

Substrate participation in forming a catalytic triad aids in antibiotic resistance by aminoglycoside N3-acetyltransferases.
Abstract
One group of enzymes that confer resistance to aminoglycoside antibiotics through covalent modification belongs to the GCN5-related N-acetyltransferase (GNAT) superfamily. We show how a unique GNAT subfamily member uses a previously unidentified noncanonical catalytic triad, consisting of a glutamic acid, a histidine, and the antibiotic substrate itself, which acts as a nucleophile and attacks the acetyl donor molecule. Neutron diffraction studies allow for unambiguous identification of a low-barrier hydrogen bond, predicted in canonical catalytic triads to increase basicity of the histidine. This work highlights the role of this unique catalytic triad in mediating antibiotic resistance while providing new insights into the design of the next generation of aminoglycosides.
INTRODUCTION
Aminoglycoside antibiotics such as kanamycin, neomycin, and tobramycin are common bactericidal agents used to treat clinically important diseases caused by Gram-positive and Gram-negative bacteria. These diseases include tuberculosis, meningitis, and listeriosis, as well as many nosocomial infections (1). However, the efficacy of aminoglycoside antibiotics has been drastically reduced because of the emergence of bacterial resistance (2).
Chemical modification is the most prevalent mechanism responsible for bacterial resistance to aminoglycoside antibiotics, which is achieved by aminoglycoside-modifying enzymes (AGMEs) (3). AGMEs represent a mechanistically diverse family of enzymes that render aminoglycosides ineffective through the transfer of various functional groups (2). Modification disrupts the requisite molecular interactions between the drug and its target, the bacterial ribosome. Aminoglycoside acetyltransferases (AACs) were the first identified members of the GCN5-related N-acetyltransferase (GNAT) superfamily of enzymes (4). Enzymes belonging to the GNAT superfamily are universally distributed in nature and catalyze the transfer of an acetyl group from acetyl–CoAs to a primary amine group on diverse acetyl-accepting substrates (5). This superfamily includes histone acetyltransferases, serotonin N-acetyltransferases, and glucosamine-6-phosphate N-acetyltransferases. Acetylation occurs through a general mechanism involving the direct nucleophilic attack of the amine group to be acetylated on the carbonyl carbon of the acetyl-CoA. Among the AACs, crystal structures of several enzymes have been reported, yet details about the kinetic and catalytic mechanisms are not available for all of them (4, 6, 7). Recently, a unique structural fold was observed in a putative GNAT acetyltransferase from Bacillus anthracis, BA2930 [Protein Data Bank (PDB) code 3E4F (8)]. The crystal structures of three additional members of this subfamily have also been reported, with all having a similar fold and conservation of proposed catalytically important residues. Yet significant knowledge about the catalytic mechanism is lacking for this pathologically important GNAT subfamily (9).
Here, we report on the aminoglycoside N3-acetyltransferase–VIa (AAC-VIa), a fifth member of this GNAT subfamily, which was isolated from a resistant clinical strain of Enterobacter cloacae (10). AAC-VIa transfers an acetyl group donated by acetyl-CoA to the N3 position on the unprimed ring of the aminoglycoside (Fig. 1). The x-ray and neutron crystal structures of AAC-VIa and its complexes with reactants allowed for the identification of a previously unidentified noncanonical catalytic triad functioning to inactivate aminoglycoside antibiotics in this GNAT subfamily, in a mechanism similar to the archetypal catalytic triad in proteases. Moreover, the neutron diffraction studies allowed for the unambiguous identification of a low-barrier hydrogen bond (LBHB), controversially predicted to function in some proteases that use a conventional catalytic triad. Direct visualization of the proton equidistant between the acceptor atoms, Oε2 of glutamic acid and the Nδ1 of histidine, is observed. Subsequent catalysis in the crystal was used to capture the product of a catalytically relevant ternary complex, resulting in a unique, postcatalysis conformational state of the acetylated sisomicin, suggesting a potential mechanism of product release. These studies allow for unprecedented chemical insights into this pathologically relevant enzyme and protein superfamily while suggesting the design requirements of the next generation of aminoglycoside antibiotics.
Fig. 1. Binary complexes of AAC-VIa.
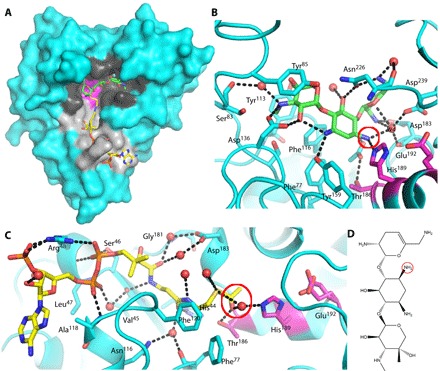
(A) Surface representation of AAC-VIa–sisomicin (green carbon atoms) binary complex superimposed on the enzyme–acetyl-CoA (yellow carbon atoms) binary complex. The AAC-VIa surface cavities that bind sisomicin and coenzyme A (CoASH) are colored black and gray, respectively, whereas the catalytic triad is shown in magenta. (B) Close-up view of sisomicin (green carbon atoms) interaction with AAC-VIa. The site of antibiotic acetylation is circled in red. (C) Close-up view of acetyl-CoA (yellow carbon atoms) interaction with AAC-VIa. The acetyl donor is circled in red. Hydrogen bonds are represented as black dashed lines, and water molecules are shown as red spheres. The active-site residues have carbon atoms colored magenta. (D) Sisomicin with the site of acetylation (the N3 position of the unprimed ring) circled.
RESULTS
Crystal structure of AAC-VIa
To gain an understanding of the structure/function relationship of AAC-VIa–mediated antibiotic inactivation, we determined crystal structures of the enzyme with and without bound substrates and reaction product (fig. S1). Crystallographic data are summarized in Table 1. The fold of AAC-VIa places it in a distinct, noncanonical GNAT subfamily, with the overall structure being essentially identical to the other four members whose structures are known to date: YokD (PDB code 2NYG), FrbF [PDB code 3SMA (9)], HMB0038 (PDB code 5HT0), and BA2930 [PDB code 3E4F (8)] (fig. S2). A large L-shaped indentation is present on the surface of AAC-VIa (fig. S1). One arm of the indentation superimposes on the acetyl-CoA binding site found in the FrbF molecular replacement search model, YokD, and the BA2930 homolog, where the donated acetyl group would be placed into the other intersecting cavity (fig. S2).
Table 1. Data collection and refinement statistics.
RMS, root mean square.
| Apo | Sisomicin | CoA | Ternary | Acetyl-CoA | Acetylated sisomicin | |
| Resolution range | 50.0 to 2.2 (2.27 to 2.20) |
50.0 to 1.8 (1.86 to 1.80) |
50.0 to 2.2 (2.28 to 2.20) |
50.0 to 2.05 (2.12 to 2.05) |
50.0 to 2.05 (2.12 to 2.05) |
50.0 to 2.2 (2.27 to 2.20) |
| Space group | C 2 | P 61 | P 61 | P 61 | P 61 | C 2 |
| Unit cell | ||||||
| a, b, and c (Å) | 89.0, 86.0, and 51.1 | 78.6, 78.6, and 83.6 | 78.1, 78.1 and 83.3 | 77.5, 77.5, and 83.9 | 78.5, 78.5, and 83.9 | 84.4, 86.9, and 50.3 |
| α, β, and γ (°) | 90, 120.0, and 90 | 90, 90, and 120 | 90, 90, and 120 | 90, 90, and 120 | 90, 90, and 120 | 90, 119.8, and 90 |
| Unique reflections | 15,393 | 26,880 | 14,077 | 18,004 | 18,240 | 14,753 |
| Multiplicity* | 2.4 (2.2) | 2.9 (2.8) | 5.6 (4.9) | 2.9 (2.7) | 2.6 (2.1) | 3.0 (2.6) |
| Completeness (%) | 90.5 (94.7) | 98.7 (96.3) | 95.1 (72.2) | 99.3 (98.2) | 98.6 (97.0) | 91.8 (97.3) |
| Mean I/σ(I) | 8.8 (2.4) | 12.0 (2.4) | 23.0 (3.8) | 11.0 (2.0) | 11.6 (2.3) | 10.4 (3.2) |
| Wilson B-factor | 37.4 | 16.54 | 32.7 | 31.58 | 23.02 | 39.2 |
| R-merge | 0.084 (0.504) | 0.089 (0.532) | 0.091 (0.484) | 0.10 (0.602) | 0.101 (0.526) | 0.094 (0.554) |
| Reflections used in refinement |
15,388 | 26,851 | 14,073 | 17,984 | 18,233 | 14,750 |
| Reflections used for R-free | 771 | 1302 | 688 | 899 | 905 | 737 |
| R-work | 0.236 (0.314) | 0.149 (0.227) | 0.176 (0.208) | 0.189 (0.289) | 0.179 (0.275) | 0.282 (0.450) |
| R-free | 0.267 (0.333) | 0.186 (0.286) | 0.212 (0.289) | 0.207 (0.278) | 0.205 (0.288) | 0.308 (0.496) |
| Number of nonhydrogen atoms |
||||||
| Macromolecules | 2028 | 2089 | 2042 | 2048 | 2043 | 2028 |
| Ligands | 1 | 35 | 48 | 79 | 51 | 35 |
| Solvent | 51 | 398 | 129 | 185 | 274 | 57 |
| RMS (bonds) | 0.002 | 0.009 | 0.002 | 0.002 | 0.002 | 0.004 |
| RMS (angles) | 0.5 | 0.9 | 0.6 | 0.5 | 0.5 | 0.9 |
| Ramachandran favored (%) | 95.1 | 97.0 | 96.0 | 96.2 | 97.0 | 96.6 |
| Ramachandran allowed (%) | 4.1 | 3.0 | 3.6 | 3.4 | 2.6 | 3.4 |
| Ramachandran outliers (%) | 0.8 | 0.0 | 0.4 | 0.4 | 0.4 | 0.0 |
| Clashscore | 7 | 5 | 3 | 2 | 2 | 6 |
| Average B-factor | 44 | 19 | 33 | 31 | 23 | 51 |
| Macromolecules | 44 | 17 | 33 | 31 | 22 | 51 |
| Ligands | 43 | 20 | 60 | 32 | 32 | 53 |
| Solvent | 42 | 30 | 36 | 34 | 29 | 49 |
| PDB code | 6BC6 | 6BC7 | 6BC5 | 6BC3 | 6BC4 | 6BC2 |
*Number in parentheses represents values in the highest-resolution shell.
Previous studies on AAC-VIa have shown that acetyl-CoA is used to modify several aminoglycoside antibiotics, such as sisomicin, tobramycin, and gentamicin (11). The binary crystal structures of acetyl-CoA–, CoASH-, and sisomicin-bound AAC-VIa were determined (Table 1). In AAC-VIa, acetyl-CoA resides in a long groove on the protein surface composed of both polar and nonpolar residues (Fig. 1A). The acetyl-CoA binding site overlaps with the binding site identified in the AAC-VIa structural homologs. The side chain of Arg48 is the only amino acid to undergo large conformational changes upon binding of the ligand. The pantetheine moiety of acetyl-CoA is bound through specific hydrogen bonds at the base of the surface cavity, ultimately terminating with a hydrogen bond between the donor acetyl carbonyl and the side chain of Thr186. A water molecule also hydrogen-bonds to the acetyl carbonyl and forms a hydrogen-bonding network with two residues, His189 and Glu192, which have been proposed in other studies to be important in the catalytic mechanism (Fig. 1) (8, 9). This water molecule is in the same location as the N3 amine of the sisomicin (Fig. 1C). The side chain of Glu192 is found in two conformations, one of which forms a short hydrogen bond with His189, interacting in a manner similar to LBHBs in catalytic triads (Fig. 1). The interactions with CoASH are essentially identical to those found in the acetyl-CoA complex. The only exception is the shifting of the terminal thiol into a conformation so that a hydrogen bonding network, similar to that formed with the terminal acetyl carbonyl, is formed with Thr186 and a water molecule (fig. S3).
Sisomicin is located in the binding site that is directly adjacent to the acetyl-CoA binding pocket and is bound by a network of polar and nonpolar interactions (Fig. 1). One specifically bound water molecule in particular (W4) is hydrogen-bonded to the site of acetylation, the N3 position of sisomicin on the deoxystreptamine ring, in addition to forming two hydrogen bonds with the adjacent sisosamine ring and three with the protein. The N3 position of the central deoxystreptamine ring is directly hydrogen-bonded to Nε2 of His189, which is flanked on an α helix by Glu192 and Thr186, the latter of which also hydrogen-bonds to the acetyl group of the acetyl-CoA.
The local environment and hydrogen-bonding interactions with the catalytically relevant functional groups in the binary reactant structures are suggestive of a catalytic mechanism involving a catalytic triad. All of the features common to catalytic triads are appropriately positioned for catalysis in the AAC-VIa active site. Glu192 hydrogen-bonds to the Nδ1 of His189, thereby making the Nε2 of His189 more basic. The Nε2 of His189 is in direct hydrogen-bonding geometry to be able to abstract a proton from the N3 position of the sisomicin, thus enabling formation of the nucleophile that would attack the acetyl-CoA. Moreover, the tetrahedral intermediate formed during the AAC-VIa–mediated acetylation reaction and the emerging oxyanion can be stabilized through hydrogen bonding to Thr186, in a manner that mimics the oxyanion hole found in the archetypal catalytic triad of trypsin and other serine proteases.
Neutron diffraction reveals the presence of an LBHB
Room temperature neutron and x-ray diffraction data were collected on deuterium-exchanged crystals of the apo-enzyme and enzyme-sisomicin complex to directly visualize the protonation state of the catalytic residues of this enzyme (Table 2). Figure 2 shows the neutron density of the sisomicin in the active site of AAC-VIa, where density for exchangeable deuterium atoms is clearly visible, as well as the interaction network of the catalytic triad. A hydrogen bond is found between Glu192 and the Nδ1 of His189, whereas the unprotonated Nε2 of His189 hydrogen-bonds to the proton to be abstracted from the neutral N3 of the sisomicin (Fig. 2). The distance between the Glu192 Oε2 oxygen and the His189 Nδ1 is 2.57 Å, which is an uncharacteristically short hydrogen bond. The length of this hydrogen bond is on the order of bond distances found in LBHBs, where the pKa values of the donor and acceptor moieties are equal, and the proton resides between the two atoms (12). The Fo-Fc neutron density map, where the deuterium atom was omitted, was used to assign the position of this hydrogen atom (Fig. 2). This atom is found 1.4 Å away from the Nε2 of His189 and 1.2 Å from the Oε2 of Glu192, consistent with this being an LBHB. In the absence of ligand, the distance between the Glu192 Oε2 oxygen and the His189 Nδ1 is 2.83 Å (Fig. 2). The formation of the binary, sisomicin-bound complex is likely the driving force for the difference in the His189 conformation between the apo-enzyme and enzyme-sisomicin complex. Superposition of the apo- and sisomicin-bound structures shows that the distance between the Nε2 of His189 and the sisomicin amino proton is essentially identical (2.2 Å). The repositioning of the His189 side chain into an LBHB with Glu192 is likely not a steric effect, yet is due to the hydrogen bond formed between the His189 Nε2 and the sisomicin, lowering the Nδ1 pKa or making the Nδ1 more basic to facilitate the LBHB with Glu192.
Table 2. Room temperature data collection and refinement statistics.
| Apo | Sisomicin | |||
| X-ray | Neutron | X-ray | Neutron | |
| Resolution range | 50.0 to 2.0 (2.07 to 2.00) | 15.0 to 2.3 (2.38 to 2.30) | 50.0-1.9 (1.97 to 1.90) | 15.0 to 2.20 (2.28 to 2.20) |
| Space group | I 2 | I 2 | ||
| Unit cell | ||||
| a, b, and c (Å) | 51.6, 86.1, and 78.8 | 50.8, 86.2, and 76.9 | ||
| β (°) | 94.0 | 94.5 | ||
| Unique reflections | 22,336 | 14,029 | 25,874 | 15,519 |
| Multiplicity* | 3.4 (3.4) | 2.8 (2.2) | 1.7 (1.6) | 2.8 (2.5) |
| Completeness (%) | 97.4 (95.3) | 91.3 (84.3) | 99.3 (97.2) | 92.5 (89.3) |
| Mean I/sigma(I) | 11.5 (3.1) | 11.6 (3.0) | 17.2 (3.1) | 10.4 (3.3) |
| R-merge | 0.069 (0.390) | 0.154 (0.287) | 0.047 (0.326) | 0.147 (0.360) |
| Reflections used in refinement | 22,336 | 14,029 | 25,867 | 15,512 |
| Reflections used for R-free | 1126 | 708 | 1286 | 763 |
| R-work (%) | 15.9 | 22.1 | 15.0 | 19.8 |
| R-free (%) | 19 | 24.6 | 18.1 | 22.0 |
| Number of non-hydrogen atoms | ||||
| Macromolecules | 2029 | 2055 | ||
| Ligands | 0 | 31 | ||
| Solvent | 88 | 126 | ||
| RMS (bonds) | 0.010 | 0.013 | ||
| RMS (angles) | 0.934 | 0.81 | ||
| Ramachandran favored (%) | 97.0 | 96.6 | ||
| Ramachandran allowed (%) | 3.0 | 3.0 | ||
| Ramachandran outliers (%) | 0.0 | 0.4 | ||
| Clashscore | 2.5 | 1.8 | ||
| Average B-factor | ||||
| Macromolecules | 55.7 | 52.9 | ||
| Ligands | 53.9 | |||
| Solvent | 58.2 | 59.1 | ||
| PDB code | 6BBR | 6BB2 | ||
*Number in parentheses represents values in the highest-resolution shell.
Fig. 2. Room temperature neutron structure of AAC-VIa.
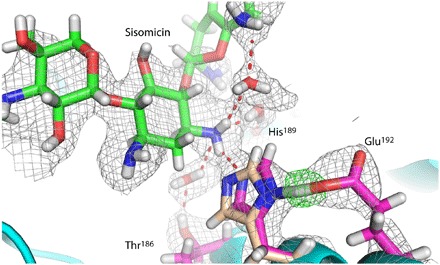
Close-up view of the neutron diffraction model and nuclear density for sisomicin-bound AAC-VIa. Sisomicin-bound active-site residues are colored with magenta carbon atoms, and sisomicin with green carbon atoms. His189 from the neutron diffraction data acquired with the apo-enzyme is shown and colored with tan carbon atoms. The 2Fo-Fc nuclear density map is shown in light gray. The Fo-Fc deuterium nuclear omit map for the proton involved in the LBHB is shown in green. Hydrogen bonds are represented as red dashed lines.
Kinetics and conservation of catalytic triad in homologs
In addition to conservation of many of the residues forming the acetyl-CoA binding site, in all members of this GNAT subfamily with known structures, the suggested catalytically important residues are conserved, including the oxyanion hole (fig. S2). Moreover, one highly promiscuous acetyl transferase, the aminoglycoside N3-acetyltransferase–IIIb (AAC-IIIb) (13–15), also shows sequence conservation of the proposed catalytic residues and binding site identified in AAC-VIa (fig. S2). To see the effect these residues have on AAC-VIa catalytic activity, we carried out activity assays of wild type and mutants of Thr186, His189, and Glu192 (fig. S2). Mutating each of the individual residues proposed to be important in the AAC-VIa catalytic mechanism decreases activity to 3 to 5% of the wild-type enzyme, suggesting the importance of these residues in this noncanonical catalytic triad. Furthermore, the E192Q mutant demonstrates the importance of the strength of the LBHB in increasing the basicity of the Nε2 of His189 in this catalytic mechanism.
Proposed catalytic mechanism
The structure of the noncatalytic, ternary enzyme-CoASH-sisomicin complex was solved to gain further insights into the catalytic mechanism (Table 1). The conformations of the bound sisomicin and CoASH in the ternary complex overlap precisely with the conformations observed in the respective binary enzyme-ligand complexes; the exception is that the pantetheine tail of the CoASH adopts a different conformation in the binary enzyme-CoASH complex while adopting the same conformation as the acetyl-CoA in the binary enzyme–acetyl-CoA complex (fig. S4). The colocalization of the CoASH in the ternary and the acetyl-CoA binary complexes suggests that this is the catalytically relevant conformation of the donor group. Therefore, a structural superposition of the ternary enzyme-CoASH-sisomicin and the binary enzyme–acetyl-CoA complexes is used to contextualize the catalytic mechanism (Fig. 3). In this modeled catalytically competent ternary complex, Glu192 forms an LBHB to the Nδ1 of His189, thereby making the Nε2 more nucleophilic to abstract a proton from the N3 position of the sisomicin (Fig. 3). This hydrogen-bonding network completes the catalytic triad and generates the nucleophilic nitrogen on the antibiotic. The N3 on sisomicin is positioned in such a manner (2.1 Å distance between atoms) that a direct in-line attack to the carbonyl carbon of acetyl-CoA is possible (Fig. 3). A nucleophilic attack on the acetyl-CoA generates a tetrahedral intermediate containing a negative charge on the oxygen atom, with the stabilization of the negative charge being achieved through the hydrogen bond to Thr186 (Fig. 3). Collapse of the tetrahedral intermediate, and abstraction of the proton from the doubly protonated His189, generates the acetylated antibiotic and CoASH while restoring the protonation state of the active site in our model based on these structural studies (Fig. 3).
Fig. 3. Proposed AAC-VIa mechanism.
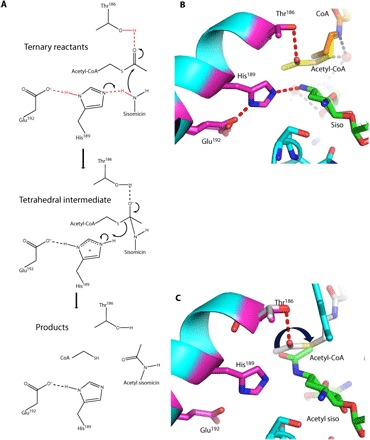
(A) Proposed catalytic mechanism of AAC-VIa–mediated acetylation of sisomicin. (B) Close-up view of the active site of the ternary enzyme-CoASH-sisomicin complex superimposed on the binary enzyme–acetyl-CoA complex. Active-site residues are colored with magenta carbon atoms, CoASH is colored with orange carbon atoms, acetyl-CoA is colored with yellow carbon atoms, and sisomicin is colored with green carbon atoms. (C) Close-up view of the active site of the enzyme–acetyl sisomicin complex superimposed on the binary enzyme–acetyl-CoA complex. Active-site residues are colored with magenta carbon atoms, acetyl-CoA is colored with gray carbon atoms, and acetylated sisomicin is colored with green carbon atoms. Hydrogen bonds are represented as red dashed lines.
Product release
Catalysis in the crystal was used to capture the product of a catalytically relevant ternary complex, resulting in a unique conformational state of the acetylated sisomicin after catalysis while suggesting a potential mechanism of product release (Table 1). Soaking of the crystals of the binary enzyme-sisomicin complex in acetyl-CoA resulted in the formation of the acetylated sisomicin product, whereas no bound acetyl-CoA or CoASH was observed. The localization of atoms in the modeled catalytically competent ternary complex would place the methyl group of the acetyl-CoA in a trans conformation (Fig. 3). However, when the tetrahedral intermediate collapses to the planar amide bond in the product crystal structure, the acetyl group, which is now on the antibiotic, rotates to a cis conformation, placing the methyl group on the apolar side of sisomicin and the carbonyl on the polar side (Fig. 3). The planarization of the tetrahedral intermediate breaks the hydrogen bond formed with the acetyl carbonyl and Thr186 while likely contributing to a higher-energy product complex and aiding in acetylated sisomicin release. Concomitant with the rotation of the acetyl group, the conformation of the sisosamine ring flips from a chair conformation in the ternary complex to a half-chair in the acetylated sisomicin product complex, potentially as a result of the formation of the amide bond geometry and subsequent planarization (Fig. 4). The flipping of the ring has several effects that also likely lead to a product state with significantly fewer bonding interactions than the starting reactant state. In the ternary complex, where sisomicin is in a chair conformation, the N21 atom is placed into the binding site, forming a hydrogen bond with Asp183, a residue conserved in the known structural homologs (fig. S2). In the ternary complex, this positions Asp183 so that it also hydrogen-bonds to two water molecules, W1 and W2, which also hydrogen-bond to the acetyl-CoA. The N21 of sisomicin forms hydrogen bonds with two water molecules in the ternary complex, W3 and W4, of which W3 also links to the acetyl-CoA. W4 has the potential to form five hydrogen bonds in the ternary complex. Three hydrogen bonds can potentially be formed with the N21, N6, and O51 atoms on sisomicin, whereas one can also be formed with the main chain of Asp183 and another with the side chain of Asp239 (Fig. 4). Upon acetylation, N21 no longer hydrogen-bonds to W3, W4, or Asp183. The side-chain rotamer of Asp183 changes as well, so that no hydrogen bonds are formed between W1 and W2. The rotameric state of Asp239 also changes upon loss of a hydrogen bond with W4 (Fig. 4). The acetylation-mediated flipping of the sisosamine antibiotic ring leads to the loss of all of these hydrogen bonds and likely contributes to product release (Fig. 4). The binary enzyme-CoASH complex lacks water molecules W1 to W4 (fig. S5A). Yet, water molecules W2 to W4 and W1 to W2 are found in the binary enzyme-sisomicin (fig. S5B) and enzyme–acetyl-CoA complexes, respectively (fig. S5C).
Fig. 4. Product-mediated cofactor release.
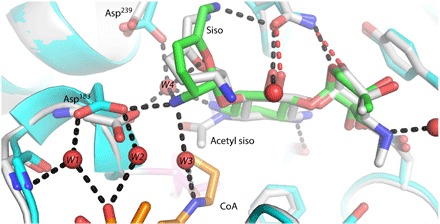
Superposition of the acetylated sisomicin (gray carbon atoms) structure in the binary enzyme–acetyl sisomicin complex on the ternary enzyme-CoASH-sisomicin (cyan protein carbon atoms, with sisomicin carbon atoms colored green and CoASH carbon atoms colored orange) structure. The acetylation of sisomicin leads to flipping of the adjacent sugar ring concomitant with active-site rearrangement that would lead to loss of CoASH product hydrogen bonds.
DISCUSSION
Overuse of antibiotics has led to a drastic increase in bacterial antibiotic resistance, which has evolved resulted in a major global health crisis. Evolution has provided bacteria with multiple routes, enabling the covalent modification of antibiotics and rendering many modern antibiotics inadequate to properly control bacterial infections. Central to the design and bactericidal nature of antimicrobial agents is a complete understanding of the enzymatic mechanisms that bacteria use to inactivate their target antibiotics.
AACs are just one type of AGMEs, but are the most frequently found variant in clinical isolates (16). The complete structure and mechanistic details of catalysis are known for only a handful of AACs, and all characterized thus far confer resistance against a broad spectrum of aminoglycosides. Our studies presented here on AAC-VIa, one of the least promiscuous AGMEs, offer direct evidence of the catalytic mechanism of this enzyme at an unprecedented, proton-level of detail. The sequence and structure similarity of AAC-VIa to two other aminoglycoside N3-acetyltransferases with high (AAC-IIIb) and moderate substrate promiscuity (AAC-IIa) suggest that this mechanism is shared in all aminoglycoside N3-acetyltransferases (14, 17).
The catalytic triad is a ubiquitous catalytic archetype, but there has been no report of a catalytic triad or conservation of the active-site residues among the GNAT family of acetyltransferases studied so far (5). Arylamine N-acetyltransferases, which are not a part of the GNAT family, catalyze the acetylation of arylamines and arylhydrazines using a catalytic triad of Cys-His-Asp, which is also used by cysteine proteases (18). Structural homologs of AAC-VIa have been characterized, and although not explicitly stated or identified, all contain the residues found in the active site of AAC-VIa. We demonstrate that the catalytic triad of AAC-VIa is a new and unique variation, where the substrate itself completes the noncanonical catalytic triad. Substrates have been reported to assist in catalysis by several different mechanisms, including general base catalysis, transition state stabilization, and orientation of water molecules (19). Substrate-assisted catalysis has been observed in many naturally occurring enzymes like Ras p21 (20), type II restriction endonucleases (21), and lysozyme (22), as well as several enzymes from the serine protease family like subtilisin (23) and trypsin (24). Many proteases that use a catalytic triad are inactivated by irreversible covalent modification of the active-site nucleophile, leading to permanent inactivation of the protein (25). However, in AAC-VIa, the nucleophile does not reside on the enzyme itself, which allows for further evasion of the facile inactivation of a nucleophile that would occur when it is located on the protein.
Among the enzymes that use a catalytic triad, the trypsin catalytic triad has been studied extensively, with neutron diffraction studies unequivocally establishing the role of the catalytic histidine and the protonated forms of active-site residues during catalysis (26). Another feature observed in the serine protease catalytic mechanism is the presence of an LBHB (27). Since first being proposed in 1993 (28), the role of LBHBs in enzyme-mediated catalysis has been a highly debated topic (29–31). Yet, the role of the LBHB in serine proteases like chymotrypsin, in aspartate proteases like HIV-1 protease, along with other enzymes like citrate synthase and triose phosphate isomerase, has been well studied and characterized (12). The strength of the LBHB functions to lower the transition state energy required for intermediate formation in these catalytic mechanisms (32).
To our knowledge, the presence of LBHBs has not been reported or postulated to play a role thus far in any of the studies on antibiotic-modifying enzymes. In the case of AAC-VIa, LBHB formation is observed when sisomicin binds and forms a hydrogen bond with the Nε2 atom of His189. This serves to increase the basic nature of the Nδ1 atom of His189, thereby facilitating abstraction of the proton from the antibiotic and subsequent formation of a tetrahedral intermediate with acetyl-CoA. This novel mechanism of antibiotic inactivation further demonstrates the role of LBHB in catalytic triad–mediated enzymatic catalysis while outlining the requirements for the next generation of aminoglycoside antibiotics that these enzymes attempt to inactivate. Antibiotics that can form the requisite polar interactions with their targets, while having sufficiently perturbed pKa values for the nucleophile, will enhance the evasion of enzymatically encoded resistance. Overall, the level of understanding of the AAC-VIa reaction mechanism afforded by neutron diffraction experiments demonstrates the functional importance of a proton level of detail in elucidation of catalytic mechanisms.
MATERIALS AND METHODS
Protein expression, purification, and kinetic assays
Wild-type AAC-VIa used for crystallography was expressed and purified as previously described (11, 13, 14). The mutants were expressed in a similar way as the wild-type enzyme and were purified in their His-tagged forms by using a Ni-Sepharose resin, followed by a Macro-Prep Q column to remove coeluting proteins. Circular dichroism was used to confirm that proteins were folded before kinetic analysis. Kinetic assays were carried out as previously described (11, 13, 14).
Crystallization and diffraction data collection
Crystals for x-ray diffraction were grown by hanging drop vapor diffusion in drops containing 2 μl of the protein solution mixed with 2 μl of the precipitant well solution. Sisomicin, CoASH, ternary sisomicin/CoASH, and acetyl-CoA crystals were grown in 17 to 25% polyethylene glycol (PEG) 4000, 0.2 M ammonium acetate, 10% glycerol, and 0.1 sodium citrate (pH 5.6) in the presence of 1 mM ligands. Crystals belonged to the P 61 space group (Table 1). The crystals of the apo-enzyme were grown from a precipitant solution containing 20 to 30% PEG 8000, 0.2 M MgCl2, and 0.1 M Tris (pH 8.5). Crystals belonged to the C 2 space group (Table 1). These crystals were subsequently soaked in 1 mM acetyl-CoA and 1 mM sisomicin for 15 min to generate the acetylated sisomicin product complex. This resulted in product crystals with a very high mosaicity (2.5°). Crystals were transferred to 35% (w/v) PEG 8000 for cryoprotection, mounted in a nylon loop, and flash-frozen in liquid nitrogen. All x-ray diffraction data were collected at 100 K on a Rigaku 007HF micromax X-ray generator with a Raxis IV++ detector. The x-ray diffraction data were scaled and indexed using HKL3000. The data collection statistics are listed in Table 1.
Crystals for neutron diffraction were grown by sitting drop vapor diffusion in drops containing 15 μl of the protein solution mixed with 15 μl of the precipitant well solution. The apo- and sisomicin-bound enzyme crystals were grown from a precipitant solution containing 20 to 30% PEG 8000, 0.2 M MgCl2, and 0.1 M Tris (pH 8.5), and sisomicin, when present, was added at a concentration of 1 mM. Crystals belonged to the C 2 space group. Once the crystals reached a volume of 0.4 mm3 (1 month), the well solution was replaced with a D2O-containing well solution to facilitate exchange of labile hydrogen atoms with deuterium over a 3-week period. Crystals were then mounted in a quartz capillary for data collection at room temperature by neutron diffraction (Table 2). Room temperature x-ray diffraction data were collected on crystals grown and mounted in an identical manner (Table 2).
Initial neutron diffraction tests were performed on the IMAGINE Instrument at the High Flux Isotope Reactor at Oak Ridge National Laboratory (ORNL). Time-of-flight neutron diffraction data were recorded using the MaNDi (33) Instrument at the Spallation Neutron Source (SNS) on-site at ORNL using all neutrons between 2.0 and 4.0 Å wavelengths. The crystal was static during data collection, whereas it was rotated on ω and φ between images to increase coverage of reciprocal space. Diffraction images were processed and integrated using the Mantid Package (34) and LAUENORM from the LAUEGEN Package (35). LAUENORM performs a wavelength normalization of the Laue data and scaling between Laue diffraction images.
Structure determination, model building, and refinement
The initial structure was solved by molecular replacement using the Phaser program (36), with the FrbF from Streptomyces rubellomurinus (3SMA) as a search model. Manual model building was carried out in Coot (37) and refined using PHENIX (38). The models exhibit good stereochemistry as determined by MolProbity (39); final refinement statistics are listed in Table 1. Atomic coordinates and structure factors for the apo-enzyme, the binary complexes with CoASH, acetyl-CoA, and sisomicin, the ternary AAC-VIa–CoASH–sisomicin complex, and the binary complex with the acetylated sisomicin have been deposited in the PDB under the accession codes of 6BC6, 6BC5, 6BC4, 6BC7, 6BC3, and 6BC2, respectively. Atomic coordinates and structure factors for the room temperature neutron and x-ray diffraction structures of apo- and sisomicin-bound AAC-VIa have been deposited in the PDB (40) under the accession codes of 6BBR and 6BB2, respectively. All molecular graphic figures were prepared by PyMOL (The PyMOL Molecular Graphics System, Version 2.0, Schrödinger LLC).
Supplementary Material
Acknowledgments
Funding: The ORNL Center for Structural Molecular Biology (FWP ERKP291) was supported by the Office of Biological and Environmental Research of the U.S. Department of Energy. Research at the SNS and High Flux Isotope Reactor of ORNL was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. This work was also supported by a grant from the NSF (MCB-1662080). Author contributions: P.K. performed the enzyme assays. P.K. and M.J.C. carried out the protein crystallization experiments, the data collection, and refinement. E.H.S and M.J.C. conceived and designed the experiments. All authors contributed to the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions of the paper are present in the paper and/or the Supplementary Materials. Diffraction data and coordinates have been deposited to the PDB. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaas8667/DC1
fig. S1. Overall structure of apo acetyltransferase AAC-VIa.
fig. S2. Kinetics and homologs of AAC-VIa.
fig. S3. The binary AAC-VIa–CoASH complex.
fig. S4. Overlay of ternary enzyme-CoASH-sisomicin complex with binary complexes of the enzyme with CoASH, acetyl-CoA, and sisomicin.
fig. S5. Sisomicin-mediated ordering of acetyl-CoA binding site.
REFERENCES AND NOTES
- 1.Moazed D., Noller H. F., Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 327, 389–394 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Davies J., Inactivation of antibiotics and the dissemination of resistance genes. Science 264, 375–382 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Shaw K. J., Rather P. N., Hare R. S., Miller G. H., Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 57, 138–163 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wybenga-Groot L. E., Draker K.-a., Wright G. D., Berghuis A. M., Crystal structure of an aminoglycoside 6′-N-acetyltransferase: Defining the GCN5-related N-acetyltransferase superfamily fold. Structure 7, 497–507 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Dyda F., Klein D. C., Hickman A. B., GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 29, 81–103 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vetting M. W., Park C. H., Hegde S. S., Jacoby G. A., Hooper D. C., Blanchard J. S., Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 47, 9825–9835 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolf E., Vassilev A., Makino Y., Sali A., Nakatani Y., Burley S. K., Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 94, 439–449 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Klimecka M. M., Chruszcz M., Font J., Skarina T., Shumilin I., Onopryienko O., Porebski P. J., Cymborowski M., Zimmerman M. D., Hasseman J., Glomski I. J., Lebioda L., Savchenko A., Edwards A., Minor W., Structural analysis of a putative aminoglycoside N-acetyltransferase from Bacillus anthracis. J. Mol. Biol. 410, 411–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bae B., Cobb R. E., DeSieno M. A., Zhao H., Nair S. K., New N-acetyltransferase fold in the structure and mechanism of the phosphonate biosynthetic enzyme FrbF. J. Biol. Chem. 286, 36132–36141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rather P. N., Mann P. A., Mierzwa R., Hare R. S., Miller G. H., Shaw K. J., Analysis of the aac(3)-VIa gene encoding a novel 3-N-acetyltransferase. Antimicrob. Agents Chemother. 37, 2074–2079 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar P., Serpersu E. H., Thermodynamics of an aminoglycoside modifying enzyme with low substrate promiscuity: The aminoglycoside N3 acetyltransferase-VIa. Proteins 85, 1258–1265 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Cleland W. W., Frey P. A., Gerlt J. A., The low barrier hydrogen bond in enzymatic catalysis. J. Biol. Chem. 273, 25529–25532 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Norris A. L., Özen C., Serpersu E. H., Thermodynamics and kinetics of association of antibiotics with the aminoglycoside acetyltransferase (3)-IIIb, a resistance-causing enzyme. Biochemistry 49, 4027–4035 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Norris A. L., Özen C., Serpersu E. H., Correction to Thermodynamics and kinetics of association of antibiotics with the aminoglycoside acetyltransferase (3)-IIIb, a resistance-causing enzyme. Biochemistry 52, 7702–7702 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Norris A. L., Serpersu E. H., Interactions of coenzyme A with the aminoglycoside acetyltransferase (3)-IIIb and thermodynamics of a ternary system. Biochemistry 49, 4036–4042 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Miller G. H., Sabatelli F. J., Hare R. S., Glupczynski Y., Mackey P., Shlaes D., Shimizu K., Shaw K. J., Bauernfeind A., Schweighart S., Shannon K., Patzer J., Molinari G., Schito G. C., GomezLus R., GomezLus S., Ferreira H., Sousa J. C., Vaz M. J. M., Collatz E., Bismuth R., Lambert T., Courvalin P., Minozzi C., Klugman K., Bilgeri Y., Giamarellou H., Petrikkos G., Akalin H., Gur D., Woloj M., Rossi A., Casellas J., Tokumoto M., Couto E., Juliet C., Pinto M. E., Zemelman R., Pedreira W., Fernandez M., Leal I., Guzman M., Murillo J., Isturiz P., Merentes A., Bremner A., Ho B., Mayer K., Ellal J., Fu W., Zhu D., Dornbusch K., Goransson E., The most frequent aminoglycoside resistance mechanisms—Changes with time and geographic area: A reflection of aminoglycoside usage patterns? Clin. Infect. Dis. 24, S46–S62 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Norris A. L., Serpersu E. H., Ligand promiscuity through the eyes of the aminoglycoside N3 acetyltransferase IIa. Protein Sci. 22, 916–928 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandy J., Mushtaq A., Holton S. J., Schartau P., Noble M. E. M., Sim E., Investigation of the catalytic triad of arylamine N-acetyltransferases: Essential residues required for acetyl transfer to arylamines. Biochem. J. 390, 115–123 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dall’Acqua W., Carter P., Substrate-assisted catalysis: Molecular basis and biological significance. Protein Sci. 9, 1–9 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schweins T., Langen R., Warshel A., Why have mutagenesis studies not located the general base in ras p21. Nat. Struct. Biol. 1, 476–484 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Horton N. C., Newberry K. J., Perona J. J., Metal ion-mediated substrate-assisted catalysis in type II restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 95, 13489–13494 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumura I., Kirsch J. F., Is aspartate 52 essential for catalysis by chicken egg white lysozyme? The role of natural substrate-assisted hydrolysis. Biochemistry 35, 1881–1889 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Carter P., Wells J. A., Engineering enzyme specificity by “substrate-assisted catalysis”. Science 237, 394–399 (1987). [DOI] [PubMed] [Google Scholar]
- 24.Corey D. R., Willett W. S., Coombs G. S., Craik C. S., Trypsin specificity increased through substrate-assisted catalysis. Biochemistry 34, 11521–11527 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Powers J. C., Asgian J. L., Ekici Ö. D., James K. E., Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 102, 4639–4750 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Kossiakoff A. A., Spencer S. A., Direct determination of the protonation states of aspartic acid-102 and histidine-57 in the tetrahedral intermediate of the serine proteases: Neutron structure of trypsin. Biochemistry 20, 6462–6474 (1981). [DOI] [PubMed] [Google Scholar]
- 27.Frey P. A., Whitt S. A., Tobin J. B., A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science 264, 1927–1930 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Gerlt J. A., Gassman P. G., An explanation for rapid enzyme-catalyzed proton abstraction from carbon acids: Importance of late transition-states in concerted mechanisms. J. Am. Chem. Soc. 115, 11552–11568 (1993). [Google Scholar]
- 29.Tamada T., Kinoshita T., Kurihara K., Adachi M., Ohhara T., Imai K., Kuroki R., Tada T., Combined high-resolution neutron and x-ray analysis of inhibited elastase confirms the active-site oxyanion hole but rules against a low-barrier hydrogen bond. J. Am. Chem. Soc. 131, 11033–11040 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Adachi M., Ohhara T., Kurihara K., Tamada T., Honjo E., Okazaki N., Arai S., Shoyama Y., Kimura K., Matsumura H., Sugiyama S., Adachi H., Takano K., Mori Y., Hidaka K., Kimura T., Hayashi Y., Kiso Y., Kuroki R., Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution x-ray and neutron crystallography. Proc. Natl. Acad. Sci. U.S.A. 106, 4641–4646 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das A., Prashar V., Mahale S., Serre L., Ferrer J.-L., Hosur M. V., Crystal structure of HIV-1 protease in situ product complex and observation of a low-barrier hydrogen bond between catalytic aspartates. Proc. Natl. Acad. Sci. U.S.A. 103, 18464–18469 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassidy C.S., Lin J., Frey P. A., A new concept for the mechanism of action of chymotrypsin: The role of the low-barrier hydrogen bond. Biochemistry 36, 4576–4584 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Coates L., Cuneo M. J., Frost M. J., He J., Weiss K. L., Tomanicek S. J., McFeeters H., Vandavasi V. G., Langan P., Iverson E. B., The macromolecular neutron diffractometer MaNDi at the Spallation Neutron Source. J. Appl. Cryst. 48, 1302–1306 (2015). [Google Scholar]
- 34.Arnold O., Bilheux J. C., Borreguero J. M., Buts A., Campbell S. I., Chapon L., Doucet M., Draper N., Leal R. F., Gigg M. A., Lynch V. E., Markvardsen A., Mikkelson D. J., Mikkelson R. L., Miller R., Palmen K., Parker P., Passos G., Perring T. G., Peterson P. F., Ren S., Reuter M. A., Savici A. T., Taylor J. W., Taylor R. J., Tolchenov R., Zhou W., Zikoysky J., Mantid—Data analysis and visualization package for neutron scattering and μ SR experiments. Nucl. Instrum. Methods Phys. Res. A 764, 156–166 (2014). [Google Scholar]
- 35.Campbell J. W., Hao Q., Harding M. M., Nguti N. D., Wilkinson C., LAUEGEN version 6.0 and INTLDM. J. Appl. Cryst. 31, 496–502 (1998). [Google Scholar]
- 36.Bunkóczi G., Echols N., McCoy A. J., Oeffner R. D., Adams P. D., Read R. J., Phaser.MRage: Automated molecular replacement. Acta. Crystallogr. D. Biol. Crystallogr. 69, 2276–2286 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaas8667/DC1
fig. S1. Overall structure of apo acetyltransferase AAC-VIa.
fig. S2. Kinetics and homologs of AAC-VIa.
fig. S3. The binary AAC-VIa–CoASH complex.
fig. S4. Overlay of ternary enzyme-CoASH-sisomicin complex with binary complexes of the enzyme with CoASH, acetyl-CoA, and sisomicin.
fig. S5. Sisomicin-mediated ordering of acetyl-CoA binding site.