

SUMMARY
Hematopoiesis, the process of mature blood and immune cell production, is functionally organized as a hierarchy, with self-renewing hematopoietic stem cells (HSCs) and multipotent progenitor (MPP) cells sitting at the very top1,2. Multiple models have been proposed as to what the earliest lineage choices are in these primitive hematopoietic compartments, the cellular intermediates, and the resulting lineage trees that emerge from them3–10. Given that the bulk of studies addressing lineage outcomes have been performed in the context of hematopoietic transplantation, current lineage branching models are more likely to represent roadmaps of lineage potential rather than native fate. Here, we utilize transposon (Tn) tagging to clonally trace the fates of progenitors and stem cells in unperturbed hematopoiesis. Our results describe a distinct clonal roadmap in which the megakaryocyte (Mk) lineage arises largely independently of other hematopoietic fates. Our data, combined with single cell RNAseq, identify a functional hierarchy of uni- and oligolineage producing clones within the MPP population. Finally, our results demonstrate that traditionally defined long-term HSCs (LT-HSCs) are a significant source of Mk-restricted progenitors, suggesting that the Mk-lineage is the predominant native fate of LT-HSCs. Our study provides evidence for a substantially revised roadmap for unperturbed hematopoiesis, and highlights unique properties of MPPs and HSCs in situ.
To probe native lineage relationships in the fully unperturbed bone marrow (BM), we used the Sleeping Beauty (SB) lineage tracing model and TARIS, an improved Tn-integration sequencing technique (Fig. 1a, and Extended Data Fig. 1 and 2)11. Our analysis relies on comparing tags across multiple differentiated populations at different time points to understand the dynamics of lineage coupling, without the need to isolate and transplant prospective progenitor populations (Fig. 1b). We pulsed adult SB mice with doxycycline (Dox) for 2 days and, at 1, 2, 4 and 8 weeks after induction, sorted Tn-labelled (DsRed+) nucleated erythroblasts (Er), megakaryocyte (Mk) progenitors, granulocytes (Gr), monocytes (Mo) and B-cell progenitors (B) (Fig. 1c). Importantly, control experiments demonstrated that negligible amounts of transposition occur 1 day after removal of Dox (Extended Data Fig. 3).
Fig. 1. Clonal analysis of hematopoietic lineage fates in the native bone marrow.
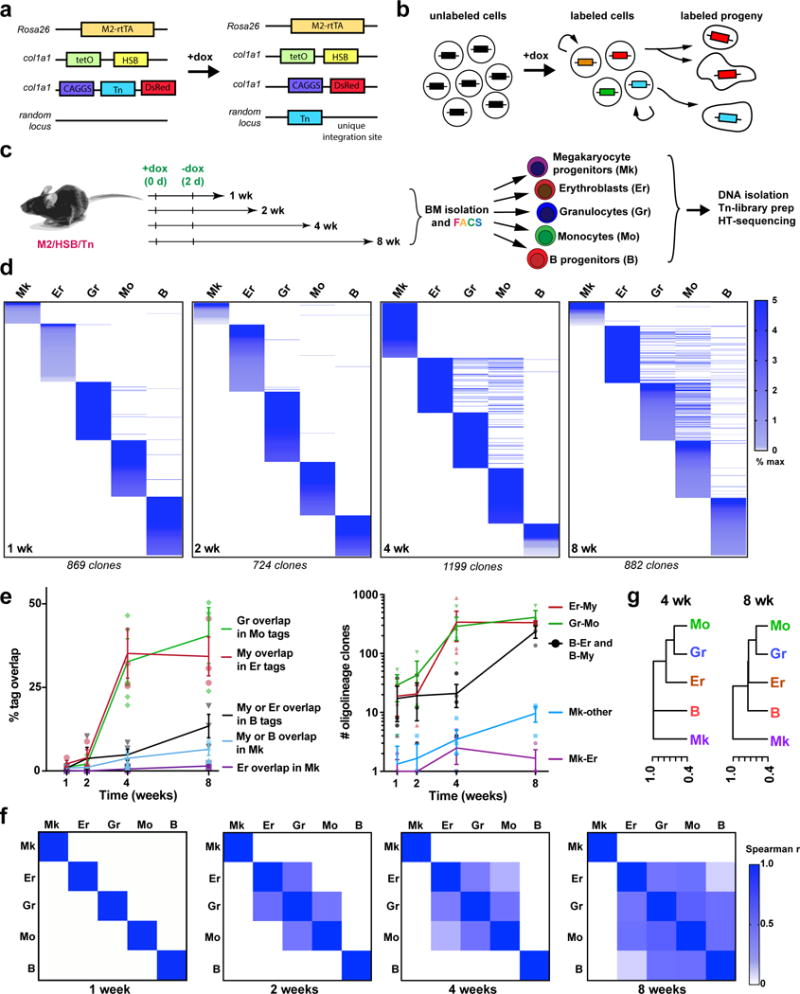
a, M2/HSB/Tn mouse model. Addition of doxycycline (Dox) induces random transposition of the Tn, and concomitant cell labelling with DsRed. The Tn insertion site is stable after removal of Dox. b, Transposon lineage tracing paradigm. Shared tags can be detected between a self-renewing progenitor stem cell and its progeny, or between two different mature cell populations. c, Experimental design. M2/HSB/Tn mice were labelled with Dox for 2 days and 5 blood lineages were isolated from BM after different periods of time. Tn-insertion tag libraries were prepared and sequenced for each population. d, Alignment of Tn tags from different lineage-committed (Lin+) blood cell populations in the BM at 1–8 weeks. Tags are coloured by frequency in each lineage, and organized by rank. Each chart is representative from 3 independent experiments. e, Percentage of clonal overlap between designated lineage pairs (left), and quantification of total number of detected bi/tri-lineage clones at 1–8 weeks (right). My refers to either Gr or Mo lineage. Mean +/− s.d. from 3 independent mice. f, Spearman correlation coefficient (ρ) matrices for all Lin+ tags at 1–8 weeks. Each matrix is average from 3 independent experiments per time point. g, Hierarchical clustering of blood lineages using (1–ρ) as the distance measure (4 and 8 weeks post-labelling).
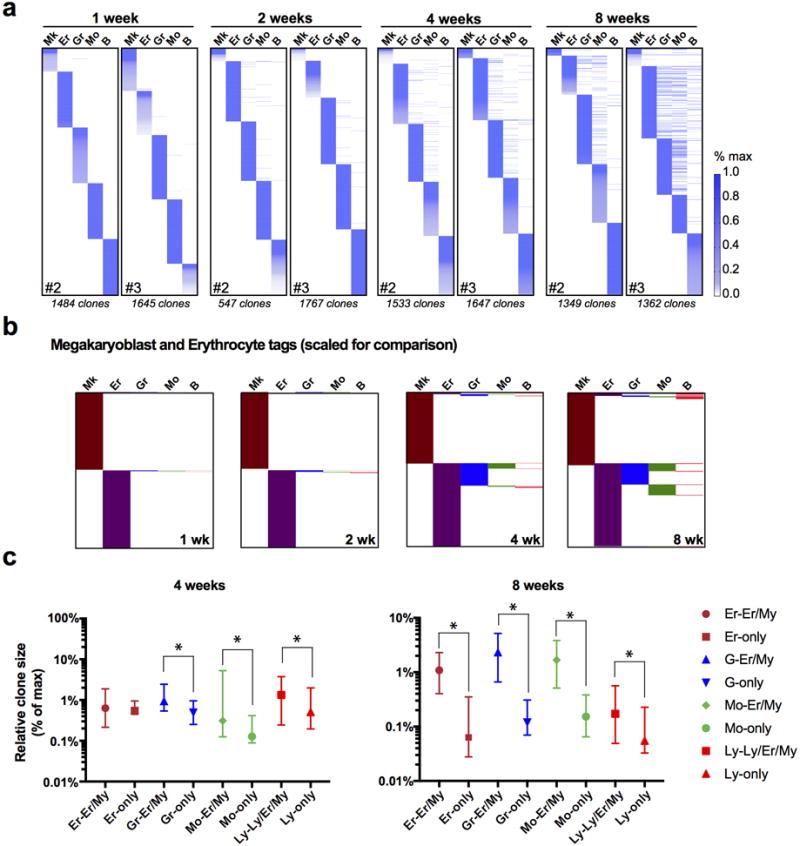
We observed that blood lineages were mostly segregated up until 4 weeks, suggesting their replacement by unilineage progenitors during this first month (Fig. 1d). At 4 weeks, we began to detect a significant number of shared tags across lineages, revealing the activity of common progenitors (Fig. 1d, Extended Data Fig. 4). At 4 weeks, 40.5%(±8.4) of all Mo detected tags (approximately 289±89 clones) were also found in the Gr compartment, confirming their well-established common origin (Fig. 1e)4. Unexpectedly, a similar proportion of Er clones were also found shared with Gr/Mo (My) tags (Fig. 1d–e), revealing a common origin for erythrocytes, granulocytes and monocytes at this stage. Remarkably, we detected virtually no Mk clones that were shared exclusively with Er cells during the whole period of observation, which would have been predicted had a megakaryocyte-erythroid progenitor (MEP)-like cell existed (Fig. 1d–e and Extended Data Fig. 4b)12,13. At 8 weeks, our analysis revealed the activity of a set of multilineage clones (239±58), with lymphoid (B), My and Er contribution, but still with no presence in Mk, indicating the existence of Mk-deficient lympho-erythromyeloid (LEM) progenitors (Fig. 1d and 1e). We did observe a very small (9.7±2.8), yet increasing, number of Mk tags shared with multiple lineages after 8 weeks (Fig. 1e and Extended Data Fig. 4a and b), suggesting that clonal Mk-lineage production can also be associated with multilineage outcomes, although at lower frequencies. Spearman rank correlation analyses of tag read distribution between lineage pairs showed a progressive association of Gr-Mo(My), Er-My and B-My progenitors, segregated from Mk progenitors (Fig. 1f–g). To address potential sampling and sensitivity limitations, we performed independent TARIS amplifications (Extended Data Fig. 5) and clone-specific PCRs (not-shown). Taken together, our results provide evidence for novel lineage couplings during unperturbed hematopoiesis, where the Mk lineage is produced largely independently from the other hematopoietic lineages, and argue for the robust activity of Er-My, Ly-My and LEM progenitor clones.
We next aimed to identify ancestral relationships by comparing the clonal repertoires of differentiated cells and previously defined progenitor populations. Classically, oligopotent progenitors reside in the common myeloid progenitor (CMP), granulocyte-monocyte progenitor (GMP) and MEP phenotypic gates (referred together as myeloid progenitors, or MyPs)4. Our data revealed largely unilineage outcomes for detected MyPs (89.0%±0.8), suggesting that these populations represent a collection of lineage restricted progenitors, functionally validating predictions from single cell expression profiling (Extended Data Fig. 6)14–16. We next focused on the MPPs, the cellular subset proposed to be upstream of MyPs. At 1 and 2 weeks, we observed a small number of ‘active’ MPP tags (overlapping with Lin+ tags), which aligned mostly with single lineages (1 wk: 75.8%±5.0, 2 wk: 66.3%±6.1), suggesting the existence of a small population of lineage-committed MPPs that rapidly produce differentiated progeny (Fig. 2a–b, Extended Data Fig. 7a). MPP output significantly increased at 4–8 weeks for all lineages (9.35%±0.6 of all MPP tags at 8 weeks), consisting mostly of oligolineage Er-My clones (79.2%±5.3 of active MPP clones). A robust number of LEM MPP clones (12±2) were detected beginning at 8 weeks (Fig. 2a), consonant with our analysis of Lin+ fractions (Fig. 1f). Although we also observed oligolineage Mk-producing MPP clones, Mk overlap was more lineage-restricted than any other lineage, even after 8 weeks (Mk: 67.8%±8.0 vs. other: 22.1%±4.6; Fig. 2a–b, Extended Data Fig. 7b), indicating that at least a subset of MPPs is responsible for a stable restricted contribution to the Mk lineage.
Fig. 2. Functional heterogeneity of MPP lineage fates in steady state hematopoiesis.
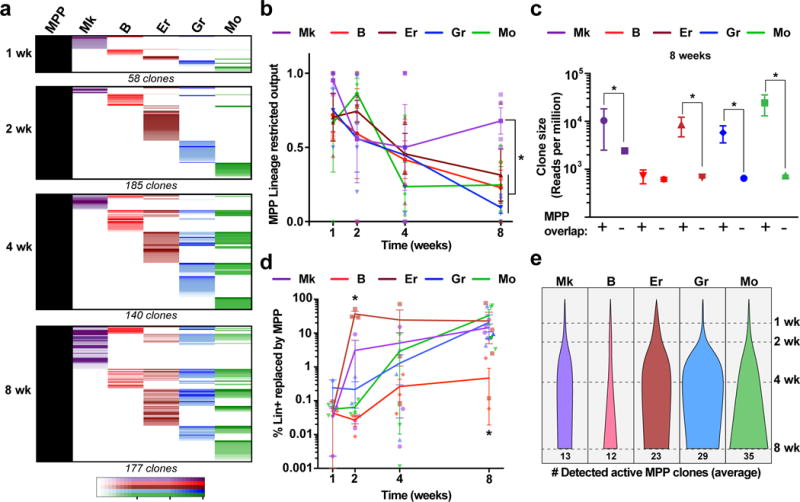
a, Chart shows the alignment of all active MPP tags together with the five analysed blood lineages at each time point (all tags collected from 3 mice per time point). LT-HSC tags were analysed in parallel and excluded from the analysis to represent only MPP behaviour. b, Fraction of active MPP tags that overlap with a single lineage (calculated independently for each lineage). Values are mean +/− s.e.m. from 3 mice. *pMk-Er=0.13, pMk-Gr=0.03, pMk-Mo=0.03, pMk-B=0.001 (8 wk). c, Distribution of Lin+ clone sizes comparing tags overlapping with MPP vs. non-overlapping at 8 wk. Values are median and interquartile ranges of all detected clones from 3 mice. *Kolmogorov-Smirnov pMk=0.03, pB=0.25, pEr=0.03, pGr=0.001, pMo=0.003. d, Fraction of each lineage replaced by MPPs calculated as the percentage of total MPP-overlapping lineage reads over time. Values are mean +/− s.e.m. from 3 independent mice. *pEr-Gr/Mo/B=0.04, pEr-Mk=0.03 (2 weeks) and pB-Er/Mk=0.03, pB-My=0.04 (8 weeks). e, Average number of detected active MPP clones per lineage per mouse at different time points (normalized for %DsRed labelling efficiency).
Our analyses also provided relative quantitative information about the dynamics of lineage replacement by MPPs. For instance, the average clone size of MPP-derived Er-My clones at 8 weeks was 18.3±7.7-fold larger when compared to non-MPP-derived clones, suggesting a significant cellular amplification, in contrast to the B lineage (1.2±0.4-fold; Fig. 2c). In addition, we found that the Er lineage was replaced at the fastest rate, with at least 35% of all Er reads overlapping with MPPs after just 2 weeks, from just a handful of Er-committed MPPs (Fig. 2d and 2e). Comparably, the Gr/Mo-producing MPPs achieved similar levels of replacement only after 2 months. Considering that our analysis cannot measure contribution of MPP clones that disappear from the MPP pool (i.e. by cell death or differentiation), our results likely underestimate the overall MPP contribution.
In order to provide further insight into the heterogeneity and hierarchy of the HSC/MPP compartment, we sorted subsets within these populations using previously described surface markers and interrogated their single cell gene expression landscape using InDrop (Fig. 3a–c)9,17. Louvain-Jaccard clustering analysis of transcriptomes resulted in 12 reproducibly distinct clusters (Fig. 3b). The majority of analysed cells (78.9% of all subsets combined) fit into one of 3 major clusters that we labelled as unprimed (“C1”, “C2”, “C3”) based on the lack of expression of lineage-restricted gene signatures (Supplementary Table 2, Extended Data Fig. 8 and 9). Intriguingly, we also identified several primed clusters (21.1% of HSC/MPPs) that formed branches defined by progressive expression of genes associated with lineage commitment (Fig. 3b–d, right). Predictably, cells indexed as LT-HSCs and MPP1s (also known as short-term HSCs) mostly fit into the “C1” (67.9%) and “C2” (78.3%) clusters, respectively. In contrast, other MPP subsets displayed different degrees of heterogeneity. MPP2s contained the largest proportion of primed cells (59.3%), and MPP4s the least (13.2%) (Fig. 3c–d). MPP2s comprised a larger number of Er-primed (18.7%) and Mk-primed (21.9%) cells, whereas MPP3s contained a larger number of My-primed cells (20.8%) (Fig. 3c–d and Extended Data Fig. 8b). Using Tn tracing, we confirmed that MPP2s presented a preference for Mk production, and generated less oligolineage output (5%±5 of all active clones) within the first week, where their immediate progeny is likely to be measured, compared to MPP3s and MPP4s (40.17%±11.4) (Fig. 3e–f). Analysis of tags not arising from upstream progenitors at 4 weeks revealed similar findings (Fig. 3g–h). On the contrary, MPP4s produced most LEM and multilineage clones (Fig. 3h) and preferentially overlapped with MPP1/ST-HSCs, suggesting that at least a fraction of MPP4s represent direct activated progeny of MPP1/ST-HSCs (Fig. 3i). Combined, our data support the notion that a functional hierarchy, consisting of progenitors at varying degrees of lineage priming, exists already within HSCs/MPPs.
Fig. 3. Transcriptional and functional hierarchy of HSC and MPP subsets.
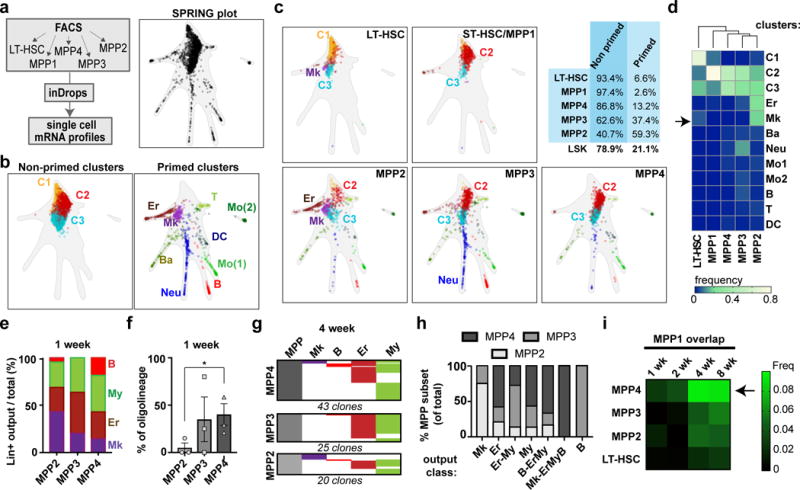
a, Experimental design for inDrops experiment (left). Transcriptional fate map of combined FACS-sorted subsets using the SPRING representation (subsampled in silico to represent proportions of the Lin−Sca1+cKit+ gate. Points represent a single HSC/MPPs distributed according to their similarity using gene expression variation. b, In silico identification of different cell populations within all combined HSC and MPP subsets. Non-primed clusters 1-3 (C1-C3, left) and lineage-primed clusters (right) are presented separated and labelled according to their primed lineage signatures: Neu, neutrophils, DC, dendritic cells, T, T-cell progenitors, B, B-cell progenitors, Ery, erythroid progenitors, Mk, megakaryocyte progenitors: Mo1 and Mo2 represent two monocyte-like signatures. c, Plots showing localization of each sorted HSC/MPP subset within the combined SPRING plot. Top right, fraction of cells from each sorted HSC/MPP subtype (and LSKs) that group within primed or non-primed clusters. d, Hierarchical clustering (Ward) of sorted HSC/MPP subsets. For each FACS sorted population, the fraction of cells corresponding to each cluster was used to analyse the similarity between subsets. The arrow points out the Mk-primed cluster within the LT-HSC gate. e, Fraction of lineage-restricted MPP-overlapping clones corresponding to each lineage, for each MPP subset at 1 week. Values are mean of 3 independent mice. f, Fraction of oligolineage output of each MPP subset after 1 week. Values are mean +/− s.e.m. of three independent mice. *Paired two-tailed t-test (MPP2 vs. MPP4) p=0.033 g, Alignment of Lin+ progeny tags of different MPP subsets (excluding tags present in HSCs/MPP1s) at 4 weeks. h, Fraction corresponding to each MPP subset for each representative lineage fate (including restricted, oligo and multilineage output) at 4 weeks (all tags detected from 4 mice). i, Frequency of MPP2/3/4 tags (and LT-HSC tags) overlapping with MPP1 at 1-8 weeks (average of 3 mice per time point).
Our single cell RNAseq data also revealed that a subset of marker-defined LT-HSCs exhibited Mk-lineage priming (Fig 3c–d, Extended Data Fig. 9). This is in line with previous reports of multipotent, yet platelet-biased subsets of LT-HSCs in the context of transplantation10,18–23. However, the physiological relevance of this observation in native hematopoiesis is unknown. With these precedents, we analysed the Lin+ Tn tag overlap of sorted LT-HSCs. While only a very small number of LT-HSC clones was active 4 weeks after labelling (5.5%±2.3), remarkably, a large majority of these clones were found exclusively in the Mk population (Fig. 4a–b and Extended Data Fig. 10a). This Mk-restricted output of LT-HSCs was more pronounced after 30 weeks post-labelling (Mk:13.3%±5.6, My-Er:3.2%±1.0) (Fig. 4c). Quantitatively, LT-HSCs accounted for replacing at least 31% of the total Mk pool, compared to just 3.8% of My-Er reads (Fig. 4d). Among all Mk cells that had a detectable tag in primitive populations, approximately half demonstrated overlap with LT-HSCs and the other half with MPPs (where no LT-HSC tag was detected) (Extended Data Fig. 10b). MPP-overlapping clones contributed to the Mk lineage to a similar extent as LT-HSCs, drastically differing from Ly-My-Er production, which is predominantly MPP driven (Fig. 4e and Extended Data Fig. 10c). Our analyses also revealed that many LT-HSC contribute to Mk in the absence of any intermediates in the MPP compartment (Fig. 4a), suggesting that at least a subset of LT-HSCs generates Mk lineage cells through a ‘direct’ pathway.
Fig. 4. Steady state megakaryocyte output from bona fide LT-HSCs.
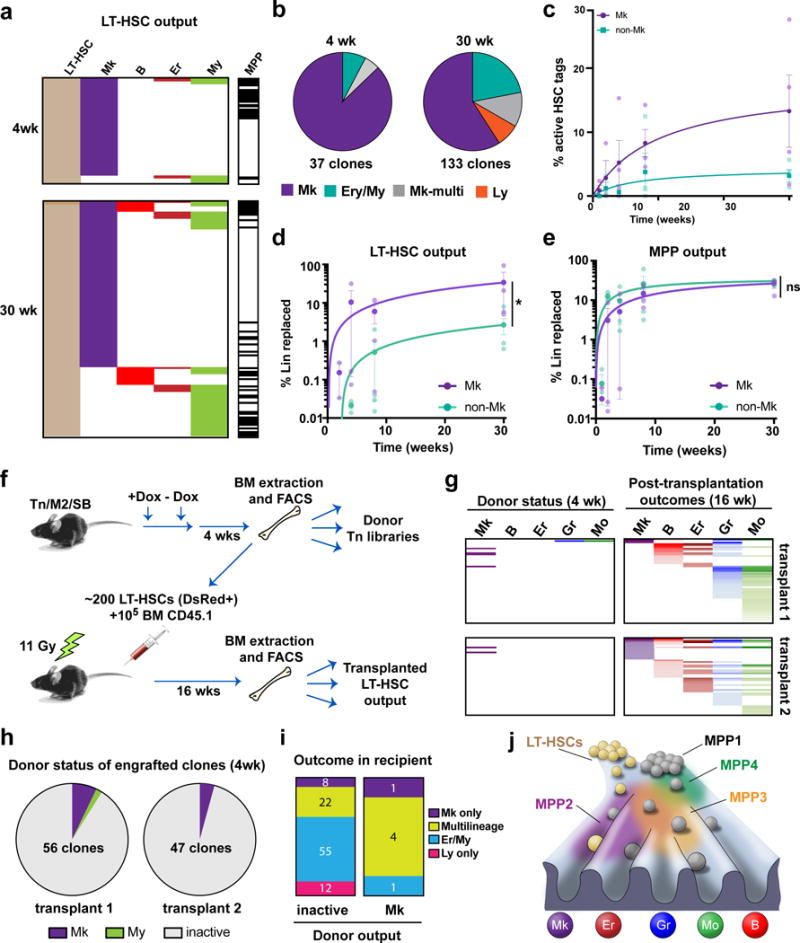
a, LT-HSCs, MPPs and Lin+ cells were purified from bone marrow at 4 and 30 wk and their Tn-tag content was analysed. Only the LT-HSC tags overlapping with detectable Lin+ progeny are shown. b, Pie-chart distribution of types of progeny detected from LT-HSCs at 4 weeks and 30 weeks after labeling. Data are pooled from 4 independent M2/HSB/Tn mice per time point. c, Percentage of labelled LT-HSC clones producing progeny at 1-8 weeks. Values are mean +/− s.e.m of 3-4 independent mice. d, Dynamics of Mk vs. non-Mk lineage replacement by LT-HSCs (measured as % of overlapping/total Lin+ reads). Values are mean +/− s.e.m. of 3-4 independent mice. Ratio paired t-test p=0.014. e, Dynamics of Mk vs. Er/My lineage replacement by MPPs (measured as % of overlapping/total Lin+ reads). Values are mean +/− s.e.m. of 3-4 independent mice. Ratio paired t-test p=0.599. f, Experimental design for parallel analysis of native vs. transplant output of LT-HSC clones. g, Alignment of all post-transplantation LT-HSC-derived lineages with unperturbed donor lineage tags. h, Pie-chart distribution of successfully engrafted LT-HSC clones by donor behaviour. Only Mk-restricted and My-restricted output was observed. Inactive means non-detectable output in the donor. i, Post-transplantation outcomes comparing donor-inactive vs. Mk-producing LT-HSC clones. j, Lineage fate landscape of unperturbed hematopoiesis. Self-renewing LT-HSCs preferentially replace Mk under steady state, and principally contribute to other blood lineages during transplantation or after injury. In contrast, MPPs take care of the majority of steady-state Ly, Er and My blood production. Different MPP sorting gates enrich for heterogeneous collections of lineage-primed and unprimed cell states within a continuum of lineage commitment pathways.
Previous studies have shown that the commonly used LT-HSC gate contains unilineage CD41+ Mk-restricted progenitors as assayed by transplant or culture10,22. To rule out potential contamination by such cells, we aimed to determine whether Mk-producing LT-HSC clones in situ had properties of classical LT-HSCs in the context of transplantation. For this, we transplanted clonally labelled LT-HSCs isolated from mice 4-weeks post induction, and at 16 weeks post-transplantation we purified mature lineages from recipients and compared their Tn repertoires with those of cells initially isolated from the donor (Fig. 4f). We observed that 6 out of 8 detected Mk-restricted LT-HSC clones in the donor were able to generate multilineage progeny in recipients (Fig. 4g–i). We reached similar conclusions when evaluating the culture potential of in situ Mk-producing LT-HSC clones (Extended Data Fig. 10d–e). Additionally, our results demonstrate that Mk-production is not exclusive to the CD41+ LT-HSC fraction (Extended Data Fig. 10f–g). Thus, we conclude that the majority of Mk-producing clones residing in the LT-HSC gate are not simply Mk-restricted progenitors, but clones that can exhibit multipotency upon transplantation.
Our work here uncovers critical features of the native hematopoietic process. In our model, as much as half of the megakaryocytic lineage is produced independently of other lineages by cells at the top of the hematopoietic ladder (Fig. 4j). A heterogeneous hierarchy of lineage-restricted and oligolineage progenitors, historically classified as MPPs, produce other hematopoietic lineages with selective lineage couplings. While our work still supports a model for progressive restriction of developmental potential, it suggests that these events are clonally heterogeneous and occur much earlier in the hematopoietic hierarchy, in line with recent data7,8,14,16. Though our data fail to provide any evidence for CMP or MEP fates in situ, many experiments have provided evidence for MEP-like cells at a clonal level4,12,13,24. We posit that while Mk-Er bipotential exists in transplant or culture setting, this fate is not substantially manifested in unperturbed conditions. Alternatively, such cellular behaviour might be too transient to be captured with our technology.
Our data demonstrate that at least a fraction of LT-HSCs behave as potent Mk-progenitors, indicating that the Mk fate is the predominant fate of HSCs in situ. However, these same cells exhibit potential for multilineage outcomes following transplantation. Thus, our findings highlight the critical differences between studying native fate versus potential in stem cell biology. Although we are unable to conclude whether a particular subset or all LT-HSCs will eventually display Mk-producing behaviour, we favour the idea that most LT-HSC clones transition through a Mk-primed state with age. Our data also suggest that an MPP population (within MPP2) is significantly involved in Mk production. It remains to be determined whether these represent two different pathways for Mk production or whether LT-HSCs are upstream of MPP2s. Finally, our results are still consonant with the idea that adult LT-HSCs have a limited lympho-myelo-erythroid output during steady-state11,25, though this finding has been debated26. Future work with second generation cell barcoding strategies27,28 in combination with Cre-based labelling will be needed to elucidate full lineage histories and determine the mechanisms of fate restriction.
METHODS
Mice
The M2/HSB/Tn mice were generated as previously described11. To induce Tn mobilization 8–10 weeks old male or female mice with the M2/HSB/Tn genotype were fed with 2 mg/ml Dox together with 5mg/ml sucrose in drinking water for 48h. Thereafter, Dox was removed and successful labelling was verified by retro-orbital sinus peripheral blood collection and analysis (70 μl) after 1 week. All animal procedures were approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee. Previous studies have estimated that most hematopoietic lineages are replaced by MPPs within 1–2 months after label25,29–31. Thus, for Lin+ lineage coupling studies, M2/HSB/Tn mice were analysed within the first 8 weeks after labelling. Since MyPs have limited self-renewal capacity and are rapidly replaced by MPPs, we performed the MyP analysis at short time points post-labelling (1 week) and only considered Tn tags not simultaneously present in MPPs.
Bone marrow preparation
After euthanasia, whole BM (excluding the cranium) was immediately isolated in 2% fetal bovine serum (FBS) in phosphate buffered saline (PBS), and erythrocytes were removed with red blood cell lysis buffer. CD45.1 (Ly5.1) mice were used as transplantation recipients (B6.SJL-Ptprca Pep3b/BoyJ, stock # 002014, the Jackson Laboratory).
Fluorescence activated cell sorting (FACS)
Lineage depletion was performed using Magnetic Assisted Cell Sorting (Miltenyi Biotec) with anti-biotin magnetic beads and the following biotin-conjugated lineage markers: CD3e, CD19, Gr1, Mac1, and Ter119. Cell populations from BM were purified through 4-way sorting using FACSAria (Becton Dickinson) and 6-way sorting using MoFlo XDP (Beckman Coulter). The following combinations of cell surface markers were used to define these cell populations: Erythroblasts: 7/4− Ly6G− Ter119+ CD71+ FSChi, Granulocytes: Ly6G+ 7-4+ B220− Ter119−, Monocytes: Ly6G− 7/4+ B220− Ter119−, pro/pre-B cells: Ly6G− B220+ IL7Ra+, Megakaryocyte progenitors: Lin− cKit+ Sca1− CD150+ CD41+, MPP1/ST-HSC: Lin− cKit+ Sca1+ Flt3− CD150− CD48−, MPP2: Lin− cKit+ Sca1+ Flt3− CD150+ CD48+, MPP3: Lin− cKit+ Sca1+ Flt3− CD150− CD48+, MPP4: Lin− cKit+ Sca1+ Flt3+ CD48+, LT-HSC: Lin− cKit+ Sca1+ Flt3− CD150+ CD48− (+/-CD41). Other populations are defined in Supplementary Table 1. Representative examples of sorted populations are shown in Supplementary Figures 1–3. Flow cytometry data were analysed with FlowJo (Tree Star). For Tn tag content extraction and analysis, we FACS-sorted all the available cells from the whole BM extract using purity modes (~98% purity) at ~75–80% efficiency. The list of antibodies (their clone number, the commercial house and concentration) was the following: Ly6B.2 FITC (7/4, Miltenyi, 1:100), Ly6G Alexa Fluor 700 (1A8, eBiosciences, 1:50), Ter119 APC (TER119, eBiosciences, 1:100), CD71 BV510 (C2, BD biosciences, 1:100), CD45R(B220) eFluor 450 (RA3-6B2, eBiosciences, 1:100), CD19 APC/Cy7 (1D3, eBiosciences, 1:50), CD127(IL-7Rα) PE/Cy7 (A7R34, Biolegend, 1:25), CD117 (cKit) FITC/APC (2B8, eBiosciences, 1:100), Ly6a (Sca1) PE/Cy7 (D7, eBiosciences, 1:100), CD135 (Flt3) APC (A2F10, Biolegend, 1:25), CD150 PE/Cy5 (TC15-12F12.2, Biolegend, 1:100), CD48 APC/Cy7 (HM48-1, BD biosciences, 1:100), CD41 BV605 (MwReg30, Biolegend, 1:100), CD3e biotin (145-2C11, eBiosciences, 1:100), CD19 biotin (MB19-1, eBiosciences, 1:100), Gr1 biotin (RB6-685, eBiosciences, 1:100), CD11b (Mac1) biotin (M1/70, eBiosciences, 1:100), Ter119 biotin (TER119, eBiosciences, 1:100), Streptavidin eFluor 450 (eBiosciences, 1:200), FcgRII/III eFluor 450 (93, eBiosciences, 1:100), CD34-FITC (RAM, eBiosciences, 1:25), CD42 APC (HIP1, Biolegend, 1:100), CD9 PE (MZ3, Biolegend, 1:200).
Transplantation assays
Whole BM cells or sort-purified LT-HSCs from M2/HSB/Tn mice were transplanted in 150 μl of αMEM (Gibco, Thermo-Fisher Scientific) through retro-orbital injection into gamma-irradiated recipient mice (split dose of 2.5+2.5 Gy for sublethal irradiation, and 5.5+5.5 Gy for lethal irradiation, with 2h interval). Donor cell engraftment and label frequency was analysed after 16 weeks using an LSRII equipment (Becton Dickinson).
HSC culture assays
1000 sort-purified LT-HSCs from M2/HSB/Tn mice were cultured together with 10,000 MS-5 stromal cells in round-bottom 96-well plates together with SCF (100 ng/ml), TPO (100 ng/ml), Flt3L (50 ng/ml), IL7 (20 ng/ml), IL3 (10 ng/ml), IL11 (50 ng/ml), and GM-CSF (20 ng/ml) in αMEM with 1% Penicillin/Streptomycin and 10% FCS (Thermo Fisher) for two weeks, changing the media 24h after sort and then every 48h (Becton Dickinson). Myeloid and lymphoid HSC progeny was FACS sorted after labelling with Gr-1, Mac-1, CD19 and B220 antibodies (eBiosciences). All growth factors and cytokines were mouse recombinant and purchased from Peprotech.
DNA isolation and amplification
Cells of interest were sorted into 1.7 ml tubes and concentrated into 5–10 μl of buffer by low speed centrifugation (700 g for 5 minutes). Samples with fewer than 10,000 cells were subjected to whole genome amplification with Phi29 kit (Epicenter/Lucigen) according to manufacturer’s instruction. Samples with more than 10,000 cells were purified by QIAamp DNA Micro kit (56304, Qiagen).
TARIS (T7-amplification mediated recovery of integration sites)
Our original technique for molecular identification of Tn integration sites was based on ligation-mediated PCRs (LM-PCR). Others and we have observed significant tag amplification biases with this method, which limit the quantitative potential of the clonal data obtained11,32,33. In order to improve the current technique, we have developed a method based on T7-polymerase linear amplification and recovery of integration sites (TARIS) (Extended Data Fig. 1). This method provided similar sensitivity levels as LM-PCR but more quantitatively and reproducibly captures the clonal composition of complex samples (Extended Data Fig. 2). For TARIS, the total purified DNA was subjected to enzymatic restriction with 10U of HindIII-HF (NEB) overnight. TARIS adaptor primer was hybridized and extended using 1U Klenow DNA polymerase (NEB) for 2h. Then, total DNA was cleaned up using AMPure XP SPRI beads (Beckman Coulter) and used as a template for a 20 μl T7 RNA polymerization reaction (NEB, High Yield Hiscribe T7 kit) overnight. Then, the template was digested with 1U of Turbo DNase (Ambion) and the RNA product was polyadenylated using 1U of polyA RNA polymerase (NEB). The polyA RNA was purified with SPRI beads, and then converted into cDNA using iScript reverse transcriptase (Biorad). TARIS cDNA was used as template for 30 PCR cycles using the HSB-transposon specific Tn-1C, the MAF-Tn-1F and the MAR-polyT primers for 30 cycles, and then 12 cycles of indexing PCR using the MP1 and ID primers (ID1-48) and the KAPA HiFi PCR kit. Solexa sequencing was carried out on HiSeq 2000 (Illumina) at the Tufts Genomics Core. Tag identification and alignment was performed as previously described11. Briefly, we extracted the Tn-containing reads from each fastq file, trimmed the adaptor and Tn sequences and aligned the integration sites to the reference mouse genome (Ensembl mm9) using bowtie 1.2. Then, reads were normalized between samples (per million reads). Sequences were always compared with at least one additional independently labelled mouse with libraries prepared in parallel and sequenced in the same HiSeq lane to account for contaminations. Tags present in the control mouse samples were filtered out (contaminating reads). Next, read frequencies were column-normalized, and graphs were coloured using a logarithmic scale. For hierarchical clustering based on Tn tag distribution, we first determined the Spearman correlation matrix for the compared populations and then performed agglomerative clustering (Single method) using (1 – correlation coefficient) as the distance metric. Curve fitting was performed with the Lowess function. All indicated statistical tests were two-tailed parametric t-tests using Welch’s s.d. correction (exceptions are mentioned where appropriate). Data visualization and statistical analysis was performed using Excel, R (v.3.3.1) and Graphpad Prism (v7). Primers used were: TARIS adaptor primer (5′-GCA TTA GCG GCC GCG AAA TTA ATA CGA CTC ACT ATA GGG AGT CTA AAG CCA TGA CAT C-3′), Tn1-C primer (5′-CTT GTG TCA TGC ACA AAG TAG ATG TCC-3′), MAF-Tn1-1F primer (5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT NNN NCG AGT TTT AAT GAC TCC AAC T-3′), and MAR-polyT primer (5′-GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT V-3′). All primers were ordered from IDT DNA technologies, at 100 nmole scale and HPLC-purified.
Single-cell RNA sequencing and low-level data processing
Transcriptome barcoding and preparation of libraries for single-cell mRNA-sequencing was performed using the most up-to-date inDrops protocol34. For our experiment, the Lin-Sca1+cKit+ BM fraction from a single BL6 mouse was labelled and FACS sorted to purify the entire LT-HSC, MPP1, MPP2, MPP3 and MPP4 fractions. Approximately 2000 cells of each fraction were encapsulated and libraries for all the populations were prepared the same day, with the same stock of primer-gels and RT-mix. Libraries were sequenced on an Illumina NextSeq 500 sequencer using a NextSeq High 75 cycle kit: 35 cycles for read 1, 6 cycle for index i7 read, and 51 cycles for read 2. Raw sequencing reads were processed using the InDrop pipeline previously described, with the following modifications: Bowtie version 1.1.1 was used with parameter –e 100; all ambiguously mapped reads were excluded from analysis; and reads were aligned to the Ensembl release 81 mouse mm10 cDNA reference.
Data visualization using SPRING
We combined mRNA count matrices from five simultaneously processed and indexed libraries (LTHSC-2A, STHSC-2A, MPP4-2A, MPP3-2A, MPP2-2A). Cells with few mRNA counts (< 1000 UMIs) and stressed cells (mitochondrial gene-set Z-score > 1) were filtered out35. The remaining high-quality cells (4248) were total-counts normalized. We next filtered genes, keeping those that were well detected (mean expression > 0.05) and highly variable (CV > 2). Finally, we reduced dimensionality by Z-scoring each gene and applying principal components analysis (PCA), retaining the top 50 PCs. The cells were then visualized using SPRING, a graph-based single-cell viewing interface36. Visual inspection of the SPRING plot revealed a strong cell cycle signature defined by high expression of genes associated with the G2/M phase (Ccnb1, Plk1, Cdc20, Aurka, Cenpf, Cenpa, Ccnb2, Birc5, Bub1, Bub1b, Ccna2, Cks2, E2f5, Cdkn2d). Hypothesizing that this cell cycle signature could affect high dimensional distances between cells in a way that obscures their segregation by lineage-specific genes, we attempted to remove it37. Specifically, we filtered from the analysis genes that were significantly correlated with the sum Z-score of G2/M genes (P < 10−4, Bonferroni corrected; 401 genes total, resulting in 28205 remaining genes). PCA and clustering analysis was repeated using the reduced gene list.
Clustering of single-cell profiles
We performed unsupervised clustering of the processed single-cell data with the Louvain-Jaccard method package from Shekhar et al38. To assess cluster stability and choose the value of k, we downsampled 85% of cells and applied the Louvain-Jaccard method using 50 Principal Components. We tested k values from 10 to 30 and for each k we compared 100 times the randomly downsampled clustering using the Jaccard-index measurement in the R package fpc (Flexible Procedures for Clustering). We considered a Jaccard-index minimum of 0.75 as sufficiently robust and selected values of k > 30, which resulted in the identification of 11-12 clusters39. Differential expression analysis was performed using the method package from Shekhar et al (results are included in Supplementary Table 2)38.
Data availability statement
The GEO accession number is: GSE90742. Additional data files will be made available upon reasonable request. SPRING plots (with and without removal of the G2/M cell cycle signature) are available for inspection at the following links:
Extended Data
Extended Data Fig. 1. TARIS.
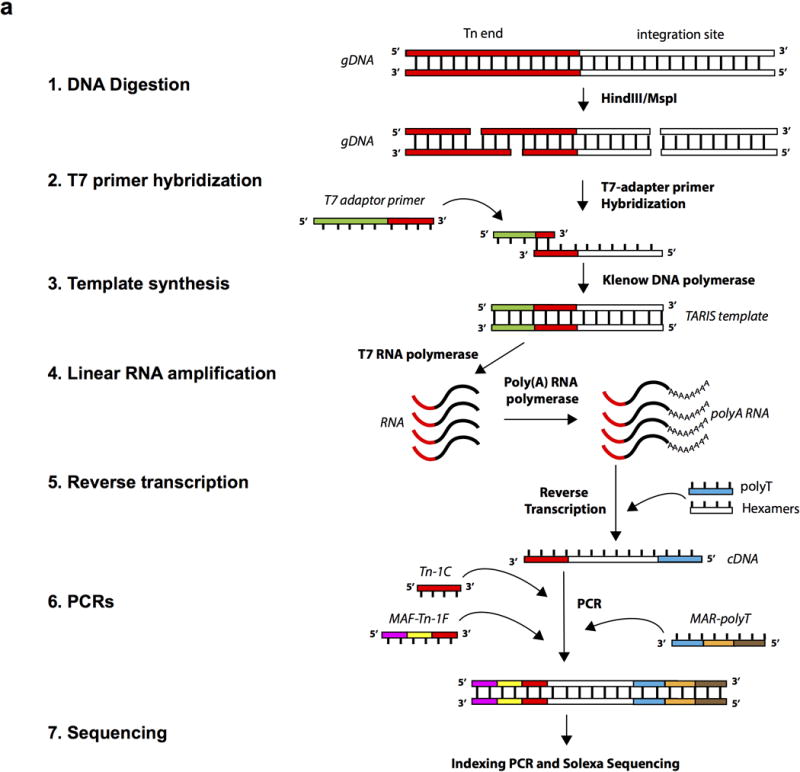
a, Illustration of the TARIS procedure. The procedure is described in detail in the Methods section of this manuscript.
Extended Data Fig. 2. Evaluation of the TARIS method.
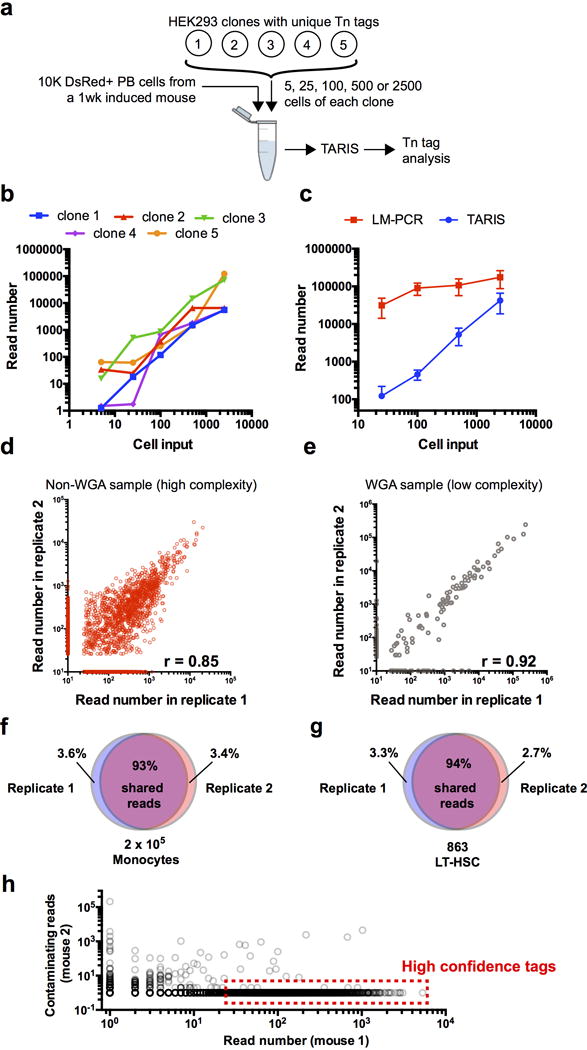
a, Design for the detection limit experiment. Spike-ins of a known number of HEK293 cells carrying unique Tn integration tags was used in a mix of 10,000 DsRed+ peripheral blood cells from a freshly induced HSB mouse. b, Detection limit chart. Values represent the read number for each clone and for each number of input cells. Both axes are in log10 scale. Values represent the sum of two independent experiments. c, Comparison of the average read number value between TARIS and the Ligation-mediated PCR (LM-PCR) method. Values represent mean +/− SD of 5 different Tn tag clones. d, Reproducibility analysis in a non-whole genome amplified sample with high complexity (2×105 BM granulocytes 4 wk after pulse). e, Reproducibility in a whole-genome amplified sample with low complexity (863 LT-HSCs 4 wk after pulse). f, Venn diagram showing overlapping Tn tag reads between two TARIS replicates from the same sample high-complexity sample (2×105 BM monocytes at 4 wk post-induction). g, Venn diagram showing overlapping Tn tag reads between two TARIS replicates from the same low-complexity sample (863 LT-HSCs at 4 wk post-induction). h, Contamination analysis between samples from two different mice. The plot represents the read numbers of tags from Lin+ populations from mouse 1, and their read number values in Lin+ populations in mouse 2. High confidence tags are selected as those tags with more than 25 reads, and at least 10-times higher read count compared to any of the samples from a separate mouse.
Extended Data Fig. 3. Analysis of residual HSB activity after doxycycline withdrawal.
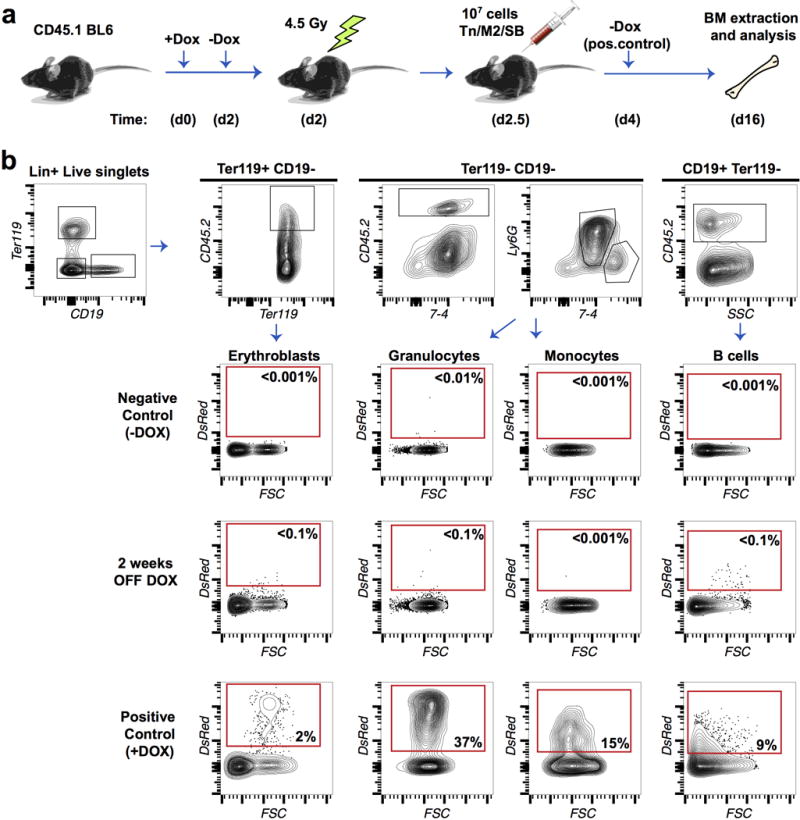
a, Experimental design. Residual HSB activity after Dox removal was assayed by transplantation into CD45.1 mice. Sub-lethally irradiated recipients were treated with Dox for 48h. Dox was removed 12h before transplantation. Ten million whole BM cells were transplanted and mice were allowed to recover for 2 weeks. As a positive control, mice were continuously treated with Dox until 48h after transplant. As a negative control, cells were transplanted into non-Dox treated mice. DsRed labelling was analysed as a proxy for HSB activity in Granulocytes, Erythroblasts, Monocytes and B cells. b, FACS plots showing the negligible labelling of CD45.2 M2/HSB/Tn cells in transplanted recipients 24h after Dox-removal.
Extended Data Fig. 4. Additional representations and analyses of Lin+ tags.
a, Lin+ clones of the second and third mice used for quantifications in Fig. 1d–g. b, Scale-adjusted binary (presence/absence) representation of all detected Mk and Er tags in the experiments from Fig. 1d–g. c, Relative quantification of scale-normalized clone sizes for each lineage, comparing unilineage vs. oligo/multilineage clones. Values are interquartile range and median from 3 independent mice at 4 week and 8 week post-induction.
Extended Data Fig. 5. Validation of My/Er and Mk-restricted tags.
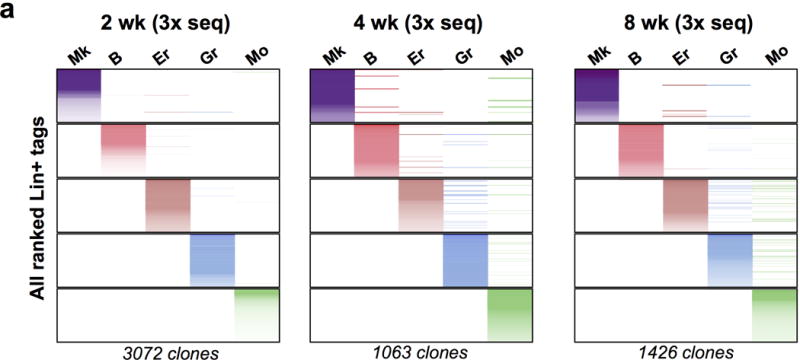
a, Three independent Tn tag libraries were prepared and sequenced from 2wk, 4wk and 8wk-chased mice. Reads from the three libraries were then pooled together for each lineage.
Extended Data Fig. 6. Lineage fate of myeloid-progenitors.
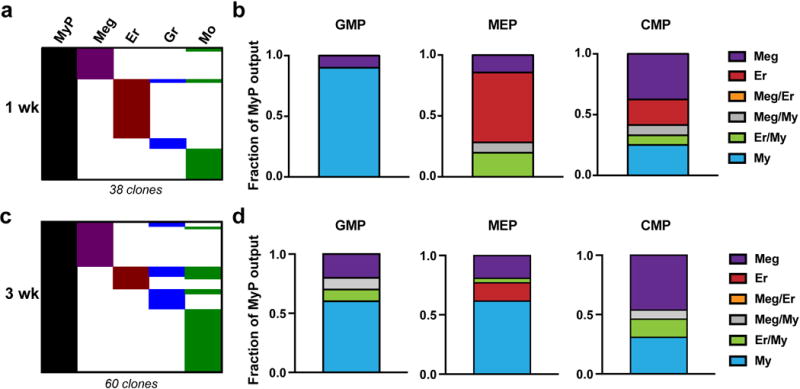
a, Two M2/HSB/Tn mice were induced and chased for 1 week, and then myeloid progenitors (GMP, MEP and CMP) and Lin+ cells were isolated from bone marrow and their Tn-tag content was analysed. Chart is a binary representation of all Lin+ tags overlapping with any myeloid progenitor tag ranked by lineage. b, Quantification of relative lineage contribution of GMPs, MEPs and CMPs as a fraction of lineage-specific/total lineage-overlapping clones for each MyP subset. Values are mean of the two analysed mice. c, An additional M2/HSB/Tn mouse was induced and chased for 3 weeks, and then processed as in (a). d, Quantification of relative lineage contribution of GMPs, MEPs and CMPs at 3 weeks post-labelling.
Extended Data Fig. 7. Additional analyses of MPP clonal outcomes.
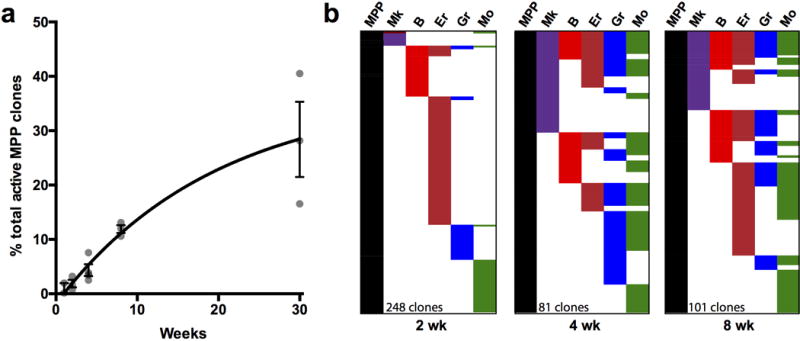
a, Quantification of the percentage of MPP clones that produced any Lin+ progeny at different time points. Values are average +/− SD from 3 mice. b, Three independent Tn tag libraries were prepared and sequenced for all the populations from one bone marrow at 2, 4 and 8 wk post-labeling. Each column in the charts represent the combined tags detected in any of the three libraries for each population.
Extended Data Fig. 8. Single cell heterogeneity of HSC/MPPs.
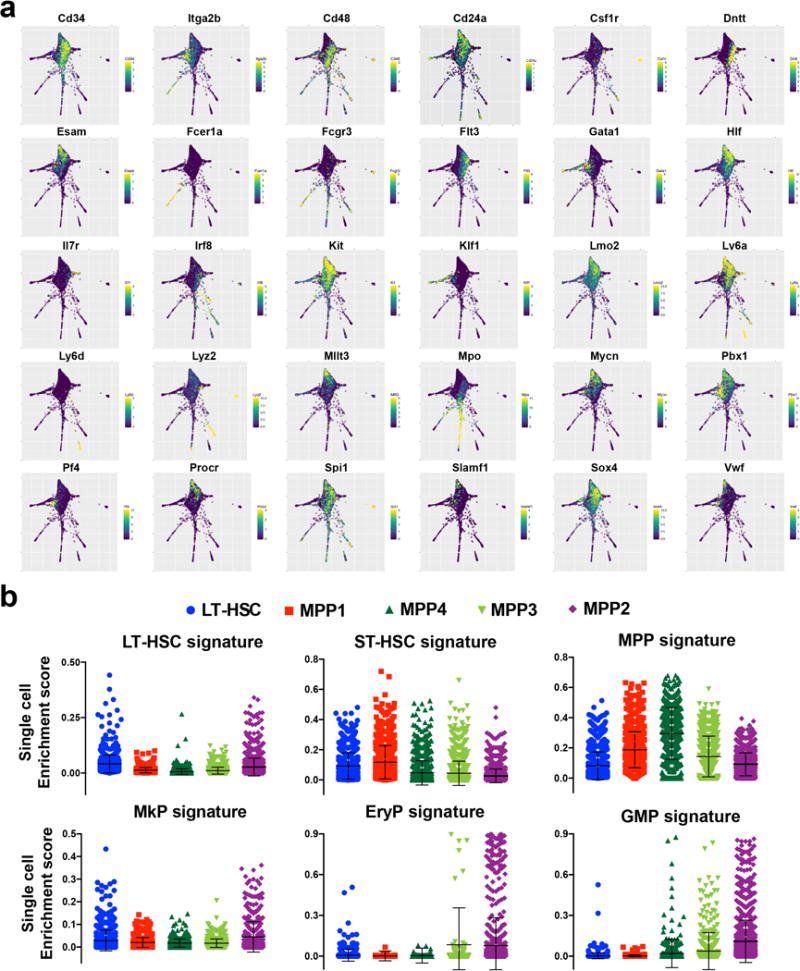
a, SPRING plots showing selected differentially expressed markers. Scale represents amount of detected mRNA copies (normalized) of each marker gene. b, Enrichment score analysis for single cells in each FACS-sorted population compared to previously obtained bulk transcriptional signatures of BM populations sorted using traditional markers (from the Immgen database).
Extended Data Fig. 9. Differentially expressed markers for clusters C1, C2, C3, and Mk.
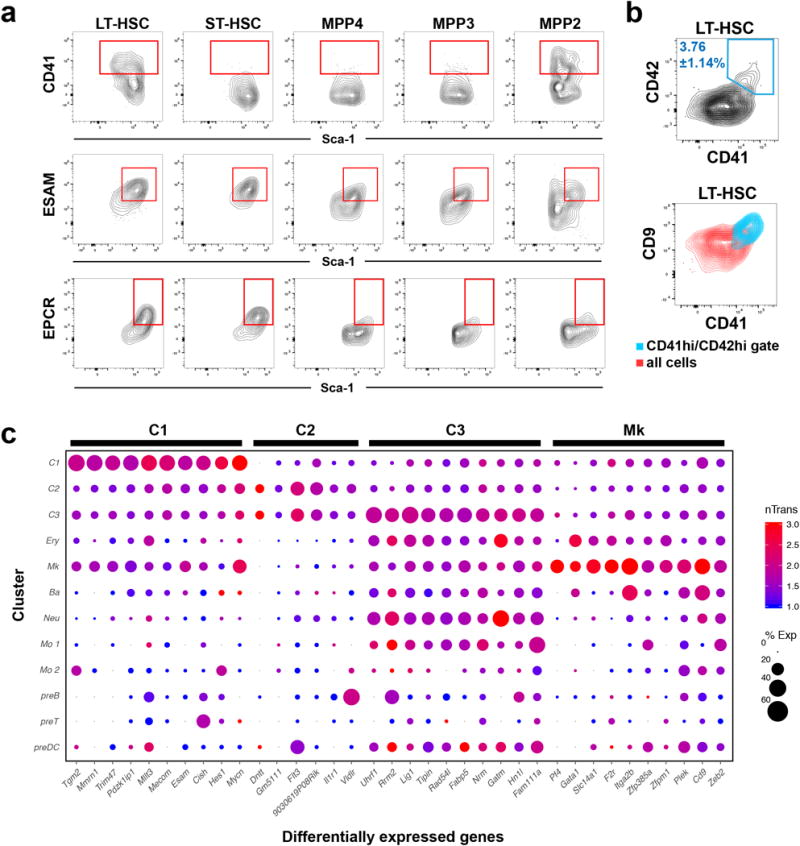
a, FACS plots showing heterogeneity in expression of cluster markers within the analysed HSC/MPP subsets. b, FACS plots showing expression of different Mk-primed cluster markers (CD41, CD42 and CD9) within the LT-HSC gate. c, The table shows the expression value (nTrans) and percentage of expressing cells from each cluster (%Exp). The top 10 highest expressed genes that distinguish each cluster are shown.
Extended Data Fig. 10. Additional data on clonal origin of Mk progenitors.
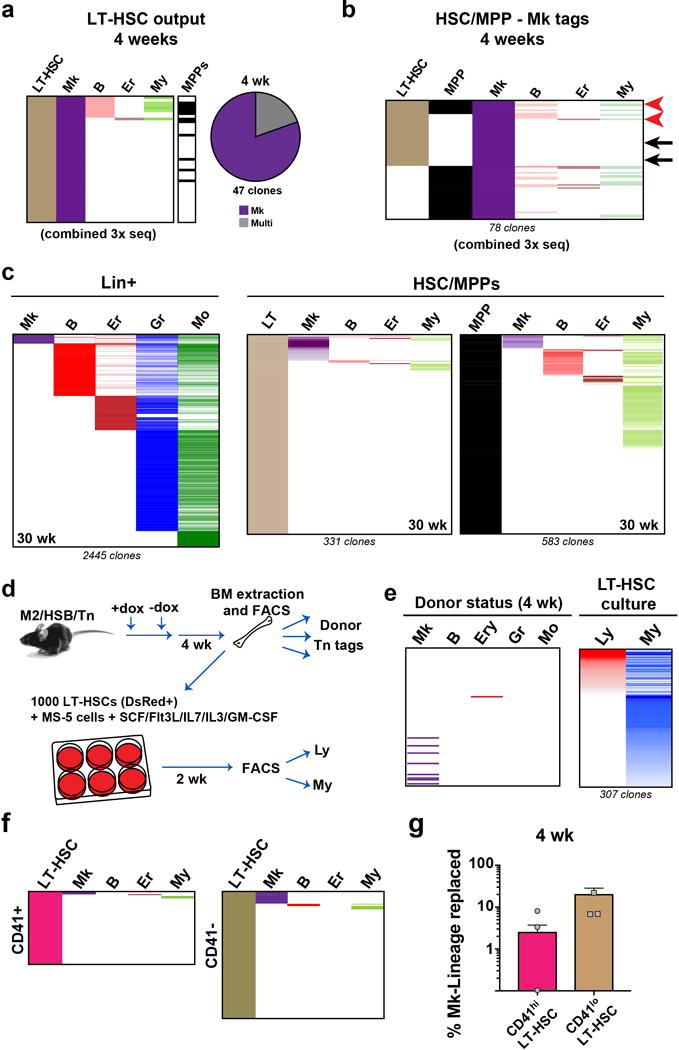
a, Three independent Tn tag libraries were prepared and sequenced for LT-HSC, MPP, and the five Lin+ populations, from one mouse at 4 weeks. Each column represents the combined tags detected from 3 amplicon libraries prepared for each population, to facilitate visualization of the smallest clones. Tags are coloured by frequency in each lineage, and organized by rank. b, Origin of Mk. Alignment of all Mk clones which had detectable tags in HSC/MPPs from a mixed library combining 3 independent sequencing reactions. Tags are coloured by frequency in each lineage (except for Mk), and organized by rank. Arrows indicate tags verified by clone-specific PCR. c, Alignment of Tn tags from all Lin+ populations, LT-HSCs and MPPs collected from 30 wk chased-mice. Tags are coloured by frequency in each lineage, and organized by rank. d, Experimental design for testing in vitro myeloid and lymphoid potential from sorted LT-HSCs. e, In vitro myeloid potential of LT-HSCs. Alignment of donor Lin+ tags with Tn tags obtained from myeloid and lymphoid cells derived from donor LT-HSCs after 2 weeks in culture. f, Clonal output of CD41hi and CD41lo LT-HSCs at 4 weeks post labeling. g, Quantification of Mk lineage replacement by CD41hi vs. CD41lo LT-HSCs (measured as % of overlapping/total Mk reads) at 4 weeks post labeling. Values are mean +/− s.e.m. of 3 independent mice.
Supplementary Material
Acknowledgments
We are grateful to members of the Camargo and Klein lab for critical comments. A.R.F. is a Merck Fellow of the Life Sciences Research Foundation and a non-stipendiary EMBO postdoctoral fellow. This work was supported by NIH grants HL128850-01A1 and P01HL13147 to F.D.C. FDC is a Leukemia and Lymphoma Society and a Howard Hughes Medical Institute Scholar.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author contribution
A.R.F and F.D.C designed the study, analysed the data and wrote the manuscript.
A.R.F. performed and analysed the experiments, assisted by M.J., S.P and J.S.
S.W., C.W., R.P. R.A.C. and A.M.K. designed and analysed InDrops experiments and transcriptome data.
F.D.C. supervised the study.
The authors declare no competing financial interests.
References
- 1.Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, Weissman IL. Identification of a lineage of multipotent hematopoietic progenitors. Development. 1997;124:1929–1939. doi: 10.1242/dev.124.10.1929. [DOI] [PubMed] [Google Scholar]
- 2.Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 3.Adolfsson J, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 5.Ceredig R, Rolink AG, Brown G. Models of haematopoiesis: seeing the wood for the trees. Nat Rev Immunol. 2009;9:293–300. doi: 10.1038/nri2525. [DOI] [PubMed] [Google Scholar]
- 6.Forsberg EC, Serwold T, Kogan S, Weissman IL, Passegue E. New evidence supporting megakaryocyte-erythrocyte potential of flk2/flt3+ multipotent hematopoietic progenitors. Cell. 2006;126:415–426. doi: 10.1016/j.cell.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 7.Notta F, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perie L, Duffy KR, Kok L, de Boer RJ, Schumacher TN. The Branching Point in Erythro-Myeloid Differentiation. Cell. 2015;163:1655–1662. doi: 10.1016/j.cell.2015.11.059. [DOI] [PubMed] [Google Scholar]
- 9.Pietras EM, et al. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell stem cell. 2015;17:35–46. doi: 10.1016/j.stem.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto R, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154:1112–1126. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Debili N, et al. Characterization of a bipotent erythro-megakaryocytic progenitor in human bone marrow. Blood. 1996;88:1284–1296. [PubMed] [Google Scholar]
- 13.Sanada C, et al. Adult human megakaryocyte-erythroid progenitors are in the CD34+CD38mid fraction. Blood. 2016;128:923–933. doi: 10.1182/blood-2016-01-693705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paul F, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163:1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Pronk CJ et al. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell stem cell. 2007;1:428–442. doi: 10.1016/j.stem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Velten L, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nature cell biology. 2017;19:271–281. doi: 10.1038/ncb3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein AM, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calaminus SD, et al. Lineage tracing of Pf4-Cre marks hematopoietic stem cells and their progeny. PLoS One. 2012;7:e51361. doi: 10.1371/journal.pone.0051361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gekas C, Graf T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood. 2013;121:4463–4472. doi: 10.1182/blood-2012-09-457929. [DOI] [PubMed] [Google Scholar]
- 20.Haas S, et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell stem cell. 2015;17:422–434. doi: 10.1016/j.stem.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Nishikii H, et al. Unipotent Megakaryopoietic Pathway Bridging Hematopoietic Stem Cells and Mature Megakaryocytes. Stem cells. 2015;33:2196–2207. doi: 10.1002/stem.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roch A, Trachsel V, Lutolf MP. Brief Report: Single-Cell Analysis Reveals Cell Division-Independent Emergence of Megakaryocytes From Phenotypic Hematopoietic Stem Cells. Stem cells. 2015;33:3152–3157. doi: 10.1002/stem.2106. [DOI] [PubMed] [Google Scholar]
- 23.Sanjuan-Pla A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502:232–236. doi: 10.1038/nature12495. [DOI] [PubMed] [Google Scholar]
- 24.Vannucchi AM, et al. Identification and characterization of a bipotent (erythroid and megakaryocytic) cell precursor from the spleen of phenylhydrazine-treated mice. Blood. 2000;95:2559–2568. [PubMed] [Google Scholar]
- 25.Busch K, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–546. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- 26.Sawai CM, et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity. 2016;45:597–609. doi: 10.1016/j.immuni.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Junker JP, et al. Massively parallel clonal analysis using CRISPR/Cas9 induced genetic scars. bioRxiv. 2017 doi: 10.1101/056499. [DOI] [Google Scholar]
- 28.Raj B, et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain by scGESTALT. bioRxiv. 2017 doi: 10.1101/205534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foudi A, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nature biotechnology. 2009;27:84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oguro H, Ding L, Morrison SJ. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell stem cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson A, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 32.Harkey MA, et al. Multiarm high-throughput integration site detection: limitations of LAM-PCR technology and optimization for clonal analysis. Stem cells and development. 2007;16:381–392. doi: 10.1089/scd.2007.0015. [DOI] [PubMed] [Google Scholar]
- 33.Wang GP, et al. DNA bar coding and pyrosequencing to analyze adverse events in therapeutic gene transfer. Nucleic acids research. 2008;36:e49. doi: 10.1093/nar/gkn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zilionis R, et al. Single-cell barcoding and sequencing using droplet microfluidics. Nature protocols. 2017;12:44–73. doi: 10.1038/nprot.2016.154. [DOI] [PubMed] [Google Scholar]
- 35.Ilicic T, et al. Classification of low quality cells from single-cell RNA-seq data. Genome biology. 2016;17:29. doi: 10.1186/s13059-016-0888-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinreb C, Wolock S, Klein A. SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. bioRxiv. 2016 doi: 10.1101/090332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buettner F, et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nature biotechnology. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 38.Shekhar K, et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell. 2016;166:1308–1323 e1330. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hennig C. Cluster validation by measurement of clustering characteristics relevant to the user. arXiv:1703.09282. 2017 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GEO accession number is: GSE90742. Additional data files will be made available upon reasonable request. SPRING plots (with and without removal of the G2/M cell cycle signature) are available for inspection at the following links: