

ABSTRACT
Branched-chain amino acid (BCAA) degradation is a major source of propionyl coenzyme A (propionyl-CoA), a key precursor of erythromycin biosynthesis in Saccharopolyspora erythraea. In this study, we found that the bkd operon, responsible for BCAA degradation, was regulated directly by PccD, a transcriptional regulator of propionyl-CoA carboxylase genes. The transcriptional level of the bkd operon was upregulated 5-fold in a pccD gene deletion strain (ΔpccD strain) and decreased 3-fold in a pccD overexpression strain (WT/pIB-pccD), demonstrating that PccD was a negative transcriptional regulator of the operon. The deletion of pccD significantly improved the ΔpccD strain's growth rate, whereas pccD overexpression repressed WT/pIB-pccD growth rate, in basic Evans medium with 30 mM valine as the sole carbon and nitrogen source. The deletion of gdhA1 and the BcdhE1 gene (genes in the bkd operon) resulted in lower growth rates of ΔgdhA1 and ΔBcdhE1 strains, respectively, on 30 mM valine, further suggesting that the bkd operon is involved in BCAA degradation. Both bkd overexpression (WT/pIB-bkd) and pccD inactivation (ΔpccD strain) improve erythromycin production (38% and 64%, respectively), whereas the erythromycin production of strain WT/pIB-pccD was decreased by 48%. Lastly, we explored the applications of engineering pccD and bkd in an industrial high-erythromycin-producing strain. pccD deletion in industrial strain S. erythraea E3 (E3pccD) improved erythromycin production by 20%, and the overexpression of bkd in E3ΔpccD (E3ΔpccD/pIB-bkd) increased erythromycin production by 39% compared with S. erythraea E3 in an industrial fermentation medium. Addition of 30 mM valine to industrial fermentation medium further improved the erythromycin production by 23%, a 72% increase from the initial strain S. erythraea E3.
IMPORTANCE We describe a bkd operon involved in BCAA degradation in S. erythraea. The genes of the operon are repressed by a TetR regulator, PccD. The results demonstrated that PccD controlled the supply of precursors for biosynthesis of erythromycin via regulating the BCAA degradation and propionyl-CoA assimilation and exerted a negative effect on erythromycin production. The findings reveal a regulatory mechanism in feeder pathways and provide new strategies for designing metabolic engineering to increase erythromycin yield.
KEYWORDS: branched-chain amino acid degradation, propionyl-CoA metabolism, precursor supply, erythromycin biosynthesis
INTRODUCTION
Branched-chain amino acids (BCAAs) leucine, isoleucine, and valine are essential amino acids that not only are building blocks for protein synthesis but also play important physiological roles. Actinobacteria convert BCAAs into acetyl coenzyme A (acetyl-CoA), propionyl-CoA, and butyryl-CoA, which are important sources of precursors for biosynthesis of a range of different polyketides (1–8). A mutant of Streptomyces avermitilis deficient in the gene encoding branched-chain α-keto acid dehydrogenase lost the ability to produce avermectins only when the culture medium was supplemented with BCAAs (9, 10). In high-erythromycin-producing strains, genes encoding key enzymes of the BCAA catabolic pathway were strongly overexpressed in Saccharopolyspora erythraea (11, 12). Gene disruption of SACE_Lrp, an efficient regulator for transporting and catabolizing branched-chain amino acids in S. erythraea, results in a 25% increase in erythromycin production, and overexpression of SACE_5387–5386, which encodes a BCAA ABC transporter, also enhanced the erythromycin production, by 36% (13).
The first step of branched-chain amino acid degradation is transamination, which generates the corresponding α-keto acids of BCAAs (BCKA) using either leucine dehydrogenase, l-leucine aminotransferase, valine dehydrogenase, l-amino-acid oxidase, or the common branched-chain amino acid aminotransferase (BCAT) (14, 15). The second step is oxidative decarboxylation of BCKA to the corresponding acyl-CoA derivative coupled with dehydrogenation, which is carried out by the branched-chain α-keto acid dehydrogenase (BCDH) complex (14, 15). The further conversion of acyl-CoA derivatives of branched-chain amino acids isoleucine, leucine, and valine is mediated by individual catabolic pathways (16–18). The enzymes catalyzing the initial two steps in the BCAA catabolic pathway are critically important for BCAA degradation and utilization. α-Ketoglutaric acid as a cosubstrate for the transamination of BCAAs is essential in the first step of BCAA degradation. Glutamate dehydrogenase plays an important role in the BCAA degradation pathway by supplying α-ketoglutaric acid. Glutamate dehydrogenase-deficient strains grew poorly in Difco nutrient broth (5 g/liter peptone and 3 g/liter beef extract), whereas glucose supplementation improved their growth (19).
BCDH is a multienzyme complex that has been studied in several bacteria, including Staphylococcus aureus (20), Bacillus subtilis (21), Pseudomonas putida (22–25), Streptomyces coelicolor (26), and S. avermitilis (27). The BCDH enzyme complex is composed of four polypeptides: a dehydrogenase (E1α), a decarboxylase (E1β), a dihydrolipoamide acyltransferase (E2), and a dihydrolipoamide dehydrogenase (E3) (20). E1α-disrupted mutant strains lost the ability to grow on solid minimal medium containing BCAAs as the sole carbon source (9, 10). In addition to BCAA catabolism, BCDH is also required for the synthesis of monomethyl branched-chain fatty acids from BCAAs. A BCDH-deficient strain of S. aureus had reduced levels of BCFAs in the membrane, which reduced its adherence to eukaryotic cells and resulted in decreased survival in a murine host (20). Genes of the BCDH enzyme complex are usually organized in a cluster (such as the bkd operon) and coregulated by regulator BkdR in bacteria (2, 21, 23–26). BkdR is a global transcriptional regulator in Escherichia coli and acts as a leucine-responsive activator or repressor (28). Transcription of the bkd operon is also negatively controlled by CodY, a global regulator of gene expression in response to nutritional conditions in B. subtilis (21). The further conversion of acyl-CoA derivatives of pathways of branched-chain amino acids leucine and isovalerate was regulated by LiuR in Pseudomonas aeruginosa (17). Nevertheless, the regulatory mechanism underlying BCAA degradation in actinobacteria remains poorly understood, especially in some important industrial antibiotic-producing actinobacteria.
Recently, we showed that PccD (SACE_3396) is a negative transcriptional regulator of propionyl-CoA carboxylase (encoded by pccBCA, SACE_3398–3400) in erythromycin-producing S. erythraea (29). In this study, we found that the transcription unit of SACE_3880–3884 associated with BCAA degradation was regulated directly by PccD. The results indicated that PccD controlled the supply of precursors for the biosynthesis of erythromycin via regulating BCAA degradation and propionyl-CoA assimilation and exerted a negative effect on erythromycin production.
RESULTS
The SACE_3880 operon is regulated directly by PccD.
In order to further explore the PccD regulon in S. erythraea, a putative PccD-binding motif (A/TTGACGG/CTGT/CTGT/A) was obtained from the protected sequence of pccBC (SACE_3398–3399) and the promoter sequence of pccA (SACE_3400) using MEME (http://meme-suite.org/) (29). Based on the predicted PccD-binding motif, we further searched for PccD-binding sites of all genes upstream in S. erythraea. Using MAST/MEME tools, we obtained 46 transcription units (TUs) with putative PccD-binding motif (see Table S1 in the supplemental material).
Among the 46 TUs, we were interested in the SACE_3880 operon, which contains five genes, SACE_3880 to SACE_3884 (SACE_3880–3884) (Fig. 1A and B), that might be involved in valine, leucine, and isoleucine degradation (here named the bkd operon). A putative PccD-binding motif (ATGTCGGTGGAGT) is located in the translation initiation region of SACE_3880 (Fig. 1C). To examine the binding activity of PccD to the upstream region of bkd operon, electrophoretic mobility shift assays (EMSAs) were performed. As shown in Fig. 1D, obvious band shifts were observed as the entire promoter region (from −300 bp to +50 bp) of the bkd operon was incubated with purified recombinant His-PccD. Reverse transcription-quantitative PCR (qRT-PCR) results showed that the overexpression of pccD (approximately 5-fold) inhibited the transcription of the bkd operon 3-fold and deletion of pccD resulted in about 5-fold upregulation of the bkd operon compared with the wild-type (WT) strain (Fig. 1E). These results demonstrated that PccD was able to specifically bind the upstream region of the bkd operon and negatively regulated its transcription. Next, we deduced a conserved sequence (a/tTGa/tCGg/cTGnt/aGt/a) from the protected sequence of pccBC, the promoter sequence of pccA, and the bkd operon using MEME (http://meme-suite.org/) (Fig. 1F).
FIG 1.

The SACE_3880 operon is regulated directly by PccD. (A) Genetic organization of the bkd operon in the S. erythraea genome. aldH (SACE_3880), gdhA1 (SACE_3882), and SACE_3884 encode aldehyde dehydrogenase (NAD+), glutamate dehydrogenase [NAD(P)+], and the 2-oxoisovalerate dehydrogenase E1 component, respectively. (B) Identification of the cotranscription of the operon (primers were designed for cross subunit genomic DNA as indicated in panel A). The cDNA was reverse transcribed from the RNA extracted from WT cultured in TSB and subjected to semiquantitative PCR. Genome DNA of S. erythraea and no reverse transcriptase PCR products were used as positive and negative controls, respectively. Semiquantitative PCR products were evaluated on 1% agarose gels. (C) Upstream promoter regions of the bkd operon. Black lines indicate the PccD-binding site. (D) EMSAs of His-PccD protein with upstream promoter regions of gdhA1. The DNA probe (about 15 ng in a 10-μl reaction system) was incubated with a protein concentration gradient (0, 1, 3, and 5 μM). Unlabeled specific probe (200-fold) or nonspecific competitor DNA (200-fold, sonicated salmon sperm DNA) was used as the control. The free probes that did not bind with protein are denoted by an arrowhead. (E) qRT-PCR analysis of the transcription profiles of SACE_3880, SACE_3882, and SACE_3884 in WT, WT/pIB-pccD and ΔpccD strains cultured in balanced Evans medium (basic Evans medium with 140 mM glucose, 100 mM NaNO3). (F) Deduction of the PccD-binding motif in S. erythraea using MEME. The standard code of the Weblogo server is shown at the top. The GTG in wireframe is the initiation codon of SACE_3880.
gdhA1 and the BcdhE1 gene in the bkd operon are involved in BCAA degradation.
The bkd operon contains five genes (Fig. 1A). Based on annotation of the KEGG database, we found that three genes may participate in BCAA degradation: SACE_3880, putatively coding for aldehyde dehydrogenase AldH; SACE_3882, putatively coding for glutamate dehydrogenase [NAD(P)+] GdhA1; and SACE_3884, putatively coding for 2-oxoisovalerate dehydrogenase E1 component BCDH E1 (Fig. 2A). Glutamate dehydrogenase (GDH) and branched-chain α-keto acid dehydrogenase (BCDH E1) play a key role in BCAA degradation, but SACE_3882 (gdhA1) and SACE_3884 (here named BcdhE1 gene) have not yet been investigated in S. erythraea. To examine the importance of the bkd operon in BCAA degradation, a gdhA1 null mutant strain (ΔgdhA1 strain) and a BcdhE1 null mutant strain (ΔBcdhE1 strain) were constructed by deletion of 958 nucleotides (nt) of gdhA1 and 1,928 nt of BcdhE1 gene, respectively, using the clustered regularly interspaced short palindromic repeat(s) (CRISPR)/Cas9-mediated genome editing method.
FIG 2.

gdhA1 and the BcdhE1 gene in the bkd operon are involved in BCAA degradation. (A) Schematic of branched-chain amino acid (taking valine as an example) catabolic pathways in S. erythraea. GDH, glutamate dehydrogenase; BCDH, branched-chain α-keto acid dehydrogenase; AldH, aldehyde dehydrogenase; PCC, propionyl-CoA carboxylase. (B) Growth curves of S. erythraea WT, ΔgdhA1, and ΔgdhA1/pIB-gdhA1 strains cultivated in basic Evans medium with 30 mM valine and growth curves of ΔgdhA1 strain cultivated in basic Evans medium with 30 mM valine plus 170 mM α-ketoglutarate. (C) Growth curves of S. erythraea WT, ΔBcdhE1, and ΔBcdhE1/pIB-BcdhE1 strains cultivated in basic Evans medium with 30 mM valine and growth curves of the ΔBcdhE1 strain cultivated in basic Evans medium with 30 mM valine plus 140 mM glucose.
Due to the lack of an α-KG regeneration pathway, the S. erythraea GDH-deficient mutant is expected to be specifically unable to grow with BCAAs as the sole carbon and nitrogen source. Indeed, the ΔgdhA1 mutant revealed a decrease in growth rate on basic Evans medium with 30 mM valine as the sole carbon and nitrogen source (Fig. 2B). As shown in Fig. 2B, either the complement of gdhA1 or the addition of α-KG was sufficient to restore the growth of the ΔgdhA1 mutant under this condition. Therefore, these results suggest that GdhA1 is involved in the regeneration of α-KG for the first step (transamination reaction) of BCAA degradation. As shown in Fig. 2C, the ΔBcdhE1 mutant revealed an obvious growth defect on 30 mM valine as the sole carbon and nitrogen source. The absence of BCDH enzyme activity is expected to cause a starvation of carbon source (Fig. 2A). Accordingly, we found that the addition of 140 mM glucose to the culture medium could restore the growth of the ΔBcdhE1 mutant (Fig. 2C). Taken together, these findings demonstrate that the bkd operon plays an important role in BCAA degradation in S. erythraea, when BCAAs are the sole carbon and nitrogen source.
PccD controls BCAA degradation.
As shown in Fig. 2A, the propionyl-CoA carboxylase (PCC) pathway is a downstream part of BCAA degradation. The regulon of PccD regulator was identified to contain three transcript units (bkd operon, pccBC operon, and pccA gene) associated with BCAA degradation. To investigate the effects of PccD on the utilization of branched-chain amino acids in S. erythraea, we examined the growth of WT, ΔpccD, ΔpccD/pIB-pccD, and WT/PIB-pccD strains cultivated in basic Evans medium with 30 mM valine as the sole nitrogen and carbon source. As shown in Fig. 3, these strains revealed different growth behaviors. The deletion of pccD (ΔpccD) significantly increased the growth rate of S. erythraea compared with the WT strain. The complement of pccD gene (ΔpccD/pIB-pccD) eliminated the increase in growth rate of the ΔpccD mutant. Overexpression of pccD (WT/PIB-pccD) obviously decreased the growth rate, and the addition of 140 mM glucose led to a partial restoration of growth (Fig. 3; see also Fig. S1 in the supplemental material). These observations indicated that PccD is a transcriptional repressor of the genes involved in BCAA degradation and inhibits the utilization of BCAAs.
FIG 3.
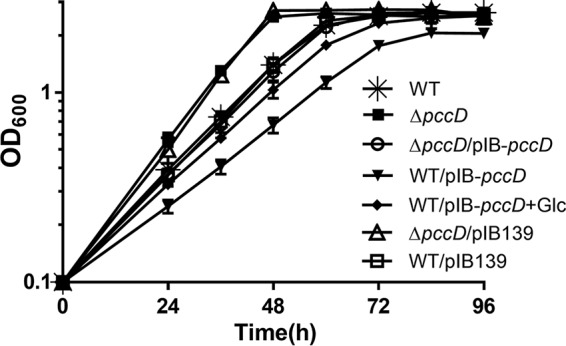
Effects of PccD on the utilization of BCAA. Growth curves of S. erythraea WT, pccD deletion (ΔpccD), pccD complementation (ΔpccD/pIB-pccD), and pccD overexpression (WT/pIB-pccD) strains grown in basic Evans medium with 30 mM valine and growth curves of WT/pIB-pccD grown in basic Evans medium with 30 mM valine plus 140 mM glucose.
Inactivation of PccD improves erythromycin production in S. erythraea.
Branched-chain amino acid degradation is a major source of propionyl-CoA, a key precursor of erythromycin biosynthesis, in S. erythraea. In the present work, it was observed that the PccD-regulated bkd operon was involved in the generation of propionyl-CoA from BCAAs. It is reasonable to propose that PccD may control the precursor supply of erythromycin biosynthesis by regulating the transcription of the bkd operon. To examine the effects of PccD and the bkd operon on erythromycin production in S. erythraea, the erythromycin concentrations of wild-type (WT), ΔpccD, ΔpccD/pIB-pccD, WT/pIB-pccD, and WT/pIB-bkd strains cultivated in industrial fermentation medium plus 30 mM valine were compared (Fig. 4). As shown in Fig. 4, pccD deletion (ΔpccD) improved the erythromycin production by 64%, and the erythromycin production of the pccD overexpression strain (WT/pIB-pccD) was decreased by 48% relative to WT. Overexpression of the bkd operon (strain WT/pIB-bkd) significantly increased erythromycin production by 38%.
FIG 4.
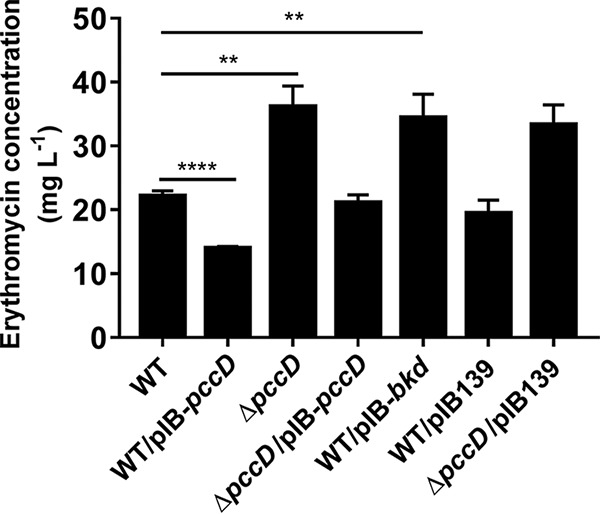
Effects of PccD on erythromycin production in S. erythraea NRRL2338. Supernatants (cultivated in industrial medium plus 30 mM valine) were collected after being cultivated for about 6 days. Erythromycin was measured by HPLC as described in Materials and Methods. Three independent replicates were used to calculate the standard deviations. **, P < 0.01; ****, P < 0.0001.
Engineering of industrial high-producing strain S. erythraea E3 for erythromycin yield increase.
In order to explore the applications of the above-described results in an industrial erythromycin-high-producing strain, we deleted the pccD gene in an industrial strain, S. erythraea E3, and then introduced the bkd overexpression plasmid pIB-bkd into it (E3ΔpccD), thus producing strain E3ΔpccD/pIB-bkd. As shown in Fig. 5, pccD deletion (E3ΔpccD) improved erythromycin production by 20%, and the deletion of pccD combined with overexpression of bkd (E3ΔpccD/pIB-bkd) increased erythromycin production by 39% compared with S. erythraea E3 in an industrial fermentation medium. The addition of 30 mM valine further improved erythromycin production by 23%, which is a 72% increase from that of the initial S. erythraea strain E3 cultivated in industrial fermentation medium.
FIG 5.
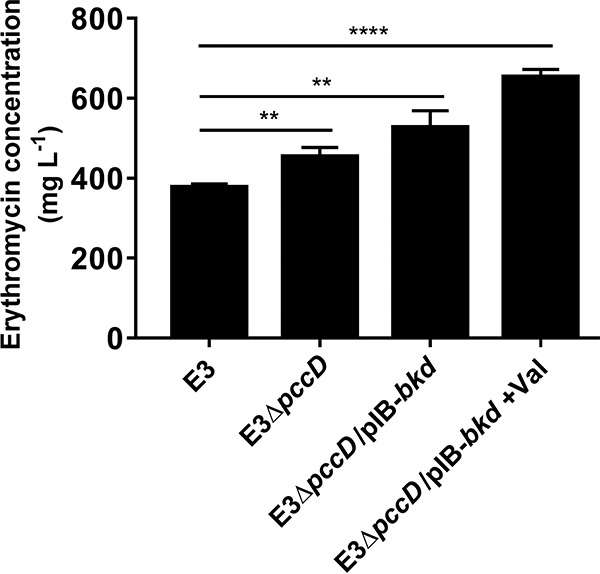
Engineering of industrial high-yielding S. erythraea strain E3 for increased erythromycin production. Supernatants (cultivated in industrial fermentation medium or industrial fermentation medium plus 30 mM valine) were collected after being cultivated for about 7 days. Erythromycin was measured by HPLC as described in Materials and Methods. Three independent replicates were used to calculate the standard deviations. **, P < 0.01; ****, P < 0.0001.
DISCUSSION
The PccD regulon in S. erythraea and its possible functions.
This study showed that the transcriptional regulator PccD binds to the upstream region of the bkd operon and acts as a repressor to directly control the transcription of the bkd operon (Fig. 1). The bkd operon contains five genes (Fig. 1A and B). Three of these genes (aldH, gdhA1, and BcdhE1 gene) in the regulon have putative functions that might be involved in BCAA degradation, based on the KEGG database. In this study, ΔgdhA1 and ΔBcdhE1 mutants demonstrated that the bkd operon plays an important role in BCAA degradation in S. erythraea. PccD was also demonstrated to negatively control the propionyl-CoA carboxylase genes (pccBCA) in S. erythraea (29). The pccBC and pccA operons were shown to have a major function in propionyl-CoA assimilation.
In most bacteria, BCAA degradation and propionyl-CoA assimilation are regulated by different transcriptional regulators (30–32). For example, BkdR (21, 23, 26) takes part in regulating BCAA degradation. LrpG (33), PrpR (34), PccR (35), and PccD (29) participated in propionyl-CoA assimilation. In Mycobacterium, acetyl/propionyl-CoA carboxylase (accD1A1) and branched-chain α-keto acid dehydrogenase (bkdABC) exist in the same operon and are regulated by a regulator, BkaR, simultaneously (36). In this study, propionyl-CoA carboxylase genes and the bkd operon were regulated by PccD in S. erythraea. Here, the physiological significance of PccD is in controlling the level of intracellular propionyl-CoA via regulation of BCAA degradation and propionyl-CoA assimilation in S. erythraea.
The PccD-regulated pathway and precursors of erythromycin biosynthesis.
Polyketides are a class of secondary metabolites produced by microorganisms and plants. They are synthesized by decarboxylative addition of malonyl thioesters, such as malonyl-CoA and methylmalonyl-CoA, to a common biosynthetic precursor (7, 8). Redirecting precursor metabolic fluxes is a useful approach in industrial fermentations to enhance metabolite production, including bioactive secondary metabolites produced by actinomycetes (37). As the starter unit of erythromycin production, the metabolic fluxes of propionyl-CoA and (S)-methyl-malonyl-CoA are very important for the production of erythromycin. In S. erythraea, the propionyl-CoA carboxylase (PCC) pathway and the methylmalonyl-CoA mutase (MCM) pathway are found to play an important role in the precursor supply (11, 38, 39). Engineering the methylmalonyl-CoA flow has already been proved to be helpful for increasing erythromycin production (38, 40, 41).
Branched-chain amino acids have long been known as an important source of a range of different polyketide precursors, such as propionyl-CoA and methylmalonyl-CoA (1–8). Some genes involved in the branched-chain amino acid synthesis and degradation pathway, such as ilvB (SACE_4565), acd (SACE_4125 and SACE_5025), and mmsA (SACE_4672), are significantly upregulated in industrial high-erythromycin-producing strains (11, 12). Thus, upregulating the metabolic pathways of BCAAs can improve the yield of erythromycin (12, 13).
In this study, we have described an operon involved in BCAA metabolism in S. erythraea. The genes of the operon are repressed by the TetR regulator PccD, which was previously shown to be a negative transcriptional regulator of propionyl-CoA carboxylase gene pccBCA. Our results demonstrated that PccD controlled the level of intracellular propionyl-CoA by regulating BCAA degradation and propionyl-CoA assimilation and exerted a negative effect on erythromycin production. These findings reveal a regulatory mechanism in feeder pathways and open the possibility of new designing strategies for metabolic engineering to increase the erythromycin yield.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. S. erythraea NRRL2338 was grown on R2YE (10.3% sucrose, 0.02% potassium sulfate, 1% magnesium chloride, 1% glucose, 0.5% yeast extract, and 0.01% Difco Casamino Acids) agar plates for 5 to 6 days at 30°C for sporulation. An agar piece of about 1 cm2 was inoculated in a 150-ml flask containing 30 ml of tryptone soya broth (TSB) medium and grown at 30°C and 200 rpm for 48 h for seed stock preparation. Next, about 0.5 ml of the seed culture was added to a 500-ml flask containing 50 ml basic Evans medium [25 mM TES (N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid), 10 mM KCl, 2 mM NaSO4, 2 mM citric acid, 0.25 mM CaCl2, 1.25 mM MgCl2, 1 mM NaMoO4, 2 mM NaH2PO4, and 0.5% Evans trace elements (40 mg liter−1 ZnCl, 200 mg liter−1 FeCl3 · 6H2O, 10 mg liter−1 CuCl2 · 2H2O, 10 mg liter−1 MnCl2 · 4H2O, 10 mg liter−1 Na2B4O7 · 10H2O, 10 mg liter−1 (NH4)6Mo7O24 · 4H2O, pH 7.0), pH 7.2] (42) supplemented with various amounts of carbon and nitrogen for transcription, phenotype, physiological, and biochemical tests.
TABLE 1.
Bacterial strains and plasmids used in the study
| Strain or plasmid | Characteristics | Source or reference |
|---|---|---|
| Strains | ||
| S. erythraea NRRL2338 | Used as parental strain, wild type | DSM 40517 |
| S. erythraea ΔpccD | S. erythraea SACE_3396 null mutant, thiostrepton resistant | 29 |
| S. erythraea WT/pIB-pccD | SACE_3396 overexpression strain, WT carrying pIB-pccD | 29 |
| S. erythraea WT/pIB139 | WT carrying vector pIB139 | This study |
| S. erythraea ΔpccD/pIB-pccD | SACE_3396 complementation strain, ΔpccD carrying pIB-pccD, which is under the control of pccD promoter | This work |
| S. erythraea ΔpccD/pIB139 | Empty plasmid control, ΔpccD carrying pIB139 | This study |
| S. erythraea ΔgdhA1 | S. erythraea SACE_3882 null mutant | This study |
| S. erythraea ΔgdhA1/pIB-gdhA1 | gdhA1 complementation strain, ΔgdhA1 carrying pIB-gdhA1 | This study |
| S. erythraea ΔgdhA1/pIB139 | Empty plasmid control, ΔgdhA1 carrying pIB139 whose ermE* promoter was replaced by SACE_3880–3884 promoter | This study |
| S. erythraea ΔbcdhE1 | S. erythraea SACE_3884 null mutant | This study |
| S. erythraea ΔbcdhE1/pIB-bcdhE1 | bcdhE1 complementation strain, ΔBcdhE1 carrying pIB-BcdhE1 gene | This study |
| S. erythraea ΔbcdhE1/pIB139 | The empty plasmid control, ΔBcdhE1 gene carrying pIB139 whose ermE* promoter was replaced by SACE_3880–3884 promoter | This study |
| S. erythraea WT/pIB-bkd | SACE_3880–3884 overexpression strain, WT carrying pIB-bkd | This study |
| E. coli Rosetta (DE3) | F− ompT hsdSB(rB− mB−) gal dcm λ(DE3)pRARE2(Camr) | Novagen |
| S. erythraea E3 | Industrial high-erythromycin-producing strain | SKLBE, ECUST |
| S. erythraea E3ΔpccD | S. erythraea E3 SACE_3396 null mutant, thiostrepton resistant | This study |
| S. erythraea E3ΔpccD/pIB-bkd | SACE_3880–3884 overexpression strain, E3ΔpccD carrying pIB-bkd | This study |
| S. erythraea WT/pKC-egfp | egfp overexpression strain, WT carrying pKC-egfp | This study |
| Plasmids | ||
| pET19b | Expression vector, Ampr | Novagen |
| pET-pccD | pET19b derivative carrying SACE_3396 | 29 |
| pKC1139 | acc(3)IV, pSG5, pBR322, oriTRK2 | 49 |
| pIB139 | E. coli-S. erythraea integrative shuttle vector containing a strong constitutive ermE* promoter, apramycin resistance | 50 |
| pIB-pccD | pIB139 carrying an extra SACE_3396 for overexpression (under the control of the ermE* promoter)/complementation (under the control of pccD promoter) | 29; this study |
| pIB-gdhA1 | pIB139 carrying an extra SACE_3882 for complementation under the control of SACE_3880–3884 promoter | This study |
| pIB-BcdhE1 | pIB139 carrying an extra SACE_3884 for complementation under the control of SACE_3880–3884 promoter | This study |
| pIB-bkd | pIB139 carrying an extra SACE_3880–3884 for gene overexpression | This study |
| pIB-egfp | pIB139 carrying an extra egfp gen for gene overexpression | This study |
| pUC-fd term | pUC57 carrying a synthetic fd terminator gene originating from bacteriophage | This study |
| pUC19-tsr | pUC19 derivative containing a 1.36-kb fragment of a thiostrepton resistance cassette in the BamHI/SmaI sites | 51 |
| pUC-pccD | pUC19-tsr, with the 1.5-kb DNA fragments upstream and downstream of the pccD gene inserted upstream and downstream of tsr correspondingly | This study |
| pKC-egfp | pKC1139 containing PermE-egfp-fd cassette for egfp overexpression | This study |
| pUC-Sencas9 | pUC57 carrying a synthetic codon-optimized cas9 gene | This study |
| pKC-cas9 | pKC1139 containing PermE-Sencas9-fd cassette | This study |
| pKC-cas9-sgRNA | pKC1139 containing PermE-Sencas9-fd cassette and PermE-sgRNA cassette | This study |
| pkC-CRISPR/Cas9 | pKC1139 containing PermE-Sencas9-fd cassette, PermE-sgRNA cassette, and homologous arm for gene deletion | This study |
Construction of gene-deficient mutant, complementation, and overexpression strains.
For an in-frame deletion of 958 nt in gdhA1 and of 1,928 nt in BcdhE1 gene, the CRISPR/Cas9-mediated genome-editing method was used as previously described (43, 44). The CRISPR/Cas9 genome-editing system used in this study is a single plasmid, named pKC-CRISPR/Cas9. The flowchart for pKC-CRISPR/Cas9 construction is shown in Fig. 2, and all the primers used for the construction of pKC-CRISPR/Cas9 are listed in Table 2. First, a Sencas9 cassette was constructed by fusing PermE, Sencas9, and fd terminator (PermE is an ermE promoter in S. erythraea; Sencas9 is a synthetic codon-optimized cas9 gene; the fd terminator gene, originating from bacteriophage, is synthesized). Second, the Sencas9 cassette was subcloned into the EcoRV/EcoRI site of pKC1139 and named pKC-cas9. Third, a synthetic genomic RNA (sgRNA) scaffold with PermE (Table 2) was synthesized and subcloned into the XbaI/HindIII site of pKC-cas9, generating pKC-cas9-sgRNA plasmid. Lastly, two homologous arms flanking the target gene were amplified, fused, and subcloned into the HindIII site of pKC-cas9-sgRNA to generate pKC-CRISPR/Cas9. The plasmid was introduced into the protoplast of S. erythraea NRRL2338 by polyethylene glycol (PEG)-mediated transformation (45). The primers used for gene deletion and identification of the gdhA1 and BcdhE1 gene deletion mutants are listed in Table 2. The gdhA1-deleted and BcdhE1 gene-deleted mutants were screened by apramycin resistance and PCR (see Fig. S3 in the supplemental material).
TABLE 2.
Oligonucleotides used in the study
| Function and name of oligonucleotide | Sequence (5′ to 3′) |
|---|---|
| Primers for PCR amplification of EMSA probe with biotin labeling | |
| EMSA3880-3884F | AGCCAGTGGCGATAAGTCGTCGCTGCACACCCGCATGTC |
| EMSA3880-3884R | AGCCAGTGGCGATAAGTGGTGTCCGGGCCGATCACTGAG |
| Primers for real-time RT-PCR | |
| RT3880F | CGGTCAACAAGGTCGGCTTCAC |
| RT3880R | ATCAGGTTGCCCAGCGAGGT |
| RT3882F | GGGTGCTGGTCATCGACAACAC |
| RT3882R | TCGTCCTCGGTCATGCCCATGT |
| RT3884F | CTCGGCTACTACAGCATCGGTTC |
| RT3884R | ATCGTGGAGGTCTGCGGGAT |
| RT8101F | GTTGCGATGCCGTGAGGT |
| RT8101R | CGGGTGTTACCGACTTTCA |
| Primers for identification of cotranscription of SACE_3880–3884 | |
| sqRT3880/3881F | TCAGCGTCAACAACTCCACCTC |
| sqRT3880/3881R | AGGCAGCCGGTAGTCCATCT |
| sqRT3881/3882F | AGATCACGCCCGACCACAAC |
| sqRT3881/3882R | GGAGACGCACACGACCTTCT |
| sqRT3882/3883F | CCGACTTCATCGCCAACGCA |
| sqRT3882/3883R | TCAGGTGGATCGGGTTGAGCAT |
| sqRT3883/3884F | GCACCGCAAGCAGAACACCAA |
| sqRT3883/3884R | CGGACGAACCGATGCTGTAGTAG |
| Guide sequence for CRISPR/Cas9-mediated deletion of gdhA1 and BcdhE1 gene | |
| 3882sgRNA | CGTGCCCGAGTCGCTCGGCGGGG |
| 3884sgRNA | GCGAAAACCCTGGTGCGCCACGG |
| Primers of homologous arm for CRISPR/Cas9-mediated deletion of gdhA1 and BcdhE1 gene | |
| 3882upHaF | ATTACCCTGTTATCCCTAAAGCTTCGGTTTCGAGTCCGTGCTCGGCACGATCGC |
| 3882upHaR | GTGTTGTCCCGGGACCGCACACGACCTTCTCCGGGCCCCACTCGT |
| 3882dwHaF | GGAGAAGGTCGTGTGCGGTCCCGGGACAACACCGCCGTCGTGCTC |
| 3882dwHaR | AACGACGGCCAGTGCCAAGCTTCTTGTGCCGTCCGGCCGAGACCGGCTC |
| 3884upHaF | ATTACCCTGTTATCCCTAAAGCTTGGACGGTCTCGACGTCGGGTACTGGCTGG |
| 3884upHaR | ATGGTCTCCTCGTCCAGACTCGTGCCCGGACGAACCGATGCTGTAGTAGC |
| 3884dwHaF | CGTCCGGGCACGAGTCTGGACGAGGAGACCATCGAGCAGGAGGCGC |
| 3884dwHaR | AACGACGGCCAGTGCCAAGCTTACGCGTCGACGCAGGTCAGTTCGGCCGACA |
| Primers for identification of gdhA1-deleted and BcdhE1 gene-deleted mutants | |
| Qc3882JLF | GCACGGTCAACAAGGTCGGCTTC |
| Qc3882JLR | GAAGCTGCACACCACGACCGAGT |
| Qc3884JLF | ATGGGCATGACCGAGGACGACG |
| Qc3884JLR | GACGAAGGCGAGCGTGAAGTGCTT |
| Primers for construction of the complementary strain | |
| 3396compF | CCAATGCATCTCCGACGGGGCGACCGAGGAGTAG |
| 3396compR | GCTCTAGACTACCCATAAGTGAATGCCCGATAGGCATCGGTGC |
| P3880-3884F | CCAATGCATGATGCGCAGCACGGTGCCACACGG |
| P3880-3884R | GGAATTCCATATGCGACATTCCACCTCTTCCTCGTCGCTGATC |
| 3882compF | GGAATTCCATATGCAGGAGATCGACGAGTGGGGCCCGGAGAAG |
| 3882compR | GCTCTAGATTACGCGGTCACGGCCTCGGG |
| 3884compF | GGAATTCCATATGAACGGCCGCGAGCCCATCGACG |
| 3884compR | GCTCTAGATCAGTGCTTGACCAGGCGCAGCG |
| Primers for construction of the overexpression strain | |
| 3880-3884overF | GGAATTCCATATGGAGTCGACGTTGAGCTCAGTGATCGGC |
| 3880-3884overR | GCTCTAGATCAGTGCTTGACCAGGCGCAGCG |
| Primers for construction of the E3ΔpccD strain | |
| pUC3396upF | CCCAAGCTTATGACCGCGAACTCCGAGACGCTCGAC |
| pUC3396upR | GCTCTAGAGTGCCGGTAGAGCGCGGAGGGC |
| pUC3396dwF | CGGGGTACCGGTTCATCGACATCCTGGTCACCGAGGTC |
| pUC3396dwR | CCGGAATTCGGCGGATGATGCCGTTCCACTCCTGC |
| Primers for E3ΔpccD strain confirmation by PCR | |
| UPF | GTACGCGGTTGAGGTGACCAGGAACTGCGG |
| Ut1 | CAGAACATACCGGTCCGCCTCATCGACTCCTCG |
| Dt1 | CGGAGAGAACGACGGGAAGGGAGAAGACGTAACC |
| DWR | CACGCCAGGTTGATGTCGGCACCGAGG |
| fd terminator sequence | CCCGGGAACCCGGCCGCGTCCGGCGCCCCCGCCGCCTTCGACGAGATCCCCGCAAAAGCGGCCTTTGACTCCCTGCAAGCCTCAGCGACCGAATATATCGGTTATGCGTGGGCGATGGTTGTTGTCATTGTCGGCGCAACTATCGGTATCAAGCTGTTTAAGAAATTCACCTCGAAAGCAAGCTGATAAACCGATACAATTAAAGGCTCCTTTTGGAGCCTTTTTTTTTGGAGATTTTCAACGTGAAAAAATTATTATTCGCAATTCCTTTAGTTGTTCCTTTCTATTCTCACTCCGCTGAAACTGTTGAAAGTTGTTTAGCAAAACCTCATACAGAAAATTCATTTACTAACGTCTGGAAAGACGACAAAACTTTAGATCTGGGGAATTC |
| Primers for construction of the PermE-egfp-fd cassette | |
| egfp-ermEF | AGCTTTGATATCGCGAGTGTCCGTTCGAGTGGCGG |
| egfp-ermER | CTCGCCCTTGCTCACCATCGCTGGATCCTACCAACCGGC |
| egfp-egfpF | GACAATCGTGCCGGTTATGGTGAGCAAGGGCGAGGAGCT |
| egfp-egfpR | CGGCCGGGTTCCCGGGTTACTTGTACAGCTCGTCCATGCCGAG |
| egfp-fdF | CGAGCTGTACAAGTAACCCGGGAACCCGGCCGC |
| egfp-fdR | CCGGAATTCCCCAGATCTAAAGTTTTGTCGTCTTTCCAG |
| Primers for construction of the PermE-Sencas9-fd cassette | |
| cas9-ermEF | AGCTTTGATATCGCGAGTGTCCGTTCGAGTGGCG |
| cas9-ermER | TGTACTTCTTGTCCATCGCTGGATCCTACCAACCGGC |
| cas9-cas9F | TTGGTAGGATCCAGCGATGGACAAGAAGTACAGCATCGGCCTG |
| cas9-cas9R | CGGCCGGGTTCCCGGGTCAGTCGCCGCCGAGCTGG |
| cas9-fdF | GCTCGGCGGCGACTGACCCGGGAACCCGGCCGC |
| cas9-fdR | CCGGAATTCCCCAGATCTAAAGTTTTGTCGTCTTTCCAG |
| Cassette sequences | |
| PermE-3882sgRNA | GCTCTAGAGCGAGTGTCCGTTCGAGTGGCGGCTTGCGCCCGATGCTAGTCGCGGTTGATCGGCGATCGCAGGTGCACGCGGTCGATCTTGACGGCTGGCGAGAGGTGCGGGGAGGATCTGACCGACGCGGTCCACACGTGGCACCGCGATGCTGTTGTGGGCTGGACAATCGTGCCGGTTCGTGCCCGAGTCGCTCGGCGGGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTAAGCTTGGG |
| PermE-3884sgRNA | GCTCTAGAGCGAGTGTCCGTTCGAGTGGCGGCTTGCGCCCGATGCTAGTCGCGGTTGATCGGCGATCGCAGGTGCACGCGGTCGATCTTGACGGCTGGCGAGAGGTGCGGGGAGGATCTGACCGACGCGGTCCACACGTGGCACCGCGATGCTGTTGTGGGCTGGACAATCGTGCCGGTTGCGAAAACCCTGGTGCGCCACGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTAAGCTTGGG |
For an insertional deletion of the pccD gene in S. erythraea E3, a previously described homologous recombination strategy was used (29). Briefly, 1.5-kb DNA fragments upstream and downstream of the pccD gene locus were amplified from S. erythraea E3 genomic DNA by PCR using the primer pairs pUC3396upF/R and pUC3396dwF/R (Table 2). The PCR products were digested with HindIII/XbaI and KpnI/EcoRI and subsequently inserted into the corresponding sites of pUC19-tsr, creating pUC-pccD knockout plasmids (Table 1). The thiostrepton resistance cassette amplified from pUC-pccD knockout plasmids by PCR using the primer pair pUC3396upF/pUC3396dwR was transferred into S. erythraea E3 by PEG-mediated transformation. The mutants were selected by thiostrepton on R3M agar {103 g liter−1 sucrose, 4 g liter−1 yeast extract, 4 g liter−1 Casamino Acids, 0.25 g liter−1 K2SO4, 4 g liter−1 tryptone, 22 g liter−1 agar, 10 g liter−1 glucose, 50 mM CaCl2, 50 mM MgCl2, 25 mM Tris-HCl [pH 7.0], 2.5 mM NaOH, 0.185 mM KH2PO4, 0.02% trace elements [40 mg liter−1 ZnCl, 200 mg liter−1 FeCl3 · 6H2O, 10 mg liter−1 CuCl2 · 2H2O, 10 mg liter−1 MnCl2 · 4H2O, 10 mg liter−1 Na2B4O7 · 10H2O, 10 mg liter−1 (NH4)6Mo7O24 · 4H2O, pH 7.0]} plates. The selected mutants were verified by PCR (see Fig. S4 in the supplemental material). Primers are listed in Table 2.
For the complementary ΔgdhA1/pIB-gdhA1 and ΔBcdhE1/pIB-BcdhE1 strains, the ermE promoter of pIB139 was first replaced by the SACE_3880–3884 promoter, which was amplified with the primer pair P3880-3884F/R (Table 2) and cloned into NsiI/NdeI sites of the pIB139. Next, the PCR products of gdhA1 and BcdhE1 gene were amplified with the primer pairs 3882compF/R and 3884compF/R, respectively (Table 2), and cloned into the NdeI/XbaI sites of the pIB139 plasmid containing the SACE_3880–3884 promoter. Their empty plasmid control ΔgdhA1/pIB139 and ΔBcdhE1/pIB139 strains were constructed by introducing the pIB139 plasmid containing the SACE_3880–3884 promoter into the protoplast of ΔgdhA1 and ΔBcdhE1 strains, respectively. The desired strains were screened by apramycin resistance.
For creating the complementary ΔpccD/pIB-pccD strain, the pccD gene was PCR amplified with the primer pair 3396compF/R (Table 2). The PCR products were then cloned into the NsiI/XbaI sites of pIB139, creating the pIB-pccD plasmid, in which pccD is under the control of its own promoter (Table 1). The complementary plasmid of pIB-pccD was introduced into the protoplast of the ΔpccD strain by PEG-mediated transformation. Its empty plasmid control strain, ΔpccD/pIB139, was constructed by the introduction of pIB139 into the protoplast of ΔpccD. The desired strains were screened by apramycin resistance.
For creating strains WT/pIB-bkd and E3ΔpccD/pIB-bkd, the bkd operon (SACE_3880–3884) was PCR amplified with the primer pair 3880–3884 over-F/R (Table 2). The PCR products were then cloned into the NdeI/XbaI sites of pIB139, creating the pIB-bkd plasmid (Table 1). The overexpression plasmids were introduced into the protoplast of S. erythraea NRRL2338 and E3ΔpccD by PEG-mediated transformation. The desired strains were screened by apramycin resistance.
RNA preparation and qRT-PCR.
Cells were cultured and harvested at the exponential growth phase (the WT strain was harvested at 36 h, and ΔpccD and ΔpccD/pIB-pccD strains were harvested at 48 h) by centrifugation at 4°C, 4,000 × g, for 10 min. Total RNA was prepared using the RNAprep Pure Cell/Bacteria kit (Tiangen Biotech, Beijing, China). Total RNA (1 μg) was reverse transcribed using the PrimeScript RT reagent kit (TaKaRa, Kusatsu, Japan). qPCR was conducted using the SYBR Premix Ex Taq GC kit (TaKaRa, Japan), and about 100 ng cDNA was added to a final PCR volume of 20 μl. PCR was performed using the primers listed in Table 2. The PCR products were evaluated on 1% agarose gels, and DNA was visualized by ethidium bromide staining. PCR assays were carried out using a CFX96 Real-Time system (Bio-Rad, CA), and the thermocycling conditions were as follows: 95°C for 5 min, followed by 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 30 s, with a final extension cycle at 72°C for 10 min. 16S_rRNA was used for the reference gene (SACE_8101). qRT-PCR validation data for amplification efficiency, calibration curves with slope and R2, and specificity (melt) are provided in Data Set S1 in the supplemental material. Analysis of the qRT-PCR (Fig. 1E) data is provided in Data Set S2 in the supplemental material. STDEV indicates the standard deviation from three independent experiment replicates.
Protein overexpression and purification.
The PccD protein was overexpressed and purified as previously described (29). Briefly, E. coli Rosetta (DE3) isolates containing pET-pccD (Table 1) were grown in 5 ml LB medium with 100 mg liter−1 ampicillin in an orbital shaker (200 rpm, 37°C) for 12 h. Next, 2.5 ml of seed culture was added to a 250-ml flask containing 50 ml TB medium (24 g liter−1 yeast extract, 12 g liter−1 tryptone, 0.4% glycerol, 17 mM KH2PO4, 72 mM K2HPO4) followed by incubation at 20°C for 20 to 24 h. His6-tagged PccD protein (His-PccD) was purified from Rosetta (DE3) isolates harboring pET-pccD as previously described (29). The purity of the purified protein was checked using SDS-PAGE. The protein concentration was determined using the Bradford assay (46).
Electrophoretic mobility shift assay.
A 300-nt probe derived from upstream of SACE_3880 was amplified by PCR using the primers listed in Table 2. The PCR products were labeled with biotin using a universal biotinylated primer (5′biotin-AGCCAGTGGCGATAAG-3′). The biotin-labeled PCR products were purified using the PCR purification kit (Shanghai Generay Biotech, China) as EMSA probes. EMSA was carried out using a chemiluminescent EMSA kit (Beyotime Biotechnology, China) as previously described (47). Biotin-labeled DNA probes (about 15 ng in a 10-μl reaction system) were incubated with a gradient concentration of proteins (1 to 5 μM) at 25°C for 20 min. For the control groups, unlabeled specific probe (200-fold) or nonspecific competitor DNA (200-fold, sonicated salmon sperm DNA) was used. Samples were separated by 6% nondenaturing PAGE gels in ice-cold 0.5× Tris-borate-EDTA at 160 V, and the bands were detected using BeyoECL Plus (Beyotime Biotechnology, China).
Fermentation and erythromycin concentration determination.
The culture conditions for laboratory-scale fermentation of erythromycin were as previously described (11). Briefly, S. erythraea NRRL23338, high-producing strain E3, and their genetically engineered strains were grown on agar plates [10 g liter−1 cornstarch, 10 g liter−1 corn steep liquor, 3 g liter−1 NaCl, 3 g liter−1 (NH4)2SO4, 2 g liter−1 CaCO3, and 2 g liter−1 agar, pH 7.2] at 30°C for sporulation. An agar piece of about 1 cm2 was inoculated in a 500-ml flask containing 50 ml of the seed medium [50 g liter−1 cornstarch, 18 g liter−1 soybean flour, 13 g liter−1 corn steep liquor, 3 g liter−1 NaCl, 1 g liter−1 (NH4)2SO4, 1 g liter−1 NH4NO3, 5 g liter−1 soybean oil, and 6 g liter−1 CaCO3, pH 6.8 to 7.0] at 34°C and 220 rpm for 48 h. Seed culture [5 ml] was inoculated in a 500-ml flask containing 60 ml of the industrial fermentation medium [40 g liter−1 cornstarch, 30 g liter−1 soybean flour, 30 g liter−1 dextrin, 2 g liter−1 (NH4)2SO4, 10 g liter−1 soybean oil, and 6 g liter−1 CaCO3 (pH 7.0 to 7.2)] with or without 30 mM valine at 30°C and 220 rpm for 6 to 7 days.
Erythromycin was extracted from the fermentation cultures as previously described (48). Erythromycin concentration was measured using an Agilent 1100 high-pressure liquid chromatography (HPLC) system with a C18 column (5 μm inner diameter, 250 by 4.6 mm), which was equilibrated with 45% solution A (K2HPO4, 30 mM, pH 8.0) and 55% solution B (acetonitrile). An isocratic program was carried out at a flow rate of 1 ml min−1, using a UV detector at 215 nm.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (31730004, 21575089, and 31700058). We have no conflict of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00049-18.
REFERENCES
- 1.Kamoun P. 1992. Valine is a precursor of propionyl-CoA. Trends Biochem Sci 17:175–176. doi: 10.1016/0968-0004(92)90258-B. [DOI] [PubMed] [Google Scholar]
- 2.Leiser A, Birch A, Robinson JA. 1996. Cloning, sequencing, overexpression in Escherichia coli, and inactivation of the valine dehydrogenase gene in the polyether antibiotic producer Streptomyces cinnamonensis. Gene 177:217–222. doi: 10.1016/0378-1119(96)00305-8. [DOI] [PubMed] [Google Scholar]
- 3.Lounes A, Lebrihi A, Benslimane C, Lefebvre G, Germain P. 1995. Regulation of valine catabolism by ammonium in Streptomyces ambofaciens, producer of spiramycin. Can J Microbiol 41:800–808. doi: 10.1139/m95-110. [DOI] [PubMed] [Google Scholar]
- 4.Tang L, Zhang YX, Hutchinson CR. 1994. Amino acid catabolism and antibiotic synthesis: valine is a source of precursors for macrolide biosynthesis in Streptomyces ambofaciens and Streptomyces fradiae. J Bacteriol 176:6107–6119. doi: 10.1128/jb.176.19.6107-6119.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang L, Zhang YX, Hutchinson CR. 1994. The genetic basis of precursor supply for the biosynthesis of macrolide and polyether antibiotics. Ann N Y Acad Sci 721:105–116. doi: 10.1111/j.1749-6632.1994.tb47382.x. [DOI] [PubMed] [Google Scholar]
- 6.Katz L. 1997. Manipulation of modular polyketide synthases. Chem Rev 97:2557–2576. doi: 10.1021/cr960025+. [DOI] [PubMed] [Google Scholar]
- 7.Hopwood DA. 1993. Genetic engineering of Streptomyces to create hybrid antibiotics. Curr Opin Biotechnol 4:531–537. doi: 10.1016/0958-1669(93)90073-6. [DOI] [PubMed] [Google Scholar]
- 8.Katz L, Donadio S. 1993. Polyketide synthesis: prospects for hybrid antibiotics. Annu Rev Microbiol 47:875–912. doi: 10.1146/annurev.mi.47.100193.004303. [DOI] [PubMed] [Google Scholar]
- 9.Hafner EW, Holley BW, Holdom KS, Lee SE, Wax RG, Beck D, McArthur HA, Wernau WC. 1991. Branched-chain fatty acid requirement for avermectin production by a mutant of Streptomyces avermitilis lacking branched-chain 2-oxo acid dehydrogenase activity. J Antibiot (Tokyo) 44:349–356. doi: 10.7164/antibiotics.44.349. [DOI] [PubMed] [Google Scholar]
- 10.Denoya CD, Fedechko RW, Hafner EW, McArthur HA, Morgenstern MR, Skinner DD, Stutzman-Engwall K, Wax RG, Wernau WC. 1995. A second branched-chain alpha-keto acid dehydrogenase gene cluster (bkdFGH) from Streptomyces avermitilis: its relationship to avermectin biosynthesis and the construction of a bkdF mutant suitable for the production of novel antiparasitic avermectins. J Bacteriol 177:3504–3511. doi: 10.1128/jb.177.12.3504-3511.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li YY, Chang X, Yu WB, Li H, Ye ZQ, Yu H, Liu BH, Zhang Y, Zhang SL, Ye BC, Li YX. 2013. Systems perspectives on erythromycin biosynthesis by comparative genomic and transcriptomic analyses of S. erythraea E3 and NRRL23338 strains. BMC Genomics 14:523. doi: 10.1186/1471-2164-14-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karnicar K, Drobnak I, Petek M, Magdevska V, Horvat J, Vidmar R, Baebler S, Rotter A, Jamnik P, Fujs S, Turk B, Fonovic M, Gruden K, Kosec G, Petkovic H. 2016. Integrated omics approaches provide strategies for rapid erythromycin yield increase in Saccharopolyspora erythraea. Microb Cell Fact 15:93. doi: 10.1186/s12934-016-0496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Chen Y, Wang W, Ren M, Wu P, Wang Y, Li C, Zhang L, Wu H, Weaver DT, Zhang B. 2017. Engineering of an Lrp family regulator SACE_Lrp improves erythromycin production in Saccharopolyspora erythraea. Metab Eng 39:29–37. doi: 10.1016/j.ymben.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Martin RR, Marshall VD, Sokatch JR, Unger L. 1973. Common enzymes of branched-chain amino acid catabolism in Pseudomonas putida. J Bacteriol 115:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazakov AE, Rodionov DA, Alm E, Arkin AP, Dubchak I, Gelfand MS. 2009. Comparative genomics of regulation of fatty acid and branched-chain amino acid utilization in proteobacteria. J Bacteriol 191:52–64. doi: 10.1128/JB.01175-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aguilar JA, Zavala AN, Diaz-Perez C, Cervantes C, Diaz-Perez AL, Campos-Garcia J. 2006. The atu and liu clusters are involved in the catabolic pathways for acyclic monoterpenes and leucine in Pseudomonas aeruginosa. Appl Environ Microbiol 72:2070–2079. doi: 10.1128/AEM.72.3.2070-2079.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forster-Fromme K, Hoschle B, Mack C, Bott M, Armbruster W, Jendrossek D. 2006. Identification of genes and proteins necessary for catabolism of acyclic terpenes and leucine/isovalerate in Pseudomonas aeruginosa. Appl Environ Microbiol 72:4819–4828. doi: 10.1128/AEM.00853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoschle B, Gnau V, Jendrossek D. 2005. Methylcrotonyl-CoA and geranyl-CoA carboxylases are involved in leucine/isovalerate utilization (Liu) and acyclic terpene utilization (Atu), and are encoded by liuB/liuD and atuC/atuF, in Pseudomonas aeruginosa. Microbiology 151:3649–3656. doi: 10.1099/mic.0.28260-0. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, Kim BG. 2016. NAD(+)-specific glutamate dehydrogenase (EC.1.4.1.2) in Streptomyces coelicolor; in vivo characterization and the implication for nutrient-dependent secondary metabolism. Appl Microbiol Biotechnol 100:5527–5536. doi: 10.1007/s00253-016-7433-8. [DOI] [PubMed] [Google Scholar]
- 20.Singh VK, Hattangady DS, Giotis ES, Singh AK, Chamberlain NR, Stuart MK, Wilkinson BJ. 2008. Insertional inactivation of branched-chain alpha-keto acid dehydrogenase in Staphylococcus aureus leads to decreased branched-chain membrane fatty acid content and increased susceptibility to certain stresses. Appl Environ Microbiol 74:5882–5890. doi: 10.1128/AEM.00882-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debarbouille M, Gardan R, Arnaud M, Rapoport G. 1999. Role of bkdR, a transcriptional activator of the sigL-dependent isoleucine and valine degradation pathway in Bacillus subtilis. J Bacteriol 181:2059–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madhusudhan KT, Huang G, Burns G, Sokatch JR. 1990. Transcriptional analysis of the promoter region of the Pseudomonas putida branched-chain keto acid dehydrogenase operon. J Bacteriol 172:5655–5663. doi: 10.1128/jb.172.10.5655-5663.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madhusudhan KT, Huang N, Sokatch JR. 1995. Characterization of BkdR-DNA binding in the expression of the bkd operon of Pseudomonas putida. J Bacteriol 177:636–641. doi: 10.1128/jb.177.3.636-641.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madhusudhan KT, Hester KL, Friend V, Sokatch JR. 1997. Transcriptional activation of the bkd operon of Pseudomonas putida by BkdR. J Bacteriol 179:1992–1997. doi: 10.1128/jb.179.6.1992-1997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madhusudhan KT, Luo J, Sokatch JR. 1999. In vitro transcriptional studies of the bkd operon of Pseudomonas putida: L-branched-chain amino acids and D-leucine are the inducers. J Bacteriol 181:2889–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sprusansky O, Stirrett K, Skinner D, Denoya C, Westpheling J. 2005. The bkdR gene of Streptomyces coelicolor is required for morphogenesis and antibiotic production and encodes a transcriptional regulator of a branched-chain amino acid dehydrogenase complex. J Bacteriol 187:664–671. doi: 10.1128/JB.187.2.664-671.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skinner DD, Morgenstern MR, Fedechko RW, Denoya CD. 1995. Cloning and sequencing of a cluster of genes encoding branched-chain alpha-keto acid dehydrogenase from Streptomyces avermitilis and the production of a functional E1 [alpha beta] component in Escherichia coli. J Bacteriol 177:183–190. doi: 10.1128/jb.177.1.183-190.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calvo JM, Matthews RG. 1994. The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiol Rev 58:466–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Z, Wang M, Ye BC. 2017. The TetR family transcriptional regulator PccD negatively controls propionyl coenzyme A assimilation in Saccharopolyspora erythraea. J Bacteriol 199:e00281-17. doi: 10.1128/JB.00281-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brock M, Buckel W. 2004. On the mechanism of action of the antifungal agent propionate. Eur J Biochem 271:3227–3241. doi: 10.1111/j.1432-1033.2004.04255.x. [DOI] [PubMed] [Google Scholar]
- 31.Gregersen N. 1981. The specific inhibition of the pyruvate dehydrogenase complex from pig kidney by propionyl-CoA and isovaleryl-Co-A. Biochem Med 26:20–27. doi: 10.1016/0006-2944(81)90026-0. [DOI] [PubMed] [Google Scholar]
- 32.Maruyama K, Kitamura H. 1985. Mechanisms of growth inhibition by propionate and restoration of the growth by sodium bicarbonate or acetate in Rhodopseudomonas sphaeroides S. J Biochem 98:819–824. doi: 10.1093/oxfordjournals.jbchem.a135340. [DOI] [PubMed] [Google Scholar]
- 33.Datta P, Shi L, Bibi N, Balazsi G, Gennaro ML. 2011. Regulation of central metabolism genes of Mycobacterium tuberculosis by parallel feed-forward loops controlled by sigma factor E (sigma(E)). J Bacteriol 193:1154–1160. doi: 10.1128/JB.00459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plassmeier J, Persicke M, Puhler A, Sterthoff C, Ruckert C, Kalinowski J. 2012. Molecular characterization of PrpR, the transcriptional activator of propionate catabolism in Corynebacterium glutamicum. J Biotechnol 159:1–11. doi: 10.1016/j.jbiotec.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Carter MS, Alber BE. 2015. Transcriptional regulation by the short-chain fatty acyl coenzyme A regulator (ScfR) PccR controls propionyl coenzyme A assimilation by Rhodobacter sphaeroides. J Bacteriol 197:3048–3056. doi: 10.1128/JB.00402-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balhana RJ, Swanston SN, Coade S, Withers M, Sikder MH, Stoker NG, Kendall SL. 2013. bkaR is a TetR-type repressor that controls an operon associated with branched-chain keto-acid metabolism in Mycobacteria. FEMS Microbiol Lett 345:132–140. doi: 10.1111/1574-6968.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olano C, Lombo F, Mendez C, Salas JA. 2008. Improving production of bioactive secondary metabolites in actinomycetes by metabolic engineering. Metab Eng 10:281–292. doi: 10.1016/j.ymben.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC, Weber JM. 2006. Effects of methylmalonyl-CoA mutase gene knockouts on erythromycin production in carbohydrate-based and oil-based fermentations of Saccharopolyspora erythraea. J Ind Microbiol Biotechnol 33:600–609. doi: 10.1007/s10295-006-0094-3. [DOI] [PubMed] [Google Scholar]
- 39.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. 2007. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
- 40.Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC, Weber JM. 2007. Engineering of the methylmalonyl-CoA metabolite node of Saccharopolyspora erythraea for increased erythromycin production. Metab Eng 9:293–303. doi: 10.1016/j.ymben.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reeves AR, Cernota WH, Brikun IA, Wesley RK, Weber JM. 2004. Engineering precursor flow for increased erythromycin production in Aeromicrobium erythreum. Metab Eng 6:300–312. doi: 10.1016/j.ymben.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Fink D, Weissschuh N, Reuther J, Wohlleben W, Engels A. 2002. Two transcriptional regulators GlnR and GlnRII are involved in regulation of nitrogen metabolism in Streptomyces coelicolor A3(2). Mol Microbiol 46:331–347. doi: 10.1046/j.1365-2958.2002.03150.x. [DOI] [PubMed] [Google Scholar]
- 43.Huang H, Zheng G, Jiang W, Hu H, Lu Y. 2015. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin (Shanghai) 47:231–243. doi: 10.1093/abbs/gmv007. [DOI] [PubMed] [Google Scholar]
- 44.Tong Y, Charusanti P, Zhang L, Weber T, Lee SY. 2015. CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol 4:1020–1029. doi: 10.1021/acssynbio.5b00038. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto H, Maurer KH, Hutchinson CR. 1986. Transformation of Streptomyces erythraeus. J Antibiot (Tokyo) 39:1304–1313. doi: 10.7164/antibiotics.39.1304. [DOI] [PubMed] [Google Scholar]
- 46.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 47.Liao CH, Yao LL, Ye BC. 2014. Three genes encoding citrate synthases in Saccharopolyspora erythraea are regulated by the global nutrient-sensing regulators GlnR, DasR, and CRP. Mol Microbiol 94:1065–1084. doi: 10.1111/mmi.12818. [DOI] [PubMed] [Google Scholar]
- 48.Tsuji K, Goetz JF. 1978. HPLC as a rapid means of monitoring erythromycin and tetracycline fermentation processes. J Antibiot (Tokyo) 31:302–308. doi: 10.7164/antibiotics.31.302. [DOI] [PubMed] [Google Scholar]
- 49.Bierman M, Logan R, O'Brien K, Seno ET, Rao RN, Schoner BE. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- 50.Wilkinson CJ, Hughes-Thomas ZA, Martin CJ, Bohm I, Mironenko T, Deacon M, Wheatcroft M, Wirtz G, Staunton J, Leadlay PF. 2002. Increasing the efficiency of heterologous promoters in actinomycetes. J Mol Microbiol Biotechnol 4:417–426. [PubMed] [Google Scholar]
- 51.Wu P, Pan H, Zhang C, Wu H, Yuan L, Huang X, Zhou Y, Ye BC, Weaver DT, Zhang L, Zhang B. 2014. SACE_3986, a TetR family transcriptional regulator, negatively controls erythromycin biosynthesis in Saccharopolyspora erythraea. J Ind Microbiol Biotechnol 41:1159–1167. doi: 10.1007/s10295-014-1449-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.