

Abstract
Erythropoiesis is a dynamic process regulated at multiple levels to balance proliferation, differentiation and survival of erythroid progenitors. Ineffective erythropoiesis is a key feature of various diseases, including β-thalassemia. The pathogenic mechanisms leading to ineffective erythropoiesis are complex and still not fully understood. Altered survival and decreased differentiation of erythroid progenitors are both critical processes contributing to reduced production of mature red blood cells. Recent studies have identified novel important players and provided major advances in the development of targeted therapeutic approaches. In this review, β-thalassemia is used as a paradigmatic example to describe our current knowledge on the mechanisms leading to ineffective erythropoiesis and novel treatments that may have the potential to improve the clinical phenotype of associated diseases in the future.
Keywords: ineffective erythropoiesis, red blood cells, thalassemia
1. Introduction
The production of adequate numbers of functional red blood cells (RBC) is necessary for tissue oxygenation and survival. In several diseases, multiple pathogenic processes result in ineffective erythropoiesis (IE) and anemia. Ongoing research continues to provide valuable insight into these pathways and their understanding drives the development of novel treatments to reverse IE and improve the anemia. In this review, we focus on β-thalassemia, which is characterized by IE, to discuss the pathological mechanisms and therapeutics that lead to IE.
2. Erythropoiesis: Steady state and stress
2.1 Normal Erythropoiesis
Erythropoiesis is a dynamic multistep process where erythroid progenitors differentiate to produce mature enucleated RBC. Hematopoietic stem cells, residing in the bone marrow (BM) niche, generate committed erythroid progenitors, which can be further functionally defined in colony assays in vitro 1–3. The early stage burst forming unit erythroid (BFU-E) generate large erythroid colonies and progress to late stage colony forming unit erythroid (CFU-E), characterized by smaller erythroid colonies. This process occurs with a concomitant decrease in proliferative capacity and increase in erythropoietin (EPO) sensitivity, noted by the appearance of erythropoietin receptors (EPOR) on the cell surface 4, 5. As erythropoiesis progresses, morphologically recognizable precursors emerge by successive mitoses 6, 7. The earliest recognizable erythroblast is the proerythroblast, which undergoes cytoplasmic maturation and nuclear changes to generate basophilic, polychromatophilic and orthrochromatic erythroblasts. Finally, the latter extrude their nuclei to become reticulocytes and, ultimately, mature RBC, which circulate in the blood stream until senescence 8. Maturation of erythroblasts is marked by progressive hemoglobinization, so that RBC contain adequate amounts of hemoglobin (Hb). Under steady state conditions homeostatic control mechanisms regulate proliferation, differentiation and destruction of erythroid precursors in order to maintain a balance between RBC production and death. This process ensures adequate oxygenation of tissues 9–11.
Multiple signaling molecules and cell-cell interactions control erythropoiesis. The master regulator is EPO, a hormone primarily produced by the interstitial cells in the renal cortex 12. In fetal life, liver is the main source of EPO while, after birth, the kidney becomes the main producer 12. EPO is released into circulation and signals through EPOR, which is downregulated following the basophilic erythroblast stage. Binding of EPO to EPOR induces the dimerization of EPOR monomers. In response to EPO stimulation and receptor reorientation, Janus kinase 2 (JAK2), found on the intracellular part of EPOR, is rapidly phosphorylated creating docking sites for the SH2 domains of several signal transduction proteins, including STAT5 (Table 1) 13, 14. Activated STAT5 translocates to the nucleus where it controls the expression of a subset of genes involved in proliferation, differentiation and survival of erythroid progenitors 15–18. Although the JAK/STAT pathway is critical in promoting erythropoiesis, another signaling cascade is also activated by EPO stimulation; the PI3K-AKT pathway. AKT kinase, the downstream target of PI3K, phosphorylates several transcription factors, including FOXOs, and regulates their activity 19, 20. FOXOs play an important role in regulating cell growth, survival and differentiation 21, 22.
Table 1.
Important mediators of erythropoiesis under different conditions.
| ERYTHROPOIESIS | ERYTHROID PATHWAYS | ERYTHROBLASTS | IRON METABOLISM |
RED BLOOD CELLS |
|---|---|---|---|---|
|
| ||||
| Steady-state | EPO-JAK2-STAT5 | BM progenitors | Hepcidin |
|
|
| ||||
| Stress | EPO-JAK2-STAT5 | Erythroid stress progenitors | ↓ Hepcidin |

|
| BMP4-SMAD5-SCF/cKIT | ||||
| GR-PPARalpha? | ||||
| Hedgehog | ||||
| Notch2 | ||||
| pIgA1/TFR-Erk/Akt | ||||
|
| ||||
| Anemia of Inflammation (iron-restricted) | EPO-JAK2-STAT5 | BM progenitors | Inflammation |
|
| ↑ Hepcidin | ||||
Expression of EPO is low under basal conditions. Specialized cells in the kidney and in the liver of the fetus and the newborn are sensitive to oxygen concentration in circulation. When local oxygen delivery to these organs decreases, EPO expression is upregulated. This effect is mediated by the activation of hypoxia inducible transcription factors (HIFs), which stimulate an enhancer element of the EPO gene 23, 24. In particular, HIF-2α is the major activator of hypoxia-induced EPO expression 24–27. EPO production can be inappropriately low in several conditions causing severe anemia. This could be due to either destruction of the EPO producing cells, as in chronic kidney disease, or repression of EPO synthesis due to circulating inflammatory cytokines, as in rheumatoid arthritis, AIDS, chronic kidney disease or cancer 28–31. On the other hand, inappropriately high EPO production may be caused by several tumors, such as renal or hepatocellular carcinoma, and results in polycythemia 32, 33.
2.2 Stress erythropoiesis
In a state of local decreased oxygen delivery (hypoxemia, anemia), a physiologic response is elicited, intended to increase oxygen delivery to tissues. This process, called stress erythropoiesis, is a uniquely defined state of erythropoiesis, that results in the production of large numbers of erythrocytes, and has distinct differences in mediators involved from steady state 34.This is a response to acute erythropoietic stress and increased EPO concentration, caused by multiple stimuli, including hemorrhage, hemolysis, high altitude and pharmacological treatment. EPO acts on EPOR and through STAT5 rapidly increases the expression of anti-apoptotic regulator BCL-XL in early erythroblasts, while suppressing the proapoptotic proteins BIM, NOXA and co-expressed tumor necrosis family receptor FAS, together with its ligand FASL 35–38. Unique to stress erythropoiesis, the phosphorylation of STAT5 produces a high intensity, graded signal, which increases with rising EPO concentration 39. The early erythroblast compartment is mainly EPO responsive, and therefore mostly affected by anti-apoptotic processes, predominantly in the spleen, where a reserve of splenic progenitors is widely expanded during hypoxia. Under steady state conditions, the majority of early erythroid progenitors in the spleen undergo apoptosis. In acute anemic stress, high EPO prevents apoptosis of this reserve erythroid progenitor population, by mechanisms mentioned earlier 40, 41. This regulatory mechanism provides a transient survival signal on erythroblasts, and remains active until suppression pathways are activated 42.
Multiple other pathways have been identified in stress erythropoiesis. In response to acute anemia, bone morphogenetic protein 4 (Bmp4) modulates the activity of the transcription factor SMAD5 to induce the expansion of a specialized population of stress erythroid progenitors in the spleen and rapidly produce a large number of erythrocytes 43, 44 (Table 1). This pool of specialized progenitors is different from steady state progenitors residing in the mouse BM, and can rapidly produce great number of RBC 43, 45–47. These cells originate from hematopoietic stem cells that migrate from the BM to the spleen. In the spleen, under stress erythropoiesis conditions, they become stress erythroid progenitors, which express different cell surface markers than steady state progenitors. These specialized progenitors acquire their characteristics due to the stress microenvironment in the spleen 40, 45, 46, 48.
Bmp4 requires hypoxia and SCF signaling, through its receptor c-KIT, to induce a response of splenic stress progenitors 44. Hypoxia has a dual pathway of action; it maximizes sensitivity of progenitors to Bmp4 and SCF while regulating expression of Bmp4 by Hif-2α. EPO has a critical role in this process, as immature splenic stress erythroid progenitors can only differentiate if EPO synthesis is increased, while hypoxia potentiates the response to EPO 46. In parallel, activation of the glucocorticoid receptor (GR) promotes self-renewal of erythroid progenitors, which is required for their rapid expansion 49. Recently, a synergistic action of peroxisome proliferator-activated receptor alpha (PPARalpha) with GR was demonstrated, but the precise role of PPARalpha in erythropoiesis remains to be elucidated50. Hedgehog signaling is also implicated in stress erythropoiesis, but studies produced controversial results 45, 51. The hedgehog pathway is an important signaling pathway for embryonic development, tissue homeostasis and stem cell biology. It can be assumed that some members of the hedgehog family induce erythroid progenitors to become stress progenitors, while others negatively regulate basal and stress erythropoiesis. Another important pathway is the Notch signaling pathway. In particular, NOTCH 2 receptor has a crucial role in erythroid differentiation and in recovery from stress erythropoiesis 52, 53.
Deficiency of NOTCH2 receptor in mice exposed to stress erythropoiesis resulted in decreased number of progenitors and inefficient stress response 52.
2.3 Erythroblastic islands
Erythroid development occurs in the presence of macrophages in structures called erythroblastic islands, which consist of a central machrophage surrounded by clusters of erythroblasts in different stages of differentiation. Adhesion interactions between erythroblasts and macrophages are necessary for erythroid development. Identification of the involved molecules has been intensively studied during the last decades in humans and mice. Some of these possible antigens include erythroblast macrophage protein (Emp), α4, α5, β1, VCAM-1, ICAM-4 and SWAP70 54, 55.
These niches are pivotal for erythropoiesis, as they create a microenvironment where cell to cell and cell to extracellular matrix interactions exert positive and negative regulatory mechanisms that control erythroid proliferation and differentiation 56. Studies in mice provide valuable insight into these pathways. In fact, macrophage depletion in steady state erythropoiesis induces a mild iron deficiency anemia with reduced number of erythroblasts in the mouse BM 57, 58. However, in stress erythropoiesis, lack of macrophages significantly impairs recovery from the anemia, which cannot be rescued by iron supplementation 54, 57, 58. These observations indicate a crucial role of macrophages on erythroid expansion and reconstitution.
3. Ineffective erythropoiesis
IE can be characterized by abnormal differentiation or maturation of erythroid progenitors, as well as increased destruction of abnormal erythroblasts 59. In IE, although proliferation and survival of erythroid progenitors is expanded, only a small proportion of cells mature, and a limited number of RBCs are produced, proportionally fewer than expected from the number of erythroblasts under normal conditions (Figure 1) 60. The combination of increased proliferation and reduced differentiation results to a net increase of erythroid progenitors. This concept was first introduced when erythrokinetic studies in patients suffering from different types of anemia revealed an imbalance between BM activity and viable RBC released in peripheral blood circulation 61–63. These observations led to the discrimination between total and effective BM activity. In β-thalassemia, on which we focus for this review, IE represents a hallmark of the disease and has been extensively studied.
Figure 1. Steady state and Ineffective Erythropoiesis.
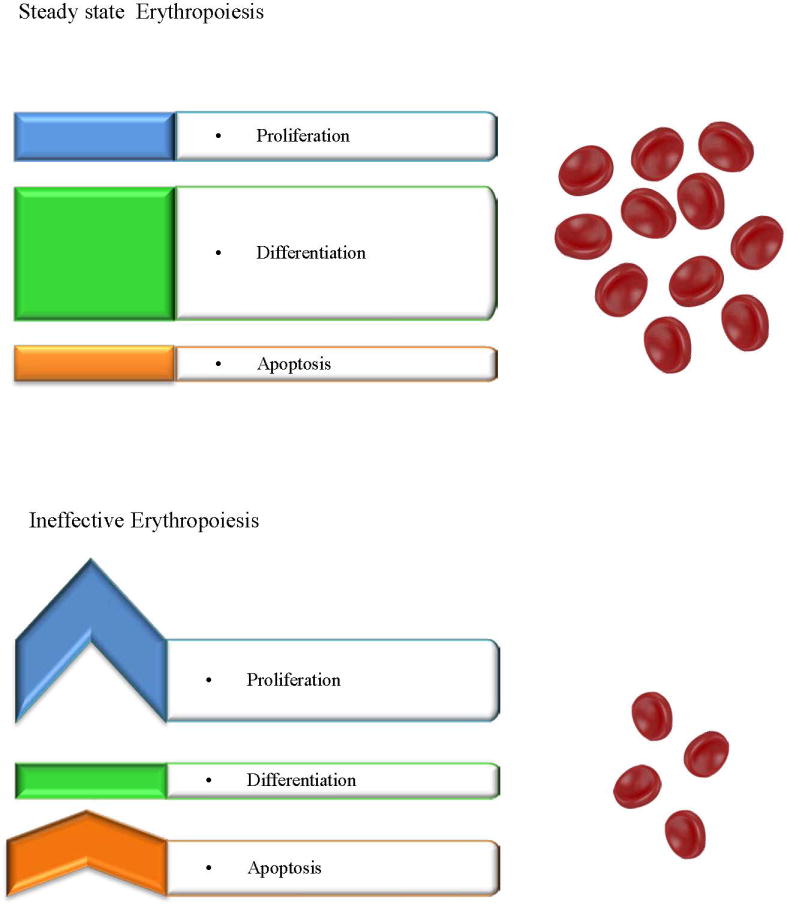
Steady state erythropoiesis is homeostatic and results in the production of red blood cells in a constant rate. In IE, decreased differentiation combined with increased proliferation and apoptosis of progenitors results in the production of limited RBCs.
3.1 Thalassemia
Thalassemias are a hereditary group of disorders characterized by insufficient or absent production of α-or β-globin chains, which are the two major components of Hb 64. Alpha thalassemia can present clinically with different phenotypes, from asymptomatic to lethal, where the severity of the anemia relates to the number of non-functional copies of the α-globin gene65. When three out of the four α-globin genes become inactivated, due to deletions with or without a nondeletion point mutation, excess β chains form β4 tetramers called hemoglobin H (HbH) 66. HbH disease is associated with moderate to severe anemia, hemolysis, splenomegaly and iron overload. Splenectomy is often recommended for these patients. The deletion of all four α-globin genes is the most severe form of the disease and usually results in hydrops fetalis during pregnancy and in-utero death of the affected embryo.
β-thalassemia is one of the most common causes of congenital anemias with a worldwide annual incidence of 1:100.000 34. It is caused by several hundred mutations in the β-globin gene or its promoter, which result in impaired production of β-globin chains 55, 67. Decreased or absent β-globin synthesis correlates with different degrees of clinical severity ranging from asymptomatic carriers to severely anemic, transfuse-dependent patients. Thalassemia major (TM) patients are diagnosed early in life with severe anemia, hepatosplenomegaly and failure to thrive. They require chronic RBC transfusions for survival and iron chelation therapy to prevent or limit iron overload. The less severe form of the disease, called thalassemia intermedia (TI), presents later in life and is non-transfusion dependent. Although patients with TI do not require RBC transfusions early on, as the disease progresses the degree of the anemia might worsen, and splenomegaly develops over time. In that case, RBC transfusions may become necessary for their survival 19. IE is a hallmark of the disease that results in progressive marrow expansion and extramedullary erythropoiesis (EMH), which occurs primarily in the spleen and the liver. The degree of expansion correlates with the severity of the anemia and can reach 30 times higher compared to normal volume 68. As a result, patients with TM can develop characteristic bone deformities 69. Finally, β-thalassemia trait patients may present with only a mild anemia and do not require any treatment 70.
The thalassemia syndromes can be alternatively classified phenotypically, based on the requirements for RBC transfusions, into two groups; transfusion-dependent thalassemias (TDTs) and not-transfusion dependent thalassemias (NTDTs). TDTs require regular RBC transfusions to survive and include TM and transfusion dependent HbH disease. NTDTs describe patients that do not require lifelong RBC transfusions and include TI and HbH disease.
Erythrokinetic and ferrokinetic studies in thalassemic patients provide an elaborative characterization of IE. Mildly to severe anemic β-thalassemia patients exhibit increased BM erythropoiesis ranging from 50% to 15%, respectively, in addition to shortened RBC survival 71. Furthermore, studies of iron incorporation into RBC, by 59Fe injections in healthy and thalassemic individuals, demonstrate increased iron absorption but decreased output of 59Fe in circulating RBC 72, 73. As a matter of fact, in healthy subjects 75% to 90% of the injected 59Fe is incorporated into newly formed RBC within 7 to 10 days, while in thalassemic patients this percentage is significantly lower, ranging from 15% to 20% 72, 74. This difference is explained by iron sequestration in organs where erythroid precursors are destructed prematurely, such as the BM. In contrast, in HbH disease peripheral hemolysis is the major pathway of anemia and IE represents a minimal pathogenetic mechanism 75, 76.
4. Mechanisms leading to ineffective erythropoiesis
Different mechanisms are involved in the pathogenesis of IE in β-thalassemia. In the last decade, our understanding of the pathologic processes implicated has significantly improved. The use of murine models of β-thalassemia has contributed extensively to this purpose. These models present with a clinical phenotype similar to that of patients with β-thalassemia. However, it should be noted that mice cannot faithfully recapitulate all aspects of human erythropoiesis. These pathways, as well as important players associated with these processes are described below.
4.1 Increased apoptosis of thalassemic erythroid precursors
The early discovery of the discrepancy between increased proliferation of erythroid progenitors in the BM of thalassemic patients and low RBC counts in circulation led to the hypothesis of enhanced apoptosis of erythroid precursors. Indeed, multiple studies in TM, using human BM samples, verify this concept by demonstrating specific ladder patterns of DNA breakdown products or by using annexin V staining analyzed by Flow cytometry and TUNEL analyses 77, 78. Moreover, two different quantitative measurements of apoptosis of erythroid progenitors, reflecting alterations in the plasma membrane and nucleus, revealed a 3- to 4-fold increase in programmed cell death in patients versus healthy controls 79. A study using an in vitro system of erythropoiesis from human TM BM cells suggested the maturation of arrest at the polychromatophilic normoblast stage 78. However, the cells were identified only morphologically and additional studies using more qualitative and quantitative assay may provide a more accurate description of this phenomenon and define at what stage thalassemic erythroid precursors undergo apoptosis.
4.2 Decreased differentiation of erythroid progenitors
Even though originally the main pathological mechanism responsible for the IE in β-thalassemia has been associated with increased apoptosis of erythroid precursors, recent observations highlight the contributing role of altered erythroid differentiation. A study using two different mouse models of β-thalassemia, i.e. TM and TI, demonstrates a significantly higher rate of proliferation of splenic erythroblasts in the presence of elevated EPO concentration, in vitro and in vivo80. Specifically, these are poorly differentiated cells expressing high levels of proteins involved in cell cycle such as EPOR, JAK2, BCL-XL, CDK2, KI-67 and CycA. Similar results were obtained when human TM peripheral hematopoietic stem cells were cultured to generate erythroblasts 81. Thalassemic erythroid progenitors had a higher absolute expression of JAK2, CYCA, BCL-XL and KI-67. Immunohistochemical analysis of these erythroid progenitors at different stages of maturation revealed upregulation of proliferative marker KI-67. In addition, spleen sections of TM patients had increased number of KI-67 positive erythroblasts 80. All these results indicate a high rate of proliferation of thalassemic erythroid progenitors. In TM and TI mice, studies of erythroid progenitors at different stages of maturation in the BM and spleen, demonstrate an increased percentage of immature progenitors with a combined reduction in the mature subpopulation. Finally, no differences were noted in apoptosis, compared to controls, when assessed by three different methodologies.
Although the mechanisms leading to decreased differentiation have not yet been elucidated, transcription factor ID1 may be an important player. ID1 is upregulated in erythroid precursors by the JAK/STAT pathway. High concentration of ID1 inhibits cell differentiation therefore continuous activation of JAK2 could hinder erythroid maturation 19, 68. An additional mediator could be the transcription factor FOXO3. Indeed, loss of Foxo3 in mice results in impaired antioxidant response and mitotic arrest in intermediate erythroid precursors, leading to decreased erythroid maturation 82. Collectively these abnormalities resemble the process in IE. Moreover, murine absence of Foxo3 induces activation of JAK/AKT/mTOR signaling pathway in immature erythroblasts 83. Hyperactivation of mTOR alters cell cycle progression and maturation rate (Figure 2). Different studies indicate the significance of this pathway in the pathogenesis of IE. Pharmacologic inhibition of mTOR signaling in thalassemic mice enhanced production of mature RBC and improved the anemia 83. In another approach, administration of resveratrol, a natural polyphenolic stilbene, in thalassemic mice activated Foxo3 and inhibited Akt signaling. After treatment, the animals demonstrated amelioration of IE and increased RBC survival 84.
Figure 2. The role of FOXO3-mTOR cross talk in β-thalassemia.
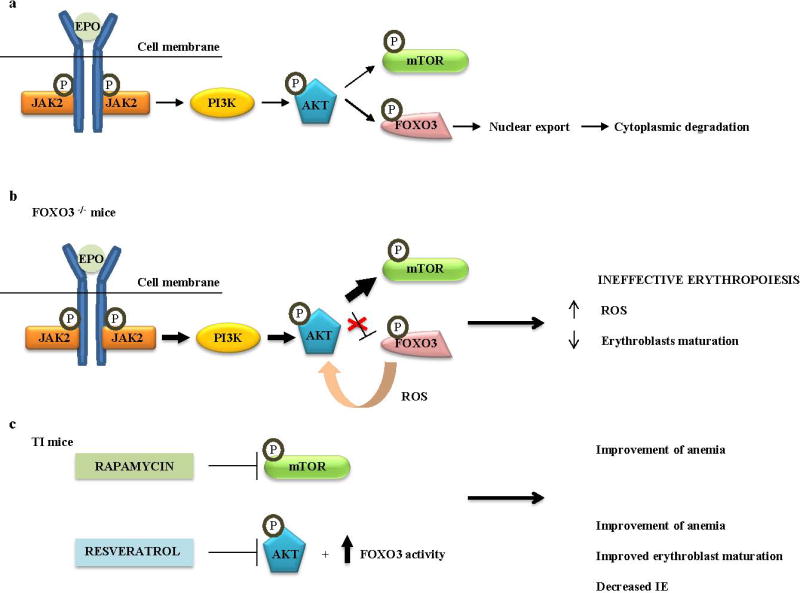
a. EPO stimulation promotes activation of phosphoinositide 3-kinase (PI3K). PI3K phosphorylates AKT kinase, which phosphorylates FOXO3 and induces its nuclear export, resulting in inhibition of FOXO3 transcriptional activity. AKT kinase also phosphorylates and regulates mammalian target of rapamycin (mTOR) kinase. b. In FOXO3 deficient mice (FOXO3 −/−) lack of FOXO3 increases activation of JAK2/AKT/mTOR signaling. Impaired antioxidant response, due to FOXO3 deficiency, increases ROS that partially mediate hyperactivation of JAK2/AKT/mTOR signaling. FOXO3 deficient erythroblasts exhibit abnormalities similar to IE. c. Inhibition of mTOR signaling by rapamycin in TI mice, improved the anemia, implicating mTOR signaling in the pathogenesis of IE. Resveratrol inhibits AKT kinase and increases activation of FOXO3. Resveratrol treatment of TI mice accelerated erythroblast maturation and decreased IE.
This model describes a vicious circle that contributes to IE and exacerbates the anemia. Hypoxia induces the production of EPO, which acts on an increasing number of erythroid progenitors through EPOR. As a result, persistent phosphorylation of JAK2 leads to survival and active proliferation and expansion of erythroid progenitors19. While the erythron increases, the spleen becomes a primary site of EMH 59, 60. Consecutively, splenomegaly can result in splenic sequestration of RBC and deterioration of the anemia 85. This mechanism is also supported by the observation that in thalassemic mice, the Hb concentration inversely correlates with weight of the spleen and degree of EMH 80.
4.3 Oxidative stress
In β-thalassemia, the defect in β-globin chain production results in the excess of α-globins. Free α-globins form aggregates that are unstable and may autoxidize, denature and precipitate as hemichromes within the cells. The result is a high production of reactive oxygen species (ROS), free heme and iron (Figure 3) 86. Free iron generates ROS through the Fenton reaction 87. In fact, increased production of ROS has been documented in RBC isolated from thalassemic patients 88, 89. ROS can oxidize lipids, nucleic acids, cellular proteins and alter the activity of transcription factors, membrane channels and signaling pathways 90. Furthermore, pathological free iron initiates self-amplifying redox reactions that induce free hemoglobin oxidation and membrane destabilization 91. As a result, extensive oxidizing damage on RBC can promote their premature death by hemolysis, thus contributing to IE.
Figure 3. Oxidative Stress in β-thalassemia.

Free α-globins form aggregates that autoxidize, denature and precipitate as hemichromes. As a result, ROS, free heme and iron are produced. ROS are also produced thought the Fenton reaction from free iron. ROS oxidize lipids, nucleic acids and cellular proteins. Hemichromes mediate the phosphorylation of the cytoplasmic domain of band-3, resulting in band-3 aggregation and disruption of ankyrin binding. Anti-band 3 antibodies, together with complement, recognize band-3/hemichrome clusters and mediate phagocytosis of erythrocytes by macrophages. Disruption of band-3 leads to decreased concentration of NADPH further inducing oxidative stress. ROS induce oxidation on protein 4.1, spektrin and actin leading to unstable and fragile RBC.
Oxidative stress can cause severe alterations in membrane stability, cellular hydration, and on cytoskeleton proteins 90. Hemichromes bound to the cytoplasmic membrane of erythroid progenitors mediate the phosphorylation of the cytoplasmic domain of band-3, an anion transmembrane transport protein 92. This results in reorientation of intramembrane and extracellular domains, band-3 aggregation and disruption of ankyrin binding 93. The newly formed band-3 clusters act as an antigen and bind with high affinity to IgG anti-band 3 antibodies and complement fragments providing a signal for removal from macrophages 94–97. Moreover, the disrupted function of band-3 results in decreased concentration of NADPH, an anti-oxidant agent and exacerbates the oxidative stress 86. Another important pathway is the oxidation of protein 4.1, a member of the cytoskeleton in erythrocytes. Defective protein 4.1 results in its reduced ability to bind with spectrin, as well as in ineffective interactions between spectrin and actin, leading to unstable and fragile RBC 90, 98, 99. α-globin aggregates form early in erythroid differentiation and colocalize with areas of defective association between spectrin and protein 4.1 in β-thalassemic erythroblasts. This could result in the oxidation of the cytoskeletal proteins, since α-globin aggregates tend to autoxidize.
4.4 Iron Metabolism
Iron overload is an important and potentially devastating complication of β-thalassemia as the excess free iron deposits in organs, particularly in the heart and liver, and contributes to the morbidity and mortality of the disease 100, 101. Although regular RBC transfusions are the main cause of iron overload in TM, dysregulated iron absorption from the gastrointestinal tract also contributes to iron accumulation in both TM and TI 68, 102, 103. In fact, studies in non-transfused patients demonstrate a 3-to 4-times fold increase in iron loading from the enterocytes than normal 59. Research in the last years has shed light on the mechanisms controlling iron regulation and on the intertwined relation between iron loading and IE (Figure 4).
Figure 4. Important players in Iron Metabolism.
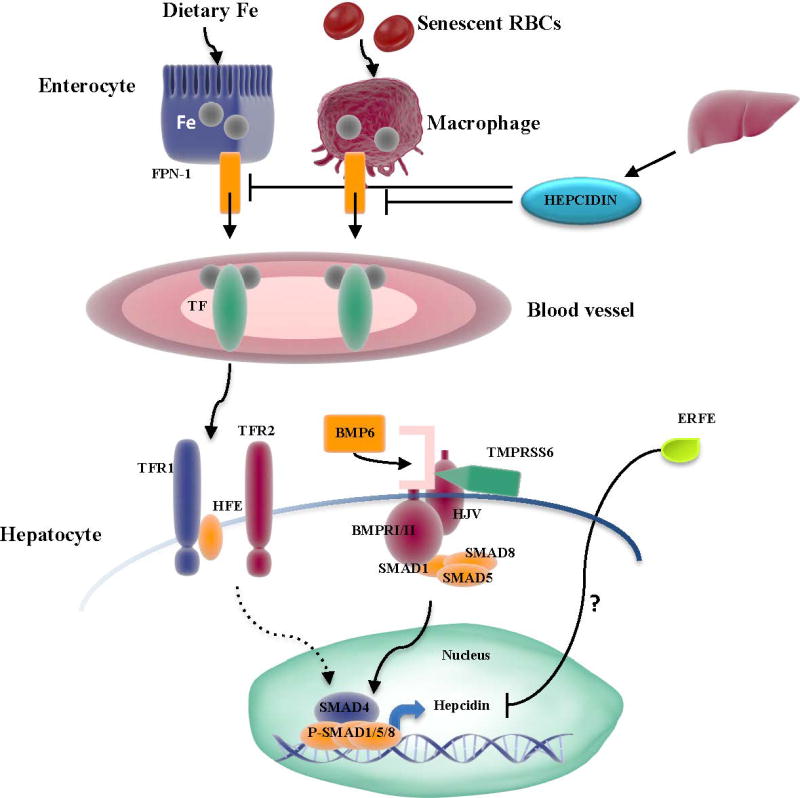
Dietary iron absorbed by the enterocytes and iron recycled from senescent RBC through macrophages gets exported through FPN-1 and successively binds to TF in blood vessels. TF-Iron complexes get transferred to hepatocytes. In this process BMP6 has a critical role. Increased intracellular iron concentration trigger BMP6 expression. In turn, BMP6 binds to type I and II receptors and co-receptor HJV that consequently activate SMAD1/5/8. In association with SMAD 4 they upregulate hepcidin expression, which inhibits iron export in the circulation. Furthermore, in hepatocytes, the relative binding of TF to TFR1 versus the HFE/TFR2 complex modulates hepcidin expression, potentially through the SMAD pathway (dotted line). On the contrary, the enzyme TMPRSS6 cleaves HJV and negatively regulates hepcidin expression. High EPO levels induce expression of the erythroid regulator ERFE in erythroblasts, which decreases hepcidin expression by a mechanism not characterized yet (indicated by a question mark).
Iron enters in circulation either by absorption of dietary iron through the enterocytes or by recycling Hb from senescent RBC through macrophages. The main regulator of systemic iron homeostasis is hepcidin, a 25-aa peptide produced predominantly in the liver 100. Under normal conditions hepcidin is released in response to plasma iron concentration and iron stores, and binds to ferroportin (FPN-1), the only currently known iron exporter found primarily on the surface of duodenal enterocytes, macrophages and hepatocytes. Binding with FPN-1 results in internalization and degradation of the latter which prevents iron export into the circulation 74, 104. Thus, in presence of high iron concentration increased expression of hepcidin reduces the amount of iron exported from enterocytes and macrophages. Both intracellular and extracellular iron concentration play a key role in hepcidin regulation. In circulation, the serum protein transferrin (TF) binds to iron and transports it to the erythron or peripheral tissues. The uptake of iron bound to TF by transferrin receptor 1 (TFR1) is the only known pathway of iron delivery for erythropoiesis. Increased iron plasma concentration is sensed by the hemochromatosis protein (HFE)/TFR1 complex and transferrin receptor 2 (TFR2) which trigger hepicidin production 105–109. Although the signaling pathways activated in this process are still not completely understood, bone morphogenetic protein (BMP)/SMAD signaling is involved. Bone morphogenetic protein 6 (BMP6) interacts with the co-receptor hemojuvelin and BMP receptors type I/II to phosphorylate and activate the SMAD1/5/8-SMAD4 complex and induce hepcidin expression 110–114. In the liver elevated intracellular iron concentration can also stimulate BMP6 expression and increase hepcidin production 115, 116. On the other hand, transmembrane protease serine 6 (TMPRSS6), an enzyme that cleaves hemojuvelin from the cell surface, is a negative regulator of hepcidin 117–120.
4.4.1 Iron Metabolism and erythropoiesis
Hepcidin expression is controlled by multiple stimuli including hypoxia, erythropoiesis and inflammation 121–125. In several diseases, including chronic inflammatory conditions, malignancies or chronic infections, hepcidin concentration is increased, causing retention of iron in macrophages and enterocytes resulting in a type of iron restricted anemia called anemia of inflammation (Table 1). In presence of elevated EPO concentration, hypoxia and increased erythropoiesis, hepcidin expression is reduced resulting in increased iron availability for the production of RBC 59, 123, 126, 127. Therefore, it is not surprising that several studies in both mice and patients affected by β-thalassemia detected decreased hepcidin concentration 103, 128–133. Downregulation of hepcidin is essential in resolving acute stress, but chronically reduced concentration will increase iron load. In fact, inappropriately suppressed production of hepcidin causes systemic iron overload even in non-transfused thalassemic patients 68. The suggested mechanisms that reduce hepcidin expression include the presence of molecules called erythroid regulators. A member of the transforming growth factor-beta superfamily (TGF-β) i.e. growth differentiation factor 15 (GDF15), and twisted-gastrulation 1 (TWSG1), a BMP binding protein, have been proposed as such molecules 106, 134–136. Nonetheless, results are inconsistent, and participation of these molecules in suppression of hepcidin has not been definitively demonstrated 106, 137–140. Recent studies suggest that the hormone erythroferrone (ERFE), whose expression is induced by EPO, may be an erythroid regulator. In a mouse model of TI high concentration of ERFE was found in the BM and spleen. Ablation of ERFE increased hepcidin concentration and decreased hepatic iron overload, suggesting that ERFE contributes to hepcidin suppression 139, 141.
In β-thalassemia, iron and IE are linked in a complicated relationship. A study using two different mouse models of β- thalassemia demonstrated that the different degrees of IE dictated the pattern of iron distribution 130. In TM mice, hepcidin concentration was very low due to the extreme IE that prevented hepcidin from sensing the iron load. In contrast, in a TI model hepcidin expression was relatively low during the first months of life but increased as iron overload worsened. This indicates that, under conditions of milder IE, hepcidin can still respond partially to iron status. Moreover, when mice suffering from severe anemia received RBC transfusions, their hematopoietic parameters and IE were improved while hepcidin expression was increased 130. This effect has also been demonstrated in human patients 103, 129, 142. All the above suggest that the expression of hepcidin is determined by the relative levels of IE and iron load.
4.5 Erythroid iron metabolism and the role of classic iron related molecules in erythropoiesis
4.5.1 The role of TFR1 and TFR2 in erythropoiesis
Iron is essential for efficient erythropoiesis and for that reason must be readily available for erythroid differentiation.. A study disrupting the Tfr1 gene in mice underlined the importance of this protein for erythropoiesis 143. Homozygous deletion of the gene was lethal and embryos were severely anemic with signs of hydrops fetalis. In addition to its canonical function, TFR1 participates in another fundamental signaling pathway. Polymeric IgA1 (pIgA1) is produced by plasma cells in hypoxia and binds to TFR1 independent of TF, activating the Erk/Akt signaling pathway 144, 145. This action of pIgA1 is additive to TF and potentiates erythroblast response to EPO, therefore promoting erythroid expansion. Recent data suggest that decreasing TFR1 concentration might ameliorate IE in β-thalassemia 146. In this study, reduced TFR1 concentration in mice affected by TI improved hematopoietic parameters and decreased α-globin precipitation in circulating RBC. These beneficial effects will need to be confirmed with further studies but indicate that targeting TFR1 may be a novel promising approach in improving IE. Other classic iron related molecules, initially discovered in the liver, might also play a role in erythropoiesis. For instance, it has been suggested that HFE is also expressed on erythroid progenitors, forming complexes with TFR1 147–151. It is then possible that HFE may compete with the overlapping sites of TFR1 for TF, regulating erythroid iron uptake 105. TFR2 is also important for normal erythropoiesis. TFR2 is expressed during erythroid differentiation and forms a complex with EPOR, possibly altering sensitivity of erythroid precursors to EPO 107, 152–157. In a recent study, lethally irradiated mice transplanted with Tfr2 deficient hematopoietic cells had increased RBC in the periphery and differentiated erythroblasts in the BM 158. The mice had normal serum EPO concentration, but erythroblasts had increased expression of EPO related genes i.e. Epor, Erfe, Bcl-X, concurring with the hypothesis of increased sensitivity to EPO. Thus, in addition to its direct role in the regulation of hepcidin in hepatocytes, Tfr2 might also contribute to the regulation of erythroid maturation.
4.5.2 The IRE/IRP regulatory system
Intracellular iron concentration regulates the expression of genes involved in iron import, export, transport, storage and usage at the post-transcriptional level. Several iron-related transcripts contain untranslated mRNA sequences called iron responsive elements (IREs) that are recognized and bound by cytoplasmic iron responsive proteins (IRPs) under low-iron conditions 159–163. IRPs can act both as translational enhancers or silencers depending on the position of the IRE on the mRNA; they promote iron storage, export and usage in presence of high iron concentration and induce iron absorption and transport in iron deficiency 159, 161, 162, 164–170. Recently, studies in mice demonstrated that deficiency of Irp1 or Irp2 resulted in different erythroid defects and revealed a new regulatory axis through Hif2α 171–173. Deficiency of Irp1 increased stabilization of Hif2α mRNA, resulting in increased EPO concentration and polycythemia. Lack of Irp2 led to microcytosis, decreased Tfr1 mRNA and increased ferritin and Alas2 concentrations 174, 175.
4.5.3 Heme and erythropoiesis
In addition to being an essential component of Hb, heme participates, as an important regulatory molecule, in erythropoiesis. It can interact with IRP2, resulting in degradation of the latter, and directly induce the transcription of specific genes, by binding to the transcriptional repressor Btb And Cnc Homology 1 (BACH1) 176–181. This pathway has been suggested to have a role in activating transcription of the globin genes 180, 182, 183. Transcription of the β-globin gene can be repressed by BACH1 or induced by nuclear factor erythroid-derived 2 (NF-E2) 184–188. In an in vitro system, inhibition of heme biosynthesis in human erythroleukemia cells decreased the concentration of α- and β-globin chains. Moreover, intracellular heme concentration was found to control BACH1 and NF-E2 recruitment 180, 182. In a separate study, it was demonstrated that heme promoted the displacement of BACH1 from the β-globin locus control region 183.
At the translational level heme can affect globin protein synthesis through the heme-regulated eIF2α kinase (HRI), which is predominantly expressed in hemoglobinized erythroblasts 189, 190. When heme concentration is low, HRI autohosphorylates and subsequently phosphorylates the α subunit of the eukaryotic translation initiation factor 2 (eIF2α) that inhibits protein synthesis 191. On the contrary, during Hb synthesis heme binds to a heme binding domain on HRI, which as a result becomes inactive, allowing the synthesis of the β-globin genes 190–192. Therefore, HRI balances the production of heme and globins to ensure that no excess globins are produced. In β-thalassemia HRI may have an effect on disease severity. In a mouse model of TI Hri deficiency was lethal; however, Hri haploinsufficency did not affect embryonic survival but led to a more severe phenotype with increased α-globin aggregates accumulation and reduced life span compared to their thalassemic counterparts 193. These results point to a critical role of HRI for erythroid progenitor survival. Further studies are needed to elucidate the potential use of HRI as a therapeutic target in β-thalassemia to decrease α-globin accumulation and ameliorate disease severity.
4.6. Molecular Chaperones
4.6.1 HSP70
Complex mechanisms promote differentiation and maturation of erythroid progenitors. The erythroid transcription factor, or GATA-binding factor 1 (GATA-1), holds a pivotal role in this process, by regulating the expression of erythroid promoters and anti-apoptotic genes, such as BCL-XL 194. In order to advance survival and differentiation of progenitors, it is necessary that specific cysteine-aspartic proteases, called caspases, are activated. As erythroid cells mature, caspases become transiently active and mediate crucial pathways of terminal differentiation, such as chromatin condensation and cell size reduction 90. Caspases have paradoxical effects on erythropoiesis. In addition to their role in erythroid maturation, they are required for programmed cell death. Two different types of caspases (initiators and effectors) finally lead to DNA damage and apoptosis 195. One of the effectors involved is caspase-3, which targets and cleaves GATA-1. It is evident that during erythroid differentiation protective mechanisms are needed to avoid cell death. Recently, the chaperone heat shock protein 70 (HSP70) was identified to protect GATA-1 in this process 11.
HSP70 is a ubiquitous chaperone that is constitutively expressed in human erythroblasts. During late stages of erythroid differentiation, when caspases become active at the basophilic stage, HSP70 colocalizes in the nucleus with GATA-1 and protects it from cleavage. EPO has a central function in this pathway by regulating cellular localization of HSP70 11, 196. In presence of low EPO concentration reduced nuclear HSP70 translocation results in less GATA-1 bound to HSP70. Free GATA-1 is prone to cleavage by caspase-3. In β-thalassemia HSP70 may be an important player that promotes IE. In vitro studies demonstrated that erythroblasts derived from TM patients contain low concentration of HSP70 and GATA-1 in the nucleus, while HSP70 is mainly localized in the cytoplasm 197. The same observations were noted in BM samples of TM patients. The decrease in concentration of nuclear HSP70 is due to interactions between HSP70 and free α-globin chains, but not Hb, in the cytoplasm. Concentration of HSP70 and GATA-1 in the nucleus decreases as maturation and hemoglobinization progress. It is hypothesized that as HSP70 forms complexes with excess free α-globin chains in the cytoplasm, GATA-1 remains unprotected and is cleaved by caspase-3. In fact, restoring nuclear concentration of HSP70 in TM erythroblasts, by lentiviral transduction, improves terminal maturation and decreases apoptosis 197. Although further research is necessary, increasing nuclear concentration of HSP70, in order to protect GATA-1, may be an alternative approach to improve IE. To this end, targeting the interactions between HSP70 and α-globin chains in the cytoplasm or decreasing HSP70 nuclear export could be beneficial for patients.
4.6.2 AHSP
The α-Hb stabilizing protein (AHSP) is an important chaperone that protects the erythroid cells from free α-globin chain toxicity. It binds to free α-globin chains, but not β chains or HbA, stabilizes them and prevents their denaturation 196, 198. AHSP is an erythroid specific chaperone primarily found in late stage erythroid precursors 196, 199. Interactions between AHSP and free α-globin chains result in the formation of a stable complex that prevents aggregation, precipitation and autoxidation of α chains. In addition, AHSP binds to α-Hb (unmatched α-globin chains bound to heme) and stabilizes it, by inhibiting hemebound iron to participate in any reactions that generate ROS. Expression of this chaperone is induced by GATA-1. It is also affected by iron status, due to the presence of a functional iron response element located in the 3’ untranslated region of the AHSP gene 200, 201.
In β-thalassemia, IE depends on the concentration of free α-Hb chains that induce oxidative stress and damage erythroid progenitors. Therefore, it can be postulated that AHSP concentration is associated with disease severity. In patients with β-thalassemia inadequate concentration of AHSP results in increased hemolysis and severe anemia that requires frequent RBC transfusions 196, 199. Studies in mice provide important information on the significance of this chaperone and its association with disease. In normal mice loss of Ahsp results in increased production of ROS, abnormal morphology and decreased life span of erythrocytes. These AHSP deficient mice demonstrate an increased proportion of immature erythroid progenitors in the spleen, maturation arrest and increased apoptosis of erythroid precursors, reflecting the setting of IE 90, 202. In a mouse model of TI, absence of Ahsp worsens the anemia and morphology of erythrocytes and exacerbates α-globin precipitation.
In contrast, in TI mice, overexpression of Ahsp failed to alleviate the severity of the disease and had no effect on hematopoietic phenotype 203. In addition, several studies in thalassemic patients did not detect an association between polymorphisms in the ASHP gene and disease severity 91, 201, 204, 205. Further research is necessary to address important questions regarding the role of AHSP as a disease modifier in β-thalassemia. This patient population is heterogeneous and studies looking at the ASHP gene sequence in relation with the patient genome could advance our current knowledge. AHSP can be potentially used in the treatment of β-thalassemia to limit α-globin precipitation, formation of ROS and ameliorate IE. For instance, a potential approach could be to target the specific mechanisms that lead to α-Hb stabilization or to produce mimetic proteins 201.
5. The erythroblastic island in β-thalassemia
Since macrophages play an important role in maintaining erythropoiesis in conditions of acute stress, it is not unexpected that their depletion in β-thalassemia, a condition of chronic stress erythropoiesis, could limit sustained erythropoiesis. This concept was tested by using clodronate-loaded liposomes to deplete macrophages in a mouse model of the disease. Treatment rapidly ameliorated the anemia, noted by increased Hb and RBC number. In addition, treated animals had decreased numbers of immature erythroid progenitors and proportionally increased differentiated erythroblasts in the BM and spleen, indicating an improvement of erythropoiesis 57. A concurrent reductive effect was also noted on spleen size and EMH. Furthermore, serum iron concentration was decreased and hepcidin expression was elevated, indicating improved iron status, which could also contribute to the amelioration of IE. Chronic use of clodronate-loaded liposomes sustained the beneficial effect in reducing IE and significantly improved the morphology and life span of RBC. In addition, α-globin chain aggregates were also decreased 57.
The effects promoted by macrophage depletion can be elucidated by better understanding macrophage-erythrocyte interactions. The effect that macrophages exert on erythroid development seems to be dependent on direct contact of the two different cell types. In vitro, when erythroblasts derived from human thalassemic patients are cultured in the presence of macrophages they proliferate in a higher rate than when cultured alone 57. This response is accompanied by a higher percentage of cells cycling and reduced numbers of differentiated enucleated cells. On the contrary, in conditions were the erythroblasts do not come in contact with the macrophages the positive effect on proliferation is hindered. These observations point to a critical role of macrophages in erythroid proliferation and survival.
In a clinical setting depletion of macrophages could be detrimental for patients, as it could compromise the immune response and increase the risk of infections. Nonetheless, targeting the interactions between erythroblasts and macrophages in the erythroblastic island could be a novel therapeutic strategy to improve IE. This approach would aim at the specific molecules involved in cellular crosstalk, while immunologic function would remain unaffected.
6. Targeting Ineffective erythropoiesis: novel potential therapies
Managing IE still remains a difficult task. Current regiments can only partially alleviate the phenotype of the disease, allowing IE to have detrimental effects on physiologic processes. The discovery of new therapeutic agents that could substantially improve erythropoiesis would reduce the need for RBC transfusions and improve quality of life of patients. Extensive research in the last years has focused on this aim, by targeting the specific mechanisms that mediate IE (Figure 5).
Figure 5. Novel treatments for β-thalassemia.
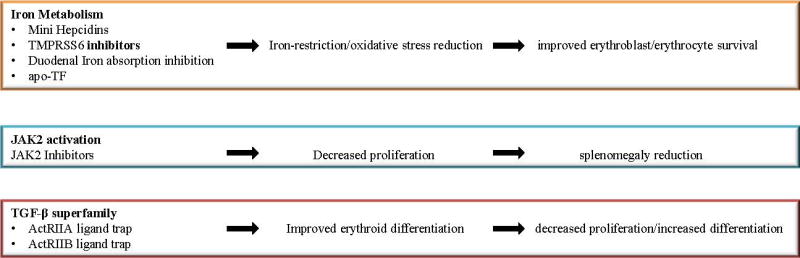
Current management of β-thalassemia with RBC transfusions and iron chelation does not efficiently address the pathophysiology of IE. As described in the text, novel drugs are being developed to target proteins that modulate erythropoiesis, iron metabolism or the activity of some of the ligands of the TGF-β superfamily. These molecules have the potential to improve the phenotype of the disease and reduce the risk of complications.
6.1 Targeting iron metabolism
Targeting iron could be beneficial for the improvement of IE. Decreased iron availability in the erythron would reduce the production of erythroid cells carrying abnormal Hb and therefore would limit the quantity of hemichromes and improve erythropoiesis. In this direction, several different therapeutic molecules have been studied. Synthetic small peptides called mini-hepcidins act like hepcidin agonists 206. These molecules can bind to FPN-1 and decrease serum iron concenration. Administration of mini-hepcidins in mouse models of hemochromatosis, a disease characterized by abnormal iron accumulation in parenchymal organs, demonstrated promising results 206, 207. The treated mice had reduced iron absorption and increased iron redistribution in splenic macrophages, which led to decreased iron concentration in the liver and heart. Furthermore, the beneficial effects of low iron on IE have been demonstrated in a mouse model of TI. Mice were either placed on low iron diet or were genetically engineered to moderately overexpress hepcidin. In both cases, limiting iron overload decreased the formation of α chain/heme aggregates and ROS while reversed IE and splenomegaly 208. The mice demonstrated improvement of anemia and amelioration of RBC structure and lifespan. In accordance with these results, when mini-hepcidins were tested in the same mouse model the same positive effects were noted on hematopoietic parameters and IE 209, 210. In addition, the mice had decreased total iron concentration in the liver, kidney and spleen.
Another promising approach is targeting the serine protease TMPRSS6, which physiologically attenuates expression of hepcidin. By decreasing the activity of TMPRSS6, iron concentration will reduce and IE should ameliorate. As a matter of fact, when Tmprss6 was deleted from a mouse model of TI the outcome was an improvement in hematologic parameters, splenomegaly and IE 211. Pharmacologic inhibition of Tmprss6, by second generation antisense oligonucleotides or lipid nanoparticle (LNP)-formulated siRNAs, demonstrated a beneficial effect in mice affected by TI. Treated mice had lower concentration of ROS, hemichromes and apoptosis along with improvement of IE, RBC survival and splenomegaly 212, 213. Recent studies suggest that a combinational therapy of iron chelators and Tmprss6 inhibitors has a superior effect in decreasing liver iron overload than each monotherapy alone or dietary iron restriction, reflected in a lower degree of IE 214, 215.
Reducing the amount of iron delivered to the erythron by each molecule of TF could ameliorate the anemia and improve the degree of IE. A study using apotransferrin (apo-TF), i.e. TF not bound to iron, in thalassemic mice demonstrated that apo-Tf administration altered the proportion of erythroid precursors to more mature versus immature and decreased α-globin precipitation and apoptosis of mature erythroid precursors 216. Moreover, chronic treatment with apo-TF normalized the anemia and iron content in the liver, heart and kidney 217. Concomitantly iron regulation was modified, noted by decreased expression of ERFE and increased concentration of hepcidin. The proposed rationale behind these observations lies in difference in TFR1-mediated iron delivery via holo-TF (with two bound iron molecules per Tf molecule) compared with monoferric-TF (with one). Not only does diferric Tf deliver twice the number of iron molecules per cycle, it binds TFR1 with greater affinity than does monoferric TF. Providing exogenous apo-TF leads to a fall in transferrin saturation and a shift in the relative population of TF molecules to the monoferric form. As such in treated mice there is decreased iron delivery to erythroid precursors, resulting in lower mean corpuscular hemoglobin (MCH) and decreased hemichrome formation 60.
All these results suggest that therapeutic strategies modulating iron metabolism can be beneficial in reducing oxidative stress and significantly improving IE. By aiming at the pathogenetic mechanism of iron overload, these treatments offer a novel approach that could produce better results than current regiments.
6.2 Use of JAK2 inhibitors
JAK2 activation is a critical process in sustaining IE and developing EMH in thalassemic patients. In addition, several studies indicate a potential role of JAK2 in iron regulation 68, 218. For instance, STAT5 controls the expression of IRP2 and TFR1 219, 220. The data suggest that activation of the JAK/STAT5 pathway induces, both directly and indirectly, iron uptake. To that end, the use of JAK2 inhibitors (JAK2i) seems as a promising therapeutic tool. Administration of JAK2i in a mouse model of TI led to a significant decrease in spleen size and IE 80. In human patients, this approach could translate to an efficient substitute of therapeutic splenectomy 19, 85. Currently, splenectomy is often the best treatment option in patients with excessive RBC transfusion needs or massive splenomegaly that prevents sufficient control of iron concentration. However, splenectomized patients have a higher risk for certain complications, such as infections and pulmonary hypertension 19. Therefore, treatment with JAK2i could be a useful alternative for this group of patients. Moreover, as spleen size decreases the degree of blood sequestration will reduce and the need for RBC transfusions will be limited 55. Data of clinical trials using JAK2i in patients suffering from myeloproliferative syndromes demonstrate a beneficial effect in reducing spleen size 221. These results have to be carefully evaluated since side effects, including anemia, were also documented 222. Such an effect could be detrimental in thalassemic patients but may be potentially avoided by a short-term use of JAK2i or with the combination of RBC transfusions. Future studies will be required to evaluate the benefits and risks of JAK2i use.
6.3 TGF-β superfamily: a new role in erythropoiesis
Recently, an innovative approach was developed to improve IE by targeting a pathway that is distinct from the EPO/JAK/STAT. This novel method focuses on regulation of erythropoiesis by members of the TGF-β superfamily, which include BMPs and growth differentiation factors (GDFs). All these ligands bind to a type II receptor and consequently phosphorylate and activate a type I receptor. Complexes containing different combinations of type I and II receptors activate SMAD signaling transcription factors, which then accumulate in the nucleus and alter gene expression 223. The interest in erythropoiesis lies in signaling through activin receptors type IIA (ActRIIA) and IIB (ActRIIB) that activate the SMAD2/3 proteins. High activation of phosphorylated SMAD2/3 has been documented in splenic erythroid progenitors of thalassemic mice, a finding that indicates a relation between activation of SMAD2/3 and IE 224–226.
Pharmacologic inhibition of this pathway has been achieved by the development of two different receptor fusion proteins that consist of the extracellular domain of either ActRIIA (Sotatercept, ACE-011 or its mouse version RAP-011) or ActRIIB (Luspatercect, ACE-536 or its mouse version RAP-536) linked to the Fc protein of IgG and act as ligand traps. Use of these two agents in mouse models of TI not only significantly improved the anemia and degree of IE, but also reduced complications of the disease 224, 226. Treatment improved erythroid differentiation with decreased immature and increased later stage erythroblasts. Furthermore, administration of RAP-011 promoted apoptosis of immature erythroid cells by inducing expression of Fas and FasL in early erythroblasts 226. It is therefore suggested that inhibition of activin receptors contributes to improvement of IE with a dual mechanism: stimulation of terminal erythroid differentiation and normalization between proliferation and differentiation. A positive effect was also noted on iron status, spleen size and oxidative stress 224, 226. The drugs reduced α-globin aggregates, ROS concentration and abnormal erythroid morphology, indicated by reduced hemoglobin debris and degree of poikilocytosis, observed in peripheral blood smears of treated animals.
In search of the possible ligands for RAP-011 and RAP-536, growth differentiation factor 11 (GDF11) has been identified as a potential candidate. The data indicate that GDF11 exerts negative regulation on late stage erythroid differentiation 226–228. Studies in mice and humans suggest that IE is associated with GDF11 overexpression in relatively immature erythroblasts mostly in the spleen where EMH takes place 226, 227. In support of this hypothesis, patients with β-thalassemia have high concentration of GDF11 in the serum 233. It is therefore proposed that these ligand traps act by binding with GDF11 and inhibiting SMAD2/3 phosphorylation. Furthermore, it is suggested that oxidative stress can induce expression of GDF11, which successively promotes ROS, resulting in an autocrine amplification loop innate to IE. These results combined, indicate that inhibiting the TGF-β signaling pathway can be an excellent therapeutic regiment for patients suffering from diseases associated with IE. Currently, ongoing phase 2 and 3 clinical trials are testing the efficacy of these agents in patients with β-thalassemia.
Practice points.
IE is a major contributor to the pathogenesis of β-thalassemia.
IE is characterized by increased numbers of erythroid progenitors in the BM and disproportionally decreased circulating RBC.
Management of β-thalassemia includes RBC transfusions and iron chelation.
Emerging treatments are under development to improve erythropoiesis and the consequent iron overload.
Research Agenda.
Assess the use of agents targeting iron metabolism such as mini-hepcidins and TMPRSS6-ASO in clinical studies.
Target iron absorption in the duodenum as a therapeutic approach in mouse models of β-thalassemia.
Better understand the role of erythroblast transferrin receptors in erythropoiesis and their potential for targeting in the treatment of β-thalassemia.
Analyze HSP70 gene sequence in a large heterogeneous patient population with different degrees of clinical severity to understand its role as a genetic modifier.
Develop approaches to protect GATA1 from cleavage in erythroid cells.
Further characterize the role of the TGF-β related molecules in erythropoiesis
Identify the specific molecules involved in macrophage-erythroblast interactions.
Evaluate the use of JAK2 inhibitors in combination with RBC transfusions.
Better understand the role of HRI as a modifier of disease severity in β-thalassemia.
Acknowledgments
Work related to this article was funded by grants from the NIH-NIDDK- R01 DK095112 and R01 DK090554 (SR). We extend special thanks to Ping La (Children’s Hospital of Philadelphia) for helpful discussion and support.
Role of the funding source
The funding sources had no role in the collection, analysis and interpretation of data or in the writing and decision to submit the manuscript for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
S.R. is a member of scientific advisory board of Ionis Pharmaceuticals. S.R. is sponsored by Ionis Pharmaceuticals.
P.R.O. declares no conflict of interest.
References
- 1.Orkin SH, Zon LI. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell. 2008;132:631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125:2605–13. doi: 10.1182/blood-2014-12-570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet. 2000;1:57–64. doi: 10.1038/35049577. [DOI] [PubMed] [Google Scholar]
- 4.Gregory CJ, Eaves AC. Three stages of erythropoietic progenitor cell differentiation distinguished by a number of physical and biologic properties. Blood. 1978;51:527–37. [PubMed] [Google Scholar]
- 5.Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB life. 2009;61:800–30. doi: 10.1002/iub.226. [DOI] [PubMed] [Google Scholar]
- 6.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 2009;106:17413–8. doi: 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Migliaccio AR. Erythroblast enucleation. Haematologica. 2010;95:1985–8. doi: 10.3324/haematol.2010.033225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gifford SC, Derganc J, Shevkoplyas SS, Yoshida T, Bitensky MW. A detailed study of time-dependent changes in human red blood cells: from reticulocyte maturation to erythrocyte senescence. Br J Haematol. 2006;135:395–404. doi: 10.1111/j.1365-2141.2006.06279.x. [DOI] [PubMed] [Google Scholar]
- 9.Testa U. Apoptotic mechanisms in the control of erythropoiesis. Leukemia. 2004;18:1176–99. doi: 10.1038/sj.leu.2403383. [DOI] [PubMed] [Google Scholar]
- 10.Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118:6258–68. doi: 10.1182/blood-2011-07-356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ribeil JA, Zermati Y, Vandekerckhove J, Cathelin S, Kersual J, Dussiot M, et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature. 2007;445:102–5. doi: 10.1038/nature05378. [DOI] [PubMed] [Google Scholar]
- 12.Jelkmann W. Erythropoietin: structure, control of production, and function. Physiol Rev. 1992;72:449–89. doi: 10.1152/physrev.1992.72.2.449. [DOI] [PubMed] [Google Scholar]
- 13.Constantinescu SN, Ghaffari S, Lodish HF. The Erythropoietin Receptor: Structure, Activation and Intracellular Signal Transduction. Trends Endocrinol Metab. 1999;10:18–23. doi: 10.1016/s1043-2760(98)00101-5. [DOI] [PubMed] [Google Scholar]
- 14.Kuhrt D, Wojchowski DM. Emerging EPO and EPO receptor regulators and signal transducers. Blood. 2015;125:3536–41. doi: 10.1182/blood-2014-11-575357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang J, Menon M, Kapelle W, Bogacheva O, Bogachev O, Houde E, et al. EPO modulation of cell-cycle regulatory genes, and cell division, in primary bone marrow erythroblasts. Blood. 2007;110:2361–70. doi: 10.1182/blood-2006-12-063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–73. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 17.Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–91. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 18.Gillinder KR, Tuckey H, Bell CC, Magor GW, Huang S, Ilsley MD, et al. Direct targets of pSTAT5 signalling in erythropoiesis. PloS One. 2017;12:e0180922. doi: 10.1371/journal.pone.0180922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melchiori L, Gardenghi S, Rivella S. beta-Thalassemia: HiJAKing Ineffective Erythropoiesis and Iron Overload. Adv Hematol. 2010;2010:938640. doi: 10.1155/2010/938640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813:1978–86. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 21.McIver SC, Kang YA, DeVilbiss AW, O'Driscoll CA, Ouellette JN, Pope NJ, et al. The exosome complex establishes a barricade to erythroid maturation. Blood. 2014;124:2285–97. doi: 10.1182/blood-2014-04-571083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang R, Camprecios G, Kou Y, McGrath K, Nowak R, Catherman S, et al. A Systems Approach Identifies Essential FOXO3 Functions at Key Steps of Terminal Erythropoiesis. PLoS Genet. 2015;11:e1005526. doi: 10.1371/journal.pgen.1005526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3' to the human erythropoietin gene. Proc. Natl. Acad. Sci. U.S.A. 1991;88:5680–4. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tojo Y, Sekine H, Hirano I, Pan X, Souma T, Tsujita T, et al. Hypoxia Signaling Cascade for Erythropoietin Production in Hepatocytes. Mol Cell Biol. 2015;35:2658–72. doi: 10.1128/MCB.00161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117:1068–77. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27:41–53. doi: 10.1016/j.blre.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapitsinou PP, Liu Q, Unger TL, Rha J, Davidoff O, Keith B, et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. 2010;116:3039–48. doi: 10.1182/blood-2010-02-270322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camacho J, Poveda F, Zamorano AF, Valencia ME, Vazquez JJ, Arnalich F. Serum erythropoietin levels in anaemic patients with advanced human immunodeficiency virus infection. Br J Haematol. 1992;82:608–14. doi: 10.1111/j.1365-2141.1992.tb06475.x. [DOI] [PubMed] [Google Scholar]
- 29.Babitt JL, Lin HY. Mechanisms of anemia in CKD. Clin J Am Soc Nephrol. 2012;23:1631–4. doi: 10.1681/ASN.2011111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baer AN, Dessypris EN, Krantz SB. The pathogenesis of anemia in rheumatoid arthritis: a clinical and laboratory analysis. Semin Arthritis Rheum. 1990;19:209–23. doi: 10.1016/0049-0172(90)90001-v. [DOI] [PubMed] [Google Scholar]
- 31.Vreugdenhil G, Wognum AW, van Eijk HG, Swaak AJ. Anaemia in rheumatoid arthritis: the role of iron, vitamin B12, and folic acid deficiency, and erythropoietin responsiveness. Ann. Rheum. Dis. 1990;49:93–8. doi: 10.1136/ard.49.2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakisaka S, Watanabe M, Tateishi H, Harada M, Shakado S, Mimura Y, et al. Erythropoietin production in hepatocellular carcinoma cells associated with polycythemia: immunohistochemical evidence. Hepatology. 1993;18:1357–62. [PubMed] [Google Scholar]
- 33.Palapattu GS, Kristo B, Rajfer J. Paraneoplastic syndromes in urologic malignancy: the many faces of renal cell carcinoma. Rev Urol. 2002;4:163–70. [PMC free article] [PubMed] [Google Scholar]
- 34.Crielaard BJ, Rivella S. beta-Thalassemia and Polycythemia vera: targeting chronic stress erythropoiesis. Int J Biochem Cell Biol. 2014;51:89–92. doi: 10.1016/j.biocel.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koulnis M, Porpiglia E, Porpiglia PA, Liu Y, Hallstrom K, Hidalgo D, et al. Contrasting dynamic responses in vivo of the Bcl-xL and Bim erythropoietic survival pathways. Blood. 2012;119:1228–39. doi: 10.1182/blood-2011-07-365346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menon MP, Karur V, Bogacheva O, Bogachev O, Cuetara B, Wojchowski DM. Signals for stress erythropoiesis are integrated via an erythropoietin receptor-phosphotyrosine-343-Stat5 axis. J Clin Invest. 2006;116:683–94. doi: 10.1172/JCI25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Pop R, Sadegh C, Brugnara C, Haase VH, Socolovsky M. Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood. 2006;108:123–33. doi: 10.1182/blood-2005-11-4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wensveen FM, Geest CR, Libregts SF, Derks IA, Ekert PG, Labi V, et al. BH3-only protein Noxa contributes to apoptotic control of stress-erythropoiesis. Apoptosis. 2013;18:1306–18. doi: 10.1007/s10495-013-0890-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porpiglia E, Hidalgo D, Koulnis M, Tzafriri AR, Socolovsky M. Stat5 signaling specifies basal versus stress erythropoietic responses through distinct binary and graded dynamic modalities. PLoS Biol. 2012;10:e1001383. doi: 10.1371/journal.pbio.1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peslak SA, Wenger J, Bemis JC, Kingsley PD, Koniski AD, McGrath KE, et al. EPO-mediated expansion of late-stage erythroid progenitors in the bone marrow initiates recovery from sublethal radiation stress. Blood. 2012;120:2501–11. doi: 10.1182/blood-2011-11-394304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koulnis M, Liu Y, Hallstrom K, Socolovsky M. Negative Autoregulation by Fas Stabilizes Adult Erythropoiesis and Accelerates Its Stress Response. PloS One. 2011;6:e21192. doi: 10.1371/journal.pone.0021192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koulnis M, Porpiglia E, Hidalgo D, Socolovsky M. Erythropoiesis: from molecular pathways to system properties. Adv Exp Med Biol. 2014;844:37–58. doi: 10.1007/978-1-4939-2095-2_3. [DOI] [PubMed] [Google Scholar]
- 43.Lenox LE, Perry JM, Paulson RF. BMP4 and Madh5 regulate the erythroid response to acute anemia. Blood. 2005;105:2741–8. doi: 10.1182/blood-2004-02-0703. [DOI] [PubMed] [Google Scholar]
- 44.Perry JM, Harandi OF, Paulson RF. BMP4, SCF, and hypoxia cooperatively regulate the expansion of murine stress erythroid progenitors. Blood. 2007;109:4494–502. doi: 10.1182/blood-2006-04-016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paulson RF, Shi L, Wu DC. Stress erythropoiesis: new signals and new stress progenitor cells. Curr Opin Hematol. 2011;18:139–45. doi: 10.1097/MOH.0b013e32834521c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiang J, Wu DC, Chen Y, Paulson RF. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood. 2015;125:1803–12. doi: 10.1182/blood-2014-07-591453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harandi OF, Hedge S, Wu DC, McKeone D, Paulson RF. Murine erythroid short-term radioprotection requires a BMP4-dependent, self-renewing population of stress erythroid progenitors. J Clin Invest. 2010;120:4507–19. doi: 10.1172/JCI41291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perry JM, Harandi OF, Porayette P, Hegde S, Kannan AK, Paulson RF. Maintenance of the BMP4-dependent stress erythropoiesis pathway in the murine spleen requires hedgehog signaling. Blood. 2009;113:911–8. doi: 10.1182/blood-2008-03-147892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang L, Prak L, Rayon-Estrada V, Thiru P, Flygare J, Lim B, et al. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature. 2013;499:92–6. doi: 10.1038/nature12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, et al. PPAR-alpha and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature. 2015;522:474–7. doi: 10.1038/nature14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lau C-i, Outram SV, Ignacio Saldaña J, Furmanski AL, Dessens JT, Crompton T. Regulation of murine normal and stress-induced erythropoiesis by Desert Hedgehog. Blood. 2012;119:4741–51. doi: 10.1182/blood-2011-10-387266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oh P, Lobry C, Gao J, Tikhonova A, Loizou E, Manent J, et al. In vivo mapping of notch pathway activity in normal and stress hematopoiesis. Cell stem Cell. 2013;13:190–204. doi: 10.1016/j.stem.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeuner A, Francescangeli F, Signore M, Venneri MA, Pedini F, Felli N, et al. The Notch2-Jagged1 interaction mediates stem cell factor signaling in erythropoiesis. Cell Death Differ. 2011;18:371–80. doi: 10.1038/cdd.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jacobsen RN, Perkins AC, Levesque JP. Macrophages and regulation of erythropoiesis. Curr Opin Hematol. 2015;22:212–9. doi: 10.1097/MOH.0000000000000131. [DOI] [PubMed] [Google Scholar]
- 55.Rivella S. beta-thalassemias: paradigmatic diseases for scientific discoveries and development of innovative therapies. Haematologica. 2015;100:418–30. doi: 10.3324/haematol.2014.114827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood. 2008;112:470–8. doi: 10.1182/blood-2008-03-077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramos P, Casu C, Gardenghi S, Breda L, Crielaard BJ, Guy E, et al. Macrophages support pathological erythropoiesis in polycythemia vera and beta-thalassemia. Nat Med. 2013;19:437–45. doi: 10.1038/nm.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med. 2013;19:429–36. doi: 10.1038/nm.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rivella S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol. 2009;16:187–94. doi: 10.1097/MOH.0b013e32832990a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ginzburg Y, Rivella S. beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118:4321–30. doi: 10.1182/blood-2011-03-283614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coleman DH, Donohue DM, Finch CA, Motulsky AG, Reiff RH. Erythrokinetics in pernicious anemia. Blood. 1956;11:807–20. [PubMed] [Google Scholar]
- 62.Giblett ER, Coleman DH, Pirzio-Biroli G, Donohue DM, Motulsky AG, Finch CA. Erythrokinetics: Quantitative Measurements of Red Cell Production and Destruction in Normal Subjects and Patients with Anemia. Blood. 1956;11:291–309. [PubMed] [Google Scholar]
- 63.Huff RL, Hennessy TG, Austin RE, Garcia JF, Roberts BM, Lawrence JH. Plasma and red cell iron turnover in normal subjects and in patients having various hematopoietic disorders. J Clin Invest. 1950;29:1041–52. doi: 10.1172/JCI102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379:373–83. doi: 10.1016/S0140-6736(11)60283-3. [DOI] [PubMed] [Google Scholar]
- 65.Piel FB, Weatherall DJ. The alpha-thalassemias. N Engl J Med. 2014;371:1908–16. doi: 10.1056/NEJMra1404415. [DOI] [PubMed] [Google Scholar]
- 66.Chui DHK, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;101:791–800. doi: 10.1182/blood-2002-07-1975. [DOI] [PubMed] [Google Scholar]
- 67.Weatherall DJ, Clegg JB. The Thalassaemia Syndromes. Fourth. Oxford, UK: Blackwell Science Ltd; 2001. pp. 733–821. [Google Scholar]
- 68.Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S12–5. doi: 10.1016/S0268-960X(12)70005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haidar R, Musallam KM, Taher AT. Bone disease and skeletal complications in patients with beta thalassemia major. Bone. 2011;48:425–32. doi: 10.1016/j.bone.2010.10.173. [DOI] [PubMed] [Google Scholar]
- 70.Guimaraes JS, Cominal JG, Silva-Pinto AC, Olbina G, Ginzburg YZ, Nandi V, et al. Altered erythropoiesis and iron metabolism in carriers of thalassemia. Eur J Haematol. 2015;94:511–8. doi: 10.1111/ejh.12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Finch CA, Sturgeon P. Erythrokinetics in Cooley's anemia. Blood. 1957;12:64–73. [PubMed] [Google Scholar]
- 72.Finch CA, Deubelbeiss K, Cook JD, Eschbach JW, Harker LA, Funk DD, et al. Ferrokinetics in man. Medicine (Baltimore) 1970;49:17–53. doi: 10.1097/00005792-197001000-00002. [DOI] [PubMed] [Google Scholar]
- 73.Cazzola M, Finch CA. Iron balance in thalassemia. Prog Clin Biol Res. 1989;309:93–100. [PubMed] [Google Scholar]
- 74.Gardenghi S, Grady RW, Rivella S. Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in beta-thalassemia. Hematol Oncol Clin North Am. 2010;24:1089–107. doi: 10.1016/j.hoc.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pootrakul P, Sirankapracha P, Hemsorach S, Moungsub W, Kumbunlue R, Piangitjagum A, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia. Blood. 2000;96:2606–12. [PubMed] [Google Scholar]
- 76.Srichaikul T, Tipayasakda J, Atichartakarn V, Jootar S, Bovornbinyanun P. Ferrokinetic and erythrokinetic studies in alpha and beta thalassaemia. Clin Lab Haematol. 1984;6:133–40. doi: 10.1111/j.1365-2257.1984.tb00535.x. [DOI] [PubMed] [Google Scholar]
- 77.Yuan J, Angelucci E, Lucarelli G, Aljurf M, Snyder LM, Kiefer CR, et al. Accelerated programmed cell death (apoptosis) in erythroid precursors of patients with severe beta-thalassemia (Cooley's anemia) Blood. 1993;82:374–7. [PubMed] [Google Scholar]
- 78.Mathias LA, Fisher TC, Zeng L, Meiselman HJ, Weinberg KI, Hiti AL, et al. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol. 2000;28:1343–53. doi: 10.1016/s0301-472x(00)00555-5. [DOI] [PubMed] [Google Scholar]
- 79.Centis F, Tabellini L, Lucarelli G, Buffi O, Tonucci P, Persini B, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood. 2000;96:3624–9. [PubMed] [Google Scholar]
- 80.Libani IV, Guy EC, Melchiori L, Schiro R, Ramos P, Breda L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112:875–85. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Forster L, Cornwall S, Finlayson J, Ghassemifar R. Cell cycle, proliferation and apoptosis in erythroblasts cultured from patients with beta-thalassaemia major. Br J Haematol. 2016 doi: 10.1111/bjh.13875. [DOI] [PubMed] [Google Scholar]
- 82.Marinkovic D, Zhang X, Yalcin S, Luciano JP, Brugnara C, Huber T, et al. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest. 2007;117:2133–44. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang X, Camprecios G, Rimmele P, Liang R, Yalcin S, Mungamuri SK, et al. FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am J Hematol. 2014;89:954–63. doi: 10.1002/ajh.23786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Franco SS, De Falco L, Ghaffari S, Brugnara C, Sinclair DA, Matte A, et al. Resveratrol accelerates erythroid maturation by activation of FoxO3 and ameliorates anemia in beta-thalassemic mice. Haematologica. 2014;99:267–75. doi: 10.3324/haematol.2013.090076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Breda L, Rivella S. Modulators of erythropoiesis: emerging therapies for hemoglobinopathies and disorders of red cell production. Hematol Oncol Clin North Am. 2014;28:375–86. doi: 10.1016/j.hoc.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Voskou S, Aslan M, Fanis P, Phylactides M, Kleanthous M. Oxidative stress in β-thalassaemia and sickle cell disease. Redox Biol. 2015;6:226–39. doi: 10.1016/j.redox.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sadrzadeh SM, Graf E, Panter SS, Hallaway PE, Eaton JW, Hemoglobin A biologic fenton reagent. J Biol Chem. 1984;259:14354–6. [PubMed] [Google Scholar]
- 88.Amer J, Goldfarb A, Fibach E. Flow cytometric measurement of reactive oxygen species production by normal and thalassaemic red blood cells. Eur J Haematol. 2003;70:84–90. doi: 10.1034/j.1600-0609.2003.00011.x. [DOI] [PubMed] [Google Scholar]
- 89.Leecharoenkiat A, Wannatung T, Lithanatudom P, Svasti S, Fucharoen S, Chokchaichamnankit D, et al. Increased oxidative metabolism is associated with erythroid precursor expansion in beta0-thalassaemia/Hb E disease. Blood Cells Mol Dis. 2011;47:143–57. doi: 10.1016/j.bcmd.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 90.Ribeil JA, Arlet JB, Dussiot M, Moura IC, Courtois G, Hermine O. Ineffective erythropoiesis in beta -thalassemia. ScientificWorldJournal. 2013;2013:394295. doi: 10.1155/2013/394295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Franceschi L, Bertoldi M, Matte A, Santos Franco S, Pantaleo A, Ferru E, et al. Oxidative Stress and -Thalassemic Erythroid Cells behind the Molecular Defect. Oxid Med Cell Longev. 2013;2013:10. doi: 10.1155/2013/985210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mannu F, Arese P, Cappellini M, Fiorelli G, Cappadoro M, Giribaldi G, et al. Role of hemichrome binding to erythrocyte membrane in the generation of band-3 alterations in beta-thalassemia intermedia erythrocytes. Blood. 1995;86:2014–20. [PubMed] [Google Scholar]
- 93.Ferru E, Giger K, Pantaleo A, Campanella E, Grey J, Ritchie K, et al. Regulation of membrane-cytoskeletal interactions by tyrosine phosphorylation of erythrocyte band 3. Blood. 2011;117:5998–6006. doi: 10.1182/blood-2010-11-317024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pantaleo A, Giribaldi G, Mannu F, Arese P, Turrini F. Naturally occurring anti-band 3 antibodies and red blood cell removal under physiological and pathological conditions. Autoimmun Rev. 2008;7:457–62. doi: 10.1016/j.autrev.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 95.Yuan J, Kannan R, Shinar E, Rachmilewitz EA, Low PS. Isolation, characterization, and immunoprecipitation studies of immune complexes from membranes of beta-thalassemic erythrocytes. Blood. 1992;79:3007–13. [PubMed] [Google Scholar]
- 96.Lutz HU, Bussolino F, Flepp R, Fasler S, Stammler P, Kazatchkine MD, et al. Naturally occurring anti-band-3 antibodies and complement together mediate phagocytosis of oxidatively stressed human erythrocytes. Proc Natl Acad Sci U S A. 1987;84:7368–72. doi: 10.1073/pnas.84.21.7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cappellini MD, Tavazzi D, Duca L, Graziadei G, Mannu F, Turrini F, et al. Metabolic indicators of oxidative stress correlate with haemichrome attachment to membrane, band 3 aggregation and erythrophagocytosis in beta-thalassaemia intermedia. Br J Haematol. 1999;104:504–12. doi: 10.1046/j.1365-2141.1999.01217.x. [DOI] [PubMed] [Google Scholar]
- 98.Advani R, Sorenson S, Shinar E, Lande W, Rachmilewitz E, Schrier SL. Characterization and comparison of the red blood cell membrane damage in severe human alpha- and beta-thalassemia. Blood. 1992;79:1058–63. [PubMed] [Google Scholar]
- 99.Shinar E, Rachmilewitz EA, Lux SE. Differing erythrocyte membrane skeletal protein defects in alpha and beta thalassemia. J Clin Invest. 1989;83:404–10. doi: 10.1172/JCI113898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Casu C, Rivella S. Iron age: novel targets for iron overload. Hematology Am Soc Hematol Educ Program. 2014;2014:216–21. doi: 10.1182/asheducation-2014.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wijarnpreecha K, Kumfu S, Chattipakorn SC, Chattipakorn N. Cardiomyopathy associated with iron overload: how does iron enter myocytes and what are the implications for pharmacological therapy? Hemoglobin. 2015;39:9–17. doi: 10.3109/03630269.2014.987869. [DOI] [PubMed] [Google Scholar]
- 102.Pippard MJ, Callender ST, Warner GT, Weatherall DJ. Iron absorption and loading in beta-thalassaemia intermedia. Lancet. 1979;2:819–21. doi: 10.1016/s0140-6736(79)92175-5. [DOI] [PubMed] [Google Scholar]
- 103.Origa R, Galanello R, Ganz T, Giagu N, Maccioni L, Faa G, et al. Liver iron concentrations and urinary hepcidin in β-thalassemia. Haematologica. 2007;92:583–8. doi: 10.3324/haematol.10842. [DOI] [PubMed] [Google Scholar]
- 104.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 105.Ramos P, Guy E, Chen N, Proenca CC, Gardenghi S, Casu C, et al. Enhanced erythropoiesis in Hfe-KO mice indicates a role for Hfe in the modulation of erythroid iron homeostasis. Blood. 2011;117:1379–89. doi: 10.1182/blood-2010-09-307462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim A, Nemeth E. New insights into iron regulation and erythropoiesis. Curr Opin Hematol. 2015;22:199–205. doi: 10.1097/MOH.0000000000000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gao J, Chen J, De Domenico I, Koeller DM, Harding CO, Fleming RE, et al. Hepatocyte-targeted HFE and TFR2 control hepcidin expression in mice. Blood. 2010;115:3374–81. doi: 10.1182/blood-2009-09-245209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Corradini E, Rozier M, Meynard D, Odhiambo A, Lin HY, Feng Q, et al. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology. 2011;141:1907–14. doi: 10.1053/j.gastro.2011.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7:205–14. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Andriopoulos B, Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41:482–7. doi: 10.1038/ng.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41:478–81. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- 112.Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–91. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q–linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 114.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–9. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 115.Rechavi G, Rivella S. Regulation of iron absorption in hemoglobinopathies. Curr Mol Med. 2008;8:646–62. doi: 10.2174/156652408786241401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823:1434–43. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lakhal S, Schodel J, Townsend AR, Pugh CW, Ratcliffe PJ, Mole DR. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signaling and iron homeostasis. J Biol Chem. 2011;286:4090–7. doi: 10.1074/jbc.M110.173096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maurer E, Gutschow M, Stirnberg M. Matriptase-2 (TMPRSS6) is directly up-regulated by hypoxia inducible factor-1: identification of a hypoxia-responsive element in the TMPRSS6 promoter region. Biol Chem. 2012;393:535–40. doi: 10.1515/hsz-2011-0221. [DOI] [PubMed] [Google Scholar]
- 119.Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8:502–11. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Truksa J, Gelbart T, Peng H, Beutler E, Beutler B, Lee P. Suppression of the hepcidin-encoding gene Hamp permits iron overload in mice lacking both hemojuvelin and matriptase-2/TMPRSS6. Br J Haematol. 2009;147:571–81. doi: 10.1111/j.1365-2141.2009.07873.x. [DOI] [PubMed] [Google Scholar]
- 121.Andrews NC. Anemia of inflammation: the cytokine-hepcidin link. J Clin Invest. 2004;113:1251–3. doi: 10.1172/JCI21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–32. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–5. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–44. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kattamis A, Papassotiriou I, Palaiologou D, Apostolakou F, Galani A, Ladis V, et al. The effects of erythropoetic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica. 2006;91:809–12. [PubMed] [Google Scholar]
- 127.Piperno A, Galimberti S, Mariani R, Pelucchi S, Ravasi G, Lombardi C, et al. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: data from the HIGHCARE project. Blood. 2011;117:2953–9. doi: 10.1182/blood-2010-08-299859. [DOI] [PubMed] [Google Scholar]
- 128.Parrow NL, Gardenghi S, Ramos P, Casu C, Grady RW, Anderson ER, et al. Decreased hepcidin expression in murine beta-thalassemia is associated with suppression of Bmp/Smad signaling. Blood. 2012;119:3187–9. doi: 10.1182/blood-2012-01-405563. [DOI] [PubMed] [Google Scholar]
- 129.Pasricha S-R, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: a longitudinal study. Blood. 2013;122:124–33. doi: 10.1182/blood-2012-12-471441. [DOI] [PubMed] [Google Scholar]
- 130.Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109:5027–35. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Weizer-Stern O, Adamsky K, Amariglio N, Rachmilewitz E, Breda L, Rivella S, et al. mRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse models. Am J Hematol. 2006;81:479–83. doi: 10.1002/ajh.20549. [DOI] [PubMed] [Google Scholar]
- 132.De Franceschi L, Daraio F, Filippini A, Carturan S, Muchitsch EM, Roetto A, et al. Liver expression of hepcidin and other iron genes in two mouse models of beta-thalassemia. Haematologica. 2006;91:1336–42. [PubMed] [Google Scholar]
- 133.Kearney SL, Nemeth E, Neufeld EJ, Thapa D, Ganz T, Weinstein DA, et al. Urinary hepcidin in congenital chronic anemias. Pediatr Blood Cancer. 2007;48:57–63. doi: 10.1002/pbc.20616. [DOI] [PubMed] [Google Scholar]
- 134.Tanno T, Porayette P, Sripichai O, Noh SJ, Byrnes C, Bhupatiraju A, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114:181–6. doi: 10.1182/blood-2008-12-195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Finkenstedt A, Bianchi P, Theurl I, Vogel W, Witcher DR, Wroblewski VJ, et al. Regulation of iron metabolism through GDF15 and hepcidin in pyruvate kinase deficiency. Br J Haematol. 2009;144:789–93. doi: 10.1111/j.1365-2141.2008.07535.x. [DOI] [PubMed] [Google Scholar]
- 136.Tanno T, Bhanu NV, Oneal PA, Goh S-H, Staker P, Lee YT, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 137.Casanovas G, Vujic Spasic M, Casu C, Rivella S, Strelau J, Unsicker K, et al. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica. 2013;98:444–7. doi: 10.3324/haematol.2012.069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Theurl I, Finkenstedt A, Schroll A, Nairz M, Sonnweber T, Bellmann-Weiler R, et al. Growth differentiation factor 15 in anaemia of chronic disease, iron deficiency anaemia and mixed type anaemia. Br J Haematol. 2010;148:449–55. doi: 10.1111/j.1365-2141.2009.07961.x. [DOI] [PubMed] [Google Scholar]
- 139.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678–84. doi: 10.1038/ng.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Frazer DM, Wilkins SJ, Darshan D, Badrick AC, McLaren GD, Anderson GJ. Stimulated erythropoiesis with secondary iron loading leads to a decrease in hepcidin despite an increase in bone morphogenetic protein 6 expression. Br J Haematol. 2012;157:615–26. doi: 10.1111/j.1365-2141.2012.09104.x. [DOI] [PubMed] [Google Scholar]
- 141.Kautz L, Jung G, Du X, Gabayan V, Chapman J, Nasoff M, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood. 2015 doi: 10.1182/blood-2015-07-658419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jenkins ZA, Hagar W, Bowlus CL, Johansson HE, Harmatz P, Vichinsky EP, et al. Iron homeostasis during transfusional iron overload in beta-thalassemia and sickle cell disease: changes in iron regulatory protein, hepcidin, and ferritin expression. Pediatr Hematol Oncol. 2007;24:237–43. doi: 10.1080/08880010701360700. [DOI] [PubMed] [Google Scholar]
- 143.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet. 1999;21:396–9. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- 144.Coulon S, Dussiot M, Grapton D, Maciel TT, Wang PH, Callens C, et al. Polymeric IgA1 controls erythroblast proliferation and accelerates erythropoiesis recovery in anemia. Nat Med. 2011;17:1456–65. doi: 10.1038/nm.2462. [DOI] [PubMed] [Google Scholar]
- 145.Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, et al. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med. 2001;194:417–25. doi: 10.1084/jem.194.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li H, Blanc L, Choesang T, Chen H, Feola M, Bao W, et al. Changes in TfR1 Expression and Trafficking Correlate with Erythroid Effectiveness in β-Thalassemic Mice. Blood. 2014;124:51. [Google Scholar]
- 147.Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, Watson N, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci U S A. 1998;95:1472–7. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Giannetti AM, Bjorkman PJ. HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. J Biol Chem. 2004;279:25866–75. doi: 10.1074/jbc.M401467200. [DOI] [PubMed] [Google Scholar]
- 149.Parkkila S, Waheed A, Britton RS, Bacon BR, Zhou XY, Tomatsu S, et al. Association of the transferrin receptor in human placenta with HFE, the protein defective in hereditary hemochromatosis. Proc Natl Acad Sci U S A. 1997;94:13198–202. doi: 10.1073/pnas.94.24.13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bennett MJ, Lebron JA, Bjorkman PJ. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature. 2000;403:46–53. doi: 10.1038/47417. [DOI] [PubMed] [Google Scholar]
- 151.Waheed A, Parkkila S, Saarnio J, Fleming RE, Zhou XY, Tomatsu S, et al. Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum. Proc Natl Acad Sci U S A. 1999;96:1579–84. doi: 10.1073/pnas.96.4.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nai A, Pellegrino RM, Rausa M, Pagani A, Boero M, Silvestri L, et al. The erythroid function of transferrin receptor 2 revealed by Tmprss6 inactivation in different models of transferrin receptor 2 knockout mice. Haematologica. 2014;99:1016–21. doi: 10.3324/haematol.2013.103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105:1803–6. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 154.Wallace DF, Summerville L, Lusby PE, Subramaniam VN. First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut. 2005;54:980–6. doi: 10.1136/gut.2004.062018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Forejtnikova H, Vieillevoye M, Zermati Y, Lambert M, Pellegrino RM, Guihard S, et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood. 2010;116:5357–67. doi: 10.1182/blood-2010-04-281360. [DOI] [PubMed] [Google Scholar]
- 156.Lee P, Hsu MH, Welser-Alves J, Peng H. Severe microcytic anemia but increased erythropoiesis in mice lacking Hfe or Tfr2 and Tmprss6. Blood Cells Mol Dis. 2012;48:173–8. doi: 10.1016/j.bcmd.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wallace DF, Secondes ES, Rishi G, Ostini L, McDonald CJ, Lane SW, et al. A critical role for murine transferrin receptor 2 in erythropoiesis during iron restriction. Br J Haematol. 2015;168:891–901. doi: 10.1111/bjh.13225. [DOI] [PubMed] [Google Scholar]
- 158.Nai A, Lidonnici MR, Rausa M, Mandelli G, Pagani A, Silvestri L, et al. The second transferrin receptor regulates red blood cell production in mice. Blood. 2015;125:1170–9. doi: 10.1182/blood-2014-08-596254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 160.Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol. 2003;39:710–5. doi: 10.1016/s0168-8278(03)00408-2. [DOI] [PubMed] [Google Scholar]
- 161.Mullner EW, Kuhn LC. A stem-loop in the 3' untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cell. 1988;53:815–25. doi: 10.1016/0092-8674(88)90098-0. [DOI] [PubMed] [Google Scholar]
- 162.Hentze MW, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, et al. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. 1987;238:1570–3. doi: 10.1126/science.3685996. [DOI] [PubMed] [Google Scholar]
- 163.Wilkinson N, Pantopoulos K. The IRP/IRE system in vivo: insights from mouse models. Front Pharmacol. 2014;5:176. doi: 10.3389/fphar.2014.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dandekar T, Stripecke R, Gray NK, Goossen B, Constable A, Johansson HE, et al. Identification of a novel iron-responsive element in murine and human erythroid delta-aminolevulinic acid synthase mRNA. EMBO J. 1991;10:1903–9. doi: 10.1002/j.1460-2075.1991.tb07716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cox TC, Bawden MJ, Martin A, May BK. Human erythroid 5-aminolevulinate synthase: promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J. 1991;10:1891–902. doi: 10.1002/j.1460-2075.1991.tb07715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Theil EC, McKenzie RA, Sierzputowska-Gracz H. Structure and function of IREs, the noncoding mRNA sequences regulating synthesis of ferritin, transferrin receptor and (erythroid) 5-aminolevulinate synthase. Adv Exp Med Biol. 1994;356:111–8. doi: 10.1007/978-1-4615-2554-7_12. [DOI] [PubMed] [Google Scholar]
- 167.Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5' untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci U S A. 1988;85:2171–5. doi: 10.1073/pnas.85.7.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gray NK, Hentze MW. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. 1994;13:3882–91. doi: 10.1002/j.1460-2075.1994.tb06699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, et al. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509:309–16. doi: 10.1016/s0014-5793(01)03189-1. [DOI] [PubMed] [Google Scholar]
- 170.Binder R, Horowitz JA, Basilion JP, Koeller DM, Klausner RD, Harford JB. Evidence that the pathway of transferrin receptor mRNA degradation involves an endonucleolytic cleavage within the 3' UTR and does not involve poly(A) tail shortening. EMBO J. 1994;13:1969–80. doi: 10.1002/j.1460-2075.1994.tb06466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Anderson SA, Nizzi CP, Chang YI, Deck KM, Schmidt PJ, Galy B, et al. The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013;17:282–90. doi: 10.1016/j.cmet.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ghosh MC, Zhang DL, Jeong SY, Kovtunovych G, Ollivierre-Wilson H, Noguchi A, et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2alpha. Cell Metab. 2013;17:271–81. doi: 10.1016/j.cmet.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wilkinson N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation. Blood. 2013;122:1658–68. doi: 10.1182/blood-2013-03-492454. [DOI] [PubMed] [Google Scholar]
- 174.Cooperman SS, Meyron-Holtz EG, Olivierre-Wilson H, Ghosh MC, McConnell JP, Rouault TA. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood. 2005;106:1084–91. doi: 10.1182/blood-2004-12-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, et al. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2) Blood. 2005;106:2580–9. doi: 10.1182/blood-2005-04-1365. [DOI] [PubMed] [Google Scholar]
- 176.Hira S, Tomita T, Matsui T, Igarashi K, Ikeda-Saito M. Bach1, a heme-dependent transcription factor, reveals presence of multiple heme binding sites with distinct coordination structure. IUBMB life. 2007;59:542–51. doi: 10.1080/15216540701225941. [DOI] [PubMed] [Google Scholar]
- 177.Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica. 2010;95:1261–8. doi: 10.3324/haematol.2009.020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001;20:2835–43. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hintze KJ, Katoh Y, Igarashi K, Theil EC. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, beta-globin, and NADP(H) quinone (oxido) reductase1. J Biol Chem. 2007;282:34365–71. doi: 10.1074/jbc.M700254200. [DOI] [PubMed] [Google Scholar]
- 180.Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun. 2004;324:77–85. doi: 10.1016/j.bbrc.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 181.Ishikawa H, Kato M, Hori H, Ishimori K, Kirisako T, Tokunaga F, et al. Involvement of Heme Regulatory Motif in Heme-Mediated Ubiquitination and Degradation of IRP2. Mol. Cell. 2005;19:171–81. doi: 10.1016/j.molcel.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 182.Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, et al. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. 2004;279:5480–7. doi: 10.1074/jbc.M302733200. [DOI] [PubMed] [Google Scholar]
- 183.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A. 2004;101:1461–6. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ney PA, Sorrentino BP, Lowrey CH, Nienhuis AW. Inducibility of the HS II enhancer depends on binding of an erythroid specific nuclear protein. Nucleic Acids Res. 1990;18:6011–7. doi: 10.1093/nar/18.20.6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Igarashi K, Hoshino H, Muto A, Suwabe N, Nishikawa S, Nakauchi H, et al. Multivalent DNA binding complex generated by small Maf and Bach1 as a possible biochemical basis for beta-globin locus control region complex. J Biol Chem. 1998;273:11783–90. doi: 10.1074/jbc.273.19.11783. [DOI] [PubMed] [Google Scholar]
- 186.Brand M, Ranish JA, Kummer NT, Hamilton J, Igarashi K, Francastel C, et al. Dynamic changes in transcription factor complexes during erythroid differentiation revealed by quantitative proteomics. Nat Struct Mol Biol. 2004;11:73–80. doi: 10.1038/nsmb713. [DOI] [PubMed] [Google Scholar]
- 187.Andrews NC, Erdjument-Bromage H, Davidson MB, Tempst P, Orkin SH. Erythroid transcription factor NF-E2 is a haematopoietic-specific basic-leucine zipper protein. Nature. 1993;362:722–8. doi: 10.1038/362722a0. [DOI] [PubMed] [Google Scholar]
- 188.Sawado T, Igarashi K, Groudine M. Activation of beta-major globin gene transcription is associated with recruitment of NF-E2 to the beta-globin LCR and gene promoter. Proc Natl Acad Sci U S A. 2001;98:10226–31. doi: 10.1073/pnas.181344198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chen JJ. Translational control by heme-regulated eIF2alpha kinase during erythropoiesis. Curr Opin Hematol. 2014;21:172–8. doi: 10.1097/MOH.0000000000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Crosby JS, Lee K, London IM, Chen JJ. Erythroid expression of the heme-regulated eIF-2 alpha kinase. Mol Cell Biol. 1994;14:3906–14. doi: 10.1128/mcb.14.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109:2693–9. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–18. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Han AP, Fleming MD, Chen JJ. Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia. J Clin Invest. 2005;115:1562–70. doi: 10.1172/JCI24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ferreira R, Ohneda K, Yamamoto M, Philipsen S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol Cell Biol. 2005;25:1215–27. doi: 10.1128/MCB.25.4.1215-1227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–67. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 196.Sumera A, Radhakrishnan A, Baba AA, George E. Review: Beta-thalassemia and molecular chaperones. Blood Cells Mol Dis. 2015;54:348–52. doi: 10.1016/j.bcmd.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 197.Arlet JB, Ribeil JA, Guillem F, Negre O, Hazoume A, Marcion G, et al. HSP70 sequestration by free alpha-globin promotes ineffective erythropoiesis in beta-thalassaemia. Nature. 2014;514:242–6. doi: 10.1038/nature13614. [DOI] [PubMed] [Google Scholar]
- 198.Khandros E, Mollan TL, Yu X, Wang X, Yao Y, D'Souza J, et al. Insights into hemoglobin assembly through in vivo mutagenesis of alpha-hemoglobin stabilizing protein. J Biol Chem. 2012;287:11325–37. doi: 10.1074/jbc.M111.313205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Favero ME, Costa FF. Alpha-Hemoglobin-Stabilizing Protein: An Erythroid Molecular Chaperone. Biochem Res Int l. 2011;2011:7. doi: 10.1155/2011/373859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.dos Santos CO, Dore LC, Valentine E, Shelat SG, Hardison RC, Ghosh M, et al. An iron responsive element-like stem-loop regulates alpha-hemoglobin-stabilizing protein mRNA. J Biol Chem. 2008;283:26956–64. doi: 10.1074/jbc.M802421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Weiss MJ, Zhou S, Feng L, Gell DA, Mackay JP, Shi Y, et al. Role of Alpha Hemoglobin-Stabilizing Protein in Normal Erythropoiesis and β-Thalassemia. Ann. N. Y. Acad. Sci. 2005;1054:103–17. doi: 10.1196/annals.1345.013. [DOI] [PubMed] [Google Scholar]
- 202.Kong Y, Zhou S, Kihm AJ, Katein AM, Yu X, Gell DA, et al. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J Clin Invest. 2004;114:1457–66. doi: 10.1172/JCI21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nasimuzzaman M, Khandros E, Wang X, Kong Y, Zhao H, Weiss D, et al. Analysis of alpha hemoglobin stabilizing protein overexpression in murine beta-thalassemia. Am J Hematol. 2010;85:820–2. doi: 10.1002/ajh.21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Viprakasit V, Tanphaichitr VS, Chinchang W, Sangkla P, Weiss MJ, Higgs DR. Evaluation of alpha hemoglobin stabilizing protein (AHSP) as a genetic modifier in patients with beta thalassemia. Blood. 2004;103:3296–9. doi: 10.1182/blood-2003-11-3957. [DOI] [PubMed] [Google Scholar]
- 205.Ranjbaran R, Okhovat MA, Mobarhanfard A, Aboualizadeh F, Abbasi M, Moezzi L, et al. Relationship between AHSP gene expression, beta/alpha globin mRNA ratio, and clinical severity of the beta-thalassemia patients. Ann Clin Lab Sci. 2014;44:189–93. [PubMed] [Google Scholar]
- 206.Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121:4880–8. doi: 10.1172/JCI57693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120:3829–36. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gardenghi S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120:4466–77. doi: 10.1172/JCI41717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Goldberg A, Nemeth E, Ganz T, Gardenghi S, MacDonald B, Rivella S. Treatment With Minihepcidin Peptide Improves Anemia and Iron Overload In a Mouse Model Of Thalassemia Intermedia. Blood. 2013;122:431. [Google Scholar]
- 210.Casu C, Oikonomidau R, Shah Y, Nemeth E, Ganz T, MacDonald B, Rivella S. Concurrent Treatment with Minhepcidin and Deferiprone Improves Anemia and Enhances Reduction of Spleen Iron in a Mouse Model of Non-Transfusion Dependent Thalassemia. Presented at 56th ASH Annual Meeting and Exposition; San Francisco, CA. December 9, 2014. [Google Scholar]
- 211.Nai A, Pagani A, Mandelli G, Lidonnici MR, Silvestri L, Ferrari G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119:5021–9. doi: 10.1182/blood-2012-01-401885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123:1531–41. doi: 10.1172/JCI66969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schmidt PJ, Toudjarska I, Sendamarai AK, Racie T, Milstein S, Bettencourt BR, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121:1200–8. doi: 10.1182/blood-2012-09-453977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Casu C, Aghajan M, Oikonomidou PR, Guo S, Monia BP, Rivella S. Combination of Tmprss6-ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica. 2015 doi: 10.3324/haematol.2015.133348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Schmidt PJ, Racie T, Westerman M, Fitzgerald K, Butler JS, Fleming MD. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of beta-thalassemia intermedia. Am J Hematol. 2015;90:310–3. doi: 10.1002/ajh.23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Li H, Rybicki AC, Suzuka SM, von Bonsdorff L, Breuer W, Hall CB, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16:177–82. doi: 10.1038/nm.2073. [DOI] [PubMed] [Google Scholar]
- 217.Gelderman MP, Baek JH, Yalamanoglu A, Puglia M, Vallelian F, Burla B, et al. Reversal of hemochromatosis by apotransferrin in non-transfused and transfused Hbbth3/+ (heterozygous B1/B2 globin gene deletion) mice. Haematologica. 2015;100:611–22. doi: 10.3324/haematol.2014.117325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.De Domenico I, Lo E, Ward DM, Kaplan J. Hepcidin-induced internalization of ferroportin requires binding and cooperative interaction with Jak2. Proc Natl Acad Sci U S A. 2009;106:3800–5. doi: 10.1073/pnas.0900453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kerenyi MA, Grebien F, Gehart H, Schifrer M, Artaker M, Kovacic B, et al. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood. 2008;112:3878–88. doi: 10.1182/blood-2008-02-138339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhu BM, McLaughlin SK, Na R, Liu J, Cui Y, Martin C, et al. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood. 2008;112:2071–80. doi: 10.1182/blood-2007-12-127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Santos FP, Verstovsek S. JAK2 inhibitors for myelofibrosis: why are they effective in patients with and without JAK2V617F mutation? Anticancer Agents Med Chem. 2012;12:1098–109. doi: 10.2174/187152012803529727. [DOI] [PubMed] [Google Scholar]
- 222.Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen PJ, Rumi E, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120:513–20. doi: 10.1002/cncr.28441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Suragani RN, Cawley SM, Li R, Wallner S, Alexander MJ, Mulivor AW, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine beta-thalassemia. Blood. 2014;123:3864–72. doi: 10.1182/blood-2013-06-511238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. 2008;112:3434–43. doi: 10.1182/blood-2008-02-139824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Dussiot M, Maciel TT, Fricot A, Chartier C, Negre O, Veiga J, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat Med. 2014;20:398–407. doi: 10.1038/nm.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20:408–14. doi: 10.1038/nm.3512. [DOI] [PubMed] [Google Scholar]
- 228.Carrancio S, Markovics J, Wong P, Leisten J, Castiglioni P, Groza MC, et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br J Haematol. 2014;165:870–82. doi: 10.1111/bjh.12838. [DOI] [PMC free article] [PubMed] [Google Scholar]