

Hyaluronic acid hydrogel alleviated inflammatory pain, altered glycosylation, and regulated matrix components for disc repair.
Abstract
Painful intervertebral disc degeneration is mediated by inflammation that modulates glycosylation and induces hyperinnervation and sensory sensitization, which result in discogenic pain. Hyaluronic acid (HA) used as a therapeutic biomaterial can reduce inflammation and pain, but the effects of HA therapy on glycosylation and pain associated with disc degeneration have not been previously determined. We describe a novel rat model of pain induced by intervertebral disc injury, with validation of the pain phenotype by morphine treatment. Using this model, we assessed the efficacy of HA hydrogel for the alleviation of pain, demonstrating that it reduced nociceptive behavior, an effect associated with down-regulation of nociception markers and inhibition of hyperinnervation. Furthermore, HA hydrogel altered glycosylation and modulated key inflammatory and regulatory signaling pathways, resulting in attenuation of inflammation and regulation of matrix components. Our results suggest that HA hydrogel is a promising clinical candidate for the treatment of back pain caused by degenerated discs.
INTRODUCTION
Discogenic low back pain is a very common problem that imposes enormous health and economic burdens on society, accounting for approximately 26 to 42% of patients with chronic low back pain (1). Primarily, intervertebral disc (IVD) degeneration is one of the causative factors of low back pain. The IVD comprises an outer ring of collagen-rich annulus fibrosus (AF), which surrounds a central core proteoglycan-rich gelatinous nucleus pulposus (NP) that mainly provides both mechanical support to absorb compressive forces and the flexibility necessary for the movements (2). Pathological insults, such as catabolic imbalance and inflammatory response, result in degradation of high–molecular mass hyaluronic acid (HA) to low–molecular mass HA, which contributes to disc degeneration through enhancement of the expression of matrix-degradative enzymes, induction of inflammatory responses, and reduction of water retention in the discs (3). The release of neurogenic mediators, such as proinflammatory cytokines [including interleukin-1β (IL-1β) and tumor necrosis factor (TNF)] (4), neurotrophins [including β-nerve growth factor (β-NGF) and brain-derived neurotrophic factor (BDNF)] (5), and neuropeptides [including calcitonin gene-related peptide (CGRP) and substance P] (6), can promote the innervation of peptidergic small neurons into aneural discs (5). These changes sensitize nociceptive terminals and facilitate the development of discogenic back pain (7).
The development of a preclinical model of discogenic pain is necessary to enable the study of the pathogenesis of painful disc degeneration and to provide a platform to determine the effectiveness of potential therapeutic interventions. However, to date, few models have been developed to investigate pain associated with disc degeneration. Currently, surgically induced disc injury, or AF injury, is commonly used for induction of anatomical disruption, because damage to the AF is also known to affect human discs. This injury model is associated with relatively rapid development of degeneration and can be precisely controlled (8). The rat tail model has also been widely adopted for the study of disc degeneration because of the ability to avoid surrounding tissue damage and the accessibility for intervention (8). However, the pain phenotype in the rat tail model has not been determined. In rodent, the reliable signs of pain (nociceptive behavioral responses) have been well characterized (9), making these animals highly appropriate for the investigational studies of painful disc degeneration. Furthermore, c-Fos expression is widely used as a marker of neuronal activity related to nociceptive processing (10), with peripheral noxious stimulation inducing rapid expression of a c-Fos–like immunoreactivity at the spinal cord and brain levels in rodents (11).
HA, or hyaluronan, is a ubiquitously expressed glycosaminoglycan (GAG) located in the extracellular matrix (ECM), in the intracellular environment, and on the cell surface (12). In physiological conditions, HA typically presents as a high–molecular mass molecule of 1 × 106 Da (13), containing around 10,000 alternating units of glucuronic acid (GlcA) and N-acetylglucosamine (GlcNAc), with β-(1,3)- and β-(1,4)-linkages (13). HA is synthesized in the cellular plasma membrane by HA synthases and secreted into the pericellular space via a multidrug resistance transporter (12). HA interacts with specific proteins, including aggrecan, versican, lymphatic vessel endothelial HA receptor 1, TNF-inducible gene 6 protein, and thrombospondin, as well as membrane receptors, such as CD44, HA-mediated motility receptor, and Toll-like receptor 4/2 (12). These interactions are vital for multiple biological processes, including morphogenesis, cell migration, cell survival, apoptosis, inflammation, and tumorigenesis (12). In IVDs, HA is aggregated with proteoglycans, such as aggrecan. Proteoglycans consist of core proteins that are modified extensively with anionic sulfated GAGs, such as chondroitin sulfate (CS) and keratan sulfate (KS). Proteoglycans enable the generation of osmotic swelling pressure during the maintenance of tissue hydration (14).
The use of HA to target multiple signaling pathways underlying painful disc degeneration is a potential therapeutic strategy for disc repair. Several different formulations of HA–poly(ethylene glycol) (PEG) hydrogels have been shown to increase the formation of multicell clusters on seeding with NP or AF cells compared with gelatin substrates (15). In a nucleotomized organ culture model, treatment with biomimetic fibrin–HA hydrogel can maintain NP cell viability at >90%, increase production of GAGs and type II collagen, and restore compressive stiffness and disc height compared with fibrin gels (16). We have demonstrated that type II collagen–4S-StarPEG–HA microgels initiate differentiation of adipose-derived stem cells toward an NP-like phenotype (17). These findings suggest that HA has a potential use as a carrier system in IVD therapeutic strategies.
Therapeutic applications of high–molecular weight HA have been shown to facilitate long-term functional improvements by attenuating inflammation and pain in a number of clinical conditions, including osteoarthritis (OA) (18). Compared with control solutions, HA solutions decrease transient receptor potential cation channel subfamily V member 1 (TRPV1)–mediated impulse firing and channel sensitization in dorsal root ganglia (DRG) cultures and reduced both heat-induced and capsaicin-induced nocifensive responses in rodents, and intra-articular injections of HA decrease capsaicin responsiveness of joint nociceptor fibers (19). In the case of IVD degeneration, we have reported therapeutic effects of a cross-linked HA-PEG hydrogel in attenuating inflammation and expression of neurotrophic factors by binding to CD44 in inflamed NP cells (20). However, the extent to and mechanisms by which HA targets inflammatory pain in disc degeneration are not currently established.
The IVD is characterized by a large carbohydrate complex, which is considered an inert material, to become hydrated and contribute to the structural scaffold. Specifically, glycans are normally in the front lines to communicate between the cells and thereby play a key role in mediating cell-cell and cell-ECM interactions from cell migration and immune response to disease progression (21). Nevertheless, the change of glycan distribution in response to IVD injury and post-implantation of any biomaterial implant is not determined.
Here, we describe the development of a novel preclinical model of pain associated with symptomatic disc degeneration in the rat tail. We have used this model to further investigate the efficacy of treatment of painful disc degeneration with an HA hydrogel. First, we hypothesized that surgical puncture–induced disc injury would elicit robust pain–related behavior in rats, induce spinal molecular markers of pain and structural disc degeneration, and differentially modulate glycosylation in AF and NP tissues. Second, we hypothesized that HA hydrogel treatment would subsequently alleviate nociceptive behavior, inhibit sensory hyperinnervation and expression of peripheral pro-nociceptive receptors TRPV1 and high-affinity NGF receptor (Trk-A) in vivo, down-regulate mRNA expression of the spinal nociception markers c-Fos and Tac1 (which encodes substance P), and regulate the glycosylation and proteomic expression to modulate inflammatory and protein regulatory pathways in both NP and AF tissues.
RESULTS
Evaluation of IVD injury–induced pain model
Rat coccygeal IVDs were surgically punctured at levels Co5–Co6 and/or Co4–Co5 (Fig. 1A, i, and C, i and ii) to induce robust pain–related behavior.
Fig. 1. Schematic representations of the experimental design and procedures.
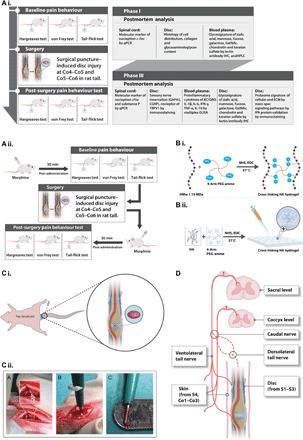
(A) Experimental design. (i and ii) The study was designed as follows: phase I, development and characterization of an IVD pain model; phase II, validation of the model using morphine; phase III, therapeutic efficacy of implanted HA hydrogel following IVD injury. (B) Cross-linking of high–molecular weight HA and four-arm PEG amine. (i) After functionalization with N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), the amine groups of HA molecules react with the succinimidyl groups of PEG amine. (ii) Hydrogels for implantation were prepared by pipetting 4 μl of a mixture of HA, PEG amine, NHS, and EDC onto a hydrophobic surface, followed by incubation at 37°C for 1 hour to complete cross-linking. (C) Surgical procedure. (i) Identification of the coccygeal disc at the Co4–Co5 level. A rubber band (marked in blue) was applied at the base of the tail. Discs were treated by injury alone or injury with implantation of HA hydrogel (marked in purple). (ii) Steps of the surgical procedure. A disc was dissected by pushing aside connective tissue and tendons until the ivory matter of the AF tissue (gray arrow) was reached. The defect was created by puncturing NP tissue through AF tissue at a diameter of 1 mm and a depth of up to 2 mm. (D) Neuroanatomy of the rat tail to define the receptive fields of the stimuli used for the pain behavior tests.
Pain phenotype
Pain phenotyping was conducted by quantitative sensory testing after surgical puncture–induced IVD injury in the rat tail.
Thermal hyperalgesia
The Hargreaves test was performed to study thermal hyperalgesia by applying a noxious thermal stimulus to the ventral base of the tail. At baseline (before any procedure), no significant difference was seen between the four experimental groups [rats receiving only isoflurane (sham-isoflurane), rats receiving a skin incision but no injury to the disc tissues (sham-incision), rats with Co4–Co5 injury, and rats with Co4–Co5 + Co5–Co6 injury]. Rats in the two sham groups had withdrawal latency comparable to baseline when evaluated on days 2, 7, 14, and 29. However, withdrawal latency was significantly shorter in both the injury groups after treatment compared with baseline or with the sham groups at each time point. No significant difference was seen between the Co4–Co5 and Co4–Co5 + Co5–Co6 injury groups, indicating that single- and adjacent-level injuries produce comparable levels of thermal hyperalgesia in this model. Thus, puncture-induced disc injury evoked thermal hyperalgesia in the rat tail, evident on postoperative days 2, 7, 14, and 29 [tested by one-way and repeated-measures analysis of variance (ANOVA), P < 0.05; n = 5 rats per group; Fig. 2A, i].
Fig. 2. Characterization of a pain model induced by IVD injury.
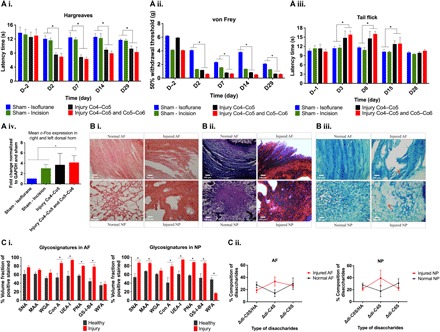
(A) Tests of pain sensitivity and their effects on gene expression. (i) The Hargreaves test demonstrated thermal hyperalgesia, with significantly lower latency times in both injury groups than in both sham control groups. (ii) The von Frey test indicated mechanical allodynia, with significantly lower 50% withdrawal threshold in the double-disc injury group than in either control group. (iii) The tail-flick test denoted hypoalgesia, with significantly higher withdrawal latency times in both injury groups compared to both sham control groups. (iv) Mean fold change of the c-Fos gene in the dorsal horn of the spinal cord was significantly higher after injury than in either control groups, with rats euthanized within 30 min of thermal stimulation at proximal rat tail at day 29. (B) Histological assessment of healthy and injured discs. (i) On postmortem histology of day 29, H&E staining showed AF tears and clustering of cells and the presence of chondroid nests in NP tissue after disc injury. (ii) Masson’s trichrome staining showed loose collagen fibers in AF tissue. (iii) Alcian blue staining showed less proteoglycans in the NP matrix. (C) Glycosignatures and sulfation patterns in response to disc injury. (i) Binding of SNA-I [to α-(2,6)–linked sialic] and MAA [to α-(2,3)–linked sialic acid] was higher in NP tissue in injured than in uninjured discs. Binding of Con A lectin (indicating mannose), UEA-I [to α-(1,2)–linked fucose], PNA [to nonsialylated Gal-β-(1,3)-GalNAc], and GS-I-B4 (to α-Gal) was higher in AF and NP tissues in injured tissues compared to uninjured discs. Binding of WFA (to α- or β-linked terminal GalNAc) was lower in NP tissues in injured compared to uninjured discs. (ii) Quantification of HA and the variously sulfated CS disaccharides by HPLC. *P < 0.05, significant difference between groups, by one-way ANOVA (A) or t test (C). n = 5 rats per group for (A), and n = 3 rats per group for (B) and (C). Data are means ± SEM. Scale bars, 100 μm.
Mechanical allodynia
von Frey filaments were applied to the same receptive field as that in the Hargreaves test to analyze mechanical allodynia. No differences between the four groups were observed at baseline. The 50% withdrawal threshold was significantly lower in the sham-incision group than in the sham-isoflurane group, and in the Co4–Co5 + Co5–Co6 injury group compared with both sham groups at days 2, 7, 14, and 29. No differences were observed between the injury groups. These results suggest that puncture-induced disc injury elicits mechanical allodynia in the rat tail until at least 29 days after operation (tested by one-way and repeated-measures ANOVA, P < 0.05; n = 5 rats per group; Fig. 2A, ii).
Thermal hypoalgesia
Tail-flick testing was conducted by applying a radiant heat stimulus to the ventral middle part of the tail. No differences in withdrawal latency were observed at baseline. No differences from baseline were observed in the sham control groups up to day 28. On days 3, 8, and 15, the latency time in the injury groups was higher than at baseline, and significantly higher than in the sham groups on the same days, suggesting hypoalgesia to a stimulus applied distal to the site of injury (tested by one-way and repeated-measures ANOVA, P < 0.05; n = 5 rats per group; Fig. 2A, iii). These results may indicate a diffuse noxious inhibitory control (DNIC)–like process operating in the model, with injury-related pain suppressing pain at a site distal from the site of injury.
Up-regulation of the c-Fos gene
We activated afferent sensory neurons by applying thermal stimulus to the distal rat tail and dissected the sacral dorsal horn in the spinal cord within 30 min following stimulation. We used this approach because transcription of c-Fos mRNA occurs rapidly, within minutes of stimulation, and its expression is transient. In our experimental groups, significant up-regulation of c-Fos mRNA expression occurred in the dorsal horn in both injury groups compared to the control groups (fold change versus sham-isoflurane was 3.7 ± 1.0 for Co4–Co5 and 4.1 ± 0.6 for Co4–Co5 + Co5–Co6 injury); however, there was no significant difference between sham-incision (3.775 ± 0.7563) and either of the injury groups (tested by one-way ANOVA, P < 0.05; n = 5 rats per group; Fig. 2A, iv). These data suggest that IVD injury induces activation of afferent neurons after noxious stimulation, which in turn results in nociceptive transmission in the central nervous system with consequent activation of second-order pain transmission neurons of the spinal cord dorsal horn, as evidenced by increased expression of the marker of neuronal activity, c-Fos.
Injured discs show microstructural disc degeneration
In the injured disc tissues at postsurgery day 29, hematoxylin and eosin (H&E) staining revealed annular rupture, mixed clustering of NP cells, and the presence of chondroid nests in the NP tissue, whereas uninjured discs had well-organized AF tissue, normal NP cellularity, and no cell clusters (Fig. 2B, i). Masson’s trichrome staining of injured discs showed loose collagen fibers in AF tissue, with fibrinous components dominating both AF and NP tissues (Fig. 2B, ii). Compared with uninjured discs, reduced Alcian blue staining in the NP matrix in injured discs indicated a reduction in expression of proteoglycans and HA in the ECM. We graded the tissue sections according to the main subcategories of histological classification (Fig. 2B, iii), recording scores of zero for uninjured discs and higher scores in all categories for injured discs. The mean total score of seven for the injured discs, according to a validated histological grading system (22), corresponded to a Thompson grade of III (Table 1). These findings indicate that IVD injury induced anatomical disruption in the disc tissue for at least a month after injury.
Table 1. Histological grading based on all staining showed microstructural disc degeneration after injury, which was clinically classified as being grade III.
| Category | Score | Histological features | Observed score | |
| Normal disc | Injured disc | |||
| AF morphology | 0 | Well-organized AF, half-ring–shaped structure, collagen lamellae | 0 | |
| 1 | Partly ruptured AF; loss of half-ring–shaped structure | 1 | ||
| 2 | Completely ruptured AF; no intact half-ring–shaped collagen lamellae | |||
| NP cellularity | 0 | Normal cellularity; no cell clusters | 0 | |
| 1 | Mixed cellularity; normal pattern with some cell clusters | |||
| 2 | Mainly clustered cellularity; chondroid nests present | 2 | ||
| NP matrix | 0 | Intense staining; blue staining dominates | 0 | |
| 1 | Reduced staining; mixture of blue and slight red staining | |||
| 2 | Faint blue staining; increased red staining | 2 | ||
| Boundary between AF and NP | 0 | Clear boundary between AF and NP tissues | 0 | |
| 1 | Boundary less clear; loss of annular-nuclear demarcation | |||
| 2 | No distinguishable boundary between AF and NP tissues | 2 | ||
| Total score | 0 | 7 | ||
Injured discs have a unique glycosignature
At the cellular and ECM levels, IVDs have been shown to have distinct glycoprofiles, as well as compositions of CS and sulfation patterns that vary from immaturity to maturity, suggesting a role for sugar moieties in modulation of age-associated disc degeneration. To investigate this role, we determined the glycoprofiles in injured and healthy IVDs with a library of lectins and carbohydrate-specific antibodies that were chosen on the basis of typical mammalian glycosylation expression.
Sialylation
Sambucus nigra agglutinin I (SNA-I) lectin binds to α-(2,6)–linked sialic acid, Maackia amurensis agglutinin (MAA) binds to α-(2,3)–linked sialic acid, and wheat germ agglutinin (WGA) binds to sialic acid and GlcNAc (Table 2). In tissues taken on day 29, we observed significantly higher binding of SNA-I and MAA in NP tissue in injured IVDs than in uninjured IVDs (Fig. 2C, i, and fig. S1, A and B). However, only slightly higher binding of SNA-I and MAA was observed in injured AF tissue than in uninjured tissue. We found no significant difference in WGA binding in both AF and NP (Fig. 3C, i, and fig. S1C). Overall, SNA-I, MAA, and WGA binding in each tissue (AF and NP) was higher in injured discs than in uninjured discs (t test, P < 0.05; n = 3; Fig. 2C). These data suggest that there is an increase in sialylation during inflammation as expected.
Table 2. Binding specificity and haptenic sugars of lectins for profiling of tissue glycosylation.
A haptenic sugar is a monosaccharide or disaccharide that can inhibit binding of a lectin.
| Lectin | Binding specificity | Haptenic sugar (100 mM) |
| SNA-I | Sialic acid-α-(2,6)–Gal(NAc)–R | Lactose |
| MAA | Sialic acid-α-(2,3)–Gal(NAc)–R | Lactose |
| WGA | Sialic acid, GlcNAc | GlcNAc |
| PNA | Gal-β-(1,3)-GalNAc (T antigen), > GalNAc > lactose > Gal, terminal β-Gal, nonsialylated | Gal |
| GS-I-B4 | Terminal α-linked Gal | Gal |
| Con A | α-Linked Man, GalNAc and GlcNAc | Man |
| UEA-I | α-(1,2)–Linked fucose | Fucose |
| WFA | GalNAc and CS | GalNAc |
Fig. 3. Alleviation of the injury-induced pain phenotype by HA hydrogel implantation and by morphine treatment.
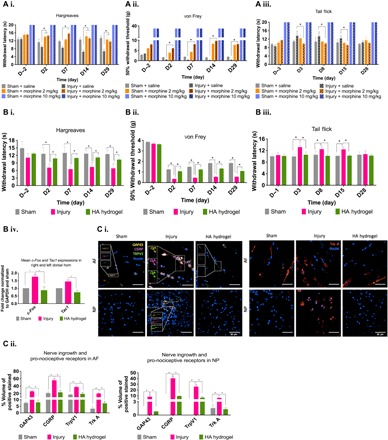
(A) (i) Alleviation of the injury-induced pain phenotype by HA hydrogel implantation and by morphine treatment. (i) Low-dose morphine (2 mg/kg) injection 30 min before the Hargreaves test resulted in significantly higher withdrawal latency times than in injured rats without morphine. (ii) Low-dose morphine injection before the von Frey test resulted in significantly higher 50% withdrawal thresholds than in injured rats or the morphine treatment. (iii) Low-dose morphine injection before the tail-flick test resulted in significantly lower withdrawal latency times than in the injured rats without morphine. (B) (i) In the Hargreaves test, withdrawal latency times were significantly higher in the HA hydrogel–treated injury group (and the control group) than in the untreated injury group. (ii) In the von Frey test, 50% withdrawal thresholds were significantly higher in the HA hydrogel–treated injury group (and the control group) compared to the untreated injury group. (iii) In the tail-flick test, withdrawal latency times were significantly lower in the HA hydrogel–treated injury group (and the control group) than in the untreated injury group. (iv) Post-mortem, expression of genes encoding molecular markers of nociception substance P (Tac1) and c-Fos was significantly lower in the HA hydrogel–treated injury group (and the control) than in the untreated injury group. (C) (i) Confocal microscopy demonstrated the presence in AF and NP tissues, particularly in the untreated injury group, of the GAP43 protein (yellow label) for nerve ingrowth, the sensory neuropeptide CGRP (purple label), and TRPV1 (green label). (ii) The NGF receptor Trk-A (red label) was evident in AF and NP tissues. (iii) The quantified percentage volume fractions of GAP43, CGRP, TRPV1, and Trk-A were significantly higher in the untreated injury group than in the sham control group or the HA hydrogel–treated injury group in both AF and NP tissues. *P < 0.05, significant difference between groups, by one-way ANOVA. n = 10 for (A) and (B), and n = 4 for (C). Data are means ± SEM. Area fraction data were normalized to the total area. Scale bars, 50 μm.
High-mannose–type glycosylation
Concanavalin A (Con A) lectin binds to mannose, which is present in all N-linked glycans (Table 2). An increase in the expression of high-mannose type N-linked structures is typically observed under cellular stress, and undifferentiated stem cells also express greater quantities of high-mannose structures than differentiated cells. In both NP and AF tissues, we detected significantly higher levels of binding of Con A in the injury groups than in the control groups (t test, P < 0.05; n = 3; Fig. 2C and fig. S1D), which may indicate an increase in cellular stress.
Fucosylation
Ulex europaeus agglutinin I (UEA-I) lectin binds to α-(1,2)–linked fucose (Table 2). In both NP and AF tissues, significantly higher levels of binding of UEA-I in the injury groups were detected compared to the control groups (t test, P < 0.05; n = 3; Fig. 2C and fig. S1E), suggesting glycosylation alterations associated with inflammation.
Galactosylation
Griffonia simplicifolia isolectin (GS-I-B4) and peanut agglutinin (PNA) lectins bind to terminal α-linked galactose (Gal) residues and Gal-β-(1,3)-GalNAc (T antigen), respectively (Table 2). In both NP and AF tissues, we detected significantly higher levels of binding of GS-I-B4 and PNA in the injury groups than in the control groups (t test, P < 0.05; n = 3; Fig. 2C and fig. S1, F and G). GS-I-B4 binding was intracellular, whereas PNA binding was seen in the ECM. Wisteria floribunda agglutinin (WFA) lectin binds to either α-linked or β-linked terminal N-acetylgalactosamine (GalNAc), including that in CS (Table 1). In both AF and NP tissues, we observed binding of WFA only in the ECM. In NP ECM, significantly lower levels of WFA binding occurred in the injury groups than in the control groups (t test, P < 0.05; n = 3; Fig. 2C and fig. S1H).
CS and HA composition
The proportions of HA and CS unsaturated disaccharides (Δdi-C0S, Δdi-C4S, and Δdi-C6S) were assessed by high-performance liquid chromatography (HPLC) in the control and injury groups (Fig. 2C, ii). The HPLC method used did not differentiate between the unsaturated HA and C0S disaccharides, likely because their peaks have a close retention time and the unsaturated HA disaccharide would be present in much greater quantity than Δdi-C0S, thereby overlapping the latter peak. Although, in both NP and AF tissues, the proportion of C4S tended to be higher in the injury groups than in the control groups, no significant differences were observed in disaccharide composition (P < 0.05). The C6S composition was lower (but not significant) in the injury groups compared to control groups of AF and NP tissues. Overall, these findings indicated IVD injury–induced matrix changes with a trend toward altered sulfation in the injured tissues. Collectively, HA-treated rats demonstrated persistent mechanical allodynia and thermal hyperalgesia, along with changes in molecular marker expression and glycosylation, and structural disc degeneration.
Validation of IVD pain model by morphine treatment
To inhibit pain in our model, we injected high (10 mg/kg) and low (2 mg/kg) doses of morphine 30 min before each nociceptive behavioral test on pre- and postoperative days, as described in Materials and Methods (Fig. 1A, ii).
Morphine reduces thermal hyperalgesia
The Hargreaves test demonstrated significantly higher withdrawal latency times in both control and injury groups with low-dose morphine compared with the saline-treated injury group (one-way ANOVA, P < 0.05; n = 5; Fig. 3A, i), indicating that morphine reduced thermal hyperalgesia in the rat tail, and that this effect persisted in the presence of injury when the thermal stimulus was close to the injury site. High-dose morphine induced a state of catalepsy, resulting in withdrawal latency times at or above the maximum for the assay (20 s).
Morphine alleviates mechanical allodynia
von Frey testing demonstrated that, on days 2, 7, and 14, the 50% withdrawal threshold was significantly higher in the injury group with low-dose morphine than in the injury group with saline treatment (one-way ANOVA, P < 0.05; n = 5; Fig. 3A, ii), indicating that morphine alleviated mechanical allodynia in the rat tail close to the site of injury. Rats that received a high dose of morphine exhibited the maximum withdrawal threshold.
Morphine reverses thermal hypoalgesia
In the tail-flick test on days 3, 8, and 15, withdrawal latency was significantly lower in the injury groups with low-dose morphine than with saline treatment (one-way ANOVA, P < 0.05; n = 5; Fig. 3A, iii), suggesting that morphine alleviated thermal hypoalgesia in the rat tail when the thermal stimulus was not close to the injury site. Rats that received a high dose of morphine were cataleptic and did not respond to the stimulus within the time frame of the test.
Therapeutic efficacy of implantable HA hydrogel in painful IVD degeneration
Our results indicate that the rat tail injury model produced a persistent pain phenotype, enabling us to investigate the effects of HA hydrogel in the modulation of pain under physiological conditions. In addition, no leakage of the implanted HA hydrogel was seen in the injured discs to the surrounding tissue.
HA hydrogel exhibits anti-hyperalgesic and anti-allodynic effects
Using this model, we performed quantitative sensory testing to observe therapeutic efficacy of HA hydrogel in the alleviation of nociceptive behavior. In injured rats that were implanted with HA hydrogel (and in sham control rats), the withdrawal latency in the Hargreaves test was significantly higher than in injured rats without HA hydrogel at each time point over a 29-day period (one-way ANOVA, P < 0.05; n = 10; Fig. 3B, i). Similarly, significant increases in the 50% withdrawal threshold in the von Frey test were observed in the HA hydrogel–treated injury group (and in the sham control group) compared with the untreated injury group (one-way ANOVA, P < 0.05; n = 10; Fig. 3B, ii). These results suggest that HA hydrogel alleviated thermal hyperalgesia and mechanical allodynia in the rat tail around the injury site. In the tail-flick test, injured rats receiving HA hydrogel treatment (and sham control rats) had significantly shorter withdrawal latency than injured rats without treatment (one-way ANOVA, P < 0.05; n = 10; Fig. 3B, iii), suggesting that HA hydrogel treatment suppresses thermal hypoalgesia in response to a stimulus applied distal to the site of injury in the rat tail.
HA hydrogel suppresses c-Fos and Tac1 expression at the spinal level
The observation that the HA hydrogel modulates pain behavior prompted us to investigate mRNA expression of the immediate-early genes c-Fos and Tac1, which encodes protachykinin-1 (a precursor protein that is cleaved into peptides including substance P), in the pain-processing neurons of the left spinal cord dorsal horn. At day 29, the dorsal horn was dissected within 30 min of application of thermal stimulus to the proximal tail. We found significantly lower c-Fos expression in the HA hydrogel–implanted injury group (and in the sham control group) compared with the untreated injury group. A similar result was obtained for Tac1 (for both c-Fos and Tac1: one-way ANOVA, P < 0.05; n = 4; Fig. 3B, iv). These results suggested that the HA hydrogel suppressed nociceptive transmission by attenuating injury-induced c-Fos and Tac1 expression in dorsal horn neurons.
HA hydrogel inhibits injury-induced peripheral sensory hyperinnervation and expression of the pro-nociceptive receptors TRPV1 and Trk-A
Using confocal fluorescence microscopy, we examined colocalization of the GAP43 protein (which is expressed during nerve ingrowth), the sensory neuropeptide CGRP, the pro-nociceptive ion channel TRPV1, and NGF receptor Trk-A in the AF and NP tissues 29 days after operation. GAP43, CGRP, and TRPV1 colocalized in AF tissue after injury (Fig. 3C, i). We also observed a projection of sensory nerve innervation with neuropeptides (GAP43 and CGRP), TRPV1, and Trk-A in NP tissue (Fig. 3C, i). We observed less GAP43, CGRP, and TrpV1 immunoreactivity in AF and NP tissues from control rats and from injured rats implanted with HA hydrogels than in tissues from untreated injured rats (Fig. 3C, i). In both AF and NP tissues, the percentage volume fractions of immunostained GAP43, CGRP, TRPV1, and Trk-A were all significantly higher in the untreated injury group than in either the HA hydrogel–treated injury group or the untreated sham control group (one-way ANOVA, n = 4). This was assessed by quantifying four microscopic views of each slide with three technical and four biological replicates (P < 0.05; Fig. 3C, ii). Overall, these findings confirmed that HA hydrogel implantation inhibited injury-induced peripheral sensory innervation and pro-nociceptive TRPV1 and Trk-A expression in AF and NP tissues.
HA hydrogel modulates sialylation, galactosylation, and GAG content
Altered sialylation has been observed in degenerative joint diseases. We observed lower SNA-I binding in AF and NP tissues of rats in the HA hydrogel–treated injury group than in the untreated injury group (one-way ANOVA, P < 0.05; n = 4; Fig. 4A, i, and fig. S2A). MAA binding showed a similar pattern of binding (one-way ANOVA, P < 0.05; n = 4; Fig. 4A, ii, and fig. S2A). No significant difference in WGA binding was observed in either AF or NP tissues of rats in the untreated injury group compared with rats in the HA hydrogel–treated injury group (Fig. 4A, iii, and fig. S2A).
Fig. 4. Effects of HA hydrogel implantation on glycosylation in the injury-induced pain model.
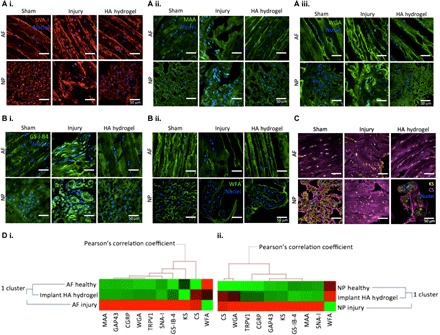
(A to C) Assessment of glycosylation on day 29 after injury through quantification of lectin binding and GAG expression (fig. S2). (A, i to iii) SNA-I (red label), MAA (green label), and WGA (green label) binding to α-(2,6)–linked sialic acid, α-(2,3)–sialylated Gal and N-acetyl-d-glucosamine, or sialic acid was observed in the sham control, injury, and HA hydrogel–treated injury groups, respectively, in AF and NP tissues. (B, i to ii) GS-I-B4 (green label) and WFA (green label) binding to α-Gal and terminal GalNAc motifs was observed in the untreated injury group in AF and NP tissues of sham control, untreated injury, and HA hydrogel–treated injury groups. (C) Expressions of CS (purple label) and KS (yellow label) were denoted in the sham control, untreated injury, and HA hydrogel–treated injury groups in AF and NP tissues. (D) Correlation between glycoprofiles and nociception markers. Clustering analysis was carried out on the quantification profiles from confocal fluorescence microscopy of glycosylation and sensory hyperinnervation and nociceptive markers in AF (i) and NP (ii) tissues for the sham control, untreated injury, and HA hydrogel–treated injury groups (n = 4). Scale bars, 50 μm.
For determination of galactosylation, intracellular GS-I-B4 binding was significantly lower in both AF and NP tissues in the HA hydrogel–treated injury group than in the untreated injury group (one-way ANOVA, P < 0.05; n = 4; Fig. 4B, i, and fig. S2B). WFA binding in the ECM of NP tissue was significantly higher in the HA hydrogel–treated injury group than in the untreated injury group (one-way ANOVA, P < 0.05; n = 4; Fig. 4B, ii, and fig. 2B), but not significantly different in AF tissue.
The CS and KS expression was determined by antibody staining. CS localized in the ECM, whereas KS localized intracellularly (Fig. 4C). CS expression was significantly higher in the untreated injury group than in the sham control group in both AF and NP tissues (one-way ANOVA, P < 0.05; n = 4; fig. S2A) and did not significantly differ between the untreated injury group and the HA hydrogel–treated injury group. KS expression was significantly higher in NP cells in the untreated injury group than in the sham control group, and was significantly lower in both AF and NP cells in the HA hydrogel–treated injury group than in the untreated injury group (one-way ANOVA, P < 0.05; n = 4; Fig. 4C and fig. S2A).
Correlation of glycosylation, innervation, and markers of nociception
Associations between binding of markers of glycosylation and innervation by nociceptive nerves in AF and NP tissues were analyzed by clustering analysis on the basis of Pearson correlation coefficient distances (Fig. 4D, i and ii). Sialylation expression (measured by binding of SNA-I, MAA, and WGA), galactosylation (GS-I-B4), KS, and CS correlated with each other and with sensory innervation (GAP43 and CGRP) and TrpV1 in AF and NP tissues. In addition, we observed a relationship between WGA and GS-I-B4 binding, and between KS and CS with GAP43, CGRP, and TRPV1, in NP tissue (Fig. 4D, i and ii).
Injury and treatment modulate the cellular and extracellular proteomic signature
Protein expression in the rat tail injury model was analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Tryptic digests of AF and NP tissues contained higher numbers of proteins than the ECM-specific proteinase K digests (fig. S3). The proteinase K digestion method identified 532 and 766 proteins specifically in the ECM of AF and NP tissues, respectively (fig. S3A). Tryptic digestion identified 2126 and 1084 cellular and extracellular proteins in AF and NP tissues, respectively (fig. S3B). Heatmaps show the relative (log2-transformed) expression levels of differentially expressed proteins (fold change >2 or <−2 for log2 expression values, P < 0.01) in the AF and NP tissues in the sham control, untreated injury, and HA hydrogel–treated injury groups (Fig. 5A, i and ii). For Ingenuity Pathway Analysis (IPA), we set the −log (P value) threshold to 1.3 to determine dysregulated signaling pathways from the data set of 389 differentially expressed proteins. The main dysregulated cellular functions, including inflammatory response innervation, neurotransmission, connective tissue and neuromuscular disorder, and ECM regulation, were analyzed in more detail with IPA, because these functions are key determinants in disc degeneration. We found that pathways involving eukaryotic initiation factor 2, serine/threonine-protein kinase mTOR (mammalian target of rapamycin), the unfolded protein response, the acute-phase response, and endothelial nitric oxide synthase were represented by the differentially expressed proteins (Fig. 5B, i). Acute-phase response signaling involves IL-1β, IL-6, TNF-α, and the glucocorticoid receptor (among many other factors) (fig. S4) and can modulate inflammation (fig. S5) and catabolic responses in AF and NP tissues. Among the upstream regulators of the cellular functions that were modified in our rat tail model, transforming growth factor β1 (TGF-β1), proto-oncogene c-Fos, SMAD family member 3 (Smad3), p38 mitogen-activated protein kinase (MAPK), and nuclear factor κB (NF-κB) were activated in AF and NP tissues in untreated and HA hydrogel–treated injury groups compared with sham controls (Fig. 5B, ii). Finally, protein synthesis, cell death and survival, connective tissue disorder, inflammatory response, and skeletal and muscular disorder were the key diseases and biological functions that had significantly higher representation in our experimental groups (Fig. 5B, iii).
Fig. 5. Proteomic analysis of IVDs.
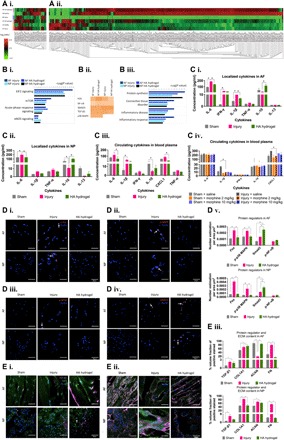
(A) Proteins were isolated from the ECM by proteinase K digestion (i) and from cells and ECM by trypsin digestion (ii). Differentially expressed proteins are shown in heatmaps of log2-transformed abundance generated by Peak Studio, with red for high expression and green for low expression. (B) IPA of the protein data set from MaxQuant analysis. (i) IPA identified canonical pathways that were likely to be involved in the response to disc injury and to HA hydrogel implantation in AF and NP tissues. (ii) Heatmap of the top upstream regulators involved in modulated cellular functions with a Z score of >1.5. (iii) Disease- and function-related pathways identified by IPA. (C) Cytokine profiling. (i and ii) In AF and NP tissues, the levels of IL-6 and IL-1β (and IFN-γ in AF) were significantly higher (and IL-10 was lower) in the untreated injury group than in the sham control or HA hydrogel–treated injury groups. (iii) In blood plasma, the levels of IL-6, CXCL1, and IL-1β were significantly higher in the untreated injury group than in the sham control or HA hydrogel–treated injury groups. (iv) Levels of IL-6, CXCL1, and IL-1β varied between saline-treated and morphine-treated groups. (D and E) Expression of the regulator proteins: (D, i and v) c-Fos, (D, ii and v) phosphorylated p38 MAPK, (D, iii and v) phosphorylated NF-κB, (D, iv and v) Smad3, (E, i and iii) TGF-β1 and type I collagen, and (E, ii and iii) aggrecan and fibronectin. *P < 0.05, significant differences between the HA-treated injury and untreated injury groups, two-way ANOVA [n = 10 for (C, iv), and n = 5 for (A), (B), (C, i and iii), (D), and (E)]; *P < 0.05, significant differences between the HA-treated injury and untreated injury groups, one-way ANOVA [n = 3 for (D) and (E)]. Data are means ± SEM. Scale bars, 50 μm.
HA hydrogel attenuates injury-induced expression of IL-6 and IL-1β
To investigate the effect on inflammatory signaling in our model, we determined the levels of IL-6, IL-1β, TNF, interferon-γ (IFN-γ), CXCL1, IL-10, and IL-13 in blood plasma and disc tissues by the multiplex enzyme-linked immunosorbent assay (ELISA) method. In AF and NP tissues and plasma, levels of proinflammatory cytokines were significantly higher (and anti-inflammatory IL-10 was lower) in the injury group than in the sham control or HA hydrogel–treated injury groups (two-way ANOVA, P < 0.05; n = 5 for AF or NP tissues for each group, n = 10 for blood plasma for each group; Fig. 5C, i to iii). Furthermore, we measured the levels of cytokines in the plasma after pain inhibition by systemic morphine injections. Plasma levels of CXCL1 and IL-1β were higher in the saline-treated injury group than in the saline-treated sham group or the morphine-treated sham and injury groups (two-way ANOVA, P < 0.05; n = 5 for each group; Fig. 5C, iv). Our results suggest that acute-phase response signaling was involved in the dysregulation of IL-6 and IL-1β in response to IVD injury, and that HA hydrogel treatment targeted these canonical pathways by down-regulating proinflammatory cytokines in both AF and NP tissues. Here, we have validated upstream regulators of c-Fos, phosphorylated p38 MAPK, and phosphorylated NF-κB, as evident in the confocal photomicrographs, all of which are involved in inflammatory pain signaling (Fig. 5D, i to iii and v).
HA hydrogel potentially regulates ECM deposition via Smad3–TGF-β1 signaling
Inflammation has a critical role in the modulation of the catabolic process during IVD degeneration. The results of our pathway analysis suggest that the Smad3 protein family was a candidate for mediation of ECM deposition via Smad3–TGF-β1 signaling after HA hydrogel treatment (Fig. 5D, iv and v, and E, i to iii). Protein analysis demonstrated higher levels of type I collagen, type II collagen, fibronectin, fibromodulin, and biglycan in NP tissue in the injury group than in the sham group or the HA hydrogel–treated injury group. In AF tissue, compared with the sham group, levels of type II collagen and fibronectin were higher and fibromodulin and biglycan were lower in the injury group. HA hydrogel treatment resulted in a lower level of type II collagen and higher level of fibromodulin compared with injury alone, but did not affect the levels of fibronectin or biglycan (fig. S6). We also observed nuclear expression of Smad3 in AF and NP tissues (Fig. 5D, iv and v).
DISCUSSION
To the best of our knowledge, this is the first study to establish a model of painful or symptomatic disc degeneration in the rat tail. We performed a surgical puncture–induced disc injury at the coccygeal level, which induced robust pain behavior in rats, persisting for at least 1 month, as shown by quantitative sensory testing. Post-mortem, we observed the induction of spinal molecular markers of nociception, structural disc degeneration, and glycosylation changes in the disc in our model. Many of the changes induced by disc injury were prevented by treatment with an HA hydrogel.
The dermatomes (skin regions) and IVD involved in the study are innervated by dorsolateral and ventrolateral caudal nerves that arise from the sacral (figs. S1 to S4) and coccygeal (Co1–Co3) segments of the spinal cord (23). Thermal and mechanical stimuli were applied to the ventral surface of the proximal tail in the Hargreaves and von Frey tests. The Hargreaves and tail-flick tests measure thermal nociception processing and can assess heat-related hyperalgesia, which is the increased sensitivity to a noxious, high-intensity thermal stimulus, suggesting activation of Aδ and C fibers of primary sensory neurons (9). The Hargreaves test identified thermal hyperalgesia close to the site of injury in the rat tail. A von Frey test, with application of different fibers, each of which produced a particular constant force (9), identified mechanical allodynia in the injured rat tail. Previously published results have also demonstrated that annular puncture in lumbar discs evokes mechanical and thermal hyperalgesia in rats (24). The tail-flick test identified thermal hypoalgesia with stimulation of a region of the rat tail distal to the site of injury, suggesting that the DNIC phenomenon occurred when noxious stimulation was applied outside of the receptive field corresponding to the injury. DNIC is supraspinally mediated and can inhibit multi-receptive wide dynamic–range neurons in the dorsal horn of the spinal cord, leading to situations in which pain inhibits pain (25). Therefore, altered endogenous descending pain modulation by DNIC has been suggested as a mechanism underlying chronic pain that could explain hypoalgesia phenomenon in the distal rat tail of this model.
Previously, physiological stimulation of afferent sensory neurons in rats has been shown to increase c-Fos–like immunoreactivity in the nuclei of dorsal horn postsynaptic neurons, indicating that c-Fos is induced by neurotransmission resulting from activation of small-diameter sensory neurons (11). We also observed up-regulation of c-Fos mRNA in the dorsal horn of the sacral segment spinal cord in disc injuries, suggesting that thermal stimulation on proximal rat tail activates nociceptive sensory neurons during the injury, and possibly inducing central sensitization in the dorsal horn of spinal cord.
The anatomical disruptions caused by disc injury resemble pathological features during disc degeneration. These changes were indicated by annular rupture, reduction in the number of collagen fibers in AF tissue, chondroid nests in NP cells, reduction in GAG content in the ECM, and formation of fibrotic tissue in both AF and NP tissues.
Glycosylation has important roles in biological processes including angiogenesis, inflammation, adhesion, differentiation, and cell-cell and cell-ECM interactions (26). Here, we observed higher levels of sialylation in injured discs, which is in line with a previous demonstration of high levels of sialylation in inflamed or degenerated human chondrocytes (27). Fully sialylated β1 subunits of voltage-gated sodium channels are known to induce uniform, hyperpolarizing shifts in channel gating, thereby being responsible for the initiation and propagation of neuronal action potentials (28). Thus, the increased sialylation is also likely to be associated with induction of hyperpolarization of the nerve action potentials. In our model, increased fucosylation, mannosylation, and galactosylation were observed in the discs. Alteration of fucosylation is one of the most common modifications associated with oligosaccharides on glycoproteins during tumorigenesis and inflammation (29). In mice, cell surface fucosylation is regulated via phospholipase Cγ by neural cell adhesion molecule L1 to modulate neurite outgrowth, cell survival, and migration (30). Previously, higher expression of nonsialylated Gal-β-(1,3)-GalNAc was demonstrated in severely degenerated human cartilage (27). GS-I-B4 (binds to α-Gal) can be used to trace non–peptide-containing sensory neurons in the discs (31). In the development of vertebrae, GalNAc has a key role in the differentiation of mesenchymal stem cells into chondroblasts (32). Our observation of lower GalNAc expression in the ECM of NP tissue after injury is consistent with previous results that showed a loss of GalNAc in neuronal degenerative disease (33).
To validate our model, we administered morphine systemically at low and high doses before testing pain behavior. Morphine is an opioid drug that targets the central and peripheral nervous systems. Morphine reduces Ca2+ influx or increases K+ efflux, thereby decreasing repolarization time and the duration of the action potential and inhibiting the release of neurotransmitters (including noradrenaline, glutamate, acetylcholine, and substance P) at presynaptic neurons, to produce an analgesic effect (34). We observed an inhibitory effect of low-dose morphine on injury-induced thermal hyperalgesia and mechanical allodynia in the rat tail close to the injury site. In contrast, high-dose morphine prevented observation of responses to these stimuli. Low-dose morphine also inhibited injury-induced thermal hypoalgesia to a stimulus applied to the tail distal to the injury site, suggesting inhibition of the DNIC phenomenon. Systemic morphine administration has previously been shown to inhibit supraspinal inhibitory control of dorsal horn convergent neurons when noxious stimuli are applied (35). We observed no tail-flick response in the rats treated with high-dose morphine, because high doses of morphine induce a state of catalepsy, which is clinically manifested by muscle rigidity, akinesia, a lack of spontaneous activity, and failure to correct an externally imposed posture (36).
At the time of surgery, we implanted HA hydrogel in one group of rats to investigate the pharmacodynamic effects in physiological conditions. Rats implanted with HA hydrogel had lower levels of thermal hyperalgesia and mechanical allodynia than those with injury alone, which corresponds to a previous observation that an HA solution reduces pain behavior in a mouse model (19). The tail-flick test showed that HA hydrogel–treated rats diminished thermal hypoalgesia, suggesting inhibition of the DNIC phenomenon, as seen with low doses of morphine. Overall, our data support the hypothesis that HA hydrogels reduce nociceptive behavioral responses to noxious or innocuous stimuli in an IVD injury pain model of the rat tail.
Hyperalgesia resulting from tissue injury and inflammation contributes to central sensitization because of the continuous activation of spinal cord excitatory neurotransmitters or neuropeptides (37). Central sensitization also increases the activity of C fibers, enhancing the excitability of postsynaptic neurons by the release of glutamate and substance P (38). We observed a lower expression of c-Fos, which is associated with the development of pain hypersensitivity and Tac1 (encodes substance P) after implantation of the HA hydrogel group, indicating that the HA hydrogel treatment suppresses injury-induced expression of spinal nociception markers in the dorsal horn.
Nociceptors are free nerve endings of fiber type Aδ or C of afferent sensory nerves that terminate in the dorsal horn of the spinal cord. Neurogenic mediators are released during hyperalgesia after tissue injury and can sensitize nociceptors, initiating nociceptive nerve transmission to the spinal cord (39). Previously, the growth of small, unmyelinated sensory nerve fibers into the inner third of the AF and into the NP has been associated with chronic low back pain; these fibers expressed GAP43 and substance P (6). The levels of expression of CGRP and Trk-A in sensory nerve fibers innervating the outer AF define these nociceptive DRG neurons as being NGF-sensitive neurons, which are small C fibers that convey a dull pain (5). Trk-A activation leads to neuronal transduction in the adult nervous system (40), and NGF has been linked to mechanically evoked pain in OA (41). Our results demonstrated the inhibitory effect of HA hydrogels on innervation, sensory neuropeptide, and pro-nociceptive receptors. This correlates with previous studies that have shown that HA lowers the excitability of the TRPV1 channel, reducing impulse activity in peripheral nociceptor endings underlying pain in cultured DRG neurons and in vivo in mice (19).
Injury-induced degenerative changes influenced glycosylation in the discs. Up-regulation of glycosyltransferase mRNA and expression of α-(2,3)/α-(2,6) sialylation have been observed in severely degenerated human cartilage, as indicated by the presence of GalNAc-β-(1,4)-GlcNAc–terminated structures and core 2 O-glycans, as well as the binding of several lectins (27). We found that HA hydrogel implantation eliminated the injury-induced elevation of sialylation and galactosylation in the discs. We observed elevation of GS-I-B4 binding and the number of sensory nerve terminals in the discs after injury, suggesting that sensory hyperinnervation (31) in our injury model mimics the processes involved in discogenic pain. This innervation seems to be inhibited by HA hydrogel implantation.
CS is composed of repeating units of GalNAc and GlcA, and GalNAc may be sulfated at the 4-position or the 6-position, which produces a high degree of structural heterogeneity (42). We found that CS expression was higher in the ECM of AF and NP tissues in the HA hydrogel treatment group, suggesting that the synthesis of sulfated GAGs may promote disc tissue restoration, because CS has also been shown to increase ECM production via the integrin-WNT5A pathway (43). In addition, we observed that CS correlated with peripheral sensory innervation and nociceptor, which may indicate that recruitment of CS deposition is required not only for tissue recovery but also to inhibit hyperinnervation and peripheral nociception, because CS from IVD-derived notochordal cells has also been shown to inhibit neurite growth in vitro (44). Fibromodulin is modified with KS and N-linked oligosaccharides, and it has a key role for the maintenance of collagen architecture in the cartilage (45). KS is composed of sulfated Gal and GlcNAc, α-(2,3)– and α-(2,6)–linked sialic acid, and α-(1,3)–linked fucose residues upon maturity (45). Up-regulation of the expression of KS, α-(2,3)– and α-(2,6)–linked sialic acid in this study, may be related to tissue reorganization during injury and remodeling after the treatment.
To elucidate the therapeutic effect of HA hydrogel in targeting signaling pathways underlying disc degeneration and pain, we identified and quantified proteins and mapped their interactions using pathway analysis. Pathway analysis revealed that acute-phase response signaling involving IL-6 and IL-1β is one of the pathways that contribute to the downstream signaling cascade that activates NF-κB, p38 MAPK, and c-Fos, which encode proinflammatory, catabolic, and acute-phase proteins that act during disc degeneration. High expression of IL-1β, IL-6, IL-8, and TNF has been shown to play an essential role in the development of pain during disc degeneration (46). However, we observed lower levels of IL-6 and IL-1β and higher levels of IL-10 in AF and NP tissues and blood plasma in the HA hydrogel treatment group, which is consistent with our previous observation that HA hydrogels target the IL-1β pathway in inflamed NP cells (20). These data suggest that HA hydrogel reduces injury-induced inflammation not only in AF and NP tissues but also systemically. The implication of this finding is that it may form the basis for the development of a biomarker/diagnostic platform for screening degenerative disc events (such as inflammation) at the systemic level. Thus, systemic levels of IL-6 have previously been shown to be elevated in patients with low back pain diagnosed with disc diseases (47).
Degenerative change commences in the ECM of NP tissue, with reductions in expression of proteoglycans, type II collagen, and water content, leading to poor hydrodynamic transfer of axial stress to the AF (1). In the later stages of IVD degeneration, dysregulated ECM synthesis has been observed, with type I collagen forming strong fibrils predominantly within the NP and inner AF, type II collagen in the outer AF, down-regulation of aggrecan, and up-regulation of fibronectin, versican, biglycan, and decorin, leading to the loss of weight-bearing capacity and disc height (1). We found that HA hydrogel treatment inhibited ECM dysregulation, reducing the development of fibrous tissue formation. Protein pathway analysis suggested that the Smad3 family regulated the ECM content, including aggrecan, type I collagen, fibronectin, and TGF-β1. We have previously shown that GlcNAc, a component of HA, promotes ECM deposition through the MAPK and SMAD pathways in ex vivo degenerated bovine discs (48).
Our findings provide the basis for a model of HA hydrogel action in reducing discogenic pain. HA hydrogel may target inflammation and nociception by attenuation of the key IL-1β and IL-6 inflammatory pathways, thereby inhibiting sensory nerve innervation and suppressing expression of peripheral and central pain markers and modulating the ECM and cellular composition. During disc injury–induced inflammation, proinflammatory cytokines, such as IL-1β, IL-6, and IFN-γ, are produced by NP and AF cells as well as by invading macrophages. IL-1β binds to its receptor IL-1R and recruits myeloid differentiation primary response protein MyD88 and IL-1R–associated kinase 4 (Irak4) to phosphorylate Irak1, enabling transient recruitment of TNF receptor–associated factor 6, dissociation of Irak1 from the receptor, and activation of Tak1 (49). Activated Tak1 (Tab1 in rats) phosphorylates the IKK complex and MAPKs to further activate and translocate c-Jun, c-Fos, CCAAT/enhancer-binding protein β, and NF-κB into the nucleus, resulting in the transcription of genes encoding acute-phase response proteins, cytokines, and degradative enzymes (49). Binding of IL-6 to IL-6R facilitates IL-6R subunit β (gp130) transphosphorylation and Janus kinase activation, which mediates recruitment of Stat3. Dimerization of phosphorylated Stat3 results in nuclear translocation and transcriptional activation of genes including those encoding acute-phase response proteins (50).
Activation of afferent unmyelinated C fibers or thinly myelinated Aδ fibers in injured discs occurs by sensitization of nociceptors, such as TrpV1, through posttranslational phosphorylation upon activation of tyrosine kinase receptors (such as Trk-A), which eventually induces membrane potential excitability and generation of the nociceptive impulse (7). The nociceptive input is transmitted from the injury site to the dorsal horn of the spinal cord. The up-regulation of presynaptic neurotransmitters, such as the amino acids aspartate and glutamate, leads to an increase in neurotransmitter release at postsynaptic neurons, which is enhanced by substance P. Synaptic impulse transmission to second-order neurons occurs (39) and subsequently activates the expression of the immediate-early gene c-Fos, thereby conveying nociceptive input to higher centers in the brain (39). The second-order neurons relay pain signals along the spinothalamic tract to the thalamus and along the spinoreticular tract to the brainstem. From these locations, the signals can be propagated to different areas of the brain, including the cerebral cortex area containing the primary and secondary somatosensory cortexes (SI and SII) and cingulate gyrus. The descending inhibitory pathways release noradrenaline and serotonin into the spinal cord to inhibit pain (39).
The possible mechanism of action of HA hydrogel involves interference in the binding of IL-1β and IL-6 to their receptors and inhibition of transcription of genes that encode proinflammatory cytokines and acute-phase proteins. HA hydrogel, possibly via the Smad3 family and TGF-β1, promotes regulation of ECM proteins. A higher expression of CS in the ECM and down-regulation of neurotrophic factors, such as NGF, mediate the inhibition of neurite outgrowth of sensory nerves in discs. Evidence for this process is provided by the suppression of the GAP43 protein, which is important for axonal growth, along with inhibition of sensory neuropeptides in the discs. The inhibition of NGF binding to its receptor, Trk-A, also interferes with posttranslational phosphorylation and can inactivate TrpV1, reducing excitability of nociceptive nerve transmission and reducing ion channel responsiveness to noxious stimuli. At the spinal level, HA hydrogel suppresses substance P in the dorsal horn and reduces c-Fos expression, thereby inhibiting nociceptive processing up to the supraspinal region of the brain to exhibit analgesic effect (Fig. 6).
Fig. 6. Possible mechanism of action of the HA hydrogel in the IVD injury–induced model of pain.
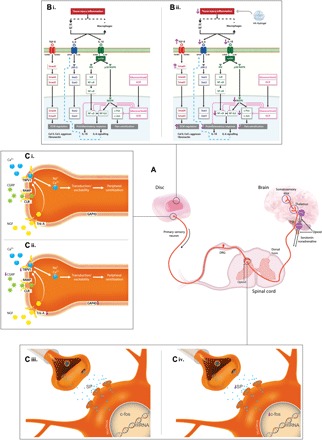
(A) Pain signaling from the discs via the dorsal horn of the spinal cord to the brain for pain processing. Descending pathways inhibit pain transmission by releasing noradrenaline and serotonin into the spinal cord. (B) (i) In injured discs, AF and NP cells (and macrophages) produce IL-1β, IL-6, and IFN-γ. Activation of IL-1β signaling facilitates the MyD88 complex to phosphorylate Irak1 and activate TGF-β–activated kinase 1 (Tak1) (Tab1 in rats). Tak1 phosphorylates IκB kinase (IKK) complex and p38 MAPK, inducing nuclear translocation of c-Jun/c-Fos, CCAAT/enhancer-binding protein β (NF-IL6), and NF-κB, for transcription of genes encoding cytokines and catabolic and acute-phase response proteins. IL-6 signaling involves transphosphorylation of the IL-6R subunit β (gp130) and activation of Janus kinase 2 (JAK-2) to facilitate nuclear translocation of signal transducer and activator of transcription 3 (Stat3) and transcription of acute-phase response genes. (ii) Implanted HA down-regulates IL-1β and IL-6 and interferes with receptor binding, inhibiting downstream cascades and gene transcription. HA hydrogel also up-regulates Smad3 via TGF-β signaling, promoting ECM synthesis. (C) (i and iii) In injured discs, sensory nerves terminate in aneural AF and NP tissues. Neurogenic mediators are released to promote nerve ingrowth, and the presence of noxious stimuli activates nociceptive fibers by sensitizing TrpV1, enabling an influx of sodium and calcium, inducing membrane potential excitability. (ii) The implanted HA hydrogel–induced IVD expression of CGRP and NGF receptor (Trk-A) is reduced compared with untreated injury, preventing hyperinnervation in discs and inhibiting pain sensitization. (iv) In the dorsal horn, the HA hydrogel may suppress nociceptive transmission by attenuating injury-induced expression of c-Fos and substance P (Tac1).
In summary, we have developed a novel IVD injury model for the study of pain associated with disc degeneration. Although this model is restricted to inflammatory pain (not radicular pain) and lack of biomechanical elements as it is adopted in non–load-bearing animals, it can provide a relatively simple platform for the investigation of potential spine interventions. Here, we have seen the therapeutic efficacy of HA hydrogel in the alleviation of the pain phenotype in this model, which may imply that the next generation of HA hydrogel may constitute an injectable system that could be translated clinically in patients with discogenic back pain due to degenerated disc.
CONCLUSIONS
The development of pain, structural degeneration, and distinct glycosylation in a rat tail model of disc degeneration were established in this study. Current analgesic treatment (morphine) reverses the pain phenotype and validates this model. Moreover, using this model, we demonstrated the anti-allodynic and anti-hyperalgesic properties of HA hydrogel treatment of disc injury. The HA hydrogel acts through reduction of nociception and inhibition of hyperinnervation and expression of nociceptive markers via alteration of glycosylation, attenuation of key inflammatory signaling molecules, and modulation of protein regulatory pathways. Our results suggest that HA hydrogel has a potential therapeutic application for the treatment of back pain associated with disc degeneration.
MATERIALS AND METHODS
All animal-related protocols were performed in accordance with national guidelines and approved by the Animal Care Research Ethics Committee at the National University of Ireland, Galway, and the Health Product Regulatory Authority, Ireland.
Animals
Experiments involved 110 adult (12-week-old) female Sprague-Dawley rats, which were housed two to three per cage in individually ventilated cages, with wood-chip bedding, enriched with a wooden stick. Food and centralized tap water were available ad libitum, lights were turned on at 06:00, the temperature was maintained at 21 ± 2°C, and the husbandry was provided by female staff. Rats were purchased from Charles River Laboratories and acclimated to the vivarium for at least 7 days before testing. For the establishment of the disc-pain model, sample sizes were n = 5 for each of the four groups [Co4–Co5 NP injury; Co4–Co5 + Co5–Co6 NP injury; sham controls receiving isoflurane, but no surgical incision (sham-isoflurane); and sham controls receiving isoflurane and a skin incision, but no injury to the disc tissues (sham-incision)] with time point day 29. To validate the disc-pain model using a reversible analgesic, sample sizes were n = 5 for each of the six groups [sham-isoflurane + saline, sham-isoflurane + low-dose morphine (2 mg/kg), sham-isoflurane + high-dose (10 mg/kg) morphine, Co4–Co5 + Co5–Co6 NP injury + saline, Co4–Co5 + Co5–Co6 NP injury + low-dose morphine, and Co4–Co5 + Co5–Co6 NP injury + high-dose morphine] with time point day 29. For the in vivo implantation of HA hydrogel, sample sizes were n = 10 for each group [sham-isoflurane, Co4–Co5 + Co5–Co6 NP injury, and Co4–Co5 + Co5–Co6 NP injury with implanted HA hydrogel (4 μl; 0.03 mg/ml)] with time point days 7 (n = 10) and 29 (n = 10).
Materials and reagents
Sodium hyaluronate (molecular mass, 1.19 × 106 Da) was purchased from Lifecore Biomedical. Four-arm PEG amine (molecular mass, 2000 Da) was purchased from JenKem Technology. Teflon tape, Superfrost Plus slides, 3-kDa centrifugal filters, and scalpel handle number 3 were purchased from Thermo Fisher Scientific. The RNeasy Mini Kit was purchased from Qiagen. Carprofen was purchased from Pfizer. Isoflurane was purchased from Chanelle Group. Morphine sulfate was purchased from Mercury Pharma Group. Saline [0.9% (w/v) sodium chloride] was purchased from B. Braun Medical. Bone-cutting forceps, teeth forceps, and iris scissors were purchased from Fine Science Tools. A 1-mm biopsy puncher with a plunger, artery forceps, and a needle holder were purchased from Medguard. Random primers, reverse transcriptase, 5× reaction buffer, 25 mM MgCl2, deoxynucleotide triphosphate (dNTP) mix, and recombinant RNasin ribonuclease (RNase) were purchased from Bioline. TaqMan FAM-labeled preprotachykinin (substance P precursor; Tac1) primers (Rn01500392_m1) and c-Fos primers (Rn00487426_g1) were purchased from Life Technologies. TaqMan VIC-labeled rodent glyceraldehyde-3-phosphate dehydrogenase (Gapdh) primer and Universal PCR Master Mix were purchased from Applied Biosystems. Lectins conjugated with fluorescein isothiocyanate (FITC) or tetramethylrhodamine isothiocyanate (TRITC) were purchased from EY Laboratories. Rabbit polyclonal anti-GAP43 (ab12274), mouse monoclonal anti-CGRP (ab81887), donkey anti-rabbit immunoglobulin G (IgG) (Alexa Fluor 555 conjugate; ab150074), rabbit monoclonal anti–Trk-A (Alexa Fluor 647 conjugate; ab194322), rabbit polyclonal anti-fibronectin (ab2413), mouse monoclonal anti–TGF-β1 (ab64715), and donkey anti-goat IgG (Alexa Fluor 488; ab150129) antibodies were purchased from Abcam. Goat polyclonal anti-TrpV1 (AF3066) and rabbit polyclonal anti-KS (NBP1-62542) antibodies were purchased from Novus Biologicals. Mouse monoclonal anti-CS antibody (C8035) was purchased from Sigma-Aldrich. Goat anti-mouse IgG (Cy5 conjugate; A10524) antibody was purchased from Thermo Fisher Scientific. FITC-conjugated rabbit polyclonal anti-COL1A1 (orb15412) and FITC-conjugated rabbit polyclonal anti-aggrecan (orb15073) antibodies were purchased from Biorbyt. Rabbit polyclonal phospho-p38 MAPK (Thr180/Tyr182; catalog no. 9211) and rabbit monoclonal phospho–NF-κB p65 (Ser536; catalog no. 93H1) antibodies were purchased from Cell Signaling Technology. 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Life Technologies. von Frey filaments were purchased from North Coast Medical Inc. An analgesic meter was purchased from IITC Life Science. Multiplex ELISA proinflammatory panel 2 was purchased from Meso Scale Discovery. Other materials and reagents were purchased from Sigma-Aldrich.
Synthesis of cross-linked HA hydrogel
HA hydrogels were fabricated by mixing 0.75% (w/v) sodium hyaluronate (1.19 × 106 Da) in 1 ml of distilled water with 75 mM four-arm PEG amine, 15% (w/v) NHS, and 9% (w/v) EDC. Spherical hydrogels were obtained by pipetting 4 μl of mixed gel solution (0.03 mg/ml) onto a hydrophobically modified glass slide (Fig. 1B). The hydrogels were then washed with phosphate-buffered saline (PBS) and sterilized under ultraviolet light for 60 min before implantation.
Quantitative behavioral nociception assays
Rats were habituated to the arena test environment for 20 min, 24 hours before the commencement of testing, which minimized locomotor activity and stress-induced analgesia during testing. The same investigator performed the scoring in all behavioral tests and was blinded to the experimental groups. The rats were assessed by the Hargreaves, von Frey, and tail-flick tests.
Hargreaves test
The test was performed 2 days before surgery (day −2) and on days 2, 7, 14, and 29 (after surgery). Rats were placed in Perspex chambers on top of a glass plate (IITC Life Science) to habituate for 20 min. Heat was introduced with a radiant light source, and latency to the response was recorded. Responses included withdrawal, flinching, licking, biting, and shaking the base of the tail. The analgesic meter test head was positioned under the base of the tail, with the beam approximately focused on the ventral surface of the tail, which was opposite to the site of injury. The test head was set to 50% of the maximum output. When the rat responded, the light source was set to “idle” intensity, and the heat source was deactivated. A cutoff of 20 s was set to prevent tissue damage. If no response occurred in this time, then the heat source was automatically returned to idle intensity. On each test day, each rat received four trials, with a minimum interval time of 3 min. Withdrawal latency for each rat was calculated as the average of the four measurements.
Tail-flick test
The tail-flick test was performed 1 day before surgery (day −1) and on days 3, 8, 15, and 28 (after surgery). On each day, the test for each rat involved four consecutive measurements. Each rat was removed from its home cage, habituated on a towel for 10 min, and then placed on the glass apparatus with its body covered with the towel as a restraint such that its tail projected out. A radiant light source was focused 5 cm from the distal end on the ventral region of the tail to introduce heat, and the latency to tail flick was recorded. A cutoff point of 20 s was assigned to prevent tissue damage. A period of at least 15 s between each test was observed to avoid sensitization of the tail. Tail-flick latency for each rat was calculated as the average of four consecutive measurements.
von Frey test
The von Frey test was carried out 2 days before surgery (day −2) and on days 2, 7, 14, and 29 (after surgery). The rats were individually placed into six-compartment rat enclosure with wire mesh floors and lids with air holes (IITC Life Science) for a 20-min habituation period to minimize exploratory activity. The test began with a 2-g filament (North Coast Medical Inc.), which was applied to the base of the tail on the ventral surface for a maximum of 6 s, with sufficient force to buckle it slightly. A positive response was considered to have occurred when the rats responded to the applied filament by flinching, licking, withdrawing, or shaking the base of the tail immediately or within the maximum of 6 s. A negative response was recorded if the animal showed no such response to the application of the filament. This process was repeated five times for the 2-g filament. If a response was obtained with the 2-g filament, then the animal was tested with progressively lower weight filaments until no response was obtained in five attempts. The test was continued with ascending filament numbers until five responses were obtained in five attempts for two consecutive filaments. The first output measurement was calculated as percentage (%) withdrawal response. This was calculated for test at each filament weight as follows
The second output measurement was the 50% withdrawal threshold. This was defined as the filament weight (or filament number) at which a 50% withdrawal response was obtained. The filament weight and the percentage response each day were tabulated for each individual animal. In XY data table of GraphPad Prism, filaments were tabulated in ascending order in the X column, and percentage response to each filament in each day was tabulated in separate Y column. A nonlinear regression curve was plotted (sigmoidal dose response, variable slope, and set bottom and top constraints as 0 and 100, respectively), and the log EC50 (log-transformed to give the logEC50) was obtained from the results sheet. A log EC50 for each animal on each day was entered into a separate table (days in the X column and groups in separate Y columns, with each rat as a separate replicate). An average for each group was computed and plotted as von Frey filament weight eliciting a 50% response versus time (days).
Implantation of HA hydrogel following surgical puncture–induced injury
On day 0, general health measurements, including body weight and temperature, were recorded before surgery. A single administration of the nonsteroidal anti-inflammatory drug carprofen (5 mg/kg, subcutaneous) was given to the rats 15 min before surgery to manage postoperative pain in the early recovery phase. The rats were briefly anesthetized with inhalation of isoflurane (5% for induction and 1.8 to 2% for maintenance). The depth of anesthesia was determined by the loss of the pedal withdrawal (toe pinch) reflex. Moisturizer was dropped into the eyes if they remained open. The surgical site of the dorsal rat tail was washed with a Betadine solution with gauze. The coccygeal discs Co4–Co5 and Co5–Co6 were identified by anatomical surface markings between two vertebral bones through the tail skin. The first disc between these two vertebral bodies counting from the base of the tail is Co4–Co5, followed by Co5–Co6. A rubber band was applied at the base of the tail not later than 30 min after identification of the discs (Fig. 1C, i). Surgery began with dissection with the aid of a binocular surgical loupe, and all surgical instruments were handled using aseptic technique. A longitudinal incision was introduced in the skin and at the connective tissue of the dorsal side of the tail covering the discs. The tendons were pushed aside until the AF tissue (ivory matter) was reached (Fig. 1C, ii). The injury was performed by punching NP tissue through the AF tissue at a diameter of 1 mm and a depth of 2 mm using a 1-mm biopsy puncture (19-gauge needle) (Fig. 1C, ii). After injury, the discs were either left alone or implanted with HA hydrogel. The tendons were layer-sutured to close the injury site, thereby covering the discs, and the animals were marked to show that the procedure had been performed. The wound was closed by suturing the skin with nylon suture using the interrupted horizontal mattress suturing method, and the rubber band was then removed from the tail to enable recovery of blood flow. For postoperative care, the rats were closely monitored until they were completely recovered from anesthesia and then placed singly in individually ventilated cages until the wounds healed satisfactorily, which took approximately 1 week. Then, the rats were housed two to three per cage in individually ventilated cages for the recovery until day 29. Wounds were examined for signs of inflammation or infection, such as redness, swelling, and/or purulent or serous discharge. General health assessment details, such as tail-skin complications, body weight, and body temperature, along with distress scores, were recorded on days 2, 7, 14, and 28 after the operation. The temperature and humidity of the housing environment were monitored, and appropriate bedding materials were provided during the studies.
Morphine treatment
In some experiments, pain was inhibited with morphine. Low-dose morphine (2 mg/kg), high-dose morphine (10 mg/kg), or vehicle (saline) was administered by subcutaneous injection, and behavioral responses were assessed 30 min after injection (Fig. 1A, ii).
Postmortem analysis
After a 29-day period post-surgery, live rats were decapitated with a guillotine, and trunk blood was collected in EDTA tubes during the procedure. Discs, AF and NP tissues, spinal cords, and brains were extracted for further tissue analysis.
Reverse transcription quantitative polymerase chain reaction
Left and right dorsal horns of the sacral segment of the spinal cord were dissected within 30 min after euthanasia, snap-frozen, and immediately placed at −80°C until further analysis. The dorsal horn of the spinal cord at the sacral and coccyx levels was homogenized in 1 ml of TRIzol reagent per 50 to 100 mg of tissue sample using TissueLyser (Qiagen). The samples were shaken at high speed in 2-ml round-bottomed microcentrifuge tubes with stainless steel beads for 15 min at room temperature. Following homogenization, the samples were centrifuged at 1200g for 5 min at 4°C, and the fatty layer was discarded. The cleared supernatant was transferred to a new microcentrifuge tube (Qiagen). For phase preparation, 200 μl of chloroform was added per 1 ml of TRIzol reagent, mixed for 15 s, and incubated for 2 min at room temperature. The mixture was then centrifuged at 13,300 rpm for 15 min at 4°C. The colorless upper aqueous phase containing RNA was transferred to a fresh tube and mixed with 600 μl of 70% molecular grade ethanol. The mixture was transferred to a Qiagen RNeasy Mini column and centrifuged at 10,000 rpm for 15 s, and flow-through was discarded. RNA bound to the column was washed with 700 μl of the RW1 buffer and centrifuged at 10,000 rpm for 15 s. Next, 500 μl of the RPE washing buffer was added to the column, followed by centrifugation at 10,000 rpm for 15 s, and then a further 500 μl of the RPE buffer and centrifugation for 2 min. RNase-free water (30 μl) was added, and the mixture was centrifuged at 10,000 rpm for 1 min to collect RNA in a new collection tube. The RNA concentration was determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific) from the ratio of absorbance at 260 and 280 nm, and the quality of the product was determined using a bioanalyzer (Agilent). A total of 5 μl of RNA (100 ng/μl) was reverse-transcribed with random primers and reverse transcriptase in a 20-μl reaction mixture consisting of 4.4 μl of 5× reaction buffer, 2.64 μl of 25 mM MgCl2, 1.1 μl of dNTPs, 1.1 μl of RNasin RNase inhibitor, and 1.1 μl of ImProm-II reverse transcriptase, with the PTC DNA Engine System (PTC-200, Peltier Thermal Cycler, MJ Research). Complementary DNA products were amplified with TaqMan gene expression assays and specific primers for Tac1, c-Fos, and Gapdh. A multiplex polymerase chain reaction (PCR) reaction was performed with TaqMan Universal PCR Master Mix, no AmpErase UNG (Applied Biosystems), and standard thermal conditions (10 min at 95°C for polymerase activation, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s) in triplicate using the StepOnePlus Real-Time PCR System (Applied Biosystems). The results were analyzed by the 2−ΔΔCt method, normalized to the endogenous Gapdh control and the sham control rats.
Lectin and immunohistochemistry
Post-mortem, discs were fixed in 4% (w/v) paraformaldehyde for 48 hours, and vertebral bones were decalcified for 2 weeks in Kristensen’s decalcifying solution containing 18% (v/v) formic acid and 3.5% (w/v) sodium formate at 4°C. All tissues were then washed under running tap water overnight and soaked in 20% (w/v) sucrose until they sank at 4°C. Tissues were embedded in optimal cutting temperature compound, snap-frozen in an isopentane bath with liquid nitrogen, and then kept at −80°C until sectioning on a cryostat (CM1850, Leica). Tissue sections (10 μm) were collected on Superfrost Plus slides (Thermo Fisher Scientific) and stored at −20°C until use. Staining was performed at room temperature, and all washes were performed three times for 5 min each time between incubations, unless otherwise stated. Three slides (one each from three different rats) were used for each lectin or antibody incubation.
For lectin histochemistry, slides were washed with tris-buffered saline supplemented with Ca2+ and Mg2+ (TBS; 20 mM tris-HCl, 100 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.2) and 0.05% (v/v) Triton X-100 (TBS-T) and then blocked with 2% (w/v) periodate–treated bovine serum albumin (BSA) (Sigma-Aldrich) in TBS for 1 hour. Inhibitory controls were carried out in parallel to verify that the lectin binding was glycan-mediated by co-incubating the lectins with 100 mM of the appropriate haptenic sugar in TBS (Table 1). Sections were washed and then incubated with eight different FITC- or TRITC-conjugated lectins [SNA-I, MAA, WGA, Con A, UEA-I, WFA, GS-IB4, and PNA (EY Laboratories)] in TBS for 1 hour (Table 1). After five washes with TBS-T, the sections were counterstained with a 1:1000 dilution of DAPI for 5 min. The slides were washed in TBS-T before coverslip mounting with ProLong Gold Antifade Mountant (Life Technologies).
For immunohistochemistry, sections were incubated with proteinase K for 15 min at 37°C, followed by 2% (w/v) BSA for 30 min at room temperature before overnight incubation at 4°C with primary antibodies. Negative control sections were incubated with PBS and washed in PBS with 0.05% (v/v) Tween 20 (PBS-T). Triple staining was performed with three selected primary antibodies [1:100 anti-GAP43 (ab12274, Abcam), 1:100 anti-TrpV1 (AF3066, Novus Biologicals), and anti-CGRP (ab81887, Abcam)] per section. Double staining was carried out with 1:100 anti-CS (C8035, Sigma-Aldrich) and 1:100 anti-KS (NBP1-62542, Novus Biologicals), 1:50 anti–TGF-β1 (ab64715, Abcam) and 1:50 anti-COL1A (orb15412, Biorbyt), or 1:50 anti-fibronectin (ab2413, Abcam) and 1:50 anti-aggrecan (orb15073, Biorbyt) antibodies. Single staining was performed with 1:50 phospho-p38 MAPK (Thr180/Tyr182) (catalog no. 9211, Cell Signaling Technology), 1:50 phospho–NF-κB p65 (Ser536) (catalog no. 93H1, Cell Signaling Technology), and 1:50 anti–Trk-A (ab194322, Abcam) antibodies. Sections were incubated with donkey anti-rabbit (Alexa Fluor 555 conjugate; ab150074, Abcam), donkey anti-goat (Alexa Fluor 488; ab150129, Abcam), and/or goat anti-mouse (Cy5 conjugate; A10524, Thermo Fisher Scientific) secondary antibodies at 1:200 dilution in PBS for 2 hours and then washed with PBS-T before counterstaining with DAPI (1:1000 in PBS) for 5 min. Sections were washed with PBS-T and coverslip-mounted, as described above. All slides were cured at 4°C in the dark for 1 day before imaging with a laser confocal microscope (Olympus FluoView 1000, Olympus America).
Histochemical image analysis and stereology quantification
Stereology quantification was adopted to calculate the percentage volume fraction of detectable lectin and antibody binding to specific glycans and antigens. Single z slice of confocal images from lectin histochemical and immunohistochemical staining was obtained from at least four microscopic views of each slide with three technical and four biological replicates using a laser confocal microscope (Olympus FluoView 1000). Volume fraction and number estimation were calculated according to a published method (51).
For volume fraction analysis, confocal images were converted to binary mode (8 bits) and adjusted to the optimal threshold of positive staining and total area using ImageJ software version 1.48 [National Institutes of Health (NIH)]. Volume fraction was calculated by quantifying the area fraction of the positively stained matrix component divided by the total area and converting into a percentage.
For the clustering analysis, data were normalized to maximal individual lectins or antibody binding across conditions, and the clustering analysis was performed using Hierarchical Clustering Explorer 3.0 (NIH) without additional normalization and with complete linkage, Euclidean distance, and Pearson correlation coefficient determination. The results are represented in heatmap format.
For the nuclear antibody staining, an estimation of positive nuclear staining was obtained by applying an unbiased counting frame to a single confocal slice in a systematic random manner. Positive nuclear staining was counted only in the guard area, without counting nuclei hit forbidden line. Number estimation was calculated by quantifying the number of positive nuclear staining per unit area (μm2).
Histology
Discs were postfixed with 4% (w/v) paraformaldehyde for 48 hours and decalcified in Kristensen’s decalcifying solution for 2 weeks and then washed for 24 hours under running tap water and paraffin-embedded. Discs were cut into transverse 5-μm sections on a microtome (Leica). Tissue sections were collected on Superfrost Plus slides and stored at room temperature until use. Staining was performed at room temperature. Sections were dewaxed in xylene in the fume hood and rehydrated through 95%, 70%, and 50% ethanol washes for 2 min each.
For H&E staining, sections were stained in Mayer’s hematoxylin for 6 min and washed under running tap water before staining in eosin for 2 min. After a quick rinse in tap water, the sections were dehydrated with 50% ethanol for 10 s, 70% ethanol for 10 s, 90% ethanol for 2 min (twice), and absolute ethanol for 2 min (twice).
For Alcian blue staining, sections were stained in Alcian blue (pH 2.5) for 30 min, washed under running tap water, and then counterstained with nuclear fast red for 10 min. After washing under running tap water, sections were dehydrated with 50% ethanol for 10 s, 70% ethanol for 10 s, 90% ethanol for 2 min (twice), and absolute ethanol for 2 min (twice).
For Masson’s trichrome staining, sections were oxidized in 0.5% (w/v) KMnO4 and 0.5% (v/v) H2SO4 for 2 min and washed in tap water before bleaching with 2% (w/v) Na2S2O5 for 2 min. Sections were washed with water and 70% ethanol and then stained with Gomori’s aldehyde fuchsin for 1 min. After quick rinses in water and 95% ethanol, sections were stained with Celestine blue for 4 min, washed, and then stained with Mayer’s hemalum stain for 4 min. Sections were washed with water, differentiated in acid alcohol for 20 s, and washed with water again before staining with Masson’s cytoplasmic stain for 1 min. Sections were quickly washed in water, differentiated in 1% (w/v) dodeca-molybdophosphoric acid (H3PO412 MoO324H2O), then counterstained with Fast Green FCF or Light Green SF, differentiated in 1% (v/v) acetic acid, and dehydrated through 50%, 70%, 95%, and absolute ethanol washes for 1 min each. All sections were cleared in two changes of xylene and covered with the distyrene-plasticizer-xylene (DPX) mounting medium and a coverslip. The sections were placed in an oven at 37°C to enable the mounting medium to solidify before imaging under a light microscope (Leica).
Histological classification of disc degeneration
Stained slides were graded with a validated histological grading system, as described previously (22). Major anatomical structures of the AF and NP were included in this classification, resulting in four subcategories. Each item was graded as zero, one, or two on the H&E and Alcian blue sections, with zero representing nondegenerative characteristics, one representing mild degenerative characteristics, and two representing severe characteristics of degeneration (Fig. 2B). The total score was the sum of the four different scoring items, resulting in a minimum score of zero, corresponding to a healthy disc, and a maximum score of eight, corresponding to an entirely degenerated disc.
HPLC analysis of HA and CS
AF and NP tissues were separated from the discs after euthanasia. Tissues were snap-frozen and immediately kept at −80°C. The samples were digested with proteinase K (0.5 mg/ml) overnight at 56°C and stored at −20°C until further analyses. HPLC was performed as described previously (52). For data analysis, the disaccharide content of each sample was identified by comparison with appropriate chromatograph standards under the same HPLC conditions as the sample, with quantification by comparison with appropriate standard curves generated by the injection of known concentrations of standards.
Proteomic analysis by LC-MS/MS
NP and AF tissues were extracted from the discs after euthanasia. Tissues were snap-frozen and immediately kept at −80°C. To extract ECM proteins, samples were digested with proteinase K (0.5 mg/ml) for <12 hours at 56°C and stored at −20°C before further analyses. To extract both cellular and ECM proteins, tissues were minced with a scalpel, transferred to a microcentrifuge tube, and incubated in 6 M urea, 10 mM dithiothreitol, and 50 mM ammonium bicarbonate (NH4HCO3) at pH 8.6 for 2 hours at room temperature with gentle agitation. To collect the supernatant, each sample was centrifuged at 3000g for 3 min, and then the total protein concentration was evaluated. The samples were digested with 2% (w/v) trypsin in 1.5 M NH4HCO3 at pH 7.8 for 18 hours at 37°C, and the enzymatic reaction was stopped with 2% (v/v) formic acid.
Samples were run on a Thermo Fisher Scientific Q Exactive mass spectrometer connected to a Dionex UltiMate 3000 RSLCnano chromatography system. Tryptic peptides were resuspended in 0.1% (v/v) formic acid. Each sample was loaded onto a fused silica emitter [75 μm internal diameter, pulled with a laser puller (Sutter Instruments P2000) and packed with Reprocil Pur C18 (1.9 μm) reversed-phase media] and separated with an increasing acetonitrile gradient over 47 min at a flow rate of 250 nl/min. The mass spectrometer was operated in positive ion mode with a capillary temperature of 320°C and a potential of 2300 V applied to the frit. All data were acquired with the mass spectrometer operating in the automatic data-dependent switching mode. A high-resolution (70,000) MS scan (300 to 1600 m/z) was performed with Q Exactive to select the eight most intense ions, before MS/MS analysis with higher-energy collisional dissociation. For protein identification, the raw data were searched against the Rattus norvegicus subset of the UniProt Swiss-Prot database using the search engine PEAKS Studio 7 (Bioinformatics Solutions) for peptides with unspecified enzymatic cleavage. Each peptide used for protein identification met specific PEAKS parameters: Only peptide scores that corresponded to a false discovery rate (FDR) of ≤1% were accepted from the PEAKS PTM database search.
Protein quantification and pathway analysis
To quantify the identified proteins, raw MS data were analyzed with MaxQuant software based on specified peptides with unspecified enzymatic cleavage for proteinase K–digested samples and semi-specific trypsin cleavage with fixed modification of carboxymethylation for tryptic samples. Each peptide used for protein identification and quantification met specific MaxQuant parameters: Only peptide scores that corresponded to an FDR of ≤1% were accepted from the MaxQuant database search. MaxQuant data were further analyzed using Perseus software for comprehensive analysis of proteomics data, including label-free quantification of the identified proteins. The list of differentially expressed proteins with a label-free quantification intensity and fold change relative to sham rats >2 or <−2 that had a P < 0.01 was uploaded into IPA (Qiagen) for assessment. The protein expression comparison between the injury and HA hydrogel groups of AF and NP tissues was conducted using an empirical test based on fold change value as counts, causal networks (such as connective tissue, inflammation, neurotransmission, ECM, and musculoskeletal), and species accordingly.
Assessment of inflammatory cytokines by ELISA
After euthanasia (on the same day), the collected blood was transferred to a 2-ml Eppendorf tube and centrifuged at 1200g for 15 min to collect blood plasma. For AF and NP proteins, tissues were minced with a scalpel and incubated in 6 M urea, 10 mM dithiothreitol, and 50 mM NH4HCO3 (pH 8.6) for 2 hours at room temperature with gentle agitation. The supernatant was centrifuged at 3000g for 3 min, and then the total protein was collected for analysis. Plasma, AF, and NP samples were analyzed with the multiplex ELISA proinflammatory panel 2 (rat) for cytokines (IL-6, IL-1β, TNF, IFN-γ, CXCL1, IL-4, IL-5, IL-13, and IL-10). The sample and reagent were prepared according to the manufacturer’s protocol. Blood plasma and AF and NP samples were diluted (fourfold) in diluent. Blocker H (150 μl) was added to each well of the ELISA plate and incubated at room temperature with fast shaking for 1 hour. The plate was then washed three times with ≥150 μl of PBS-T. A 50-μl sample volume, including the calibrators, was added to each well and further incubated at room temperature with shaking for 2 hours. After three washes with PBS-T, 25 μl of 1× detection antibody solution was added to each well and incubated at room temperature with shaking for 2 hours. The plate was washed three times, and an additional 150 μl of 2× read buffer T was added to each well before plate reading detection based on electrochemiluminescence using MES QuickPlex SQ 120 (Meso Scale Discovery).
Statistical analysis
Statistical differences were analyzed by GraphPad Software using repeated-measures ANOVA or one-way ANOVA for analyses of the Hargreaves, von Frey, and tail-flick tests; one-way ANOVA for the PCR, HPLC, and lectin and antibody staining and two-way ANOVA for the ELISA in experiments involving HA hydrogel; and Student’s t test for the lectin analysis in the validation of the injury model. All ANOVAs were further evaluated with Bonferroni post-hoc analysis, and P < 0.05 was deemed statistically significant. All error bars indicate SEM.
Supplementary Material
Acknowledgments
We thank M. Doczyk for assistance in drawing the schematics presented in this article. We acknowledge the editorial assistance of A. Sloan and K. Feerick. We acknowledge technical help from O. Carroll, K. Wynne, N. Solovyova, E. Jennings, and D. Tiernan in this study. We also acknowledge support from the Mass Spectrometry Resource, Conway Institute of Biomolecular and Biomedical Research, University College Dublin, and Centre for Molecular Biology Facility and Bioresources Unit, National University of Ireland, Galway. Funding: This project has received funding from the Majlis Amanah Rakyat. This publication has also emanated from research conducted with the financial support of Science Foundation Ireland and is co-funded under the European Regional Development Fund under grant number 13/RC/2073. Author contributions: I.L.M.I. designed, performed, and analyzed all the experiments and surgeries and wrote the manuscript. S.A.A. assisted in animal surgeries. M.K. contributed to and discussed the glycomics study. D.S. inspired rat tail model. P.D. guided the stereology quantifications and discussed the imaging results. D.P.F. designed all the animal studies and postmortem-related inflammation and pain experiments and discussed the results. A.P. directed the project, designed all the experiments, and discussed all the results. All authors contributed toward the manuscript preparation and approved the final manuscript. Competing interests: I.L.M.I. and A.P. are inventors on two patent applications related to this work (PCT/EP2017/075041, filed 3 October 2017, and UK Patent No. 1616849.4, filed 6 October 2016). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaaq0597/DC1
fig. S1. Confocal microphotographs of glycan expressions in IVD at post-injury day 29.
fig. S2. Glycosignatures of IVD post-implantation of HA hydrogel at day 29.
fig. S3. Number of extracted proteins in the disc.
fig. S4. IPA revealed canonical “acute-phase signaling” in AF and NP tissues from the data set of differentially expressed proteins in our experimental groups.
fig. S5. Inflammation network in the injured disc analyzed by IPA.
fig. S6. Dysregulation of ECM proteins in the disc.
REFERENCES AND NOTES
- 1.Peng B.-G., Pathophysiology, diagnosis, and treatment of discogenic low back pain. World J. Orthop. 4, 42–52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raj P. P., Intervertebral disc: Anatomy-physiology-pathophysiology-treatment. Pain Pract. 8, 18–44 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Quero L., Klawitter M., Schmaus A., Rothley M., Sleeman J., Tiaden A. N., Klasen J., Boos N., Hottiger M. O., Wuertz K., Richards P. J., Hyaluronic acid fragments enhance the inflammatory and catabolic response in human intervertebral disc cells through modulation of toll-like receptor 2 signalling pathways. Arthritis Res. Ther. 15, R94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee J. M., Song J. Y., Baek M., Jung H.-Y., Kang H., Han I. B., Kwon Y. D., Shin D. E., Interleukin-1β induces angiogenesis and innervation in human intervertebral disc degeneration. J. Orthop. Res. 29, 265–269 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Freemont A. J., Watkins A., Le Maitre C., Baird P., Jeziorska M., Knight M. T. N., Ross E. R. S., O’Brien J. P., Hoyland J. A., Nerve growth factor expression and innervation of the painful intervertebral disc. J. Pathol. 197, 286–292 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Freemont A. J., Peacock T. E., Goupille P., Hoyland J. A., O’Brien J., Jayson M. I. V., Nerve ingrowth into diseased intervertebral disc in chronic back pain. Lancet 350, 178–181 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Zhang X., Huang J., McNaughton P. A., NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 24, 4211–4223 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alini M., Eisenstein S. M., Ito K., Little C., Kettler A. A., Masuda K., Melrose J., Ralphs J., Stokes I., Wilke H. J., Are animal models useful for studying human disc disorders/degeneration? Eur. Spine J. 17, 2–19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Bars D., Gozariu M., Cadden S. W., Animal models of nociception. Pharmacol. Rev. 53, 597–652 (2001). [PubMed] [Google Scholar]
- 10.Harris J. A. Using c-fos as a neural marker of pain. Brain Res. Bull. 45, 1–8 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Hunt S. P., Pini A., Evan G., Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature 328, 632–634 (1987). [DOI] [PubMed] [Google Scholar]
- 12.Dicker K. T., Gurski L. A., Pradhan-Bhatt S., Witt R. L., Farach-Carson M. C., Jia X., Hyaluronan: A simple polysaccharide with diverse biological functions. Acta Biomater. 10, 1558–1570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maharjan A. S., Pilling D., Gomer R. H., High and low molecular weight hyaluronic acid differentially regulate human fibrocyte differentiation. PLOS ONE 6, e26078 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith L. J., Nerurkar N. L., Choi K.-S., Harfe B. D., Elliott D. M., Degeneration and regeneration of the intervertebral disc: Lessons from development. Dis. Model. Mech. 4, 31–41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeong C. G., Francisco A. T., Niu Z., Mancino R. L., Craig S. L., Setton L. A., Screening of hyaluronic acid–poly(ethylene glycol) composite hydrogels to support intervertebral disc cell biosynthesis using artificial neural network analysis. Acta Biomater. 10, 3421–3430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z., Kaplan K. M., Wertzel A., Peroglio M., Amit B., Alini M., Grad S., Yayon A., Biomimetic fibrin–hyaluronan hydrogels for nucleus pulposus regeneration. Regen. Med. 9, 309–326 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Fontana G., Thomas D., Collin E., Pandit A.. Microgel microenvironment primes adipose-derived stem cells towards an NP cells-like phenotype. Adv. Healthc. Mater. 3, 2012–2022 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Radaeva I. F., Kostina G. A.. Use of hyaluronic acid for the treatment of various pathologic states. Pharm. Chem. J. 32, 492–494 (1998). [Google Scholar]
- 19.Caires R., Luis E., Taberner F. J., Fernandez-Ballester G., Ferrer-Montiel A., Balazs E. A., Gomis A., Belmonte C., de la Peña E., Hyaluronan modulates TRPV1 channel opening, reducing peripheral nociceptor activity and pain. Nat. Commun. 6, 8095 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isa I. L. M., Srivastava A., Tiernan D., Owens P., Rooney P., Dockery P., Pandit A., Hyaluronic acid based hydrogels attenuate inflammatory receptors and neurotrophins in interleukin-1β induced inflammation model of nucleus pulposus cells. Biomacromolecules 16, 1714–1725 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Griffin M. E., Hsieh-Wilson L. C., Glycan engineering for cell and developmental biology. Cell Chem. Biol. 23, 108–121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rutges J. P. H. J., Duit R. A., Kummer J. A., Bekkers J. E. J., Oner F. C., Castelein R. M., Dhert W. J. A., Creemers L. B., A validated new histological classification for intervertebral disc degeneration. Osteoarthritis Cartilage 21, 2039–2047 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Schaumburg H. H., Zotova E., Raine C. S., Tar M., Arezzo J., The rat caudal nerves: A model for experimental neuropathies. J. Peripher. Nerv. Syst. 15, 128–139 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Lai A., Moon A., Purmessur D., Skovrlj B., Winkelstein B. A., Cho S. K., Hecht A. C., Iatridis J. C., Assessment of functional and behavioral changes sensitive to painful disc degeneration. J. Orthop. Res. 33, 755–764 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Wijk G., Veldhuijzen D. S., Perspective on diffuse noxious inhibitory controls as a model of endogenous pain modulation in clinical pain syndromes. J. Pain 11, 408–419 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Haltiwanger R. S., Lowe J. B., Role of glycosylation in development. Annu. Rev. Biochem. 73, 491–537 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Toegel S., Bieder D., André S., Altmann F., Walzer S. M., Kaltner H., Hofstaetter J. G., Windhager R., Gabius H.-J., Glycophenotyping of osteoarthritic cartilage and chondrocytes by RT-qPCR, mass spectrometry, histochemistry with plant/human lectins and lectin localization with a glycoprotein. Arthritis Res. Ther. 15, R147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson D., Montpetit M. L., Stocker P. J., Bennett E. S.. The sialic acid component of the β1 subunit modulates voltage-gated sodium channel function. J. Biol. Chem. 279, 44303–44310 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Li J., Hsu H.-C., Ding Y., Li H., Wu Q., Yang P., Luo B., Rowse A. L., Spalding D. M., Bridges S. L. Jr, Mountz J. D., Inhibition of fucosylation reshapes inflammatory macrophages and suppresses type II collagen-induced arthritis. Arthritis Rheumatol. 66, 2368–2379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y.-l., Wu G.-z., Dawe G. S., Zeng L., Cui S.-s., Loers G., Tilling T., Sun L., Schachner M., Xiao Z.-C., Cell surface sialylation and fucosylation are regulated by L1 via phospholipase Cγ and cooperate to modulate neurite outgrowth, cell survival and migration. PLOS ONE 3, e3841 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujimoto K., Miyagi M., Ishikawa T., Inoue G., Eguchi Y., Kamoda H., Arai G., Suzuki M., Orita S., Kubota G., Sakuma Y., Oikawa Y., Kuniyoshi K., Ochiai N., Kishida S., Nakamura J., Aoki Y., Toyone T., Takahashi K., Ohtori S., Sensory and autonomic innervation of the cervical intervertebral disc in rats: The pathomechanics of chronic discogenic neck pain. Spine 37, 1357–1362 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Taheri M. M. H., Bideskan A. R. E., Miri M. R., Regulatory changes of N-acetylgalactosamine terminal sugar in early mouse embryonic paraxial mesenchyme. Cell J. 14, 130–141 (2012). [PMC free article] [PubMed] [Google Scholar]
- 33.Baig S., Wilcock G. K., Love S., Loss of perineuronal net N-acetylgalactosamine in Alzheimer’s disease. Acta Neuropathol. 110, 393–401 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Chahl L. A., Opioids—Mechanisms of action. Aust. Prescr. 19, 63–65 (1996). [Google Scholar]
- 35.Le Bars D., Chitour D., Kraus E., Clot A. M., Dickenson A. H., Besson J. M., The effect of systemic morphine upon diffuse noxious inhibitory controls (DNIC) in the rat: Evidence for a lifting of certain descending inhibitory controls of dorsal horn convergent neurones. Brain Res. 215, 257–274 (1981). [DOI] [PubMed] [Google Scholar]
- 36.Afify E. A., Daabees T. T., Gabra B. H., Abou Zeit-Har M. S., Role of nitric oxide in catalepsy and hyperthermia in morphine-dependent rats. Pharmacol. Res. 44, 533–539 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Mayer D. J., Mao J., Holt J., Price D. D., Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc. Natl. Acad. Sci. U.S.A. 96, 7731–7736 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann S., Doubell T. P., Leslie T., Woolf C. J., Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature 384, 360–364 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Steeds C. E. The anatomy and physiology of pain. Surgery 34, 55–59 (2016). [Google Scholar]
- 40.Huang E. J., Reichardt L. F., Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Lane N. E., Schnitzer T. J., Birbara C. A., Mokhtarani M., Shelton D. L., Smith M. D., Brown M. T., Tanezumab for the treatment of pain from osteoarthritis of the knee. N. Engl. J. Med. 363, 1521–1531 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayes A. J., Tudor D., Nowell M. A, Caterson B., Hughes C. E., Chondroitin sulfate sulfation motifs as putative biomarkers for isolation of articular cartilage progenitor cells. J. Histochem. Cytochem. 56, 125–138 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhattacharjee M., Chawla S., Chameettachal S., Murab S., Bhavesh N. S., Ghosh S., Role of chondroitin sulphate tethered silk scaffold in cartilaginous disc tissue regeneration. Biomed. Mater. 11, 025014 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Purmessur D., Cornejo M. C., Cho S. K., Roughley P. J., Linhardt R. J., Hecht A. C., Iatridis J. C., Intact glycosaminoglycans from intervertebral disc-derived notochordal cell-conditioned media inhibit neurite growth while maintaining neuronal cell viability. Spine J. 15, 1060–1069 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lauder R. M., Huckerby T. N., Nieduszynski I. A.. Lectin affinity chromatography of articular cartilage fibromodulin: Some molecules have keratan sulphate chains exclusively capped by α(2, 3)-linked sialic acid. Glycoconj. J. 28, 453–461 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Wuertz K., Haglund L., Inflammatory mediators in intervertebral disk degeneration and discogenic pain. Global Spine J. 3, 175–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber K. T., Alipui D. O., Sison C. P., Bloom O., Quraishi S., Overby M. C., Levine M., Chahine N. O., Serum levels of the proinflammatory cytokine interleukin-6 vary based on diagnoses in individuals with lumbar intervertebral disc diseases. Arthritis Res. Ther. 18, 3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murab S., Samal J., Shrivastava A., Ray A. R., Pandit A., Ghosh S., Glucosamine loaded injectable silk-in-silk integrated system modulate mechanical properties in bovine ex-vivo degenerated intervertebral disc model. Biomaterials 55, 64–83 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Braddock M., Quinn A., Targeting IL-1 in inflammatory disease: New opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 3, 330–339 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Nishimoto N., Kishimoto T., Interleukin 6: From bench to bedside. Nat. Clin. Pract. Rheumatol. 2, 619–626 (2006). [DOI] [PubMed] [Google Scholar]
- 51.V. Howard, M. G. Reed, Unbiased Stereology. Three-Dimensional Measurement in Microscopy (BIOS Scientific Publishers, 1998). [Google Scholar]
- 52.Collin E. C., Kilcoyne M., White S. J., Grad S., Alini M., Joshi L., Pandit A. S., Unique glycosignature for intervertebral disc and articular cartilage cells and tissues in immaturity and maturity. Sci. Rep. 6, 23062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaaq0597/DC1
fig. S1. Confocal microphotographs of glycan expressions in IVD at post-injury day 29.
fig. S2. Glycosignatures of IVD post-implantation of HA hydrogel at day 29.
fig. S3. Number of extracted proteins in the disc.
fig. S4. IPA revealed canonical “acute-phase signaling” in AF and NP tissues from the data set of differentially expressed proteins in our experimental groups.
fig. S5. Inflammation network in the injured disc analyzed by IPA.
fig. S6. Dysregulation of ECM proteins in the disc.