

Abstract
Many intrinsically disordered proteins (IDPs) form fuzzy complexes upon binding to their targets. Although many IDPs are weakly bound in fuzzy complexes, some IDPs form high-affinity complexes. One example is the nonstructural protein 1 (NS1) of the 1918 Spanish influenza A virus, which hijacks cellular CRKII through the strong binding affinity (Kd ∼10 nM) of its proline-rich motif (PRMNS1) to the N-terminal Src-homology 3 domain of CRKII. However, its molecular mechanism remains elusive. Here, we examine the interplay between structural disorder of a bound PRMNS1 and its long-range electrostatic interactions. Using x-ray crystallography and NMR spectroscopy, we found that PRMNS1 retains substantial conformational flexibility in the bound state. Moreover, molecular dynamics simulations showed that structural disorder of the bound PRMNS1 increases the number of electrostatic interactions and decreases the mean distances between the positively charged residues in PRMNS1 and the acidic residues in the N-terminal Src-homology 3 domain. These results are analyzed using a polyelectrostatic model. Our results provide an insight into the molecular recognition mechanism for a high-affinity fuzzy complex.
Introduction
There is growing interest in understanding molecular recognition mechanisms between proline-rich motifs (PRMs) and their cognate domains such as Src-homology 3 (SH3) domains because PRM is one of the most common linear motifs in the eukaryotic proteome (1, 2). PRMs are also highly enriched in intrinsically disordered proteins (IDPs) or intrinsically disordered regions (IDRs) (3). Although they lack stable conformations, IDPs/IDRs mediate protein-protein interactions in signal transduction or transcription regulation (4, 5, 6). Hence, it is of importance to understand the molecular mechanisms determining the binding affinity and selectivity of IDRs. Recent studies have shown that some IDPs/IDRs retain substantial conformational flexibility even in a complex with their binding partner. This mode of complex was described as a fuzzy complex (7). Given the growing importance of fuzzy complexes in biological processes, elucidating their molecular recognition mechanism is essential.
The nonstructural protein 1 (NS1) of influenza A viruses (IAVs) plays an important role in suppressing the antiviral immune responses of host cells (8, 9). Unlike common seasonal IAVs, the 1918 Spanish IAV uses NS1 to hijack cellular signaling adaptor CRKII (10). Recently, we have shown that the viral hijacking occurs because a PRM in the NS1 binds to the N-terminal SH3 (nSH3) domain of CRK with an exceptionally high affinity (11). The binding affinity of the nSH3 domain with PRMNS1 is significantly higher than its interactions with other cellular binding partners. For example, the nSH3 domain binds PRMNS1 with ∼3000-fold higher affinity than the PRM of JNK1 (11, 12). It was suggested that the high affinity is due to long-range electrostatic interactions between positively charged residues in PRMNS1 and a negatively charged binding interface in the nSH3 domain (11). However, the molecular mechanism by which long-range electrostatic interactions increase the affinity of PRMNS1 to the nSH3 domain remains elusive.
Long-range electrostatic interactions play important roles in the molecular recognition in fuzzy complexes (13, 14). Borg et al. (15) proposed a polyelectrostatic model, in which multiple charges in an IDP increase its binding affinity to a rigid partner through nonspecific, long-range electrostatic interactions as a binding mechanism of a fuzzy complex. In this model, fuzzy complexes undergo fast conformational exchanges between multiple conformations, interacting with a folded partner through long-range electrostatic interactions.
Here, we provide evidence that PRMNS1 retains its conformational flexibility when complexed with the nSH3 domain, indicating a fuzzy complex. In addition, we present the contribution of long-range electrostatic interactions and a detailed structural mechanism by which long-range electrostatics can increase the binding affinity of PRMNS1 to the nSH3 domain. We obtained these results using a combination of x-ray crystallography, NMR dynamics experiments, and molecular dynamics (MD) simulation. We find that structural disorder of the bound PRMNS1 plays a key role in mediating the long-range, nonspecific electrostatic interactions with the nSH3 domain.
Materials and Methods
Protein and peptides
All protein samples for crystallization, fluorescence, and NMR experiments were prepared as described elsewhere (12). Synthetic peptides were purchased in a crude form and further purified using reverse-phase high-performance liquid chromatography in our laboratory. The amino acid sequence of PRMNS1 is YG210RPPLPPKQKRK221. The N- and C-termini of peptides were acetylated and amidated, respectively. The peptide concentration was determined by measuring the ultraviolet absorption at 280 nm of a single tyrosine at the N-terminal end of the peptide. The isotope-labeled PRMNS1 contained an additional Gly residue at the C-terminus to prevent proteolysis during bacterial expression and purification. Based on our structure, we assumed that one additional Gly at the C-terminus does not affect the structure and dynamics of PRMNS1 substantially.
Crystallization and structure determination
Four millimolar nSH3 was mixed with 5 mM PRMNS1 for the crystallization trials. The sample was crystallized by the hanging drop vapor diffusion method in 0.1 M sodium acetate (pH 4.6), 30% PEG 2000, and 0.2 M ammonium sulfate, which is the duplicate of the previous crystallization condition (Protein Data Bank (PDB): 5UL6) for the nSH3:PRMNS1 complex. A 1.75 Å resolution data set was collected at 120 K using an R-AXIS IV2+ image plate detector mounted on a Rigaku MicroMax 007HF x-ray generator (Rigaku, The Woodlands, TX). The data were processed using iMosflm in the CCP4 package (16). The structure was determined using the nSH3 domain model (PDB: 5UL6) as a search model and the Phenix software suite (17). The NS1 PRM peptide was modeled into the difference map manually in COOT (18) and refined with Phenix. The electrostatic potential surface of nSH3 domain was calculated using APBS and PDB: 2PQR (19, 20, 21).
NMR spectroscopy
All NMR experiments were conducted using protein samples in 20 mM sodium phosphate (pH 6.1), 80 mM NaCl, 0.02% sodium azide, 1 mM EDTA, 10 μM DSS (4,4-dimethyl-4-silapentane-sulfonate), and 10% D2O at 25°C. NMR data for PRMNS1 were acquired using a sample containing isotope-labeled PRMNS1 (490 μM) and nonlabeled nSH3 domain (500 μM). NMR data for the nSH3 domain were acquired using a sample containing isotope-labeled nSH3 domain (320 μM) and nonlabeled PRMNS1 (350 μM). The temperature-dependent heteronuclear single quantum coherence (HSQC) spectra were acquired using a Bruker 500 MHz spectrometer (Bruker, Billerica, MA). Each spectrum was acquired with 64 scans per t1 point. The temperatures of NMR samples were calibrated using deuterated methanol-d4 (22). The assignment of backbone 1H, 13C, 15N resonances was carried out using Bruker 600 and 800 MHz spectrometers, as described elsewhere (12). NMR spectra were processed with NMRPipe (23) and analyzed with NMRViewJ (One Moon Scientific, Westfield, NJ) and CARA (24).
Measurement of NMR relaxation parameters
All relaxation parameters were measured at 25°C using a Bruker 600 MHz spectrometer. For the heteronuclear nuclear Overhauser effect (NOE) measurements, a recycle delay of 12 s was used in the reference experiment. The saturation of proton during the steady state was performed by applying 180° pulses for 4 s (25). For R2 measurement, a recycle delay of 2 s was used between transitions. Errors of the relaxation parameters were estimated using duplicated measurements.
MD simulation
Simulations were performed using CHARMM version 40a1 with param36 all-atom force field (26, 27). An initial energy optimization was carried out for 1600 steps (with 400 steps using the steepest descent method and 1200 steps using the adopted basis Newton-Raphson method) in the generalized Born with a simple switching implicit solvent (28). The protein complex was solvated using the TIP3P water model and electrically neutralized with Cl− and Na+ ions. The system was heated from 0 to 300 K for 100 ps and then equilibrated for 160 ps under the constant number, pressure, temperature ensemble at 1 atm and 300 K. Production runs were performed under the constant number, volume, temperature ensemble at 300 K.
Cluster analysis
With backbone heavy atoms in the nSH3 domain as a positional reference, PRMNS1 conformations from the MD trajectories were analyzed using a cluster analysis (29). Agglomerative hierarchical clustering was used based on the similarity between different conformations, which is measured as the squared Euclidean distance of the backbone heavy atoms in PRMNS1. The average linkage was chosen to measure intercluster similarity, which is the average of all pairwise similarities between observations in two clusters. The representative conformation of each cluster was chosen to have the smallest root-mean-square deviation from the calculated average structure for the cluster.
Binding assay
The dissociation constants (Kd) of nSH3:PRM complexes were measured by monitoring the change of tryptophan fluorescence signal. The excitation wavelength was 295 nm. All binding assays were performed in a stirred 1-cm-pathlength cuvette using a PTI QM-400 fluorimeter. The protein concentration used for the fluorescence-based binding assays was 0.1 μM. The measurements were done in 20 mM sodium phosphate (pH 6.1) and either 80 mM or 1 M NaCl at 25°C. The Kd was calculated by assuming a 1:1 complex and by the global fitting of the repeatedly measured fluorescence intensities to Eq. 1:
| (1) |
where ΔF and ΔFmax are the change and the maximum amplitude of signal change, respectively. Pt is the total protein concentration and Lt is the total ligand concentration at each titration point. The reported Kd values are the average of two repeated measurements.
Results and Discussion
Long-range electrostatic interaction increases the binding affinity of nSH3:PRMNS1 complex
It was previously suggested that positively charged residues, particularly in the C-terminal region of PRMNS1, interact with a large negatively charged surface of the nSH3 domain through long-range electrostatic interactions (11). To directly test the contribution of long-range electrostatics in increasing the binding affinity, we measured Kd values of the nSH3:PRMNS1 complex in the presence of 80 mM and 1 M NaCl. The Kd increased by more than 100-fold in the presence of 1 M NaCl, compared to that in the presence of 80 mM NaCl (Fig. 1).
Figure 1.
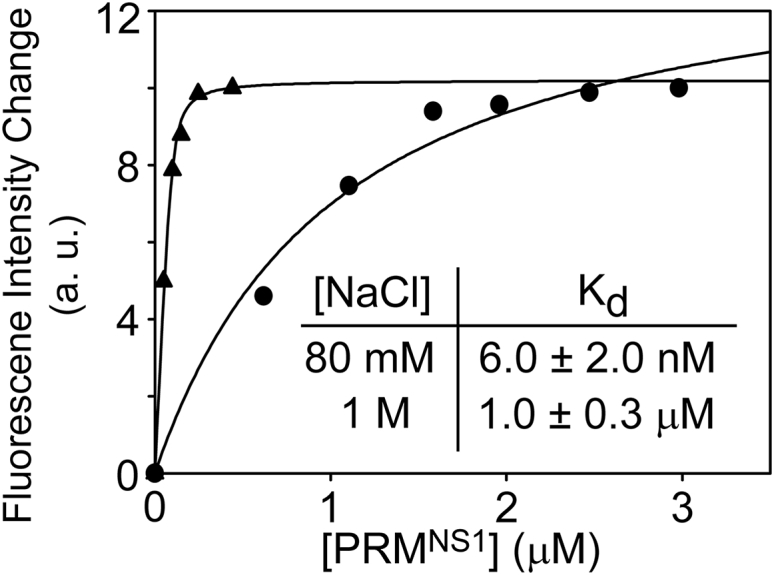
Fluorescence-based measurement of binding affinity between the nSH3 domain and PRMNS1 in the presence of 80 mM (triangles) and 1 M (circles) NaCl. The inset shows Kd values from repeated measurements.
We attributed the large decrease in the affinity to the screening of long-range electrostatics in the complex. The Debye screening lengths are 10.7 and 3.0 Å in the solution, with ionic strengths of 80 mM and 1 M, respectively. Hence, long-range interactions are screened in a high ionic strength solution, whereas the short-range electrostatic interactions within 3 Å remain effective under the same condition. Indeed, our previous crystal structure indicated that the distances of all short-range electrostatic interactions in the nSH3:PRMNS1 are within 3 Å (Fig. S1). Moreover, all these short-range electrostatic interactions in the nSH3:PRMNS1 complex are well-conserved in the complexes with much weaker affinities (12, 30), indicating that these specific salt bridges are not responsible for the unusually high affinity of PRMNS1.
We further examined the effect of the positive charges in PRMNS1 on the binding affinity using mutagenesis. We replaced all the C-terminal positively charged residues (K219, R220, and K221) with Gly. We chose Gly to minimize the effect of other types of amino acids on the intrinsic conformational propensity of PRMNS1 and to retain the flexibility of the C-terminal region. As expected, the mutant PRMNS1 showed significantly lower affinity to the nSH3 domain. Its Kd is 0.9 μM, which is similar to that shown in the presence of 1 M NaCl (Fig. S2). Taken together, these results show that the long-range electrostatic interactions play an important role in increasing the stability of the nSH3:PRMNS1 complex.
New crystal structure displays alternative binding mode of bound PRMNS1
In this study, we determined a new—to our knowledge—crystal structure of the nSH3:PRMNS1 complex that displays a different binding mode of PRMNS1 from that of the previous structure (Fig. 2; Fig. S3; Table S1). To distinguish the two conformations of PRMNS1, the previous and the current structures are labeled as PRMNS1A and PRMNS1B, respectively. The two complex structures showed similar changes in overall solvent-accessible surface area upon complexation: 923 and 1080 Å2 for nSH3:PRMNS1A and nSH3:PRMNS1B, respectively. The two bound PRMNS1 showed virtually identical structures with respect to the N-terminal region and core PPLPPK motif (Fig. 2 B), including all intermolecular short-range electrostatic interactions mediated by K217.
Figure 2.

Comparison of crystal structures between the nSH3:PRMNS1A (green, PDB: 5UL6) and nSH3:PRMNS1B (cyan, PDB: 6ATV) complexes. (A) shows the overlaid structure of the nSH3 domains. (B) shows the overlaid structures of PRMNS1A and PRMNS1B. To see this figure in color, go online.
In contrast, the C-terminal regions of bound PRMNS1 showed a large conformational difference (Fig. 2 B). This is due to large changes in the backbone φ/ψ angles of Q218, from −82.0°/−27.5° in PRMNS1A to −122.9°/176.9° in PRMNS1B (Fig. 2 B). This results in drastic changes in the intermolecular interactions mediated by positively charged residues in the C-terminal end of Q218 in PRMNS1 (Fig. S4). The most notable change is the exchange of positions between R220 and K219. In PRMNS1A, K219 is the major residue interacting with acidic residues in the RT-loop of the nSH3 domain. In PRMNS1B, R220 occupies the position and K219 is fully exposed to solvent (Fig. 3, A and B). Another noticeable change is the hydrogen bonds mediated by E166 in the nSH3 domain. E166 forms a hydrogen bond with the backbone amide of Q218 in PRMNS1A, but it forms an additional hydrogen bond with the side chain NHε of R220 in PRMNS1B (Fig. 3, C and D). These results suggest that the bound PRMNS1 may undergo conformational exchange between the two alternative binding modes in solution state.
Figure 3.

Conformational difference in the C-terminal region of bound PRMNS1. The changes in the positions of K219 and R220 in PRMNS1A and PRMNS1B are displayed in (A) and (B). Hydrogen bonds between E166 of the nSH3 domain (gray) and Q218 in PRMNS1A are shown in (C), and those between Q218 and R220 in PRMNS1B are shown in (D). To see this figure in color, go online.
Structural heterogeneity of PRMNS1 probed by NMR spectroscopy
Using NMR spectroscopy, we examined whether the alternative binding of PRMNS1 observed by crystallography is populated in solution state. The NMR 1H-15N HSQC spectrum of 15N-PRMNS1 bound to the 14N-nSH3 showed that the backbone HN resonance of Q218 shifted significantly downfield (10.47 ppm) because of the hydrogen bond with E166 of the nSH3 as shown in the crystal structures (Figs. 3 C and 4 A). However, the peak broadened significantly at 25°C, indicating a chemical exchange between alternative conformations in intermediate NMR timescale. When we lowered the temperature of the NMR sample to 6°C, the backbone amide peak of Q218 was split into two peaks, major and minor, indicating that the exchange between the two conformations is in the slow-exchange regime (Fig. 4 A). In contrast, other C-terminal residues showed one set of peaks at both 6 and 25°C (Fig. S5).
Figure 4.

1H-15N HSQC spectra of bound PRMNS1. (A) displays the backbone amide resonances of Q218 and (B) the side-chain HNε resonances of R220 at 25 (black) and 6°C (blue). To see this figure in color, go online.
To examine whether these split peaks correspond to the two crystallographic conformations of PRMNS1, we compared the calculated backbone 1H/15N chemical shifts of Q218 in PRMNS1A and PRMNS1B with those of the major and minor peaks observed at 6°C. We used SHIFTX2 for the calculation of chemical shifts from the crystal structures (31). The calculated 1H/15N chemical shifts of Q218 were 9.64/122.63 and 9.24/121.58 ppm for PRMNS1A and PRMNS1B, respectively. The differences in the calculated chemical shifts between PRMNS1A and PRMNS1B are comparable to those between the major and minor peaks, although the absolute chemical shifts differ between experimental and calculated values. This indicates that PRMNS1A and PRMNS1B might correspond to minor (downfield-shifted) and major (upfield-shifted) peaks, respectively (blue peaks in Fig. 4 A).
We also observed that the side-chain NHε peak of R220 in the bound PRMNS1 was split into at least two peaks at 6°C (Fig. 4 B). Although we noticed another minor peak besides the two peaks, its intensity was too low for assignment. Hence, it was not considered in this study. The major peak (Hε = 7.53 ppm) shifted more noticeably downfield than the minor peak (Hε = 7.34 ppm). The SHIFTX2 calculation also yielded noticeably different chemical shifts for Hε-R220 in the two crystal structures: 7.26 and 8.09 ppm for PRMNS1A and PRMNS1B, respectively. This is reasonable because the interaction between Hε-R220 and E166 of the nSH3 is present in PRMNS1B, but not in PRMNS1A. However, it should be noted that the chemical shift differences only allow indirect structural inference, and further structural characterization will be required for unambiguous conclusion.
For further comparison, we analyzed the intermolecular NOESY spectrum between PRMNS1 and the nSH3 domain using 15N-labeled PRMNS1 bound to nonlabeled nSH3 domain (Fig. S6). However, the C-terminal residues only showed short-range intramolecular (i.e., within PRMNS1) crosspeaks, which are not characteristic enough for structural comparison with the crystal structures. This result indicates that the C-terminal positively charged residues in PRMNS1 remain flexible when undergoing conformational exchange in the slow NMR timescale at 6°C.
Dynamic disorder of PRMNS1 probed by NMR spectroscopy and MD simulation
We characterized the conformational flexibility of bound PRMNS1 using NMR relaxation experiments. The {1H}-15N heteronuclear NOE reports on backbone dynamics in a picosecond timescale (32). Higher (>0.7) NOE values correspond to ordered regions, and lower (<0.7) or negative NOE values indicate high conformational flexibility in the picosecond to nanosecond timescale. Overall, NOE values for PRMNS1 were lower than those for the nSH3 domain (Figs. 5 A and S7). Interestingly, even the residues in the PRM-binding surface of the nSH3 domain showed higher NOE values than the bound PRMNS1. This feature has been observed in other fuzzy complexes (15, 33) in which a bound IDP/IDR retains high conformational flexibility on the surface of a rigid protein. Although the residues in the core PPLPPK sequence showed relatively high NOE values (∼0.65), the N- and C-terminal residues were highly disordered (NOE < 0.4). These results indicate that the nSH3:PRMNS1 complex is a partial fuzzy complex, in which the bound PRMNS1 is relatively rigid in the core region but highly flexible in the terminal regions.
Figure 5.
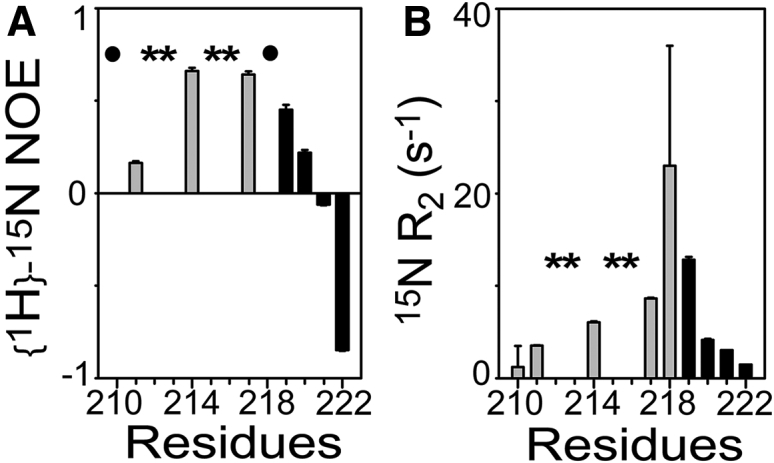
(A) {1H}-15N heteronuclear NOE and (B) 15N R2 of PRMNS1 in the complexed state. The asterisks represent the positions of Pro. The solid circles represent the positions of the residues whose peak intensities are too weak to measure. Black bars correspond to the C-terminal residues showing different conformations in the two crystal structures.
We also observed that the bound PRMNS1 is flexible in microsecond to millisecond timescales. At 25°C, the 15N NMR R2 values of K217, Q218, and K219 of PRMNS1 were noticeably elevated compared to those of other residues. The elevated R2 value indicates the presence of conformational dynamics of bound PRMNS1 in microsecond to millisecond timescales (Fig. 5 B) (34). In particular, the linewidth of Q218 was severely broadened such that accurate measurement of its R2 value was not possible. This is consistent with the structural heterogeneity of this region observed in the crystal structures. Hence, exchange between the two conformations PRMNS1A and PRMNS1B might be responsible for the elevated R2 values.
The elevated R2 values are not due to the on-off process of PRMNS1 to the nSH3 domain because R2 values of the PRMNS1 binding sites in the nSH3 domain are not affected (Fig. S6). Our previous study has shown that the binding-unbinding processes of PRMNS1 induces significant NMR line broadening in the nSH3 domain (11). However, we cannot exclude the possibility that unidentified conformations are involved in mediating the conformational exchange processes. These results indicate that conformation of PRMNS1 is highly dynamic over broad timescales ranging from picoseconds to milliseconds.
To further elucidate the conformational dynamics of PRMNS1 in the bound state, we performed two all-atom 100 ns MD simulations, each using one of the crystal structures as the starting structure (Fig. 6). The two simulations did not converge well with each other during the 100 ns simulation time, indicating that the conversion between the two PRMNS1 conformations is slower than the simulation timescale. This is because the conversion requires large changes in the φ/ψ angles of Q218 associated with breakage and reformation of multiple interactions, such as E166nSH3-Q218PRMNS1 and E149nSH3-R220PRMNS1. Consistent with this, R220 formed a stable hydrogen bond with E149nSH3 with 89% occupancy in the simulation of PRMNS1B, whereas it did not form in the simulation of PRMNS1A. The occupancy of E166nSH3-Q218PRMNS1 interaction was 17% and 64% in the simulation of PRMNS1A and PRMNS1B, respectively. These results are consistent with our NMR data, indicating that N- and C-terminal regions undergo conformational exchanges in the microsecond to millisecond timescale.
Figure 6.

MD simulations of the nSH3:PRMNS1 complexes. In (A), representative PRMNS1 structures were selected from cluster analysis (see Materials and Methods) of the MD trajectories using PRMNS1A (green) and PRMNS1B (cyan). The positively and negatively charged residues are shown in blue and red, respectively. Side chains were omitted in (B) for clarity. To see this figure in color, go online.
Effects of structural disorder of bound PRM on long-range electrostatic interactions with the nSH3 domain
It was proposed that the polyelectrostatic model explains the thermodynamic contribution of long-range electrostatic interactions in a fuzzy complex (15). According to the model, Kd decreases exponentially as the net charge of a disordered ligand increases. We have previously shown that the binding affinity between the nSH3 domain and PRMs depends exponentially on the net charge of PRM (11). However, the bound PRMNS1 is partially fuzzy in that its terminal regions are highly flexible and its core PxxP sequence is relatively rigid. Thus, it remains to be determined whether the partial fuzziness of the bound PRMNS1 increases the multiplicity of long-range electrostatic interactions with the nSH3 domain as assumed in the polyelectrostatic model. This is an important question because many fuzzy complexes might be partially fuzzy.
However, it is difficult to test whether the structural disorder increases the number of long-range electrostatic interactions because there is no straightforward experimental method to tune the conformational flexibility of the bound ligand. As an alternative approach, we compared the number of long-range electrostatic interactions between MD trajectories and crystal structures that represent mobile and static states of the bound PRMNS1, respectively.
We first identified acidic residues in the nSH3 domain that are involved in electrostatic interactions with the positive charges in the bound PRMNS1 by measuring changes in the side-chain carboxyl carbon chemical shifts (13Cγ/δO) of all acidic residues in the nSH3 domain between the free and complexed states. This analysis identified six acidic residues (D142, D147, E149, D150, D163, and E166) (Fig. 7 A) that are located in and around the PRMNS1-binding interface in the nSH3 domain (Fig. 7 B). Next, we measured the mean pairwise distances between the acidic residues in the nSH3 domain and positively charged residues in PRMNS1 in our MD simulations and in the two crystal structures (Table S2). In this analysis, K217 in PRMNS1 was excluded because it is involved in well-defined short-range electrostatics, which are also present in other nSH3:PRM complexes. Interestingly, the mean pairwise distances calculated from the MD trajectories were considerably shorter than those calculated from the crystal structures (Table 1; Table S2). One obvious exception in Table 1 was the distance between K221 in PRMNS1 and D163 in the nSH3 domain, which is shorter in the crystal structures because of the lattice contacts around K221. The distance would be longer without the lattice contacts. These results indicate that partial disorder of the bound PRMNS1 increases the number of long-range electrostatic interactions, as was assumed in the polyelectrostatic model.
Figure 7.

Detection of long-range electrostatic interactions between the nSH3 domain and PRMNS1 using NMR spectroscopy. (A) shows the 13Cγ/δ chemical shift perturbations of all acidic residues in the nSH3 domain upon the binding of PRMNS1. (B) gives the positions of acidic residues whose 13Cγ/δ chemical shifts are perturbed noticeably. Overlaid structures of PRMNS1A (green) and PRMNS1B (cyan) are shown. To see this figure in color, go online.
Table 1.
Differences in Average Pairwise Distances between MD and Crystal Structures
| Residues in PRMNS1 | Difference in Average Pairwise Distances (MDa–Crystalb) (Å) |
|||||
|---|---|---|---|---|---|---|
| Acidic Residues in the nSH3 Domain | ||||||
| D142 | D147 | E149 | D150 | D163 | E166 | |
| R211 | negative 0.9 | –c | –c | –c | –c | –c |
| K219 | –c | negative 2.3 | negative 1.3 | negative 2.2 | 0.9 | negative 0.5 |
| R220 | –c | negative 1.0 | negative 0.4 | negative 0.8 | negative 1.1 | negative 2.2 |
| K221 | –c | negative 3.4 | negative 0.9 | negative 1.5 | 3.8 | negative 0.4 |
Average distances of MD simulations of PRMNS1A and PRMNS1B.
Average distances of two crystal structures (PRMNS1A and PRMNS1B).
These distances were longer than 15 Å and were not included in the calculation.
Although the polyelectrostatic model was developed to understand the binding mechanism of a fully fuzzy complex, its validity for partially fuzzy complex has not rigorously tested. Many IDP/IDR mediated complexes may be partially rather than fully disordered. Therefore, our results provide mechanistic insights into the role of nonspecific, long-range electrostatic interactions in other partially fuzzy complexes as well.
Conclusions
Many viral proteins interact with host modular binding domains such as SH3 domains by mimicking the cellular linear binding motifs (35, 36). Although cellular PRMs bind to their cognate SH3 domains with weak affinities (Kd ∼10 μM) (11, 12, 37), viral PRMs often show significantly higher affinities to the target SH3 domains (11, 38, 39). Both viral and cellular PRMs contain conserved core PxxP motif, but peripheral sequences in viral PRMs contain more charged residues than the ones in the cellular PRMs (38, 39, 40). Our study highlights that the pandemic IAV NS1 hijacks cellular CRK by exploiting the long-range electrostatic interactions mediated by the disordered peripheral region of its PRM. Recently, it was indicated that the amphiphysin-2 SH3 domain and PRM in NS3 of the Chikungunya virus bind via polyelectrostatic interactions mediated by the positively charged disordered region in the PRM (38). Interestingly, the binding affinity of this complex was also high (Kd = 24 nM). It is of interest that both viral proteins employ a similar strategy to hijack host signaling proteins containing SH3 domains. Therefore, it is likely that conformational dynamics within a complex are closely related to the functional roles of many SH3:PRM complexes (41). We anticipate that elucidating the molecular mechanisms underlying the binding affinity and selectivity of SH3:PRM interactions will help us understand host protein-protein interactions and design an inhibitor of viral infections.
Author Contributions
J.-H.C. conceived the research. Q.S., D.Z., B.Z., P.L., and J.-H.C. conducted experiments. J.S. and W.H. conducted MD simulations. J.-H.C. and W.H. wrote the manuscript. All authors reviewed the manuscript.
Acknowledgments
Work in the Cho lab was supported by start-up funds from Texas A&M University.
Editor: Monika Fuxreiter.
Footnotes
Qingliang Shen and Jie Shi contributed equally to this work.
Seven figures and two tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(18)30190-5.
Contributor Information
Wonmuk Hwang, Email: hwm@tamu.edu.
Jae-Hyun Cho, Email: jaehyuncho@tamu.edu.
Supporting Material
References
- 1.Rubin G.M., Yandell M.D., Lewis S. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li S.S.-C. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem. J. 2005;390:641–653. doi: 10.1042/BJ20050411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theillet F.-X., Kalmar L., Uversky V.N. The alphabet of intrinsic disorder: I. Act like a Pro: On the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. Proteins. 2013;1:e24360. doi: 10.4161/idp.24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 5.Mittag T., Kay L.E., Forman-Kay J.D. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010;23:105–116. doi: 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- 6.Uversky V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015;282:1182–1189. doi: 10.1111/febs.13202. [DOI] [PubMed] [Google Scholar]
- 7.Tompa P., Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Krug R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr Opin Virol. 2015;12:1–6. doi: 10.1016/j.coviro.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finkelstein D.B., Mukatira S., Naeve C.W. Persistent host markers in pandemic and H5N1 influenza viruses. J. Virol. 2007;81:10292–10299. doi: 10.1128/JVI.00921-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heikkinen L.S., Kazlauskas A., Saksela K. Avian and 1918 Spanish influenza a virus NS1 proteins bind to Crk/CrkL Src homology 3 domains to activate host cell signaling. J. Biol. Chem. 2008;283:5719–5727. doi: 10.1074/jbc.M707195200. [DOI] [PubMed] [Google Scholar]
- 11.Shen Q., Zeng D., Cho J.H. The molecular mechanisms underlying the hijack of host proteins by the 1918 Spanish influenza virus. ACS Chem. Biol. 2017;12:1199–1203. doi: 10.1021/acschembio.7b00168. [DOI] [PubMed] [Google Scholar]
- 12.Bhatt V.S., Zeng D., Cho J.H. Binding mechanism of the N-terminal SH3 domain of CrkII and proline-rich motifs in cAbl. Biophys. J. 2016;110:2630–2641. doi: 10.1016/j.bpj.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shammas S.L., Crabtree M.D., Clarke J. Insights into coupled folding and binding mechanisms from kinetic studies. J. Biol. Chem. 2016;291:6689–6695. doi: 10.1074/jbc.R115.692715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H.X., Pang X., Lu C. Rate constants and mechanisms of intrinsically disordered proteins binding to structured targets. Phys. Chem. Chem. Phys. 2012;14:10466–10476. doi: 10.1039/c2cp41196b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borg M., Mittag T., Chan H.S. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc. Natl. Acad. Sci. USA. 2007;104:9650–9655. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 17.Adams P.D., Afonine P.V., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Dolinsky T.J., Czodrowski P., Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolinsky T.J., Nielsen J.E., Baker N.A. PDB2PQR: an automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Findeisen M., Brand T., Berger S. A 1H-NMR thermometer suitable for cryoprobes. Magn. Reson. Chem. 2007;45:175–178. doi: 10.1002/mrc.1941. [DOI] [PubMed] [Google Scholar]
- 23.Delaglio F., Grzesiek S., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 24.Keller R. CANTINA Verlag; Goldau, Switzerland: 2004. The Computer Aided Resonance Assignment Tutorial. [Google Scholar]
- 25.Ferrage F., Reichel A., Ghose R. On the measurement of 15N-1H nuclear Overhauser effects. 2. Effects of the saturation scheme and water signal suppression. J. Magn. Reson. 2010;207:294–303. doi: 10.1016/j.jmr.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart K., Foloppe N., Mackerell A.D.J., Jr. Optimization of the CHARMM additive force field for DNA: Improved treatment of the BI/BII conformational equilibrium. J. Chem. Theory Comput. 2012;8:348–362. doi: 10.1021/ct200723y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Im W., Lee M.S., Brooks C.L., 3rd Generalized born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 29.Shao J., Tanner S.W., Cheatham T.E. Clustering molecular dynamics trajectories: 1. characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 30.Wu X., Knudsen B., Kuriyan J. Structural basis for the specific interaction of lysine-containing proline-rich peptides with the N-terminal SH3 domain of c-Crk. Structure. 1995;3:215–226. doi: 10.1016/s0969-2126(01)00151-4. [DOI] [PubMed] [Google Scholar]
- 31.Han B., Liu Y., Wishart D.S. SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cavanagh J., Fairbrother W.J., Skelton N.J. Elsevier Academic Press; Burlington, MA: 2007. Protein NMR Spectroscopy. [Google Scholar]
- 33.Sharma R., Raduly Z., Fuxreiter M. Fuzzy complexes: specific binding without complete folding. FEBS Lett. 2015;589(19 Pt A):2533–2542. doi: 10.1016/j.febslet.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 34.Palmer A.G., 3rd, Kroenke C.D., Loria J.P. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 35.Davey N.E., Travé G., Gibson T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011;36:159–169. doi: 10.1016/j.tibs.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Duro N., Miskei M., Fuxreiter M. Fuzziness endows viral motif-mimicry. Mol. Biosyst. 2015;11:2821–2829. doi: 10.1039/c5mb00301f. [DOI] [PubMed] [Google Scholar]
- 37.Ladbury J.E., Arold S. Searching for specificity in SH domains. Chem. Biol. 2000;7:R3–R8. doi: 10.1016/s1074-5521(00)00067-3. [DOI] [PubMed] [Google Scholar]
- 38.Tossavainen H., Aitio O., Permi P. Structural basis of the high affinity interaction between the alphavirus nonstructural protein-3 (nsP3) and the SH3 domain of amphiphysin-2. J. Biol. Chem. 2016;291:16307–16317. doi: 10.1074/jbc.M116.732412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shelton H., Harris M. Hepatitis C virus NS5A protein binds the SH3 domain of the Fyn tyrosine kinase with high affinity: mutagenic analysis of residues within the SH3 domain that contribute to the interaction. Virol. J. 2008;5:24. doi: 10.1186/1743-422X-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arold S., Franken P., Dumas C. The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure. 1997;5:1361–1372. doi: 10.1016/s0969-2126(97)00286-4. [DOI] [PubMed] [Google Scholar]
- 41.Stollar E.J., Lin H., Forman-Kay J.D. Differential dynamic engagement within 24 SH3 domain: peptide complexes revealed by co-linear chemical shift perturbation analysis. PLoS One. 2012;7:e51282. doi: 10.1371/journal.pone.0051282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.