

SUMMARY
The neuronal gene Arc is essential for long-lasting information storage in the mammalian brain, mediates various forms of synaptic plasticity, and has been implicated in neurodevelopmental disorders. However, little is known about Arc’s molecular function and evolutionary origins. Here we show that Arc self-assembles into virus-like capsids that encapsulate RNA. Endogenous Arc protein is released from neurons in extracellular vesicles that mediate the transfer of Arc mRNA into new target cells where it can undergo activity-dependent translation. Purified Arc capsids are endocytosed and are able to transfer Arc mRNA into the cytoplasm of neurons. These results show that Arc exhibits similar molecular properties of retroviral Gag proteins. Evolutionary analysis indicates that Arc is derived from a vertebrate lineage of Ty3/gypsy retrotransposons, which are also ancestral to retroviruses. These findings suggest that Gag retroelements have been repurposed during evolution to mediate intercellular communication in the nervous system.
Graphical abstract
INTRODUCTION
Brains have evolved to process and store information from the outside world through synaptic connections between interconnected networks of neurons. Despite the fundamental importance of information storage in the brain, we still lack a detailed molecular and cellular understanding of the processes involved and their evolutionary origins. Eukaryotic genomes are littered with DNA of viral or transposon origin, which comprise about half of most mammalian genomes (Smit, 1999). There is growing appreciation that the sequences encoded by these elements can provide raw material for the emergence of new functions and regulatory elements (Chuong et al., 2017). In vertebrates, these include dozens of protein-coding genes derived from sequences previously encoded by transposons (Feschotte and Pritham, 2007) or retroviruses (Kaneko-Ishino and Ishino, 2012). Interestingly, many of these transposon-derived genes are expressed in the brain, but their molecular functions remain to be elucidated.
The neuronal gene Arc contains structural elements found within viral Group-specific antigen (Gag) polyproteins that may have originated from the Ty3/gypsy retrotransposon family (Campillos et al., 2006; Shepherd, 2017; Zhang et al., 2015), although the role these Gag elements play in Arc function has not been explored. Arc is a master regulator of synaptic plasticity in mammals and is required for protein synthesis-dependent forms of long-term potentiation (LTP) and depression (LTD) (Bramham et al., 2010; Shepherd and Bear, 2011). Arc can regulate synaptic plasticity through the trafficking of AMPA-type glutamate receptors (AMPARs) via the endocytic machinery (Chowdhury et al., 2006). This endocytic pathway maintains levels of surface AMPARs in response to chronic changes in neuronal activity through synaptic scaling, thus contributing to neuronal homeostasis (Shepherd et al., 2006). Arc’s expression in the brain is highly dynamic; its transcription is tightly coupled to encoding of information in neuronal circuits in vivo (Guzowski et al., 1999). Arc mRNA is transported to dendrites and becomes enriched at sites of local synaptic activity where it is locally translated into protein (Steward et al., 1998; Waung et al., 2008). Intriguingly, aspects of Arc mRNA regulation resemble some viral RNAs, as Arc contains an internal ribosomal entry site (IRES) that allows cap-independent translation (Pinkstaff et al., 2001). In vivo, Arc is required to transduce experience into long-lasting changes in visual cortex plasticity (McCurry et al., 2010) and for long-term memory (Guzowski et al., 2000; Plath et al., 2006). In addition, Arc has been implicated in various neurological disorders that include Alzheimer’s disease (Wu et al., 2011), neurodevelopmental disorders such as Angelman (Greer et al., 2010; Pastuzyn and Shepherd, 2017) and Fragile-X Syndromes (Park et al., 2008), and schizophrenia (Fromer et al., 2014; Manago et al., 2016; Purcell et al., 2014). Thus, precise regulation of Arc expression and activity in the nervous system seems essential for normal cognition.
Despite its importance, little is known about Arc protein biochemistry and molecular function. Here we uncover a potential role for Arc in mediating intercellular communication via extracellular vesicles (EVs). Synaptic communication is modulated by many other communication pathways that include glia-neuron interactions, and emerging evidence suggests that EVs mediate intercellular signaling in the nervous system (Budnik et al., 2016; Zappulli et al., 2016). EVs can be broadly divided into two groups, microvesicles and exosomes, which are defined both by size and subcellular origin. Microvesicles pinch off from the plasma membrane directly and are usually 100–300nm in diameter, whereas exosomes are derived from intraluminal vesicles that originate from multivesicular bodies (MVBs) and are usually <100nm in size. EVs can transport cargo that do not readily cross the plasma membrane, such as membrane proteins and various forms of RNA. The observation that EVs can function in the intercellular transport of these molecules within the nervous system opens an entirely new perspective on intercellular communication in the brain.
Here, we find that Arc protein self-assembles into oligomers that resemble virus capsids and exhibits several other biochemical properties seen in retroviral Gag proteins such as RNA binding. Moreover, Arc is released from neurons in EVs and is able to transfer its own mRNA into neurons. The Drosophila Arc homologue, dArc1, also forms capsids and mediates intercellular transfer of its own mRNA at the fly neuromuscular junction, despite originating from a distinct retrotransposon lineage. These data suggest that co-option of retroviral-like Gag elements may have provided an evolutionary pathway for novel mechanisms that mediate intercellular signaling and have been intricately involved in the evolution of synaptic plasticity and animal cognition.
RESULTS
Fly and Tetrapod Arc Genes Originated Independently from Distinct Lineages of Ty3/gypsy Retrotransposons
To shed light into Arc’s evolutionary origins, we performed phylogenomic analyses (Figure 1A, S1A). Highly conserved, unique orthologs of the murine Arc genes were identified throughout the tetrapods (mammals, birds, reptiles, amphibians), but were conspicuously absent from all fish lineages and other deuterostomes examined (94 species). The closest relatives of Arc in the coelacanth, zebrafish, and carp genomes were encoded by prototypical Ty3/gypsy retrotransposons, with indications of recent transposition activity. Similarly, orthologs and paralogs of Drosophila Arc (darc1, darc2) were identified in all schizophoran (true) flies represented in the database but were not detected in any other dipteran (e.g., mosquitoes) or protostome species (286 species; Figure S1B). The closest retrotransposon relatives of the fly Arc genes were found in the genomes of the silkworm and Argentine ant. Interestingly, while Arc appears to be a single-copy gene in all tetrapods examined, the gene has experienced multiple rounds of duplication during schizophoran evolution (Figure S1B). Phylogenetically, tetrapod Arc genes cluster with Ty3/gypsy retrotransposons from fish, while the fly Arc homologs group with a separate lineage of Ty3/gypsy retrotransposons from insects (Figure 1A). These results indicate that the tetrapod and fly Arc genes originated independently from distinct lineages of Ty3/gypsy retrotransposons but still share significant homology in the retroviral Gag domain.
Figure 1. Arc forms virus-like capsids via a conserved retroviral Gag CA domain.
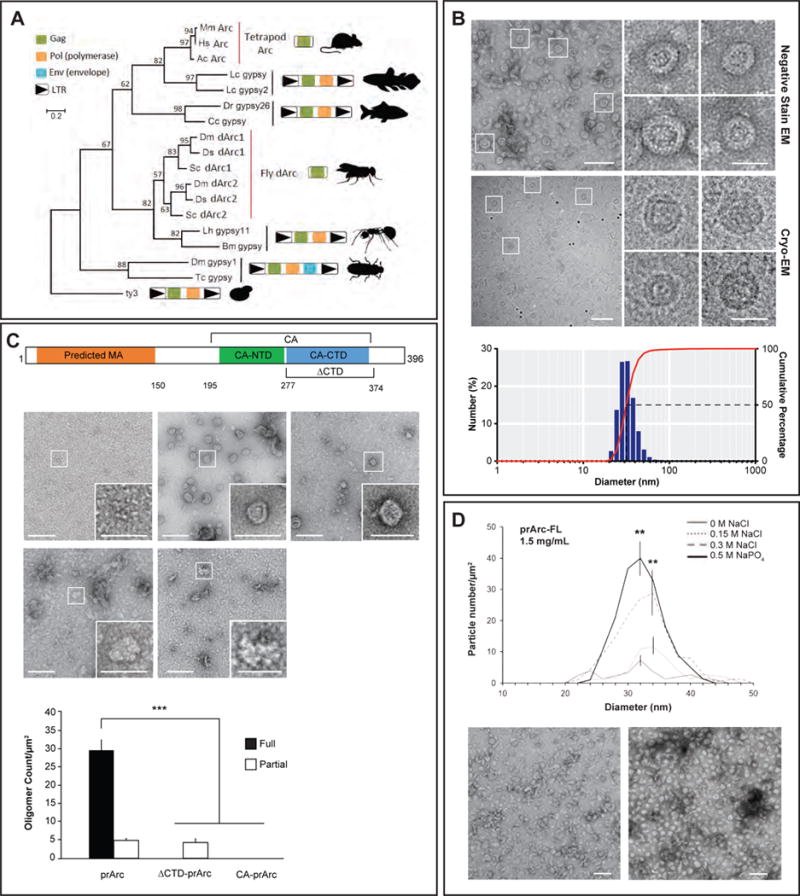
(A) Maximum likelihood phylogeny based on an amino acid alignment of tetrapod Arc, fly dArc1, and Gag sequences from related Ty3/gypsy retrotransposons. Schematics of Gag-only Arc genes and Ty3/gypsy elements are included to the right of the tree. In lineages without Arc genes, the most closely related sequences to Arc are Gag-pol poly-proteins flanked by long terminal repeats (LTRs) as expected in bona fide Ty3/gypsy retrotransposons.
(B) (top) Representative negative stain EM images of full-length purified rat Arc (prArc) protein (1mg/mL, 42,000x). (i–iv) Magnified view of boxed particles. Scale bars=30nm. Representative cryo-EM images of prArc (2mg/mL, 62,000x). (v–vii) Magnified images of Arc capsids showing the double-layered capsid shell. Scale bars=30nm. (bottom) Dynamic light scattering analysis of prArc capsids. The weighted size distribution profile is represented as a histogram of the number of particles.
(C) Schematic of Arc protein with the predicted matrix (MA) (orange), CA-NTD (green), and CA-CTD (blue) domains. Also depicted: ΔCTD deletion mutant and the CA domain constructs. Representative negative stain EM images of purified GST, prArc, the Drosophila Arc homologue dArc1, prArc-ΔCTD, and CA-prArc (all 1mg/mL, 20,000x). Inset scale bars=50nm. (bottom) Quantification of capsid formation. Fully formed capsids include spherical particles that are between 20–60nm and have clear double shells, while partially formed capsids do not have clear double shells (scale bars=100nm). Data is the average of 3 independent experiments±SEM using 3 different prArc preparations. ***p<0.001, two-way ANOVA with post hoc t-tests.
(D) (top) To determine properties of Arc capsid stability, prArc was exchanged into buffers with increasing molar concentrations of salt and examined by negative stain EM. Arc capsids were counted manually and quantified in each buffer condition at a protein concentration of 1.5mg/mL. Data is the average of 3 independent experiments±SEM using different prArc preparations. **p<0.01, Student’s t-test. (bottom) Representative EM images of prArc under 0M NaCl and 0.5M NaPO4 conditions.
Arc Proteins Self-assemble into Virus-like Capsids
Ty3 retrotransposons can form oligomeric particles that resemble retroviral capsids (Hansen et al., 1992), and Arc also has a propensity to oligomerize (Myrum et al., 2015). Retroviral capsid formation is essential for infectivity and is primarily mediated by the Gag polyprotein, which in HIV contains four main functional domains: matrix/MA, capsid/CA, nucleocapsid/NC, and p6 (Freed, 2015). Arc has both primary sequence (Campillos et al., 2006) and structural similarity to CA of HIV and Foamy Virus Gag polyproteins (Taylor et al., 2017; Zhang et al., 2015), suggesting that Arc may share functional similarities to Gag proteins. To characterize the biochemical properties of Arc protein, we expressed rat Arc in bacteria as a Glutathione S-transferase (GST) fusion protein. The expressed protein was purified by affinity and size exclusion chromatography, and the GST tag was removed by proteolysis (Figure S2A, B). Purified preparations of rat Arc (prArc) were analyzed using negative stain electron microscopy (EM) and cryo-EM. These experiments revealed that prArc spontaneously forms oligomeric structures that resemble virus-like capsids (Figure 1B). prArc capsids exhibited a double-shell structure with a mean diameter of 32±0.2nm. Similarly, bacterially-expressed and purified dArc1 (Figure S2C), the Drosophila Arc homologue, also self-assembled into capsid-like structures (Figure 1C). Purified Arc protein that was expressed in an insect cell expression system also assembled into similar virus-like capsids (data not shown), indicating that oligomerization was not an artifact of bacterial expression. Immature retroviral capsids are formed by the uncleaved Gag polyprotein, and the major stabilizing interactions are made by the C-terminal domain (CTD) of the CA region (Mattei et al., 2016). To test whether the putative Arc CA CTD is also required for self-assembly, we expressed and purified a rat Arc mutant protein that lacked this domain (prArc-ΔCTD, missing aa277–374, Figure 1C and S2A, B) (Zhang et al., 2015). EM analyses revealed that prArc-ΔCTD was unable to form double-shelled capsids, although intermediate irregular structures were occasionally observed (Figure 1C). To test whether the Arc CA domain was sufficient for capsid assembly we created a mutant Arc protein that contained aa195–364 (CA-prArc, Figure 1C and Figure S2A). CA-prArc was not sufficient to form capsid-like structures. Arc capsids exhibit other properties similar to HIV capsids, including sensitivity to salt and phosphate levels (Purdy et al., 2008); increasing concentrations of NaCl from 0mM to 300mM resulted in stable prArc capsids and high NaPO4 further stabilized capsid formation (Figure 1D).
To test whether Arc forms oligomers in cells, we expressed Arc in HEK293 cells, which lack endogenous Arc, and performed chemical crosslinking to test for the presence of oligomeric species. Arc proteins crosslinked in situ formed higher molecular weight species with the SDS-PAGE mobility expected for dimer and trimer subunits (Figure S2D), which is reminiscent of HIV Gag subunits using a similar crosslinking assay (Campbell and Rein, 1999). In contrast, transfected GFP did not form higher molecular weight crosslinks under the same conditions.
Arc Binds and Encapsulates RNA
Retroviral encapsulation of viral genomic RNA is a complex process mediated by a network of interactions between Gag, RNA and lipid membranes (Mailler et al., 2016). HIV Gag contains zinc-finger knuckle motifs in the NC domain that mediate viral RNA binding and selection (Carlson et al., 2016), but in the absence of viral RNA, Gag can also bind cellular mRNAs, which may reflect nonspecific RNA interactions with the basic MA and NC domains (Comas-Garcia et al., 2016). Interestingly, Foamy Virus Gags do not contain zinc-finger domains and bind RNA through C-terminal glycine-arginine-rich patches (Hamann and Lindemann, 2016), indicating that distinct Gag domains from different viral families have evolved to perform similar biochemical processes. Like Foamy Virus Gag, Arc does not appear to contain zinc-finger domains but may bind RNA through ionic interactions in its N-terminus. We observed that prArc appeared to co-purify with RNA or other nucleic acids, as the preparations had a higher A260/280 spectrophotometric ratio than would be expected for a pure recombinant protein (prArc 1.04±0.024; Endophilin3A 0.55±0.006; n=3, p<0.01; Figure S2B). We therefore hypothesized that Arc might bind and encapsulate RNA. To ascertain whether prArc capsids contain mRNA, we determined levels of Arc mRNA and a highly abundant bacterial mRNA, asnA (Zhou et al., 2011), using qRT-PCR. We detected both Arc and asnA mRNA (Figure 2A). However, Arc mRNA levels were ~10-fold higher than asnA. Bacterial cell lysate contained ~15-fold higher Arc mRNA levels than asnA (Figure 2A), suggesting that prArc capsids show little specificity for a particular mRNA, but encapsulate abundant RNA according to stoichiometry. If mRNA is encapsulated in capsids, it should be resistant to ribonuclease (RNase) treatment. RNase did not degrade Arc or asnA mRNA, but significantly degraded exogenous free GFP mRNA (Figure 2B), indicating that Arc and asnA mRNA were protected from RNase degradation.
Figure 2. Arc protein interacts with mRNA.
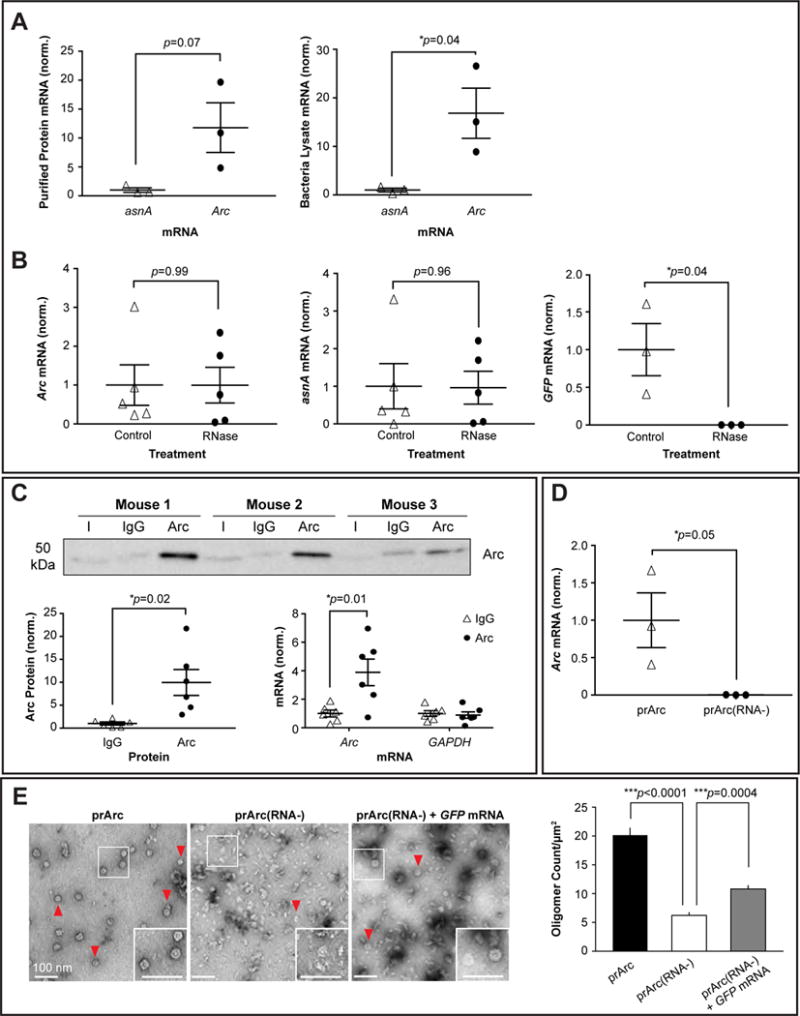
(A) (left) qRT-PCR of Arc mRNA and the bacterial mRNA asnA from prArc. (right) qRT-PCR of Arc and asnA mRNA from total bacteria lysate. Data presented as the mean±SEM normalized to the average of the asnA group (Student’s t-test, n=3 independent protein preparations, *p<0.05).
(B) Protein preparations were treated with or without RNase A for 15 min and qRT-PCR was performed. RNase treatment did not affect Arc and asnA mRNA levels (paired t-test, n=5 independent protein samples), but significantly degraded exogenous/free GFP mRNA (paired t-test, n=3 independent samples, *p<0.05). Data presented as ±SEM normalized to the average of the untreated group.
(C) (top) Representative Western blots of Arc protein that was immunoprecipitated (IP) from WT mouse cortical tissue using an Arc or IgG antibody. Input (I)=10% total lysate. (bottom, left) Quantification of Arc protein IP, showing significant enrichment of Arc protein using an Arc antibody. (bottom, right) qRT-PCR was performed on the eluted fractions from the IP. Arc mRNA was specifically pulled down in the IP (two-way ANOVA with repeated measures and Sidak’s multiple comparisons: Arc+Arc vs. Arc+IgG, p=0.01; Arc+Arc vs. GAPDH+Arc, p=0.013; Arc+Arc vs. GAPDH+IgG, p=0.011). Data presented as the mean±SEM normalized to the average of the IgG group.
(D) qRT-PCR of Arc mRNA from prArc and prArc(RNA−). There was significantly less Arc mRNA in the prArc(RNA−) preparations. Presented as the mean±SEM normalized to the average of the prArc group (Student’s t-test, n=3 independent samples, *p=0.05).
(E) (left) Representative negative stain EM images of prArc, prArc(RNA−), and prArc(RNA−) incubated with 7.3% (w/w) GFP mRNA at RT for 2h (0.25mg/mL, 15,000x). Fully formed capsids are indicated by red arrows (scale bars=100nm). (right) Capsids were quantified as in Figure 1C. Data is presented as the average of 6 images from each condition ±SEM. ***p<0.001, unpaired t-test.
We tested whether Arc protein associates with Arc mRNA in vivo by immunoprecipitating Arc protein from mouse cortical lysate, followed by qRT-PCR (Figure 2C). Arc mRNA was found to selectively immunoprecipitate (IP) with Arc protein, while GAPDH was not enriched in Arc IPs. These results suggest that Arc protein and its mRNA form a complex in neurons in vivo.
Arc Capsid Assembly Requires RNA
To form the immature viral capsid, HIV Gag must bind RNA (Mailler et al., 2016). To test whether Arc capsid formation requires RNA, we purified full-length Arc protein as above and then stripped bound nucleic acids (“prArc(RNA−)”, Figure S3A) as previously performed on HIV Gag (Ganser et al., 1999). This procedure reduced the A260/280 ratio significantly (prArc(RNA−) 0.68±0.03, prArc 1.04±0.024; n=3, p<0.05) and we were unable to detect Arc mRNA association by qRT-PCR (Figure 2D). Stripping RNA resulted in significantly fewer fully-formed capsids (Figure 2E), suggesting that Arc capsids require RNA for normal assembly. To show directly that RNA facilitated Arc capsid assembly, we exogenously added GFP mRNA to prArc(RNA−) (7.3% w/w), which resulted in significantly more fully-formed Arc capsids.
Arc Protein and Arc mRNA are Released by Neurons in Extracellular Vesicles
Retroviral capsids and EVs are released from cells using similar cellular machinery, such as the MVB pathway (Nolte-’t Hoen et al., 2016). Since Arc exhibits many of the biochemical properties of a viral Gag protein, we tested whether Arc protein might also be released from cells. We harvested media from Arc-transfected HEK293 cells and purified the EV fraction. This fraction contained vesicular structures that were <100nm and resembled exosomes (Figure S3B). Arc protein was detected in the EV fraction, which was also positive for the EV marker ALIX, but lacked actin (Figure 3A). Conversely, Arc-ΔCTD-transfected HEK cells exhibited little expression in the EV fraction (Figure 3B), suggesting that proper Arc capsid assembly may be required for Arc release via EVs. We performed qRT-PCR on the EV fraction from HEK cell media and detected Arc mRNA that was resistant to RNase treatment (Figure 3C).
Figure 3. Arc is released from cells in extracellular vesicles.
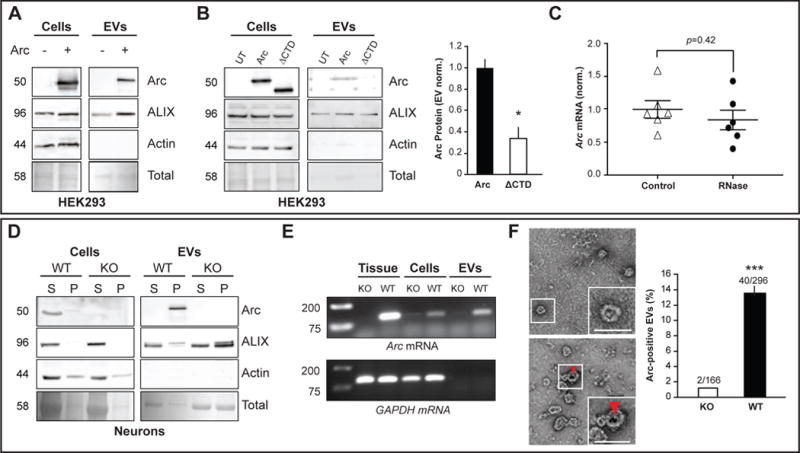
(A) HEK cells in 10-cm dishes were transfected with full-length rat WT myc-Arc and media collected 24h later. Representative Western blots (n=3 independent experiments) show Arc protein in total cell lysates (cells) and the EV fraction purified from cell media in Arc transfected (+) and untransfected (−) cells. ALIX was used as an EV fraction marker. Ponceau stain was used to visualize the total amount of protein in each lane.
(B) HEK293 cells were transfected with myc-Arc-WT or myc-Arc-ΔCTD and media collected 24h later. Representative Western blots (n=3 independent experiments) show Arc protein in total cell lysates (cells) and the EV fraction from cell media. Arc levels in the EV fraction were normalized to Arc protein levels in the cell lysate for each experiment and data is presented normalized to WT levels (n=3). *p<0.05, Student’s t-test.
(C) HEK EV fractions were untreated (control) or treated with RNase (n=6 independent cultures) prior to RNA extraction. qRT-PCR was used to measure Arc mRNA levels and data is presented as the mean±SEM normalized to the average of the untreated group. Paired t-test.
(D) Media was harvested from DIV15 cultured cortical neurons obtained from WT and Arc KO mice after 24h incubation and the EV fraction was purified from collected media. Blots indicate levels of Arc, ALIX, and actin from supernatant (S)/soluble fraction and pellet (P)/insoluble fraction for total cellular lysate (cells). (S)/last wash of the ultracentrifugation purification protocol and final pellet (P)/EV fraction for purified EV fraction (EVs). 2.5% of S and P were loaded for cellular lysates. 5% of S and P were loaded for the EV fraction.
(E) RT-PCR using Arc and GAPDH primers was performed on WT or KO mouse cortical tissue, mouse cortical DIV15 WT or KO neurons (cells), and EVs purified from media collected from WT or KO cultured neurons. Arc mRNA was present in all three preparations, while GAPDH mRNA was absent from EVs.
(F) (top) Immunogold labeling for Arc in EVs obtained from the same Arc KO or WT cultured neuronal media in (D). Red arrow indicates a 10nm immunogold particle (20,000x). (bottom) Quantification of EVs (vesicular structures <100nm) that were Arc-positive±SEM, using immunogold labeling (n=3 independent experiments/EV preparations). ***p<0.001, Student’s t-test.
Native Arc protein was also found in the EV fraction prepared from media harvested from DIV15 cultured cortical mouse neurons (Figure 3D). Since Arc mRNA associates with Arc protein in brain lysate, we used RT-PCR to show that Arc mRNA is also present in EVs purified from neurons (Figure 3E). Arc protein in EVs was resistant to trypsin digestion (Figure S3C), indicating that Arc protein and RNA was protected or bound in a complex within EVs. To directly determine whether Arc protein is present in EVs, we conducted immunogold-labeling of endogenous Arc in the EV fraction from cultured neurons and found that Arc is present in a subpopulation of EVs (Figure 3F). To test whether Arc release in EVs is activity-dependent, we purified the EV fraction from media collected from untreated or KCl-treated WT cultured cortical neurons (Figure S3D). KCl treatment, which increases neuronal activity, resulted in significantly more Arc released into the media.
Arc Mediates Intercellular Transfer of mRNA in Extracellular Vesicles
Virus particles are able to infect cells through complex interactions of the viral envelope and host cell membrane, while EVs can also transfer cargo such as RNAs cell-to-cell (Valadi et al., 2007). We predicted that Arc might be able to transfer mRNA, either directly via mRNA encapsulated in prArc or in Arc-containing EVs. We transfected GFP/myc-Arc or nuclear-GFP into HEK (donor) cells and collected media from these cells after 18h, which was then incubated with untransfected, naïve HEK (recipient/”transferred”) cells for 24h. We observed high Arc expression in a sparse population of naïve HEK cells (Figure 4A), while cells incubated with media from cells transfected with nuclear-GFP alone did not express nuclear-GFP. Fluorescent in situ hybridization (FISH) for Arc mRNA revealed high levels of Arc mRNA in recipient cells. Uptake of Arc protein and mRNA was endocytosis-dependent, as application of Dynasore (a potent inhibitor of clathrin-dependent endocytosis (Macia et al., 2006)) significantly blocked transfer of Arc protein (Figure S4A). Since encapsulation of RNA by Arc capsids is nonspecific in vitro, we tested whether Arc could co-transfer highly abundant mRNAs. Donor HEK cells were transfected with myc-Arc and/or a membrane-bound GFP (mGFP) and media was collected after 24h. Recipient HEK cells showed clear transfer of both GFP protein and mRNA when donor cells contained Arc (Figure 4B). No transfer was observed from cells transfected only with mGFP. This data suggests that Arc EVs released from HEK cells are capable of transferring highly abundant mRNAs cell-to-cell.
Figure 4. Arc extracellular vesicles mediate intercellular transfer of protein and mRNA in HEK293 cells.
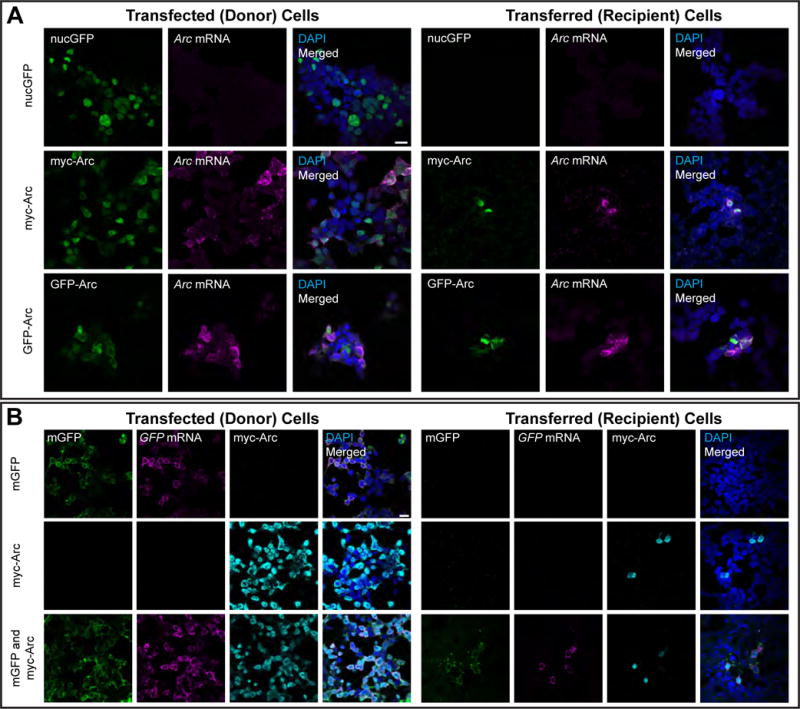
(A) Donor HEK cells in 10-cm dishes were transfected with GFP-Arc, myc-Arc, or nuclear GFP (nucGFP) for 6h. Culture media containing plasmid DNA and transfection reagents was then removed and replaced with fresh culture media. 18h later, this media was removed and used to replace media on naïve recipient HEK cells on coverslips in 12-well plates. 24h later, these cells were fixed and combined FISH for Arc mRNA and ICC for Arc protein was performed. (left) Representative images of HEK cells grown on coverslips and transfected with the same protocol as in 10-cm dishes, showing Arc protein (ICC) and Arc mRNA (FISH). (Right) Representative images of recipient HEK cells showing Arc mRNA and protein were present in cells that received media from GFP-Arc- and myc-Arc-transfected cells, but not nucGFP-transfected cells. Scale bar=20μm. Representative of 7 independent experiments and cultures. (B) Donor HEK cells in 10-cm dishes were transfected as in (A) with membrane GFP (mGFP), myc-Arc, or both constructs together. The media was replaced after 6h, and 18h later, transferred to naïve recipient HEK cells in 12-well plates. 24h later, cells were fixed and combined FISH/ICC for GFP mRNA and Arc protein was performed. (left) Representative images of transfected HEK cells grown on coverslips, showing mGFP fluorescence, Arc protein and GFP mRNA. (right) Representative images of recipient HEK cells that show co-transfer of GFP protein and mRNA with Arc protein. No GFP transfer was observed in the mGFP only group. Scale bar=20μm. Representative of 3 independent experiments and cultures.
To test whether Arc capsids can transfer Arc mRNA into neurons, we incubated cultured hippocampal neurons from Arc KO mice with prArc. Since the Arc KO line contains GFP knocked into the Arc locus (Wang et al., 2006), we imaged Arc in the red channel and were unable to detect GFP fluorescence in the green channel (Figure S4B). We observed uptake of Arc protein into KO neurons above antibody background levels (see Figure S4C for antibody specificity) within 1h of protein incubation, which peaked around 4h of incubation (Figure 5A). To directly determine whether Arc capsids can transfer Arc mRNA into neurons, we measured Arc mRNA levels in Arc KO neurons incubated with prArc. Arc FISH showed robust and high levels of transferred Arc mRNA after 4h of incubation with prArc (Figure 5B). RNase treatment of prArc prior to incubation had no effect on mRNA transfer (Figure S5A), further suggesting that Arc capsids are able to protect and encapsulate Arc mRNA. Blocking endocytosis using Dynasore prevented uptake of both prArc protein and Arc mRNA (Figure S5B). Transferred mRNA and protein were evident both in early endosomes (marked by Rab5) and non-endosome compartments in dendrites (Figure S5C). Both uptake and transfer of purified prArc-ΔCTD and CA-prArc protein and mRNA was significantly less than the full-length protein, indicating that capsid formation is required for uptake into neurons (Figure 5C, D). Lack of protein uptake was not due to poor detection by the custom-made Arc polyclonal antibody (Figure S5D). Strikingly, prArc(RNA−) was unable to be taken up but instead coated the outside of neurons (Figure S6), further suggesting that intact Arc capsids are required for uptake and transfer.
Figure 5. Arc capsids transfer Arc mRNA into neurons.
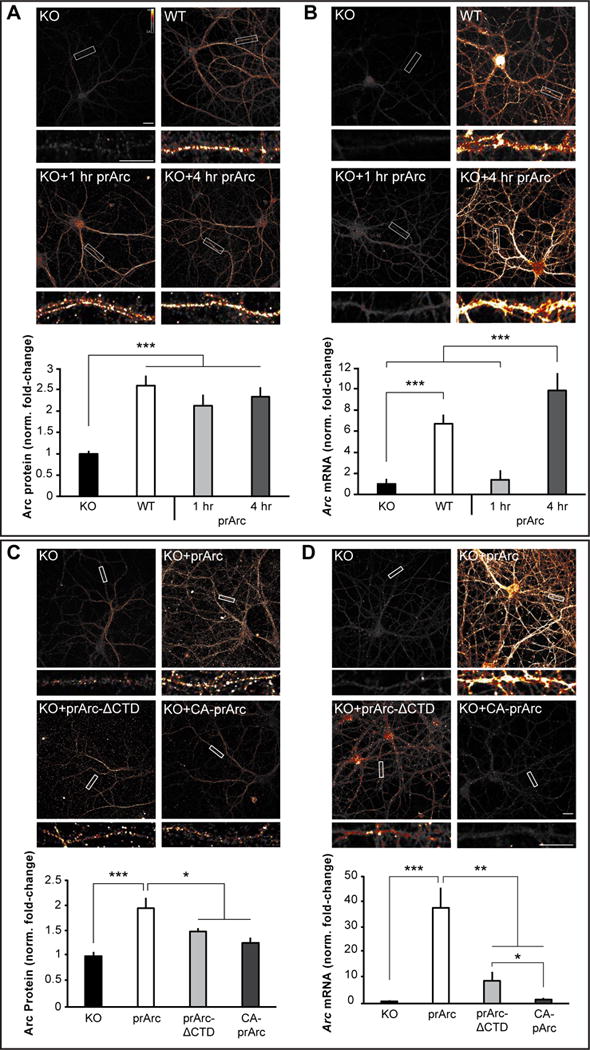
(A) Representative images of Arc ICC from DIV15 cultured hippocampal Arc KO neurons treated for 1 or 4h with 4μg prArc or WT control neurons. prArc-treated neurons showed increased dendritic Arc levels in untreated KO neurons. (B) Neurons were treated as in (A); representative images of Arc mRNA (FISH) are shown. 4h of prArc treatment significantly increased dendritic Arc mRNA levels. (C) Representative images of Arc ICC from DIV15 cultured hippocampal KO neurons treated with 4μg prArc, prArc-ΔCTD, or CA-prArc for 4h. KO neurons treated with prArc-ΔCTD and CA-prArc showed lower levels of Arc protein than prArc. (D) Neurons were treated as in (C); representative images of Arc mRNA are shown. Neurons treated with prArc-ΔCTD and CA-prArc showed lower levels of Arc mRNA than prArc. Dendritic segments boxed in white are shown magnified beneath each corresponding image. 30-μm segments of two dendrites/neuron were analyzed for integrated density measurements in all groups (n=10 neurons). Arc mRNA and Arc protein levels were normalized to untreated KO neurons and displayed as fold-change±SEM. Student’s t-test: *p<0.05. **p<0.01. ***p<0.001. Scale bars=10μm. Images are false-colored with the Smart LUT from ImageJ. All data are representative of 3–7 independent experiments using different protein preparations and cultures.
To test whether endogenous Arc can transfer mRNA, we incubated Arc KO cultured hippocampal neurons with purified EVs prepared from media from WT or KO cortical neurons. Arc KO neurons incubated with WT EVs showed a clear increase in dendritic Arc levels, while KO neurons incubated with EVs derived from KO cells exhibited no increase in dendritic Arc levels (Figure 6A). In addition, FISH showed that Arc mRNA in WT EVs was transferred into KO neurons (Figure 6B). Uptake of Arc mRNA was not significantly affected by prior treatment of EVs with RNase (Figure S7A), indicating that uptake was not due to free or unbound Arc mRNA in the EV fraction. Blocking endocytosis with Dynasore prevented the uptake of Arc protein and mRNA from EVs (Figure S7B). Notably, transferred Arc mRNA expression exhibited cell-wide localization in both early endosomes and non-endosome compartments (Figure S7C) and was virtually indistinguishable from Arc mRNA distribution in WT neurons. These data indicate that endogenous Arc released via EVs is able to transfer Arc mRNA neuron-to-neuron.
Figure 6. Endogenous Arc transfers Arc mRNA into neurons via extracellular vesicles.
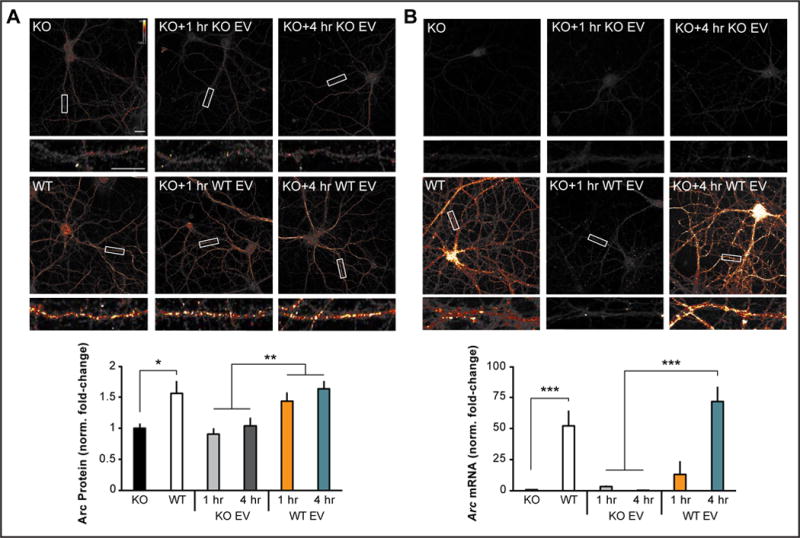
(A) Representative images of Arc ICC from DIV15 cultured hippocampal Arc KO neurons treated for 1 or 4h with 10μg of the EV fraction prepared from 10-cm dishes of DIV15 high-density cortical WT or Arc KO neurons. 1 and 4h treatment with KO EVs did not increase dendritic Arc levels, whereas 1 and 4h of treatment with WT EVs significantly increased dendritic Arc protein levels. (B) Neurons were treated as in (A); representative images of Arc mRNA (FISH) are shown. 1 and 4h treatment with KO EV did not increase dendritic Arc mRNA levels. 1h of treatment with WT EV did not significantly increase dendritic Arc levels, while 4h increased dendritic Arc mRNA levels. 30-μm segments of two dendrites/neuron were analyzed for integrated density measurements in all groups (n=10 neurons). Arc mRNA and Arc protein levels were normalized to untreated KO neurons and displayed as fold-change±SEM. Student’s t-test: *p<0.05. **p<0.01. ***p<0.001. Scale bars=10μm. Representative of 6 independent experiments using different EV preparations and cultures.
Transferred Arc mRNA Can Undergo Activity-Dependent Translation
If Arc mRNA associated with Arc capsids is transferred into the cytoplasm of neurons, we predicted that we would observe an increase in dendritic Arc protein by inducing translation of Arc mRNA through activation of the group 1 metabotropic glutamate receptor (mGluR1/5) by the agonist DHPG, as previously shown for endogenous Arc (Waung et al., 2008). As predicted, Arc protein levels were significantly increased in dendrites of Arc KO neurons after DHPG (5 min; 100μM) application in cells incubated with prArc (Figure 7A). This increase was not evident if a protein synthesis inhibitor (cycloheximide; 180μM) was applied prior to DHPG application. KO neurons incubated with WT EVs for 4h and then treated with DHPG exhibited an increase in dendritic Arc levels that was also dependent on protein synthesis (Figure 7B). Although these experiments cannot definitively distinguish de novo translated Arc from protein that was taken up, these data suggest that Arc capsids or EVs are capable of transferring Arc mRNA between neurons and that this mRNA is available in the cytoplasm of dendrites for activity-dependent translation.
Figure 7. Arc capsid- and EV-transferred Arc mRNA is accessible for activity-dependent translation.
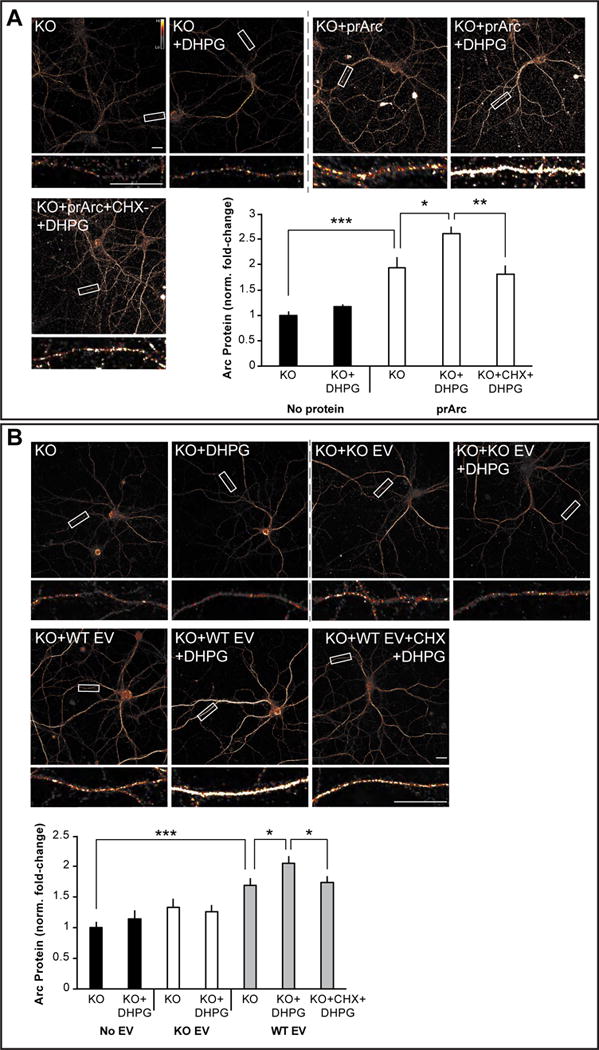
(A) Representative images of Arc ICC from DIV15 cultured hippocampal Arc KO neurons treated for 4h with 4μg prArc. To induce translation of Arc mRNA, 30 min prior to fixation, neurons were treated with the mGluR1/5 agonist DHPG (100μM) for 5 min and then washed out. 1h prior to fixation, a subset of neurons was pretreated with cycloheximide (CHX; 180μM) to block protein translation. prArc significantly increased dendritic Arc expression in KO neurons and DHPG treatment further increased dendritic Arc levels, which was blocked by pretreatment with CHX. DHPG had no effect on untreated KO neurons. (B) Representative images of Arc ICC from DIV15 hippocampal Arc KO neurons treated for 4h with 10μg of the EV fraction prepared from 10-cm dishes of DIV15 high density cortical WT or Arc KO neurons. A subset of neurons was treated with DHPG and CHX as in (A). WT EVs significantly increased dendritic Arc expression in KO neurons, while KO EVs had no effect. DHPG treatment had no effect on dendritic Arc expression in untreated KO neurons or KO EV-treated KO neurons. However, DHPG treatment significantly increased dendritic Arc levels in WT EV-treated KO neurons, which was blocked by pretreatment with CHX. 30-μm segments of two dendrites/neuron were analyzed for integrated density measurements in all groups (n=10 neurons). Arc mRNA and Arc protein levels were normalized to untreated KO neurons and displayed as fold-change±SEM. Student’s t-test: *p<0.05. **p<0.01. ***p<0.001. Scale bars=10μm. Representative of 3 independent experiments using different EV/protein preparations and cultures.
DISCUSSION
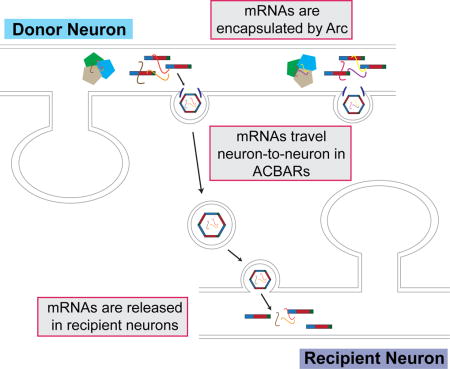
Here we show that mammalian Arc protein exhibits many hallmarks of Gag proteins encoded by retroviruses and retrotransposons: self-assembly into capsids, RNA encapsulation, release in EVs, and intercellular transmission of RNA. These data suggest that Arc can mediate intercellular trafficking of mRNA via Arc EVs (which we term ACBARs for Arc Capsids Bearing Any RNA), revealing a novel molecular mechanism by which genetic information may be transferred between neurons.
Arc Functions as a Repurposed Gag Protein
Our data show a remarkable conservation of viral Gag properties in Arc. Since Arc shows structural homology to the Gag CA domain (Zhang et al., 2015), the capability of self-assembly into oligomeric capsids is perhaps not too surprising. However, Arc seems to retain other important biochemical properties of Gag that are not intuitive from its sequence. Despite lacking clear zinc-finger RNA binding domains such as in HIV Gag, Arc encapsulates RNA, and RNA binding seems critical for capsid formation. This is reminiscent of Foamy Virus Gags, which have evolved different RNA-binding motifs to HIV Gag (Hamann and Lindemann, 2016) and also structurally resemble Arc (Taylor et al., 2017). HIV Gag-RNA interactions are complex and involve multiple components of Gag, including the MA domain, and are regulated by host cellular factors (Mailler et al., 2016). Gag MA-RNA interactions are also critical for virus particle formation at membranes (Kutluay et al., 2014). Moreover, if viral RNA is not present, Gag encapsulates host RNA, and any single-stranded nucleic acid longer than ~20–30 nucleotides can support capsid assembly (Campbell and Rein, 1999), indicating a general propensity to bind abundant RNA. Indeed, precisely how viral RNA is preferentially packaged into Gag capsids in cells remains an intensive area of investigation (Comas-Garcia et al., 2016).
The uptake and transfer of RNA by purified Arc protein is surprising as this occurs in the absence of an “envelope” or lipid bilayer. Uptake of both purified Arc capsids and endogenous EVs occurs through endocytosis. While EVs and exosomes are easily taken up through the endosomal pathway, it remains unclear how RNA can cross the endosomal membrane without membrane fusion proteins (Tkach and Thery, 2016). Our data suggests that, like non-enveloped viruses, Arc protein itself contains the ability to transfer RNA across the endosomal membrane. While it remains unclear how non-enveloped capsids transfer RNA into the cytoplasm, some studies suggest this could occur through specific receptor-capsid interactions, or via a pH-dependent conformational change of the capsid that allows either pore formation or lytic degradation of membranes (Tsai, 2007). We speculate that Arc protein may interact with the endosomal membrane to allow transfer of mRNA into the cytoplasm as the capsid is disassembled. This is reflected in the lag between protein uptake and mRNA expression seen in our experiments, which may be a result of the time it takes for mRNA to become accessible to our FISH probes. In vivo, the lipid membrane around ACBARs may dictate targeting and uptake, while the Arc capsid within protects and allows transfer of RNA. Intriguingly, prArc that lacks RNA is unable to form capsids that cannot be taken up, suggesting uptake may be a regulated process that requires properly-formed capsids. Since Arc seems to regulate a naturally occurring mechanism of RNA transfer, we believe that harnessing this pathway may allow new means of genetic engineering or RNA delivery into cells, using ACBARs, that may avoid the hurdle of immune activation.
Arc’s Gag Homology Reveals a New Signaling Pathway in Neurons
Exosome and EV signaling has emerged as a critical mechanism of intercellular communication, especially in the immune system and in cancer biology (Becker et al., 2016). However, the role of intercellular signaling through EVs in the nervous system has only recently been investigated, with studies suggesting that these pathways may play important roles in synaptic plasticity (Budnik et al., 2016; Zappulli et al., 2016). Canonical exosomes are formed in MVBs, which are derived from the endosomal pathway and usually require the ESCRT complex to be released (Raposo and Stoorvogel, 2013), although the biogenesis of EVs in general is more varied. HIV Gag is able to form virions independent of the MVB pathway, although the ESCRT machinery is still required for particle release; thus, Arc may form ACBARs independent of the canonical exosome pathway. These pathways are not mutually exclusive and elucidating the biogenesis of ACBARs within neurons will require further investigation.
Since Arc is rapidly synthesized locally in dendrites (Park et al., 2008; Waung et al., 2008), it is conceivable that high local concentrations of Arc protein promote capsid assembly in dendrites where encapsulation of dendritically-localized mRNAs could occur. Since Arc capsids do not seem to show specificity in RNA binding in vitro and Arc EVs can transfer highly abundant mRNAs, we speculate that the specificity of ACBAR cargo is conferred by the precise spatial and temporal expression of Arc protein in neurons (Figure S7D). Consistent with the identification of Arc mRNA associated with Arc protein from brain, Arc mRNA levels are highly and uniquely abundant in dendrites in vivo after bouts of neuronal activity or experience (de Solis et al., 2017). Gag-RNA interactions are regulated by host cellular proteins such as Staufen (Mouland et al., 2000), a protein that is also a critical regulator of dendritic mRNA trafficking in neurons, including Arc mRNA (Heraud-Farlow and Kiebler, 2014). The parallels between dendritic mRNA regulation and virus-RNA interactions are striking, suggesting that cellular factors may play an important role in ACBAR biogenesis and RNA packing. Many questions remain: What other cargo do ACBARs contain? What are the docking mechanisms for ACBARs? Is there spatial/temporal specificity of intercellular signaling in the brain?
Our data also indicates that Arc may mediate intercellular signaling to control synaptic function and plasticity in a non-cell autonomous manner. Although there is a paucity of data on neuronal EVs, previous studies have shown that EVs can be secreted in an activity-dependent manner and include AMPARs as cargo (Faure et al., 2006). Since Arc has previously been implicated in AMPAR trafficking at synapses and spine elimination (Chowdhury et al., 2006; Mikuni et al., 2013) at weak synapses (Okuno et al., 2012), a potential role for ACBARs may be to eliminate synaptic material. Arc also regulates homeostatic forms of plasticity such as AMPAR scaling (Shepherd et al., 2006) and cross-modal plasticity across different brain regions (Kraft et al., 2017), which could be regulated at the circuit level in a non-cell autonomous manner. We favor the idea that released Arc functions to carry intercellular cargo that alters the state of neighboring cells required for cellular consolidation of information.
Previous studies have shown that Drosophila neuromuscular junction plasticity requires trans-synaptic signaling mediated through the Wnt pathway in exosomes (Korkut et al., 2009). Interestingly, the Drosophila Arc homologue dArc1 exhibits similar properties of intercellular transfer of mRNA in the fly nervous system and is one of the most abundant proteins in Drosophila EVs (Lefebvre et al., 2016), suggesting a remarkable convergence of biology despite a large evolutionary divergence of these species. A recent study has also implicated Arc in the mammalian immune system (Ufer et al., 2016), where it controls dendritic cell-dependent T-cell activation, expanding the potential repertoire and importance of Arc-dependent intercellular signaling beyond the nervous system. Moreover, EVs have been implicated in the pathology of various neurodegenerative disorders, as several pathogenic proteins such as prions, β-amyloid peptide, and α-synuclein are released from cells in association with EVs (Zappulli et al., 2016). In Alzheimer’s disease (AD), immunohistochemical analysis in brain sections from patients with AD showed enrichment of the exosomal marker ALIX around neuritic plaques (Rajendran et al., 2006). This suggests that EVs may provide a significant source of extracellular Aβ peptide. Arc regulates the activity-dependent cleavage of APP and β-amyloid production through interactions with presenilin (Wu et al., 2011), suggesting that ACBARs may also be involved in Alzheimer’s disease pathogenesis.
Evolution of synaptic plasticity and cognition
Ty3/gypsy retrotransposons are ancient mobile elements that are widely distributed and often abundant in eukaryotic genomes and are considered ancestral to modern retroviruses (Malik et al., 2000). There is evidence that coding sequences derived from Ty3/gypsy and other retroviral-like elements have been repurposed for cellular functions repeatedly during evolution (Feschotte and Gilbert, 2012). For instance, multiple envelope genes of retroviral origins have been co-opted during mammalian evolution to promote cell-cell fusion and syncytiotrophoblast formation in the developing placenta (Cornelis et al., 2015). There are more than one hundred Gag-derived genes in the human genome alone (Campillos et al., 2006), and genetic knockouts of their murine orthologs have revealed that some, like Arc, are essential for cognition (Irie et al., 2015). However, the molecular function of these Gag-derived proteins has been poorly characterized, and whether they were co-opted to serve similar cellular processes remains an open question. This study and the accompanying article now reveal that two distantly related Gag-derived genes have been independently co-opted in fly and tetrapod ancestors to participate in a similar process of EV-dependent intercellular trafficking of RNA in the nervous system. Given the plethora of retroelements populating eukaryotic genomes, we speculate that many other Gag proteins have been repurposed for cellular processes that await discovery in a variety of organisms.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Jason Shepherd (jason.shepherd@neuro.utah.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T cells were purchased from ATCC (#CRL-11268). Cells were maintained at 37°C with 5% CO2 in DMEM media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA) and passaged every 3–4 days at 70% confluency. For transfections and transfer experiments, HEK cells were seeded to 10-cm dishes or collagen-coated glass coverslips in 12-well plates.
Mouse models
Wild-type and Arc knock-out mice
C57BL/6 Arc knock-out (KO) mice (C57BL/6-Arctm1Stl/J, a kind gift from Dr. Kuan Wang, NIH) have GFP knocked in to the Arc ORF (Wang et al., 2006). Arc KO and wild-type (WT) mice used in these studies were littermates from heterozygous (Arc+/−) crosses. Both male and female mice were used. No differences between sexes in the experiments conducted in this study were noted, and data from both sexes were therefore grouped together. Mice were housed in breeding pairs, or group-housed with littermates of the same sex after weaning (2–5 mice/cage), on a 12:12 h day:night cycle, with food and water provided ad libitum. Hippocampal and cortical primary neuron cultures were prepared from E18 embryos, while brain lysates were taken from P30–50 mice. Mice were test- and procedure-naïve before terminal experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Utah.
Primary neuron culture
Primary neuron cultures were prepared from male and female E18 Arc KO or WT mouse cortex and hippocampus as previously described (Shepherd et al., 2006). Tissue was dissociated in DNase (0.01%; Sigma-Aldrich, St. Louis, MO) and papain (0.067%; Worthington Biochemicals, Lakewood, NJ), and then triturated with a fire-polished glass pipette to obtain a single-cell suspension. Cells were pelleted at 500×g for 4 min, the supernatant removed, and cells resuspended and counted with a TC-20 cell counter (Bio-Rad, Hercules, CA). Neurons were plated on glass coverslips (Carolina Biological Supply, Burlington, NC) coated with poly-L-lysine (0.2 mg/mL; Sigma-Aldrich) in 12-well plates (Greiner Bio-One, Monroe, NC) at 90,000 cells/mL, or in 10-cm plastic dishes at 800,000 cells/mL. Neurons were initially plated in Neurobasal media containing 5% horse serum, 2% GlutaMAX, 2% B-27, and 1% penicillin/streptomycin (Thermo Fisher Scientific) in a 37°C incubator with 5% CO2. On DIV4, neurons were fed via half media exchange with astrocyte-conditioned Neurobasal media containing 1% horse serum, GlutaMAX, and penicillin/streptomycin, 2% B-27, and 5 μM cytosine β-D-arabinofuranoside (AraC) (Sigma-Aldrich). Neurons were fed with astrocyte-conditioned media every three days thereafter.
METHOD DETAILS
Plasmids
The open reading frame (ORF) of full-length rat Arc (NP_062234.1) cDNA was subcloned from pRK5-myc-Arc. The insert was amplified by PCR, digested with BamH1 and Xho1, and ligated into the pGEX-6p1 (GE Healthcare, Little Chalfont, UK) expression vector between the BamH1 and Xho1 restrictions sites. The GST-Arc ORF was similarly amplified and cloned into the pFastBac1 vector (Thermo Fisher Scientific) between the BamH1 and Xho1 restriction sites. prArc-ΔCTD was generated by blunt end cloning after PCR amplification of the Arc ORF from pGEX-6p1-Arc, excluding sequence coding amino acids 277–374. Amino acids 195–364 of the Arc ORF (CA-prArc) was similarly cloned into the pET11a vector, which contained a His tag. pBluescript-SKII-GFP was generated by restriction digest of mEGFP (BBA16881.1) from pGL4.11-arc7000-mEGFP-ArcUTRs (generously provided by Dr. Haruhiko Bito, University of Tokyo) and subsequent ligation into the KpnI and SacI restriction sites flanking the insert in pBluescript-SKII-ArcUTRs plasmid (generously provided by Dr. Kristen Keefe, University of Utah). The pGEX-4T-1 Drosophila Arc1 (NP_610955.1) construct was provided by Dr. Mark Metzstein, University of Utah. EGFP-C3-Arc and pRK5-myc-Arc were generously provided by Dr. Kimberly Huber (UT Southwestern) and Dr. Paul Worley (Johns Hopkins University), respectively. All protein expression constructs were transformed into DH5α E. coli cells and individual colonies were screened by Sanger Sequencing (GeneWiz, South Plainfield, NJ) sequencing services, using primers synthesized by Integrated DNA Technologies (Coralville, IA). Trace files were analyzed using A Plasmid Editor (APE) freeware available from the University of Utah. Sequenced verified constructs were then transformed into BL21-DE3 bacterial cells for protein expression. See supplemental table 1 for specific oligo primer sequences.
Protein purification
Starter bacteria cultures for protein expression were grown overnight at 37°C in LB supplemented with ampicillin and chloramphenicol. Starter cultures were used to inoculate large-scale 500 mL cultures of ZY auto-induction media. Large-scale cultures were grown to OD600 of 0.6–0.8 at 37°C at 150 rpm and then shifted to 19°C at 150 rpm for 16–20 h. Cultures were then pelleted at 5000×g for 15 min at 4°C and cell pellets were resuspended in 30 mL lysis buffer (500 mM NaCl, 50 mM Tris, 5% glycerol, 1 mM DTT, pH 8.0 at room temperature (RT) for Arc constructs and GST; 300 mM KCl, 50 mM Tris, 1% Triton X-100, 1 mM DTT, pH 7.4 at RT for Endophilin3A) and flash frozen in liquid nitrogen. Frozen pellets were thawed quickly at 37°C and brought to a final volume of 1 g pellet:10 mL lysis buffer, supplemented with DNase, lysozyme, aprotinin, leupeptin, PMSF, and pepstatin. Lysates were then sonicated for 8–10×45 s pulses at 90% duty cycle and pelleted for 45 min at 21,000×g. For GST-tagged constructs, cleared supernatants were then passed through a 0.45 μm filter and incubated with pre-equilibrated GST Sepharose 4B affinity resin in a gravity flow column overnight at 4°C. Bound protein was then washed twice with two column volumes (20 resin bed volumes each) of lysis buffer, re-equilibrated with 150 mM NaCl, 50 mM Tris, 1 mM EDTA, 1 mM DTT, pH 7.2 at RT, and cleaved on-resin overnight at 4°C with PreScission Protease (GE Healthcare) for the GST-tagged constructs, or thrombin (Sigma-Aldrich) for dArc1. Cleaved proteins were then buffer exchanged to 150 mM NaCl, 50 mM Tris, pH 7.4 at RT to kill protease activity, run on an S200 size exclusion column to separate the cleaved protein, and peak fractions were pooled. GST was affinity-purified as described above using Sepharose 4B resin and eluted directly using 15 mM reduced L-glutathione, 10 mM Tris, pH 7.4 at RT. His-tagged CA-prArc was affinity-purified as described above using Ni+ resin (Roche, Basel, Switzerland) and eluted directly using 250 mM imidazole, 10 mM Tris, pH 7.4 at RT. GST and CA-prArc were then buffer exchanged to 150 mM NaCl, 50 mM Tris, pH 7.4 at RT. To strip Arc protein of nucleic acids for prArc(RNA−) preparations, cell pellets were lysed in 20 mM NaCl, 50 mM Tris, 5% glycerol, 2 mM MgCl2, 1 mM DTT, pH 8.0 at RT as described above. Nucleic acids were precipitated from cell supernatants by dropwise addition of 10% PEI, pH 8.0 to a final concentration of 0.1% followed by incubation at 4°C for 20 min and pelleting for 20 min at 27,000×g. The resulting supernatant was then precipitated by addition of saturated ammonium sulfate to a final concentration of 30%. Precipitated protein was pelleted at 10,000×g for 10 min, resuspended in 60 mL lysis buffer, and affinity purified. The cleaved affinity-purified product was then dialyzed to Q-column buffer A (Q-A; 20 mM NaCl, 50 mM Tris, pH 7.4 at RT) overnight. Dialyzed protein was then subjected to anion exchange chromatography (HiTrap Q, GE Healthcare) with a gradient of Q-A buffer to Q-B buffer (1 M NaCl, 50 mM Tris, pH 7.4). Average yields for purified proteins were 10.5 mg (8–13 mg) per liter of cell culture.
Electron microscopy
Negative stain
For all negative stain specimens, copper 200-mesh grids coated with Formvar and carbon (Electron Microscopy Sciences or Ted Pella, Redding, CA) were glow discharged for 20–45 s in a vacuum chamber at 30mA. 3.5 μL sample was then applied to the grid for 35–45 s and excess sample was wicked away using filter paper. Grids were then immediately washed 2–4× for 5 s with 30 μL water droplets, then once with 1% uranyl acetate (UA) on parafilm. Excess water/UA was wicked away and then a final droplet of UA was applied for 30 s. Excess UA was wicked away and grids were air dried for 30–60 s. Imaging was performed using either an FEI T12, FEI Tecnai Spirit microscope operated at 120 kV equipped with a Gatan Orius SC200B CCD camera or JEOL 1400 electron microscope.
Cryo-EM
Purified Arc protein was dialysed into 300 mM NaCl, 50 mM Tris, pH 7.4 and concentrated twice using Amicon 100 MWCO centrifugal filters (Millipore, Burlington, MA) to yield a final protein concentration of ~2 mg/mL. 10 nm diameter gold beads were added to the sample. Degassed 2/2-3C C-flat grids (Electron Microscopy Sciences, Hatfield, PA) were glow discharged for 45 s at 30 mA. Sample was applied to the grid 2 times for 30 s, and the grid was plunge frozen in liquid ethane using a FEI Vitrobot Mark IV. Micrographs were acquired using a FEI Tecnai G2 F20 microscope operated at 200 kV, equipped with a FEI Falcon II direct detector. The nominal defocus was 1.3 μm.
EM quantification
Grids were surveyed visually to check for uniformity of sample application. For each experiment, six images were taken from randomly selected grid squares. Full and partially formed particles between 20–40 nm were then counted manually using ImageJ. Counts were then divided by the image field of view (2.07 μm2) and data presented as oligomer count/μm2.
Arc capsid assembly assay:
GFP mRNA was added to prArc(RNA−) (5 mg/mL in low salt buffer: 20 mM NaCl, 50 mM Tris, pH 7.4 at RT) at a nucleic acid:protein ratio of 7.3% (w/w) (corresponding to 1 molecule of Arc to 10 nucleotides). Reactions were then diluted to 1 mg/mL of prArc(RNA−) by dropwise addition of low salt buffer or capsid assembly buffer (500 mM NaPO4, 50 mM Tris, 0.5 mM EDTA, pH 7.5 at RT) and incubated for 2 h at RT. Following incubation, negative stain EM grids were prepared of each reaction at 0.25 mg/mL and capsid formation was quantified by manual counting of 6 images. Fully formed capsids included spherical particles between 20–50 nm with clear double shells. Similar results were seen in three independent protein preparations.
Dynamic light scattering
Purified Arc protein was subjected to dynamic light scattering measurements on a Malvern Zetasizer Nano ZSP instrument. The scattering was carried out at 25°C and at a fixed angle of 173° (backward scattering). The scattered intensity is represented as number of particles under the assumption that the scattering intensity from spherical particles is proportional to the size to the sixth power.
Phylogenetic reconstruction
NCBI genome sequence databases were queried using the human or Drosophila melanogaster Arc protein sequence using tBLASTn. Repbase was also queried using the CENSOR program to identify known repeat families with high sequence similarity to mammalian or brachyceran Arc genes, respectively. The following sequence IDs were used for analysis: (Genbank locus) Mm ARC—AHBB01089569; Hs ARC—LIQK02016549; Ac ARC—AAWZ02020354; Lc gypsy2—AFYH01030203; CC gypsy—LHQP01046008; Dm ARC1—JSAE01000572; Ds ARC1—CAKG01020471; Sc ARC1—LDNW01019671; Dm ARC2—JXOZ01003752; Ds ARC2—AWUT01001000; Sc ARC2—LDNW01019670; Bm gypsy—BABH01046987; Tc gypsy—AAJJ02003810. Repbase: Lc gypsy—Gypsy2-1-I_Lch; Dr gypsy26—Gypsy-26-I_DR; Lh gypsy11—Gypsy-11_LH-I; Dm gypsy1—Gypsy1-I_DM; ty3—TY3. Protein (Arc and Gag) sequences that were found to have high similarity to Arc proteins and Gags of other related Ty3/gypsy elements were aligned using the MUSCLE program. Trimmed Arc/Gag alignments were uploaded to MEGA7 for subsequent maximum likelihood phylogenetic reconstruction using default parameters, and 500 bootstrap iterations were performed to generate a lineage tree. Drosophila melanogaster dArc1 and dArc2 protein sequences were used to query schizophoran fly protein databases using BLASTp. More hits were observed than expected if darc1 were present in one-to-one orthologs in the species examined. Protein FASTA sequences were aligned using MUSCLE and a maximum likelihood phylogram was generated using MEGA.
HEK cell experiments
Transfections
HEK cells were transfected using polyethyleneimine (PEI) at a ratio of 3 μg PEI:1 μg DNA diluted in Opti-MEM (Thermo Fisher Scientific). Cells were transfected at approximately 60–70% confluency. For EV isolation and media transfer experiments, culture media was exchanged 4–6 h post-transfection to remove PEI and DNA, and media was harvested 24 h later.
Transfection and transfer
Media from transfected HEK cells was harvested 24 h post-transfection and centrifuged at 500×g for 4 min to remove dead cells and debris. Media from untransfected, naïve cells was removed and replaced with the cleared transfected media and incubated for an additional 24 h. Following incubation, cells were fixed and combined immunocytochemistry/fluorescence in situ hybridization (ICC/FISH) for Arc or GFP protein and RNA was performed as described below.
Endocytosis blockade
To block endocytosis, a group of naïve HEK cells plated on coverslips in 12-well plates that were receiving media from GFP-Arc-transfected HEK cells were treated at the same time with 80 μM Dynasore (Abcam, Cambridge, MA) for the first 6 h, then the media was removed and replaced with fresh HEK media. 18 h later, Dynasore-treated and untreated HEK cells were fixed. The entire 18-mm coverslip was viewed with a 20x objective and the number of clusters of GFP-Arc-transferred cells was manually counted. Representative images were obtained using a 20X objective on an Olympus FV1000 confocal microscope (Tokyo, Japan).
Neuron transfer experiments
DIV15 cultured neurons were used for all neuronal experiments. For purified Arc protein incubation experiments, neurons were treated with 4 μg of purified prArc, prArc-ΔCTD, CA-prArc, or prArc(RNA−) protein in normal neuronal feeding media and incubated for 1 or 4 h. For extracellular vesicle (EV) incubation experiments, neurons were treated with 10 μg protein from the purified EV fraction obtained from eight 10-cm dishes of DIV15 cultured cortical neurons in which E18 WT cortical neurons had been plated at 800,000 cells/mL (see “Cell Culture” methods), and incubated for 1 or 4 h. A subset of neurons in the purified protein- and EV-treated experiments was treated with 100 μM of the group 1 mGluR agonist dihydroxyphenylglycine ((S)-3,5-DHPG; Tocris Bioscience, Bristol, UK) for 5 min, which was then washed out and replaced with previously conditioned neuronal media, and neurons were allowed to rest for 25 min before fixation. To block protein translation during DHPG treatment, a subset of neurons was pretreated with 180 μM cycloheximide (CHX, Sigma-Aldrich) 30 min before DHPG. CHX was left in the media for 1 h total. To block endocytosis, neurons were pretreated with 80 μM Dynasore (Abcam, Cambridge, MA) for 30 min before adding purified protein . For RNase treatments, a sample of either prArc or WT EV was incubated with RNase A (1:1000; Omega Bio-tek, Norcross, GA) for 15 min, then SUPERase-In RNase Inhibitor (1 U/μL; Thermo Fisher Scientific) immediately before being added to neurons. The treated samples were then added to neurons and incubated for 4 h.
Immunocytochemistry
After treatments, neurons were washed twice with 37°C 4% sucrose/1X phosphate-buffered-saline (PBS; 10X: 1.4 M NaCl, 26.8 mM KCl, 62 mM Na2HPO4, 35.3 mM KH2PO4, pH 7.4), then fixed for 15 min with 4% sucrose/4% formaldehyde (Thermo Fisher Scientific) in 1X PBS. Neurons were washed 3 × 5 min with 1X PBS, permeabilized for 10 min with 0.2% Triton X-100 (Amresco, Solon, OH) in 1X PBS, and blocked for 30 min in 5% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) in 1X PBS. Neurons were then incubated in primary antibody diluted in block for 1 h at RT, washed 3 × 5 min in 1X PBS, and incubated in secondary antibody diluted in block for 1 h at RT. Neurons on coverslips were mounted on glass slides in Fluoromount (Thermo Fisher Scientific) and dried overnight at RT. Primary antibodies used were: rabbit anti-Arc (1:1000; custom-made; ProteinTech, Rosemont, IL); rabbit anti-Arc (1:1000; Synaptic Systems, Goettingen, Germany); chicken anti-MAP2 (1:5000; ab5392; Abcam); mouse anti-Rab5 (1:1000; BD Biosciences, San Jose, CA); DAPI nuclear stain (Molecular Probes, Thermo Fisher Scientific). Secondary antibodies used were: Alexa Fluor 405, 488, 555, or 647 for the appropriate animal host (1:750; Thermo Fisher Scientific or Jackson ImmunoResearch).
Combined FISH/ICC in neurons and HEK cells
The fluorescent in situ hybridization (FISH) procedure for Arc and GFP was based on a previously published protocol (Daberkow et al., 2007). We used a full-length rat Arc ribonucleotide probe (rat and mouse Arc are 99% identical at the amino acid level) or EGFP (see cloning strategy above in “Plasmids”) as in the published protocol, but modified the protocol for use in cultured neurons and HEK cells instead of brain sections. Arc and GFP plasmids were linearized with Not1 and purified via standard phenol/chloroform extraction. The linearized antisense Arc or GFP were used to make a ribonucleotide probe that had DIG-UTP incorporated using a T7 DIG RNA labeling kit (Sigma-Aldrich), then purified with a G-50 spin column (GE Healthcare). Cells were washed once with 37°C 4% sucrose/1X PBS, then fixed for 15 min with 4% sucrose/4% formaldehyde in 1X PBS. Cells were washed 3 × 5 min with 1X PBS, permeabilized in 0.2% Triton X-100 for 10 min, washed 2 × 5 min in 1X PBS, then 5 min with 2X saline-sodium citrate (SSC; 20X: 3 M NaCl, 300 mM citric acid trisodium salt dihydrate, pH 7). Cells were prehybridized in 1X prehybridization solution (Sigma-Aldrich) for 30 min. The DIG-labeled Arc or GFP ribonucleotide probe was diluted 1:3 with ddH2O, denatured at 90°C for 5 min, put on wet ice for 2 min, then mixed with RNA hybridization buffer (23.75 mM Tris-HCl, 1.19 mM EDTA, 357 mM NaCl, 11.9% dextran sulfate, 1.19X Denhardt’s solution (Thermo Fisher Scientific), 2.5% nuclease-free water, 60% formamide (Fisher Scientific, Hampton, NH)). The Arc probe (1:500) or GFP probe (1:750) was hybridized to the cultured cells at 56°C for 16 h. The following day, cells underwent a series of washes to decrease background signal: 3 × 5 min 2X SSC, 15 min in RNase A (1:1000; Omega Bio-tek) at 37°C, 10 min 2X SSC at RT, 10 min 0.2X SSC at RT, 15 min 0.2X SSC at 56°C, 10 min 0.2X SSC at RT, 5 min TNT (0.1 M Tris-HCl, 0.15 M NaCl, 0.05% Tween-20, pH 7.5). Cells were then blocked in TNB (0.1 M Tris-HCl, 0.15 M NaCl, 0.5% w/v blocking reagent (Sigma-Aldrich), pH 7.5) with 2.5% sheep serum (Jackson ImmunoResearch) and 2.5% donkey serum for 30 min. In the primary antibody step, a DIG-HRP (1:1000; Sigma-Aldrich) and either MAP2 (1:2500; Abcam), Arc (1:500; custom-made), or Rab5 (1:500; BD Biosciences) antibody were diluted together in TNB with 2.5% sheep serum and 2.5% donkey serum and incubated on the cells for 1 h. After 3 × 5 min washes in TNT, the DIG-HRP signal was developed using a TSA Plus Cyanine 3 kit (1:50; PerkinElmer, Waltham, MA) for 30 min. Cells were washed for 5 min in TNT and 5 min in 1X PBS, then secondary antibody was diluted 1:750 in 5% donkey serum and 1X PBS and incubated on the cells for 1 h to detect MAP2, Arc, or Rab5. Nuclei were stained with DAPI (Thermo Fisher Scientific), then coverslips were mounted on glass slides with Fluoromount and dried overnight at RT.
Cell imaging and analysis
Imaging
Coverslips were imaged using a 60X oil objective on an Olympus FV1000 confocal microscope (Tokyo, Japan) and images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). Neurons included for analysis were selected in an unbiased manner by looking at MAP2 dendritic morphology for cell health. Coverslips were viewed blind to find the brightest immunofluorescence in each independent experiment, and this value was then used to set the image acquisition settings for that experiment. Images from all coverslips in that experiment were then acquired using the exact same settings.
Analysis of dendritic Arc protein and mRNA expression
During analysis, images were blindly thresholded (to remove background fluorescence and to ensure images were analyzed in the linear range) to the brightest immunofluorescence in an individual experiment, and the same threshold was applied to all other images in that experiment. Integrated density (average pixel intensity × area) of two 30-μm dendritic segments/neuron was measured from each coverslip. In general, thick proximal dendritic branches were avoided in our analysis to control for potential differences in dendritic volume. The KO control group in each experiment, whether ICC or FISH, was set as “1”, and the integrated density values in the other groups were normalized to this and are displayed in the graphs as fold-change ± SEM. For representative images in the figures, the Smart look-up table (LUT) in ImageJ was applied to highlight differences in Arc expression between groups.
Analysis of Arc/Rab5 colocalization
Two 30-μm dendritic segments/neuron were selected for analysis of Arc protein or mRNA colocalization with Rab5 protein. The Arc channel and Rab5 channel were thresholded to the same value across all images. Using ImageJ, a mask was made of the thresholded section of dendrite for both Rab5 and Arc. The Arc mask was applied to the Rab5 mask and the number of overlapping puncta was quantified. The number of Arc particles overlapping Rab5 was divided by the total number of Arc particles in the stretch of dendrite to determine the Arc/Rab5 colocalization.
Western blots
Immunoblotting and analysis
Western blot samples were mixed with 4X Laemlli buffer (40% glycerol, 250 mM Tris, 4% SDS, 50 mM DTT, pH 6.8) and heated at 70°C for 5 min. SDS-P AGE gel electrophoresis was used to separate protein samples. Separated samples were transferred to a nitrocellulose membrane (GE Healthcare). Following transfer, membranes were briefly stained with 0.1% Ponceau stain, then destained with 1% acetic acid to remove background, for imaging of total protein. Membranes were blocked in 5% milk + 1X tris-buffered saline (TBS; 10X: 152.3 mM Tris-HCl, 46.2 mM Tris base, 1.5 M NaCl, pH 7.6) for 30 min at RT, then incubated in primary antibody in 1X TBS for either 1 h at RT or overnight at 4°C. Membranes were washed 3 × 10 m in in 1X TBS, then incubated in an HRP-conjugated secondary antibody (Jackson ImmunoResearch) in block for 1 h at RT. After 3 × 10 min in 1X TBS, a chemiluminescent kit (Bio-Rad, Hercules, CA) was used to detect the protein bands, and the membranes were imaged on an Azure c300 gel dock (Azure Biosystems, Dublin, CA). Blots were analyzed and quantified using the Gel Analysis plugin in ImageJ.
Antibodies
Antibodies were used at the following concentrations: Arc (1:000; mouse monoclonal, Santa Cruz), Arc (1:000; rabbit polyclonal, custom, Protein Tech), ALIX (1:500; rabbit polyclonal, custom, provided by Dr. Wesley Sundquist), actin (1:1000; HRP-conjugated, Abcam), GFP (1:1000; chicken polyclonal, Aves). All secondary antibodies were used at a dilution of 1:10,000 (HRP-conjugated goat anti-rabbit, goat anti-mouse, goat anti-chicken, Jackson ImmunoResearch).
Coomassie gels
Samples for analysis via SDS-PAGE were mixed with 4X Laemlli buffer and heated at 70°C for 5 min. Protein samples were separated on 10% SDS gels. Gels were then stained with 0.1% Coomassie blue stain (0.1% w/v Coomassie blue, 50% methanol, 10% acetic acid, 40% water) for 30 min and destained overnight in destain solution (50% methanol, 10% acetic acid, 40% water). Gels were visualized using an Azure c300 gel dock under the auto-exposure setting on the visible channel. Gel exposures were analyzed and quantified using the Gel Analysis plugin in ImageJ.
Immunoprecipitation
WT and Arc KO cortices were dissected out and homogenized in 150 mM NaCl, 50 mM Tris, 1% Triton X-100, 0.5% sodium deoxycholate, 0.05% SDS, pH 7.4 (IP lysis buffer), with protease inhibitor added fresh (Roche). Homogenates were pelleted at 200×g for 5 min at 4°C to remove tissue debris. Supernatants were removed, diluted from 2 mL to 4 mL, and rocked at 4°C for 10 min before being pelleted at 17,000×g for 10 min at 4°C to remove insoluble material. Cleared supernatants were removed, a small aliquot was taken as the input, and the remainder used for immunoprecipitation. Supernatants were immunoprecipitated with either Arc antibody (rabbit polyclonal, custom-made; Protein Tech) or normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA) at 1 μg/500 μL lysate for 2 h at 4°C with gentle rocking. Following antibody incubation, a 10% volume of washed 50/50 Protein A bead slurry (Thermo Fisher Scientific) was added to the antibody/lysate mixture and incubated for an additional hour at 4°C with rocking. Bead-antibody complexes were then pelleted briefly at low speed, supernatants were removed, and beads were washed three times with IP buffer. Washed beads were then resuspended in 200 μL IP buffer. With half of the bead slurry, protein was eluted from the beads with 17 μL 4X Laemlli buffer for 5 min at RT, then 50 μL IP buffer was added and the solution was removed from the beads into a new tube and heated at 70°C for 5 min. The input (10% lysate volume) and 30 μL each of the IgG and antibody elutions were separated by SDS-PAGE on a 10% acrylamide gel and immunoblotted as described above. The bands for the input and IgG and Arc elutions were analyzed using the Gel Analysis plugin in ImageJ, and the data were represented graphically as a ratio of the signal from each elution over the input signal from each individual mouse. With the other half of the bead slurry, the IP buffer was adjusted to 1% SDS and 0.8 mg Proteinase K (New England Biolabs, Ipswich, MA) was added. Samples were then incubated at RT for 30 min with rocking and total RNA was extracted as described below.
Chemical crosslinking of Arc proteins in situ
Transfected HEK cells expressing myc-Arc-WT or a GFP control were briefly trypsinized, quenched with DMEM (Thermo Fisher Scientific), and pelleted. Media was removed and pelleted cells were then crosslinked with 0.4% formaldehyde in PBS for 10 min with rocking at RT. Cell suspensions were immediately quenched with Tris to a final concentration of 50 mM and repelleted. Supernatants were removed and cell pellets were then lysed with 150 mM NaCl, 50 mM Tris, 1% Triton X-100, pH 7.4 (lysis buffer) for 20 min at 4°C with rocking. Lysates were cleared by centrifugation at 21,000×g for 10 min at 4°C and cleared supernatants were then run on a 4–8% gradient gel and analyzed via Western blot with antibodies for Arc (mouse monoclonal, Santa Cruz) and GFP (chicken polyclonal, Aves).
RNA extraction
For all samples, total RNA was extracted using TRIzol (Thermo Fisher Scientific). TRIzol-extracted samples were mixed 5:1 with chloroform, incubated at RT for 3 min, and pelleted at 12,000×g at 4°C for 10 min. The resulting aqueous phase was taken and mixed 1:1 with isopropanol, incubated at RT, and pelleted at 12,000×g at 4°C for 10 min. The resulting supernatant was removed and pellet washed with cold 75% ethanol. Washed pellets were then repelleted at 7500×g for 5 min at 4°C. The supernatant was removed and dried pellets were resuspended in ddH2O.
RT-PCR
Total RNA concentrations were measured by A260/280 on a Nanodrop (Thermo Scientific). Reverse transcription reactions were carried out using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) with 100–200 ng of RNA as template. Resulting cDNAs were amplified using rat Arc, GAPDH primer sets for 35 cycles with a 60°C annealing temperature. Resulting PCR products were analyzed on 1.5% agarose gels stained with SYBR Safe (Thermo Fisher Scientific). Rat Arc primers: Fwd, ACCATATGACCACCGGCGGC; Rev, TCCAGCATCTCAGCTCGGCAC. GAPDH primers: Fwd, CATGGCCTTCCGTGTTCCTA; Rev, GCCTGCTTCACCACCTTCTT. RT-PCR gels were quantified using the ImageJ gel analyzer tool.
qRT-PCR
To determine the amount of RNA associated with Arc protein, quantitative RT-PCR was performed on mRNA prepared from 1: whole mouse cortices immunoprecipitated with Arc and IgG protein, 2: EV fractions prepared from HEK cells (see below, “Extracellular vesicle purification”), and 3: lysate and purified protein from bacteria (BL21, Thermo Fisher Scientific) transfected with rat Arc plasmid (pGEX-GST-ArcFL). Some samples were treated with RNase (25 μg, RNase A, Thermo Fisher Scientific) to determine if the mRNA associated with Arc protein was protected from degradation relative to exogenously added GFP antisense RNA (generating using T7 RNA polymerase from linearized pBluescript-SKII-GFP). Preparation 1: Mice were sacrificed after 24 h of dark-housing and 2 h of enriched environment. Whole cortices were dissected and homogenized in IP lysis buffer as described above. After immunoprecipitation, bead slurry was incubated in guanidine thiocyanate containing RLT lysis buffer and column purification of RNA was performed using Qiagen RNeasy Micro Kit (Qiagen, Hilden, Germany). Total eluate was used for reverse transcription using High Capacity cDNA Reverse Transcription Kit with 50 U of Multiscribe Reverse Transcriptase and random oligo primers (Thermo Fisher Scientific). Preparations 2 and 3: total RNA was extracted using TRIzol (Thermo Fisher Scientific) as described above (“RNA extraction”). Reverse transcription reactions (25°C for 10 min, 37°C for 2 h, 85°C for 5 min) were carried out using a High Capacity cDNA Reverse Transcription Kit. Resulting cDNA was prepared for qPCR using PowerUp SYBRgreen Master Mix (Thermo Fisher Scientific) in a 96-well plate with primers against rat Arc, GAPDH and asnA (see above, “RT-PCR”; asnA primers: Fwd, GCGTGGATGCCGACACGTTG; Rev, ATACCGCCGCCGATGGTCTG). qPCR was performed on a QuantStudio 3 Real Time PCR System (Thermo Fisher Scientific) using the following protocol: Pre-incubation: 50°C for 2 min, 95°C for 2 min. Amplification: 40 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 1 min. Melt curve: 95°C for 1 s, 60°C for 20 s, continuous ramp at 0.1 5°C/s up to 95°C. Ct values of greater than 30 were considered undetectable. Differences in expression were determined using the standard curve method, where a standard DNA sample was serially diluted (10-fold), analyzed for the gene of interest, and the linear equation calculated. The resulting linear equation was used to determine where the Ct values of test samples fell within the standard curve and the result was transformed (log10) to reflect the dilution of the standard sample. Differences were calculated measuring the fold-change from the average of the control values for any given group (test/average control).
Extracellular vesicle purification
Extracellular vesicles (EVs) were purified from HEK cell and primary neuronal cultures as previously described (Lachenal et al., 2011). Media was spun successively at 2,000 and 20,000×g to remove dead cells and debris, and then at 100,000×g to pellet EVs. The crude EV pellet following the initial high-speed spin was resuspended in cold PBS and repelleted at 100,000×g for 1 h at 4°C in an SW41 rotor. The washed EV pellet was further purified by centrifugation over a 10–20% sucrose-PBS gradient at 100,000×g overnight at 4°C. The resulting pellet was washed in cold PBS to remove excess sucrose and then repelleted at 100,000×g for 1 h at 4°C. The final, washed pellet was resuspended in PBS and used for downstream analysis with EM, Western blotting, and neuron treatments.
Trypsin digestion and RNase assays
Trypsin was added to prArc and EVs at 0.05 mg/mL for 30 min at RT followed by addition of 1 mM PMSF for 10 min to inactivate trypsin. Untreated and trypsin-treated samples were then analyzed by Western blot. RNase A was added to WT neuron lysates and EVs at 50 μg/mL for 15 min at 37°C. Untreated and RNase-treated samples for RT-PCR were then directly extracted with TRIzol.
Immunogold labelling
Immunogold labeling was performed with modifications as previously described (Korkut et al., 2013). Samples were fixed overnight in 2% formaldehyde at 4°C with gentle rocking. Samples were then applied to glow discharged Formvar copper mesh grids (Ted Pella) and allowed to adhere at room temperature for 10 min. Samples were then quenched by 3 washes of 0.1 M Tris, pH 7.4. Samples were then permeabilized for 10 min at RT, blocked, and stained for Arc (1:500; custom-made). 5 nm gold-conjugated secondary antibodies were used for staining without silver enhancement. Following antibody labeling, grids were negative stained as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-way ANOVA with or without repeated measures (with post hoc Sidak’s tests) or two-tailed unpaired t-tests were performed using GraphPad Prism (GraphPad Software, San Diego, CA) or JMP Pro statistical software (SAS; Cary, NC). Significance was set at p < 0.05. All data shown are representative of at least two experimental replicates. Details of the statistics (N, number of experimental replicates, description of how the data are displayed) can be found in figure legends and/or the Results section.
Supplementary Material
Figure S1. Alignment of primary amino acid sequences of Ty3/Gag elements and origin of dipteran Arc genes relating to Figure 1.
(A) Translated genomic DNA sequences corresponding to Arc or gypsy Gag proteins were aligned using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/). Aligned sequences were shaded using the boxshade plot server (http://www.ch.embnet.org/software/BOX_form.html), using default parameters (50% sequences sharing amino acid identity for shading). Note: the alignments only contain fragments of Arc genes, not the full-length sequences with start sites. Species included: Mm—Mus musculus, house mouse; Hs—Homo sapiens, human; Ac—Anolis carolinensis, Carolina anole lizard; Lc—Latimeria chalumnae, West Indian Ocean coelacanth; Dr—Danio rerio, zebrafish; Cc—Cyprinus carpio, common carp; Dm—Drosophila melanogaster, common vinegar fly; Ds—Drosophila suzukii, spotted-wing fly; Sc—Stomoxys calcitrans, stable fly, Lh—Linepithema humile, Argentine ant; Bm—Bombyx mori, silkworm; Tc—tribolium castaneum, red flour beetle.
(B) (left) Maximum likelihood phylogenetic analysis of Arc homologs found in Drosophilidae, Muscidae, and Tephritidae flies. Multiple copies of darc1 were observed throughout tephritid flies and in the house fly, Musca domestica. For each sequence, the Genbank accession number is given after the abbreviated species names. Tephritidae: RZ—Rhagoletis zephyria; CC—Ceratitis capitate; BD—Bactrocera dorsalis. Muscidae: MD—Musca domestica; SC—Stomoxys calcitrans. (right) Putative duplication history of dArc in schizophoran flies as inferred from the phylogenetic analysis in (A). Since all schizophoran flies examined possess a homolog of darc1 and darc2, the duplication of the ancestral dArc must have occurred prior to the divergence of these species (blue triangle). This ancestral duplication event was followed by multiple rounds of duplication of darc1 (green triangles) in some of the lineages: two duplication events in the common ancestor of the Tephritidae, one additional duplication in the lineage of Ceratitis capitata, and one additional duplication in the lineage of Bactrocera dorsalis. Independently, darc1 experienced three rounds of duplication in the lineage of Musca domestica. By contrast, darc2 has apparently remained a single copy gene in the species examined.
Figure S2. Recombinant protein purification and experiments relating to Figure 2.
A) (left to right) Representative Coomassie gel of affinity purifications of full-length rat Arc (prArc), prArc-ΔCTD, CA-prArc, GST, and Endo3A showing similar expression levels to that of prArc. prArc-ΔCTD and Endo3A were prepared in the same manner as prArc. GST was directly eluted from the affinity resin using 15mM L-glutathione. His-tagged CA-prArc was eluted from Ni2+ affinity resin using 250mM imidazole. All proteins were then buffer exchanged into 150mM NaCl, 50mM Tris, pH 7.4 following GST-tag cleavage by Precision Protease or elution. Buffer conditions were adjusted for all proteins for each experiment: 500mM NaPO4, 50mM Tris, pH 7.4 for capsid stability. Analyses showing the partitioning of bacterially-expressed protein into soluble (sup) and insoluble (pellet) fractions (lanes 1, 2), capture of the protein on a GST or Ni2+ affinity matrices (lanes 3–5 show the flow through (FT), wash and captured protein, respectively). This panel demonstrates the protein expression levels and the efficacy and efficiency of affinity capture.
(B) Representative Coomassie gels of peak fractions of prArc, prArc-ΔCTD, and Endo3A eluted from S200 size exclusion columns. Peak fractions were pooled and concentrated to a final stock concentration of 1mg/mL. prArc was concentrated to 1mg/mL from each purification for use in all biochemistry/EM experiments, unless noted. For cell biology experiments, prArc was diluted to 0.4mg/mL and 4 μg total protein was used per condition.
(C) Representative Coomassie gel of affinity purification of Drosophila dArc1 from BL21 bacteria lysates demonstrating similar expression levels to rat prArc.
(D) HEK293 cells in 12-well plates were transfected with full-length rat WT Arc or GFP plasmids using Lipofectamine at equal DNA concentrations and subjected to formaldehyde crosslinking in situ. Cell lysates were blotted with anti-GFP or anti-Arc antibodies. Note that higher molecular species corresponding to Arc dimers and trimers can be observed in the crosslinked Arc sample, but not in the GFP sample.
Figure S3. RNA binding experiments related to Figure 2 and properties of Arc EVs related to experiments in Figure 3.
(A) Representative Coomassie gels of nucleotide stripping of prArc. (left) Cells were lysed in 20mM NaCl, 50mM Tris, 2mM MgCl2, 5% glycerol, 1mM DTT, pH 8.0. Fractions shown are supernatant and pellet fractions of cellular lysis after pelleting at 21,000×g for 45 min. The supernatant from this step was treated with 0.1% PEI to precipitate nucleic acids. This treatment resulted in a shift in the A260/280 ratio from 1.71±0.018 to 1.29±0.023, indicating a drop in nucleic acid content. The sample was pelleted at 27,000×g for 20 min and the resulting supernatant was treated with ammonium sulfate (AmSulf) precipitation to concentrate Arc and pelleted at 10,000×g for 10 min. The AmSulf pellet containing Arc was then subjected to affinity purification as above. (right) Representative Coomassie gel of peak fractions of cleaved, affinity purified PEI treated Arc from an anion exchange column. This chromatography step further stripped bound nucleic acids from Arc. Peak fractions were concentrated to 1mg/mL and the final measured A260/280 ratio for these fractions was 0.68±0.03 (n=3), indicating that PEI-treated prArc was largely free of nucleic acids.
(B) (left) Representative negative stain EM images of purified EVs from Arc-transfected HEK293 cell media collected for 24 h used for Western blot analysis. (right) Representative negative stain EM images of purified EVs from WT cultured neuron media collected for 24 h used for Western blot analysis. Red arrows indicate purified EVs.
(C) (left) Western blot of Arc in untreated EVs or EVs treated with trypsin (0.05mg/mL) for 30 min. prArc was used as a positive control for trypsin activity. (right) Quantification of Arc Western blot normalized to total protein. Trypsin degraded prArc but had no effect on Arc protein present in neuronal EVs.
(D) Activity dependence of Arc secretion. Purified EV fraction from media collected from DIV15 cortical neurons in 10-cm dishes from untreated WT neurons was compared with treatment with KCl. Media was freshly exchanged with basal media or media supplemented with KCl to a final concentration of 50mM. Following media exchange, cells were incubated for 1 h and media was collected and the EV fraction was purified. (left) Western blots of Arc and total protein from the purified EV fraction from cultured neuronal media. (right) Quantification of Arc protein levels, normalized to total protein. KCl treatment resulted in significantly more Arc released into the media (n=2; p<0.05).
Figure S4. HEK cell experiments related to Figure 4 and custom-made Arc antibody control experiments.
(A) HEK293 cells in 10-cm dishes were transfected with GFP-Arc as in Fig. 4. 18 h later, media from GFP-Arc-transfected HEK cells in 10-cm dishes was transferred to naïve HEK cells in 12-well plates, and 80μM Dynasore was added in one group at the same time to block endocytosis. After 6 h, the Dynasore-treated media was exchanged for fresh HEK media. 18 h later, cells were fixed and clusters of GFP-Arc-expressing cells over an entire 18mm coverslip were manually counted through a 20X objective (n=3 coverslips/group). (left) Representative images of one 20X field of view. (right) Dynasore significantly reduced the number of clusters of GFP-Arc-positive cells over the entire coverslip. Student’s t-test: *p<0.05. Scale bar=50 μm. Representative of three independent experiments and cultures with similar results. (B) DIV15 cultured hippocampal Arc KO and WT neurons were immunostained for the dendritic protein MAP2 with Alexa Fluor 555 only (top row), or both MAP2 (Alexa 555) and Arc (Alexa 488; bottom two rows). Imaging settings for Arc were determined based on Arc immunostaining in WT neurons (bottom row). No GFP fluorescence from GFP knocked in to the Arc locus in the KO neurons was visible under these imaging conditions. Example of two independent experiments. (C) DIV15 cultured hippocampal Arc KO and WT neurons were fixed and immunostained with either a custom-made rabbit polyclonal Arc antibody (ProteinTech) or the commercially available Synaptic Systems rabbit polyclonal Arc antibody, as well as the dendritic protein MAP2. All groups were imaged with the same acquisition settings. 30-μm segments of two dendrites/neuron were analyzed in all groups (n=10 neurons) and were chosen using MAP2 staining. Both antibodies were able to detect a difference between Arc KO and WT neurons, although the signal:noise was better using the custom-made antibody. Arc in the soma/nucleus varies widely from neuron to neuron with both antibodies, under basal conditions. Student’s t-test: **Arc KO vs. WT with custom antibody, p<0.01. #Arc KO vs. WT with Synaptic Systems (“SySy”) antibody, p<0.05. Arc images are false-colored with the Smart LUT in ImageJ to better display differences in Arc expression. Example of two independent experiments.
Figure S5. RNase and uptake experiments relating to Figure 5.
(A) To test whether Arc mRNA is protected in prArc capsids, samples were subjected to 15 min treatment with RNase A, then RNase inhibitor (1U/μl) to quench activity, prior to incubation with neurons. (left) Representative images of Arc mRNA in DIV15 cultured hippocampal Arc KO neurons incubated with the treated or untreated prArc samples for 4 h. (right) prArc treatment resulted an increase in dendritic Arc mRNA levels in Arc KO neurons. prArc treated with RNase did not affect Arc mRNA transfer. (B) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 4 μg prArc. In one group, 30 min before prArc was added, neurons were pretreated with 80μM Dynasore to block endocytosis. (left) Representative images of Arc protein and mRNA levels. (right) Pretreatment with Dynasore significantly blocked uptake/transfer of prArc protein and Arc mRNA. Student’s t-test: *p<0.05. ***p<0.001. Example of three independent experiments (A, B). Scale bars in all panels=10 μm. (C) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 4 μg prArc. Either combined FISH/ICC for Arc mRNA and Rab5 protein, or ICC for Arc and Rab5 protein, was performed. (left) Representative images of dendrites showing Arc mRNA plus Rab5 protein or Arc and Rab5 protein. (right) Arc protein and mRNA showed around 50% colocalization in dendrites with Rab5. White arrowheads indicate Arc alone, and yellow arrowheads indicate Arc/Rab5 colocalization. Example of two independent experiments. Scale bar=10 μm. (D) Purified protein samples of prArc, prArc(RNA−), prArc-ΔCTD, and CA-prArc were separated by SDS-PAGE, and the resulting Western blot was immunostained for Arc using our custom-made Arc antibody. The antibody successfully detected all of the mutant constructs, suggesting that the lack of Arc immunostaining observed in transfer experiments was not a result of an inability of the antibody to detect the mutants. “Total” is Ponceau stain for total protein for each sample.
Figure S6. Purified Arc stripped of nucleic acids cannot be taken up by neurons.
DIV15 cultured hippocampal Arc KO neurons were treated with 4 μg prArc or prArc(RNA−) for 4 h before being fixed. One group from each treatment was not permeabilized during the immunocytochemistry procedure for Arc and MAP2. prArc-treated neurons that were non-permeabilized showed little to no MAP2 and Arc immunostaining. However, prArc(RNA−)-treated neurons showed no difference in Arc immunostaining between permeabilized and non-permeabilized conditions, although MAP2 immunostaining was still absent in the non-permeabilized condition, suggesting that prArc(RNA−) accumulates on the outside of the neurons. Dendritic segments boxed in white are shown magnified beneath each corresponding image. Scale bars=10 μm. Example of three independent experiments. Arc images are false-colored with the Smart LUT from ImageJ to highlight differences in Arc expression. Merged images have MAP2 immunostaining in magenta and Arc in green.
Figure S7. RNase and uptake experiments relating to Figure 6.
(A) To test whether Arc mRNA is protected in neuronal EVs, EVs prepared from 10-cm dishes of DIV15 cultured WT cortical neurons were subjected to 15 min treatment with RNase A, then RNase inhibitor (1U/μl) to quench activity, prior to incubation with neurons. DIV15 cultured hippocampal Arc KO neurons were incubated with 10 μg of the treated or untreated WT EV samples for 4 h. (left) Representative images of Arc mRNA levels in neurons. (right) WT EV treatment resulted an increase in dendritic Arc mRNA levels in Arc KO neurons. WT EV treated with RNase did not affect Arc mRNA transfer. (B) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 10 μg of the EV fraction harvested from the media of 10-cm dishes containing DIV15 high-density cultured cortical WT or Arc KO neurons. In one group, 30 min before EVs were added, neurons were pretreated with 80μM Dynasore to block endocytosis. (top) Representative images of Arc protein levels (left) or Arc mRNA levels (right). (bottom) Pretreatment with Dynasore significantly blocked uptake of Arc protein and mRNA from WT EVs. Arc protein and mRNA expression was normalized to Arc KO and is displayed as fold-change±SEM. Dendritic segments boxed in white are shown magnified beneath each corresponding image. 30-μm segments of two dendrites/neuron were analyzed in all groups (n=10 neurons) and were chosen using MAP2 staining. Student’s t-test: ***p<0.001. Scale bars=10 μm. Example of three independent experiments. Images are false-colored with the Smart LUT from ImageJ to highlight differences in Arc expression. (C) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 10 μg of WT EVs, then fixed. Either combined FISH/ICC for Arc mRNA and Rab5 protein, or ICC for Arc and Rab5 protein, was performed. (left) Representative images of dendrites showing Arc mRNA plus Rab5 protein or Arc and Rab5 protein. (right) Arc mRNA and protein showed 30–40% colocalization in dendrites with Rab5. White arrowheads indicate Arc alone, and yellow arrowheads indicate Arc/Rab5 colocalization. Example of two independent experiments. Scale bar=10 μm.
(D) Model: Comparison of HIV Gag and Arc capsid life cycle. (top) HIV Gag protein self-assembles (via the CA domain) in the cytosol and at the plasma membrane (by myristoylation of the MA domain), while the capsid encapsulates viral RNA (via the NC domain). The immature HIV capsid is released from the cell in an ESCRT-dependent manner (via the p6 domain) with membrane that contains the viral envelope protein (Env). The mature virus particles bind host cells through surface receptors (such as CD4) and membrane fusion occurs. Alternatively, in some cell types virus particles are first endocytosed prior to fusion and particles released into the cell after full fusion occurs in the endosome. Viral RNA is released and then reversed transcribed into viral DNA that is integrated into the host genome. (bottom) Arc mRNA is trafficked out into dendrites in RNA granules that contain a selection of different mRNAs. Local translation of Arc mRNA takes place in dendrites in response to neuronal activity. High concentrations of Arc protein self-assemble and form Arc capsids, which encapsulate select mRNAs that are spatially proximal, including Arc mRNA. Arc capsids are released from dendrites in Arc Capsids Bearing Any RNA (ACBARs) and transfer of mRNA and other putative cargo takes place in neighboring dendrites.
HIGHLIGHTS.
The neuronal gene Arc encodes a protein that forms virus-like capsids
Arc protein exhibits similar biochemical properties as retroviral Gag proteins
Endogenous Arc protein is released from neurons in extracellular vesicles (EVs)
Arc EVs and capsids can mediate intercellular transfer of Arc mRNA in neurons
Acknowledgments
We thank Dr. Adam Frost and Raghav Kalia for technical assistance in the initial stages of this project. We thank Dr. Wesley Sundquist for helpful discussions and the Sundquist/Bass labs for experimental help and support. We thank Dr. Vivian Budnik for helpful discussions and sharing of unpublished data. We thank Dr. Kristen Keefe for experimental help with the FISH assay. We thank Jenifer Einstein and all members of the Shepherd lab for technical help and support. This work was supported by the NIH (R00 NS076364, R01 MH112766 to J.D.S.; R01 GM77582, R01 GM112972 to C.F.) and startup funds from the University of Utah (J.D.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
E.D.P. performed immunoprecipitation, cultured neuron, immunocytochemistry, and fluorescent in situ hybridization experiments. C.E.D. and R.B.K. performed biochemistry and electron microscopy experiments. A.V.T. and M.K.S. performed qPCR experiments. D.B. assisted with electron microscopy experiments. J.M. and C.F. performed phylogenomic analysis. S.E., D.R.M., and J.A.B. conducted cryo-EM and DLS experiments. N.Y. performed initial biochemical and electron microscopy experiments. E.D.P., C.E.D., and J.D.S. conceived and designed experiments. J.D.S. wrote the manuscript; all authors discussed results and edited the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell. 2016;30:836–848. doi: 10.1016/j.ccell.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Alme MN, Bittins M, Kuipers SD, Nair RR, Pai B, Panja D, Schubert M, Soule J, Tiron A, et al. The Arc of synaptic memory. Exp Brain Res. 2010;200:125–140. doi: 10.1007/s00221-009-1959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Ruiz-Canada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. 2016;17:160–172. doi: 10.1038/nrn.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Rein A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J Virol. 1999;73:2270–2279. doi: 10.1128/jvi.73.3.2270-2279.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campillos M, Doerks T, Shah PK, Bork P. Computational characterization of multiple Gag-like human proteins. Trends Genet. 2006;22:585–589. doi: 10.1016/j.tig.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Carlson LA, Bai Y, Keane SC, Doudna JA, Hurley JH. Reconstitution of selective HIV-1 RNA packaging in vitro by membrane-bound Gag assemblies. Elife. 2016;5 doi: 10.7554/eLife.14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 2017;18:71–86. doi: 10.1038/nrg.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas-Garcia M, Davis SR, Rein A. On the Selective Packaging of Genomic RNA by HIV-1. Viruses. 2016;8 doi: 10.3390/v8090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis G, Vernochet C, Carradec Q, Souquere S, Mulot B, Catzeflis F, Nilsson MA, Menzies BR, Renfree MB, Pierron G, et al. Retroviral envelope gene captures and syncytin exaptation for placentation in marsupials. Proc Natl Acad Sci U S A. 2015;112:E487–496. doi: 10.1073/pnas.1417000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daberkow DP, Riedy MD, Kesner RP, Keefe KA. Arc mRNA induction in striatal efferent neurons associated with response learning. Eur J Neurosci. 2007;26:228–241. doi: 10.1111/j.1460-9568.2007.05630.x. [DOI] [PubMed] [Google Scholar]
- de Solis CA, Morales AA, Hosek MP, Partin AC, Ploski JE. Is Arc mRNA Unique: A Search for mRNAs That Localize to the Distal Dendrites of Dentate Gyrus Granule Cells Following Neural Activity. Front Mol Neurosci. 2017;10:314. doi: 10.3389/fnmol.2017.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, Grange J, Schoehn G, Goldberg Y, Boyer V, et al. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci. 2006;31:642–648. doi: 10.1016/j.mcn.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet. 2012;13:283–296. doi: 10.1038/nrg3199. [DOI] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO. HIV-1 assembly, release and maturation. Nat Rev Microbiol. 2015;13:484–496. doi: 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704–716. doi: 10.1016/j.cell.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2:1120–1124. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- Hamann MV, Lindemann D. Foamy Virus Protein-Nucleic Acid Interactions during Particle Morphogenesis. Viruses. 2016;8 doi: 10.3390/v8090243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen LJ, Chalker DL, Orlinsky KJ, Sandmeyer SB. Ty3 GAG3 and POL3 genes encode the components of intracellular particles. J Virol. 1992;66:1414–1424. doi: 10.1128/jvi.66.3.1414-1424.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heraud-Farlow JE, Kiebler MA. The multifunctional Staufen proteins: conserved roles from neurogenesis to synaptic plasticity. Trends Neurosci. 2014;37:470–479. doi: 10.1016/j.tins.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie M, Yoshikawa M, Ono R, Iwafune H, Furuse T, Yamada I, Wakana S, Yamashita Y, Abe T, Ishino F, et al. Cognitive Function Related to the Sirh11/Zcchc16 Gene Acquired from an LTR Retrotransposon in Eutherians. PLoS Genet. 2015;11:e1005521. doi: 10.1371/journal.pgen.1005521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko-Ishino T, Ishino F. The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front Microbiol. 2012;3:262. doi: 10.3389/fmicb.2012.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkut C, Ataman B, Ramachandran P, Ashley J, Barria R, Gherbesi N, Budnik V. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell. 2009;139:393–404. doi: 10.1016/j.cell.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkut C, Li Y, Koles K, Brewer C, Ashley J, Yoshihara M, Budnik V. Regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron. 2013;77:1039–1046. doi: 10.1016/j.neuron.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AW, Mitra A, Bauer AQ, Snyder AZ, Raichle ME, Culver JP, Lee JM. Visual experience sculpts whole-cortex spontaneous infraslow activity patterns through an Arc-dependent mechanism. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1711789114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutluay SB, Zang T, Blanco-Melo D, Powell C, Jannain D, Errando M, Bieniasz PD. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell. 2014;159:1096–1109. doi: 10.1016/j.cell.2014.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachenal G, Pernet-Gallay K, Chivet M, Hemming FJ, Belly A, Bodon G, Blot B, Haase G, Goldberg Y, Sadoul R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol Cell Neurosci. 2011;46:409–418. doi: 10.1016/j.mcn.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Lefebvre FA, Benoit Bouvrette LP, Perras L, Blanchet-Cohen A, Garnier D, Rak J, Lecuyer E. Comparative transcriptomic analysis of human and Drosophila extracellular vesicles. Sci Rep. 2016;6:27680. doi: 10.1038/srep27680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Mailler E, Bernacchi S, Marquet R, Paillart JC, Vivet-Boudou V, Smyth RP. The Life-Cycle of the HIV-1 Gag-RNA Complex. Viruses. 2016;8 doi: 10.3390/v8090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S, Eickbush TH. Poised for contagion: evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res. 2000;10:1307–1318. doi: 10.1101/gr.145000. [DOI] [PubMed] [Google Scholar]
- Manago F, Mereu M, Mastwal S, Mastrogiacomo R, Scheggia D, Emanuele M, De Luca MA, Weinberger DR, Wang KH, Papaleo F. Genetic Disruption of Arc/Arg3.1 in Mice Causes Alterations in Dopamine and Neurobehavioral Phenotypes Related to Schizophrenia. Cell Rep. 2016;16:2116–2128. doi: 10.1016/j.celrep.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei S, Schur FK, Briggs JA. Retrovirus maturation-an extraordinary structural transformation. Curr Opin Virol. 2016;18:27–35. doi: 10.1016/j.coviro.2016.02.008. [DOI] [PubMed] [Google Scholar]
- McCurry CL, Shepherd JD, Tropea D, Wang KH, Bear MF, Sur M. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat Neurosci. 2010;13:450–457. doi: 10.1038/nn.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuni T, Uesaka N, Okuno H, Hirai H, Deisseroth K, Bito H, Kano M. Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron. 2013;78:1024–1035. doi: 10.1016/j.neuron.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouland AJ, Mercier J, Luo M, Bernier L, DesGroseillers L, Cohen EA. The double-stranded RNA-binding protein Staufen is incorporated in human immunodeficiency virus type 1: evidence for a role in genomic RNA encapsidation. J Virol. 2000;74:5441–5451. doi: 10.1128/jvi.74.12.5441-5451.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrum C, Baumann A, Bustad HJ, Flydal MI, Mariaule V, Alvira S, Cuellar J, Haavik J, Soule J, Valpuesta JM, et al. Arc is a flexible modular protein capable of reversible self-oligomerization. Biochem J. 2015 doi: 10.1042/BJ20141446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte-’t Hoen E, Cremer T, Gallo RC, Margolis LB. Extracellular vesicles and viruses: Are they close relatives? Proc Natl Acad Sci U S A. 2016;113:9155–9161. doi: 10.1073/pnas.1605146113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, Akashi K, Ishii Y, Yagishita-Kyo N, Suzuki K, Nonaka M, Kawashima T, Fujii H, Takemoto-Kimura S, Abe M, et al. Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIbeta. Cell. 2012;149:886–898. doi: 10.1016/j.cell.2012.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastuzyn ED, Shepherd JD. Activity-Dependent Arc Expression and Homeostatic Synaptic Plasticity Are Altered in Neurons from a Mouse Model of Angelman Syndrome. Frontiers in Molecular Neuroscience. 2017;10 doi: 10.3389/fnmol.2017.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkstaff JK, Chappell SA, Mauro VP, Edelman GM, Krushel LA. Internal initiation of translation of five dendritically localized neuronal mRNAs. Proc Natl Acad Sci U S A. 2001;98:2770–2775. doi: 10.1073/pnas.051623398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006;52:437–444. doi: 10.1016/j.neuron.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy JG, Flanagan JM, Ropson IJ, Rennoll-Bankert KE, Craven RC. Critical role of conserved hydrophobic residues within the major homology region in mature retroviral capsid assembly. J Virol. 2008;82:5951–5961. doi: 10.1128/JVI.00214-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD. Arc - An endogenous neuronal retrovirus? Semin Cell Dev Biol. 2017 doi: 10.1016/j.semcdb.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14:279–284. doi: 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Stoye JP, Taylor IA. A comparative analysis of the foamy and ortho virus capsid structures reveals an ancient domain duplication. BMC Struct Biol. 2017;17:3. doi: 10.1186/s12900-017-0073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, Thery C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell. 2016;164:1226–1232. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- Tsai B. Penetration of nonenveloped viruses into the cytoplasm. Annu Rev Cell Dev Biol. 2007;23:23–43. doi: 10.1146/annurev.cellbio.23.090506.123454. [DOI] [PubMed] [Google Scholar]
- Ufer F, Vargas P, Engler JB, Tintelnot J, Schattling B, Winkler H, Bauer S, Kursawe N, Willing A, Keminer O, et al. Arc/Arg3.1 governs inflammatory dendritic cell migration from the skin and thereby controls T cell activation. Science Immunology. 2016;1:eaaf8665–eaaf8665. doi: 10.1126/sciimmunol.aaf8665. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- Wang KH, Majewska A, Schummers J, Farley B, Hu C, Sur M, Tonegawa S. In vivo two-photon imaging reveals a role of arc in enhancing orientation specificity in visual cortex. Cell. 2006;126:389–402. doi: 10.1016/j.cell.2006.06.038. [DOI] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Petralia RS, Kurushima H, Patel H, Jung MY, Volk L, Chowdhury S, Shepherd JD, Dehoff M, Li Y, et al. Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell. 2011;147:615–628. doi: 10.1016/j.cell.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulli V, Friis KP, Fitzpatrick Z, Maguire CA, Breakefield XO. Extracellular vesicles and intercellular communication within the nervous system. J Clin Invest. 2016;126:1198–1207. doi: 10.1172/JCI81134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wu J, Ward MD, Yang S, Chuang YA, Xiao M, Li R, Leahy DJ, Worley PF. Structural basis of arc binding to synaptic proteins: implications for cognitive disease. Neuron. 2015;86:490–500. doi: 10.1016/j.neuron.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Zhou L, Lim Q, Zou R, Stephanopoulos G, Too HP. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol Biol. 2011;12:18. doi: 10.1186/1471-2199-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Alignment of primary amino acid sequences of Ty3/Gag elements and origin of dipteran Arc genes relating to Figure 1.
(A) Translated genomic DNA sequences corresponding to Arc or gypsy Gag proteins were aligned using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/). Aligned sequences were shaded using the boxshade plot server (http://www.ch.embnet.org/software/BOX_form.html), using default parameters (50% sequences sharing amino acid identity for shading). Note: the alignments only contain fragments of Arc genes, not the full-length sequences with start sites. Species included: Mm—Mus musculus, house mouse; Hs—Homo sapiens, human; Ac—Anolis carolinensis, Carolina anole lizard; Lc—Latimeria chalumnae, West Indian Ocean coelacanth; Dr—Danio rerio, zebrafish; Cc—Cyprinus carpio, common carp; Dm—Drosophila melanogaster, common vinegar fly; Ds—Drosophila suzukii, spotted-wing fly; Sc—Stomoxys calcitrans, stable fly, Lh—Linepithema humile, Argentine ant; Bm—Bombyx mori, silkworm; Tc—tribolium castaneum, red flour beetle.
(B) (left) Maximum likelihood phylogenetic analysis of Arc homologs found in Drosophilidae, Muscidae, and Tephritidae flies. Multiple copies of darc1 were observed throughout tephritid flies and in the house fly, Musca domestica. For each sequence, the Genbank accession number is given after the abbreviated species names. Tephritidae: RZ—Rhagoletis zephyria; CC—Ceratitis capitate; BD—Bactrocera dorsalis. Muscidae: MD—Musca domestica; SC—Stomoxys calcitrans. (right) Putative duplication history of dArc in schizophoran flies as inferred from the phylogenetic analysis in (A). Since all schizophoran flies examined possess a homolog of darc1 and darc2, the duplication of the ancestral dArc must have occurred prior to the divergence of these species (blue triangle). This ancestral duplication event was followed by multiple rounds of duplication of darc1 (green triangles) in some of the lineages: two duplication events in the common ancestor of the Tephritidae, one additional duplication in the lineage of Ceratitis capitata, and one additional duplication in the lineage of Bactrocera dorsalis. Independently, darc1 experienced three rounds of duplication in the lineage of Musca domestica. By contrast, darc2 has apparently remained a single copy gene in the species examined.
Figure S2. Recombinant protein purification and experiments relating to Figure 2.
A) (left to right) Representative Coomassie gel of affinity purifications of full-length rat Arc (prArc), prArc-ΔCTD, CA-prArc, GST, and Endo3A showing similar expression levels to that of prArc. prArc-ΔCTD and Endo3A were prepared in the same manner as prArc. GST was directly eluted from the affinity resin using 15mM L-glutathione. His-tagged CA-prArc was eluted from Ni2+ affinity resin using 250mM imidazole. All proteins were then buffer exchanged into 150mM NaCl, 50mM Tris, pH 7.4 following GST-tag cleavage by Precision Protease or elution. Buffer conditions were adjusted for all proteins for each experiment: 500mM NaPO4, 50mM Tris, pH 7.4 for capsid stability. Analyses showing the partitioning of bacterially-expressed protein into soluble (sup) and insoluble (pellet) fractions (lanes 1, 2), capture of the protein on a GST or Ni2+ affinity matrices (lanes 3–5 show the flow through (FT), wash and captured protein, respectively). This panel demonstrates the protein expression levels and the efficacy and efficiency of affinity capture.
(B) Representative Coomassie gels of peak fractions of prArc, prArc-ΔCTD, and Endo3A eluted from S200 size exclusion columns. Peak fractions were pooled and concentrated to a final stock concentration of 1mg/mL. prArc was concentrated to 1mg/mL from each purification for use in all biochemistry/EM experiments, unless noted. For cell biology experiments, prArc was diluted to 0.4mg/mL and 4 μg total protein was used per condition.
(C) Representative Coomassie gel of affinity purification of Drosophila dArc1 from BL21 bacteria lysates demonstrating similar expression levels to rat prArc.
(D) HEK293 cells in 12-well plates were transfected with full-length rat WT Arc or GFP plasmids using Lipofectamine at equal DNA concentrations and subjected to formaldehyde crosslinking in situ. Cell lysates were blotted with anti-GFP or anti-Arc antibodies. Note that higher molecular species corresponding to Arc dimers and trimers can be observed in the crosslinked Arc sample, but not in the GFP sample.
Figure S3. RNA binding experiments related to Figure 2 and properties of Arc EVs related to experiments in Figure 3.
(A) Representative Coomassie gels of nucleotide stripping of prArc. (left) Cells were lysed in 20mM NaCl, 50mM Tris, 2mM MgCl2, 5% glycerol, 1mM DTT, pH 8.0. Fractions shown are supernatant and pellet fractions of cellular lysis after pelleting at 21,000×g for 45 min. The supernatant from this step was treated with 0.1% PEI to precipitate nucleic acids. This treatment resulted in a shift in the A260/280 ratio from 1.71±0.018 to 1.29±0.023, indicating a drop in nucleic acid content. The sample was pelleted at 27,000×g for 20 min and the resulting supernatant was treated with ammonium sulfate (AmSulf) precipitation to concentrate Arc and pelleted at 10,000×g for 10 min. The AmSulf pellet containing Arc was then subjected to affinity purification as above. (right) Representative Coomassie gel of peak fractions of cleaved, affinity purified PEI treated Arc from an anion exchange column. This chromatography step further stripped bound nucleic acids from Arc. Peak fractions were concentrated to 1mg/mL and the final measured A260/280 ratio for these fractions was 0.68±0.03 (n=3), indicating that PEI-treated prArc was largely free of nucleic acids.
(B) (left) Representative negative stain EM images of purified EVs from Arc-transfected HEK293 cell media collected for 24 h used for Western blot analysis. (right) Representative negative stain EM images of purified EVs from WT cultured neuron media collected for 24 h used for Western blot analysis. Red arrows indicate purified EVs.
(C) (left) Western blot of Arc in untreated EVs or EVs treated with trypsin (0.05mg/mL) for 30 min. prArc was used as a positive control for trypsin activity. (right) Quantification of Arc Western blot normalized to total protein. Trypsin degraded prArc but had no effect on Arc protein present in neuronal EVs.
(D) Activity dependence of Arc secretion. Purified EV fraction from media collected from DIV15 cortical neurons in 10-cm dishes from untreated WT neurons was compared with treatment with KCl. Media was freshly exchanged with basal media or media supplemented with KCl to a final concentration of 50mM. Following media exchange, cells were incubated for 1 h and media was collected and the EV fraction was purified. (left) Western blots of Arc and total protein from the purified EV fraction from cultured neuronal media. (right) Quantification of Arc protein levels, normalized to total protein. KCl treatment resulted in significantly more Arc released into the media (n=2; p<0.05).
Figure S4. HEK cell experiments related to Figure 4 and custom-made Arc antibody control experiments.
(A) HEK293 cells in 10-cm dishes were transfected with GFP-Arc as in Fig. 4. 18 h later, media from GFP-Arc-transfected HEK cells in 10-cm dishes was transferred to naïve HEK cells in 12-well plates, and 80μM Dynasore was added in one group at the same time to block endocytosis. After 6 h, the Dynasore-treated media was exchanged for fresh HEK media. 18 h later, cells were fixed and clusters of GFP-Arc-expressing cells over an entire 18mm coverslip were manually counted through a 20X objective (n=3 coverslips/group). (left) Representative images of one 20X field of view. (right) Dynasore significantly reduced the number of clusters of GFP-Arc-positive cells over the entire coverslip. Student’s t-test: *p<0.05. Scale bar=50 μm. Representative of three independent experiments and cultures with similar results. (B) DIV15 cultured hippocampal Arc KO and WT neurons were immunostained for the dendritic protein MAP2 with Alexa Fluor 555 only (top row), or both MAP2 (Alexa 555) and Arc (Alexa 488; bottom two rows). Imaging settings for Arc were determined based on Arc immunostaining in WT neurons (bottom row). No GFP fluorescence from GFP knocked in to the Arc locus in the KO neurons was visible under these imaging conditions. Example of two independent experiments. (C) DIV15 cultured hippocampal Arc KO and WT neurons were fixed and immunostained with either a custom-made rabbit polyclonal Arc antibody (ProteinTech) or the commercially available Synaptic Systems rabbit polyclonal Arc antibody, as well as the dendritic protein MAP2. All groups were imaged with the same acquisition settings. 30-μm segments of two dendrites/neuron were analyzed in all groups (n=10 neurons) and were chosen using MAP2 staining. Both antibodies were able to detect a difference between Arc KO and WT neurons, although the signal:noise was better using the custom-made antibody. Arc in the soma/nucleus varies widely from neuron to neuron with both antibodies, under basal conditions. Student’s t-test: **Arc KO vs. WT with custom antibody, p<0.01. #Arc KO vs. WT with Synaptic Systems (“SySy”) antibody, p<0.05. Arc images are false-colored with the Smart LUT in ImageJ to better display differences in Arc expression. Example of two independent experiments.
Figure S5. RNase and uptake experiments relating to Figure 5.
(A) To test whether Arc mRNA is protected in prArc capsids, samples were subjected to 15 min treatment with RNase A, then RNase inhibitor (1U/μl) to quench activity, prior to incubation with neurons. (left) Representative images of Arc mRNA in DIV15 cultured hippocampal Arc KO neurons incubated with the treated or untreated prArc samples for 4 h. (right) prArc treatment resulted an increase in dendritic Arc mRNA levels in Arc KO neurons. prArc treated with RNase did not affect Arc mRNA transfer. (B) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 4 μg prArc. In one group, 30 min before prArc was added, neurons were pretreated with 80μM Dynasore to block endocytosis. (left) Representative images of Arc protein and mRNA levels. (right) Pretreatment with Dynasore significantly blocked uptake/transfer of prArc protein and Arc mRNA. Student’s t-test: *p<0.05. ***p<0.001. Example of three independent experiments (A, B). Scale bars in all panels=10 μm. (C) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 4 μg prArc. Either combined FISH/ICC for Arc mRNA and Rab5 protein, or ICC for Arc and Rab5 protein, was performed. (left) Representative images of dendrites showing Arc mRNA plus Rab5 protein or Arc and Rab5 protein. (right) Arc protein and mRNA showed around 50% colocalization in dendrites with Rab5. White arrowheads indicate Arc alone, and yellow arrowheads indicate Arc/Rab5 colocalization. Example of two independent experiments. Scale bar=10 μm. (D) Purified protein samples of prArc, prArc(RNA−), prArc-ΔCTD, and CA-prArc were separated by SDS-PAGE, and the resulting Western blot was immunostained for Arc using our custom-made Arc antibody. The antibody successfully detected all of the mutant constructs, suggesting that the lack of Arc immunostaining observed in transfer experiments was not a result of an inability of the antibody to detect the mutants. “Total” is Ponceau stain for total protein for each sample.
Figure S6. Purified Arc stripped of nucleic acids cannot be taken up by neurons.
DIV15 cultured hippocampal Arc KO neurons were treated with 4 μg prArc or prArc(RNA−) for 4 h before being fixed. One group from each treatment was not permeabilized during the immunocytochemistry procedure for Arc and MAP2. prArc-treated neurons that were non-permeabilized showed little to no MAP2 and Arc immunostaining. However, prArc(RNA−)-treated neurons showed no difference in Arc immunostaining between permeabilized and non-permeabilized conditions, although MAP2 immunostaining was still absent in the non-permeabilized condition, suggesting that prArc(RNA−) accumulates on the outside of the neurons. Dendritic segments boxed in white are shown magnified beneath each corresponding image. Scale bars=10 μm. Example of three independent experiments. Arc images are false-colored with the Smart LUT from ImageJ to highlight differences in Arc expression. Merged images have MAP2 immunostaining in magenta and Arc in green.
Figure S7. RNase and uptake experiments relating to Figure 6.
(A) To test whether Arc mRNA is protected in neuronal EVs, EVs prepared from 10-cm dishes of DIV15 cultured WT cortical neurons were subjected to 15 min treatment with RNase A, then RNase inhibitor (1U/μl) to quench activity, prior to incubation with neurons. DIV15 cultured hippocampal Arc KO neurons were incubated with 10 μg of the treated or untreated WT EV samples for 4 h. (left) Representative images of Arc mRNA levels in neurons. (right) WT EV treatment resulted an increase in dendritic Arc mRNA levels in Arc KO neurons. WT EV treated with RNase did not affect Arc mRNA transfer. (B) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 10 μg of the EV fraction harvested from the media of 10-cm dishes containing DIV15 high-density cultured cortical WT or Arc KO neurons. In one group, 30 min before EVs were added, neurons were pretreated with 80μM Dynasore to block endocytosis. (top) Representative images of Arc protein levels (left) or Arc mRNA levels (right). (bottom) Pretreatment with Dynasore significantly blocked uptake of Arc protein and mRNA from WT EVs. Arc protein and mRNA expression was normalized to Arc KO and is displayed as fold-change±SEM. Dendritic segments boxed in white are shown magnified beneath each corresponding image. 30-μm segments of two dendrites/neuron were analyzed in all groups (n=10 neurons) and were chosen using MAP2 staining. Student’s t-test: ***p<0.001. Scale bars=10 μm. Example of three independent experiments. Images are false-colored with the Smart LUT from ImageJ to highlight differences in Arc expression. (C) DIV15 cultured hippocampal Arc KO neurons were treated for 4 h with 10 μg of WT EVs, then fixed. Either combined FISH/ICC for Arc mRNA and Rab5 protein, or ICC for Arc and Rab5 protein, was performed. (left) Representative images of dendrites showing Arc mRNA plus Rab5 protein or Arc and Rab5 protein. (right) Arc mRNA and protein showed 30–40% colocalization in dendrites with Rab5. White arrowheads indicate Arc alone, and yellow arrowheads indicate Arc/Rab5 colocalization. Example of two independent experiments. Scale bar=10 μm.
(D) Model: Comparison of HIV Gag and Arc capsid life cycle. (top) HIV Gag protein self-assembles (via the CA domain) in the cytosol and at the plasma membrane (by myristoylation of the MA domain), while the capsid encapsulates viral RNA (via the NC domain). The immature HIV capsid is released from the cell in an ESCRT-dependent manner (via the p6 domain) with membrane that contains the viral envelope protein (Env). The mature virus particles bind host cells through surface receptors (such as CD4) and membrane fusion occurs. Alternatively, in some cell types virus particles are first endocytosed prior to fusion and particles released into the cell after full fusion occurs in the endosome. Viral RNA is released and then reversed transcribed into viral DNA that is integrated into the host genome. (bottom) Arc mRNA is trafficked out into dendrites in RNA granules that contain a selection of different mRNAs. Local translation of Arc mRNA takes place in dendrites in response to neuronal activity. High concentrations of Arc protein self-assemble and form Arc capsids, which encapsulate select mRNAs that are spatially proximal, including Arc mRNA. Arc capsids are released from dendrites in Arc Capsids Bearing Any RNA (ACBARs) and transfer of mRNA and other putative cargo takes place in neighboring dendrites.