcription in wastewater treatment bioreactors. The NiCE chip can provide a high-throughput format to quantify and prepare sequence libraries for multiple N cycle functional genes. While there is room for future improvement, this tool should significantly advance our ability to explore the N cycle in various environmental samples.
KEYWORDS: nitrogen cycle, quantitative PCR, microfluidic chip, amplicon sequencing, wastewater treatment
INTRODUCTION
Microorganisms play key roles in the nitrogen (N) cycle (1, 2). For example, nitrifiers oxidize ammonium (NH4+) to nitrite (NO2−) and/or nitrate (NO3−) under oxic conditions (3, 4). Under oxygen-limited conditions, denitrifiers reduce oxidized nitrogenous compounds, such as NO3− and NO2−, to nitric oxide (NO), nitrous oxide (N2O), and dinitrogen gas (N2) (5). N2 gas can also be produced via an anaerobic ammonium oxidation (anammox) reaction, in which NH4+ oxidation and NO2− reduction are coupled under anoxic conditions (6).
Products of these N-transforming reactions have a great impact on ecosystem structure and function. For example, nitrification, denitrification, and anammox reactions have been used to remove N from wastewater (7, 8). In agricultural soils, NO3− and NO2− produced via nitrification can be leached from fields to contaminate groundwater, rivers, lakes, and oceans to cause negative health (e.g., blue-baby syndrome) and ecological (e.g., eutrophication) consequences (9). Nitrification and denitrification can also produce N2O, which is considered a strong greenhouse gas and a significant contributor to the destruction of the ozone layer (10). To mitigate these globally important environmental problems, we need to identify microbes responsible for these N cycle reactions.
Culture-independent approaches such as quantitative PCR (qPCR) and PCR amplicon sequencing are powerful tools to identify, quantify, and analyze the sequence diversity of the genes related to the N cycle (e.g., see references 11–14). Because PCR is a sensitive technique, we can amplify genes present at low concentrations. However, since there are many functional genes for various N cycle reactions (see Fig. S1 in the supplemental material), we need to run many PCRs to quantify and sequence all of these genes. In addition, even when a single functional gene is targeted, several PCR runs may be necessary to cover the diversity of the gene sequences present, since sequences with a few base pair mismatches to the primers may not amplify the target genes. Many researchers have developed primers by incorporating degenerate bases, in an attempt to amplify target genes from diverse taxa; however, it is still difficult to design “universal” primers (e.g., see references 15–17).
PCR-independent genomic approaches, such as metagenomics and metatranscriptomics, are free of PCR bias and therefore are useful for the identification of functional gene sequences (e.g., see references 18 and 19). However, functional genes of interest are present in small proportions relative to the total number of metagenome sequences (20). Due to this obstacle, we may need to execute multiple next-generation sequencing (NGS) runs to obtain functional gene sequences sufficient for statistical analysis. This process can become costly, which can limit the number of samples ultimately sequenced. A microarray-based approach has been successfully applied to detect genes related to biogeochemical cycles, e.g., GeoChip (21–23) and MicroTOOLs (24). However, this process requires >0.5 μg DNA or cDNA to obtain reliable results (23), and reactions need to be done on many replicates, which is often difficult for many environmental samples. Whole-community genome amplification and whole-community RNA amplification have also been applied to increase DNA and RNA amounts, respectively (25, 26); however, amplification bias is still of concern (27). In addition, it is often difficult to distinguish true-positive results from nonspecific background noise due to cross-hybridization to nontarget genes (28). We cannot verify the microarray results by recovering and sequencing the DNA/cDNA fragments hybridized with specific probes; therefore, complementary approaches such as qPCR and NGS are often used in combination with microarray experiments (29).
One promising approach to overcome these issues is the use of a high-throughput microfluidic qPCR platform in which different qPCR assays are run simultaneously in nanoliter-volume chambers present on a chip. The microfluidic qPCR chip system can greatly reduce the amounts of time, labor, and reagent required compared with conventional qPCR systems (12), and the sensitivity, specificity, and quantitative performance of the chip are comparable to those of conventional qPCR (30). In addition, PCR amplicons can be recovered and used for NGS (31, 32).
The objectives of this study are to (i) simultaneously quantify multiple N cycle functional genes (Fig. S1) as well as taxon-specific 16S rRNA gene markers by using microfluidic qPCR chip technology (nitrogen cycle evaluation [NiCE] chip), (ii) identify key microbes contributing to the N cycle by sequencing the PCR amplicons recovered from the NiCE chip (Fig. 1), and (iii) apply the NiCE chip amplicon sequencing approach for analysis of mixed-culture samples and environmental samples.
FIG 1.

Pipeline of the NiCE chip assay. (a) Quantitative PCR is performed by using a 48.48 AccessArray (AA) chip with EvaGreen chemistry. (b) Recovered PCR amplicons are tagged with an Illumina index and adaptors (Illumina) and used for MiSeq 300-bp paired-end sequencing.
RESULTS
Specificity and sensitivity of the PCR assays.
The specificity and sensitivity of the PCR assays used in this study were examined by using the NiCE chip and conventional PCR. We used 33 previously validated PCR assays to detect amoA, hao, nxrB, hzs, hdh, narG, napA, nirK, nirS, norB, nosZ, nrfA, and nifH (see Table S1 in the supplemental material). Genomic DNA from 16 pure bacterial strains, including nitrifiers, denitrifiers, N2-fixing bacteria, and dissimilatory nitrite reduction to ammonium (DNRA) bacteria, and those from two enrichment cultures of ammonia-oxidizing archaea (AOA) and anammox bacteria were used (Table 1).
TABLE 1.
Pure bacterial strains, enrichment culture, and environmental samples used in this study
| Bacterial strain or enrichment culture | N cycle functional gene(s) | GenBank accession no. or reference(s) |
|---|---|---|
| Nitrosomonas europaea NBRC 14298 | amoA, hao, nirK, norB | NC_004757 |
| AOA enrichment culturea | amoA | H. Fujitani, unpublished |
| Nitrobacter vulgarisa | nxrB | H. Fujitani, unpublished |
| Nitrospira japonica J1a | nxrB | 50 |
| Nitrospira sp. ND1a | nxrB | 49 |
| Bradyrhizobium sp. TSA1 | napA, nirS, nirK, norB, nosZ | LFJC00000000 |
| Cupriavidus necator H16 | narG, napA, nirS, norB, nosZ | NC_008313, NC_008314, NC_005241 |
| Noviherbaspirillum autotrophicum TSA66T | narG, napA, nirS, norB, nosZ | JWJG00000000 |
| Paracoccus denitrificans JCM 21484T | napA, nirS, norB, nosZ | BBFH00000000 |
| Pseudogulbenkiania sp. strain 2002 | narG, nirS, norB, nosZ | NZ_ACIS00000000 |
| Pseudogulbenkiania sp. strain NH8B | narG, nirS, norB, nosZ | NC_016002 |
| Pseudomonas aeruginosa PAO1 | narG, napA, nirS, norB, nosZ | NC_002516 |
| Anaeromyxobacter dehalogenans 2CP-C | nrfA, norB, nosZ | NC_007760 |
| Campylobacter jejuni JCM 2013 | napA, nrfA | |
| Escherichia coli K-12 MG1655 | narG, napA, nrfA | NC_000913 |
| Azospirillum brasilense Sp7 | napA, nirK, norB, nosZ, nifH | NZ_CP012914–NZ_CP012919 |
| Bradyrhizobium diazoefficiens USDA110T | napA, nirK, norB, nosZ, nifH | NC_004463 |
| Herbaspirillum seropedicae JCM 21448T | narG, norB, nifH | NZ_CP011930 |
| “Candidatus Brocadia sinica” enrichment | hao, hzs, nxrB, nrfA | BAFN01000001–BAFN01000003 |
Provided by Hirotsugu Fujitani.
The outcomes of PCR amplification on the NiCE chip are summarized in Fig. 2. Functional genes for the N cycle and the 16S rRNA gene were simultaneously amplified on the NiCE chip. For instance, all N cycle functional genes present in the Nitrosomonas europaea genome (amoA, hao, nirK, and norB) were successfully amplified on the NiCE chip (Fig. 2). Sequence analysis verified that these amplicons were bona fide amoA, hao, nirK, and norB genes from N. europaea; therefore, the functional genes were specifically amplified on the NiCE chip. Similarly, nxrB and archaeal amoA were correctly amplified from nitrite-oxidizing bacteria (NOB) and AOA, respectively, and hzs, hao-hdh, nxrB, and nrfA were correctly amplified from an anammox bacterium, “Candidatus Brocadia sinica” (Fig. 2 and Table S2). Amplifications of functional genes other than hzs, hao-hdh, nxrB, and nrfA were also found in the enrichment culture of “Ca. Brocadia sinica” (i.e., bacterial amoA, nirK, nirS, and nifH), which were probably derived from the genomic DNA of nonanammox microorganisms coexisting in the enrichment culture, such as nitrifiers (positive for amoA and nirK), denitrifiers (positive for nirK and nirS), and sulfate-reducing bacteria (SRB) (positive for nifH).
FIG 2.

Heat map showing the ranges in qPCR CT values on the NiCE chip for detection of N cycle functional genes and the 16S rRNA gene. The amplicon size, strain identification used to construct standard DNA, and CT values obtained from the nontemplate controls are shown at the bottom. The box with a diagonal line indicates the occurrences of nonspecific amplification. 1, primers 341F and 805R; 2, primers Archaea-F KO and Archaea-R KO; 3, primers amoA_1F and amoA_2R; 4, primers amoA_1F and amoA_F1_R2; 5, primers Gamo172 F1 and Gamo172 F1_R1; 6, primers Gamo172 F1 and Gamo172 F1_R2; 7, primers Gamo172 F2 and Gamo172 F2_R1; 8, primers Arch-amoAF and Arch-amoAR; 9, primers Arch-amoAFA and Arch-amoAR; 10, primers Arch-amoAFB and Arch-amoAR; 11, primers Arch-amoA-for and Arch-amoA-rev; 12, primers hzocl1F1 and hzocl1R2; 13, primers haoF4 and haoR2; 14, primers hzsA_1597F and hzsA1857R; 15, primers NxrB 1F and NxrB 1R; 16, primers nxrB169f and nxrB638r; 17, primers W9F and T38R; 18, primers narG1960f and narG2650r; 19, primers nrfAF2aw and nrfAR1; 20, primers V66 and V67; 21, primers V17m and napA4r; 22, primers FlaCu and R3Cu; 23, primers nirK876 and nirK1040; 24, primers nirK_166F and NxrB 1F; 25, primers nirSCd3aF and nirSR3cd; 26, primers norB2 and norB6; 27, primers cnorB-2F and cnorB-6R; 28, primers qnorB2F and qnorB7R; 29, primers qnorB2F and qnorB5R; 30, primers nosZ1F and nosZ1R; 31, primers nosZ-II-F and nosZ-II-R; 32, primers nifHF and nifHR; 33, primers IGK3 and DVV. a, strains used only to construct standard DNA: T, Pseudomonas sp. strain JCM 20650; U, Rhodobacter sphaeroides JCM 6121T; V, Gemmatimonas aurantiaca JCM 11422T; X, synthetic DNA fragment containing amoA from Nitrosopumilus maritimus SCM1 (GenBank accession no. CP000866; locus tag Nmar_1500).
Nonspecific amplification was also detected on the NiCE chip. For example, amplifications of amoA and nirK were detected in the Escherichia coli genome (represented by boxes with a slanted line in Fig. 2), although E. coli does not possess these genes. Based on blastn analysis, these false-positive sequences were amplified from nontarget regions in the E. coli genome, probably due to the nonspecific binding of the primers. Nonspecific sequences were also detected in other bacterial samples, and they occupied up to 27% of the total sequence reads (Table S3). The proportion of nonspecific amplicons was larger in environmental samples. These nonspecific sequences were removed for downstream analyses.
In addition to NiCE chip amplification, conventional PCR was also performed with the same reagents and annealing temperature as those for the NiCE chip assay to further examine the specificity of the PCR assays (Fig. S2). The results of the NiCE chip and conventional PCR assays were similar. Most results (91%) out of 576 reactions (i.e., 32 assays × 18 samples) were identical between the NiCE chip and the conventional PCR assays. Nonspecific amplification was commonly observed by the two methods in specific assay-sample combinations. Nonspecific amplification was frequently observed in assays done with primers Gamo172F1 and Gamo172F1_R1, primers nirSCd3aF and nirSR3cd, and primers nifHF and nifHR. These results suggest that nonspecific amplification was due to the nature of the PCR assays.
In addition to specificity, the sensitivity of the PCR assays was examined by using a serial dilution of plasmid DNA containing the target gene fragment (102 to 107 copies/μl). When we had standard DNA, we detected target genes at levels as low as 103 copies/μl by using this assay (Fig. S3). Slopes of the standard curves were relatively large (>4) due to the qPCR conditions used in this study (i.e., condition B to halt amplification). Some assays were not sensitive enough to detect 105 copies/μl (e.g., assays for narG and nrfA), probably due to the long amplicon size, high GC content, or base mismatches in the primer annealing sites.
Analysis of mock community samples.
The applicability of the NiCE chip approach for environmental samples was examined by using artificially mixed DNA samples (i.e., mock communities). Sequencing analysis of the NiCE chip amplicons showed that the relative abundances of sequence reads for a specific bacterium increased as their proportion increased in the mock community (Fig. 3). For example, the relative abundances of the napA, nrfA, and E. coli 16S rRNA gene sequences increased as the proportion of E. coli bacteria increased in mock community A (Fig. 3a). Accordingly, the relative abundances of the nirS, norB, nosZ, Pseudomonas aeruginosa 16S rRNA gene, amoA, hao, and Nitrosomonas europaea 16S rRNA gene sequences decreased. The proportion of the N. europaea 16S rRNA gene was much smaller than the proportion of N. europaea genomic DNA in the mock community A, most likely due to differences in the rRNA gene copy numbers in the genomes (i.e., 7, 4, and 1 copy per genome of E. coli, P. aeruginosa, and N. europaea, respectively).
FIG 3.

NiCE chip results for the mock community samples. (a) Mixture of a nitrifier, a denitrifier, and a DNRA bacterium (N. europaea strain NBRC 14298, Pseudomonas aeruginosa strain PAO1, and E. coli K-12 strain MG1655); (b) mixture of various denitrifying bacteria (P. aeruginosa strain PAO1, Bradyrhizobium sp. strain TSA1, Noviherbaspirillum autotrophicum strain TSA66T); (c) mixture of N2-fixing bacteria (Bradyrhizobium diazoefficiens strain USDA110T, Herbaspirillum seropedicae strain JCM 21448T, and Azospirillum brasilense strain Sp7T).
Environmental samples.
NiCE chip amplicon sequencing was used to examine the quantities and the diversities of N cycle functional gene and 16S rRNA gene sequences in anaerobic granular sludge collected from two upflow anaerobic sludge blanket (UASB) reactors (designated UASB-M and UASB-L) and aerobic granular sludge in a partial nitrification (PN) reactor. While DNA was extracted from the two UASB anaerobic granular sludge samples, RNA was extracted from aerobic PN granule samples. When we used the RNA samples, we did not include the 16S rRNA gene assays in the NiCE chip assay, because otherwise, rRNA sequence reads could overwhelm the amplicon sequence libraries.
The 16S rRNA gene sequencing results suggested that bacteria affiliated with the phyla Proteobacteria, Chloroflexi, Caldiserica, Firmicutes, and Bacteroidetes and archaea affiliated with the phylum Euryarchaeota were the dominant members in the UASB-M and UASB-L samples (see Fig. S4 in the supplemental material). These microorganisms included methanogens (phylum Euryarchaeota) and SRB (class Deltaproteobacteria). The 16S rRNA gene sequences from methanogens in the family Methanosaetaceae accounted for 2.3% and 2.1% of the UASB-M and UASB-L samples, respectively. The 16S rRNA gene sequences from SRB in the families Desulfovibrionaceae, Desulfobulbaceae, and Desulfobacteraceae accounted for 0.096%, 0.051%, and 0.03% of the UASB-M sample and 2.0%, 1.7%, and 0.57% of the UASB-L sample, respectively. In addition to NiCE chip amplicon sequencing, MiSeq analysis of the 16S rRNA gene amplicons was done with a conventional library preparation protocol to analyze the microbial community structure of the UASB-L and UASB-M samples. In the NiCE chip amplicon sequencing approach, primers 341F and 805R and primers Archaea-F KO and Archaea-R KO were used to amplify bacterial and archaeal 16S rRNA genes, respectively, while only primers 341F and 805R were used for the conventional library preparation protocol. Therefore, the archaeal 16S rRNA gene was more abundantly detected by the NiCE chip approach. To eliminate this bias, we compared only the bacterial community structures assessed by NiCE chip amplicon sequencing and MiSeq done with the conventional library preparation protocol. Similar bacterial taxonomic compositions were obtained by the two methods (Fig. S4). The relative proportions of each phylum were not significantly different (P > 0.05) between the two methods based on the chi-square (χ2) test and Fisher's exact test.
nxrB, napA, nirK, nirS, norB, nosZ, nrfA, and nifH sequences were obtained from both the UASB-M and UASB-L samples (Fig. 4 and Table S4). Among these sequences, nifH sequences were the most abundantly detected sequences, followed by napA. Shannon and inverse Simpson indices of diversity were calculated for each functional gene (Table S5). Phylogenetic analysis showed great diversity within nifH (Fig. S5) and napA (Fig. S6) sequences. The most abundantly detected nifH operational taxonomic units (OTUs) showed similarities to the nifH genes of methanogens (e.g., Methanosaeta concilii [GenBank accession no. WP_013718497] and Methanobacterium sp. strain 42_16 [GenBank accession no. KUK73267]) and of SRB (e.g., Desulfobacca acetoxidans [GenBank accession no. WP_013705354] and Desulfobulbus propionicus [GenBank accession no. ADW19185] and Desulfomonile tiedjei [GenBank accession no. WP_014813165]). The most dominant OTU of the napA sequences obtained from the UASB-M and UASB-L samples showed similarities to napA from Magnetospira sp. strain QH2 (GenBank accession no. WP_046020782) and Arcobacter butzleri (GenBank accession no. WP_020847347), respectively (Fig. S5). The hzs and narG sequences were not detected.
FIG 4.
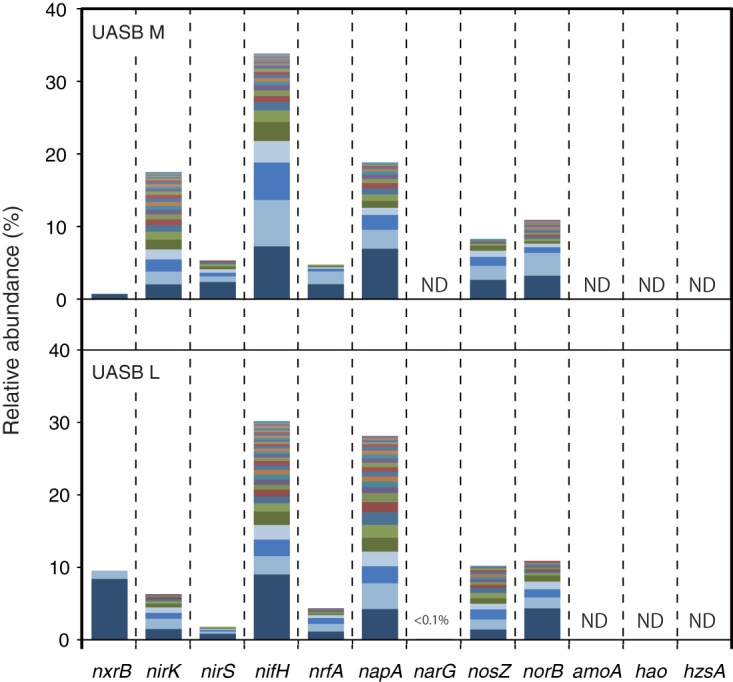
Relative abundances of the N cycle functional gene sequences detected in the anaerobic granular sludge samples (UASB-M and UASB-L). The numbers of total sequence reads were 41,059 and 60,333 for UASB-L and UASB-M, respectively. OTUs determined based on 95% sequence similarity are shown in different colors. See Table S4 in the supplemental material for detail information for each OTU. ND, not detected.
The NiCE chip approach was further applied to analyze the active transcription of N cycle functional genes (i.e., transcriptome analysis). Whole transcripts were extracted from aerobic granules (triplicate samples) collected at nine different time points during one sequencing batch cycle (4 h) from a PN reactor (10). That previous study was done to identify the microorganisms responsible for N2O production. The same cDNA samples were previously used for conventional qPCR to quantify amoA, narG, nirK, nirS, norB, and nosZ transcripts and for NGS to analyze norB sequence diversity (10). These conventional qPCRs were done under the optimal thermal conditions reported previously. Therefore, we could compare the quantitative and qualitative data obtained by using the NiCE chip with those obtained by the conventional approach.
Similar to the previous study (10), transcripts of AOB amoA, N. europaea- and N. eutropha-specific nirK, nirS, cnorB, and nosZ clade I were abundantly detected in the aerobic PN granules (Fig. 5), whereas narG, general nirK, qnorB, and nosZ clade II were below the detection limit. The absence of AOA, NOB, and anammox bacteria was also confirmed, similar to the previous study (33). Levels of transcription of nirS and cnorB significantly changed over time (P < 0.05), most likely in response to the availability of acetate (10). Strong correlations (P < 0.05 by Pearson, Spearman, and Kendall tests) were observed between the quantities measured by using the NiCE chip and those measured by conventional qPCR for all gene transcripts shown in Fig. 5, except for transcripts from N. europaea- and N. eutropha-specific nirK (Table S6). The weak correlation for this gene transcript may be due to the relatively large error bars.
FIG 5.

Quantities of gene transcripts of amoA (A), N. europaea- and N. eutropha-specific nirK (B), nirS (C), cnorB (D), and nosZ clade I (E), obtained from the nitrification-denitrification granules as measured by the NiCE chip assay. Means ± standard deviations of data from triplicate granule samples are shown.
All PCR amplicons recovered from the NiCE chip were sequenced by MiSeq, which verified the amplification of the target molecules. Here, we focused on analyzing the diversity of cnorB transcripts to identify the microorganisms responsible for N2O production (i.e., cNorB reduces nitric oxide to produce N2O). A total of 223,843 reads were identified as cnorB sequences in the MiSeq results. Four major OTUs were identified when a 5% sequence dissimilarity was used as a cutoff for OTU clustering. Each of the four OTUs contained the sequences representing the OTUs identified in the previous study: 2A_63087, 3B_67163, 7A_13064, and 7A_78700 (10). The OTU represented by sequence 2A_63087 was closely related to cnorB from Nitrosomonas spp. and Nitrosococcus spp., whereas the other three OTUs were related to cnorB from Rhodocyclales bacteria such as Azoarcus spp., Dechloromonas spp., and Thauera spp. (10). While these major OTUs collectively occupied 82 to 100% of the sequence libraries, the relative proportions of these OTUs changed over time (Fig. 6A). For example, the proportion of the OTU represented by sequence 3B_67163 increased 10 and 20 min after aeration and decreased thereafter. A similar trend was also observed when 454 pyrosequencing was done with a conventional library preparation (Fig. 6B). The relative proportions of each of the four major OTUs were not significantly different (P > 0.05) between the use of the NiCE chip followed by MiSeq analysis and 454 pyrosequencing done with a conventional library preparation protocol based on the chi-square test and Fisher's exact test.
FIG 6.

Relative abundances of the cnorB transcript sequences obtained from the nitrification-denitrification granules as measured by using the NiCE chip (A) and 454 pyrosequencing done with a conventional library preparation protocol (10) (B). Four major OTUs determined based on 95% sequence similarity are shown in different patterns. ■, 2A_63087 (Nitrosomonas-related norB); ▨, 7A_78700;  , 7A_13064; □, 3B_67163;
, 7A_13064; □, 3B_67163;  , other OTUs. Phylogenetic positions of these OTUs were previously reported (10).
, other OTUs. Phylogenetic positions of these OTUs were previously reported (10).
DISCUSSION
Various enzymatic reactions are involved in the N cycle. Accordingly, multiple PCR primers are necessary to quantify and analyze the diversities of N cycle functional genes. Microfluidic qPCR, which allows us to simultaneously quantify multiple N cycle functional genes and recover PCR amplicons for downstream sequencing analysis, is a suitable approach to evaluate the N cycle in environmental samples.
Here, we report that the NiCE chip approach can provide high-throughput quantitative data for N cycle functional genes. Amplicons recovered from the NiCE chip can be used for MiSeq sequencing analysis. In addition to the N cycle functional genes, the NiCE chip can also amplify taxon-specific 16S rRNA gene markers, which allows us to examine microbial community structure. Previously, the microfluidic DynamicArray chip was used to quantify multiple virulence factor genes (12, 30, 34); however, the amplicons were not analyzed for sequencing. In contrast, the microfluidic AccessArray chip was used to prepare sequencing libraries targeting multiple taxon-specific 16S rRNA genes (31) and those targeting highly variable regions of pathogen-specific genes (32); however, those studies did not analyze the quantitative performance of the AccessArray chip. This study showed that the quantitative detection of multiple genes is possible by using the AccessArray chip, and the amplicons can be used for downstream sequencing analysis.
N cycle functional genes were detected at a limit of quantification as low as 103 copies/μl by using the NiCE chip, which might be higher than that of conventional qPCR (10) but sufficient for many environmental samples such as the wastewater samples examined in this study. The reason for the relatively high limit of quantification for the NiCE chip is the small volume (35 nl) in the reaction chamber. The sample premix (5 μl) contains 1 μl of a DNA sample. Therefore, 7 nl out of 35 nl originates from the DNA sample. This means that only 7 copies of the target gene can be dispensed into each chamber when the DNA sample contains 1,000 copies/μl of the target gene molecule. If necessary, the sensitivity of detection can be increased by using a preamplification reaction such as specific target amplification prior to microfluidic qPCRs (12, 30, 34).
The selection of primers is critically important for both NiCE chip and conventional qPCR assays because both the specificity and sensitivity of qPCR depend largely on the primers used. Ideally, the melting temperatures of the primers should be similar to each other, as qPCR is performed under the same reaction conditions as those for the microfluidic qPCR format, including the AccessArray chip used in this study. Because we used previously validated PCR assays with primer melting temperatures ranging from 50°C to 78°C, the specificity of each assay may not be optimum in the NiCE chip format (i.e., annealing temperature of 50°C). The generation of nonspecific bands observed in this study may be due, in part, to the relatively low annealing temperature used in the NiCE chip run. Designing new primers that have almost identical melting temperatures should improve the specificity of the assays (e.g., see reference 30). For this purpose, peptide nucleic acid or locked nucleic acid (LNA) can be used to increase the melting temperature and the specificity of primers. LNA is a modified RNA analog and forms LNA/DNA hybrids as well as DNA/DNA hybrids (35, 36). The LNA/DNA hybrid shows higher thermal stability and a higher duplex melting temperature than the DNA/DNA hybrid, resulting in an increase of the specificity of PCR amplification (37, 38). In addition, to minimize the occurrence of nonspecific amplification, it might be helpful to decrease the number of degenerate bases in the primers. Many previously designed PCR primers contain degenerate bases to amplify target N cycle functional genes from diverse taxa. However, the chance of obtaining nonspecific amplicons is increased by using degenerate bases (39). Instead of the use of primers with degenerate bases, we could use multiple primers, each of which targets a subgroup of target genes (40). By using high-throughput microfluidic qPCR, these multiple qPCRs can be run simultaneously.
In addition to the primer melting temperature, amplicon size is also important to consider. Short amplicons (i.e., <150-bp target fragments, which correspond to a <250-bp final fragment, including Illumina adapters) usually have higher amplification efficiencies and therefore higher detection sensitivities than long amplicons (30). In addition, short amplicons can form clusters on a MiSeq flow cell more efficiently than can long amplicons, thereby dominating the resulting sequence reads. Long amplicons (i.e., >950 bp, including the Illumina adapter) are not well sequenced for the same reason. PCR amplicons recovered from the NiCE chip include a mixture of all amplicons generated from a single sample. Therefore, we cannot adjust the ratio of the PCR products from each assay after the NiCE chip run. Because of this amplicon size limitation, we decided to include assays that generate amplicons with a size of 250 to 950 bp on the NiCE chip and therefore could not include several assays that previously performed well in the literature (e.g., amoA assays reported in reference 39).
The amplification efficiency during qPCR can also cause different amplicon yields. In general, short amplicons are more efficiently amplified during the PCR than are long amplicons. To overcome this issue, we used two amplification conditions during a single NiCE chip run: condition A (95°C for 15 s, 50°C for 30 s, and 72°C for 1 min) and condition B (95°C for 15 s, 80°C for 30 s, 50°C for 30 s, and 72°C for 1 min). Incubation at 80°C under condition B may allow the reassociation of some of the single-stranded DNA to form double-stranded DNA (dsDNA). Short PCR amplicons are more likely to reassociate than are long PCR amplicons and genomic DNA, as the reassociation kinetics is a function of the fragment length (41). Therefore, condition B can facilitate the amplification of long fragments while halting the amplification of short fragments and thereby can increase the presence of long fragments in the resulting sequence libraries. It is important to note that condition B is not necessary if quantification is the sole purpose of the use of the chip, i.e., when the NiCE chip is used to quantify multiple N cycle functional genes and not to prepare libraries for sequencing.
NiCE chip amplicon sequencing was successfully applied for the quantification and analysis of the diversities of the N cycle functional genes and their transcripts in environmental samples. A significant correlation between the quantities of the N cycle functional gene transcripts in aerobic PN granule samples measured by using the NiCE chip and those measured by conventional qPCRs that were run under optimal thermal conditions was observed, supporting the good quantitative performance of the NiCE chip method. Sequencing of the PCR amplicons recovered from the NiCE chip confirmed that the target genes were successfully amplified. In addition, the proportions of norB genotypes identified by the NiCE chip followed by MiSeq were not significantly different from those identified by 454 pyrosequencing done with a conventional library preparation protocol. To eliminate the effect of different sequencing technologies (i.e., MiSeq versus 454 pyrosequencing), we also compared the MiSeq results for the 16S rRNA gene amplicon libraries prepared by using the NiCE chip approach to those of libraries prepared by the conventional PCR-based method. The MiSeq results obtained by the two library preparation methods were not significantly different by chi-square and Fisher's exact tests. These results suggest that the NiCE chip amplicon sequencing approach can generate relative-abundance data with a quality similar to that of data generated by the conventional amplicon sequencing approach. Therefore, the NiCE chip amplicon sequencing approach might be useful for targeting multiple N cycle functional genes. Previously, Herbold et al. (42) reported multiplex amplicon sequencing by using a flexible barcoding approach, which can be applied to analyses of N and S cycle functional genes. Although that approach is useful, separate PCRs need to be performed for each target gene, which is laborious, especially when many genes are analyzed. In contrast, multiple target genes are amplified in a single NiCE chip run, greatly reducing the time and labor needed for library preparation.
The NiCE chip is useful for evaluation of the occurrence of N-transforming reactions in environmental samples. In this study, we found napA, norB, nirK, nirS, nosZ, nrfA, nxrB, and nifH in anaerobic granular sludge collected from the UASB system. Most of the sequences showed similarities only to environmental clones, suggesting that microorganisms involved in N-transforming reactions in anaerobic granular sludge have not yet been well characterized. It is noteworthy that the nifH sequences related to methanogens and SRB were abundantly detected in both the UASB-M and UASB-L samples. While N2 fixation by methanogens (43, 44) and SRB (45, 46) was previously demonstrated in batch culture, it is still unknown whether these microorganisms fix N2 in a UASB reactor or not. Therefore, the activity of N2-fixing microorganisms identified by the NiCE chip needs to be examined in the future.
In conclusion, the NiCE chip amplicon sequencing approach can provide a high-throughput format to quantify and prepare sequencing libraries for multiple N cycle functional genes. While nonspecific amplification occurred in some assays with the NiCE chip, this issue can be overcome by designing new primers or by using confirmatory approaches (e.g., conventional qPCR or digital PCR). Nonetheless, the high-throughput nature of this “nice” approach should advance our ability to explore the occurrences of multiple N-transforming reactions in various environmental samples.
MATERIALS AND METHODS
Bacterial strains, enrichment culture, and standard DNA samples.
The specificity of NiCE chip amplicon sequencing analysis was examined by using genomic DNA extracted from pure bacterial strains and enrichment cultures, as shown in Table 1. The bacterial strains were provided by the National Institute of Technology and Evaluation (NITE) and the Japan Collection of Microorganisms (JCM) and cultured as recommended by the suppliers. An enrichment culture of the anammox bacterium “Ca. Brocadia sinica” was collected from an anaerobic membrane bioreactor in which “Ca. Brocadia sinica” cells accounted for more than 90% of the total biomass (47, 48). Genomic DNA from Nitrospira japonica J1 and Nitrospira sp. strain ND1 (49, 50) and an AOA enrichment culture were provided from Yoshitsugu Fujitani (Waseda University).
Artificially mixed DNA samples (i.e., mock communities) were prepared by mixing genomic DNAs at known proportions. Mock community A was composed of N. europaea strain NBRC 14298 (a nitrifying bacterium), Pseudomonas aeruginosa strain PAO1 (a denitrifying bacterium), and E. coli K-12 strain MG1655 (a DNRA bacterium). Mock community B was composed of various denitrifying bacteria: P. aeruginosa strain PAO1, Bradyrhizobium sp. strain TSA1 (51), and Noviherbaspirillum autotrophicum strain TSA66T (52). Mock community C was composed of various N2-fixing bacteria: Bradyrhizobium diazoefficiens strain USDA110T, Herbaspirillum seropedicae strain JCM 21448T, and Azospirillum brasilense strain Sp7T. These bacterial genomic DNAs were mixed at different ratios, 1:1:1, 8:1:1, 90:9:1, or 99:0.9:0.1.
Linearized plasmids containing target gene sequences were synthesized or prepared as previously described (12) and used as standard DNA for the NiCE chip and conventional qPCR assays.
Environmental samples.
Aerobic granular sludge was collected from a PN reactor (33). Reactor operational conditions and the microbial community structure of the granular sludge were previously described (33). The transcription level of norB was examined by reverse transcription-qPCR, and norB transcript pyrosequencing analysis was carried out with a Roche 454 FLX pyrosequencer (10). In this study, we used the same cDNA samples as the ones used previously (10) to compare the results obtained by norB pyrosequencing and those obtained by NiCE chip amplicon sequencing.
Anaerobic granular sludge was collected from two UASB reactors, designated UASB-M and UASB-L. UASB-M (diameter of 56 cm and height of 470 cm) and UASB-L (diameter of 6 cm and height of 30 cm) were fed with domestic sewage at 1 and 4 kg m−3 day−1 of chemical oxygen demand (COD) loads, respectively. UASB-M was operated without temperature control (water temperature ranging from 10.1°C to 27.3°C), while UASB-L was operated at 20°C. The typical composition of the domestic sewage supplied to the reactors was as follows: 269 ± 73 mg liter−1 COD, 21 mg N liter−1 for NH4+, 0.03 mg N liter−1 for NO3−, 0.18 mg N liter−1 for NO2−, and 14 ± 17 mg S liter−1 for SO42−. During reactor operation, the production of methane and sulfide was monitored. The methane concentration was measured by using a GC-2014 gas chromatograph equipped with a thermal conductivity detector as previously described (53). The concentration of sulfide in the liquid phase was determined by a titration method using iodine and sodium thiosulfate solutions. In the UASB-L and UASB-M reactors, 10 to 60 ml/day of methane and 1 to 5 mg S liter−1 of sulfide were produced.
DNA and RNA extraction.
Genomic DNA was extracted from pure culture samples by using a DNeasy blood and tissue kit (Qiagen). DNA was extracted from enrichment cultures and environmental samples by using a PowerSoil DNA isolation kit (Mo Bio Laboratories). DNA concentrations were determined by using a Quant-iT PicoGreen dsDNA assay kit (ThermoFisher Scientific). Total RNA was extracted from aerobic PN granule samples by using a RiboPure Bacteria kit (Invitrogen). After DNase I treatment, cDNA was synthesized as previously described (10).
Oligonucleotide primers.
Previously validated oligonucleotide primers were used for the NiCE chip (see Table S1 in the supplemental material). These primers target the amoA, nxrB, narG, napA, nirS, nirK, nosZ (both clade I and type II nosZ), hzs, hao-hdh, norB (both cnorB and qnorB), nrfA, nifH, and 16S rRNA genes. The 5′ ends of the forward and reverse primers contained Illumina tag sequences (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′ and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′, respectively).
Conventional PCR.
Conventional PCR was carried out as previously described. The PCR mixture (20 μl) contained 1× FastStart HiFi reaction buffer (Roche), 4.5 mM MgCl2, 5% (vol/vol) dimethyl sulfoxide (DMSO), 0.2 mM each deoxynucleoside triphosphate (dNTP), 0.2 μM each primer, 0.5 U FastStart HiFi enzyme blend (Roche), and 1 μl template DNA (ca. 10 ng). PCR was performed by using a Veriti thermal cycler (Applied Biosystems) under the following cycling conditions: 35 cycles of 30 s at 95°C, 30 s at 50°C, and 1 min at 72°C and a final extension step for 10 min at 72°C. The size of the PCR amplicon was analyzed by agarose gel electrophoresis.
Microfluidic qPCR.
Microfluidic qPCR was carried out by using a BioMark HD reader (Fluidigm) with a 48.48 AccessArray chip (Fluidigm). The 20× primer solutions contained 1× AccessArray loading reagent (Fluidigm) and 4 μM each forward and reverse primers. The presample master mix (5 μl) contained 1× FastStart HiFi reaction buffer (Roche), 1× AccessArray loading reagent (Fluidigm), 4.5 mM MgCl2, 5% (vol/vol) DMSO, 0.2 mM each dNTP, 0.05 U FastStart HiFi enzyme blend (Roche), 1× EvaGreen (Biotium), 0.5× 5-carboxyl-X-rhodamine (ROX), and 1 μl template DNA (ca. 10 ng). The 20× primer solutions and the presample master mix were dispensed onto the AccessArray chip and mixed by using an IFC controller AX instrument (Fluidigm), according to the manufacturer's instructions. All primers listed in Table S1 in the supplemental material were used in the microfluidic qPCR run, except when RNA (cDNA) samples were used; the 16S rRNA gene assays were not included when RNA samples were used.
qPCR was performed under the following thermal conditions: 50°C for 120 s, 70°C for 20 min, and 95°C for 10 min, followed by 10 cycles of condition A (95°C for 15 s, 50°C for 30 s, and 72°C for 1 min), 2 cycles of condition B (95°C for 15 s, 80°C for 30 s, 50°C for 30 s, and 72°C for 1 min), 8 cycles of condition A, 2 cycles of condition B, 8 cycles of condition A, 5 cycles of condition B, and melting curve analysis from 60°C to 95°C. The detection of PCR products was achieved by monitoring the fluorescence intensity of EvaGreen dye. The threshold cycle (CT) was defined as the cycle at which the EvaGreen signal became significantly higher than the background level and was determined by using Real-Time PCR Analysis Software version 3.0.2 (Fluidigm). The standard curves were generated by linear regression analysis of the CT values versus the amounts of the template DNA (log copies per microliter), as described previously (30). Amounts of gene transcripts in the aerobic PN granule samples were normalized based on the quantity of 16S rRNA that was measured previously by conventional qPCR (10). PCR products (i.e., amplicon mixture from 48 assays) were recovered from the chip for each sample by using the AccessArray Harvest reagent and the IFC controller AX instrument according to the manufacturer's instructions.
Sequencing library preparation and MiSeq run.
The recovered PCR products were tagged with a sample-unique index and Illumina adapter sequences at their 5′ ends (Nextera XT index kit v2; Illumina) by PCR. The PCR mixture (10 μl) contained 1× Kapa HiFi HS ReadyMix (Kapa Biosystems), 2 μl each forward and reverse primers, and 1 μl of the recovered PCR products. PCR was performed under the following cycling conditions: 95°C for 3 min; 15 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and 72°C for 5 min. After agarose gel electrophoresis, PCR products (200 to 1,000 bp) were excised from the gel and purified by using a NucleoSpin gel and PCR cleanup kit (TakaraBio). Tagged amplicons were pooled and sequenced by using the Illumina MiSeq platform in 300-bp paired-end sequencing reaction mixtures with the v3 reagent kit (Illumina) according to the manufacturer's instructions.
Bioinformatics analysis.
The generated reads were processed for the removal of adapter sequences using cutadapt and for quality trimming using prinseq (54, 55). We discarded reads that contained <50 bp or that were associated with an average Phred-like quality score of <25. Paired-end sequence reads were assembled in the paired-end assembler for the Illumina sequence by using FLASH-1.2.11 (56). Nucleic acid sequences with ≥95% similarity were grouped into an OTU by usearch8 (57). The sequence reads of functional genes representing each OTU were subjected to blastn and blastx searches against reference sequences of the fungene (58) and KEGG databases, respectively. In the present study, the applicability of NiCE chip amplicon sequencing was validated by using genomic DNA of pure bacterial strains and enrichment cultures. The presence and absence of the functional genes involved in nitrogen transformation were examined by a blastn search (threshold E value of <10−6) of the previously determined genome sequences (Table 1). Amino acid sequences of the following genes were used as the query sequences for the blastn search: N. europaea amoA, hao, nirK, and norB; “Ca. Brocadia sinica” hzs and nxrB; E. coli narG, napA, and nrfA; P. aeruginosa nirS, norB, and nosZ; and A. brasilense nifH. For NOB and AOA, the blastn search was not applicable because genome sequences have not yet been determined. Phylogenetic trees were generated based on the deduced NifH and NapA amino acid sequences by the maximum likelihood method and the Jones-Taylor-Thornton model using MEGA 6.06 software (59). Phylogenetic affiliations of the OTUs based on the 16S rRNA gene sequences were identified by using a blastn search against reference sequences in the Greengenes version 13_5 database (60) and the nr database (National Center for Biotechnology Information). The 16S rRNA gene sequences obtained by NiCE chip amplicon sequencing and those obtained by MiSeq analysis done by using a conventional library preparation method were analyzed by using Qiime version 1.9.1 (61), with equal numbers of sequences (n = 14,000) across samples and library preparation methods.
Statistical analysis.
The statistical significance of gene or transcript quantities across samples was assessed by analysis of variance (ANOVA), and pairwise comparisons among means were performed by using Tukey's honestly significant difference (HSD) test, at an α value of 0.05. Pearson's correlation, Spearman's rank correlation, and Kendall's rank correlation coefficients were calculated for quantitates obtained by using the NiCE chip and those obtained by conventional qPCR. Differences in the relative proportions of sequence reads obtained by the two sequencing methods (NiCE chip followed by MiSeq analysis versus 454 pyrosequencing or MiSeq analysis done by using a conventional library preparation protocol) were analyzed by using a χ2 test. All statistical analyses were done by using R version 3.3.2.
Inverse Simpson and Shannon diversity indices were calculated by using the equations 1/∑i(ni/n)2 for the inverse Simpson index and −∑i(ni/n)ln(ni/n) for the Shannon index, where ni is the number of sequence reads grouped into an OTU and n is the total number of sequence reads affiliated with a particular N cycle functional gene.
Accession number(s).
Sequence data were deposited in the NCBI nucleotide sequence database under accession numbers LC230165 to LC257534.
Supplementary Material
ACKNOWLEDGMENTS
We thank Reiko Hirano, Akiyoshi Ayumi, and Nora Powers for technical assistance and Hirotsugu Fujitani and Yuga Hirakata for providing AOA and NOB cultures and anaerobic granular sludge. We also thank Kelly Duhn for scientific editing of the manuscript.
This work was supported in part by Mitsui Co. & Ltd., the Environment Fund (to S.I.), the MnDRIVE Initiative of the University of Minnesota (to S.I.), grants-in-aid for scientific research (no. 26281017 and 17H01854) from the Japan Society for the Promotion of Science (JSPS) (to T.S.), and the National Institute of Polar Research (NIPR) through general collaboration project no. 27-29 (to M.O.). Part of the computations was performed by using a supercomputer maintained by the National Institute of Genetics (NIG), Research Organization of Information and Systems (ROIS), Japan.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02615-17.
REFERENCES
- 1.Canfield DE, Glazer AN, Falkowski PG. 2010. The evolution and future of Earth's nitrogen cycle. Science 330:192–196. doi: 10.1126/science.1186120. [DOI] [PubMed] [Google Scholar]
- 2.Vlaeminck SE, Hay AG, Maignien L, Verstraete W. 2011. In quest of the nitrogen oxidizing prokaryotes of the early Earth. Environ Microbiol 13:283–295. doi: 10.1111/j.1462-2920.2010.02345.x. [DOI] [PubMed] [Google Scholar]
- 3.Arp DJ, Chain PSG, Klotz MG. 2007. The impact of genome analyses on our understanding of ammonia-oxidizing bacteria. Annu Rev Microbiol 61:503–528. doi: 10.1146/annurev.micro.61.080706.093449. [DOI] [PubMed] [Google Scholar]
- 4.van Kessel MAHJ, Speth DR, Albertsen M, Nielsen PH, Op den Camp HJM, Kartal B, Jetten MSM, Lücker S. 2015. Complete nitrification by a single microorganism. Nature 528:555–559. doi: 10.1038/nature16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zumft WG. 1997. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61:533–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oshiki M, Satoh H, Okabe S. 2016. Ecology and physiology of anaerobic ammonium oxidizing (anammox) bacteria. Environ Microbiol 18:2784–2796. doi: 10.1111/1462-2920.13134. [DOI] [PubMed] [Google Scholar]
- 7.Lackner S, Gilbert EM, Vlaeminck SE, Joss A, Horn H, van Loosdrecht MCM. 2014. Full-scale partial nitritation/anammox experiences—an application survey. Water Res 55:292–303. doi: 10.1016/j.watres.2014.02.032. [DOI] [PubMed] [Google Scholar]
- 8.Ali M, Okabe S. 2015. Anammox-based technologies for nitrogen removal: advances in process start-up and remaining issues. Chemosphere 141:144–153. doi: 10.1016/j.chemosphere.2015.06.094. [DOI] [PubMed] [Google Scholar]
- 9.Huffman RL, Fangmeier DD, Elliot WJ, Workman SR, Schwab GO. 2013. Soil and water conservation engineering, 7th ed American Society of Agricultural and Biological Engineers, St. Joseph, MI. [Google Scholar]
- 10.Ishii S, Song Y, Rathnayake L, Tumendelger A, Satoh H, Toyoda S, Yoshida N, Okabe S. 2014. Identification of key N2O production pathways in aerobic partial nitrifying granules. Environ Microbiol 16:3168–3180. doi: 10.1111/1462-2920.12458. [DOI] [PubMed] [Google Scholar]
- 11.Nunoura T, Nishizawa M, Kikuchi T, Tsubouchi T, Hirai M, Koide O, Miyazaki J, Hirayama H, Koba K, Takai K. 2013. Molecular biological and isotopic biogeochemical prognoses of the nitrification-driven dynamic microbial nitrogen cycle in hadopelagic sediments. Environ Microbiol 15:3087–3107. doi: 10.1111/1462-2920.12152. [DOI] [PubMed] [Google Scholar]
- 12.Ishii S, Kitamura G, Segawa T, Kobayashi A, Miura T, Sano D, Okabe S. 2014. Microfluidic quantitative PCR for simultaneous quantification of multiple viruses in environmental water sample. Appl Environ Microbiol 80:7505–7511. doi: 10.1128/AEM.02578-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pester M, Maixner F, Berry D, Rattei T, Koch H, Lücker S, Nowka B, Richter A, Spieck E, Lebedeva E, Loy A, Wagner M, Daims H. 2014. NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira. Environ Microbiol 16:3055–3071. doi: 10.1111/1462-2920.12300. [DOI] [PubMed] [Google Scholar]
- 14.Segawa T, Ishii S, Ohte N, Akiyoshi A, Yamada A, Maruyama F, Li Z, Hongoh Y, Takeuchi N. 2014. The nitrogen cycle in cryoconites: naturally occurring nitrification-denitrification granules on a glacier. Environ Microbiol 16:3250–3262. doi: 10.1111/1462-2920.12543. [DOI] [PubMed] [Google Scholar]
- 15.Throbäck IN, Enwall K, Jarvis Å, Hallin S. 2004. Reassessing PCR primers targeting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol Ecol 49:401–417. doi: 10.1016/j.femsec.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 16.Gaby JC, Buckley DH. 2012. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One 7:e42149. doi: 10.1371/journal.pone.0042149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helen D, Kim H, Tytgat B, Anne W. 2016. Highly diverse nirK genes comprise two major clades that harbour ammonium-producing denitrifiers. BMC Genomics 17:155. doi: 10.1186/s12864-016-2465-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR, Malmstrom RR, Chisholm SW, DeLong EF. 2010. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci U S A 107:16420–16427. doi: 10.1073/pnas.1010732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llorens-Marès T, Yooseph S, Goll J, Hoffman J, Vila-Costa M, Borrego CM, Dupont CL, Casamayor EO. 2015. Connecting biodiversity and potential functional role in modern euxinic environments by microbial metagenomics. ISME J 9:1648–1661. doi: 10.1038/ismej.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu D, He Y, Yue H, Wang Q. 2015. Microbial structures and community functions of anaerobic sludge in six full-scale wastewater treatment plants as revealed by 454 high-throughput pyrosequencing. Bioresour Technol 186:163–172. doi: 10.1016/j.biortech.2015.03.072. [DOI] [PubMed] [Google Scholar]
- 21.He Z, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC, Huang Z, Wu W, Gu B, Jardine P, Criddle C, Zhou J. 2002. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J 1:67–77. doi: 10.1038/ismej.2007.2. [DOI] [PubMed] [Google Scholar]
- 22.Chan Y, Van Nostrand JD, Zhou J, Pointing SB, Farrell RL. 2013. Functional ecology of an Antarctic dry valley. Proc Natl Acad Sci U S A 110:8990–8995. doi: 10.1073/pnas.1300643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tu Q, Yu H, He Z, Deng Y, Wu L, Van Nostrand JD, Zhou A, Voordeckers J, Lee YJ, Qin Y, Hemme CL, Shi Z, Xue K, Yuan T, Wang A, Zhou J. 2014. GeoChip 4: a functional gene-array-based high-throughput environmental technology for microbial community analysis. Mol Ecol Resour 14:914–928. doi: 10.1111/1755-0998.12239. [DOI] [PubMed] [Google Scholar]
- 24.Shilova IN, Robidart JC, Tripp HJ, Turk-Kubo K, Wawrik B, Post AF, Thompson AW, Ward B, Hollibaugh JT, Millard A, Ostrowski M, Scanlan DJ, Paerl RW, Stuart R, Zehr JP. 2014. A microarray for assessing transcription from pelagic marine microbial taxa. ISME J 8:1476–1491. doi: 10.1038/ismej.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu L, Liu X, Schadt CW, Zhou J. 2006. Microarray-based analysis of subnanogram quantities of microbial community DNAs by using whole-community genome amplification. Appl Environ Microbiol 72:4931–4941. doi: 10.1128/AEM.02738-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao H, Yang ZK, Gentry TJ, Wu L, Schadt CW, Zhou J. 2007. Microarray-based analysis of microbial community RNAs by whole-community RNA amplification. Appl Environ Microbiol 73:563–571. doi: 10.1128/AEM.01771-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Van Nostrand JD, Wu L, He Z, Li G, Zhou J. 2011. Microarray-based evaluation of whole-community genome DNA amplification methods. Appl Environ Microbiol 77:4241–4245. doi: 10.1128/AEM.01834-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou J, He Z, Yang Y, Deng Y, Tringe SG, Alvarez-Cohen L. 2015. High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. mBio 6:e02288-. doi: 10.1128/mBio.02288-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yergeau E, Bokhorst S, Kang S, Zhou J, Greer CW, Aerts R, Kowalchuk GA. 2012. Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J 6:692–702. doi: 10.1038/ismej.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishii S, Segawa T, Okabe S. 2013. Simultaneous quantification of multiple food- and waterborne pathogens by use of microfluidic quantitative PCR. Appl Environ Microbiol 79:2891–2898. doi: 10.1128/AEM.00205-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hermann-Bank ML, Skovgaard K, Stockmarr A, Larsen N, Mølbak L. 2013. The Gut Microbiotassay: a high-throughput qPCR approach combinable with next generation sequencing to study gut microbial diversity. BMC Genomics 14:788. doi: 10.1186/1471-2164-14-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ison SA, Delannoy S, Bugarel M, Nagaraja TG, Renter DG, den Bakker HC, Nightingale KK, Fach P, Loneragan GH. 2016. Targeted amplicon sequencing for single-nucleotide-polymorphism genotyping of attaching and effacing Escherichia coli O26:H11 cattle strains via a high-throughput library preparation technique. Appl Environ Microbiol 82:640–649. doi: 10.1128/AEM.03182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song Y, Ishii S, Rathnayake L, Ito T, Satoh H, Okabe S. 2013. Development and characterization of the partial nitrification aerobic granules in a sequencing batch airlift reactor. Bioresour Technol 139:285–291. doi: 10.1016/j.biortech.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi N, Oshiki M, Ito T, Segawa T, Hatamoto M, Kato T, Yamaguchi T, Kubota K, Takahashi M, Iguchi A, Tagawa T, Okubo T, Uemura S, Harada H, Motoyama T, Araki N, Sano D. 2017. Removal of human pathogenic viruses in a down-flow hanging sponge (DHS) reactor treating municipal wastewater and health risks associated with utilization of the effluent for agricultural irrigation. Water Res 110:389–398. doi: 10.1016/j.watres.2016.10.054. [DOI] [PubMed] [Google Scholar]
- 35.Koshkin AA, Rajwanshi VK, Wengel J. 1998. Novel convenient syntheses of LNA [2.2.1]bicycle nucleosides. Tetrahedron Lett 39:4381–4384. doi: 10.1016/S0040-4039(98)00706-0. [DOI] [Google Scholar]
- 36.Obika S, Nanbu D, Hari Y, Andoh JI, Morio KI, Doi T, Imanishi T. 1998. Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methyleneribonucleosides. Tetrahedron Lett 39:5401–5404. doi: 10.1016/S0040-4039(98)01084-3. [DOI] [Google Scholar]
- 37.Simeonov A, Nikiforov TT. 2002. Single nucleotide polymorphism genotyping using short, fluorescently labeled locked nucleic acid (LNA) probes and fluorescence polarization detection. Nucleic Acids Res 30:e91. doi: 10.1093/nar/gnf090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikenaga M, Sakai M. 2014. Application of locked nucleic acid (LNA) oligonucleotide-PCR clamping technique to selectively PCR amplify the SSU rRNA genes of bacteria in investigating the plant-associated community structures. Microbes Environ 29:286–295. doi: 10.1264/jsme2.ME14061q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meinhardt KA, Bertagnolli A, Pannu MW, Strand SE, Brown SL, Stahl DA. 2015. Evaluation of revised polymerase chain reaction primers for more inclusive quantification of ammonia-oxidizing archaea and bacteria. Environ Microbiol Rep 7:354–363. doi: 10.1111/1758-2229.12259. [DOI] [PubMed] [Google Scholar]
- 40.Wei W, Isobe K, Nishizawa T, Zhu L, Shiratori Y, Ohte N, Koba K, Otsuka S, Senoo K. 2015. Higher diversity and abundance of denitrifying microorganisms in environments than considered previously. ISME J 9:1954–1965. doi: 10.1038/ismej.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wetmur JG, Davidson N. 1968. Kinetics of renaturation of DNA. J Mol Biol 31:349–370. doi: 10.1016/0022-2836(68)90414-2. [DOI] [PubMed] [Google Scholar]
- 42.Herbold CW, Pelikan C, Kuzyk O, Hausmann B, Angel R, Berry D, Loy A. 2015. A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front Microbiol 6:731. doi: 10.3389/fmicb.2015.00731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murray PA, Zinder SH. 1984. Nitrogen fixation by a methanogenic archaebacterium. Nature 312:284–286. doi: 10.1038/312284a0. [DOI] [Google Scholar]
- 44.Leigh JA. 2000. Nitrogen fixation in methanogens: the archaeal perspective. Curr Issues Mol Biol 2:125–131. [PubMed] [Google Scholar]
- 45.Riederer-Henderson MA, Wilson PW. 1970. Nitrogen fixation by sulphate-reducing bacteria. J Gen Microbiol 61:27–31. doi: 10.1099/00221287-61-1-27. [DOI] [PubMed] [Google Scholar]
- 46.Bertics VJ, Sohm JA, Treude T, Chow CET, Capone DG, Fuhrman JA, Ziebis W. 2010. Burrowing deeper into benthic nitrogen cycling: the impact of bioturbation on nitrogen fixation coupled to sulfate reduction. Mar Ecol Prog Ser 409:1–15. doi: 10.3354/meps08639. [DOI] [Google Scholar]
- 47.Oshiki M, Shimokawa M, Fujii N, Satoh H, Okabe S. 2011. Physiological characteristics of the anaerobic ammonium-oxidizing bacterium ‘Candidatus Brocadia sinica’. Microbiology 157:1706–1713. doi: 10.1099/mic.0.048595-0. [DOI] [PubMed] [Google Scholar]
- 48.Oshiki M, Awata T, Kindaichi T, Satoh H, Okabe S. 2013. Cultivation of planktonic anaerobic ammonium oxidation (anammox) bacteria by using membrane bioreactor. Microbes Environ 28:436–443. doi: 10.1264/jsme2.ME13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ushiki N, Fujitani H, Aoi Y, Tsuneda S. 2013. Isolation of Nitrospira belonging to sublineage II from a wastewater treatment plant. Microbes Environ 28:346–353. doi: 10.1264/jsme2.ME13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujitani H, Ushiki N, Tsuneda S, Aoi Y. 2014. Isolation of sublineage I Nitrospira by a novel cultivation strategy. Environ Microbiol 16:3030–3040. doi: 10.1111/1462-2920.12248. [DOI] [PubMed] [Google Scholar]
- 51.Ishii S, Ashida N, Otsuka S, Senoo K. 2011. Isolation of oligotrophic denitrifiers carrying previously uncharacterized functional gene sequences. Appl Environ Microbiol 77:338–342. doi: 10.1128/AEM.02189-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishii S, Ashida N, Ohno H, Segawa T, Yabe S, Otsuka S, Yokota A, Senoo K. 2017. Noviherbaspirillum denitrificans sp. nov., a denitrifying bacterium isolated from rice paddy soil and Noviherbaspirillum autotrophicum sp. nov., a denitrifying, facultatively autotrophic bacterium isolated from rice paddy soil and proposal to reclassify Herbaspirillum massiliense as Noviherbaspirillum massiliense comb. nov. Int J Syst Evol Microbiol 67:1841–1848. doi: 10.1099/ijsem.0.001875. [DOI] [PubMed] [Google Scholar]
- 53.Hirakata Y, Oshiki M, Kuroda K, Hatamoto M, Kubota K, Yamaguchi T, Harada H, Araki N. 2016. Effects of predation by protists on prokaryotic community function, structure, and diversity in anaerobic granular sludge. Microbes Environ 31:279–287. doi: 10.1264/jsme2.ME16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 55.Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 58.Fish JA, Chai B, Wang Q, Sun Y, Brown CT, Tiedje JM, Cole JR. 2013. FunGene: the functional gene pipeline and repository. Front Microbiol 4:291. doi: 10.3389/fmicb.2013.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.