

Abstract
Although BRAF inhibitor monotherapy yields response rates >50% in BRAFV600-mutant melanoma, only ~5% with BRAFV600E colorectal cancer (CRC) respond. Preclinical studies suggest that lack of efficacy in BRAFV600E CRC is due to adaptive feedback reactivation of MAPK signaling, often mediated by EGFR. This clinical trial evaluated BRAF and EGFR inhibition with dabrafenib (D) + panitumumab (P) ± MEK inhibition with trametinib (T) to achieve greater MAPK suppression and improved efficacy in 142 patients with BRAFV600E CRC. Confirmed response rates for D+P, D+T+P, and T+P were 10%, 21%, and 0%, respectively. Pharmacodynamic analysis of paired pre- and on-treatment biopsies found that efficacy of D+T+P correlated with increased MAPK suppression. Serial cell-free DNA analysis revealed additional correlates of response and emergence of KRAS and NRAS mutations on disease progression. Thus, targeting adaptive feedback pathways in BRAFV600E CRC can improve efficacy, but MAPK reactivation remains an important primary and acquired resistance mechanism.
Keywords: BRAF, colorectal cancer, EGFR, MEK
INTRODUCTION
Activating gene mutations in the mitogen-activated protein kinase (MAPK) pathway are frequently observed in cancer and promote tumor cell migration, proliferation, and survival (1, 2). The serine/threonine protein kinase BRAF belongs to the RAF family of kinases (1, 2) (including ARAF and CRAF [RAF1]), which are normally activated by RAS family members (KRAS, NRAS, and HRAS), typically in response to signals from receptor tyrosine kinases (RTKs) (2, 3). BRAFV600 mutations lead to constitutive, RAS-independent activation of BRAF kinase activity and MAPK pathway signaling through downstream activation of MEK (MEK1 and MEK2) and ERK (ERK1 and ERK2) kinases (2, 3).
Oncogenic BRAFV600E mutations are present in ≈10% of colorectal cancers (CRCs) (2, 4) and ≈50% of melanomas (5). In CRC, BRAFV600E mutations confer a poor prognosis, resulting in nearly a 2-fold increase in mortality relative to wild-type BRAF in the metastatic setting (1, 6, 7). BRAFV600E mutation in CRC is associated with a right-sided primary site, advanced age, female sex, high tumor grade, and precursor sessile serrated adenomas (8). BRAFV600E CRC is also associated with the CpG island methylator phenotype (i.e., hypermethylated phenotype), which may result in the epigenetic inactivation of MLH1, inducing a mismatch repair (MMR) deficiency and consequently a microsatellite instability (MSI) phenotype (9). Among patients harboring BRAFV600E metastatic CRC, ≈20% exhibit deficient MMR deficiency (8). RAF inhibitors, such as vemurafenib and dabrafenib, selectively inhibit RAF monomers and have produced dramatic response rates >50% in metastatic melanoma, leading to their US Food and Drug Administration (FDA) approval for this indication (10, 11). However, single-agent BRAF inhibitors have demonstrated a surprising and striking lack of efficacy in patients with CRC harboring the same BRAFV600E mutation (12–16). Indeed, an initial study of vemurafenib in patients with the BRAFV600E mutation had a response rate of only 5% (16).
Preclinical studies have suggested that a primary reason for the differential sensitivities of BRAFV600E melanoma and CRC is that CRCs harbor robust adaptive feedback signaling networks that lead to reactivation of MAPK signaling following BRAF inhibitor treatment (12, 15). In this proposed model, inhibition of BRAFV600E leads to an initial reduction in MAPK signaling, causing a loss of expression of ERK-dependent negative feedback mediators that act to constrain MAPK pathway activation (Fig. 1A) (12). Loss of negative feedback leads to an induction of RAS activity and activation of other RAF kinases (such as CRAF), which bypass the effects of the BRAF inhibitor by generating BRAF inhibitor-resistant RAF dimers and restore MAPK pathway signaling (12). Increased RAS activity following BRAF inhibition is thought to be driven primarily by RTK signaling, which is present to a greater degree in CRC than in melanoma, and preclinical studies have suggested that 1 RTK in particular—the epidermal growth factor receptor (EGFR)—may play a dominant role in mediating MAPK reactivation in many BRAFV600E CRCs (12, 15). Indeed, the combination of BRAF and EGFR inhibition was found to produce improved MAPK suppression and lead to tumor regression in BRAFV600E CRC xenografts (12, 15).
Figure 1.
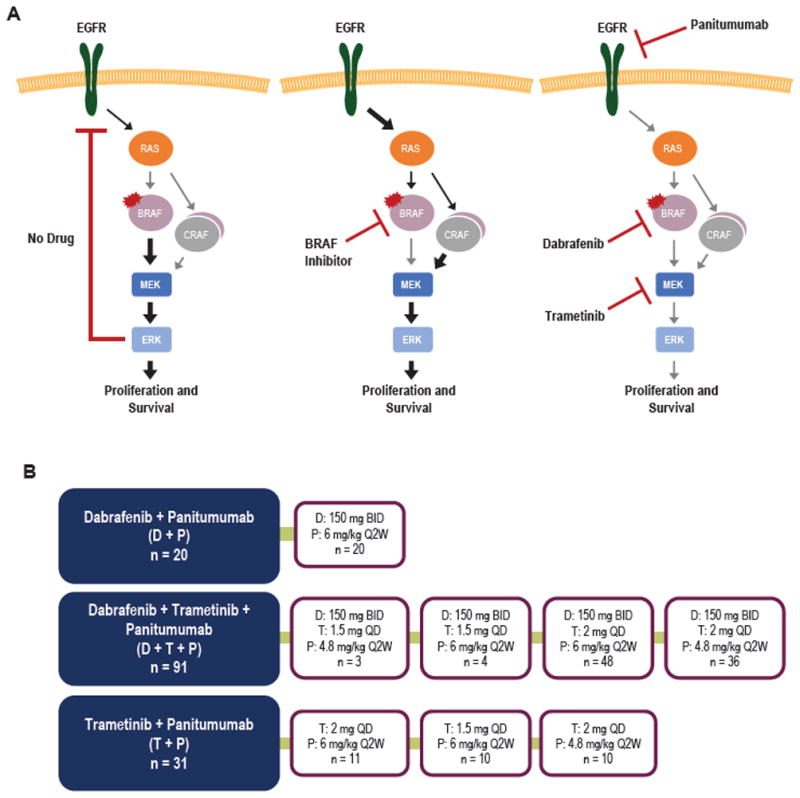
Targeting adaptive feedback signaling in BRAFV600E CRC. A, Model of adaptive feedback signaling in BRAFV600E CRC. Left, In the absence of drug, MAPK activity is driven by mutant BRAF, and ERK-dependent negative feedback signals constrain RTK-mediated activation of RAS. Center, BRAF inhibitor alone leads to transient inhibition of MAPK signaling and loss of ERK-dependent negative feedback signals, allowing RTK-mediated reactivation of the MAPK pathway through RAF dimers (including BRAF and CRAF). Right, Combined inhibition of BRAF, EGFR, and MEK is hypothesized to prevent adaptive feedback reactivation and maintain MAPK pathway suppression. B, Trial schematic showing treatment arms and dosing cohorts for treatment of patients with BRAFV600E CRC. Note that patients treated at doses of dabrafenib 150 mg BID, trametinib 2 mg QD, and panitumumab at 6 mg/kg or dabrafenib 150 mg BID, trametinib 2 mg QD, and panitumumab at 4.8 mg/kg were enrolled into the dose escalation and dose expansion phases of the trial.
Thus, these data suggest that therapies capable of blocking feedback reactivation may produce more robust inhibition of MAPK signaling, resulting in improved efficacy in BRAFV600E CRC. As an initial test of this hypothesis in BRAFV600E CRC, we previously performed a clinical trial of combined BRAF and MEK inhibition with dabrafenib and trametinib that demonstrated improved pathway suppression in preclinical models of BRAFV600E CRC (17). Indeed, this strategy has been successful in BRAFV600E/K melanoma and BRAFV600E non-small cell lung cancer, improving outcomes in patients who received the combination of dabrafenib and trametinib vs dabrafenib alone, leading to FDA approval for this combination in these indications (18–21). Combined BRAF and MEK inhibition led to a modestly improved response rate of 12% in 43 patients with BRAFV600E-metastatic CRC, but analysis of paired pretreatment and on-treatment biopsy specimens suggested that MAPK pathway suppression remained suboptimal (17). Therefore, we hypothesized that targeting EGFR as a key mediator of feedback signaling in combination with a BRAF inhibitor, with or without a MEK inhibitor, may optimize MAPK pathway suppression and lead to improved efficacy in BRAFV600E CRC (17).
Here, we report the results of a clinical trial of combined BRAF and EGFR inhibition, combined MEK and EGFR inhibition, and combined BRAF, EGFR, and MEK inhibition in patients with metastatic BRAFV600E CRC. Paired pretreatment and on-treatment biopsy specimens were collected and analyzed to assess the pharmacodynamic effects of each therapy. Serial plasma specimens were obtained, and cell-free DNA (cfDNA) was analyzed to provide correlates of response and to identify mechanisms of acquired resistance.
RESULTS
Patient Characteristics
Between December 2012 and the time of data cutoff for this interim analysis (May 6, 2016), 142 patients with metastatic BRAFV600E CRC were enrolled in 1 of 3 treatment arms, as outlined in Fig. 1B: 1) combined BRAF and EGFR inhibition with dabrafenib and panitumumab (D+P, n = 20); 2) the “triplet” combination of BRAF, MEK, and EGFR inhibition with dabrafenib, trametinib, and panitumumab (D+T+P, n = 91); and 3) combined MEK and EGFR inhibition with trametinib and panitumumab (T+P, n = 31). Patient characteristics are shown in Table 1. In general, patient characteristics were well-balanced across groups.
Table 1.
Patient demographics across treatment arms
| D + P (n = 20) | T + P (n = 31) | D + T + P (n = 91) | |
|---|---|---|---|
|
| |||
| Age, median (range), years | 58.0 (42–84) | 57.0 (39–74) | 60.0 (28–83) |
|
| |||
| Female, n (%) | 11 (55) | 18 (58) | 58 (64) |
|
| |||
| ECOG performance status at baseline, n (%) | |||
| 0 | 13 (65) | 17 (55) | 47 (52) |
| 1 | 7 (35) | 14 (45) | 44 (49) |
|
| |||
| Prior lines of therapy, n (%) | |||
| 0 | 4 (20) | 1 (3) | 21 (23) |
| 1 | 8 (40) | 14 (45) | 27 (30) |
| 2 | 7 (35) | 11 (35) | 33 (36) |
| 3 | 1 (5) | 4 (13) | 9 (10) |
| 4 | 0 | 1 (3) | 1 (1) |
| 5 | 0 | 0 | 0 |
|
| |||
| Prior anti-EGFR therapy, n (%) | |||
| Yes | 1 (5) | 10 (32) | 13 (14) |
| No | 19 (95) | 21 (68) | 78 (86) |
|
| |||
| Primary tumor location, n (%) | |||
| Colon | 18 (90) | 26 (84) | 76 (84) |
| Left side | 4 (22) | 10 (38) | 19 (25) |
| Right side | 14 (78) | 16 (62) | 57 (75) |
| Rectum | 2 (10) | 5 (16) | 15 (16) |
Dose Determination and Safety
The initial dose assessment began with the evaluation of D+P at their full labeled doses (dabrafenib 150 mg orally twice daily [BID] and panitumumab 6 mg/kg intravenously [IV] every 2 weeks [Q2W]). No dose-limiting toxicities (DLTs) were observed, and a total of 20 patients were treated at this dose level. D+P was well tolerated, and the majority of events were grade 1 or 2; 45% of patients had a grade 3/4 event. The most common adverse events (AEs) of all grades were dermatitis acneiform (60%), nausea (50%), fatigue (50%), and diarrhea (45%); none were grade 3/4 (Table 2). Only one grade 3/4 AE (hypophosphatemia: n = 2 [10%]) occurred in >1 patient in the D+P group.
Table 2.
Adverse events occurring in > 30% of patients in any treatment arma
| AE, n (%) | D + P (n = 20) | T + P (n = 51)b | D + T + P (n = 91) | |||
|---|---|---|---|---|---|---|
| Total | Grade 3/4 | Total | Grade 3/4 | Total | Grade 3/4 | |
| Any event | 20 (100) | 9 (45) | 50 (98) | 34 (67) | 91 (100) | 64 (70) |
| Diarrhea | 9 (45) | 0 | 37 (73) | 1 (2) | 59 (65) | 6 (7) |
| Dermatitis acneiform | 12 (60) | 0 | 27 (53) | 9 (18) | 54 (59) | 9 (10) |
| Nausea | 10 (50) | 0 | 18 (35) | 1 (2) | 51 (56) | 2 (2) |
| Dry skin | 7 (35) | 1 (5) | 17 (33) | 3 (6) | 49 (54) | 2 (2) |
| Fatigue | 10 (50) | 0 | 13 (25) | 0 | 45 (49) | 6 (7) |
| Pyrexia | 7 (35) | 0 | 20 (39) | 0 | 44 (48) | 4 (4) |
| Vomiting | 6 (30) | 0 | 15 (29) | 1 (2) | 39 (43) | 2 (2) |
| Decreased appetite | 5 (25) | 0 | 12 (24) | 0 | 36 (40) | 2 (2) |
| Rash | 3 (15) | 0 | 16 (31) | 3 (6) | 28 (31) | 10 (11) |
| Hypomagnesemia | 8 (40) | 1 (5) | 12 (24) | 2 (4) | 26 (29) | 1 (1) |
| Constipation | 7 (35) | 1 (5) | 7 (14) | 0 | 17 (19) | 1 (1) |
Safety data were based on the most recent interim analyses (data cutoff May 6, 2016). The median follow-up time (defined as time in months from study start to last contact or death) for patients treated with D + P was 10.6 months (2.1–22 months), for patients treated with D + T + P was 6.2 months (1.5–47.2 months), and for patients with a BRAFV600E mutation treated with T + P was 6.4 months (0.4–18.6 months).
Safety data for the T + P arm are for all patients, including those with BRAF wild type (n = 20) and BRAFV600E (n = 31).
Dose escalation to the full label doses of each of the triplet agents, D+T+P, was completed (dabrafenib 150 mg orally BID, trametinib 2 mg orally daily, and panitumumab 6 mg/kg IV Q2W). A total of 48 patients were enrolled at the highest dose, and the spectrum of AEs was similar to that with D+P. Diarrhea (65% all grades, 7% grade 3/4), nausea (56% all grades, 2% grade 3/4), and dermatitis acneiform (59% all grades, 10% grade 3/4) were the most frequent AEs among all patients treated with D+T+P. However, a greater incidence and severity of AEs was observed with D+T+P than with D+P, and 70% of patients had a grade 3 or 4 AE (Table 2). A corresponding increase in AEs that led to dose reductions, interruptions, or discontinuations was observed in the D+T+P arm vs the D+P arm (Supplementary Table S1). In the D+T+P arm, 18% of patients had an AE that resulted in study therapy discontinuation, 54% had an AE that resulted in dose reduction, and 71% of patients had an AE that led to dose interruption or delay. In an effort to reduce the dermatologic toxicity observed, 32 patients were enrolled to a D+T+P arm with a reduced panitumumab dose of 4.8 mg/kg IV every 2 weeks. Although no clear difference in AEs was noted (Supplementary Table S2), the rate of serious AEs (SAEs) in general and AEs leading to discontinuation were lower in the panitumumab 4.8-mg/kg arm than in the 6-mg/kg arm (SAEs: 15/32 [47%] vs 16/24 [67%]; AEs leading to discontinuation: 4/32 [13%] vs 7/24 [29%]) despite longer follow-up in the 4.8-mg/kg arm. However, note that the number of patients in the 4.8-mg/kg panitumumab arm who experienced dose interruptions (26/32, 81%) was higher than that in the 6-mg/kg arm (16/24, 67%); no differences in the rate of dose reduction were observed.
The remaining “doublet” of T+P was evaluated, starting at the full label dose of each agent (trametinib 2 mg orally daily and panitumumab 6 mg/kg IV every 2 weeks). However, in the absence of dabrafenib, these agents were not tolerated in combination due to excessive dermatologic toxicity (18% grade 3/4 dermatitis acneiform). The most common AEs among all patients (n = 51; includes patients with wild type BRAF) who received T+P were diarrhea (73% all grades, 2% grade 3/4), dermatitis acneiform (53% all grades, 18% grade 3/4), and pyrexia (39% all grades, 0% grade 3/4). Additional de-escalated doses of trametinib and panitumumab were evaluated (Fig. 1B; trametinib 1.5 mg once daily + panitumumab 6 mg/kg Q2W; trametinib 2 mg once daily + panitumumab 4.8 mg/kg Q2W), but dermatologic toxicity remained a challenge.
Two fatal SAEs occurred in patients enrolled in the D+T+P arm. One event was due to hemorrhage, and the other was death due to an unknown cause; however, neither event was considered to be related to the study drugs (Supplementary Table S1).
Efficacy
Efficacy measures for the 3 treatment arms are also based on a data cutoff date of May 6, 2016 (Fig. 2, A–C). Two patients (10%) in the D+P arm had a confirmed complete response (CR) or partial response (PR), and 16 patients (80%) had stable disease; disease control was 90% overall. In the T+P arm, no patients achieved CR/PR and 17 patients (55%) had stable disease. The D+T+P arm resulted in a confirmed CR/PR in 19 patients (21%), stable disease in 59 patients (65%), and an overall disease control rate of 86%. Duration of response (DOR) in the D+T+P arm was estimable but not mature, with a median of 7.6 months (95% CI, 2.9-not evaluable months) (Table 3).
Figure 2.

Efficacy of D+P, T+P, and D+T+P in patients with BRAFV600E CRC. A-C, Waterfall plots showing best response by RECIST in the D+P (A), T+P (B), and D+T+P (C) cohorts. Dotted lines represent the 30% threshold for PR. Bar color represents the best confirmed response by RECIST. D, PFS for the D+P, T+P, and D+T+P cohorts. Median PFS with 95% CIs are shown for each treatment arm.
Table 3.
Summary of efficacy by treatment cohort (investigator review)
| Assessment | D + T + P (n = 91) | T + P (n = 31) | D + P (n = 20) | D + T(n = 43)a |
|---|---|---|---|---|
| Best confirmed response, n (%) | ||||
| CR | 1 (1) | 0 | 1 (5) | 1 (2) |
| PR | 18 (20) | 0 | 1 (5) | 2 (5) |
| SD | 59 (65) | 17 (55) | 16 (80) | 24 (56) |
| PD | 8 (9) | 12 (39) | 2 (10) | 10 (23) |
| NE | 5 (5) | 2 (6) | 0 | 6 (14) |
| ORR (CR + PR), n (%) [95% CI] | 19 (21)[13.1–30.7] | 0[0–11.2] | 2 (10)[1.2–31.7] | 3 (7) |
| DOR (95% CI), months | 7.6 (2.9-NR) | 0 | 6.9 (5.9–8.0) | -- |
| DCR (CR + PR + SD), % | 86 | 55 | 90 | 68 |
| Median PFS, months | 4.2 | 2.6 | 3.5 | 3.5 |
| Unconfirmed CR + PR, n (%) | 29 (32) | 1 (3) | 3 (15) | 5 (12) |
DCR, disease control rate; NE, not evaluable; NR, not reached; ORR, overall response rate; PD, progressive disease; SD, stable disease.
Key efficacy measures are shown across treatment arms. Efficacy data for patients treated with D + T (Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol 2015;33(34):4023-31; ref 17) are shown for comparison.
The median progression-free survival (PFS) was 3.5 months (95% CI, 2.8–5.8 months) in the D+P arm, 2.6 months (95% CI, 1.4–2.8 months) in the T+P arm, and 4.2 months (95% CI, 4.0–5.6 months) in the D+T+P arm (Fig. 2D). Median overall survival (OS) was 13.2 months (95% CI, 6.7–22.0 months) in the D+P arm, 8.2 months (95% CI, 6.5–9.4 months) in the T+P arm, and 9.1 months (95% CI, 7.6–20.0 months) in the D+T+P arm (estimable but not mature; Supplementary Fig. S1).
Target Engagement—Pharmacodynamic Analysis of Paired Tumor Biopsy Specimens
Per the protocol, paired fresh tumor biopsy specimens obtained before treatment (within 3 weeks of treatment start) and on day 15 of treatment were required for all patients enrolled. Pharmacodynamic markers were analyzed in 10, 21, and 26 paired biopsy specimens collected from patients in the D+P, T+P, and D+T+P arms, respectively. The effect of each therapy on MAPK signaling output (assessed as the change in phosphorylated ERK [pERK] levels by immunohistochemistry from the day 15 on-treatment biopsy specimen), relative to the pretreatment biopsy, was evaluated. Values were compared with paired biopsy specimens from patients with BRAFV600E CRC treated in our previous trial of BRAF + MEK inhibition with dabrafenib and trametinib (17) and with patients with BRAFV600-mutant melanoma treated with BRAF inhibition (dabrafenib) alone (22) (Fig. 3). A significant reduction in pERK levels was seen between the baseline and on-treatment biopsy specimens with the T+P doublet and D+T+P triplet (P = 0.002 for both), but not with the D+P doublet (P = 0.5) (Fig. 3A). The D+T+P triplet, which demonstrated the greatest efficacy, also resulted in the greatest amount of pERK inhibition (60%) compared with T+P (41%), D+T (37%) (17), and D+P (23%) (Fig. 3B); however, a statistically significant correlation between pERK inhibition and response was not observed. The D+T+P triplet also produced the greatest suppression of phosphorylated ribosomal protein S6 (pS6), which is regulated by ERK activity in BRAF-mutant cancers, and represents a potential mechanistic/pharmacodynamic marker of responsiveness (23) (Supplementary Fig. S2). However, none of the therapies produced as robust a degree of pERK inhibition as did the previously published data for dabrafenib monotherapy in melanoma samples (84%) (22) (Fig. 3B). Taken together, these findings provide a likely explanation for why even the D+T+P triplet in CRC still falls short of the >50% response rate observed with the single-agent BRAF inhibitor in BRAFV600E-mutant melanoma and supports the hypothesis that inadequate MAPK suppression due to robust and complex adaptive feedback in BRAFV600E CRC limits clinical benefit.
Figure 3.
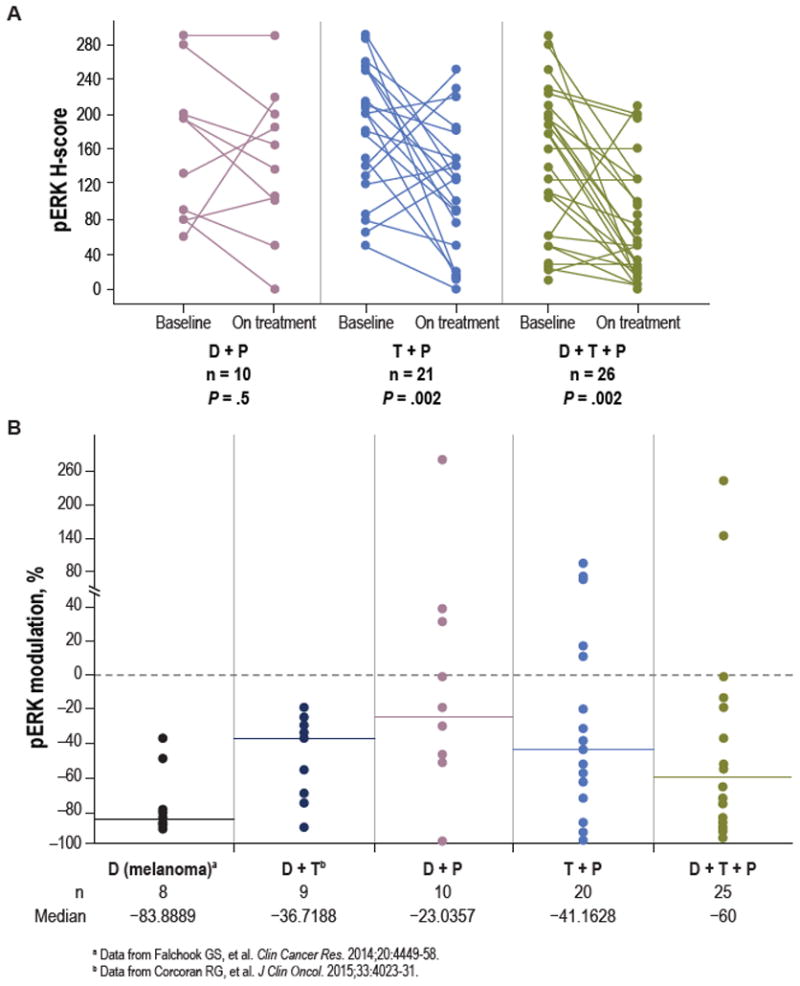
Pharmacodynamic analysis of paired tumor biopsy specimens. A, H-scores for pERK in paired baseline and day 15 on-treatment tumor biopsy specimens from patients treated with D+P, T+P, and D+T+P. P values represent paired t test. B, The percentage change in pERK H-score in the on-treatment tumor biopsy specimen relative to the baseline biopsy specimen in individual patients according to treatment. The percentage change in pERK H-score in paired on treatment biopsy specimens for patients with BRAFV600E CRC treated with D+T and BRAFV600-mutant melanoma treated with dabrafenib alone are shown for comparison. Horizontal bars represent the median.
Clinical Factors, MSI Status, and Response to D+T+P
The relationship between response rate and several clinical factors (including prior anti-EGFR therapy and panitumumab dose) was evaluated in patients treated with D+T+P (Supplementary Fig. S3).
MSI is frequently associated with BRAFV600E mutation in CRC (24), with MSI/MMR status previously reported to affect prognosis in patients with BRAFV600E CRC (8, 25). MSI/MMR status was available for 78 patients (86%) treated with D+T+P and who had evaluable best clinical response and PFS data (Supplementary Fig. S4A). In the 11 of 78 patients (14%) whose tumors were MSI-high/MMR deficient (dMMR), the response rate was 46% (5 of 11; 95% CI, 17%–77%) compared with 27% (18 of 67; 95% CI, 17%–39%) in patients whose tumors were microsatellite stable (MSS)/MMR proficient (pMMR), which was not statistically significant (Supplementary Fig. S4B). However, a trend toward a statistically significant increase in PFS (HR, 2.624; 95% CI, 0.997–6.907; log-rank test, P = 0.0449) was noted in patients with MSI receiving D+T+P, although it is not possible to determine whether this effect is predictive or prognostic (Supplementary Fig. 4C). None (0/67) of the MSS/pMMR patients with CRC remained on study for >1 year, whereas 3 of 11 (27%) of the MSI-high/dMMR patients with CRC remained on study for >1 year. Of these 3 patients, 1 achieved a PR lasting >24 months, and another patient demonstrated a CR lasting >26 months. Of note, the 1 patient treated with D+P who achieved CR was MSS/pMMR.
Analysis of Cell-Free DNA and Response to D+T+P
We used a highly sensitive method for the detection of tumor-derived mutations in cfDNA termed BEAMing (Beads, Emulsion, And Magnetics) to monitor changes in the levels of BRAFV600E in blood during treatment (26). BRAFV600E levels were analyzed in plasma from 85 patients treated with D+T+P: 71 of 85 patients had BRAF mutations detected by BEAMing at baseline (83.5%). A marked decrease in BRAFV600E levels in cfDNA from baseline was noted by 4 weeks in patients achieving a PR or CR with D+T+P, with all but 1 patient exhibiting reductions of ≥95%. The decrease in BRAFV600E levels was significantly greater in patients with responses than in patients with stable or progressive disease (P = 0.004) and was correlated significantly with the best percentage tumor change (P = 0.001, R = 0.414) (Fig. 4A, B). These results suggest that serial monitoring of BRAFV600E levels in cfDNA at baseline and on treatment may be a clinically useful marker of tumor response.
Figure 4.

Serial cfDNA analysis to define correlates of response and resistance. A, Percentage change in BRAFV600E mutation levels in cfDNA (week 4 vs baseline) or CEA levels (week 6 vs baseline) for patients achieving CR/PR, stable disease, or progressive disease (PD). CEA analysis was limited to patients with baseline levels above the upper limit of normal. P values represent CR/PR vs stable disease/PD by 2-tailed t test. B, Scatterplot of correlation between change in BRAFV600E mutation levels in cfDNA (week 4 vs baseline) or CEA levels (week 6 vs baseline) vs best percentage tumor change. Color of dots indicates the level of response achieved. C, Spider plots showing BRAFV600E mutation levels in cfDNA or CEA levels during therapy for patients achieving CR/PR, stable disease, or PD. D, Three representative patients treated with D+T+P with serial cfDNA monitoring of BRAFV600E mutation levels and hot spot KRAS and NRAS mutations at baseline, at week 4 of therapy, and at time of PD, showing emergence of 1 or more KRAS or NRAS mutations.
We compared the predictive value of BRAFV600E levels in cfDNA with serum levels of carcinoembryonic antigen (CEA), which is commonly used as a blood-based tumor marker in patients with CRC as part of standard clinical practice. The BRAFV600E mutation was detectable in 71 of 85 (84%) evaluable patients; however, elevated CEA levels were detected in only 68 of 126 (54%) evaluable patients across arms and in 43 of 81 (53%) evaluable patients in the D+T+P arm. In contrast with BRAFV600E levels in cfDNA, the change in CEA levels by 6 weeks of treatment was not statistically significant between patients who achieved CR/PR and those with stable or progressive disease (Fig. 4A). In serial blood collections obtained throughout therapy, a consistent rebound in BRAFV600E levels was observed in cfDNA at the time of disease progression, whereas a consistent pattern was not observed with CEA levels (Fig. 4C). Taken together, these data suggest that monitoring BRAFV600E levels in cfDNA during therapy correlates well with response, and disease trajectory in patients with BRAFV600E-mutant CRC was more informative than CEA—the standard clinical tumor marker for CRC.
cfDNA analysis can also be an effective tool for identifying and detecting mechanisms of acquired resistance to therapy (27–31). Prior studies have revealed that acquired resistance to BRAF-directed therapy in patients with BRAFV600E CRC is frequently driven by genomic alterations (e.g., RAS mutations), which lead to reactivation of MAPK signaling (28, 32, 33). We used a BEAMing panel to detect the presence of 11 common hot spot mutations in KRAS and NRAS (see Methods for further details) in cfDNA before treatment, during treatment, and at disease progression. We observed that, of the 29 evaluable patients who achieved a response (CR or PR) or stable disease with D+T+P and had cfDNA data available at the time of progression, 14 patients (48%) developed ≥1 detectable KRAS or NRAS mutation in cfDNA at the time of disease progression, which was not detectable at baseline. As shown in Fig. 4D, the initial decrease in BRAFV600E mutation levels after initiation of therapy in these patients was followed by an eventual rebound in BRAFV600E levels on disease progression, accompanied by the emergence of ≥1 KRAS or NRAS mutation. In 6 of 29 patients (33%), >1 subclonal RAS mutation was observed on disease progression, suggesting the potential for tumor heterogeneity in the context of acquired resistance to therapy.
DISCUSSION
We present the results of a clinical trial of combined BRAF and EGFR inhibition, with or without MEK inhibition in BRAFV600E CRC. The trial was designed to target the key adaptive feedback pathways driving primary resistance to BRAF inhibition alone. Both combined BRAF and EGFR inhibition (with D+P) and combined BRAF, EGFR, and MEK inhibition (with D+T+P) were tolerated at the full label doses of all agents. However, the frequency and severity of AEs was greater in the D+T+P arm than in the D+P arm, most notably in terms of dermatologic toxicity. Remarkably, while all three agents were tolerated together at full dose, combined EGFR and MEK inhibition only (T+P) was not tolerated at full dose, due to dermatologic toxicity. Although this may be considered counterintuitive, it highlights the unique biology of the MAPK pathway and its key implications for therapy. Although BRAF inhibitors effectively suppress MAPK signaling by mutant BRAFV600E monomers in tumor cells, they do not inhibit the MAPK pathway in normal cells, where RAF signals as a RAS-depdendent dimer and paradoxically activates MAPK signaling (34–36). This activation underlies the frequent development of MAPK-driven tumors (eg, proliferative skin lesions and secondary cutaneous malignancies) in patients receiving BRAF inhibitor monotherapy (37). Thus, BRAF inhibitors exhibit greater selectivity than other MAPK pathway inhibitors, allowing a greater degree of specific tumor MAPK suppression with less systemic toxicity; conversely, agents that inhibit MAPK signaling in all cells (such as MEK inhibitors) have greater systemic toxicity, limiting the achievable dose in patients and resulting in suboptimal MAPK inhibition in tumor cells. Moreover, the potential opposing effects of BRAF and MEK or EGFR inhibitors in normal cells likely counteract the effects on the MAPK pathway, providing a mechanistic explanation for the decreased toxicity seen with the triplet regimen in this trial. Taken together, these data illustrate how the therapeutic window advantages offered by BRAF inhibitors make them key components of therapeutic combinations for BRAFV600E cancers.
Modest clinical activity was seen in the D+P arm, compared with reported response rates with BRAF inhibitor monotherapy; the confirmed response rate was 10%, while 15% were unconfirmed. These data are consistent with the efficacy reported for similar BRAF/EGFR inhibitor combinations (13, 38–40). Notably, a recent update of a study evaluating cetuximab + irinotecan with or without the BRAF inhibitor vemurafenib demonstrated that in patients treated with the triple combination, response rate was 16% (n = 44 evaluable patients), with a median PFS of 4.3 months among all patients in this arm (n = 49) (40). Despite preclinical studies supporting EGFR as the primary driver of MAPK reactivation in BRAFV600E CRC (12, 15), these data suggest that EGFR may be a critical mediator of resistance; however, many patients may harbor other redundant mechanisms of adaptive MAPK reactivation. Consistent with this hypothesis, we observed that D+P led to MAPK suppression in on-treatment tumor biopsy specimens in only a subset of patients, suggesting that EGFR-independent mechanisms of MAPK reactivation play an important role in this disease. In support of this, some BRAFV600E CRCs do not express elevated levels of EGFR, and BRAFV600E CRC cell lines have been identified in which MAPK reactivation and resistance are driven by RTKs other than EGFR, such as MET (12, 41). Collectively, these data support the need to inhibit both EGFR-dependent and -independent feedback signals in BRAFV600E CRC.
Combined BRAF, MEK, and EGFR inhibition with D+T+P demonstrated increased efficacy, with a confirmed and unconfirmed response rate of 21% and 32%, respectively — these figures being one of the highest response rates observed with any regimen to date in BRAFV600E-mutant CRC (16, 17). Consistent with the potential importance of inhibiting EGFR-dependent and -independent feedback signals, D+T+P produced the greatest degree of MAPK pathway suppression in on-treatment biopsy specimens. However, D+T+P still produced suboptimal MAPK suppression when compared with dabrafenib alone in BRAFV600-mutant melanoma, providing a possible explanation for why the efficacy of this triplet in CRC still falls short of BRAF inhibitors alone in melanoma. This observation may also support the existence of adaptive feedback signals capable of overcoming the D+T+P triplet to drive MAPK reactivation and primary resistance to therapy. Therefore, developing therapeutic strategies that can overcome these signals and optimize MAPK pathway inhibition will be key.
In addition to driving primary resistance, our data also suggest that MAPK reactivation is a key mechanism of secondary or acquired resistance to therapy in BRAFV600E CRC. Previously, we and others reported that acquired resistance to BRAF inhibitor combinations in BRAFV600E CRC can be driven by an array of alterations in MAPK pathway components and lead to pathway reactivation, including RTK amplification, RAS mutation or amplification, BRAFV600E amplification, and MEK mutations. This finding also highlights the critical importance of MAPK signaling in these cancers (28, 32, 33). Here, in a larger cohort of patients, we observed that almost half of patients (48%) demonstrated emergence of KRAS or NRAS mutations in cfDNA at the time of disease progression. MAPK pathway alterations may be present in an even larger percentage of patients, because the cfDNA panel used detects only a limited number of mutations in KRAS and NRAS; therefore, other MAPK pathway alterations known to drive resistance, such as other KRAS or NRAS mutations, RAS or BRAF amplifications, and MEK mutations, would not be detected. Furthermore, many (33%) of these patients exhibited emergence of multiple subclonal RAS mutations at progression, suggesting the potential for tumor heterogeneity in the context of acquired resistance to therapy. Indeed, a previous study by Kopetz and colleagues suggested that many BRAFV600E CRCs may harbor pre-existing tumor subclones with 1 or more RAS mutation prior to therapy, leading to the potential for rapid emergence of heterogeneous resistant subclones (16).
Collectively, these observations raise an important conceptual issue: even though the D+T+P combination contains a MEK inhibitor, many of the resistance signals driving resistance occur upstream of MEK, including RTK-driven feedback in primary resistance and MAPK pathway alterations upstream of MEK in acquired resistance. Theoretically, these signals should still be intercepted by the MEK inhibitor and should not lead to MAPK reactivation. In targeted therapy paradigms, resistance alterations almost always occur at the level of or downstream of the drug target, not upstream. This finding highlights a key vulnerability of MEK inhibitors, i.e., increased upstream pathway flux can lead to MEK hyperactivation and a reduced ability of MEK inhibitors to maintain pathway suppression, which has been demonstrated in preclinical studies (28, 42). This also suggests that alternative strategies or agents capable of maintaining profound blockade of MAPK signaling may be key to enhancing activity in BRAFV600E CRC. Previously, we reported that ERK inhibitors, which act immediately downstream of MEK, can more effectively maintain MAPK suppression and can overcome many of the upstream resistance mechanisms to which MEK inhibitors are vulnerable (28, 32). Thus, investigating ERK inhibitors or other agents that might achieve more robust and complete MAPK blockade may be key future strategies for BRAFV600E CRC.
Overall, our study provides an example of how identifying and targeting key adaptive feedback signals can overcome resistance and improve response in BRAFV600E CRC, although further optimization is needed. We observed MAPK reactivation as a consistent mechanism of both primary and acquired resistance, underscoring the MAPK pathway as a critical target in this disease. However, despite improvements in the response rate, the DOR is poor and median PFS is only 4.2 months. Our data suggest that rapid emergence of resistant subclones harboring MAPK-activating alterations may be a major driver of treatment failure and that future strategies aimed at suppressing or overcoming these resistance mechanisms may help to sustain clinical benefit. Such strategies might include next-generation targeted combinations or combinations with other classes of agents, such as cytotoxic chemotherapy, as was recently reported (40).
Prior studies, including The Cancer Genome Atlas, have demonstrated frequent associations between BRAFV600E mutation and MSI in CRC (24), with MSI status reported to affect prognosis in patients with BRAFV600E CRC (25). In the current study, many of the small group of patients who achieved prolonged benefit for >1 year while on therapy (including 3 patients who had a DOR ≥20 months) were noted to have MSI-high tumors. Similarly, the tumor from the 1 patient from our prior trial of dabrafenib and trametinib in BRAFV600E CRC who maintained a CR for >4 years was also MSI (17). Given recent data supporting the increased immunogenicity of MSI CRC and increased responsiveness to immune checkpoint inhibition (43–45), this observation suggests a potential role for the immune system in promoting durable response. Indeed, as data from melanoma and KRAS-mutant CRC suggest a potential synergy between MAPK inhibition and immune checkpoint inhibition (46, 47), combining optimal MAPK inhibition with immunotherapy may be a promising future strategy. Collectively, we hope that identifying and targeting key resistance mechanisms in BRAFV600E CRC will continue to lead to important improvements in clinical outcome for patients with this poor-prognosis molecular subtype of CRC.
METHODS
Study Design
This trial was an open-label, phase I study to investigate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of trametinib and dabrafenib when administered in combination with the anti-EGFR antibody panitumumab in patients with BRAFV600E-mutation positive metastatic CRC (NCT01750918). Patients were enrolled to receive D+P, T+P, or D+T+P (Fig. 2) in initial dose-escalation studies to identify the optimal dosing strategy, followed by expansion cohorts to investigate the safety and clinical activity of each of the combination treatments. The appropriate ethics committee or institutional review board at each study center approved the study protocol. The study was conducted in accordance with Guidelines for Good Clinical Practice and the ethical principles described in the Declaration of Helsinki following all applicable local regulations.
Study Population
Eligible patients were required to have histologically or cytologically confirmed advanced or metastatic BRAFV600E-mutation positive CRC with measurable disease as per Response Evaluation Criteria In Solid Tumors (RECIST) v1.1. BRAFV600E mutation status was determined by local testing. Patients were required to be aged ≥18 years, have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, have adequate baseline organ function (as determined by laboratory parameters), and be of non-child-bearing potential or agree to use contraception as outlined in the protocol. Key exclusion criteria included history of prior malignancy (other than CRC), BRAF mutation other than V600E, any serious or unstable pre-existing medical condition, active hepatitis B or C infection, and prior exposure to a BRAF or MEK inhibitor. All patients provided written informed consent before enrollment.
Study Treatment
The study began with dose-escalation cohorts for all 3 drug combinations (D+P, D+T+P, and T+P) using a standard 3 + 3 enrollment scheme. Expansion cohorts were then enrolled to investigate the safety and clinical activity of the combinations. Patients in the D+P doublet arm were started in a dose-escalation cohort at the full monotherapy doses of dabrafenib (150 mg BID) and panitumumab (6 mg/kg Q2W) (Fig. 1B). No dose de-escalations were required. Once the D+P dose was confirmed at the full dose of both agents, another cohort of patients was assigned to the D+T+P triplet arm. In the initial cohort, dabrafenib was started at full dose of 150 mg orally BID, a trametinib starting dose of 1.5 mg once daily, and panitumumab starting dose of 4.8 mg/kg IV Q2W. Dose escalation continued until the maximum tolerated dose (MTD) was determined, and the full dose of all 3 agents was tested in the final cohort: dabrafenib 150 mg BID, trametinib 2 mg orally daily, and panitumumab 6 mg IV Q2W. The DLT observation period was 28 days, and no DLTs were identified in the D+T+P cohort; the MTD was declared as the labeled dose of all 3 agents. Patients in the T+P arm, which included patients with BRAFV600E metastatic CRC and BRAF wild-type metastatic CRC with anti-EGR therapy acquired resistance, received a starting dose of trametinib 2 mg once daily and panitumumab 6 mg/kg IV Q2W. No DLTs were identified in this cohort, but patients experienced delayed dermatologic toxicity with long-term dosing. Thus, sub-MTD doses were explored: trametinib 1.5 mg once daily and panitumumab 6 mg/kg IV Q2W; trametinib 2 mg once daily and panitumumab 4.8 mg/kg IV Q2W. Approximately 20 patients were then enrolled into expansion cohorts for each arm (including dose-escalation patients from selected dose groups). To further optimize the dose for the D+T+P arm, the protocol was later amended to explore the additional patients at 2 doses of panitumumab: 4.8 mg/kg IV vs 6 mg IV Q2W. At the time of radiological disease progression, patients in the D+P and T+P arms had the option of crossing over to the D+T+P arm.
Study Assessments
The primary endpoint was the safety of each of the drug combinations. Secondary endpoints included investigator-assessed overall response rate, DOR, PFS, overall survival, and the pharmacokinetics and pharmacodynamics of the drug combinations.
All patients treated with the T+P combination (n = 51) were evaluated for safety, and the full safety data set for these patients was derived from this population. However, only 31 patients treated with T+P were BRAF mutant, and efficacy is reported only for this subset.
Patients received study therapy until disease progression, unacceptable toxicity, death, or discontinuation for any other reason. Patients were assessed weekly for the first 28 days of dosing and then every 4 weeks throughout the continuation period. Follow-up visits were conducted at 14 days, 4 weeks, and 8 weeks after study drug discontinuation and then subsequently every 8 weeks for survival follow-up. Safety was monitored throughout the study for all patients across cohorts via physical examinations, laboratory evaluations, vital sign and weight measurements, performance status evaluations, ocular and dermatologic examinations, concomitant medication monitoring, electrocardiograms, echocardiograms, and AE monitoring (characterized and graded per Common Terminology Criteria for Adverse Events, v4.0). AEs were recorded using standard Medical Dictionary for Regulatory Activities coding. Dose interruptions, reductions, and discontinuations for all of the study drugs were monitored.
Tumors were assessed using investigator-read computed tomography or MRI at baseline, every 6 weeks until week 24, and then every 8 weeks until progression or death. Response determination was based on RECIST v1.1. In addition to imaging, the CEA tumor marker was collected. For the subset of patients who showed a confirmed CR or PR, DOR was defined as the time in weeks from the first documented evidence of CR or PR (the first response prior to confirmation) until the time of documented disease progression or death due to any cause, whichever was first. PFS was defined as the time in weeks between the first dose and the date of disease progression or death due to any cause. Finally, overall survival was defined as the time in weeks from the first dose of study drug until death due to any cause.
Serial blood samples for assessment of pharmacokinetic parameters were collected predose and postdose on days 1 and 15 and predose on day 21 in the first 28 days of dosing. In the continuation period, blood samples were collected every 4 weeks up to and including week 20 on study.
Statistical Methods
The all-treated population was used for analysis of clinical activity, which included all patients who received ≥1 dose of study medication. Patients evaluable for efficacy were defined as those who had ≥1 adequate postbaseline radiological disease assessment. The pharmacokinetics population included all treated patients for whom a blood sample for pharmacokinetics analysis was available. The biomarker population was defined as the participants in the all-treated population for whom a tumor biopsy/tissue sample was obtained and analyzed. Analysis of patients who received an intrapatient dose escalation or who transferred from doublet to triplet therapy were included in the crossover population.
Dose-escalation phases of the study followed a 3 + 3 dose-escalation procedure. Evaluation of safety data from ≥3 patients who had completed 28 days of dosing on study was required prior to defining a new dose and starting the next cohort. To facilitate dose-escalation/de-escalation decisions, an adaptive Bayesian logistic regression model (BLRM) was used to predict the probability of DLTs at the dose levels yet to be tested. Specifically, an 8-parameter BLRM for combination treatment was fitted on the DLT data (i.e., absence or presence of DLT) accumulated throughout the dose-escalation phase to model the dose-toxicity relationship of D+T+P when given in combination (48).
Prior distributions for trametinib were calculated based on the toxicity data observed in the first-time-in-human study MEK111054, in which trametinib was administered alone. Similarly, prior distributions for dabrafenib were determined based on data observed in the first-time-in-human study BRF112680, in which dabrafenib was administered alone. Prior distributions of the parameter trametinib-dabrafenib interaction was based on data observed in study BRF113220, in which trametinib and dabrafenib was administered in combination. A noninformative prior was assumed for the other combination of the 2 or 3 compounds with panitumumab. The model was used only as a guide for what further doses to study in the presence of DLTs along with the 3 + 3 results.
The expansion phases of the study used a Bayesian predictive adaptive design that allowed the trial to be monitored more frequently at multiple stages (48). The criterion was based on a historically unimportant response rate of 15% vs a response rate of interest of 30%.
Biomarker Analyses
Pharmacodynamic Analyses
Fresh predose (baseline) and paired on-treatment (day 15) tumor biopsy specimens were collected and analyzed to assess the pharmacodynamic effects of each therapy. The MAPK pathway activation status was determined via immunohistochemistry assessment of pERK levels (Cell Signaling; MOS075, clone 20G11). In addition, pS6 (Cell Signaling; MOS341, clone D68F8) was also analyzed in a subset of the available fresh biopsy specimens at a sponsor-designated laboratory. For pERK and pS6, the H-score was derived as follows: [1 × (% cells 1+) + 2 × (% cells 2+) + 3 × (% cells 3+)]. Nonparametric P values for the median differences between pretreatment and day 15 (±2) H-scores were derived for comparisons within and across arms.
Microsatellite Instability Analyses
Genomic DNA was isolated from tumor and nontumor regions of tissue, and paired normal and tumor DNA were analyzed for MSI with 5 markers: BAT-25, BAT-26, NR-21, NR-24, and MONO-27. DNA was amplified by PCR. Fragment size distribution analysis was performed using high-resolution capillary electrophoresis with fluorescence detection. Fragment size distributions from tumor and nontumor tissue for each of the 5 markers were compared, and the stability or instability in size distribution patterns was determined. Significant changes in a marker indicate instability and imply a phenotypic decrease in tumor MMR activity. MSI status was reported as stable or high. In positive cases, 2 of 5 loci need to show instability. Instability was defined as variation of ≥3 bp PCR product size at the specific locus between nontumor and tumor samples. In a subset of samples, no sufficient normal DNA was available; MLH1, MSH2, MSH6, and PMS2 were analyzed immunohistochemically. If all markers stained positive, the tumor was considered to be MSS. If 1 of the markers was negative, the tumor was considered to be MSI.
We combined the confident calls that passed the quality-control criteria for MSI/MSS from both of the platforms. The box-plot comparisons across MSI/MSS were statistically assessed using the nonparametric Kruskal-Wallis P values. Time-to-event models stratifying based on MSI status were built, and Kaplan-Meier survival plots were assessed between MSI/MSS status using HR and 95% CIs and log-rank P values.
Cell-Free DNA Analyses
Plasma samples were collected at baseline, at week 4, and at progression. Baseline cfDNA and serial cfDNA collections were analyzed for the presence of mutations to provide correlates of response and to identify mechanisms of acquired resistance. Mutations were assessed in plasma cfDNA using BEAMing technology (Sysmex Inostics) and a predefined targeted hot spot mutation panel: BRAFV600E, KRAS (G12S, G12R, G12C, G12D, G12A, G12V, G13D), NRAS (Q61K, Q61R, Q61L, Q61H), and PIK3CA (E542K, E545K, H1047R, H1047L). The BEAMing assay uses emulsion PCR on magnetic beads and flow cytometry to quantify the fraction of mutation-positive DNA to wild-type DNA. The mutant fraction (MF)—defined by the ratio of the mutant beads to the sum of wild-type, mixed, and mutant beads—was used to compare mutation hot spot levels in cfDNA.
The BRAFV600E MF ratio between week 4 and baseline was defined as follows: log10 (MF at week 4 + 1E-05) – log10 (MF at baseline + 1E-05). The BRAFV600E MF ratio between “at progression” and baseline was defined as follows: log10 (MF at progression + 1E-05) – log10 (MF at baseline + 1E-05). Nonparametric Kruskal-Wallis P values were derived to compare the BRAFV600E MF ratios between week 4 and baseline across response groups. Pearson correlation was used to measure the linear correlation between the change in BRAFV600E levels in cfDNA and the best percentage tumor change.
CEA Analyses
Serum intensity (SI) levels of CEA (or, CEACAM5), which is commonly used as a blood-based tumor marker in patients with CRC as part of standard clinical practice, were used to profile the patients from this trial. We limited our CEA-related analyses to only patients’ samples with baseline SI levels above the upper normal range as derived per the clinical protocol. The changes in SI level between week 6 and baseline were calculated as the log ratio: log10 (SI at week 6) – log10 (SI at baseline). Nonparametric Kruskal-Wallis P values were derived to compare SI ratios between week 6 and baseline across response groups.
Study Oversight
This study was designed, conducted, and analyzed by the funder (Novartis) in conjunction with the authors. All authors had full access to the study data and share final responsibility for the content of the manuscript and the decision to submit for publication.
Supplementary Material
SIGNIFICANCE.
This trial demonstrates that combined BRAF + EGFR + MEK inhibition is tolerable, with promising activity in patients with BRAFV600E CRC. Our findings highlight the MAPK pathway as a critical target in BRAFV600E CRC and the need to optimize strategies inhibiting this pathway to overcome both primary and acquired resistance.
Acknowledgments
This study was supported by GlaxoSmithKline. As of March 2, 2015, dabrafenib and trametinib are assets of Novartis AG. R.B. Corcoran acknowledges support from a Damon Runyon Clinical Investigator Award, NIH/NCI P50 CA127003 and R01CA208437. Research supported by a Stand Up to Cancer Colorectal Cancer Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT22-17). Stand Up to Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
The authors acknowledge Yiquin Yan for biomarker statistical analyses, Kohinoor Dasgupta for clinical statistical analyses, and Ilona Tala for biomarker sample collection. Medical writing assistance was provided by William Fazzone, PhD (ArticulateScience LLC), funded by Novartis Pharmaceuticals Corporation.
Footnotes
Disclosure of Potential Conflicts of Interest
R.B. Corcoran is a consultant/advisory board member for Amgen, Astex Pharmaceuticals, Avidity Biosciences, BMS, Genentech, Merrimack, N-of-one, Roche, Shire, and Taiho and has received research funding from AstraZeneca and Sanofi.
T. André reports a scientific advisory role/consultant for Amgen, Bristol-Myers Squibb, MSD Oncology, Sanofi, Servier, Roche and Xbiotech and has received honoraria from Amgen, BMS, Bayer, Baxter, Celgene, Lilly, Novartis, Roche, and Yakult.
C. Atreya reports reseach grants from GlaxoSmithKline, Merck, and Novartis and is a consultant/advisory board member of Bayer Diagnostics and Genentech.
J.H.M. Schellens reported an advisory role for AstraZeneca, Sotio, Roche, Merck and MerckSerono and stock in Modra Pharmaceuticals bv.
T. Yoshino reports a research grant from GlaxoSmithKline K.K. and a grant from Boehringer Ingelheim GmbH outside the submitted work.
Y. Humblet has been invited by Merck KGaA and Sanofi to international oncology meetings, and his hospital has received money for patient follow-up compensation.
A.J. McRee is an advisory board member for Merck.
S. Siena reports a scientific advisory role for Amgen, Bayer, Eli Lilly, Ignyta, Merck, Novartis, Roche, and Sanofi.
G. Middleton reports research grants from AstraZeneca, GemVax & Kael, and Merck, Sharpe, and Dohme, reports equity ownership in PhosImmune, and has received honoraria from BMS and Eli Lilly.
K. Muro reports receipt of honoraria from Chugai, Merck Serono, Taiho, Takeda, and Yakult, and reports a scientific advisory role for Eli Lilly and Ono.
M.S. Gordon reports consultancy, involvement in speakers bureau, and a scientific advisory role for, equity ownership in, and research funding support, honoraria, and patents and royalties from, GlaxoSmithKline.
J. Tabernero reports a scientific advisory role for Amgen, Bayer, Boehringer Ingelheim, Celgene, Chugai, Genentech, Inc., Lilly, MSD, Merck Serono, Novartis, Pfizer, F. Hoffmann-La Roche Ltd, Sanofi, Symphogen, Taiho, and Takeda.
R. Yaeger reports consulting or advisory roles for Advaxis and GlaxoSmithKline.
P. J. O’Dwyer has received consulting fees from Genentech, BMS, Boehringer Ingelheim, and clinical trials support from Genentech, BMS, AZ, Celgene, Merck, Syndax, GSK, Abbvie, Incyte, Minneamrata, Pharmacyclics, Five Prime, and Fortyseven.
A.S. Jung is an employee of and owns stock in Amgen.
J.C. Brase is an employee of Novartis.
S Jaeger was an employee of Novartis during the conduct of this study and writing of the report.
S. Bettinger is an employee of Novartis.
B Mookerjee is an employee of Novartis and owns stock in GlaxoSmithKline and Novartis.
F. Rangwala is an employee of Novartis.
E.Van Cutsem reports consulting or advisory roles for Bayer, Lilly, Roche, and Servier and has received research funding from Amgen, Bayer, Boehringer Ingelheim, Lilly, Novartis, Roche, Sanofi, Celgene, Ipsen, Merck, Merck KGaA, and Servier.
The remaining authors declare no potential conflicts of interest.
References
- 1.Safaee AG, Jafarnejad SM, Tan L, Saeedi A, Li G. The prognostic value of BRAF mutation in colorectal cancer and melanoma: A systematic review and meta-analysis. PLoS One. 2012;7(10):e47054. doi: 10.1371/journal.pone.0047054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barras D. BRAF mutation in colorectal cancer: An update. Biomark Cancer. 2015;7(Suppl 1):9–12. doi: 10.4137/BIC.S25248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20(20):5322–30. doi: 10.1158/1078-0432.CCR-14-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, et al. Survival in BRAF V600-mutant advanced melanoma treated wtih vemurafenib. N Engl J Med. 2013;366(8):707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris V, Overman MJ, Jiang ZQ, Garrett C, Agarwal S, Eng C, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer. 2014;13(3):164–71. doi: 10.1016/j.clcc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 8.Cohen R, Cervera P, Svrcek M, Pellat A, Dreyer C, de Gramont A, et al. BRAF-mutated colorectal cancer: What is the optimal strategy for treatment? Curr Treat Options Oncol. 2017;18(2):9. doi: 10.1007/s11864-017-0453-5. [DOI] [PubMed] [Google Scholar]
- 9.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 10.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–88. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 12.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–35. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Jr, Dayyani F, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657–67. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prahallad A, Sun C, Huang S, Di NF, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 16.Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–8. doi: 10.1200/JCO.2015.63.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol. 2015;33(34):4023–31. doi: 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 19.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386(9992):444–51. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 20.Long GV, Grob J, Nathan P, Ribas A, Robert C, Schadendorf D, et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: A pooled analysis of individual patient data from randomised trials. Lancet Oncol. 2016;17(12):1743–54. doi: 10.1016/S1470-2045(16)30578-2. [DOI] [PubMed] [Google Scholar]
- 21.Planchard D, Besse B, Groen HJ, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984–93. doi: 10.1016/S1470-2045(16)30146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau HT, Brown MP, et al. Dose selection, pharmacokinetics, and pharmacodynamics of BRAF inhibitor dabrafenib (GSK2118436) Clin Cancer Res. 2014;20(17):4449–58. doi: 10.1158/1078-0432.CCR-14-0887. [DOI] [PubMed] [Google Scholar]
- 23.Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Piris A, Nazarian RM, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med. 2013;5(196):196ra98. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamane LS, Scapulatempo-Neto C, Alvarenga L, Oliveira CZ, Berardinelli GN, Almodova E, et al. KRAS and BRAF mutations and MSI status in precursor lesions of colorectal cancer detected by colonoscopy. Oncol Rep. 2014;32(4):1419–26. doi: 10.3892/or.2014.3338. [DOI] [PubMed] [Google Scholar]
- 25.Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013;105(15):1151–6. doi: 10.1093/jnci/djt173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3(7):551–9. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 27.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahronian LG, Sennott EM, Van Allen EM, Wagle N, Kwak EL, Faris JE, et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 2015;5(4):358–67. doi: 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):827–827b. doi: 10.1038/nm0715-827b. [DOI] [PubMed] [Google Scholar]
- 30.Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016;6(2):147–53. doi: 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252–63. doi: 10.1158/2159-8290.CD-16-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oddo D, Sennott EM, Barault L, Valtorta E, Arena S, Cassingena A, et al. Molecular landscape of acquired resistance to targeted therapy combinations in BRAF-mutant colorectal cancer. Cancer Res. 2016;76(15):4504–15. doi: 10.1158/0008-5472.CAN-16-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pietrantonio F, Oddo D, Gloghini A, Valtorta E, Berenato R, Barault L, et al. MET-driven resistance to dual EGFR and BRAF blockade may be overcome by switching from EGFR to MET inhibition in BRAF-mutated colorectal cancer. Cancer Discov. 2016;6(9):963–71. doi: 10.1158/2159-8290.CD-16-0297. [DOI] [PubMed] [Google Scholar]
- 34.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 35.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207–15. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Geel RMJM, Tabernero J, Elez E, Bendell JC, Spreafico A, Schuler M, et al. A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 2017;7(6):610–9. doi: 10.1158/2159-8290.CD-16-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21(6):1313–20. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kopetz S, McDonough SL, Lenz H, Magliocco AM, Atreya CE, Diaz LA, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406) J Clin Oncol. 2017;35(suppl) doi: 10.1200/JCO.20.01994. abstract 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whittaker SR, Cowley GS, Wagner S, Luo F, Root DE, Garraway LA. Combined pan-RAF and MEK inhibition overcomes multiple resistance mechanisms to selective RAF inhibitors. Mol Cancer Ther. 2015;14(12):2700–11. doi: 10.1158/1535-7163.MCT-15-0136-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3(149):ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;17(4):1206. doi: 10.1016/j.celrep.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bendell JC, Hubbard JM, O’Neil BH, Jonker DJ, Starodub A, Peyton JD, et al. Phase 1b/II study of cancer stemness inhibitor napabucasin (BBI-608) in combination with FOLFIRI +/− bevacizumab (bev) in metastatic colorectal cancer (mCRC) patients (pts) J Clin Oncol. 2017;35(suppl) abstract 3529. [Google Scholar]
- 47.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014;2(7):643–54. doi: 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JJ, Liu DD. A predictive probability design for phase II cancer clinical trials. Clin Trials. 2008;5(2):93–106. doi: 10.1177/1740774508089279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.