

Efficient CO production realizing Wächtershäuser’s scenario was demonstrated in simulated early ocean hydrotherm systems.
Abstract
Wächtershäuser’s proposal of the autotrophic origin of life theory and subsequent laboratory demonstrations of relevant organic reactions have opened a new gate for the exploration of the origin of life. However, this scenario remains controversial because, at present, it requires a high pressure of CO as a source of carbon and reducing energy, although CO must have been a trace C species on the Hadean Earth. We show that, simulating a geoelectrochemical environment in deep-sea hydrothermal fields, CO production with up to ~40% Faraday efficiency was attainable on CdS in CO2-saturated NaCl solution at ≤–1 V (versus the standard hydrogen electrode). The threshold potential is readily generated in the H2-rich, high-temperature, and alkaline hydrothermal vents that were probably widespread on the early komatiitic and basaltic ocean crust. Thus, Wächtershäuser’s scenario starting from CO2 was likely to be realized in the Hadean ocean hydrothermal systems.
INTRODUCTION
Since the first publication of Wächtershäuser’s theory of the autotrophic origin of life from primordial carbon fixation within a sulfide-rich hydrothermal vent (1, 2), a number of laboratory simulations for prebiotic organic synthesis have been performed within this scenario. Representative examples were published in Science more than a decade ago by Huber and Wächtershäuser: Under mildly hot, anaerobic, and alkaline aqueous conditions in the presence of metal sulfides (for example, FeS and NiS), they demonstrated the C–C bond formation that reminds us of the reductive acetyl–coenzyme A pathway (3), the reductive polymerization of HCN to α-hydroxy and α-amino acids (4), and the polymerization of α-amino acids to short peptides (5). Nevertheless, the significance of the autotrophic scenario has been questioned (6), because the proposed sulfide-pulled CO2 reduction has never been realized: Huber-Wächtershäuser’s organic syntheses required a high pressure of CO (1 bar or more) as a source of carbon and reducing energy. The CO concentrations in the atmosphere and volcanic emissions on the Hadean Earth were likely to be trace (7) unless there was a huge supply of meteorite-derived reducing power (8) that would have inevitably destroyed the surface environment. Although Wächtershäuser maintained a life’s origin in a CO-rich volcanic environment in a later addition to his theory (9), the setting is likely incompatible with the conditions crucial to the organic synthesis (3–5) because volcanic environments are inherently acidic to neutral (10). Russell and colleagues (11–13) and, more recently, Lane and Martin (14, 15) have proposed an alternative theory that the primordial carbon assimilation took place in the Hadean ocean alkaline hydrothermal systems through the direct coupling of seawater CO2 and hydrothermal H2 on metal sulfides, but no clear experimental support has yet been provided (16, 17).
A different mechanism of abiotic CO2 reduction in the early ocean hydrothermal systems has been suggested (18–20), for which redox, proton, and thermal gradients between the hydrothermal fluids and seawater across the sulfide deposits provide a continuous supply of reductive energy for inorganic carbon fixation. Using a sulfide chimney block obtained from a black smoker vent in the southern Lau Basin, Nakamura et al. (18) observed a long-distance electric conduction across the rock [0.2 to 9.1 ohms cm–1 at 300 K; (21)] and its catalytic function for redox reactions at the mineral-seawater interface. Yamamoto et al. (19) verified the electricity generation in the deep-sea hydrothermal environment using a fuel cell coupling the oxidation of hydrothermal H2S and the reduction of seawater O2 in an artificial hydrothermal vent. In addition, the in situ electrochemical surveys of the Okinawa Trough hydrothermal fields (20) gave multiple lines of evidence for natural and widespread electricity generation via sulfide deposits. These laboratory and field investigations strongly suggest that geoelectrochemical energy and reactive surfaces are ubiquitous in the present-day hydrothermal systems and were even more abundant and widespread across the early ocean floor.
Here, we present a laboratory demonstration of efficient electroreduction of CO2 on metal sulfides and discuss its relevance to the autotrophic scenario of the origin of life. We prepared low-crystalline sulfides of various metals (Ag, Cd, Co, Cu, Fe, Mn, Ni, Pb, or Zn) by mixing an alkaline solution of sodium sulfide and an acidic solution of the corresponding metal chloride. This mixing-induced sulfide formation likely occurred in the early alkaline hydrothermal vent environments and could comprise the main body of hydrothermal mineral deposits as the geoelectrochemical reactors (13, 22). Notice that the following experiments considerably augment recent studies by our group (23) and others (24), revealing the catalytic capability of some crystalline sulfides (for example, greigite). Here, we also studied molybdenum and tungsten sulfides (MoS2 and WS2) because of their importance in microbial metabolic reactions (25–27).
RESULTS
The CO2 electrolysis was conducted in an H-type electrochemical cell that had two compartments separated by a proton exchange membrane, with a Ag/AgCl reference electrode and a carbon working electrode placed at one side, and a Pt counter electrode at the other side (fig. S1). The metal sulfide–deposited carbon electrode was exposed to a constant potential for 24 hours in 100 mM NaCl saturated with 1 atm CO2 at room temperature (~25°C). Liquid-phase products were characterized by high-performance liquid chromatography (HPLC) and proton nuclear magnetic resonance (1H NMR) spectroscopy, and gas-phase products were characterized by gas chromatography (GC). Figure 1 shows the Faraday efficiencies (FEs) of CO2 reduction to CO (CO2 + 2H+ + 2e– → CO + H2O) and to formate (CO2 + H+ + 2e– → HCOO–) at –0.8, –1.0, and –1.2 V [versus the standard hydrogen electrode (SHE)]. CO and formate were the sole carbon products observed with the FE > 0.1%, along with H2 that evolved from the competing proton reduction (2H+ + 2e– → H2). The CO formation occurred effectively on CdS and Ag2S at –1.0 and –1.2 V (versus SHE), whereas negligible amounts of CO were produced in other experimental conditions or in the absence of sulfide. Additional experiments with varied potential (Fig. 1C) revealed that CO was generated at an electric potential below –0.8 V (versus SHE) on both sulfides. The FE shifted positively with decreasing potential and attained around 40% on CdS and 30% on Ag2S at ≤–1.0 and ≤–1.1 V (versus SHE), respectively. For formate, active formation was observed only on CuS (FE ~4% at –1.0 and –1.2 V versus SHE; Fig. 1B). These product characteristics were largely different from those on the corresponding pure metals (28), indicating the significance of sulfur to the surface-CO2-electron interactions.
Fig. 1. Electrochemical CO2 reduction on various metal sulfides in 0.1 M NaCl saturated with 1 atm CO2 at room temperature (~25°C).
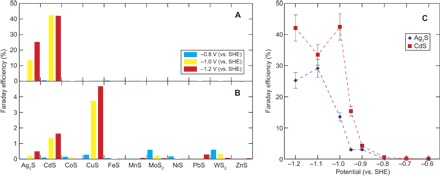
(A and B) FEs for CO2 reduction to (A) CO (CO2 + 2H+ + 2e– → CO + H2O) and (B) HCOO– (CO2 + 2H+ + 2e– → HCOO–) at –0.8, –1.0, and –1.2 V versus the SHE. (C) CO production efficiencies on CdS and Ag2S as a function of electric potential. Error bars correspond to the overall reproducibility of the experimental data that was evaluated from three independent experiments.
To examine whether the electrochemically produced CO serves as a driving force for the subsequent abiotic organic synthesis, we collected the reductive gas generated on CdS at –1.0 V (versus SHE) that was primarily composed of CO (ca. 40%) and H2 (ca. 60%) (Fig. 1C), sealed it in a serum bottle containing a solution of simple carbon compounds (methanethiol, potassium cyanide, or glycine) and NiS, and incubated it in a similar manner as was reported by Huber and Wächtershäuser (3–5) (see the Supplementary Materials). Our experiments largely reproduced the original reports, although there were smaller yields with fewer varieties of products owing to the lower initial CO concentration (fig. S2). We did not carry out a “one-pot” organic synthesis in an electrochemical cell because dissolved neutral species inevitably penetrate into the counter electrode side and undergo oxidation at uncontrollable potential, which makes data interpretation difficult. The occurrence and combination of multiple reactions are likely to be more realistic than single-pot reaction in natural hydrothermal vent environments. The complexity of the geochemical system could also allow the accumulation of organic compounds (29) that are generated under a continuous supply of CO by the sulfide-catalyzed CO2 electroreduction.
DISCUSSION
Were the experimental conditions examined above geologically and geochemically feasible in the Hadean ocean hydrothermal systems? Our experiments revealed that CO production on metal sulfides occurs at electric potentials below –0.8 V (versus SHE). In submarine hydrothermal vents, electrons are catalytically provided by the oxidation of reductive chemicals at the fluid-mineral interface (20). If H2S is assumed to be the sole electron source in the fluid, the potential is always higher than the threshold (–0.8 V versus SHE) in a wide range of temperature and pH (Fig. 2, A and B). However, if H2 is present at a concentration of 1 mmol kg–1 or higher (Fig. 2, C and D), H2 oxidation in hot and alkaline pH conditions can provide the potential well below the desirable value [for example, –0.98 V (versus SHE) at 200°C and pH 12 in the presence of 1 mmol kg–1 H2]. The H2-enriched hydrothermal fluid systems have been hypothesized to occur abundantly on the Hadean and early Archean ocean floor; two representative systems are a komatiite-hosted high-temperature type at oceanic ridges and plateaus (Fig. 3A) (30) and a low-temperature type hosted by komatiite or peridotite at ridge flank regions (13). A recent hydrothermal experiment of komatiite in CO2-saturated NaCl solution demonstrated that high concentrations of CO2 suppress H2 generation during low-temperature serpentinization (<250°C) owing to the formation of Fe2+-bearing dolomite, whereas the high-temperature serpentinization (>300°C) can generate extraordinarily high concentrations of H2 even with CO2-saturated seawater (31). Under such high-temperature conditions, the dehydration of epidote to anorthite followed by the precipitation of calcite controls the pH and makes it highly alkaline (pH ≥12), which has been demonstrated by a hydrothermal alteration experiment of basalt in CO2-rich seawater (32). It seems likely that alkaline hydrothermal systems were widespread across the early ocean crust and floor that were dominated by basalt and/or komatiite (33). Meanwhile, the early basalt-hosted hydrothermal systems with CO2-rich seawater may have also generated a variety of hydrothermal fluids with different H2 concentrations and pH values. With a total large surface area of basalt-seawater reaction in the hydrothermal circulation from recharge to discharge, the seawater CO2 would be quickly removed, and the CO2-depleted water/rock reaction could generate acidic to neutral, metal-rich (and occasionally H2-rich) black smoker fluids as observed today (Fig. 3B) (32).
Fig. 2. Thermodynamically predicted electric potentials of fluids from deep-sea hydrothermal systems.
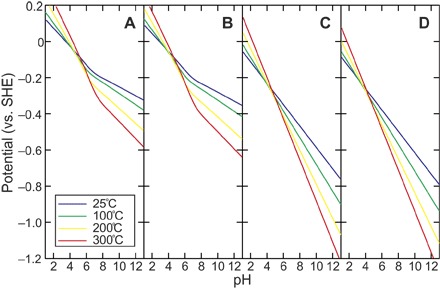
Redox potentials of (A) H2S (1 mmol kg–1), (B) H2S (10 mmol kg–1), (C) H2 (1 mmol kg–1), and (D) H2 (10 mmol kg–1) as a function of pH at 25°, 100°, 200°, and 300°C calculated assuming the half reactions of S + 2H+ (or H+) + 2e– → H2S (or HS–) [for (A) and (B)] and 2H+ + 2e– → H2 [for (C) and (D)]. S (solid sulfur) was chosen as the H2S (or HS–) oxidation product because the H2S/S redox couple provides a major potential control on the sulfide-rich hydrothermal vent environments (19). See the Supplementary Materials for the calculation procedure.
Fig. 3. Abiotic carbon fixation in the primitive hydrothermal system.

(A) The hot and active mantle convection in the primitive Earth induced a lot of massive upwelling of komatiite/basaltic melts beneath the ocean floor (30), (B) where interactions between the solidified (ultra)mafic rocks and the CO2-rich seawater with different water/rock ratios led to a variety of end-member fluid chemistry, including the H2-rich, high-temperature (T), and alkaline type and the metal-rich, high-temperature, and acidic (or neutral) ones (31, 32). (C) On the ocean floor, mixing of the hydrothermal fluids and seawater generated sulfide-rich chimneys (22), and the potential gradient across the chimney drove a continuous electron flow. The electric potential at the chimney-seawater interface could reach less than –1 V (versus SHE) in alkaline hydrothermal vent environments. The low potential, in the presence of sulfides rich in Cd2+ and Ag+, allowed the electrochemical CO2 reduction to CO with the FE as high as dozens of percent, together with H2 evolution. The produced CO served as a driving force for the subsequent abiotic organic synthesis that preceded the origin of life as schematically indicated in the figure.
Metals from the present-day black smoker vents have been observed to emanate over thousands-of-kilometers scale across hydrothermal plumes under soluble and/or colloidal forms with a limited vertical diffusion (34). Wider and denser metal distributions in the early deep ocean are envisioned (35) because of the anaerobic, Fe-rich, and slightly acidic character of ancient seawater (36, 37) with much greater global thermal convection than the present level (38). Cd2+ is a trace but ubiquitous hydrothermal dissolved metal ion (39) and has a very poor solubility in alkaline sulfide conditions (40). Therefore, Cd2+ and CdS dispersed from the early basalt-hosted hydrothermal systems would have precipitated and incorporated in the sulfide deposits in the closely localized alkaline hydrothermal vent environments.
There is no definitive constraint on the dissolved sulfide flux from the early ocean alkaline hydrothermal vents. If the fluid sulfide concentrations were within or lower than the micromolal level, the fluid-seawater mixing is expected to precipitate Cd, Zn, and Cu selectively against Ni, Co, and Mn owing to their difference in sulfide solubility (40). FeS precipitation would not have occurred even when a high seawater Fe2+ level (50 μmol kg–1) had been attained (40). In the present-day and Paleozoic hydrothermal sulfide deposits, Cd is observed primarily as an isomorphous impurity in sphalerite (ZnS), rather than a single CdS crystal, with the Cd/Zn substitution ratio of up to 13.2 weight % (41, 42). The Cd/Zn ratio tends to increase at higher precipitation temperature and lower concentrations of dissolved sulfide and chloride (43, 44); hence, hot and alkaline hydrothermal systems could favor the formation of Cd-enriched ZnS. Naturally generated multiple metal-sulfide precipitates in some cases exhibit higher catalytic efficiencies for the electroreduction of CO2 than the individual sulfides (23). Thus, some of the hydrothermal sulfide deposits of the H2-rich, high-temperature, and alkaline hydrothermal systems that were probably widespread in the early komatiitic and basaltic ocean floor (Fig. 3, A and B) are considered to provide favorable conditions for electrochemical CO production. This environmental situation is fully consistent with that assumed in Wächtershäuser’s abiotic organic synthesis (Fig. 3C) (3–5). Therefore, this study provides the first experimental evidence that the presence of the key player in Wächtershäuser’s scenario for the autotrophic origin of life, CO, was possible under a geologically and geochemically plausible setting on the primitive Earth.
The geoelectrochemical CO production demonstrated in this study could also enhance the theory for the origin of membrane bioenergetics (14). A number of microorganisms, in both archaea and bacteria, are known to use CO and formate as energy and carbon sources (45, 46). With the redox potential of a CO/CO2 pair (E0 = –520 mV), which is even lower than that for H2/H+ (E0 = –414 mV), CO can solely reduce ferredoxin (E0 = –400 mV) and generates a larger quantity of adenosine triphosphate (ATP) in the course of the autotrophic carbon assimilation than the H2/CO2 pair does (46). The energetic advantage of the CO metabolism would have brought an extra benefit for the origin and early evolution of chemolithotrophic systems in the early ocean hydrothermal vent environments.
At least two issues remain to be addressed: Why only Ag and Cd sulfides can effectively reduce CO2 to CO, and whether methanethiol and cyanide were really available. Ag2S and CdS are characterized by a relatively high energy conduction band edge (47) and by a relatively weak metal-S bond strength (48) among the sulfides examined in this study. The former property likely facilitates electron transfer from the charged surface to the adsorbed oxidants (H2O and CO2) (49). The latter energetically favors the C–S bond formation through reconnecting the metal-S bond, facilitating the CO2 conversion to CO by stabilization of the reaction intermediate (50). Although these advantages qualitatively explain the high CO productivities of CdS and Ag2S (Fig. 1), a definitive understanding of our results requires detailed structural and electronic study of adsorbed species. The availability of methanethiol and cyanide anion remains controversial (51, 52), although several mechanisms have been proposed for their abiotic formation in hydrothermal systems (53, 54). Because electrochemistry is a frequently used process to synthesize organic compounds with C–N and C–S bonds (55), the geoelectrochemical settings could have favored the production of reactive C1 compounds from naturally ubiquitous carbon, nitrogen, and sulfur sources (for example, CO2, N2O, and SO2) under naturally feasible conditions.
MATERIALS AND METHODS
Preparation of metal sulfides
All metal sulfides except for WS2 and MoS2 were prepared by a dropwise addition of 100 mM Na2S(aq) [or 50 mM Na2S(aq) for the case of Ag2S] into a 100 mM aqueous solution of the corresponding metal chloride (AgCl, CdCl2, CoCl2, CuCl2, FeCl2, MnCl2, NiCl2, PbCl2, or ZnCl2) under vigorous stirring with a final volume ratio of 1:1. The valence states of metals are all stable under the estimated Hadean ocean redox condition [0.432 V (versus SHE)] (56). Solid precipitates were then separated from the supernatant by centrifugation and were dried under vacuum. To prevent oxidation by atmospheric O2, the sample preparation was conducted in a globe box filled with N2 gas (>99.9999%), with 4% H2 being added (the COY system). In many cases, the synthesized sulfides showed broad x-ray diffraction signals, with low intensity indicating low crystallinity (fig. S3). All chemicals were purchased from Wako as reagent grade. Deaerated Milli-Q water (18.2 megohms) was used as the solvent. WS2 and MoS2 were obtained from Sigma-Aldrich (99%, ~90 nm) and were used without further purification.
Electrochemical experiments
Figure S1 schematically illustrates the electrochemical cell. For each experiment, about 50 mg of metal sulfide catalyst was deposited on the carbon working electrode (5.7 cm2), deaerated 100 mM NaCl(aq) was poured into the cell (60 ml to the working electrode side and 10 ml to the opposite side), and pure CO2 gas (>99.995%) was allowed to flow (20 ml min–1) into the solution for at least 1 hour before each experiment. While keeping the CO2 flowing, a constant potential was applied on the carbon electrode by using a multi-potentiostat (PS-08; Toho Technical Research). CO2 buffered the solution slightly acidic (pH = 5.75 ± 0.25) during the electrolysis. All potentials were measured against an Ag/AgCl reference electrode in saturated KCl and converted to the SHE scale using the following equation
| (1) |
After 24 hours of duration, the sample solution was filtered with a polytetrafluoroethylene (PTFE) membrane filter (pore size, 0.22 μm) and was analyzed by HPLC and 1H NMR spectroscopy. The gas-phase product was collected in a polypropylene gas trap immersed in the solution and was analyzed by GC. The gas sampling was carried out for a certain fixed period (note that the sampling period was varied depending on the activity of the reductive gas generation), for which bubbles generated on the metal sulfides were accumulated into the gas trap while intrusion of the flowed CO2 was carefully avoided. The gas products collected after various durations of the electrochemical experiment showed mutually similar compositional characteristics over the first 24 hours (within the overall reproducibility; ±10%). The FE was calculated from the following equations
| (2) |
for the liquid-phase products, and
| (3) |
for the gas-phase products.
Organic synthesis experiments
Three types of organic synthesis experiments were conducted according to the methods reported by Huber and Wächtershäuser (3–5), with the electrochemically generated reductive gas on CdS at –1 V (versus SHE) replacing pure CO gas.
(i) NiSO4 ⋅ 6H2O (131 mg, 0.5 mmol) was deaerated in a serum bottle (100 ml) sealed with a butyl rubber cap and an aluminum stopper, and 2.5 ml of aqueous solution containing Na2S (39 mg, 0.5 mmol), CH3SNa (1.8 mg, 25 μmol), and NaOH was added for the in situ precipitation of NiS. The bottle was then filled with the reductive gas (80 ml, 1 atm) and was heated at 100°C for 7 days. The sample suspension was then filtered with a PTFE membrane filter and analyzed by liquid chromatography–mass spectrometry (LC-MS) for identification of organic acid products and by HPLC for their quantification. The filtrate had a pH of 8.6, which was measured by a portable pH meter (Seven2Go Pro, Mettler Toledo).
(ii) A serum bottle (100 ml) was charged with Ca(OH)2 (0.5 g, 6.7 mmol), closed, deaerated, and then charged with 2.5 ml of aqueous solution containing NiSO4 (77 mg, 0.5 mmol) and KCN (33 mg, 0.5 mmol) together with the reductive gas (80 ml, 1 atm). After heating at 100°C for 5 days, the sample suspension was filtered and analyzed for organic acid and amino acid products. The filtrate showed a pH of 12.6.
(iii) A serum bottle (100 ml) was charged with NiSO4 ⋅ 6H2O (131 mg, 0.5 mmol), closed, deaerated, and then charged with 2.5 ml of aqueous solution containing Na2S (78 mg, 1 mmol) and glycine (9.4 mg, 125 μmol) together with the reductive gas (80 ml, 1 atm). After heating at 100°C for 1 day, the sample suspension was filtered and analyzed for glycine peptides. The filtrate showed a pH of 7.9.
Sample analysis
Organic acid products were quantified by using a Shimadzu HPLC system equipped with an electric conductivity detector and an anion exchange column (Shim-pack SCR-102H, Shimadzu) set at 40°C. The p-toluenesulfonic acid solution (5 mM) was used as the eluent at a rate of 1.6 ml min–1.
1H NMR spectra were acquired using a Bruker Avance III spectrometer (400 MHz) at the sample temperature of 303.0 K. Typically, 0.45 ml of sample solutions was mixed with 0.05 ml of D2O (99.9%; Merck Millipore) containing 5 mM 3-(trimethylsilyl)-1-propanesulfonic acid-d6 sodium (DSS-d6; Sigma-Aldrich) and was placed in an NMR tube (5 mm outside diameter; Wilmad-LabGlass). DSS-d6 was used for the calibration of the 0-ppm (parts per million) position and to quantify the product concentrations as an internal standard. A solvent suppression was run to minimize the solvent signal. The obtained spectra of sample solutions after the CO2 electrolysis (fig. S4) showed an intense signal of HCOO– at 8.45 ppm when CuS was used as a catalyst, whereas no significant peak except for H2O and DSS-d6 appeared on the other spectral regions and in the other sample spectra. The quantified concentrations of HCOO– coincided well with the corresponding HPLC results (within ±10%); therefore, the FE was calculated using the HPLC data and was presented in Fig. 1B.
The GC measurements were conducted using a GC-4000 (GL Sciences) equipped with a HayeSep Q column (GL Sciences) and a Shincarbon-ST column (Shinwa). He (>99.99995%) was used as the carrier gas at a flow rate of 24 ml min–1. The column temperature was initially kept at 40°C for 6 min, raised to 200°C at a rate of 20°C min–1, kept for 6 min, raised to 250°C at a rate of 50°C min–1, and then kept for 20 min. H2, CO, CH4, CO2, C2H6, and C3H8 were observed with the retention times of ca. 2.1, 5.0, 8.8, 11.8, 16.7, and 28.0 min, respectively. A pulsed discharge detector was used to quantify the concentrations of CO, CH4, C2H6, and C3H8, and a thermal conductivity detector for the H2 concentration. Some gas samples were also analyzed by a gas chromatograph mass spectrometer (GCMS-QP2020; Shimazu) equipped with MICROPACKED-ST column (Shinwa). Results obtained by the different systems were consistent with each other within the error (±10%).
Amino acid and peptide products were analyzed using a Jasco HPLC system equipped with post-column derivatization with o-phthalaldehyde and a fluorescence detector operated at 345 nm for excitation and at 455 nm for emission. Five citrate buffer solutions of different citrate concentrations and pH values were used as eluents in a stepwise condition. A cation-exchange column (AApak Na II-S2, Jasco) was used at 50°C.
LC-MS measurements were conducted with a Xevo Qtof MS platform (Waters) combined with a UPLC system (Acquity, Waters). To enhance the ionization of small acids while suppressing the interference with inorganic salts, positive-mode electrospray ionization combined with reversed-phase LC was performed after derivatization with 2-nitrophenylhydrazine (57). In the LC separation using a CORTECS UPLC C18 column (Waters), 0.1% trifluoroacetate and acetonitrile were used as the eluents for binary gradient elution with the flow rate at 0.3 ml min−1 and the column temperature at 35°C. The acetonitrile volumetric ratio was initially held at 10%, increased linearly to 40% and then to 90% for 2 to 6 min and 6 to 6.7 min, respectively, and held at 15% for 7 min. The time-of-flight analyzer was set to a sensitivity mode with a resolving power of 20,000 at the mass/charge ratio (m/z) of 556 corresponding to protonated leucine encephalin, and the m/z range of 20 to 1500 was calibrated with sodium formate.
Thermodynamic calculation
The redox potentials of H2S and H2 as a function of temperature and pH (Fig. 2) were calculated, respectively, assuming the following half reactions
| (4) |
| (5) |
using the equations
| (6) |
| (7) |
In these equations, T, R, and F stand for temperature in kelvin, the gas constant (8.31447 J mol–1 K–1), and the Faraday constant (96485 J mol–1 V–1), respectively. αi represents the activity of the species i, and x signifies the mole fraction of H2S (; and denote the molality of H2S and HS–, respectively). In addition, ΔfGo(i) represents the standard partial molal Gibbs free energy of formation of the species i at desired temperature and pressure, which were calculated according to the revised Helgeson-Kirkham-Flowers (HKF) equations of state (58) together with the thermodynamic data and the revised HKF parameters reported by Shock et al. for H2 and H2S (59) and HS– (60). The temperature and pressure dependences of ΔfGo for S (solid sulfur) were calculated as follows
| (8) |
where and , respectively, represent the standard molal Gibbs energy and entropy at the reference temperature (Tr = 298.15 K) and pressure (Pr = 1 bar). represents the standard molal heat capacity at Pr, and denotes the standard molal volume at the temperature of interest. In the present calculation, the values of , , and as a function of temperature for S were taken from Robie et al. (61), whereas the value of was assumed to be constant in the range of temperature and pressure of our interest. The value of x is expressed as
| (9) |
where γi represents the activity coefficient of the species i (αi = Mi × γi) and is the dissociation constant of H2S (H2S → HS– + H+), whose value was calculated as
| (10) |
In all calculations, the values of γi were calculated by using the extended Debye-Hückel equation (62) with the ionic strength of 0.1 (NaCl). The pressure was set to 100 bar. S2– and the ion pair NaHS were not considered in this calculation because these are expected to be minor S and/or Na species in the examined aqueous conditions (63, 64).
Supplementary Material
Acknowledgments
We thank Y. Hongo and Y. Suna for their support in the organic product analysis. Funding: This research was supported by Japan Society for the Promotion of Science KAKENHI (grant nos. 16H04074, 16K13906, 17H06105, and JP2610s6003) and the Astrobiology Center Program of the National Institutes of Natural Sciences (grant no. AB292004). Author contributions: N.K., R.N., M.Y., K.T., and Y.O. designed this study. N.K., R.N., M.Y., Y.L., and A.Y. established the setup for electrochemical experiment. M.Y. and K.T. provided information for the present and ancient deep-sea hydrothermal systems. N.K. and A.G. performed sample analysis. Y.U. and N.Y. provided analytical instruments. All authors contributed to writing the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaao7265/DC1
fig. S1. A schematic of the electrochemical cell.
fig. S2. Abiotic organic synthesis driven by the electrochemically generated reductive gas on CdS at –1.0 V (versus SHE) in 100 mM NaCl saturated with 1 atm of CO2.
fig. S3. X-ray diffraction patterns of metal sulfides.
fig. S4. 1H NMR spectra of the CO2-saturated 0.1 M NaCl after applying –1.2 V (versus SHE) for 24 hours in the presence of metal sulfides.
table S1. Summary of total current densities and FEs for CO2 reduction on metal sulfides.
table S2. Peak list of x-ray diffraction patterns of metal sulfides.
REFERENCES AND NOTES
- 1.Wächtershäuser G., Before enzymes and templates: Theory of surface metabolism. Microbiol. Rev. 52, 452–484 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wächtershäuser G., Pyrite formation, the first energy source for life: A hypothesis. Syst. Appl. Microbiol. 10, 207–210 (1988). [Google Scholar]
- 3.Huber C., Wächtershäuser G., Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 276, 245–247 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Huber C., Wächtershäuser G., α-Hydroxy and α-amino acids under possible Hadean, volcanic origin-of-life conditions. Science 314, 630–632 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Huber C., Wächtershäuser G., Peptides by activation of amino acids with CO on (Ni,Fe)S surfaces: Implications for the origin of life. Science 281, 670–672 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Keefe A. D., Miller S. L., McDonald G., Bada J., Investigation of the prebiotic synthesis of amino acids and RNA bases from CO2 using FeS/H2S as a reducing agent. Proc. Natl. Acad. Sci. U.S.A. 92, 11904–11906 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trail D., Watson E. B., Tailby N. D., The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 480, 79–82 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Zahnle K., Schaefer L., Fegley B., Earth’s earliest atmosphere. Cold Spring Harb. Perspect. Biol. 2, a004895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wächtershäuser G., From volcanic origins of chemoautotrophic life to Bacteria, Archaea and Eukarya. Philos. Trans. R. Soc. London B Biol. Sci. 361, 1787–1806 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulkidjanian A. Y., Bychkov A. Y., Dibrova D. V., Galperin M. Y., Koonin E. V., Origin of first cells at terrestrial, anoxic geothermal fields. Proc. Natl. Acad. Sci. U.S.A. 109, E821–E830 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russell M. J., Hall A. J., The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. London 154, 377–402 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Russell M. J., Martin W., The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 29, 358–363 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Russell M. J., Hall A. J., Martin W., Serpentinization as a source of energy at the origin of life. Geobiology 8, 355–371 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Lane N., Martin W. F., The origin of membrane bioenergetics. Cell 151, 1406–1416 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Lane N., Bioenergetic constraints on the evolution of complex life. Cold Spring Harb. Perspect. Biol. 6, a015982 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herschy B., Whicher A., Camprubi E., Watson C., Dartnell L., Ward J., Evans J. R. G., Lane N., An origin-of-life reactor to simulate alkaline hydrothermal vents. J. Mol. Evol. 79, 213–227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson J. B., The “Origin-of-Life Reactor” and reduction of CO2 by H2 in inorganic precipitates. J. Mol. Evol. 85, 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Nakamura R., Takashima T., Kato S., Takai K., Yamamoto M., Hashimoto K., Electrical current generation across a black smoker chimney. Angew. Chem. Int. Ed. 49, 7692–7694 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto M., Nakamura R., Oguri K., Kawagucci S., Suzuki K., Hashimoto K., Takai K., Generation of electricity and illumination by an environmental fuel cell in deep-sea hydrothermal vents. Angew. Chem. Int. Ed. 52, 10758–10761 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto M., Nakamura R., Kasaya T., Kumagai H., Suzuki K., Takai K., Spontaneous and widespread electricity generation in natural deep-sea hydrothermal fields. Angew. Chem. Int. Ed. 56, 5725–5728 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Ang R., Khan A. U., Tsujii N., Takai K., Nakamura R., Mori T., Thermoelectricity generation and electron–magnon scattering in a natural chalcopyrite mineral from a deep-sea hydrothermal vent. Angew. Chem. Int. Ed. 54, 12909–12913 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Russell M. J., Barge L. M., Bhartia R., Bocanegra D., Bracher P. J., Branscomb E., Kidd R., McGlynn S., Meier D. H., Nitschke W., Shibuya T., Vance S., White L., Kanik I., The drive to life on wet and icy worlds. Astrobiology 14, 308–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaguchi A., Yamamoto M., Takai K., Ishii T., Hashimoto K., Nakamura R., Electrochemical CO2 reduction by Ni-containing iron sulfides: How is CO2 electrochemically reduced at bisulfide-bearing deep-sea hydrothermal precipitates? Electrochim. Acta 141, 311–318 (2014). [Google Scholar]
- 24.Roldan A., Hollingsworth N., Roffey A., Islam H.-U., Goodall J. B. M., Catlow C. R. A., Darr J. A., Bras W., Sankar G., Holt K. B., Hogarth G., de Leeuw N. H., Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions. Chem. Commun. 51, 7501–7504 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Nitschke W., Russell M. J., Hydrothermal focusing of chemical and chemiosmotic energy, supported by delivery of catalytic Fe, Ni, Mo/W, Co, S and Se, forced life to emerge. J. Mol. Evol. 69, 481–496 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Döring A., Schulzke C., Tungsten’s redox potential is more temperature sensitive than that of molybdenum. Dalton Trans. 39, 5623–5629 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Schoepp-Cothenet B., van Lis R., Philippot P., Magalon A., Russell M. J., Nitschke W., The ineluctable requirement for the trans-iron elements molybdenum and/or tungsten in the origin of life. Sci. Rep. 2, 263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noda H., Ikeda S., Oda Y., Imai K., Maeda M., Ito K., Electrochemical reduction of carbon dioxide at various metal electrodes in aqueous potassium hydrogen carbonate solution. Bull. Chem. Soc. Jpn. 63, 2459–2462 (1990). [Google Scholar]
- 29.Baaske P., Weinert F. M., Duhr S., Lemke K. H., Russell M. J., Braun D., Extreme accumulation of nucleotides in simulated hydrothermal pore systems. Proc. Natl. Acad. Sci. U.S.A. 104, 9346–9351 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takai K., Nakamura K., Suzuki K., Inagaki F., Nealson K. H., Kumagai H., Ultramafics-Hydrothermalism-Hydrogenesis-HyperSLiME (UltraH3) linkage: A key insight into early microbial ecosystem in the Archean deep-sea hydrothermal systems. Paleontol. Res. 10, 269–282 (2006). [Google Scholar]
- 31.Ueda H., Shibuya T., Sawaki Y., Saitoh M., Takai K., Maruyama S., Reactions between komatiite and CO2-rich seawater at 250 and 350°C, 500 bars: Implications for hydrogen generation in the Hadean seafloor hydrothermal system. Prog. Earth Planet. Sci. 3, 35 (2016). [Google Scholar]
- 32.Shibuya T., Yoshizaki M., Masaki Y., Suzuki K., Takai K., Russell M. J., Reactions between basalt and CO2-rich seawater at 250 and 350°C, 500 bars: Implications for the CO2 sequestration into the modern oceanic crust and the composition of hydrothermal vent fluid in the CO2-rich early ocean. Chem. Geol. 359, 1–9 (2013). [Google Scholar]
- 33.Barley M. E., Pickard A. L., An extensive, crustally-derived, 3325 to 3310 Ma silicic volcanoplutonic suite in the eastern Pilbara Craton: Evidence from the Kelly Belt, McPhee Dome and Corunna Downs Batholith. Precambrian Res. 96, 41–62 (1999). [Google Scholar]
- 34.Resing J. A., Sedwick P. N., German C. R., Jenkins W. J., Moffett J. W., Sohst B. M., Tagliabue A., Basin-scale transport of hydrothermal dissolved metals across the South Pacific Ocean. Nature 523, 200–203 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Mloszewska A. M., Pecoits E., Cates N. L., Mojzsis S. J., O’Neil J., Robbins L. J., Konhauser K. O., The composition of Earth’s oldest iron formations: The Nuvvuagittuq Supracrustal Belt (Québec, Canada). Earth Planet. Sci. Lett. 317–318, 331–342 (2012). [Google Scholar]
- 36.Halevy I., Alesker M., Schuster E. M., Popovitz-Biro R., Feldman Y., A key role for green rust in the Precambrian oceans and the genesis of iron formations. Nat. Geosci. 10, 135–139 (2017). [Google Scholar]
- 37.Tosca N. J., Guggenheim S., Pufahl P. K., An authigenic origin for Precambrian greenalite: Implications for iron formation and the chemistry of ancient seawater. Geol. Soc. Am. Bull. 128, 511–530 (2016). [Google Scholar]
- 38.Lowell R. P., Keller S. M., High-temperature seafloor hydrothermal circulation over geologic time and Archean banded iron formations. Geophys. Res. Lett. 30, 1391 (2003). [Google Scholar]
- 39.Tivey M. K., Generation of seafloor hydrothermal vent fluids and associated mineral deposits. Oceanography 20, 50–65 (2007). [Google Scholar]
- 40.Saito M. A., Sigman D. M., Morel F. M. M., The bioinorganic chemistry of the ancient ocean: The co-evolution of cyanobacterial metal requirements and biogeochemical cycles at the Archean–Proterozoic boundary? Inorg. Chim. Acta 356, 308–318 (2003). [Google Scholar]
- 41.Cook N. J., Ciobanu C. L., Pring A., Skinner W., Shimizu M., Danyushevsky L., Saini-Eidukat B., Melcher F., Trace and minor elements in sphalerite: A LA-ICPMS study. Geochim. Cosmochim. Acta 73, 4761–4791 (2009). [Google Scholar]
- 42.Maslennikov V. V., Maslennikova S. P., Large R. R., Danyushevsky L. V., Herrington R. J., Ayupova N. R., Zaykov V. V., Lein A. Y., Tseluyko A. S., Melekestseva I. Y., Tessalina S. G., Chimneys in Paleozoic massive sulfide mounds of the Urals VMS deposits: Mineral and trace element composition with modern black, grey, white and clear smokers. Ore Geol. Rev. 85, 64–106 (2017). [Google Scholar]
- 43.Bazarkina E. F., Pokrovski G. S., Zotov A. V., Hazemann J.-L., Structure and stability of cadmium chloride complexes in hydrothermal fluids. Chem. Geol. 276, 1–17 (2010). [Google Scholar]
- 44.Wen H., Zhu C., Zhang Y., Cloquet C., Fan H., Fu S., Zn/Cd ratios and cadmium isotope evidence for the classification of lead-zinc deposits. Sci. Rep. 6, 25273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Windman T., Zootova N., Schwandner F., Shock E. L., Formate as an energy source for microbial metabolism in chemosynthetic zones of hydrothermal ecosystems. Astrobiology 7, 873–890 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Diender M., Stams A. J. M., Sousa D. Z., Pathways and bioenergetics of anaerobic carbon monoxide fermentation. Front. Microbiol. 6, 1275 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Y., Schoonen M. A. A., The absolute energy positions of conduction and valence bands of selected semiconducting minerals. Am. Mineral. 85, 543–556 (2000). [Google Scholar]
- 48.Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies (CRC Press, 2007). [Google Scholar]
- 49.Schoonen M. A. A., Xu Y., Bebie J., Energetics and kinetics of the prebiotic synthesis of simple organic acids and amino acids with the FeS-H2/FeS2 redox couple as reductant. Orig. Life Evol. Biosph. 29, 5–12 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Varley J. B., Hansen H. A., Ammitzbøll N. L., Grabow L. C., Peterson A. A., Rossmeisl J., Nørskov J. K., Ni–Fe–S cubanes in CO2 reduction electrocatalysis: A DFT study. ACS Catal. 3, 2640–2643 (2013). [Google Scholar]
- 51.Proskurowski G., Lilley M. D., Seewald J. S., Fruh-Green G. L., Olson E. J., Lupton J. E., Sylva S. P., Kelley D. S., Abiogenic hydrocarbon production at Lost City hydrothermal field. Science 319, 604–607 (2008). [DOI] [PubMed] [Google Scholar]
- 52.McCollom T. M., Donaldson C., Generation of hydrogen and methane during experimental low-temperature reaction of ultramafic rocks with water. Astrobiology 16, 389–406 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Kamaluddin, Yanagawa H., Egami F., Formation of molecules of biological interest from formaldehyde and hydroxylamine in a modified sea medium. J. Biochem. 85, 1503–1507 (1979). [DOI] [PubMed] [Google Scholar]
- 54.Heinen W., Lauwers A. M., Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig. Life Evol. Biosph. 26, 131–150 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Feroci M., Orsini M., Rossi L., Sotgiu G., Inesi A., Electrochemically promoted C–N bond formation from amines and CO2 in ionic liquid BMIm–BF4: Synthesis of carbomates. J. Org. Chem. 72, 200–203 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Shibuya T., Russell M. J., Takai K., Free energy distribution and hydrothermal mineral precipitation in Hadean submarine alkaline vent systems: Importance of iron redox reactions under anoxic conditions. Geochim. Cosmochim. Acta 175, 1–19 (2016). [Google Scholar]
- 57.Hou D., Fan J., Han L., Ruan X., Feng F., Liu W., Zheng F., Determination of small halogenated carboxylic acid residues in drug substances by high performance liquid chromatography-diode array detection following derivatization with nitro-substituted phenylhydrazines. J. Chromatogr. A 1438, 46–56 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Shock E. L., Oelkers E. H., Johnson J. W., Sverjensky D. A., Helgeson H. C., Calculation of the thermodynamic properties of aqueous species at high pressures and temperatures. Effective electrostatic radii, dissociation constants and standard partial molal properties to 1000°C and 5 kbar. J. Chem. Soc. Faraday Trans. 88, 803–826 (1992). [Google Scholar]
- 59.Shock E. L., Helgeson H. C., Sverjensky D. A., Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Standard partial molal properties of inorganic neutral species. Geochim. Cosmochim. Acta 53, 2157–2183 (1989). [Google Scholar]
- 60.Shock E. L., Sassani D. C., Willis M., Sverjensky D. A., Inorganic species in geologic fluids: Correlations among standard molal thermodynamic properties of aqueous ions and hydroxide complexes. Geochim. Cosmochim. Acta 61, 907–950 (1997). [DOI] [PubMed] [Google Scholar]
- 61.R. A. Robie, B. S. Hemingway, J. R. Fisher, Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 Bar (105 Pascals) Pressure and at Higher Temperatures (U.S. Geological Survey Bulletin 1452, U.S. Geological Survey 1978). [Google Scholar]
- 62.Helgeson H. C., Kirkham D. H., Flowers G. C., Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: IV. Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600°C and 5 kB. Am. J. Sci. 281, 1249–1516 (1981). [Google Scholar]
- 63.Migdisov A. A., Williams-Jones A. E., Lakshtanov L. Z., Alekhin Y. V., Estimates of the second dissociation constant of H2S from the surface sulfidation of crystalline sulfur. Geochim. Cosmochim. Acta 66, 1713–1725 (2002). [Google Scholar]
- 64.Stefánsson A., Seward T. M., Experimental determination of the stability and stoichiometry of sulphide complexes of silver(I) in hydrothermal solutions to 400°C. Geochim. Cosmochim. Acta 67, 1395–1413 (2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaao7265/DC1
fig. S1. A schematic of the electrochemical cell.
fig. S2. Abiotic organic synthesis driven by the electrochemically generated reductive gas on CdS at –1.0 V (versus SHE) in 100 mM NaCl saturated with 1 atm of CO2.
fig. S3. X-ray diffraction patterns of metal sulfides.
fig. S4. 1H NMR spectra of the CO2-saturated 0.1 M NaCl after applying –1.2 V (versus SHE) for 24 hours in the presence of metal sulfides.
table S1. Summary of total current densities and FEs for CO2 reduction on metal sulfides.
table S2. Peak list of x-ray diffraction patterns of metal sulfides.