

Short abstract
Nonerythroid αII-spectrin is a structural protein whose roles in the nucleus have just begun to be explored. αII-spectrin is an important component of the nucleoskelelton and has both structural and non-structural functions. Its best known role is in repair of DNA ICLs both in genomic and telomeric DNA. αII-spectrin aids in the recruitment of repair proteins to sites of damage and a proposed mechanism of action is presented. It interacts with a number of different groups of proteins in the nucleus, indicating it has roles in additional cellular functions. αII-spectrin, in its structural role, associates/co-purifies with proteins important in maintaining the architecture and mechanical properties of the nucleus such as lamin, emerin, actin, protein 4.1, nuclear myosin, and SUN proteins. It is important for the resilience and elasticity of the nucleus. Thus, αII-spectrin’s role in cellular functions is complex due to its structural as well as non-structural roles and understanding the consequences of a loss or deficiency of αII-spectrin in the nucleus is a significant challenge. In the bone marrow failure disorder, Fanconi anemia, there is a deficiency in αII-spectrin and, among other characteristics, there is defective DNA repair, chromosome instability, and congenital abnormalities. One may speculate that a deficiency in αII-spectrin plays an important role not only in the DNA repair defect but also in the congenital anomalies observed in Fanconi anemia , particularly since αII-spectrin has been shown to be important in embryonic development in a mouse model. The dual roles of αII-spectrin in the nucleus in both structural and non-structural functions make this an extremely important protein which needs to be investigated further. Such investigations should help unravel the complexities of αII-spectrin’s interactions with other nuclear proteins and enhance our understanding of the pathogenesis of disorders, such as Fanconi anemia , in which there is a deficiency in αII-spectrin.
Impact statement
The nucleoskeleton is critical for maintaining the architecture and functional integrity of the nucleus. Nonerythroid α-spectrin (αIISp) is an essential nucleoskeletal protein; however, its interactions with other structural and non-structural nuclear proteins and its functional importance in the nucleus have only begun to be explored. This review addresses these issues. It describes αIISp’s association with DNA repair proteins and at least one proposed mechanism of action for its role in DNA repair. Specific interactions of αIISp with other nucleoskeletal proteins as well as its important role in the biomechanical properties of the nucleus are reviewed. The consequences of loss of αIISp, in disorders such as Fanconi anemia, are examined, providing insights into the profound impact of this loss on critical processes known to be abnormal in FA, such as development, carcinogenesis, cancer progression and cellular functions dependent upon αIISp’s interactions with other nucleoskeletal proteins.
Keywords: Nonerythroid alpha spectrin, DNA interstrand cross-link repair, telomere function and dysfunction, Fanconi anemia, nucleoskeleton, peripheral nucleoskeletal proteins
Introduction
The nucleus houses not only the genome but also a complex group of proteins that associates with the nucleoskeleton and which contributes to the structural and functional complexity of the nucleus. This network of proteins is essential for the structural integrity of the nucleus as well as for proper regulation of DNA metabolism, DNA replication and transcription and DNA repair.1–9 Among these proteins is nonerythroid alpha spectrin (αIISp).7,10–14 In the nucleus, αIISp is best known for its role in DNA repair and chromosome stability.12–19 It directly interacts with damaged DNA and with proteins involved in DNA repair. In addition, it associates with structural proteins in the nucleoskeleton such as lamin, emerin, actin, protein 4.1, and nuclear myosin7,19–21 and through these interactions is thought to make an important contribution to the structural integrity of the nucleus as well as its elasticity.7,22
αIISp consists of an extended array of triple α-helical repeats which contributes to the elasticity of the nucleoskeleton with which it associates.21,23–26 It contains a SH3 domain, which is a site that plays an important role in protein–protein interactions and assembly of protein networks involved in intracellular signaling, signal transduction, and protein synthesis.27–31 We have shown that the Fanconi anemia protein, FANCG, binds directly to the SH3 domain of αIISp and have hypothesized that this binding is important in maintaining the stability of αIISp in the nucleus.32 We have demonstrated that αIISp is important in repair of DNA interstrand cross-links (ICLs) in both genomic (non-telomeric) and telomeric DNA and plays a critical role in maintaining genomic stability after DNA damage.12–19 Based on these studies, we have proposed that αIISp acts as a scaffold to recruit DNA repair proteins to sites of damage.12,18
In addition to αIISp’s interaction with DNA repair proteins, it also interacts with a number of other functionally important groups of proteins in the nucleus (Figure 1). These include structural proteins, such as lamin, emerin, actin, nuclear myosin, proteins important in chromatin remodeling, Fanconi anemia (FA) proteins, and proteins involved in transcription and RNA processing (Figure 1).7,19–21 Thus, αIISp’s interaction with a diverse group of proteins in the nucleus indicates that it has multiple roles in various nuclear processes (Figure 1). This review will concentrate on the functional importance of the interaction of αIISp in the nucleus of human cells with proteins involved in DNA repair, particularly those involved in the repair of DNA ICLs, and with structural proteins associated with the nucleoskeleton. This is an area of expanding research as new proteins interacting with αIISp are identified and new roles for these interactions are demonstrated.
Figure 1.

Overview of the localization of αIISp in normal human cells. αIISp is present in the cytoplasm where it is an important component of the cytoskeleton and interacts with a number of proteins including βII spectrin, actin and a number of accessory proteins.23–25,33–39 In the nucleus, it is present in the peripheral nucleoskeleton, where it interacts with lamins, emerin, actin, protein 4.1, nuclear myosin and LINC complex proteins.7,19–21 αIISp is also present in the inner nucleoskeleton where it interacts with DNA repair proteins, FA proteins, chromatin remodeling proteins, transcription and RNA processing proteins, and βIVΣ5 spectrin.19
αIISp interacts with DNA repair proteins and is linked to DNA repair
αIISp has long been recognized as an important component of the cytoskeletal network.23–25 It interacts with β-spectrins in the cytoplasm to form a tetramer of α/β spectrin which plays a role in cell architecture and structural support of the plasma membrane, trafficking of proteins, vesicles and organelles, signal transduction, synaptic transmission in neurons, cell division and proliferation, cell adhesion, and cell development and morphogenesis (Figure 1).23–25,33–39 αIISp is also present in the nucleus but a similar association with β-spectrins has not, as yet, been demonstrated.19
In the nucleus, αIISp has been shown to play an important role in DNA repair.12–19 It interacts with proteins involved in DNA interstrand cross-link repair, nucleotide excision repair (NER), homologous recombination repair (HRR), and nonhomologous end joining, (NHEJ).19 Its role in repair of DNA ICLs has been extensively studied.11–19 DNA ICL repair is a complex process and can occur in both replicating and non-replicating DNA.33–36 It involves a number of steps which include damage recognition and incision, translesion DNA synthesis, homologous recombination, and NER.45–51
Multiple lines of evidence have demonstrated an involvement of αIISp in the repair of DNA ICLs: (1) Purified bovine brain αIISp binds directly to a DNA substrate containing 4,5′,8-trimethylpsoralen (TMP) ICLs as does αIISp from HeLa cell nuclei.12 Based on the crystal structure of αIISp, we have proposed that αIISp could interact with the DNA in its minor groove via hydrogen bonding between polar residues present on the surface of αIISp’s α-helical repeat units and the N3 of purines and O2 of pyrimidines in DNA.12 After ICL damage, the minor groove would open up due to formation of an ICL. This would enhance the ability of αIISp to bind to DNA after ICL damage.12 (2) αIISp interacts with XPF, an endonuclease involved in ICL repair and production of incisions at the site of an ICL.14,45,48,52 After ICL damage, αIISp co-localizes with XPF in nuclear foci (Figure 2(a)).13,16,17 Since αIISp binds directly to DNA containing ICLs, this indicates that these foci represent co-localization of these proteins at sites of ICLs. (3) The time course for formation of αIISp and XPF foci in the nucleus is the same, indicating that they are involved in the same steps in ICL repair (Figure 3).13 (4) αIISp is needed for the recruitment of XPF to sites of DNA ICLs and for production of incisions produced by XPF at these sites.15,17 Knockdown of αIISp by siRNA leads to a loss in localization of XPF to nuclear foci after damage with a DNA ICL agent.15,17 (5) Incisions produced by XPF at a site-specific TMP ICL on a DNA substrate are inhibited by a monoclonal antibody against αIISp from normal human lymphoblastoid cells which specifically recognizes and binds to αIISp.12 Purified αIISp enhances incisions produced by XPF at sites of ICLs on this substrate.12 These studies have led us to propose that αIISp acts as a scaffold to aid in the recruitment of XPF to sites of ICLs; in its absence, XPF is not recruited and there is loss of incisions at these sites12,18 (6) αIISp is critical for chromosomal stability and cell survival after DNA ICL damage.15,18 Knock-down of αIISp by siRNA leads to decreased chromosomal stability and decreased cell survival after treatment with a DNA ICL agent.15 Thus, αIISp plays an important role in DNA ICL repair and its interaction with specific DNA repair proteins is critical for this process.
Figure 2.
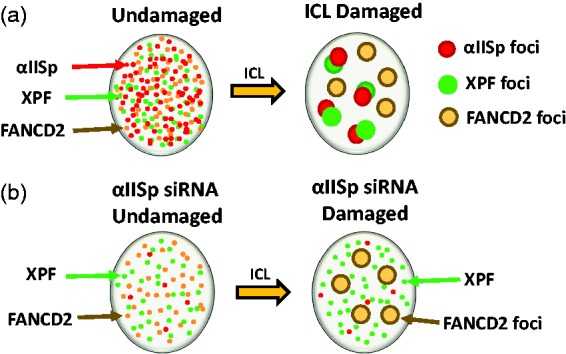
Diagrammatic representation of the formation of αIISp, XPF, and FANCD2 foci in the nuclei of normal human cells after damage with a DNA ICL agent. (a) In undamaged cells, αIISp, XPF, FANCD2 are localized diffusely in the nucleus; 16 h after damage with either 8-MOP plus UVA light or mitomycin C (MMC), αIISp and XPF are co-localize in nuclear foci at sites of ICLs.13 FANCD2 has also formed nuclear foci; however, these foci do no colocalize with αIISp foci and it is inferred from this that they do not co-localize with XPF foci.64 Since, after DNA ICL damage, FANCD2 foci form before those of αIISp or XPF foci (at 2 h vs. 8 h) and follow a different time course for formation, we have proposed that FANCD2 acts upstream of αIISp and XPF.64 Thus, at the same point in time after ICL damage, FANCD2 do not localize on the same sites on DNA as αIISp and XPF, hence the different localization of FANCD2 foci compared to those of αIISp and XPF. (b) In normal human cells in which αIISp has been knocked-down by siRNA, XPF and FANCD2 are localized diffusely in the nucleus; however, levels of αIISp are only ∼30% of those found in non-treated cells; 16 h after ICL damage, FANCD2 is localized in nuclear foci; however, XPF does not form nuclear foci.13,64 Thus, αIISp is needed for formation of XPF nuclear foci and localization of XPF to sites of DNA ICLs, but it is not needed for the localization of FANCD2 to sites of damage.64
Figure 3.

Compiled time course for appearance of αIISp, XPF, FANCA, FANCD2, and γ-H2AX nuclear foci after damage with a DNA ICL agent. After treatment of normal human cells with a DNA ICL agent, 8-MOP plus UVA light, and, at the indicated time after DNA damage, the percentage of nuclei showing multiple αIISp, XPF, FANCA, FANCD2, and γ-H2AX foci was determined at time points from 0 to 72 h after treatment. At each time point, nuclear foci for 100 nuclei were counted. Nuclei were counted as positive if they contained four or more foci. Error bars represent SEM. (Modified from Zhang et al.64 with permission from Wiley Periodicals, Inc. Notations of XPF and FANCA foci from Sridharan et al.13).
αIISp interacts with FA proteins; Its association with FANCA
FA is an inherited bone marrow failure disorder. One of the identifying hallmarks of this disorder is a deficiency in ability to repair DNA ICLs.46,47,51–60 Clinically, it is characterized by bone marrow failure, developmental abnormalities, chromosomal instability, and a high predisposition to development of cancer.54–56,59–63 We have shown that there is a deficiency in αIISp in cells from a number of FA complementation groups.11,15,16 This deficiency has been shown to correlate with a defect in DNA ICL repair, as demonstrated by decreased unscheduled DNA synthesis (UDS) and decreased cell survival after damage.14 FA thus serves as an excellent model for elucidation of the effects of loss of αIISp on cellular processes in human cells.
A core complex of FA proteins is involved in the preincision steps in the ICL repair process.46,47,57,60,63 This complex is critical for the monoubiquitination of the FA proteins, FANCD2, and FANCI.46,47,57,60,63 One of the proteins in this core complex is FANCA. In addition to its role as a component of the core complex, we have shown that FANCA also interacts with αIISp and XPF at the site of damage.11–13 It co-localizes with both αIISp and XPF in nuclear foci in cells at sites of ICLs created by psoralen plus UVA light (Figure 4(a)) and co-immunoprecipitates with both αIISp and XPF.13 It also binds to a synthesized DNA substrate containing a psoralen interstrand cross-link.12 The time course for formation of FANCA nuclear foci is the same as that for both αIISp and XPF (Figure 3).13,64 These three proteins co-localize in nuclear foci from 10 to 22 h after ICL damage after which time they are no longer observed (Figure 3).13 These studies thus demonstrate that αIISp associates with both XPF and FANCA at sites of DNA ICLs, indicating their involvement in the ICL repair process. The exact role of FANCA in this step is not yet clear.
Figure 4.
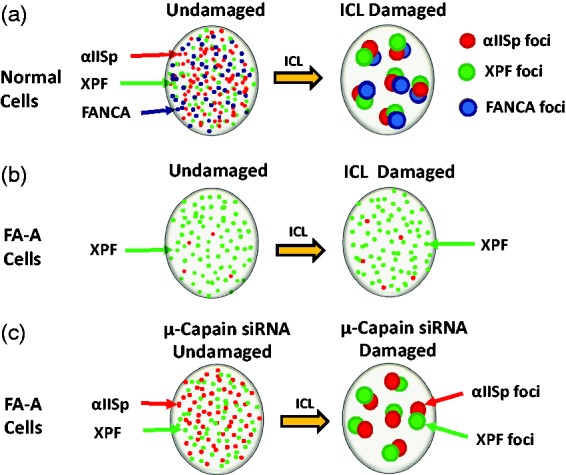
Diagrammatic representation of the formation of αIISp, XPF, and FANCA foci in the nuclei of normal human cells and FA-A cells after damage with a DNA ICL agent. (a) In undamaged cells, αIISp, XPF, and FANCA are localized diffusely in the nucleus; 16 h after damage with either 8-MOP plus UVA light or MMC, αIISp, XPF, and FANCA co-localize with each other in nuclear foci at sites of ICLs.13 (b) In FA-A cells, in which levels of αIISp are ∼30% of normal levels, XPF is localized diffusely in the nucleus. FANCA is not discernable and there are very low levels of αIISp; 16 h after ICL damage, XPF does not form nuclear foci and there are few αIISp foci.13,64 This indicates that αIISp is needed for formation of XPF nuclear foci and localization of XPF to sites of DNA ICLs.64 (c) In FA-A cells, after treatment with µ-calpain siRNA, levels of αIISp are restored to normal and both αIISp and XPF are localized diffusely in the nucleus.16 Sixteen hours after damage with an ICL agent, MMC, both αIISp and XPF foci form and these foci co-localize at sites of damage. 16 This indicates that αIISp is needed for the formation of XPF foci at sites of DNA ICLs.
αIISp and FANCD2 dissociate from each other after DNA ICL damage
αIISp associates with another FA protein, FANCD2, in the nucleus of normal human cells before DNA ICL damage.64 However, after FANCD2 is monoubiquitinated in the initial steps of DNA ICL repair, FANCD2 and αIISp dissociate from each other.64 We have shown that following ICL damage, FANCD2 localizes to nuclear foci which form before those of αIISp. αIISp and FANCD2 do not co-localize in the same foci (Figure 2(a)).64 Formation of FANCD2 foci follows a different time course than those αIISp, XPF, and FANCA (Figure 3).64 FANCD2 localizes to foci that appear 2 h after damage with the DNA ICL agent, 8-MOP plus UVA light, plateau at 16 h and are still present at 72 h after damage.64 In contrast, αIISp, XPF, and FANCA foci are first observed at 10 h after damage, peak at 16 h and are no longer observed by 24 h after ICL damage (Figure 3). This indicates that FANCD2 is recruited to sites of damage before αIISp, XPF, and FANCA and acts upstream of all three proteins.64 Results similar to these have been obtained using Xenopus extracts.48 These studies showed that FANCD2 is recruited to sites of damage before XPF.48 Collectively, these studies indicate that following DNA ICL damage and monoubiquitination of FANCD2, αIISp, XPF, and FANCA co-localize to sites of damage and act downstream of FANCD2 and that αIISp and FANCA play a role with XPF in the incision step in ICL repair. They also indicate that FANCD2 is involved in an earlier step in this process as well as in subsequent steps in the ICL repair process, such as homologous recombination, as has been proposed.41
Our studies in normal human cells have also shown that αIISp is not needed for the monoubiquitination of FANCD2.64 Knock-down of αIISp has no effect on the localization of FANCD2 to nuclear foci or chromatin.64 This indicates that αIISp is not needed for the functioning of monoubiquitinated FANCD2 (FANCD2-Ub), which further indicates that it acts downstream of FANCD2-Ub. Studies using Xenopus extracts have similarly shown that XPF is not required for the monoubiquitination of FANCD2.48 Thus, two proteins shown to play a role in DNA ICL repair, αIISp, and XPF are targeted to the same site after ICL damage and this site is different from that of FANCD2-Ub and is downstream of that of FANCD2.
FANCG binds to both αIISp and XPF-ERCC1
We have demonstrated, using yeast two-hybrid analysis as well as co-immunoprecipitation, that FANCG has strong binding affinity for αIISp.32 It has a consensus sequence that binds to the Src-homology 3 (SH3) domain of αIISp.32 SH3 domains are modular domains that are involved in protein–protein interactions and assembly of protein networks involved in intracellular signaling, protein synthesis, and cellular organization.29–31 Three major classes of protein ligands bind to SH3 domains: class I, class II, and class 1@.31,65–69 The SH3 domain of αIISp preferentially binds to class 1@ ligands.69 A number of FA proteins have motifs with consensus sequences that can bind to SH3 domains.32 These motifs represent another important class of motifs in FA proteins that could interact with cellular proteins containing SH3 domains, such as those involved in signal transduction and intracellular signaling.32 FANCG contains a class 1@ consensus sequence.32 Of particular interest, we have shown that FANCG specifically binds to the SH3 domain of αIISp via this consensus sequence.32 FANCG also has binding affinity to XPF-ERCC1.32 It contains seven tetratricopeptide repeat (TPR) motifs, which are motifs involved in protein–protein interactions.70–73 We have shown that TPRs 1, 2, 3, and 6 are important for binding of full length FANCG with the central domain of ERCC1 (residues 120–220).74 ERCC1 binds to XPF via its C-terminal domain (residues 220–297), which is different from the nuclease domain of XPF that is involved in production of incisions at sites of damage.75–77 Thus, we propose that FANCG, through its binding to both αIISp and XPF/ERCC1, acts as a link between αIISp and XPF/ERCC1 at sites of damage.
Model for αIISp’s role in DNA ICL repair and its mechanism of action
Based on evidence described above that αIISp is critical for repair of DNA ICLs, we have proposed a model for the role of αIISp in DNA ICL repair (Figure 5). According to this model, when DNA replication is blocked at the site of an ICL, a FA core complex comprising FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and associated proteins, in conjunction with FANCM, is recruited to the damaged DNA.46,47,60,78–80 The FA core complex, through its E2-ubiquitin conjugating activity and E3-ubiquitin ligase activity, leads to the monoubiquitination of FANCD2 (FANCD2-Ub) and FANCI (FANCI-Ub) to form a heterodimer known as ID2.46,47,79,81 The ID2 heterodimer localizes to the damaged DNA.46,47,79,81 αIISp is recruited and binds to DNA at the site of the ICL downstream of and at a different site than ID2.12,64 It is not known whether FANCD2-Ub or ID2 is involved in recruitment of αIISp to sites of ICLs. FANCG binds to αIISp and recruits XPF-ERCC1 which binds to FANCG.32,74 Thus, αIISp, via its binding to FANCG, aids in the recruitment of XPF-ERCC1 to the site of an ICL. XPF-ERCC1 is then involved in the incision step of ICL repair in conjunction with the nuclease/protein complex, SLX4-SLX1, leading to unhooking of the ICL.45–49,80 Other nucleases may also be involved in this step.46,47,49,80 The importance of αIISp in recruitment of XPF-ERCC1 is demonstrated by the loss of localization of XPF to sites of ICLs when αIISp is knocked down (Figure 2(b)).15 Translesion DNA synthesis then takes place by a multi-subunit translesion polymerase.46,47,60,61,80 In the later stages of ICL repair, the broken DNA duplex is repaired by homologous recombination by the cooperative functioning of a series of downstream proteins and the adducted DNA base is removed.46,47,57,80 The additional FA proteins and other repair proteins involved in these later steps, as well as in earlier steps in the repair process, are described in a number of reviews43,46,58,60,80 and are not shown in this model since the emphasis has been put on where αIISp fits into the ICL repair process.
Figure 5.

A model for αIISp’s role in DNA ICL repair and a mechanism of action proposed by our laboratory. When DNA replication is stalled at the site of an ICL, the FA core complex, in conjunction with FANCM, is recruited to the DNA.46,60,78–80 The core complex monoubiquitinates FANCD2 and FANCI (FANCD2-Ub and FANCI-Ub), which form a heterodimer (ID2) that binds to the damaged DNA.46,47,79,81 αIISp is recruited to the DNA at the site of the ICL and binds to the DNA downstream from the ID2 complex and at a different site.12,64 FANCG binds to the SH3 domain of αIISp via a consensus sequence that recognizes SH3 domains.32 FANCG in turn recruits XPF-ERCC1 to the site of damage.74 FANCG binds to the central domain of ERCC1; the TPR motifs of FANCG are involved in this interaction.74 Thus, αIISp, via its binding to FANCG, aids in the recruitment of XPF-ERCC1 to the site of an ICL. XPF-ERCC1 is then involved in the incision of DNA at this site in conjunction with other nucleases such as SLX4-SLX1.46–49,80 This leads to unhooking of the ICL. Translesion DNA synthesis then takes place by a translesion DNA polymerase (TLS pol).46,47,60,61,80 In subsequent steps, the DNA duplex is repaired by homologous recombination (HR) via a series of downstream proteins and the adducted base is removed by nucleotide excision repair (NER).46,47,57,80 Additional FA proteins and other repair proteins involved in these steps are not shown in this model since the emphasis has been put on where αIISp fits into the ICL repair process.
Our combined studies thus propose a mechanism of action for αIISp in its role in DNA repair acting as a scaffold to recruit proteins to sites of damage: αIISp directly binds to DNA at the site of a DNA ICL; it directly binds, via its SH3 domain, to FANCG, and FANCG in turn directly binds to ERCC1. XPF, through its binding , is thus recruited, via αIISp, to the site of damage where it can incise the DNA.
Regulation of αIISp stability by FA proteins
αIISp is an essential protein in cells and its depletion can lead to cell death.15,37,38,82,83 Therefore, maintaining its stability in cells is critical. We have evidence that FA proteins aid in this process and help maintain cellular levels of αIISp.16 We have proposed that this is accomplished by regulation of cleavage of αIISp by the protease, µ-calpain.16 In cells, µ-calpain cleaves αIISp into distinct cleavage products and this process is important for a number of functions mediated by αIISp.84–87
Increased activity of µ-calpain can lead to enhanced cleavage of αIISp which has deleterious effects on a number of cellular processes.84–87 We have shown that in cells from a number of FA complementation groups, FA-A, FA-C, FA-D2, FA-F, and FA-G, there is a 2.5–3.5 fold increase in µ-calpain activity compared to normal cells.16 This correlates with increased cleavage of αIISp in these cells.16 We have proposed that FA proteins play a critical role in maintaining αIISp stability in cells and preventing its cleavage by µ-calpain.16,18 We have developed a model to explain the role of FA proteins in this process.
Model for the role of FA proteins in maintaining αIISp stability
There are several possible mechanisms by which FA proteins could reduce cleavage of αIISp by µ-calpain. One mechanism we propose is that in normal cells, an FA protein such as FANCG binds to the SH3 domain of αIISp and this in turn leads to decreased ability of µ-calpain to cleave αIISp (Figure 6).16 In support of this, a number of FA proteins contain motifs with a consensus sequence that recognizes SH3 domains.32 We have shown that FANCG has a consensus sequence that specifically recognizes and binds to the SH3 domain of αIISp.32 An equilibrium would exist among the FA protein (i.e. FANCG), low-molecular weight phosphotyrosine phosphatase (LMW-PTP), and the kinase, c-Src for binding to the SH3 domain16 When c-Src binds to the SH3 domain, it phosphorylates Try1176 at the µ-calpain cleavage site and prevents cleavage of αIISp by µ-calpain.86,88 When LMW-PTP binds to the SH3 domain, Tyr1176 becomes dephosphorylated, enabling µ-calpain to cleave αIISp (Figure 6).86,88 Binding of the FA protein to the SH3 domain would then prevent binding of LMW-PTP to this site, and inhibit its ability to dephosphorylate Tyr1176, thus inhibiting cleavage of αIISp by µ-calpain.16 Of particular interest, at least 18 patient-derived mutations have been identified in FANCG which would result in the FANCG protein either missing or having a defect in the motif that binds to the SH3 domain of αIISp.32 This would increase binding of LMW-PTP to the SH3 domain of αIISp leading to increased cleavage by µ-calpain. Thus, we propose that maintenance of normal levels of FA proteins in cells is critical for maintenance of normal levels of uncleaved αIISp.
Figure 6.

A model proposed for the regulation of αIISp stability by FA proteins. This model proposes that FA proteins play a critical role in maintaining the stability of αIISp in the nucleus of normal human cells by regulating its cleavage by the protease, µ-calpain. There are several possible mechanisms by which this can be accomplished; one is presented here. In this figure, a segment of αIISp, containing repeats 8–10, is shown. FANCG is used to illustrate one potential role of an FA protein in this process in normal human cells. An equilibrium exists among c-Src, LMW-PTP, and FANCG for binding to αIISp at its SH3 domain.16 Binding of c-Src to the SH3 domain leads to phosphorylation of Tyr1176 (Y1176) at the µ-calpain cleavage site in repeat 10 of αIISp, preventing cleavage of αIISp by µ-calpain.86,88 When LMW-PTP binds to the SH3 domain, Tyr1176 becomes dephosphorylated, enabling µ-calpain to cleave αIISp.86,88 Binding of FANCG to the SH3 domain blocks binding of LMW-PTP to the SH3 domain and prevents cleavage of αIISp by µ-calpain.16 An FA protein may also inhibit αIISp cleavage by binding to µ-calpain and inhibiting its ability to cleave αIISp.16 In FA cells (for example, FA-G cells), the absence of FANCG would lead to a greater probability that LMW-PTP would bind to the SH3 domain of αIISp, leading to dephosphorylation of Tyr1176 and cleavage of αIISp by µ-calpain. FANCG would also not be present to additionally bind to µ-calpain and inhibit its cleavage of αIISp. In other FA complementation groups, similar events could occur. (Modified from Zhang et al.16 with permission from the American Chemical Society).
Another mechanism by which FA proteins may inhibit αIISp cleavage by µ-calpain is by binding to µ-calpain and inhibiting its activity. In support of this view are our yeast two-hybrid data showing that FANCA and FANCG bind directly to µ-calpain.16 A third mechanism by which FA proteins could potentially regulate αIISp cleavage would be by modulating binding of calmodulin to αIISp. When calmodulin binds to a site on αIISp adjacent to µ-calpain’s cleavage site, this leads to enhanced cleavage of αIISp by µ-calpain.89–91 An FA protein could potentially bind to calmodulin inhibiting its binding to αIISp and thus could decrease µ-calpain activity and αIISp cleavage. Thus, there are a number of ways in which FA proteins could regulate αIISp stability and reduce its cleavage by µ-calpain. This suggests another potential role for FA proteins in the cell, that of maintenance of αIISp stability.
Consequences of loss of FA proteins
In FA cells, deficiency in a specific FA protein correlates with reduced levels of αIISp and reduced localization of a repair protein such as XPF to sites of damage (Figure 4(b)).11 When levels of these FA proteins are restored by transfection of FA cells with the appropriate FA c-DNA, αIISp levels in these corrected cells (FA-A+, FA-C+, FA-G+) are similar to normal.11,16 Thus, there is a correlation between αIISp level in cells and the presence of a specific FA protein. These studies further strengthen the view that FA proteins are important in maintaining αIISp stability.16–18 Different FA proteins could have different roles in modulating αIISp stability and reducing its cleavage by µ-calpain. This could occur through their interactions, either directly or indirectly, with αIISp, µ-calpain or possibly calmodulin.
Knock-down of µ-calpain in FA cells restores αIISp levels to normal
We have proposed that if the increased activity of µ-calpain in FA cells is reduced, then levels of αIISp could be returned to normal and DNA repair should be enhanced.16 In support of this view, we have shown that knock-down of µ-calpain by siRNA in FA-A cells leads to an increase in αIISp levels to those similar in normal cells and to correction of chromosomal instability, to formation of XPF foci at sites of DNA damage (Figure 4(c)), to ability to repair DNA ICLs and to increased cell survival after damage.16 Thus, by knocking down µ-calpain and restoring levels of αIISp to normal in FA-A cells, we were able to restore a number of the phenotypic characteristics of these FA-A cells to normal.16 Levels of the FANCA protein, however, were not increased and there was still a deficiency in this protein in FA-A cells.16 We have proposed that when restoration of αIISp levels in FA cells is achieved by an alternate means (i.e. knocking-down µ-calpain), these cells are then able to carry out functions they could otherwise not accomplish such as DNA repair and maintenance of chromosome stability and telomere function after ICL damage.16,18 Thus, in these FA cells, the presence of a specific FA protein becomes less critical, since an endpoint of its function (i.e. maintenance of αIISp stability) has been achieved by another means.
αIISp prevents telomere dysfunction after DNA ICL damage
αIISp interacts with telomeres
Telomeres are specialized nucleoprotein structures at the ends of chromosomes which are critical for preserving genomic integrity.92–94 Damage to telomeric DNA, if left unrepaired, can lead to telomere dysfunction and chromosomal instability.92–94 A multiprotein complex, shelterin, binds to telomeres and helps protect them and prevent telomere dysfunction.92,94–96 Repair of damage to telomeric DNA is thus essential for insuring the integrity of telomeric DNA and maintenance of chromosome stability. We have shown that αIISp is important in this repair process.17 It is recruited to telomeres after DNA ICL damage where it co-localizes with two proteins in the shelterin complex, TRF1, and TRF2.17 αIISp is also needed for the recruitment of XPF to telomeres after ICL damage and it co-localizes with XPF at these sites of damage.17 Knock-down of αIISp by siRNA results in loss of localization of XPF to telomeres after damage.17 These studies thus demonstrate that αIISp plays a role in the recruitment of XPF to sites of DNA damage in telomeres and in repair of DNA ICLs at these sites.
Telomeres replicate in S phase.92–94,97 Since production of ICLs in DNA can lead to stalled replication forks,41–44 it is thus essential that, at the time of replication, damage occurring in telomeric DNA is repaired so that DNA replication can be re-initiated. Of particular interest, αIISp specifically localizes to telomeres in S phase indicating its importance in the DNA repair response at the site of a stalled replication fork.17
αIISp is needed for telomere function after DNA ICL damage
αIISp is also critical for maintenance of telomere function after ICL damage.17 We have demonstrated that knock-down of αIISp in normal human cells leads to telomere dysfunction.17 This can be assessed by examination for the presence of telomere dysfunction-induced foci (TIF) after ICL damage, which can be determined by quantitation of γ-H2AX foci.17 These foci represent sites of DNA double-strand breaks (DSBs) that arise when replication forks are stalled at sites of ICLs and fail to restart.98 They are used as an indicator of dysfunctional telomeres.98 In normal human cells, loss of αIISp by knock-down leads to an increased number of TIF positive cells after ICL damage.17 Another indicator of telomere dysfunction after ICL damage is an increase in chromosomal aberrations. Knock-down of αIISp in normal cells leads to an increase in chromosomal aberrations, particularly sister chromatid end-to-end fusions.17 A third indicator of telomere dysfunction is loss of telomeres. This is proposed to occur when telomeres are stalled at replication forks and the replication forks collapse.92,93 DSBs then form in DNA, leading to breakage of telomeres and their loss. We have demonstrated that, after knock-down of αIISp in normal human cells and exposure of these cells to an ICL agent, there is a catastrophic loss of telomeres (Figure 7(a) and (b)).17 We hypothesize that reduced levels of αIISp in these cells prevent effective repair of DNA ICLs in telomeric DNA during S phase and that this results in stalling of the replication fork, formation of DSBs in telomeric DNA, and a significant loss of telomeres.17 Thus, these studies collectively demonstrate that αIISp plays a critical role in maintenance of telomere function after DNA ICL damage and provides evidence for another important role for αIISp in the nucleus.
Figure 7.
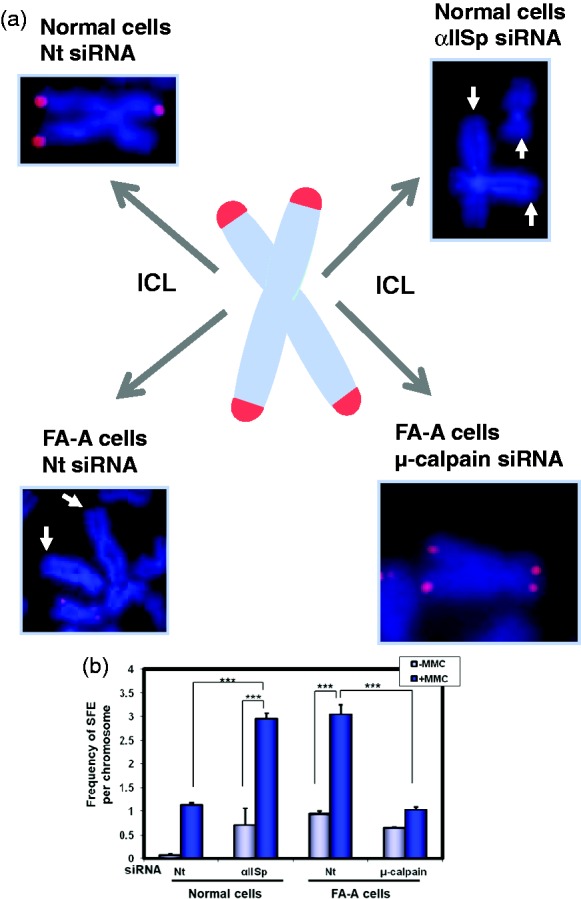
A deficiency in αIISp results in loss of telomeres after DNA ICL damage. (a) In normal human and FA-A cells, after treatment with MMC, metaphase chromosomes were stained with DAPI (blue).17 Telomeric DNA was identified using FISH with a Cy3-labeled telomere specific PNA probe (red).17 In normal cells transfected with αIISp siRNA, so as to knock-down αIISp, and in FA-A cells transfected with non-target (Nt) siRNA, levels of αIISp, in both groups of cells, are ∼30% of those in normal cells. After treatment of these cells with MMC, there is an increased loss of telomeres (or the presence of signal free ends (SFEs)) in telomeres.17 Arrows point to signal free ends (SFEs) in telomeres. After FA-A cells are transfected with µ-calpain siRNA, so as to increase levels of αIISp to those present in normal cells, and subsequently treated with MMC, the frequency of SFE in chromosomes was reduced to levels similar to those in normal cells transfected with Nt siRNA. (Chromosome inserts are reproduced from Zhang et al.17 with permission of Oxford University Press) (b) Quantitation of the frequency of SFEs per chromosome for normal and FA-A cells was carried out. Means of five independent experiments are shown in which 4600 chromosomes per experiment were scored for telomeric signals on each of the chromatids. These results indicate that αIISp plays a critical role in maintenance of telomere function after DNA ICL damage.17 Error bars are S.E.M. *** P< 0.0001. (Reproduced from Zhang et al.17 with permission from Oxford University Press).
αIISp loss in FA cells correlates with telomere dysfunction after ICL damage
Cells from patients with FA serve as an excellent model for the effects of loss of αIISp on telomere function. We have shown that there is a significant increase in telomere dysfunction in FA-A cells after DNA ICL damage.17 Two important findings support this view: First there is an increase in the number of TIF positive cells in FA cells (FA-A) after ICL damage as well as a significant loss of telomeres (Figure 7(a) and (b)).17 Importantly, when levels of αIISp are returned to normal by knocking down µ-calpain, the number of TIF positive cells is reduced to normal levels as is the number of chromosomes with a loss of telomeres (Figure 7(a) and (b)).17 Second, in FA-A cells, co-localization of XPF with telomeres (TRF1) is not observed after ICL damage.17 However, after restoration of αIISp levels to normal by knocking down µ-calpain, normal levels of XPF localize to telomeres.17 These studies using FA-A cells again importantly demonstrate that αIISp plays a critical role in telomere maintenance after ICL damage.
αIISp is essential for chromosome stability
Interaction of αIISp with FA and DNA repair proteins is essential for maintenance of its stability and the role it plays in repair of ICLs during DNA replication in both genomic and telomeric DNA.13,16,21 These processes are critical for maintenance of genomic stability. We have shown that loss of αIISp from cells leads to increased chromosomal aberrations after DNA ICL damage.15,17 This is clearly observed in normal human cells in which αIISp has been knocked-down by siRNA.15,17 These aberrations include sister chromatid end-to-end fusions, chromatid breaks, chromosome exchanges, and radials.15,17 Similar chromosomal aberrations have been observed in FA cells after ICL damage.54–56 Since levels of αIISp in FA cells are 35–40% of normal and are similar to levels in normal cells in which αIISp has been knocked down, this indicates that a reduction in levels of αIISp in cells can have a significant effect on chromosome stability after DNA ICL damage. These changes correlate with loss of repair of DNA ICLs that occurs in both genomic and telomeric DNA.15,17 Of importance, the chromosomal aberrations observed in FA-A cells after ICL damage are corrected when µ-calpain is knocked down resulting in return of αIISp levels in the nucleus to normal.16 Studies have also shown that translational repression of SPTAN1, the gene which codes for αII spectrin, by miR-128–3p results in chromosome instability, acceleration of cell cycle arrest, and a reduced DNA damage response after MMC treatment.99 These studies, collectively, further accentuate the critical role αIISp plays in maintenance of chromosome stability after ICL damage.
Nuclear beta spectrins
In the cytoplasm, αII-spectrin interacts with β-spectrins to form a tetramer which, as mentioned above, is important in maintaining cell architecture and plasma membrane stability as well as in a number of other cellular functions.23–25,33–39 This type of interaction has not as yet been observed in the nucleus. However, several β-spectrins have been described in the nucleus. Two isoforms of βIV-spectrin have been identified in the nucleus of a variety of mammalian cell types: a truncated, 77 kDa major isoform, SpβIVΣ5, and a full-length minor isoform, SpβIVΣ1.100 An antibody developed by Tse et al., which recognizes both isoforms of βIV spectrin, shows that, on Western blot analysis, SpβIVΣ5 is the major band observed and SpβIVΣ1 is present in very faint bands.98 Indirect immunofluorescence studies show that SpβIVΣ5 co-localizes in the nuclear matrix with promyelocytic leukemia (PML) bodies.100 The function of SpβIVΣ5 in the nucleus is not yet clear; however, evidence suggests that it contributes to the formation of PML bodies.100 SpβIVΣ1 is diffusely distributed throughout the nuclear matrix.100 It has a conserved actin/4.1-binding domain, which is hypothesized to bind to actin and protein 4.1 in the nucleus.100 SpβIVΣ1 is further hypothesized to contribute to the formation of the nuclear skeleton.100 Our studies, using normal human lymphoblastoid cells and the antibody developed by Tse et al.100 against SpβIV, have shown that SpβIV localizes in major foci, presumed to be PML bodies, and in a fine reticular pattern throughout the nuclear matrix (unpublished data), in agreement with the findings of Tse et al.100 We have additionally shown that αII-spectrin co-immunoprecipitates with SpβIVΣ5 and with PML protein from nuclear extracts.19 These studies, combined with those of Tse et al.,100 suggest that both αII spectrin and SpβIVΣ5 play a role in PML body formation/function. Since SpβIVΣ1 is a full-length protein with actin/4.1-binding sites and is postulated to be a component of the nuclear matrix,100 we hypothesize that the interaction we have demonstrated between αII spectrin, actin and protein 4.1 in the nucleus19 could be via association of αII spectrin with SpβIVΣ1. This suggests exciting new possibilities regarding the interaction αII spectrin with β-spectrins in the nucleus which need to be further explored.
Another β-spectrin, termed β2-spectrin (β2SP, gene Sptbn1),101–103 which is a TGF-β/Smad3/4 adaptor protein and transcriptional cofactor that can regulate TGF-β signaling, has been shown to undergo nuclear translocation and have nuclear functions in mouse embryonic fibroblasts (MEFs).101–104 Studies indicate a role for β2SP in genomic stability and DNA repair.101–104 Sptbn1-homozygous-null mouse embryonic fibroblasts, following exposure to different genotoxic agents such as ionizing radiation, mitomycin C, hydroxyurea, and ethanol-derived acetaldehyde, exhibit a significant increase in chromosomal aberrations and reduced cell survival compared to wild type MEFs.101–104 Studies suggest that these β2SP depleted cells have a defect in DNA double-strand break repair by homologous recombination and that this repair defect is specific for S-phase.101–104 Thus, β2SP may have an important role in the DNA damage response just as has been demonstrated for αIISp. Since βII-spectrin in the cytoplasm has binding sites for actin and for protein 4.1 and also binds to αII-spectrin,23–25 it could be hypothesized that, in the nucleus, αII-spectrin similarly binds to βII-spectrin and that it is through this interaction that it achieves a functional association with actin and protein 4.1. There is as yet, however, no reported evidence for an interaction between αII-spectrin and βII-spectrin in the nucleus of human cells. Collectively, the studies described above point to the possibility of association of αII-spectrin with full length β-spectrins in the nucleus and that more than one form of β-spectrin may be involved. Such an association could be an important component of αII-spectrin’s structural role in the nucleus.
αIISp interacts with structural proteins in the nucleoskeleton
In addition to its interaction with proteins in the nucleus involved in DNA repair and its role in the DNA repair response and in chromosome stability, αIISp also has a role in functioning of the nucleoskeleton.6,7,19–22 It interacts with structural proteins in the nucleoskeleton, which constitutes a network of interacting components including the lamins (types A and B), actin, emerin, protein 4.1, nesprin (spectrin-repeat domains), myosins, kinesins, titan, and βSpIV.7,19–21 Of these nucleoskeletal proteins, αIISp has been shown, thus far, to interact with lamin A, actin, emerin, protein 4.1, nuclear myosin 1c, and βSpIVΣ5.19–21 The potential functional importance of the interaction of αIISp with these specific nucleoskeletal proteins will be discussed below.
Major proteins in the nucleoskeleton and their function
The nucleoskeleton is a structurally and functionally complex network of proteins that confers mechanical properties and functionality to the nucleus and the genome.6,7,20,21,105 It is composed of two major components: a peripheral component concentrated near the nuclear envelope and an internal component that extends throughout the interior of the nucleus and consists of more loosely associated proteins.7,21 There are functional differences between these two components of the nucleoskeleton. Numerous reviews have been written regarding the array and localization of proteins comprising the nucleoskeleton and their role in nuclear function.6,7,20,21 This section will briefly review those proteins which have, thus far, been shown to associate with αIISp.
Among the foremost of these nucleoskeletal proteins are the lamins. A-type and B-type lamins concentrate underneath the inner nuclear membrane where they form the nuclear lamina network.7 They are also distributed loosely throughout the nucleoplasm.3,7,106–110 They are essential components of the nuclear architecture and are critical for many aspects of normal nuclear function and maintenance of nuclear integrity.3,107–110 They have a broad range of functions due to their interactions with proteins that function in a variety of cellular pathways.3,7,20,109,110 Lamin networks make an important contribution to mechanical support and stiffness of the nucleus, to mechanoresponsive functions of the nucleus and to mechanotransduction.3,105,107,108,111 Lamins interact with numerous proteins in the nucleoskeleton including actin, emerin, protein 4.1, spectrin, myosin 1c, SUN2, kinesin, titin, and the nuclear mitotic apparatus.7,19–21 They regulate and support protein complexes involved in gene expression, DNA replication, transcription, chromatin organization, differentiation, DNA repair, epigenetic regulation, and signaling.3,7,107,108 Lamins thus contribute, either mechanically or non-mechanically, to a variety of cellular functions.
Actin, another protein associated with the nucleoskeleton, is present both in a monomeric or globular form (G-actin) and in a polymerized filamentous form (F-actin).7,112–115 Actin interacts functionally with lamin complexes, which may aid in actin filament formation.7,12,116,117 It also binds to emerin, protein 4.1, and αII spectrin.19,20 Perinuclear actin and nuclear lamins help mediate the nuclear response to mechanical stress and contribute to nuclear shape.20,21,118,119 Actin has many diverse roles in the nucleus which include transcription, nuclear export, chromatin remodeling, chromosome relocation, gene expression, mRNA processing, nuclear envelope assembly, and DNA repair.7,20,112,114,116 Thus, like the lamins, it has both structural and non-structural roles.
Emerin is a nuclear membrane protein associated with the nucleoskeleton.20,109,120 It localizes primarily in the inner membrane of the nuclear envelope and binds directly to lamins.20,109,120 Emerin helps connect the nuclear lamina to the nuclear envelope and stabilizes the lamin network, contributing to nuclear lamina structure and function.20,109,120–123 Purification of emerin containing complexes from HeLa cell nuclei has revealed a protein complex which includes αII-spectrin, actin, nuclear myosin, lamins A and B, and SUN2.20,21 It has been suggested that an important function of emerin is to interlink actin-nuclear myosin-spectrin complexes with the A- type and B- type lamins at the nuclear envelope and to help maintain the structural integrity of the nucleus.20,21 Emerin has roles in chromatin organization and tethering chromosomes to the nuclear envelope, promoting formation of nuclear actin filaments, nuclear assembly, gene regulation, mitosis, development, signaling, and mechanotransduction.20,21,109,120–122 Protein 4.1, another component of the nucleoskeleton, is enriched at the nuclear envelope.7,121,124 It is thought to be an important component of the cortical network, connecting actin, αII-spectrin, emerin, and A-type lamin to the nuclear envelope and in doing so has an impact upon nuclear structure and architecture.7,20,121,124 Thus, the nucleoskeleton is important for maintaining the shape and functional integrity of the nucleus and it is the interaction between these, as well as other nucleoskeletal proteins, which plays a critical role in these processes.
αII-spectrin’s interaction with nucleoskeleton proteins
αIISp interacts with a number of these nucleoskeletal proteins. It is present throughout the nucleus in both the peripherial and internal nucleoskeleton.7,13,19–21,64 In the peripheral nucleoskeleton, it is observed as an intense band just beneath the nuclear envelope (Figure 8(a))13,64 where it is associated with other nucleoskeletal proteins (Figure 8(b)).7,20,21 It has been proposed that spectrin forms a cortical network with lamin and actin filaments that is anchored by emerin and protein 4.1 to the nuclear envelope and is important for its support.7,20,21 Its co-immunoprecipitation with lamin A, actin, emerin, protein 4.1 and βSpIVΣ5,19 as well as its co-purification in a complex with actin, emerin, nuclear myosin 1c, lamins A and B and SUN2,20,21 indicates that it is a significant component of the nucleoskeleton (Figure 8(b)).
Figure 8.
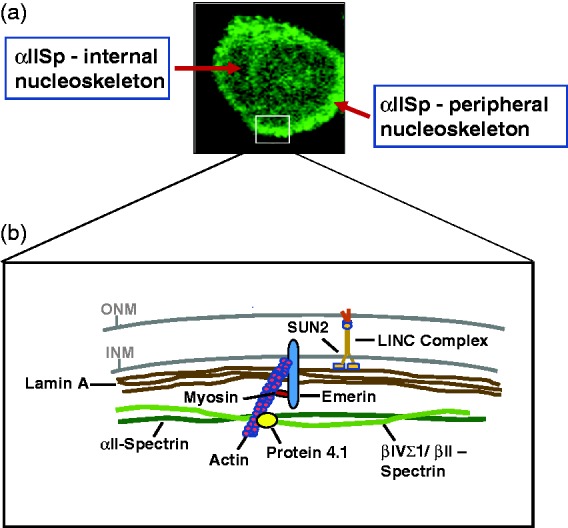
αIISp in normal human cells is present in both the peripheral and internal nucleoskeleton. (a) In normal human lymphoblastoid cells, using indirect immunofluorescence and staining with anti- αIISp, αIISp is observed in both the internal nucleoskeleton and the peripheral nucleoskeleton. (Frame reproduced from Zhang et al.64 with permission from Wiley). (b) This is a diagram of the proposed interaction of αIISp with some of the proteins in the peripheral nucleoskeleton. αII-spectrin is proposed to interact with β-spectrin (β-II or βIVΣ1) and form a cortical network with lamin A and actin that is anchored by emerin, nuclear myosin and protein 4.1 to the nuclear envelope7,19,20. This is supported by studies which show that αIISp co-immunoprecipitates with lamin A, actin, protein 4.1, emerin and βIV-spectrin19 and co-purifies with lamin A, actin, emerin, nuclear myosin 1c and SUN2, a component of the LINC complex.20,21 βII-spectrin has been shown to be present in the nucleus101–104 and though it has not, as yet, been demonstrated to interact with αII-spectrin, it has been included in the diagram. Thus, αIISp may have an influence on the interaction of nuclear lamins with the LINC complex and may play a role in the biomechanical properties of the nucleus and mechanical coupling of the nucleus and cytoplasm in either undamaged and/or DNA damaged cells.
In the cytoplasm, αII-spectrin interacts with βII-spectrin to form a heterodimer important in cytoskeletal structure and function.23–25,36 Though both the long form of βIV-spectrin, i.e., SpβIVΣ1, and βII-spectrin have been identified in the nucleus of nonerythoid cells,22–24,98 no definitive association between αII spectrin and these β-spectrins has been identified. As mentioned above, αII-spectrin’s interaction with actin and protein 4.1 in the nucleus could be via an association with a β-spectrin, as it is in the cytoplasm (Figure 8(b)). Such an association could play an important role in nuclear architecture and structure and function of the nucleoskeleton. Both SpβIVΣ1 and βII-spectrin have sites that can bind actin and protein 4.1.23–25,100 αII-spectrin co-immunoprecipites with SpβIV and, though it associates mainly with the truncated SpβIVΣ5,19 it has some association with SpβIVΣ1 (unpublished data). SpβIVΣ1 is present in the nuclear matrix.100 Thus, potentially, αII-spectrin could form an association with SpβIVΣ1 in the nucleus which would be important in its interaction with actin and protein 4.1. It could also potentially form such an interaction with βII-spectrin. Such interaction of αII-spectrin with either βIV-spectrin or βII-spectrin has been shown to occur in different locations in the cytoplasm in neurons.125–128 Thus, this type of interaction could potentially take place in the nucleus under different circumstances. The existence and nature of the interaction of αII-spectrin with these as well as other nucleoskeletal proteins remain a largely unexplored area which needs to be more fully investigated. It will be extremely important to determine this so that the functional significance a deficiency in αII spectrin has on these interactions may be determined.
Thus, αIISp has both structural and non-structural roles in the nucleus. In its structural role, together with other nucleoskeletal proteins, it is thought to make an important contribution to nuclear architecture and the organization, stability, elasticity, and mechanical properties of the nucleus.7,20,30 In its non-structural role, it is best known for its involvement in DNA repair, where it plays an important role in the repair process during S-phase of the cell cycle, indicating its importance in DNA repair at the time of DNA replication.12–19 In addition, it could play a role in other functional activities in the nucleus such as chromatin remodeling, transcription, and RNA processing.19 Thus, deficiencies in αIISp in a cell could have far reaching consequences and may impact a number of important cellular functions.
Additional potential roles for αIISp in the nucleus
Another very interesting finding regarding α-spectrin function has been described by Goodman et al.40,129,130 They have demonstrated that in human erythrocytes, α-spectrin has chimeric E2/E3 ubiquitin conjugating/ligating enzymatic activity.39,129,130 Post-translational modification of proteins by ubiquitin involves a multi-enzymatic system consisting of: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin protein ligase (E3).131–134 Such modification can affect functional regulation of proteins, cellular regulatory mechanisms, and signal transduction and DNA repair pathways.40,131–134 In human erythrocytes, it has been demonstrated that αI-spectrin is monoubiquitinated at two sites by its own E2/E3 enzymatic activity.40,133,135 One of these sites, in the N-terminus, represents the location where αI-spectrin makes contact with βI-spectrin to form a heterodimer.133,135,136 Goodman et al. showed that ubiquitination of αI-spectrin does not regulate the rate of heterodimer formation; however, ubiquitination of αI-spectrin increases the rate of dissociation of actin and protein 4.1 from βI-spectrin.133,136,137 These studies are of interest since they indicate that ubiquitination of αI-spectrin could potentially affect cytosksletal organization.
Whether, in non-erythroid cells, αII-spectrin could have similar E2/E3 ubiquitinating activity is a very interesting question. Based on sequence similarity between erythroid and non-erythroid α-spectrin and conservation of sequences in these proteins throughout evolution, Goodman et al.40 have suggested that αII-spectrin could serve as an E2/E3 ubiquitin conjugating/ligating enzyme in non-erythroid cells. They have shown that αSpIIΣ1 is ubiquitinated in hippocampal neurons, which supports this hypothesis.138 Whether αII-spectrin in mammalian nuclei could also have E2/E3 chimeric enzyme activity, and whether it undergoes self-ubiquitination, is unknown. Also, whether it could potentially ubiquitinate other proteins in the nucleus, as has been shown for α1-spectrin in erythroid cells,40 has not been reported. We have shown that knockdown of αIISp in normal human cells (to levels approximately 35–40% of normal) does not affect the monoubiquitination of FANCD2.64 This indicates that αIISp is not involved in the monoubiquitination of FANCD2 and that it acts downstream of monoubiquitinated FANCD2 in the initial steps of the ICL repair process.64 Whether αIISp has E2/E3 enzymatic activities that would act in the later steps in the repair process or could be involved in monoubiquitination of other structural or non-structural proteins in the nucleus is an area which certainly needs to be investigated.
Another possibility is that if αIISp in the nucleus has E2/E3 enzymatic activity which can lead to its self-ubiquitination, this increased level of ubiquitination could potentially lead to decreased association of actin and protein 4.1 with β-spectrin, as has been shown to occur in erythroid cells.136,137 Increased self-ubiquitination activity, as well as factors affecting such an activity, could have a significant influence on nucleoskeletal structure and function in both mature normal human cells and in human cells during development. This raises a number of interesting questions regarding αIISp’s potential E2/E3 enzymatic activity and the effect this could have on nuclear architecture and on nuclear functional activity.
αIISp’s involvement in biomechanical properties of the nucleus
Biomechanical studies have shown that the nucleus displays complex mechanical properties which include compressibility and elasticity.7,22,105,120,139 These studies point to involvement of the nucleoskeleton and proteins associated with it in this process.3,22,105,139 Lamins in the nucleoskeleton have been shown to stabilize the nuclear architecture and influence nuclear responses to mechanical signals.3,100,120,139 They are the major mechanical elements of the nucleus and make a critical contribution to the mechanical stiffness of the nucleus.7,20,108,139 Lamins are important for mechanotransduction (i.e. translating mechanical changes or stimuli a cell receives into biochemical signals) and have an important role in mechanically coupling the cytoskeleton to the nucleoskeleton.3,7,111,119,120,139
It has been proposed that αIISp forms a network in the nucleus which has a mechanical role.7,20,22 Studies in which levels of αII-spectrin in the nucleus of HeLa cells were knocked-down to approximately 50% of those found in control cells have demonstrated that there is a loss in nuclear recovery from compression due to a reduction in elasticity of the nucleus.22 This has been attributed to reduction in the αII-spectrin network in the nucleoskeleton which has a negative impact on the resilience of the nucleus to compression.22 Disruption of the αII-spectrin network thus causes a reduction in the elasticity of the nucleus but has no effect on the stiffness.22 These results importantly indicate that αII-spectrin has a mechanical role in the nucleus at the inner nuclear membrane where it contributes to the resilience of the nucleus and its recovery after compression, providing elasticity to the cell nucleus.22 Thus, within the nucleoskeleton, there are two important networks that provide mechanical stability to the nucleus: the lamin network, which makes important contributions to the mechanical stiffness of the nucleus, and the spectrin network, which is important for providing elasticity of the nucleus.7,20,22,111,139
Mechanical coupling between the nucleus and cytoplasm is critical for cellular function and has been shown to involve LINC (links the nucleoskeleton and cytoskeleton) complexes which span the nuclear envelope.140–144 Lamin A can bind directly to LINC complex proteins SUN1 and SUN2.143 Association of A-type lamins and emerin with the LINC complexes may help mediate mechanotransduction signaling and nucleoskeletal-cytoskeletal coupling.7,143 αII-Spectrin has been shown, in HeLa cell nuclei, to form a protein complex with emerin, actin, nuclear myosin, lamins A and B, and SUN2,20,21 It is possible that it could have an influence on the interaction of nuclear lamins with the LINC complex. αII-spectrin may be important in LINC complex mechanical coupling of the nucleoskeleton and cytoskeleton, in undamaged cells and/or in cells which have undergone DNA damage. These are studies which still need to be carried out and which could be of great functional importance. This is of particular interest since the LINC complex proteins, SUN1 and SUN2, have been shown to play a role in the DNA damage response, in addition to the diverse other functions in which they are involved.145–149 Studies suggest that the LINC complex promotes homologous recombination at sites of DSBs in DNA and in this way may also function in DNA interstrand cross-link repair, where homologous recombination takes place in the latter steps of the repair process.147,149
Thus, interactions between lamin A, actin, emerin, spectrin, protein 4.1, and nuclear myosin may be important for the normal biomechanical properties of the nucleoskeleton and for maintenance of tension, stiffness, and elasticity of the nuclear envelope.7,21,22 A deficiency in one or more of these proteins could thus affect the stability of the nuclear architecture and mechanotransduction through LINC complexes at the nuclear envelope.7 Whether such a deficiency would affect the biomechanical properties of the nucleoskeleton after cells are treated with a DNA damaging agent is a very intriguing question. Since αIISp plays a role in DNA repair, it will be important to determine whether loss or a deficiency of αIISp and the resulting diminished DNA repair has an effect on these biomechanical properties after DNA damage.
Effects of loss of αIISp on cellular morphology
αIISp in the cytoplasm has been shown to play a role in cellular morphology and its loss can lead to changes in cell shape, cell proliferation, cell adhesion, and cell spreading capabilities. We have shown that, after knock-down of αIISp in normal human lymphoblastoid cells to the levels found in FA cells, which are 35–40% of normal levels, these cells lose their pleomorphic shape; they are smaller and rounded with few if any pseudopodia, resembling FA lymphoblastoid cells in culture.15 These studies suggest a role for αIISp in cell structure. Similarly, studies on depletion of αII-spectrin in Jurkat T-cells have demonstrated that αII-spectrin is important for cell adhesion and formation of actin-rich lamellipodia and filopodia which are involved in immunological synapse formation upon T-cell activation.150 Depletion of αII-spectrin leads to changes in the dynamics of the actin skeleton and decreased actin accumulation in areas of cell contact, indicating the important role of αII-spectrin in actin organization and function in the cytoskelaton.150 In addition, studies have shown that, after knock-down of αII-spectrin by siRNA in a human melanoma cell line, the cells became rounded, decreased in size and had defects in cell proliferation, cell adhesion and spreading and displayed cell cycle arrest.37 They also showed disorganization of the actin skeleton and loss of actin stress fibers.37 These studies suggest that αII-spectrin may play a role in actin network formation.37 Of interest, investigations on macrophages from Fancc−/− mice have shown that these cells are round in shape, lack the multiple protrusions seen in wild type macrophages, and have deficiencies in cell migration.151 These cells showed impaired filamentous actin rearrangements.151 Since we have found that there is a deficiency in αIISp in bone marrow cells from Fancc−/− mice (unpublished results), we have speculated that the morphological changes observed in macrophages from Fancc−/− mice are due to reduced levels of αIISp in these cells which in turn leads to rearrangements of the actin filaments.15 These studies, collectively, suggest that αIISp plays a role in actin skeletal organization in the cytoplasm and that a deficiency in αIISp affects cellular morphology, in part through its effects on actin filament organization. Since we and others have shown that αIISp interacts with actin in the nucleus,7,19,20 it is possible that αIISp has a similar role in organization of actin in the nucleus, resulting in similar consequences when a deficiency in nuclear αIISp occurs. This is an exciting area of investigation that needs to be explored further.
Clinical significance of loss of αIISp
αIISp is an essential cellular protein. Total loss of αIISp leads to cell death.15,37,82,83 Thus, loss or dysfunction of αIISp in cells is of profound clinical relevance. In FA patient’s cells, a deficiency in αIISp correlates with phenotypic changes occurring after exposure to DNA ICL agents (i.e. chromosomal aberrations, cell cycle defects, defects in repair of DNA ICLs, and telomere dysfunction).15–17 Our studies, which demonstrate that restoration of αIISp levels to normal in FA cells corrects a number of the phenotypic deficiencies occurring after ICL damage, are of particular interest.16 In these studies, knockdown of µ-calpain led to an increase of αIISp to normal levels but had little effect on cell viability.16 Studies using a mouse model have shown that decreasing levels of µ-calpain by siRNA knockdown has no effect on development.152 Thus, developing methodologies that target µ-calpain and reduce its levels or activity in FA cells, so as to reduce cleavage of αIISp, could be of clinical importance, possibly in conjunction with other modalities, and suggests a potential new direction that could be explored for therapeutic intervention in FA.
Studies indicate that there may be a link between loss of αIISp and the pathogenesis of neoplastic bone marrow disorders such as leukemia.152,153 It has been shown that, in bone marrows examined from acute myeloid leukemia (AML) patients, 44% had a loss of αIISp, which suggests a possible role for αIISp in leukemogenesis.153,154 This is of particular interest since FA patients develop bone marrow failure and have a strong predisposition to develop AML.54,155 If αIISp loss plays a role in leukemogenesis, this could be of significant importance in the etiopathogenesis of these disorders.
Since αIISp is an important component of the nucleoskeleton and its levels are decreased in cells from FA patients,11,13,18 an extremely interesting question is what effect does a deficiency in αIISp have on various cellular processes such as development. In mammalian cells, αIISp is expressed throughout all stages of development and is critical in many developmental processes.38,156–158 Studies have shown that, in αII-spectrin knockout mice, embryos with a homozygous deletion of the Spna2 gene (the gene coding for αII-spectrin) displayed prominent cardiac, craniofacial and neural tube abnormalities and died between day 12.5 and day 16.5.38 Loss of αII-spectrin also reduced the steady state protein levels but not the transcriptional levels of βII- and βIII-spectrin, leading to redistribution of both βII- and βIII-spectrin as well as of ankyrin.38 Cultured embryonic fibroblasts from Spna2−/−mice displayed impaired growth and spreading and sparse lamellipodia which lacked cortical actin.38 Thus, loss of αII-spectrin disturbs the levels and distribution of its associated proteins (i.e. β-spectrins, actin, and ankyrin).38 These studies indicate that αIISp is required for the stability and organization of these proteins and for cell spreading, tissue patterning, and organ development in vertebrates.38 They highlight the important effect αIISp has on cellular morphology and development.
FA patients show a number of congenital abnormalities, such as radial ray deformities, absent radii, urogenital malformations, renal anomalies, and ear malformations.159 It may be hypothesized that a deficiency in αIISp, which has both structural and non-structural roles, may have an important influence on developmental processes and is a significant factor leading to many of the congenital abnormalities observed in FA patients. According to this hypothesis, a deficiency in αIISp in cells such as those from FA patients leads not only to defects in DNA repair and chromosome instability but also to development abnormalities; these abnormalities may be due wholly or in part to loss of αIISp and its interaction with other nucleoskeletal proteins. Thus, development of technologies which restore levels of αIISp to normal in cells such as those from FA patients may potentially provide an important target for therapeutic regimes.
There is evidence that αII-spectrin plays critical roles in brain development and epileptic encephalopathy.128,160–162 Mutations in SPTAN1, the gene encoding αII-spectrin, have been shown to cause early infantile epileptic encephalopathy type 5 (EIEE5) leading to severe neurodevelopment impairment and progressive brain atrophy.128,160–162These mutations in SPTAN1 lead to severely altered neuronal morphology.128,160 αII-spectrin is a critical structural protein in neurons.24,34,128,138,163 In EIEE5, the most severe mutations in SPTAN1 lead to defects in binding of αII-spectrin to β-spectrins (II-IV).128 Binding of αII-spectrin to βII or βIV spectrin depends upon their location in the neuron or the stage of development of the neuron.160,161,163 This binding is critical for neuronal development and structure.24,160,161 The importance of this interaction is clearly demonstrated in EIEE5 where deficiencies in this binding lead to severe neurological abnormalities128. Whether this type of interaction between αII-spectrin and β-spectrins also takes place in the nucleus, though this has been proposed, is not yet entirely clear. However, this raises the interesting possibility that a deficiency in interaction of αII-spectrin and β-spectrins in the nucleus could lead to identification of another group of pathological disorders.
As described above, a deficiency or loss of αII-spectrin in cells can be due to a number of different causes and can lead to a number of different pathological manifestations, depending on the disorder. In FA, for example, reduced levels of αII-spectrin in cells (35–40% of normal) are due to increased cleavage by µ-calpain at its cleavage site.16 In this disorder, it is the FA gene products, the FA proteins, which are proposed to play a role in maintaining the stability of αII-spectrin in the cell by limiting its cleavage by µ-calpain.16 This disorder is characterized by developmental abnormalities, bone marrow failure, a predisposition to cancer, and DNA repair defects.54–65,59–63 In another disorder, EIEE5, reduced levels of αII-spectrin are due to mutations in the αII-spectrin gene, SPTAN1, which leads to loss of αII-spectrin in neurons.128,160–162 This disorder is characterized by severe neurodevelopmental deficiencies and progressive brain atrophy.128,160–162 Thus, severe clinical pathologies are observed when there are losses in αII-spectrin due either to its increased breakdown or to its reduced expression in cells.
An important factor that needs to be kept in mind when examining cells for levels of αII-spectrin in the nucleus is that this is a labile protein. It can break down easily, especially when samples are being prepared for analysis, such as for Western blots.11,12 Thus, care needs to be taken in sample preparation. Further investigations into levels of αII-spectrin in various pathological disorders could thus potentially reveal that there is a loss of αII-spectrin, due either to its enhanced breakdown or to its decreased expression, which could provide important insights into the etiopathology of these disorders.
Conclusions and perspectives
αIISp, an essential cellular protein and an important component of the nucleoskeleton, has structural and non-structural functions, both of which are important. The best documented role of αIISp in human cell nuclei is in DNA repair.12–19 Our laboratory has shown that αIISp is critical for repair of ICLs both in genomic and, separately, in telomeric DNA and is necessary for telomere function and for chromosome stability after DNA ICL damage.11–19 We have proposed that αIISp functions as a scaffold aiding in the recruitment of repair proteins, such as XPF-ERCC1, to sites of damage and that it acts downstream from monoubiquitinated FANCD2.12,15,64 Based on our studies, we have hypothesized that, mechanistically, αIISp binds to DNA at sites of damage and recruits XPF-ERCC1 to these sites via FANCG, which acts as a link joining αIISp to XPF-ERCC1, enabling incisions to be made at the site of damage.32,74 Thus, αIISp contributes to an essential component of the ICL repair process, which involves a multitude of other proteins. We have proposed that FA proteins play an important role in maintaining the stability of αIISp in the cell and have suggested several mechanisms as to how this may occur.16,18 Additionally, αIISp’s interactions with other proteins in the nucleus, such as those involved in other types of DNA repair, chromatin remodeling, transcription and RNA processing, indicate that it has a number of other non-structural roles in the nucleus.18
αIISp also has a structural role in the nucleus that has been far less extensively examined. It associates with the peripheral nucleoskeleton and co-immunoprecipitates and/or co-purifies with a number of the peripheral nucleoskeletal proteins.19–21 These include lamin, emerin, actin, protein 4.1, nuclear myosin, βIV-spectrin, and SUN proteins.19–21 Through its functional interactions with these other structural proteins, αIISp may have a significant role in the architecture and mechanical properties of the nucleus.7,20–22 It has been shown to make an important contribution to the resilience and elasticity of the nucleus.7,20,21 However, the effect DNA damage has on these functions has not yet been investigated and this clearly warrants further investigation.
The finding that αIISp has both structural and non-structural roles in the nucleus contributes to our understanding of the significance a loss in αIISp can have on cell function. In a disorder such as FA, in which we documented a marked deficiency in αIISp in the nucleus, the importance this deficiency has on the phenotypic characteristics observed is clearly demonstrated by studies which show that restoration of αIISp levels to normal corrects these defects.16,17 FA patients, however, also have a number of congenital abnormalities. An important question is whether a deficiency in αIISp in FA cells is related to these defects. In mouse models, loss of αIISp has been shown to lead to developmental anomalies.38 It is thus possible that αIISp plays an important role in human development. A deficiency in αIISp in FA could be involved in the developmental anomalies observed, such as failure of digit formation and renal abnormalities. This is a totally unexplored area which would be exceedingly interesting to investigate and could lead to determination of whether a deficiency in αIISp is an important factor in these developmental abnormalities.
Loss of αIISp could be of clinical significance in other disorders. An important question is whether such a loss plays a role in the pathogenesis of bone marrow diseases such a leukemia. This is of particular interest since a study has shown that there is a loss of αIISp in bone marrow from a sizable portion of AML patients.153,154 Other investigations have demonstrated that specific mutations in the SPTAN1 gene coding for αII-spectrin, which affect the ability of αII-spectrin to bind to β-spectrins in neurons, lead to a group of infantile epileptic encephalopathies (EIEE5).128,160–162 These findings further confirm the importance of interaction of αII- and β-spectrins for neuronal cytoskeletal structure and function.128,160–162 It will be of considerable interest to determine whether such a relationship between these spectrins is also present in the nucleus and whether deficiencies in this interaction contribute to specific pathological conditions.
Thus, a deficiency in αIISp in cells has the potential to affect a number of different systems encompassing both structural and non-structural elements of cell function. Obtaining a better knowledge of αIISp’s functional interaction with other nuclear proteins will increase our understanding of the clinical significance loss of αIISp has on critical cellular systems and enhance our understanding of the pathogenesis of disorders, such as FA and EIEE5, in which there is a deficiency in αIISp.
Author’s contribution
ML wrote the manuscript and prepared the figures.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by NHLBI, National Institutes of Health, Grant RO1 HL054860.
References
- 1.Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Gene Dev 2002; 16:533–47 [DOI] [PubMed] [Google Scholar]
- 2.Taddei A, Hediger F, Neumann FR, Gasser SM. The function of nuclear architecture: a genetic approach. Annu Rev Genet 2004; 38:305–45 [DOI] [PubMed] [Google Scholar]
- 3.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 2008; 22:832–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redwood AB, Gonzales-Suarez I, Gonzalo S. Regulating the levels of key factors in cell cycle and DNA repair: new pathways revealed by lamins. Cell Cycle 2011; 10:3652–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol 2009; 10:243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson KL, Dawson SC. Functional evolution of nuclear structure. J Cell Biol 2011; 195:171–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon DN, Wilson KL. The nucleoskeleton as a genome-associated dynamic network of networks. Nat Rev Mol Cell Biol 2011; 12:695–708 [DOI] [PubMed] [Google Scholar]
- 8.Liu N-A, Sun J, Kono K, Horikoshi Y, Ikura T, Tong X, Haraguchi T, Tashiro S. Regulation of homologous recombinational repair by lamin B1 in radiation-induced DNA damage. FASEB J 2015; 29:2514–25 [DOI] [PubMed] [Google Scholar]
- 9.Gibbs-Seymour I, Markiewicz EW, Bekker-Jensen S, Mailand N, Hutchison CJ. Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging Cell 2015; 14:162–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brois DW, McMahon LW, Ramos NL, Anglin LM, Walsh CE, Lamabert MW. A deficiency in a 230 kDA DNA repair protein in Fanconi anemia complementation group A cells is corrected by the FANCA cDNA. Carcinogenesis 1999; 20:1845–53 [DOI] [PubMed] [Google Scholar]
- 11.McMahon LW, Walsh CE, Lambert MW. Human α spectrin II and the Fanconi anemia proteins FANCA and FANCC interact to form a nuclear complex. J Biol Chem 1999; 274:32904–8 [DOI] [PubMed] [Google Scholar]
- 12.McMahon LW, Sangerman J, Goodman SR, Kumaresan K, Lambert MW. Human alpha spectrin II and the FANCA, FANCC, and FANCG proteins bind to DNA containing psoralen interstrand cross-links. Biochemistry 2001; 40:7025–34 [DOI] [PubMed] [Google Scholar]
- 13.Sridharan D, Brown M, Lambert WC, McMahon LW, Lambert MW. Nonerythroid alpha II spectrin is required for recruitment of FANCA and XPF to nuclear foci induced by DNA interstrand cross-links. J Cell Sci 2003; 116:823–5 [DOI] [PubMed] [Google Scholar]
- 14.Kumaresan K, Sridharan D, McMahon L, Lambert MW. Deficiency in incisions produced by XPF at the site of a DNA interstrand cross-link in Fanconi anemia cells. Biochemistry 2007; 46:14359–68 [DOI] [PubMed] [Google Scholar]
- 15.McMahon LW, Zhang P, Sridharan DM, Lefferts JA, Lambert MW. Knockdown of αII spectrin in normal human cells by siRNA leads to chromosomal instability and decreased DNA interstrand cross-link repair. Biochem Biophys Res Commun 2009; 381:288–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang P, Sridharan D, Lambert MW. Knockdown of µ-calpain in Fanconi anemia, FA-A, cells by siRNA restores αII spectrin levels and corrects chromosomal instability and defective DNA interstrand cross-link repair. Biochemistry 2010; 49:5570–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang P, Herbig U, Coffman F, Lambert MW. Non-erythroid alpha spectrin prevents telomere dysfunction after DNA interstrand cross-link damage. Nucleic Acids Res 2013; 41:5321–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert MW. Functional significance of nuclear α spectrin. J Cell Biochem 2015; 116:1816–30 [DOI] [PubMed] [Google Scholar]
- 19.Sridharan DM, McMahon LW, Lambert MW. αII-Spectrin interacts with five groups of functionally important proteins in the nucleus. Cell Biol Intl 2006; 30:866–78 [DOI] [PubMed] [Google Scholar]
- 20.Holaska JM, Wilson KL. An emerin ‘proteome’: purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signalling, mechano-sensing and nuclear architecture. Biochemistry 2007; 46:8897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zohn Z, Wilson KL, Dahl KN. Beyond lamins: other structural components of the nucleoskeleton. Methods Cell Biol 2010; 98:97–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armiger TJ, Spagnol ST, Dahl KN. Nuclear mechanical resilience but not stiffness is modulated by αII-spectrin. J Biomech 2016; 49:3983–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkelmann JC, Forget BG. Erythroid and nonerythroid spectrins. Blood 1993; 81:3173–85 [PubMed] [Google Scholar]
- 24.Goodman SR, Zimmer WE, Clark MB, Zagon IS, Barker JE, Bloom ML. Brain spectrin: of mice and men. Brain Res Bull 1995; 36:593–606 [DOI] [PubMed] [Google Scholar]
- 25.de Matteis MA, Morrow JS. Spectrin tethers and mesh in the biosynthetic pathway. Cell Sci 2000; 113:2331–43 [DOI] [PubMed] [Google Scholar]
- 26.Brown JW, Bullitt E, Sriswasdi S, Harper S, Speicher DW, McKnight CJ. The physiological molecular shape of spectrin: a compact supercoil resembling a Chinese finger Trap. PLOS Comput Biol 2015; 11:e1004302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musacchio A, Gibson T, Lehto VP, Saraste M. SH3 – an abundant protein domain in search of a function. FEBS Lett 1992; 307:55–61 [DOI] [PubMed] [Google Scholar]
- 28.Cohen GB, Ren R, Baltimore D. Modular binding domains in signal transduction proteins. Cell 1995; 80:237–48 [DOI] [PubMed] [Google Scholar]
- 29.Kuriyan J, Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct 1997; 26:259–88 [DOI] [PubMed] [Google Scholar]
- 30.McPherson PS. Regulatory role of SH3 domain-mediated protein-protein interactions in synaptic vesicle endocytosis. Cell Signal 1999; 11:229–38 [DOI] [PubMed] [Google Scholar]
- 31.Mayer BJ. SH3 domains: complexity in moderation. J Cell Sci 2001; 114:1253–63 [DOI] [PubMed] [Google Scholar]
- 32.Lefferts JA, Wang C, Sridharan D, Baralt M, Lambert MW. The SH3 domain of αII spectrin is a target for the Fanconi anemia protein, FANCG. Biochemistry 2009; 48:254–63 [DOI] [PubMed] [Google Scholar]
- 33.Goodman SR, Zagon IS, Kulikowski RR. Identification of a spectrin-like protein in nonerythroid cells. Proc Natl Acad Sci USA 1981; 78:7570–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zagon IS, Higbee R, Riederer BM, Goodman SR. Spectrin subtypes in mammalian brain: an immunoelectron microscopic study. J Neurosci 1986; 6:2977–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gascard P, Mohandas N. New insights into functions of erythroid proteins in nonerythroid cells. Curr Opin Hematol 2000; 7:123–9 [DOI] [PubMed] [Google Scholar]
- 36.Bennet V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 2001; 81:353–92 [DOI] [PubMed] [Google Scholar]
- 37.Metral S, Machnicka B, Bigot S, Colin Y, Dhermy D, Lecomte M-C. αII-Spectrin is critical for cell adhesion and cell cycle. J Biol Chem 2009; 284:2409–18 [DOI] [PubMed] [Google Scholar]
- 38.Stankewich MC, Cianci CD, Stabach PR, Ji L, Nath A, Morrow J. Cell organization, growth, and neural and cardiac development require αII-spectrin. J Cell Sci 2011; 124:3956–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Machnicka B, Grochowalska R, Boguslawska DM, Sikorski AF, Lecomte MC. Spectrin-based skeleton as an actor in cell signaling. Cell Mol Life Sci 2012; 69:191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodman SR, Chapa RP, Zimmer WE. Spectrin’s chimeric E2/E3 enzymatic activity. Exp Biol Med 2015; 240:1039–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akkari YM, Bateman R, Reifsteck CA, Olson SB, Grompe MDNA. replication is required to elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol Cell Biol 2000; 20:8283–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 2008; 134:969–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009; 326:1698–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Legerski RJ. Repair of DNA interstrand cross-links during S phase of the mammalian cell cycle. Environmen Mol Mutagen 2010; 51:540–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumaresan KR, Hang B, Lambert MW. Human endonucleolytic incision of DNA 3’ and 5’ to a site-directed psoralen monoadduct and interstrand cross-link. J Biol Chem 1995; 270:30709–16 [DOI] [PubMed] [Google Scholar]
- 46.Kim H, D’andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 2012; 26:1393–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013; 493:356–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klein DD, Boon RACM, Long DT, Szypowska AA, Raschle M, Walter JC, Knipscheer PXPF-ERCC1. acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 2014; 54:460–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014; 19:135–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haynes B, Saadat N, Myung B, Shekhar MP. Crosstalk between translesion synthesis, Fanconi anemia network, and homologous recombination repair pathways in interstrand DNA crosslink repair and development of chemoresistance. Mutat Res Rev Mutat Res 2015; 763:258–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopez-Martinez D, Liang C-C, Cohn MA. Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci 2016; 73:3097–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumaresan KR, Lambert MW. Fanconi anemia, complementation group A, cells are defective in ability to produce incisions at sites of psoralen interstrand cross-links. Carcinogenesis 2000; 21:741–51 [DOI] [PubMed] [Google Scholar]
- 53.Lambert MW, Tsongalis JT, Lambert WC, Parrish DD. Correction of the DNA repair defect in Fanconi anemia complementation groups A and D cells. Biochem Biophys Res Commun 1997; 230:687–91 [DOI] [PubMed] [Google Scholar]
- 54.Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006; 25:5875–84 [DOI] [PubMed] [Google Scholar]
- 55.de Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat Res 2009; 668:11–9 [DOI] [PubMed] [Google Scholar]
- 56.Moldovan G-L, D’andrea A. How the Fanconi anemia pathway guards the genome. Rev Genet Ann 2009; 43:223–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Ann Rev Biophys 2014; 43:257–78 [DOI] [PubMed] [Google Scholar]
- 58.Cantor SB, Brosh RM., Jr. What is wrong with Fanconi anemia cells? Cell Cycle 2014; 13:3823–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brosh RM, Jr, Bellani M, Liu Y, Seidman MN. Fanconi anemia: a DNA repair disorder characterized by accelerated decline of the hematopoietic system cell compartment and other features of aging. Aeging Res Rev 2017; 33:67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mamrak NE, Shimamura A, Howlell NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev 2017; 31:93–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheung RS, Taniguchi T. Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Intl. J Hematol 2017; 106:335–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood 2002; 101:822–6 [DOI] [PubMed] [Google Scholar]
- 63.Swuec P, Ranault L, Borg A, Shah F, Murphy VJ, van Twest S, Snijders AP, Deans AJ, Costa A. The FA core complex contains a homo-dimeric catalytic module for symmetric mono-ubiquitination of FANCI-FANCD2. Cell Rep 2017; 18:611–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang P, Sridharan D, Lambert MW. Nuclear α spectrin differentially affects monoubiquitinated versus non-ubiquitinated FANCD2 function after DNA interstrand cross-link damage. J Cell Biochem 2016; 117:671–83 [DOI] [PubMed] [Google Scholar]
- 65.Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science 1993; 259:1157–61 [DOI] [PubMed] [Google Scholar]
- 66.Fen S, Chen JK, Yu H, Simon JA, Schreiber SL. Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3-ligand interactions. Science 1994; 266:1241–7 [DOI] [PubMed] [Google Scholar]
- 67.Lim WA, Richards FM, Fox RO. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 1994; 372:375–9 [DOI] [PubMed] [Google Scholar]
- 68.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J 2000; 14:231–41 [PubMed] [Google Scholar]
- 69.Cesareni G, Panni S, Nardelli G, Castagnoli L. Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett 2002; 513:38–44 [DOI] [PubMed] [Google Scholar]
- 70.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 1999; 21:932–9 [DOI] [PubMed] [Google Scholar]
- 71.D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci 2003; 28:655–62 [DOI] [PubMed] [Google Scholar]
- 72.Blom E, van de Vrugt HJ, de Vries Y, de Winter JP, Arwert F, Joenje H. Multiple TPR motifs characterize the Fanconi anaemia FANCG protein. DNA Repair 2004; 3:77–84 [DOI] [PubMed] [Google Scholar]
- 73.Hussain S, Wilson JB, Blom W, Thompson LH, Sung P, Gordon SM, Kupfer GM, Joenje H, Mathew CG, Jones NJ. Tetratricopeptide-motif-mediated interaction of FANCG with recombination proteins XRCC3 and BRCA2. DNA Repair 2006; 5:629–40 [DOI] [PubMed] [Google Scholar]
- 74.Wang C, Lambert MW. The Fanconi anemia protein, FANCG, binds to the ERCC1-XPF endonuclease via its tetratricopeptide repeats and the central domain of ERCC1. Biochemistry 2010; 49:5560–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Laat WL, Sijbers AM, Odijk H, Jaspers NG, Hoeijmakers JH. Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res 1998; 26:4146–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tripsianes K, Folkers GE, Ab E, Das D, Odijk H, Jaspers NGJ, Hoeijmakers JHJ, Kapten R, Boelens R. The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure 2005; 13:1849–58 [DOI] [PubMed] [Google Scholar]
- 77.Choi YJ, Ryu KS, Ko YM, Chae YK, Pleton JG, Wemmer DE, Choi BS. Biophysical characterization of the interaction domains and mapping of the contact residues in the XPF-ERCC1 complex. J Biol Chem 2005; 280:28644–52 [DOI] [PubMed] [Google Scholar]
- 78.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 2007; 8:735–48 [DOI] [PubMed] [Google Scholar]
- 79.Seki S, Ohzeki M, Uchiada A, Hirano S, Matsushita N, Kitao H, Oda T, Yamashita T, Kashihara N, Tsubahara A, Takata M, Ishiai M. A requirement of FancL and FancD2 monoubiquitination in DNA repair. Genes Cells 2007; 12:299–310 [DOI] [PubMed] [Google Scholar]
- 80.Bhattachavjee S, Nandi S. DNA damage response and cancer therapeutics through the lens of the Fanconi anemia DNA repair pathway. Cell Commun Signal 2017; 15:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liang CC, Li Z, Lopez-Martinez D, Nicholson WV, Venien-Bryan C, Cohn MA. The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat Commun 2016; 7:12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee J, Coyne RS, Dubreuil RR, Goldstein LS, Branton D. Cell shape and interaction defects in alpha-spectrin mutants of Drosophila melanogaster. J Cell Biol 1993; 123:1797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Norman KR, Moerman DG. αSpectrin is essential for morphogenesis and body wall muscle formation in Caenorhabditis elegans. J Cell Biol 2002; 157:665–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huh GY, Glant SB, Je S, Morrow JS, Kim JH. Calpain proteolysis of αII-spectrin in the normal adult human brain. Neurosci Lett 2001; 316:41–4 [DOI] [PubMed] [Google Scholar]
- 85.Pike BR, Flint J, Dutta S, Johnson E, Wang KKW, Hayes RL. Accumulation of non-erythroid αII-spectrin and calpain-cleaved αII-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J Neurochem 1297; 78:1297–306 [DOI] [PubMed] [Google Scholar]
- 86.Nicolas G, Fournier CM, Galand C, Malbert-Colas L, Bournier O, Kroviarski Y, Bourgeois M, Camonis JH, Dhermy D, Grandchamp B, Lecomte MC. Tyrosine phosphorylation regulates alpha II spectrin cleavage by calpain. Mol Cell Biol 2002; 22:3527–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Czogalla A, Sikorski AF. Spectrin and calpain: a ‘target’ and ‘sniper’ in the pathology of neuronal cells. Cell Mol Life Sci 2001; 62:1913–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nedrelow JH, Cianci CD, Morrow JSC-S. binds αII spectrin’s Src homology 3 (SH3) domain and blocks calpain susceptibility by phosphorylating Tyr1176. J Biol Chem 2003; 278:7725–41 [DOI] [PubMed] [Google Scholar]
- 89.Rotter B, Kroviarski Y, Nicholas G, Dhermy D, Lecomte MC. AlphaII-spectrin is an in vitro target for caspase-2, and its cleavage is regulated by calmodulin binding. Biochem J 2004; 378:161–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harris AS, Croall DE, Morrow JS. Calmodulin regulates fodrin susceptibility to cleavage by calcium-dependent protease I. J Biol Chem 1989; 264:17401–8 [PubMed] [Google Scholar]
- 91.Simonovic M, Zhang Z, Cianci CD, Steitz TA, Morrow JS. Structure of the calmodulin αII-spectrin complex provides insight into the regulation of cell plasticity. J Biol Chem 2006; 281:34333–40 [DOI] [PubMed] [Google Scholar]
- 92.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19:2100–10 [DOI] [PubMed] [Google Scholar]
- 93.Murnane JP. Telomere dysfunction and chromosome instability. Mutat Res 2012; 730:28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet 2008; 42:301–34 [DOI] [PubMed] [Google Scholar]
- 95.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 2010; 11:171–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science 2012; 336:593–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wright WE, Tesmer VM, Liao ML, Shay JW. Normal human telomeres are not late replicating. Exptl Cell Res 1999; 251:492–9 [DOI] [PubMed] [Google Scholar]
- 98.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol 2003; 13:1549–56 [DOI] [PubMed] [Google Scholar]
- 99.Zhang R, Liu C, Niu Y, Jing Y, Zhang H, Wang J, Yang J, Zen K, Zhang J, Zhang C-Y, Li D. MicroRNA-128-3p regulates mitomycin C-induced DNA damage response in lung cancer cells through repressing SPTAN1. Oncotarget 2016; 8:58098–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tse WT, Tang J, Jin O, Korsgren C, John KM, Kung AL, Gwynn BB, Peter LL, Lux SEA. new spectrin beta IV, has a major truncated isoform that associates with promyelocytic leukemia protein nuclear bodies and the nuclear matrix. J Biol Chem 2001; 276:2397485. [DOI] [PubMed] [Google Scholar]
- 101.Thenappan A, Shukkla V, Abdul Khalek FJ, Li Y, Shetty K, Liu P, Li L, Johnson RL, Johnson L, Mishra L. Loss of TGFβ adaptor protein β2SP leads to delayed liver regeneration in mice. Hepatology 2011; 53:1641–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Horikoshi N, Pandita RK, Mujoo K, Hambarde S, Sharma D, Mattoo AR, Chakraborty S, Charaka V, Hunt CR, Pandita TK. β2-spectrin depletion impairs DNA damage repair. Oncotarget 2016; 7:33557–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen J, Shukla V, Farci P, Andricovich J, Jogunoori W, Kwong LN, Katz LH, Sheety K, Rashid A, Su X, White J, Li L, Wang AY, Blechacz B, Raju GS, Davila M, Nguyen B-N, Stroehlein JR, Chen J, Kim SS, Levin H, Machida K, Tsukamoto H, Michaely P, Tzatsos A, Mishra B, Amber R, Mishra L. Loss of the transforming growth factor-β effector β2-spectrin promotes genomic instability. Hepatology 2017; 65:678–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tang Y, Katuri V, Dillner A, Mishra B, Deng C-X, Mishra L. Disruption of transforming growth factor-β signalling in ELF β2-spectrin-deficient mice. Science 2003; 299:576–7 [DOI] [PubMed] [Google Scholar]
- 105.Dhal KN, Kalinowski A. Nucleoskeleton mechanics at a glance. J Cell Sci 2011; 124:675–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol 2010; 2:a000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gerace L, Huber MD. Nuclear lamina at the crossroads of the cytoplasm and nucleus. J Struct Biol 2012; 177:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Davidson PNM, Lammerding J. Broken nuclei–lamins, nuclear mechanics and disease. Trends Cell Biol 2014; 24:247–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gruenbaum Y., Medalia O. Lamins: the structure and protein complexes. Curr Opin Cell Biol 2015; 32:7–12 [DOI] [PubMed] [Google Scholar]
- 110.Barton LJ, Soshnev AA, Geyer PK. Networking in the nucleus: a spotlight on LEM-domain proteins. Curr Opin Cell Biol 2015; 34:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lammerding J, Schulze PC, Takahaski T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 2004; 113:370–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Visa N, Percipalle P. Nuclear functions of actin. Cold Spring Harb Perspect Biol 2010; 2:a000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hofmann WA. Cell and molecular biology of nuclear actin. Int Rev Cell Mol Biol 2009; 273:219–63 [DOI] [PubMed] [Google Scholar]
- 114.Gieni RS, Hendzel MJ. Actin dynamics and functions in the interphase nucleus: moving toward an understanding of nuclear polymeric actin. Biochem Cell Biol 2009; 87:283–306 [DOI] [PubMed] [Google Scholar]
- 115.Virtanen JA, Vartianinen MK. Diverse functions for different forms of nuclear actin. Curr Opin Cell Biol 2017; 46:33–8 [DOI] [PubMed] [Google Scholar]
- 116.Castano E, Philimonenko VV, Kahle M, Fukalova J, Kalendova A, Yildirim S, Dzijak R, Dingova-Krasna H, Hozak P. Actin complexes in the cell nucleus: new stones in an old fields. Histochem Cell Biol 2010; 133:607–27 [DOI] [PubMed] [Google Scholar]
- 117.Kristo I, Bajusz I, Bajusz C, Borkuti P, Vilmos P. Actin, actin-binding proteins, and actin-related proteins in the nucleus. Histochem Cell Biol 2016; 145:373–88 [DOI] [PubMed] [Google Scholar]
- 118.Khatau SB, Hale CM, Stewart-Hutchinson J, Patel MS, Stewart CL, Searson PC, Hotzic D, Wirtz D. A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci USA 2009; 106:19017–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miroshnikova YA, Nava MN, Wickstrom SA. Emerging roles of mechanical forces in chromatin regulation. J Cell Sci 2017; 130:2243–50 [DOI] [PubMed] [Google Scholar]
- 120.Bengtsson L, Wilson KL. Multiple and surprising new functions for emerin, a nuclear membrane protein. Curr Opin Cell Biol 2004; 16:73–9 [DOI] [PubMed] [Google Scholar]
- 121.Holaska JM, Kowalski AM, Wilson SL. Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the inner nuclear membrane. PLoS Biol 2004; 2:1354–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lammerding J, Hsiao J, Schulze PC, Kazlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol 2005; 170:781–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Berk JM, Tifft KE, Wilson KL. The nuclear envelope LEM-domain protein emerin. Nucleus 2013; 4:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Meyer AJ, Almendrala DK, Go MM, Krauss SW. Structural protein 4.1R is integrally involved in nuclear envelope protein localization, centrosome-nucleus association and transcriptional signaling. J Cell Sci 2010; 124:1433–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Komada M, Soriano P. [Beta]IV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol 2002; 156:337–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang Y, Ogawa Y, Hedstrom KL, Rasband MN. BetaIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrin G. J Cell Biol 2007; 176:508–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Galiano MR, Jha S, Ho TS, Zhang C, Ogawa Y, Chang KJ, Stankewich MC, Mohler PJ, Rasband MN. A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell 2012; 149:1125–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang Y, TJ, Nelson AD, Glanowska K, Murphy GC, Jenkins PM, Parent JM. Critical roles of αII spectrin in brain development and epileptic encephalopathy. J Clin Invest 2018; 128:760–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kakhniashvilli DG, Chaudhary T, Zimmer WE, Bencsath FA, Jardine L, Goodman SR. Erythrocyte spectrin is an E2 ubiquitin conjugating enzyme. Biochemistry 2001; 40:1163–42 [DOI] [PubMed] [Google Scholar]
- 130.Hsu YJ, Zimmer WE, Goodman SR, Erythrocyte spectrin’s chimeric ED2/E3 ubiquitin conjugating/ligating activity. Cell Mol Biol, 2005;51:187–93. [PubMed] [Google Scholar]
- 131.Hochstrasser M. Protein degradation or regulation: UB the judge. Cell 1996; 84:813–5 [DOI] [PubMed] [Google Scholar]
- 132.Schwartz AL, Ciechanover A. The ubiquitin proteasome pathway and pathgenesis of human diseases. Annu Rev Med 1999; 50:57–74 [DOI] [PubMed] [Google Scholar]
- 133.Galluzi L, Nicholas G, Paiardini M, Magnani M, Lecomte MC. Identification of ubiquitinated repeats in human erythroid α-spectrin. Eur J Biochem 2000; 267:2812–8 [DOI] [PubMed] [Google Scholar]
- 134.Dwane L, Gallagher WM, Chonghaile TN, O’connor DP. The emerging role of non-traditional ubiquitination in oncogenic pathways. J Biol Chem 2017; 292:3543–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Galluzi L, Paiardini M, Lecomte MC, Magnani M. Identification of the main ubiquitination site in human erythroid α-spectrin. FEBS Lett 2001; 489:254–8 [DOI] [PubMed] [Google Scholar]
- 136.Riahi MH, Kakhniashvili DG, Goodman SR. Ubiquitination of red blood cell alpha-spectrin does not affect heterodimer formation. Amer J Hematol 2005; 78:281–7 [DOI] [PubMed] [Google Scholar]
- 137.Ghatpande SG, Goodman SR. Ubiquitination of spectrin regulates the erythrocyte spectrin-protein 4.1-actin ternary complex dissociation: implications for sickle cell membrane skeleton. Cell Mol Biol 2004; 50:67–74 [PubMed] [Google Scholar]
- 138.Sangerman J, Killiea A, Chronister R, Pappolla N, Goodman SR. Alpha-spectrins are major ubiquitinated proteins in rat hippocampal neurons and components of ubiquitinated inclusions in neurodegenerative disorders. Brain Res Bull 2001; 54:405–11 [DOI] [PubMed] [Google Scholar]
- 139.Dahl KN, Riberio AJS, Lammerding J. Nuclear shape, mechanics and mechanotransduction. Circ Res 2008; 102:1307–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Martins RP, Finan JD, Guilak F, Lee DA. Mechanical regulation of nuclear structure and function. Annu Rev Biomed Eng 2012; 14:431–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.King MC, Lusk PC. A model for coordinating nuclear mechanics and membrane remodelling to support nuclear integrity. Curr Opin Cell Biol 2016; 41:9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Crisp M, Liu Q, Rous K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and the cytoplasm: role of the LINC complex. J Cell Biol 2005; 172:41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry CM, Trembath RC, Shackleton SSUN1. Interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 2006; 26:3738–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lombardi ML, Jaalouk DE, Shanhan MC, Burke B, Roux KJ, Lammerding J. The interaction between nesprins and Sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J Biol Chem 2011; 286:26743–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oza P, Jaspersen SL, Miel A, Dekker J, Peterson CL. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 2009; 23:912–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lei K, Zhu X, Xu C, Shao C, Xu T, Zhuang Y, Han M. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr Biol 2012; 22:1609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Swartz RK, Rodriquez EC, King MC. A role for nuclear envelope-bridging complexes in homology-directed repair. Mol Biol Cell 2014; 25:2461–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lottersberger F, Karssemeijer N, Dimitrova N, de Lange T. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell 2015; 163:880–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lawrence KS, Tapley EC, Cruz VE, Li Q, Aung K, Hart KC, Schwartz TU, Starr DA, Engebrecht JLINC. complexes promote homologous recombination in part through inhibition of nonhomologous end joining. J Cell Biol 2016; 215:801–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Meissner JM, Sikorski AF, Nawara T, Grzesiak J, Marycz KM, Bogustawska DM, Michalczyk I, Lecomte M-C, Machnicka B. α-spectrin in T cells is involved in the regulation of cell-cell contact leading to immunological synapse formation. Plos One 2017; 12:e0189545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu Y, Ballman K, Li D, Khan S, Derr-Yellin E, Shou W, Haneline LS. Impaired function of Fanconi anemia type C-deficient macrophages. J Leukoc Biol 2012; 91:333–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Goll DE, Thompson VF, Ki H, Wei W, Cong J. The calpain system. Physiol Rev 2003; 83:731–801 [DOI] [PubMed] [Google Scholar]
- 153.Wolqust LP, Cannizzarro LA, Rameshjk H, Xue X, Wang D, Bhattacharuya PK, Gong JZ, McMahon C, Albanese JM, Sunkara JL, Ratech H. Differential expression in normal hematopoiesis and alterations in neoplastic bone marrow disorders. Am J Clin Path 2011; 136:300–8 [DOI] [PubMed] [Google Scholar]
- 154.Gorman EB, Chen L, Albanese J, Ratech H. Pattern of spectrin expression in B-cell lymphomas: loss of spectrin isoforms is associated with nodule-forming and germinal center-related lymphomas. Mod Pathol 2007; 20:1245–52 [DOI] [PubMed] [Google Scholar]
- 155.Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer 2003; 97:425–40. [DOI] [PubMed] [Google Scholar]
- 156.Riederer BM, Zagon IS, Goodman SR. Brain spectrin(240/235) and brain spectrin(240/235E): differential expression during mouse brain development. J Neurosci 1987; 7:864–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zagon IS, Riderer BM, Goodman SR. Spectrin expression during mammalian brain ontogeny. Brain Res Bull 1987; 18:799–807 [DOI] [PubMed] [Google Scholar]
- 158.Zimmer WE, Ma Y, Zagon IS, Goodman SR. Developmental expression of brain beta-spectrin isoform messenger RNAs. Brain Res 1992; 594:75–83 [DOI] [PubMed] [Google Scholar]
- 159.Glanz A, Fraser FC. Spectrum of anomalies in Fanconi anaemia. J Med Genet 1982; 19:412–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Saitsu H, Tohyama J, Kumada T, Egawa K, Hamada K, Okada I, Mizuguchi T, Osaka H, Miyata R, Furukawa T, Haginoya K, Hoshino H, Goto T, Hachiya Y, Yamagata T, Saitoh S, Nagai T, Nishiyama K, Nishimura A, Miyake N, Komada M, Hayashi K, Hirai S, Ogata K, Kato M, Fukuda A, Matsumoto N. Dominant-negative mutations in α-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet 2010; 86:881–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tohyama J, Nakashima M, Nabatame S, Gaik-Siew C, Miyata R, Rener-Primec Z, Kato M, Matsumoto N, Saitsu H. SPTAN1. encephalopathy: distinct phenotypes and genotypes. J Human Genet 2015; 60:167–73 [DOI] [PubMed] [Google Scholar]
- 162.Syrbe S, Harms FL, Parrini E, Montomoli M, Mutze U, Helbig KL, Polster T, Albrecht B, Bernbeck U, van Binsbergen E, Biskup S, Burglen L, Denecke J, Heron B, Heyne HO, Hoffmann GF, Hornemann F, Matsushige T, Matsuura R, Kato M, Korenke GC, Kuechler A, Lammer C, Merkenschlager A, Mignot C, Ruf S, Nakashima M, Saitsu H, Stamberger H, Pisano T, Tohyama J, Weckhuysen S, Werckx W, Wickeart J, Mari F, Verbeek NE, Moller RS, Koeleman B, Matsumoto N, Dobyns WB, Battaglia D, Lemke JR, Kutsche K, Guerrini R. Delineating SPTAN1 associated phenotypes: from isolated epilepsy to encephalopathy with progressive brain atrohy. Brain 2017; 140:2322–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Riederer BM, Zagon IS, Goodman SR. Brain spectrin (240/235) and brain spectrin (240/235E): two distinct spectrin subtypes with different locations within mammalian neural cells. J Cell Biol 1986; 102:2088–97 [DOI] [PMC free article] [PubMed] [Google Scholar]