

Abstract
Background
The economic value of ginseng in the global medicinal plant trade is estimated to be in excess of US$2.1 billion. At the same time, the evolutionary placement of ginseng (Panax ginseng) and the complex evolutionary history of the genus is poorly understood despite several molecular phylogenetic studies. In this study, we use a full plastome phylogenomic framework to resolve relationships in Panax and to identify molecular markers for species discrimination.
Results
We used high-throughput sequencing of MBD2-Fc fractionated Panax DNA to supplement publicly available plastid genomes to create a phylogeny based on fully assembled and annotated plastid genomes from 60 accessions of 8 species. The plastome phylogeny based on a 163 kbp matrix resolves the sister relationship of Panax ginseng with P. quinquefolius. The closely related species P. vietnamensis is supported as sister of P. japonicus. The plastome matrix also shows that the markers trnC-rps16, trnS-trnG, and trnE-trnM could be used for unambiguous molecular identification of all the represented species in the genus.
Conclusions
MBD2 depletion reduces the cost of plastome sequencing, which makes it a cost-effective alternative to Sanger sequencing based DNA barcoding for molecular identification. The plastome phylogeny provides a robust framework that can be used to study the evolution of morphological characters and biosynthesis pathways of ginsengosides for phylogenetic bioprospecting. Molecular identification of ginseng species is essential for authenticating ginseng in international trade and it provides an incentive for manufacturers to create authentic products with verified ingredients.
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1160-y) contains supplementary material, which is available to authorized users.
Keywords: Barcoding, Genome, Ginseng, Marker, mPTP, NGS, Panax, Phylogenomics, Plastid
Background
Ginseng has been used in traditional medicine in China for thousands of years [1], but it was not until early 18th century that long-term, intense harvest nearly extirpated Panax ginseng C.A.Mey. from the wild [2]. Demand for ginseng roots in the 18th century also fuelled a subsequent boom in wild-harvesting American ginseng (P. quinquefolius L.) that decimated wild populations in North America [3]. Today wild P. ginseng occurs in only a few localities in Russia and China, with the largest distribution in the southern part of the Sikhote-Alin mountain range [4]. P. ginseng is Red-Listed in Russia, and roots and parts thereof from Russian populations are CITES Appendix II/NC listed [5]. Many other Asian ginseng species are also endangered but preliminary data is only available for wild-harvesting and conservation of P. assamicus R.N.Banerjee (synonym of P. bipinnatifidus var. angustifolius (Burkill) J.Wen) [6], P. japonicus (T.Nees) C.A.Mey. [7] and P. pseudoginseng Wall. [8, 9].
Elucidating the evolutionary relationships among species in the genus is essential to understand evolution of this Holarctic disjunct genus, but also evolution of derived secondary metabolite pathways. In addition, a phylogenetic framework can be used to develop accurate molecular identification of Panax, and enable identification of ginseng material in trade, both crude drugs and derived products, which is essential for conservation efforts and protection of the remaining wild populations of P. ginseng and related Panax species, since all may be under the pressure of illegal harvesting and international trade [10]. Furthermore, identification of Panax species and authentication of derived products is of great commercial importance as authentic ginseng is costly and the incentive for substitution is significant.
The phylogeny of Panax has been studied using several molecular markers, but lack of variation in the most commonly used markers highlight an important limitation of the method. The nuclear ribosomal ITS yields insufficient resolution for accurate species assignment [11] and even using multiple markers in combination, matK, trnD, psbK-psbI, rbcL and ycf1 have a limited accuracy in identification of Panax species [12, 13]. The mutation rate of the studied markers does not allow a fine scale resolution, and is insufficient for identification of all Panax species and cultivars. The question of what species are in trade remains a mystery. Aside from phylogenetic approaches, a multitude of molecular and chemical analysis approaches have been developed and applied, including Arbitrarily Primed Polymerase Chain Reaction (AP-PCR) [14], PCR-Restriction Fragment Length Polymorphism (PCR-RFLP) and Mutant Allele Specific Amplification (MASA) [15], Random Amplified Polymorphic DNA (RAPD) and High Performance Liquid Chromatography [16], Fourier Transformed-Infrared Spectroscopy (FT-IR) [17], Two-Dimensional Correlation Infrared Spectroscopy (2D-IR) [17], Multiplex Amplification Refractory Mutation System-PCR (MARMS) [18, 19], Microchip Electrophoresis Laser-Induced Fluorescence Detection [20], and microsatellite markers [21]. Most methods have focused on either positive identification of P. ginseng, or distinguishing P. ginseng and P. quinquefolius L., but most have limited resolution in detecting infraspecific or interspecific substitution, especially with poorly known congeneric species.
Suitability of molecular markers is often measured in interspecific distance using distance methods to estimate the number of variable sites or pairwise distances between sequences. Most current methods are based on the Refined Single Linkage (RESL) algorithm implemented in Barcode of Life Database (BOLD) [22] or clustering on distance matrices (Crop [23], OBITools [24], UCLUST [25], and Vsearch [26]) and ideally set a threshold to distinguish between intraspecific and interspecific variation, sometimes referred to as the “barcoding gap” [27]. Several programs and software packages determine and visualize barcoding gaps, including Automatic Barcode Gap Discovery (ABGD) [28] and Spider [29]. These distance-based methods are fast and suitable for large datasets, but they are not always biologically meaningful, especially when the species groups have complex evolutionary histories, including incomplete lineage sorting, and hybridization [30, 31]. As an alternative, tree-based methods offer several advantages compared to distance based methods. First, these methods do not work with a specified threshold (% variation, no barcoding gap) and second, these accommodate evolutionary processes, making them particularly suitable for species delimitation and identification. Several studies have shown that these methods are also more sensitive and more powerful for accurate species discrimination [32]. Recently proposed methods include the Generalized Mixed Yule Coalescent (GMYC) [33], Bayesian species identification using the multispecies coalescent (MSC) model [34], and Poisson Tree Processes (PTP, mPTP) [25, 32]. Despite constant methodological improvements, there is no silver bullet for species delimitation and concerns have been raised that species delimitation approaches are sensitive to the structure of the data tested [35]. Species delimitation methods assess speciation and coalescent processes but also the data structure of the selected markers [35]. From a marker development perspective, tree based methods provide an opportunity to increase the quality of the selection process of the barcoding markers. Here we use the mPTP approach [32] to test if speciation processes are supported by the barcoding markers and accordingly choose the best markers for delimitation of Panax species. mPTP method has the advantage of being computationally efficient, while at the same time accommodating better to population-specific and sampling characteristics than PTP and GYMC [32].
Evolution and phylogenetics of Panax
Previous phylogenetic studies of the Araliaceae family have identified four monophyletic groups (the Asian Palmate group, the Polyscias-Pseudopanax group, the Aralia-Panax group, and the greater Raukaua group) [36, 37]. However deep nodes are not well-supported to date [36, 37], and a broad sampling within Aralioideae is necessary to obtain an accurate placement of the Aralia-Panax group. Monophyly of the genus Panax (Araliaceae) is well supported by morphological synapomorphies, such as palmately compound leaves, a whorled leaf arrangement, a single terminal inflorescence, valvate petals in floral buds, and a bi- or tricarpellate ovary, as well as by several molecular phylogenies [12, 38]. A number of species have emerged from the complex of subspecies of P. pseudoginseng in the 1970s, and taxonomic studies have resulted in the description of various new species [38–40]. Currently 13 species of ginseng are recognized with broad consensus [38, 41], but publication of new taxa at species, subspecies and variety level are common [42, 43].
Previous molecular phylogenies support P. stipuleatus H.T.Tsai & K.M.Feng and P. trifolius L. as the sister group of all other ginseng species. Nevertheless the placement of several other species still remains unclear (e.g., P. binnatifidus, P. ginseng, P. japonicus, P. quiquefolius, P. vietnamensis Ha & Grushv., P. wangianus S.C.Sun, P. zingiberensis C.Y.Wu & Feng). Species delimitation within the genus is problematic due to species of tetraploid origin (e.g., P. bipinnatifidus, P. ginseng, P. japonicus, and P. quinquefolius [44]), recent speciation events [12], high intraspecific morphological variation (e.g., P. pseudoginseng Wall.) and ancient genome duplication events [41, 45].
Phylogenetic studies have explored evolutionary relationships in Araliaceae with standard phylogenetic markers, such as the nuclear ribosomal ITS [11, 36, 38, 41, 44–46] and several plastid markers [11–13, 41]. More recently, an attempt with seven nuclear genes was tested with moderate results (PGN7, W8, W28, Z7, Z14, Z15, Z16) [12]. The topologies obtained were conflicting and non-consistent with previous evolutionary inferences of the genus, which is likely a result of multiple copies of nuclear genes and ancient whole genome duplication events [47]. Whole genome data have also been used to design microsatellites for species identification, but these have found limited application [21, 48–52]. Extensive population genetic studies have been done only on P. quinquefolius [53–59] and P. ginseng [60, 61] due to their major economic importance.
Developments in high throughput sequencing have provided new approaches for genome sequencing: increasing outputs and decreasing costs have made this a cost-effective alternative to Sanger-based amplicon sequencing [62, 63]. Full plastid genome sequencing, i.e. plastome sequencing, has been proposed as an augmented approach to DNA barcoding [64, 65], and is a straightforward method that recovers all standard barcodes plus the full plastome. The limited costs of shotgun sequencing and the availability of a number of Araliaceae reference plastomes facilitates the study of relationships in the family. Plastome phylogenies have helped disentangle evolutionary relationship in a number of plant clades [66], including Poales [67], magnoliids [68], Pinus [69], Amborella [67], Equisetum [70], and Camellia [71]. Single-copy nuclear genes have corroborated the robustness of plastome phylogenies [72–75], however plastome phylogenies reflect only maternal inheritance, and as such will not always be representative species trees. An advantage of plastome data for phylogenetic studies is the low mutation rate of plastid sequences, the abundance of plastid DNA in most material [76] and the low cost of generating whole plastid genomes with high throughput sequencing.
In total DNA, the proportion of plastid DNA typically constitutes only ~ 0.01–13% depending on the size of the nuclear genome, tissue and season [77–79]. Shot-gun sequencing studies might have relatively low efficacy in plastid genome recovery due to the small proportion of plastid DNA in the total DNA. Ginseng species have a large genome size of 5–10 Gb [80, 81], and one can expect a proportion of plastid DNA of 1–5% in the gDNA [79], which makes shotgun sequencing relatively ineffective in obtaining full plastome data. Several methods have been developed for enriching plastid content prior to sequencing (for a discussion see Du et al. [82]. We apply a new plastid enrichment method to improve the shotgun sequencing efficacy, that utilizes the low methylation of the plastid genome compared to the nuclear genome [83]. The method uses the methyl-CpG-binding domain (MBD2) to partition fragments of genomic DNA into a methylation-poor fraction (e.g. enriched for plastid) and a methylation-rich fraction (e.g. depleted in plastid) [84]. This method has the advantage that it uses a small quantity of dry material (below 40 mg) and is suitable for non-model organisms.
This study has four main aims: (1) to construct a well-supported phylogeny of the genus Panax, while testing if the full plastome data yield sufficient variation to support and resolve phylogenetic relations in Panax, and specifically the position of the economically important P. ginseng; (2) to test if MBD2 can be used to fractionate Panax DNA into eukaryotic nuclear (methyl-CpG-rich) vs. organellar (methyl-CpG-poor) elements, and subsequently sequence the MBD2 depleted DNA to optimize plastome read yield; (3) to determine if the plastid genome can be used for molecular identification of traded species; and 4) to make a case for the need of a resolved plastome phylogeny to be used to design short markers for Panax species identification from processed ginseng products.
Methods
Sampling
Fresh material of three species, P. bipinnatifidus, P. stipuleanatus, and P. vietnamensis (2), was sampled in Vietnam (Table 1, Additional file 1: Table S1) and 57 selected Araliaceae plastid genomes from across the Araliaceae family were downloaded from open data repositories (Additional file 2: Table S2) [12, 85–97]. Plant samples were collected in public land and no specific permits were required. At least two individuals or species were selected per genus, but for Panax we used 38 plastid genomes from eight species. Hydrocotyle verticillata was selected as outgroup based on its early divergence within Araliaceae [44].
Table 1.
Summary information for the four assembled plastome genomes
Library preparation and sequencing
We extracted total DNA from two individuals of those sample collected in Vietnam, using a Qiagen DNeasy plant extraction kit with the provided protocol. The total DNA was quantified prior to library preparation to assess DNA quantity, fragmentation and fragment length distribution on a Fragment Analyzer (Advanced Analytical Technologies, Inc., Ankeny, USA) using the High Sensitivity genomic DNA Reagent Kit (50–40,000 bp) (Additional file 3: Figure S1). We selected one individual per extracted sample based on the yield and fragment size of the total DNA. The selected samples had average fragment sizes in excess of 10 kbp and a minimum DNA concentration of 4.77 ng/μl (Additional file 3: Figure S1).
We used a NEBNext Microbiome DNA Enrichment Kit (New England Biolabs, Ipswich, Massachusetts, USA) that uses IgG1 fused to the human methyl-CpG-binding domain (together “MBD2-Fc”) to pull down a methyl-CpG-enriched fraction from a bead-associated element, leaving a methyl-depleted fraction in the supernatant. About 400 ng template DNA extract was used per sample and the manufacturers recommendations were respected with the following exceptions. The non-methylated DNA fractions were purified using 0.9X AMpure XP beads (Beckman Coulter, Brea, CA, USA) and eluted in 40 μl 1X TE buffer. To capture the methylated DNA, we followed the manufacturer’s protocol. Quality control in terms of size, purity and molar concentration (nmol/l) of both the methylated and the non-methylated fractions were measured using a Fragment Analyzer (Advanced Analytical Technologies Inc., USA) with a DNF-488-33 HS dsDNA Reagent Kit. The DNA was subsequently sheared to ~ 400 bp fragments using a M220 Focused Ultrasonicator (Covaris Inc., Woburn, MA, USA) using microTUBES-50 (Covaris Inc.). We used the NEBNext Fast DNA Library Prep Set for Ion Torrent (NEB) for end repair and adapter ligation of the sheared DNA. The samples were indexed using the IonXpress Barcode Adapter kit (ThermoFischer, Waltham, MA, USA). For each of the four samples both fractions, methyl-CpG-enriched and methyl-CpG-depleted, were indexed and sequenced. After adapter ligation, the four methyl-CpG-enriched fractions were pooled in one library and the four methyl-CpG-depleted fractions were pooled in another library. The adapter-ligated libraries were size selected (450–540 bp) using a BluePippin (Sage Science, Beverly, MA, USA), and subsequently amplified using the NEBNext Fast DNA Library Prep Set for Ion Torrent kit using 12 PCR cycles. The amplified libraries were purified twice using 0.7X AMpure XP beads. The purified amplified libraries were loaded on the sequencing chips using an Ion Chef (LT) and sequenced on an Ion Torrent Personal Genome Machine (LT) using Ion 318 v2 chips (LT) and the Ion PGM Sequencing 400 kit (LT).
Bioinformatic analyses and assembly
Sequencing reads were demultiplexed into FASTQ files using Flexbar version 3.0.3. Trimmomatic version 0.36 [98] was used for adapter trimming and quality filtering of reads using a sliding window of 15 bp and an average Phred threshold of 20. Low-end quality bases below a Phred score of 20 were removed, and only reads longer than 100 bp were retained. MITOBim version 1.7 [99] was used for assembly of the single-end Ion Torrent reads using iterative mapping with in silico baiting using the following reference plastomes, P. vietnamensis (KP036470) and P. stipuleanatus (KX247147).
Inverted repeats and ambiguous portions of the assembly were resequenced using Sanger sequencing. Specific primers were designed and used for DNA amplification of interest regions. PCR was performed on a Mastercycler® Pro (Eppendorf, USA) in a 20 μl final volume containing 2.5 μM of each primer, 1 mM of each dNTP, 10X DreamTaq Buffer, 0.75 U DreamTaq DNA polymerase (ThermoFisher Scientific, USA) and deionized water. The PCR cycling conditions included a sample denaturation step at 94 °C for 2 min followed by 35 cycles of denaturation at 94 °C for 30 s, primer annealing at 50–55 °C for 30 s and primer extension at 72 °C for 1 min, followed by a final extension step at 72 °C for 5 min. PCR products were then purified using GeneJET PCR Purification Kit (ThermoFisher Scientific, USA). Sanger sequencing was performed on an ABI 3500 Genetic Analyzer system using BigDye Terminator v3.1 Cycle Sequencing Kit. Cycle sequencing was performed on a Veriti Thermal Cycler (Applied Biosystems, USA) using 3.2 μM of each primer, 200 ng purified PCR product, 5X BigDye Sequencing Buffer, 2.5X Ready Reaction Premix and deionized water in a 20 μl final volume. The thermocycling conditions included 1 min at 96 °C followed by 25 cycles of denaturation at 96 °C for 1 min, primer annealing at 50 °C for 5 s and primer extension at 60 °C for 4 min, followed by a holding step at 4 °C. Extension products were purified using ethanol/EDTA precipitation with 5 μl of EDTA 125 mM, 60 μl of absolute ethanol. Purified products were denatured at 95 °C for 5 min using 10 μl Hi-Di Formamide. DNA electrophoresis was performed in 80 cm × 50 μ capillary with POP-4 polymer (Applied Biosystems, USA).
In order to test the efficacy of the NEBNext Microbiome DNA Enrichment Kit the proportion of reads belonging to the plastome was estimated for both the methylated and the non-methylated fraction. The P. ginseng whole genome sequencing SRR19873 experiment was used to estimate the starting proportion of plastome reads, by mapping the reads against the plastid genome of P. ginseng (NC_006290) using Bowtie 2. Association of reads to their taxonomic identification and organelles, was made using a tailored database of Panax plastome data representing the same data as that downloaded from public repositories for the phylogenetic analyses. For the mitochondrial data, all angiosperm mitochondrion genomes available on NCBI were used, and for the microbiome all remaining reads were blasted against the full NCBI database. Taxonomic identifications were retrieved using the lowest common ancestor (LCP) algorithm in Megan version 5.11.3, with minimum read length of 150 bp and at least 10 reads for each taxon identified with an e-value of 1e-20 or less. The proportion of plastid DNA in the gDNA was estimated using Bowtie2 by mapping the proportion of reads belonging to the plastid genome for P. ginseng (following SRR experiment SRR1181600).
The plastid genomes were annotated using Geneious version 6.1, and annotations of exons and introns were manually checked by alignment with their respective genes in the same annotated species genome. Representative maps of the chloroplast genomes were created using OGDraw (Organellar Genome Draw, [100]).
Phylogenomics
The matrix for phylogenomic analyses consisted of complete aligned plastid genomes, and the global alignment was done using MAFFT version 7.3 [101] with local re-alignment using MUSCLE version 3.8.31 [102], and manual adjustments where necessary. Aligned DNA sequences have been deposited in the Open Science Framework (OSF) directory (https://osf.io/ryuz6). The final matrix has a total length of 163,499 bp for a total of 61 individuals with no missing data. Single nucleotide polymorphisms (SNPs) were visualized using Circos version 0.69 [103]. Relationships from the nucleotide matrix were inferred using Maximum Likelihood (ML) and Bayesian inference. First, an un-partitioned phylogenetic analysis was performed to estimate a single nucleotide substitution model and branch length parameters for all characters. Next, the data was partitioned in coding regions, introns and intergenic spacers, and a best-fit partitioning scheme for the combined dataset was determined using PartitionFinder version 2.1.1 [104] using the Bayesian Information Criterion (Additional file 4: Table S3). Branch lengths were linked across partitions.
The dataset was analyzed using RAxML version 8.2.10 [105] and mrBayes version 3.2.6 [106]. RAxML and Bayesian searches used the partition model determined by PartitionFinder. For the ML analyses, tree searches and bootstrapping were conducted simultaneously with 1000 bootstrap replicates. Bayesian analysis were started using a random starting tree and were run for a total of ten million generations, sampling every 1000 generations. Four Markov runs were conducted with eight chains per run. We used AWTY to assess the convergence of the analyses [107]. Conflicting data within ML and Bayesian analyses were visualized and explored using the R package phangorn using the consensusNet function [108].
Barcoding - mPTP
Suitable barcoding markers were selected by extracting the SNP density over the plastid genome alignment of all Panax species and individuals included in this study (matrix available as supplementary data on OSF). We used SNP-sites version 2.3.2 [109] to extract the SNP positions from the alignment of a matrix containing only the Panax species, and created bins every 800 bp using Bedtools version 2.26.0 [110] (script available on OSF) and plotted the SNP density using Circos [103] (Fig. 1). The coordinates of each annotation on the aligned Panax species matrix were found using a reference consisting of the four annotated genomes produced in this study, and subsequently exported to Circos. We selected the most variable regions and designed suitable primers for these regions (Fig. 5, Additional file 5: Table S4). From the matrix used for the Aralioideae, we extracted 15 plastid markers (Fig. 5) and download ITS sequences for the Aralia-Panax group (Figs. 3 and 5) (Additional file 2: Table S2). We performed maximum likelihood analyses on individual and concatenated matrices using RAxML. mPTP analyses were performed using the ML trees from the individual and concatenated markers, and using the Markov chain Monte Carlo (MCMC) algorithm with two chains and the Likelihood Ratio Test set to 0.01.
Fig. 1.
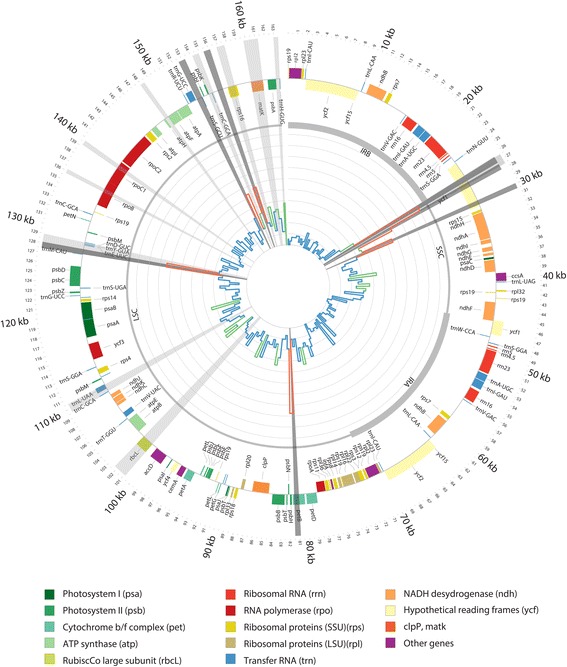
Plastid genome representation of the 38 aligned Panax genomes. The internal histogram plot represents the SNPs density over the alignment of the plastid genomes of Panax genus. The colours indicate when the standard deviation of the bin falls in different interval compare to the average standard deviation, between 0 and 1 in blue (low variation), between 1 to 2 in green (moderate variation) and over two in red (highly variable). Inverted repeats A and B (IRA and IRB), large single copy (LSC) and small single copy (SSC) are shown in the inner circle by different line weights. Genes shown outside the outer circle are transcribed clockwise, and those inside are transcribed counter clockwise. Genes belonging to different functional groups are color-coded. Radial grey highlights show the regions in focus of study, light grey previously used barcodes, in dark grey newly developed barcodes
Fig. 5.
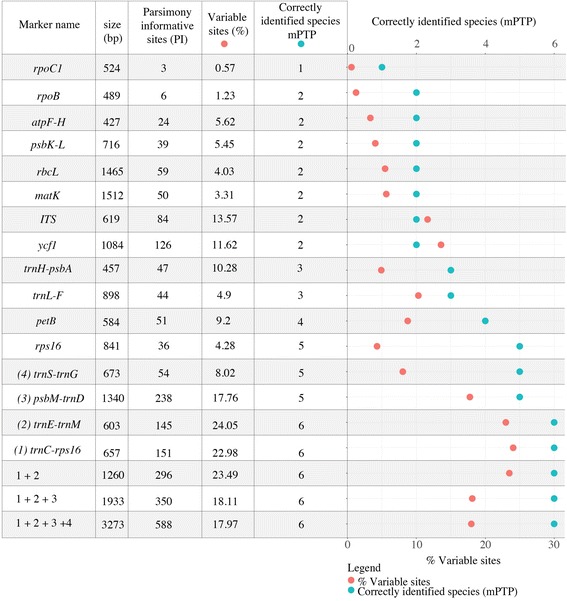
Percentage of variable site (orange) and successful species identified with the mPTP analyses (blue), for each marker and the concatenated matrices
Fig. 3.
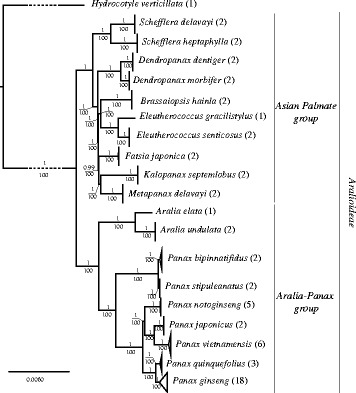
Bayesian tree inferred from the 61 plastid genomes from the Aralioideae tribe. Numbers above branches indicate posterior probabilities and below the ML bootstrap values
Results
Ion torrent sequencing
After filtering out low-quality reads, 1.9 out of 3.3 and 3.3 out of 4.9 million reads were retained for the pooled MDB2 depleted and enriched fractions respectively. The chloroplast assemblies covered the entire circular plastid genome for all four accessions for the MDB2 depleted fraction (Additional file 6: Figure S2, Additional file 7: Figure S3, Additional file 8: Figure S4, Additional file 9: Figure S5; Table 1). The Sanger generated plastid sequences confirmed the genome assemblies in 18 regions, and also confirmed sequences of the inverted repeat. Complete lengths of the four plastid genomes ranged from 156,036 bp to 156,302 bp (Table 1). All four plastid genomes had the same genome structure and gene arrangement as that of the already assembled Panax plastid genomes.
Methylation enrichment
The Fragment Analyzer results showed that DNA quantity and fragmentation differed for the four DNA samples (Additional file 3: Figure S1), and the results were used to normalize concentrations for subsequent capture. DNA concentrations after capture and fragment size selection are much lower for the methyl-depleted fraction compare to the methyl-CpG-enriched fraction (Fig. 2). The success of the fragment size selection was relatively poor for one of the P. vietnamensis. and resulted in a poorer quality in the sequencing and enrichment due to the excessive abundance of short DNA fragments. The shorter reads for P. vietnamensis. yielded a lower coverage for its genome assembly (16.9 X) (Table 1).
Fig. 2.
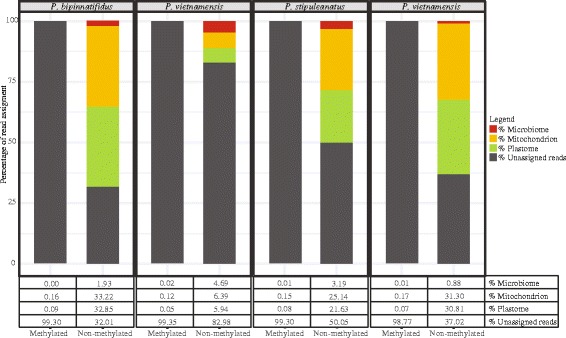
Proportion of mitochondrial, plastid and microbiome reads from the methylated and non-methylated fraction for the four enriched libraries. These results show that the enrichment procedure successfully capture more than 99.5% of the plastid sequences contained in the total DNA
The enrichment and depletion of methylated DNA by pulling down a methyl-CpG-enriched fraction and leaving a methyl-depleted fraction drastically increased the proportion of organellar DNA within the depleted fraction. P. ginseng SRR experimental data had 5.63% plastid genome reads. In the methylation-depleted fraction, we found a variation of plastome reads ranging from 6 to 33%. In the methylation-enriched fraction, less than 1% of the reads are from the plastome. The enrichment also increased microbiome contamination in the depleted fraction from 0.8 to 4%. Overall, one of the P. vietnamensis samples was the least successful sample in the enrichment and yielded fewer and shorter reads.
Phylogenetic analyses
Alignment of the plastid genomes for phylogenetic analyses were consistent in length throughout the dataset. Based on the alignment, average plastome pairwise identity for the Araliaceae family is 83% and 99.2% for the Panax clade. The percentage of identical sites is 83.9% and 96.8% respectively. The global plastome alignment has a matrix length of 163,499 bp. Coding regions, introns and intergenic spacers represented 259 original partition schemes, and the best-fit partitioning scheme from PartitionFinder divided the data into 73 partitions (Additional file 4: Table S3).
Inspection of the posterior probabilities calculated using AWTY, yielded an estimated burnin of 10% for the Bayesian analysis. Phylogenetic analyses revealed significant divergence between major clades of the Araliaceae family. The ML and Bayesian trees showed strongly supported clades for all genera of the family (Fig. 3). Furthermore, the tree shows maximum support for each species of Panax included in the analyses. All intergeneric and infrageneric relationships were strongly supported (Fig. 3).
The basal node segregates two clades, one clade includes two genera, Aralia and Panax. The second clade includes Schefflera, Fatsia, Eleutherococcus, Kalopanax, Metapanax, Brassaiopsis, and Dendropanax. All species included in the study are monophyletic and have maximum support in both Bayesian and ML analyses.
The Araliaceae clade
The Araliaceae clade showed maximum support in the phylogeny except for the Fatsia clade, where the support is 99.6%. Schefflera is sister to the rest of the clade, followed by Dendropanax, then a clade with Brassaiopsis/Eleutherococcus and finally a clade with Fatsia/Kalopanax/Metapanax. A comparison of the partitioned and non-partitioned analyses shows no differences in topology and support in the Aralia-Panax clade, but does in the remaining Araliaceae clade.
The Aralia-Panax clade
The genus Panax is monophyletic and Aralia, represented by two species, A. elata and A. undulata, is the sister group to the genus Panax. Panax stipuleatus and P. binnatifidus form a distinct clade sister to a clade consisting of P. notoginseng and its sister group of P. vietnamensis and P. japonicus, which as a whole is sister to P. quinquefolius and P. ginseng.
The consensus network was computed from the two Bayesian runs after discarding 10% burnin (Fig. 4). The network analysis shows two main conflicts in the data, one within the P. ginseng clade and another within the P. vietnamensis clade. Both clades have very little intraspecific variation (soft incongruence), and more variable markers are needed to segregate the different individuals correctly for these two species.
Fig. 4.
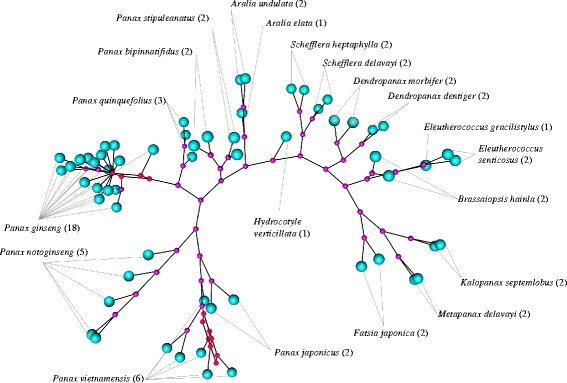
Consensus network from Bayesian runs with 10% burn in. This network shows two main conflicts in the data, one on the P. ginseng clade, where there is very little intraspecific variation and one with the clade of P. vietnamensis. The nodes are represented in pink and the tips in blue. The red nodes show the two splits within the data
Barcoding analyses
The SNP density analyses retrieved 2052 SNPs over the full plastid alignment. We identified three regions (Figs. 1 and 5) that are suitable barcoding markers. Each of these regions has on average of 83 SNPs within Panax (Fig. 5). Individual marker phylogenies of these regions are suitable to segregate most of the species clades (Additional file 10: Figure S7, Additional file 11: Figure S8, Additional file 12: Figure S9, Additional file 13: Figure S10, Additional file 14: Figure S11). The exceptions are the two sister pairs, P. quinquefolius and P. ginseng, and P. binnatifidus and P. stipuleatus, where the bootstrap supports are weaker, leading to inference of single clades. The ML phylogeny of the concatenated markers, fully supports all species clades, except P. binnatifidus and P. stipuleatus (Fig. 5, Additional file 14: Figure S11).
In the mPTP analysis for the full plastid dataset, the Average Support Value (ASV) assesses the congruence of support values with the ML delimitation. The analyses return an ASV of 97.9%, suggesting a high confidence for the given species delimitation scheme. Species delimitation recognized 21 distinct entities out of 20 species (Additional file 15: Figure S6). Over-representation and intraspecific variation of the P. ginseng samples has resulted in oversplitting this clade into two discrete entities. The P. stipuleatus / P. binnatifidus clade has lower data structure and the analyses does not strongly support the group as two independent mPTP entities (PP = 0.68). P. quinquefolius has been also divided into two subgroups, but the posterior probability of the subdivision is low (PP = 0.4).
The result of mPTP analyses for all previously used and the newly proposed markers are described in Fig. 5 and the supported nodes for the speciation events have been added to the phylogenetic tree (Additional file 10: Figure S7, Additional file 11: Figure S8, Additional file 12: Figure S9, Additional file 13: Figure S10, Additional file 14: Figure S11). Out of the 15 analysed markers only four can be used to discriminate most species. Figure 5 also shows that regions with the highest density of parsimony informative sites are not necessarily the most efficient for species discrimination, and both skewed aggregated mutations as well as homoplasy can obscure phylogenetic patterns.
Discussion
Evolution of Araliaceae and ginsengs
The evolution of the Asian palmate group (Fig. 3) is concordant with previously published articles that show Schefflera at the base of the group. The paraphyletic genus Dendropanax was usually the most divergent in the group, but is now basal to the rest of the group. This position might be due to low sampling within the Asian palmate group. Results for Brassaiopsis, Eleutherococcus, Fatsia, Kalopanax and Metapanax, correspond with previously published phylogenies. Early radiations with interlineage hybridizations and genome doubling have been reported in the group [111] and this could explain the short internal branches. Further phylogenomic and biogeographical studies should be conducted to better understand the radiation of the Araliaceae.
In the Aralia-Panax group, Aralia is sister to Panax, and we find that P. stipuleatus forms a well-supported clade with P. binnatifidus, whereas previous studies have often reported that P. binnatifidus groups with P. omeiensis, P. wangianus, P. zingiberensis and P. major [11, 12, 38, 41], all four of which are however missing here. Due to the difficulty in obtaining material of P. vietnamensis, only three studies have included P. vietnamensis in a phylogeny [13, 96, 112]. The study by Lee et al. [112] using the plastid marker trnC–trnD does not resolve the position of P. vietnamesis in the phylogeny, but does identify a distinct clade consisting of P. notoginseng, P. japonicus and P. vietnamensis, which is also supported by our data. Komatsu et al. [13] recover a clade consisting of P. vietnamensis along with P. japonicus and P. pseudoginseng subsp. himalaicus, a synonym of P. bipinnatifidus. Inferring P. japonicus to belong to this clade is contradictory to previous studies that have found a clade consisting of P. quinquefolius, P. ginseng and P. japonicus [12, 38, 41, 112]. The plastome phylogeny supports a sister-relationship of P. ginseng and P. quinquefolius, the two economically most important species of ginseng. Although this full plastome phylogeny significantly differs from previously published molecular phylogenies, the new evolutionary pattern is strongly supported by bootstrap values and posterior probabilities.
Incongruence between markers from different origin
Full length plastid genome data are a major improvement for the Panax phylogeny, and the addition of a bigger dataset has a strong influence on the phylogenetic hypothesis. However, discrepancies between full-length plastid genome phylogenies and nrDNA phylogenies are common in plants. nrDNA has been widely used for phylogenetic studies of Panax [11, 38, 41, 46], but the limitations of this approach have been extensively reviewed in [113]. Drawbacks of nrDNA include difficulties in aligning, and its limited use for phylogenetic inference between closely related and/or recently diverged taxa. It is also a challenge to determine the orthology and the paralogy of nrDNA sequences in the case of hybridization events or incomplete lineage sorting [114–116]. Bailey et al. [114] emphasise that despite valuable phylogenetic information from nrDNA, it might not the optimal choice to assess species trees, especially in case of allopolyploids or tetrapolypoids. Since this is also the case in Panax, we argue that nrDNA may be inappropriate to reconstruct the evolutionary history of this genus.
Phylogenetic congruence as well as incongruence of nuclear genomic and plastid marker data is well documented [117–119]. In the case of Panax, two of the nuclear markers used by [12] support the clade of P. ginseng and P. quinquefolius (Z14, Z8). However, our topology is incongruent for the remaining clades. Incongruences between the maternally inherited plastid genome and the biparentally inherited nuclear genes can be expected in genera with allopolyploid hybrids, like Panax [12]. Plastid phylogenies are not always representative of the species tree and might conflict with hypotheses of parsimonious morphological evolution [116, 120, 121]. Incongruences between plastome and nuclear gene trees have been reported in wide ranging groups of plants, such as Asclepia [72], Helianthus [122] and Silene [120].
Enrichment
The novel method based on methylation-based enrichment increased the concentration of plastid DNA by 30% which is in the range found by a previous pilot study [84]. It is a suitable method for enriching the organellar genome before sequencing. The methylated fraction shows extremely low amounts of organellar DNA, meaning that we removed more than 99% of the non-methylated DNA from the total DNA. The P. vietnamensis sample had originally more degraded DNA and as a result shows a less successful enrichment. Using MBD2 to increase the concentration of organellar DNA in the total DNA allows multiplexing a larger number of samples. This method is appropriate for building plastid reference genome databases for barcoding projects. In case of degraded samples, we recommend removal of shorter DNA fragments before the enrichment.
Selecting markers for molecular Panax identification
In DNA barcoding and plant product identification and authentication projects it is common to work with degraded DNA substrates for which it might be difficult to use methylation enrichment or the full plastid genome as a barcoding strategy. However, alternatives such as target enrichment and amplicon sequencing are possible [64, 123–125]. Here we have identified four variable regions that possess sufficient variation and genetic structure to discriminate most ginseng species. The identification of ginseng species is relatively complex because of the recent evolution and hybridization events. P. ginseng and P quinquefolius have recently diverged plastid genomes, and so do P. binnatifidus and P. stipuleatus [47]. Species delimitation using mPTP shows that for such species complexes traditional barcoding markers do not have enough structure for delimiting species. However, if carefully selected, some regions highlight specific structural patterns that enable the discrimination of species. The trnC-rps16 region seems to be particularly promising, as it has enough variation to discriminate most species (Additional file 15: Figure S6). If plastid markers are to be used for barcoding, it is more relevant to use a combination of markers because mPTP analyses are better suited for multi-marker analyses [32]. A concatenated matrix with two, three or four markers combined improves the efficacy in segregating all the Panax species and specifically also those in closely related complexes. Our results suggest that a combination of the following markers: trnC-rps16, trnE-trnM and psbM-trnD (Fig. 5) enables confident identification of the main traded species P. ginseng, P. quinquefolius and P. vietnamensis. For further development, a complete sampling of all Panax species with multiple accessions per taxon should be made to confirm the observed variation in the selected markers.
In order to design accurate markers to monitor the trade of the medicinal species, it is necessary to understand the evolution of the targeted group. Many studies are based on the generic barcodes suggested by iBOL (International Barcode of Life) (rbcL and matK) without having strong evidence for the evolutionary hypotheses of the targeted group and a limited idea a fortiori of the discriminatory power of the used markers. Nonetheless, when a barcoding study targets a specific plant group or genus, and the barcode markers fail to yield a supported phylogeny, then one should aim to construct robust phylogenies with new markers to achieve species discrimination. If the phylogenetic hypothesis is not robust, or if the data are weak in structure as it is often the case with the standard barcoding markers, rbcL and matK, the resulting identifications might be misleading because of inaccurate species delimitation hypotheses [31].
Conclusion
The addition of genomic data for the phylogeny of Panax radically changes what is known about the evolution of the genus. The implications in terms of phylogeography are still unclear due to missing taxa, and the addition of population data and additional species should improve our insight into the evolutionary history of the genus. The development of species delimitation methods changes perspectives in molecular identification and DNA barcoding by incorporating evolution hypotheses at the species level. The newly proposed molecular markers allow for accurate identification of Panax species and enable authentication of ginseng and derived products and monitoring of the ginseng trade, while ultimately aiding conservation of wild ginseng.
Additional files
Table S1. Voucher specimens. (DOCX 14 kb)
Table S2. Araliaceae species used for this study and their accession numbers. (PDF 385 kb)
Figure S1. Fragment analyzer DNA report of P. bipinnatifidus, P. sp. (puxailaileng), P. stipuleanatus, P. vietnamensis samples, for the genomic DNA (gDNA), for the non-methylated and methylated fractions. (PDF 410 kb)
Table S3. Partition finder scheme. (DOCX 29 kb)
Table S4. selected markers and their primer sequences. (DOCX 19 kb)
Figure S2. Annotated plastid genome for P. binnatifidus (PDF 442 kb)
Figure S3. Annotated plastid genome for P. sp. (puxailaileng). (PDF 359 kb)
Figure S4. Annotated plastid genome for P. vietnamensis. (PDF 456 kb)
Figure S5. Annotated plastid genome for P. stipuleanatus. (PDF 440 kb)
Figure S7. ML phylogeny for marker trnC-rps16. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 108 kb)
Figure S8. ML phylogeny for marker trnE-trnM. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 85 kb)
Figure S9. ML phylogeny of marker trnS-trnG. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 116 kb)
Figure S10. ML phylogeny of the marker psbM-trnD. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 102 kb)
Figure S11. ML phylogeny for the concatenated matrix with the four markers, trnC-rps16, trnS-trnG, trnE-trnM and psbM-trnD. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 108 kb)
Figure S6. Results of the mPTP species delimitation analysis on the full plastid genome matrix. The red lines illustrate the branches representing speciation and the brown lines the branches representing coalescence processes. The numbers on the branches represent the Bayesian posterior probabilities for the delimited species. (PDF 145 kb)
Acknowledgments
The authors wish to thank the following people and organizations, Jarl Andreas Anmarkrud for his assistance for the enrichment, members of de Boer group for their helpful discussions and feedback during manuscript preparation. This work was performed on the Abel Cluster, owned by the University of Oslo and the Norwegian metacentre for High Performance Computing (NOTUR), and operated by the Department for Research Computing at USIT, the University of Oslo IT-department. http://www.hpc.uio.no/. We would like to acknowledge the support of Vietnam Academy of Science and Technology (VAST).
Funding
This project was supported by Vietnam Academy of Science and Technology (grant No. VAST02.01/16–17) and the European Union’s Seventh Framework Programme for research, technological development and demonstration under the Grant agreement no. 606895 to the FP7-MCA-ITN MedPlant, “Phylogenetic Exploration of Medicinal Plant Diversity”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The raw sequence data from the P. bipinnatifidus, P. stipuleanatus, and P. vietnamensis samples have been to submitted to GenBank on the following accessions: SRR5725242, SRR5725240, SRR5725505, SRR5725492, SRR5738925, SRR5738922, SRR5738920, SRR5738920, SRR5738927.The DNA matrix used for the phylogenomic analyses are available on Open Science Framework, (10.17605/OSF.IO/Z7RWE). The plastome sequences of P. bipinnatifidus, P. stipuleanatus, and P. vietnamensis (2) have been submitted to NCBI GenBank, (MF377620, MF377621, MF377622, MF377623).
Abbreviations
- 2D-IR
Two-Dimensional Correlation Infrared Spectroscopy
- ABGD
Automatic Barcode Gap Discovery
- AP-PCR
Arbitrarily Primed Polymerase Chain Reaction
- ASV
Average Support Value
- AWTY
Are We There Yet
- BOLD
Barcode of Life Database
- dNTP
Deoxynucleotide
- FT-IR
Fourier Transformed-Infrared Spectroscopy
- gDNA
Genomic DNA
- GMYC
Generalized Mixed Yule Coalescent
- iBOL
International Barcode Of Life
- LCP
Lowest Common ancestor
- MARMS
Multiplex Amplification Refractory Mutation System-PCR
- MASA
Mutant Allele Specific Amplification
- MBD2
Methyl-CpG-Binding Domain
- MBD2-Fc
Methylated CpG-specific Binding protein (MBD2), fused to the Fc fragment of human IgG
- MCMC
Markov Chain Monte Carlo
- ML
Maximum Likelihood
- mPTP
Multi-rate Poisson Tree Processes
- MSC
Multispecies Coalescent
- nrDNA
Nuclear Ribosomal DNA
- OSF
Open Science Framework
- PCR
Polymerase Chain Reaction
- PCR-RFLP
PCR-Restriction Fragment Length Polymorphism
- PTP
Poisson Tree Processes
- RAPD
Random Amplified Polymorphic DNA
- RESL
Refined Single Linkage
- SNP
Single-Nucleotide Polymorphism
Authors’ contributions
The project was conceived and designed by HdB, HLTT, NVH, and VM. NNL performed the laboratory work. VM performed data analysis. AK, VM and HdB drafted the manuscript. All other authors gave useful contribution on the analysis of data and text of the manuscript. All authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1160-y) contains supplementary material, which is available to authorized users.
Contributor Information
V. Manzanilla, Email: vincent.manzanilla@nhm.uio.no
A. Kool, Email: anneleen.kool@nhm.uio.no
L. Nguyen Nhat, Email: linhnn@igr.ac.vn
H. Nong Van, Email: vhnong@igr.ac.vn
H. Le Thi Thu, Email: hienlethu@igr.ac.vn
H. J. de Boer, Email: hugo.deboer@nhm.uio.no
References
- 1.Robbins CS. American ginseng: the root of North America's medicinal herb trade: Traffic North America; 1998.
- 2.Millspaugh CF. American medicinal plants: an illustrated and descriptive guide to plants indigenous to and naturalized in the United States which are used in medicine: Dover Publications; 1892.
- 3.Kimmens AC. Tales of the ginseng: Morrow; 1975.
- 4.Zhuravlev YN, Koren OG, Reunova GD, Muzarok TI, Gorpenchenko TY, Kats IL, Khrolenko YA. Panax ginseng natural populations: their past, current state and perspectives. Acta Pharmacol Sin. 2008;29(9):1127–1136. doi: 10.1111/j.1745-7254.2008.00866.x. [DOI] [PubMed] [Google Scholar]
- 5.programme SotcoitiesowffUNe . CITES (convention on international trade in endangered species) handbook: convention on species of wild fauna and flora, July 2016. 2016. [Google Scholar]
- 6.Basnet D, Dey K. Studies on seed germination of an Indian ginseng (Panax assamicus Ban. spec. nov.) for successful cultivation and conservation. Indian J For. 2008.
- 7.Zhang S, Wang R, Zeng W, Zhu W, Zhang X, Wu C, Song J, Zheng Y, Chen P. Resource investigation of traditional medicinal plant Panax japonicus (T. Nees) CA Mey and its varieties in China. J Ethnopharmacol. 2015;166:79–85. doi: 10.1016/j.jep.2015.02.051. [DOI] [PubMed] [Google Scholar]
- 8.Joshi G, Tiwari K, Tiwari R, Uniyal M. Conservation and large scale cultivation strategy of Indian ginseng- Panax pseudoginseng wall. Indian Forester. 1991;117(2):131–134. [Google Scholar]
- 9.Jain A. Vulnerable and threatened plants of economic value: Panax pseudo-ginseng wall. (The Himalayan Ginseng) MFP News. 1994;4:21. [Google Scholar]
- 10.Blundell AG, Mascia MB. Discrepancies in reported levels of international wildlife trade. Conserv Biol. 2005;19(6):2020–2025. doi: 10.1111/j.1523-1739.2005.00253.x. [DOI] [Google Scholar]
- 11.Zuo Y, Chen Z, Kondo K, Funamoto T, Wen J, Zhou S. DNA barcoding of Panax species. Planta Med. 2011;77(02):182–187. doi: 10.1055/s-0030-1250166. [DOI] [PubMed] [Google Scholar]
- 12.Shi F-X, Li M-R, Li Y-L, Jiang P, Zhang C, Pan Y-Z, Liu B, Xiao H-X, Li L-F. The impacts of polyploidy, geographic and ecological isolations on the diversification of Panax (Araliaceae) BMC Plant Biol. 2015;15(1):297. doi: 10.1186/s12870-015-0669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komatsu K, Zhu S, Fushimi H, Qui TK, Cai S, Kadota S. Phylogenetic analysis based on 18S rRNA gene and matK gene sequences of Panax vietnamensis and five related species. Planta Med. 2001;67(05):461–465. doi: 10.1055/s-2001-15821. [DOI] [PubMed] [Google Scholar]
- 14.Yap KY-L, Chan SY, Lim CS. Infrared-based protocol for the identification and categorization of ginseng and its products. Food Res Int. 2007;40(5):643–652. doi: 10.1016/j.foodres.2006.11.009. [DOI] [Google Scholar]
- 15.Li Y-M, Sun S-Q, Zhou Q, Qin Z, Tao J-X, Wang J, Fang X. Identification of American ginseng from different regions using FT-IR and two-dimensional correlation IR spectroscopy. Vib Spectrosc. 2004;36(2):227–232. doi: 10.1016/j.vibspec.2003.12.009. [DOI] [Google Scholar]
- 16.Mihalov JJ, Marderosian AD, Pierce JC. DNA identification of commercial ginseng samples. J Agric Food Chem. 2000;48(8):3744–3752. doi: 10.1021/jf000011b. [DOI] [PubMed] [Google Scholar]
- 17.Liu D, Li Y-G, Xu H, Sun S-Q, Wang Z-T. Differentiation of the root of cultivated ginseng, mountain cultivated ginseng and mountain wild ginseng using FT-IR and two-dimensional correlation IR spectroscopy. J Mol Struct. 2008;883:228–235. doi: 10.1016/j.molstruc.2008.02.025. [DOI] [Google Scholar]
- 18.Zhu S, Fushimi H, Cai S, Komatsu K. Species identification from ginseng drugs by multiplex amplification refractory mutation system (MARMS) Planta Med. 2004;70(02):189–192. doi: 10.1055/s-2004-815502. [DOI] [PubMed] [Google Scholar]
- 19.Park M-J, Kim MK, In J-G, Yang D-C. Molecular identification of Korean ginseng by amplification refractory mutation system-PCR. Food Res Int. 2006;39(5):568–574. doi: 10.1016/j.foodres.2005.11.004. [DOI] [Google Scholar]
- 20.Qin J, Leung FC, Fung Y, Zhu D, Lin B. Rapid authentication of ginseng species using microchip electrophoresis with laser-induced fluorescence detection. Anal Bioanal Chem. 2005;381(4):812–819. doi: 10.1007/s00216-004-2889-2. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Jo BH, Lee KL, Yoon E, Ryu GH, Chung KW. Identification of new microsatellite markers in Panax ginseng. Mol Cells. 2007;24(1):60. [PubMed] [Google Scholar]
- 22.Ratnasingham S, Hebert PD. A DNA-based registry for all animal species: the Barcode Index Number (BIN) system. PLoS One. 2013;8(7):e66213. doi: 10.1371/journal.pone.0066213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao X, Jiang R, Chen T. Clustering 16S rRNA for OTU prediction: a method of unsupervised Bayesian clustering. Bioinformatics. 2011;27(5):611–618. doi: 10.1093/bioinformatics/btq725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyer F, et al. Obitools: a unix–inspired software package for DNA metabarcoding. Molecular Ecology Resources. 2016;16(1):176–82. [DOI] [PubMed]
- 25.Zhang J, Kapli P, Pavlidis P, Stamatakis A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 2013;29(22):2869–2876. doi: 10.1093/bioinformatics/btt499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebert PD, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. PLoS Biol. 2004;2(10):e312. doi: 10.1371/journal.pbio.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puillandre N, Lambert A, Brouillet S, Achaz G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol. 2012;21(8):1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x. [DOI] [PubMed] [Google Scholar]
- 29.Brown SD, Collins RA, Boyer S, Lefort MC, Malumbres-Olarte J, Vink CJ, Cruickshank RH. Spider: an R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol Ecol Resour. 2012;12(3):562–565. doi: 10.1111/j.1755-0998.2011.03108.x. [DOI] [PubMed] [Google Scholar]
- 30.Meier R, Zhang G, Ali F. The use of mean instead of smallest interspecific distances exaggerates the size of the “barcoding gap” and leads to misidentification. Syst Biol. 2008;57(5):809–813. doi: 10.1080/10635150802406343. [DOI] [PubMed] [Google Scholar]
- 31.Ross HA, Murugan S, Sibon Li WL. Testing the reliability of genetic methods of species identification via simulation. Syst Biol. 2008;57(2):216–230. doi: 10.1080/10635150802032990. [DOI] [PubMed] [Google Scholar]
- 32.Kapli P, et al. Multi-rate poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics. 2017;33(11):1630–638. [DOI] [PMC free article] [PubMed]
- 33.Fujisawa T, Barraclough TG. Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: a revised method and evaluation on simulated data sets. Syst Biol. 2013;62(5):707–24. [DOI] [PMC free article] [PubMed]
- 34.Yang Z, Rannala B. Bayesian species identification under the multispecies coalescent provides significant improvements to DNA barcoding analyses. Mol Ecol. 2017;26(11):3028–36. [DOI] [PubMed]
- 35.Sukumaran J, Knowles LL. Multispecies coalescent delimits structure, not species. Proc Natl Acad Sci. 2017;114(7):1607–1612. doi: 10.1073/pnas.1607921114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wen J. Evolution of the Aralia–Panax complex (Araliaceae) as inferred from nuclear ribosomal ITS sequences. Edinb J Bot. 2001;58(02):243–257. doi: 10.1017/S0960428601000610. [DOI] [Google Scholar]
- 37.Plunkett GM, Wen J, Lowry Ii PP. Infrafamilial classifications and characters in Araliaceae: insights from the phylogenetic analysis of nuclear (ITS) and plastid (trnL-trnF) sequence data. Pl Syst Evol. 2004;245(1):1–39. [Google Scholar]
- 38.Wen J, Zimmer EA. Phylogeny and biogeography of Panax L. (the ginseng genus, Araliaceae): inferences from ITS sequences of nuclear ribosomal DNA. Mol Phylogenet Evol. 1996;6(2):167–177. doi: 10.1006/mpev.1996.0069. [DOI] [PubMed] [Google Scholar]
- 39.Hara H. On the Asiatic species of the genus Panax. J Jpn Bot. 1970.
- 40.Zhou J, Huang W, Wu M, Yang C, Feng K, Wu Z. Triterpenoids from Panax Linn. and their relationship with taxonomy and geographical distribution. Acta Phytotaxonomica Sin. 1975;13(2):29–45. [Google Scholar]
- 41.Choi H-K, Wen J. A phylogenetic analysis of Panax (Araliaceae): integrating cpDNA restriction site and nuclear rDNA ITS sequence data. Pl Syst Evol. 2000;224(1):109–120. doi: 10.1007/BF00985269. [DOI] [Google Scholar]
- 42.Kumar Sharma S, Krishan Pandit M. A new species of Panax L. (Araliaceae) from Sikkim Himalaya, India. Syst Bot. 2009;34(2):434–438. doi: 10.1600/036364409788606235. [DOI] [Google Scholar]
- 43.Duy NV, Trieu LN, Chinh ND, Tran VT. A new variety of Panax (Araliaceae) from Lam Vien Plateau, Vietnam and its molecular evidence. Phytotaxa. 2016;277(1):12. doi: 10.11646/phytotaxa.277.1.4. [DOI] [Google Scholar]
- 44.Yi T, Lowry PP, Plunkett GM. Chromosomal evolution in Araliaceae and close relatives. Taxon. 2004;53(4):987–1005. doi: 10.2307/4135565. [DOI] [Google Scholar]
- 45.Choi HI, Kim NH, Lee J, Choi BS, Do Kim K, Park JY. Evolutionary relationship of Panax ginseng and P. quinquefolius inferred from sequencing and comparative analysis of expressed sequence tags. Genet Resour Crop Evol. 2013;60:1377–1387. doi: 10.1007/s10722-012-9926-3. [DOI] [Google Scholar]
- 46.Wen J, Plunkett GM, Mitchell AD, Wagstaff SJ. The evolution of Araliaceae: a phylogenetic analysis based on ITS sequences of nuclear ribosomal DNA. Syst Bot. 2001;26(1):144–167. [Google Scholar]
- 47.Kim NH, Choi HI, Kim KH, Jang W, Yang TJ. Evidence of genome duplication revealed by sequence analysis of multi-loci expressed sequence tag-simple sequence repeat bands in Panax ginseng Meyer. J Ginseng Res. 2014;38:130–135. doi: 10.1016/j.jgr.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang P, Shi F-X, Li Y-L, Liu B, Li L-F. Development of highly transferable microsatellites for Panax ginseng (Araliaceae) using whole-genome data. Appl Plant Sci. 2016;4(11):1600075. doi: 10.3732/apps.1600075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma K-H, Dixit A, Kim Y-C, Lee D-Y, Kim T-S, Cho E-G, Park Y-J. Development and characterization of new microsatellite markers for ginseng (Panax ginseng CA Meyer) Conserv Genet. 2007;8(6):1507–1509. doi: 10.1007/s10592-007-9284-4. [DOI] [Google Scholar]
- 50.Van Dan N, Ramchiary N, Choi SR, Uhm TS, Yang T-J, Ahn I-O, Lim YP. Development and characterization of new microsatellite markers in Panax ginseng (CA Meyer) from BAC end sequences. Conserv Genet. 2010;11(3):1223–1225. doi: 10.1007/s10592-009-9924-y. [DOI] [Google Scholar]
- 51.Choi H-I, Kim N-H, Kim J-H, Choi B-S, Ahn I-O, Lee J-S, Yang T-J. Development of reproducible EST-derived SSR markers and assessment of genetic diversity in Panax ginseng cultivars and related species. J Ginseng Res. 2011;35(4):399–412. doi: 10.5142/jgr.2011.35.4.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu H, Xia T, Zuo Y-J, Chen Z-J, Zhou S-L. Development and characterization of microsatellite markers for Panax notoginseng (Araliaceae), a Chinese traditional herb. Am J Bot. 2011;98(8):e218–e220. doi: 10.3732/ajb.1100043. [DOI] [PubMed] [Google Scholar]
- 53.Joly S, et al. Genetic structure of the American ginseng (Panax quinquefolius L.) in Eastern Canada using reduced-representation high-throughput sequencing. Botany. 2016;95(4):429–34.
- 54.Cruse-Sanders JM, Hamrick J. Genetic diversity in harvested and protected populations of wild American ginseng, Panax quinquefolius L.(Araliaceae) Am J Bot. 2004;91(4):540–548. doi: 10.3732/ajb.91.4.540. [DOI] [PubMed] [Google Scholar]
- 55.Bai D, Brandle J, Reeleder R. Genetic diversity in North American ginseng (Panax quinquefolius L.) grown in Ontario detected by RAPD analysis. Genome. 1997;40(1):111–115. doi: 10.1139/g97-015. [DOI] [PubMed] [Google Scholar]
- 56.Schluter C, Punja ZK. Genetic diversity among natural and cultivated field populations and seed lots of American ginseng (Panax quinquefolius L.) in Canada. Int J Plant Sci. 2002;163(3):427–439. doi: 10.1086/339512. [DOI] [Google Scholar]
- 57.Cruse-Sanders J, Hamrick J. Spatial and genetic structure within populations of wild American ginseng (Panax quinquefolius L., Araliaceae) J Hered. 2004;95(4):309–321. doi: 10.1093/jhered/esh046. [DOI] [PubMed] [Google Scholar]
- 58.Grubbs HJ, Case MA. Allozyme variation in American ginseng (Panax quinquefolius L.): variation, breeding system, and implications for current conservation practice. Conserv Genet. 2004;5(1):13–23. doi: 10.1023/B:COGE.0000014064.44592.bc. [DOI] [Google Scholar]
- 59.Boehm C, Harrison H, Jung G, Nienhuis J. Organization of American and Asian ginseng germplasm using randomly amplified polymorphic DNA (RAPD) markers. J Am Soc Hortic Sci. 1999;124(3):252–256. [Google Scholar]
- 60.Li S, Li J, Yang X-L, Cheng Z, Zhang W-J. Genetic diversity and differentiation of cultivated ginseng (Panax ginseng CA Meyer) populations in North-East China revealed by inter-simple sequence repeat (ISSR) markers. Genet Resour Crop Evol. 2011;58(6):815–824. doi: 10.1007/s10722-010-9618-9. [DOI] [Google Scholar]
- 61.Reunova GD, Koren OG, Muzarok TI, Zhuravlev YN. Microsatellite analysis of Panax ginseng natural populations in Russia. Chin Med. 2014;5(04):231. doi: 10.4236/cm.2014.54028. [DOI] [Google Scholar]
- 62.Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotech. 2012;30(5):434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 63.Glenn TC. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 2011;11(5):759–769. doi: 10.1111/j.1755-0998.2011.03024.x. [DOI] [PubMed] [Google Scholar]
- 64.Coissac E, et al. From barcodes to genomes: extending the concept of DNA barcoding. Mol Ecol. 2016;25(7):1423–28. [DOI] [PubMed]
- 65.Nock CJ, Waters DL, Edwards MA, Bowen SG, Rice N, Cordeiro GM, Henry RJ. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol J. 2011;9(3):328–333. doi: 10.1111/j.1467-7652.2010.00558.x. [DOI] [PubMed] [Google Scholar]
- 66.Jansen RK, Ruhlman TA. Plastid genomes of seed plants. Genomics of chloroplasts and mitochondria: Dordrecht: Springer; 2012. p. 103–26.
- 67.Givnish TJ, Ames M, McNeal JR, McKain MR, Steele PR, Graham SW, Pires JC, Stevenson DW, Zomlefer WB, Briggs BG. Assembling the tree of the monocotyledons: plastome sequence phylogeny and evolution of Poales1. Ann Mo Bot Gard. 2010;97(4):584–616. doi: 10.3417/2010023. [DOI] [Google Scholar]
- 68.Cai Z, Penaflor C, Kuehl JV, Leebens-Mack J, Carlson JE, Boore JL, Jansen RK. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: implications for the phylogenetic relationships of magnoliids. BMC Evol Biol. 2006;6(1):77. doi: 10.1186/1471-2148-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parks M, Cronn R, Liston A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009;7(1):84. doi: 10.1186/1741-7007-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karol KG, Arumuganathan K, Boore JL, Duffy AM, Everett KD, Hall JD, Hansen SK, Kuehl JV, Mandoli DF, Mishler BD. Complete plastome sequences of Equisetum arvense and Isoetes flaccida: implications for phylogeny and plastid genome evolution of early land plant lineages. BMC Evol Biol. 2010;10(1):321. doi: 10.1186/1471-2148-10-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang H, Shi C, Liu Y, Mao S-Y, Gao L-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14(1):151. doi: 10.1186/1471-2148-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weitemier K, Straub SCK, Cronn RC, Fishbein M, Schmickl R, McDonnell A, Liston A. Hyb-Seq: combining target enrichment and genome skimming for plant phylogenomics. Appl Plant Sci. 2014;2(9):1400042. doi: 10.3732/apps.1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmickl R, et al. Phylogenetic marker development for target enrichment from transcriptome and genome skim data: the pipeline and its application in southern African Oxalis (Oxalidaceae). Mol Ecol Resour. 2016;16(5):1124–35. [DOI] [PubMed]
- 74.Mandel JR, Dikow RB, Funk VA. Using phylogenomics to resolve mega-families: an example from Compositae. J Syst Evol. 2015;53(5):391–402. doi: 10.1111/jse.12167. [DOI] [Google Scholar]
- 75.Mandel JR, Dikow RB, Funk VA, Masalia RR, Staton SE, Kozik A, Michelmore RW, Rieseberg LH, Burke JM. A target enrichment method for gathering phylogenetic information from hundreds of loci: an example from the Compositae. Appl Plant Sci. 2014;2(2):1300085. doi: 10.3732/apps.1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Särkinen T, Staats M, Richardson JE, Cowan RS, Bakker FT. How to open the treasure chest? Optimising DNA extraction from herbarium specimens. PLoS One. 2012;7(8):e43808. doi: 10.1371/journal.pone.0043808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steele PR, Hertweck KL, Mayfield D, McKain MR, Leebens-Mack J, Pires JC. Quality and quantity of data recovered from massively parallel sequencing: examples in Asparagales and Poaceae. Am J Bot. 2012;99(2):330–348. doi: 10.3732/ajb.1100491. [DOI] [PubMed] [Google Scholar]
- 78.Straub SCK, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot. 2012;99(2):349–364. doi: 10.3732/ajb.1100335. [DOI] [PubMed] [Google Scholar]
- 79.Twyford AD, Ness RW. Strategies for complete plastid genome sequencing. Mol Ecol Resour. 2017;17(5):858–68. [DOI] [PMC free article] [PubMed]
- 80.Obae GS. Nuclear DNA, content and genome size of American ginseng. J Med Plant Res. 2012;6.
- 81.Pan YZ, Zhang YC, Gong X, Li FS. Estimation of genome size of four Panax species by flow cytometry. Plant Diversity Res. 2014;36
- 82.Du FK, Lang T, Lu S, Wang Y, Li J, Yin K. An improved method for chloroplast genome sequencing in non-model forest tree species. Tree Genet Genomes. 2015;11(6):114. doi: 10.1007/s11295-015-0942-2. [DOI] [Google Scholar]
- 83.Feng S, Cokus SJ, Zhang X, Chen P-Y, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci. 2010;107(19):8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yigit E, Hernandez DI, Trujillo JT, Dimalanta E, Bailey CD. Genome and metagenome sequencing: using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2014;2(11):1400064. doi: 10.3732/apps.1400064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim K, Nguyen VB, Dong J, Wang Y, Park JY, Lee S-C, Yang T-J. Evolution of the Araliaceae family inferred from complete chloroplast genomes and 45S nrDNAs of 10 Panax-related species. Sci Rep. 2017;7:4917. doi: 10.1038/s41598-017-05218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bock DG, Kane NC, Ebert DP, Rieseberg LH. Genome skimming reveals the origin of the Jerusalem artichoke tuber crop species: neither from Jerusalem nor an artichoke. New Phytol. 2014;201(3):1021–1030. doi: 10.1111/nph.12560. [DOI] [PubMed] [Google Scholar]
- 87.Li R, Ma P-F, Wen J, Yi T-S. Complete sequencing of five Araliaceae chloroplast genomes and the phylogenetic implications. PLoS One. 2013;8(10):e78568. doi: 10.1371/journal.pone.0078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yao X, Liu Y-Y, Tan Y-H, Song Y, Corlett RT. The complete chloroplast genome sequence of Helwingia himalaica (Helwingiaceae, Aquifoliales) and a chloroplast phylogenomic analysis of the Campanulidae. PeerJ. 2016;4:e2734. doi: 10.7717/peerj.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang L, Du X-J, Li X-F. The complete chloroplast genome sequence of the evergreen plant Dendropanax dentiger (Araliaceae) Mitochondrial DNA Part A. 2016;27(6):4193–4194. doi: 10.3109/19401736.2014.1003909. [DOI] [PubMed] [Google Scholar]
- 90.Kim K-J, Lee H-L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004;11(4):247–261. doi: 10.1093/dnares/11.4.247. [DOI] [PubMed] [Google Scholar]
- 91.Yang J-B, Yang S-X, Li H-T, Yang J, Li D-Z. Comparative chloroplast genomes of Camellia species. PLoS One. 2013;8(8):e73053. doi: 10.1371/journal.pone.0073053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen Q, Feng X, Li M, Yang B, Gao C, Zhang L, Tian J. The complete chloroplast genome sequence of Fatsia japonica (Apiales: Araliaceae) and the phylogenetic analysis. Mitochondrial DNA Part A. 2016;27(4):3050–3051. doi: 10.3109/19401736.2015.1063129. [DOI] [PubMed] [Google Scholar]
- 93.Kim K, Lee S-C, Lee J, Lee HO, Joh HJ, Kim N-H, Park H-S, Yang T-J. Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLoS One. 2015;10(6):e0117159. doi: 10.1371/journal.pone.0117159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao Y, Yin J, Guo H, Zhang Y, Xiao W, Sun C, Wu J, Qu X, Yu J, Wang X, et al. The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Front Plant Sci. 2015;5:696. doi: 10.3389/fpls.2014.00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dong W, Liu H, Xu C, Zuo Y, Chen Z, Zhou S. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 2014;15(1):138. doi: 10.1186/s12863-014-0138-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nguyen B, Kim K, Kim Y-C, Lee S-C, Shin JE, Lee J, Kim N-H, Jang W, Choi H-I, Yang T-J. The complete chloroplast genome sequence of Panax vietnamensis Ha et Grushv (Araliaceae) Mitochondrial DNA Part A. 2017;28(1):85–86. doi: 10.3109/19401736.2015.1110810. [DOI] [PubMed] [Google Scholar]
- 97.Zong X, Song J, Lv J, Wang S. The complete chloroplast genome sequence of Schefflera octophylla. Mitochondrial DNA Part A. 2016;27(6):4685–4686. doi: 10.3109/19401736.2015.1106502. [DOI] [PubMed] [Google Scholar]
- 98.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–120. [DOI] [PMC free article] [PubMed]
- 99.Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic acids research. 2013;41(13):e129–e129. [DOI] [PMC free article] [PubMed]
- 100.Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52(5):267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- 101.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lanfear R, Calcott B, Ho SY, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- 105.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 106.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 107.Nylander JA, Wilgenbusch JC, Warren DL, Swofford DL. AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics. 2008;24(4):581–583. doi: 10.1093/bioinformatics/btm388. [DOI] [PubMed] [Google Scholar]
- 108.Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27(4):592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, Harris SR. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genomics. 2016;2(4):e000056. doi: 10.1099/mgen.0.000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Quinlan AR: BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;11–12. [DOI] [PMC free article] [PubMed]
- 111.Valcárcel V, Fiz-Palacios O, Wen J. The origin of the early differentiation of Ivies (Hedera L.) and the radiation of the Asian Palmate group (Araliaceae) Mol Phylogenet Evol. 2014;70:492–503. doi: 10.1016/j.ympev.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 112.Lee C, Wen J. Phylogeny of Panax using chloroplast trnC–trnD intergenic region and the utility of trnC–trnD in interspecific studies of plants. Mol Phylogenet Evol. 2004;31(3):894–903. doi: 10.1016/j.ympev.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 113.Álvarez I, Wendel JF. Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol. 2003;29(3):417–434. doi: 10.1016/S1055-7903(03)00208-2. [DOI] [PubMed] [Google Scholar]
- 114.Bailey C. Characterization of angiosperm nrDNA polymorphism, paralogy, and pseudogenes. Mol Phylogenet Evol. 2003;29 [DOI] [PubMed]
- 115.Fehrer J, Gemeinholzer B, Chrtek J, Bräutigam S. Incongruent plastid and nuclear DNA phylogenies reveal ancient intergeneric hybridization in Pilosella hawkweeds (Hieracium, Cichorieae, Asteraceae) Mol Phylogenet Evol. 2007;42(2):347–361. doi: 10.1016/j.ympev.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 116.Soltis DE, Kuzoff RK. Discordance between nuclear and chloroplast phylogenies in the Heuchera group (Saxifragaceae). Evolution. 1995;49(4):727–42. [DOI] [PubMed]
- 117.Heyduk K, Trapnell DW, Barrett CF, Leebens-Mack J. Phylogenomic analyses of species relationships in the genus Sabal (Arecaceae) using targeted sequence capture. Biol J Linn Soc. 2016;117(1):106–120. doi: 10.1111/bij.12551. [DOI] [Google Scholar]
- 118.Manzanilla V, Bruneau A. Phylogeny reconstruction in the Caesalpinieae grade (Leguminosae) based on duplicated copies of the sucrose synthase gene and plastid markers. Mol Phylogenet Evol. 2012;65(1):149–162. doi: 10.1016/j.ympev.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 119.Novikova PY, et al. Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism. Nat Genet. 2016;48(9):1077. [DOI] [PubMed]
- 120.Popp M, Oxelman B. Inferring the history of the polyploid Silene aegaea (Caryophyllaceae) using plastid and homoeologous nuclear DNA sequences. Mol Phylogenet Evol. 2001;20(3):474–481. doi: 10.1006/mpev.2001.0977. [DOI] [PubMed] [Google Scholar]
- 121.Wendel JF, Doyle JJ. Phylogenetic incongruence: window into genome history and molecular evolution. Molecular systematics of plants II. Boston: Springer; 1998. p. 265–96.
- 122.Stephens JD, Rogers WL, Mason CM, Donovan LA, Malmberg RL. Species tree estimation of diploid Helianthus (Asteraceae) using target enrichment. Am J Bot. 2015;102(6):910–920. doi: 10.3732/ajb.1500031. [DOI] [PubMed] [Google Scholar]
- 123.Raclariu AC, Mocan A, Popa MO, Vlase L, Ichim MC, Crisan G, Brysting AK, de Boer H. Veronica officinalis product authentication using DNA metabarcoding and HPLC-MS reveals widespread adulteration with Veronica chamaedrys. Front Pharmacol. 2017;8:378. doi: 10.3389/fphar.2017.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Raclariu AC, Paltinean R, Vlase L, Labarre A, Manzanilla V, Ichim MC, Crisan G, Brysting AK, de Boer H. Comparative authentication of Hypericum perforatum herbal products using DNA metabarcoding, TLC and HPLC-MS. Sci Rep. 2017;7(1):1291. doi: 10.1038/s41598-017-01389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Veldman S, et al. High-throughput sequencing of African chikanda cake highlights conservation challenges in orchids. Biodivers Conserv. 2017;26(9):2029–46.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Voucher specimens. (DOCX 14 kb)
Table S2. Araliaceae species used for this study and their accession numbers. (PDF 385 kb)
Figure S1. Fragment analyzer DNA report of P. bipinnatifidus, P. sp. (puxailaileng), P. stipuleanatus, P. vietnamensis samples, for the genomic DNA (gDNA), for the non-methylated and methylated fractions. (PDF 410 kb)
Table S3. Partition finder scheme. (DOCX 29 kb)
Table S4. selected markers and their primer sequences. (DOCX 19 kb)
Figure S2. Annotated plastid genome for P. binnatifidus (PDF 442 kb)
Figure S3. Annotated plastid genome for P. sp. (puxailaileng). (PDF 359 kb)
Figure S4. Annotated plastid genome for P. vietnamensis. (PDF 456 kb)
Figure S5. Annotated plastid genome for P. stipuleanatus. (PDF 440 kb)
Figure S7. ML phylogeny for marker trnC-rps16. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 108 kb)
Figure S8. ML phylogeny for marker trnE-trnM. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 85 kb)
Figure S9. ML phylogeny of marker trnS-trnG. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 116 kb)
Figure S10. ML phylogeny of the marker psbM-trnD. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 102 kb)
Figure S11. ML phylogeny for the concatenated matrix with the four markers, trnC-rps16, trnS-trnG, trnE-trnM and psbM-trnD. The bootstrap values are represented in italic on the branches. The red branches represent supported species delimitation. (PDF 108 kb)
Figure S6. Results of the mPTP species delimitation analysis on the full plastid genome matrix. The red lines illustrate the branches representing speciation and the brown lines the branches representing coalescence processes. The numbers on the branches represent the Bayesian posterior probabilities for the delimited species. (PDF 145 kb)
Data Availability Statement
The raw sequence data from the P. bipinnatifidus, P. stipuleanatus, and P. vietnamensis samples have been to submitted to GenBank on the following accessions: SRR5725242, SRR5725240, SRR5725505, SRR5725492, SRR5738925, SRR5738922, SRR5738920, SRR5738920, SRR5738927.The DNA matrix used for the phylogenomic analyses are available on Open Science Framework, (10.17605/OSF.IO/Z7RWE). The plastome sequences of P. bipinnatifidus, P. stipuleanatus, and P. vietnamensis (2) have been submitted to NCBI GenBank, (MF377620, MF377621, MF377622, MF377623).