

SUMMARY
Excess plasma triglycerides (TG) are a key component of obesity-induced metabolic syndrome. We have shown that γ-secretase inhibitor (GSI) treatment improves glucose tolerance due to inhibition of hepatic Notch signaling, but find additional, Notch-independent reduction of plasma TG-rich lipoproteins (TRLs) in GSI-treated, as well as hepatocyte-specific γ-secretase knockout (L-Ncst) mice, which suggested a primary effect on hepatocyte TRL uptake. Indeed, we found increased VLDL and LDL particle uptake in L-Ncst hepatocytes and Ncst-deficient hepatoma cells, in part through reduced γ-secretase-mediated low-density lipoprotein receptor (LDLR) cleavage and degradation. To exploit this novel finding, we generated a liver-selective Nicastrin ASO, which recapitulated glucose and lipid improvements of L-Ncst mice, with increased levels of hepatocyte LDLR. Collectively, these results identify the role of hepatic γ-secretase to regulate LDLR, and suggest that liver-specific γ-secretase inhibitors may simultaneously improve multiple aspects of the metabolic syndrome.
eTOC blurb
Kim et al. discover that hepatic γ-secretase inhibition improves glucose homeostasis, and simultaneously reduces plasma triglycerides and non-HDL cholesterol, in part through blocking LDLR cleavage and degradation
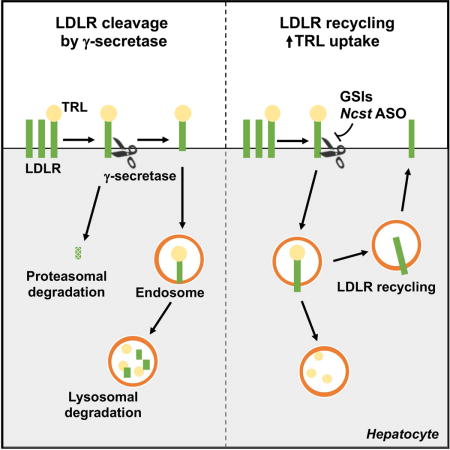
INTRODUCTION
One of the core components of the metabolic syndrome is excess plasma triglycerides (TG), or hypertriglyceridemia, which is an independent risk factor for coronary heart disease (CHD), above and beyond other insulin resistance-related complications (i.e., high apolipoprotein B (ApoB), low high-density lipoprotein (HDL) cholesterol levels, hypertension and Type 2 Diabetes) (Brunzell, 2007; Nordestgaard et al., 2007). This is noteworthy, as despite great strides in the so-called “statin era” to reduce LDL cholesterol in high-risk patients, cardiovascular risk remains disproportionately elevated in people with the metabolic syndrome. Unfortunately, hypertriglyceridemia is often not effectively treated, leading to a significant unmet therapeutic need in an increasingly obese population (Brunzell and Ayyobi, 2003).
Hypertriglyceridemia is associated with an overproduction and secretion of triglyceride-rich lipoproteins (TRLs), due to increased liver lipid substrate availability (Adiels et al., 2005; Choi and Ginsberg, 2011), and/or reduced catabolism of TRLs and their remnants, due to reduced lipoprotein lipase (LPL) activity, insufficient hepatic remnant receptors or competition of dietary and hepatic-derived lipoproteins for a common clearance pathway (Ayyobi and Brunzell, 2003; Bishop et al., 2008; Mamo et al., 2001). Abnormal TRL remnant catabolism may also be related to various apolipoproteins found on TRLs – of these, the best-studied is apolipoprotein C3 (ApoC3) (Jong et al., 1999). People with complete absence of ApoC3 have very low TG levels associated with rapid plasma TG clearance (Ginsberg et al., 1986; Norum et al., 1982), and decreased risk of coronary heart disease in Amish and Ashkenazi Jewish populations has been observed with genetic variants that confer partial ApoC3 deficiency (Pollin et al., 2008). Similarly, ApoC3 knockout mice demonstrate markedly lower plasma TG levels (Maeda et al., 1994) while ApoC3 transgenic mice show hypertriglyceridemia (Ito et al., 1990) and increased atherosclerosis (Masucci-Magoulas et al., 1997; Zheng, 2014). But as plasma TG and ApoC3 are highly correlated (Le et al., 1988; Schonfeld et al., 1979), the causal factor in altered atherosclerosis risk in mouse and man cannot be disentangled – in fact, these data have led to a renewed push to identify novel therapeutic targets to reduce CHD risk in hypertriglyceridemic patients.
The γ-secretase is a multiprotein complex consisting of redundant catalytic (Presenilin 1 or 2) and regulatory (Aph-1a or -1b) subunits, as well as unique targeting (Nicastrin) and enhancer (PEN2) components that regulate intramembrane proteolysis of Type 1 transmembrane proteins (Wolfe, 2006). As γ-secretase mediates the pathologic cleavage of Alzheimer’s precursor protein (APP) to generate amyloid β-protein (Aβ), γ-secretase inhibitors (GSIs) have been proposed as Alzheimer Disease (AD) therapeutics (Selkoe, 2001). Unfortunately, lack of efficacy has plagued GSIs in clinical trials for AD (Doody et al., 2013), but their antagonistic effects on Notch receptors have led to efforts to repurpose these therapeutics as antineoplastic agents (De Jesus-Acosta et al., 2014; Wei et al., 2010), and more recently, for metabolic disease (Bi and Kuang, 2015; Pajvani et al., 2011; Sparling et al., 2016). For instance, we found that GSI treatment of diet-induced or genetic mouse models of obesity improved hepatic insulin sensitivity, likely through inhibition of Notch co-activation of FoxO1-mediated hepatic glucose production (Pajvani et al., 2011).
Here, we describe our finding that GSIs reduce plasma TG and non-HDL cholesterol, independent of liver Notch signaling. To elucidate the mechanism of this unexpected result, we created hepatocyte-specific γ-secretase knockout (albumin-Cre:Nicastrinflox/flox, henceforth L-Ncst) mice, which similar to GSI treatment, also show improved glucose tolerance and reduced plasma TG due to increased hepatocyte TRL uptake. These data suggest that liver-specific γ-secretase inhibition has therapeutic potential to protect from hypertriglyceridemia, without known GI toxicity of GSIs (Real et al., 2009). To exploit this potential, we developed liver-selective Ncst antisense oligonucleotides (ASOs), and found that Ncst ASO-treated mice also show lower plasma TG. These parallel pharmacologic and genetic approaches suggest a non-Notch, hepatocyte γ-secretase target that regulates plasma TG. In fact, beyond APP and Notch, an increasing number of additional putative γ-secretase Type 1 transmembrane protein targets have been identified (De Strooper, 2003; Shih Ie and Wang, 2007; Wolfe, 2006; Wolfe and Kopan, 2004). To this end, an unbiased proteomics screen (Hemming et al., 2008) identified but did not experimentally validate a potential candidate for the γ-secretase effect on hepatocyte TRL uptake, the LDL receptor (LDLR). Indeed, we find that Nicastrin binds the C-terminal domain of LDLR, targeting LDLR for γ-secretase-mediated cleavage, which in turn induces LDLR lysosomal degradation. Thus, Ncst ASO treatment fails to lower plasma TG in Ldlr−/− or Ldlr ASO-treated mice. These data uncover the novel role of hepatic γ-secretase to regulate LDLR, and highlight the potential of liver-specific γ-secretase inhibitors to simultaneously ameliorate obesity-induced glucose intolerance and hypertriglyceridemia.
RESULTS
γ-secretase inhibitors (GSIs) reduce plasma TG
Treatment of obese mice with the γ-secretase inhibitor (GSI), dibenzazepine, improves glucose tolerance and reduces hepatocyte glucose production, likely by inhibiting hepatic Notch activity (Pajvani et al., 2011). As genetic (hepatocyte-specific deletion of Rbpj; hence L-Rbpj mice (Pajvani et al., 2013)) or more specific pharmacologic Notch inhibition, and conversely, adenoviral transduction of constitutively-active Notch1 did not affect plasma TG or cholesterol levels (Figure S1A–S1D), we were surprised to note that GSI treatment of normal chow- or high fat diet (HFD)-fed wild-type and L-Rbpj mice led to a significant reduction in plasma TG, VLDL-TG, and non-HDL cholesterol (Figure 1A, 1B and S1E). GSI-mediated reduction of VLDL-TG was even more dramatic in obese, dyslipidemia-prone (Nishina et al., 1994), leptin-deficient (ob/ob) mice (Figure 1C and 1D).
Figure 1. γ-secretase inhibitors (GSIs) reduce plasma TG.
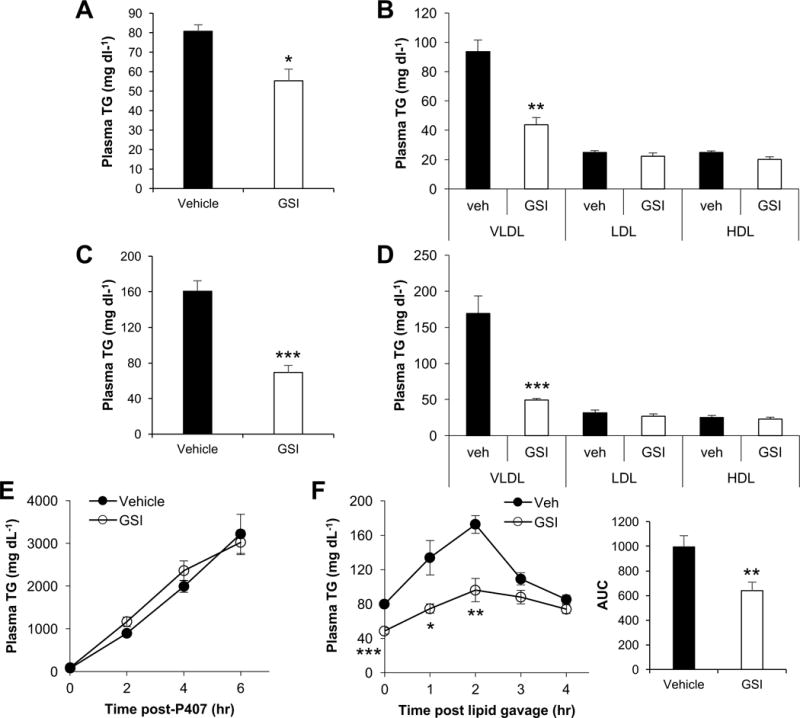
(A–D) Plasma TG (A and C) and lipoprotein-associated TG analyzed by density gradient ultracentrifugation (B and D) in vehicle or GSI-treated, wild-type or leptin-deficient ob/ob mice (C and D) (n=6 per group). (E) Plasma TG following i.p. injection with poloxamer-407 (1g per kg body weight), or (F) after oral olive-oil gavage in 5 h-fasted vehicle- or GSI-treated mice (n=6–7 per group). *P < 0.05, **P < 0.01, ***P<0.001 as compared to the indicated control by two-way ANOVA. All data are shown as the means ± s.e.m.
We did not detect differences in body or adipose weight with GSI treatment (Sparling et al., 2016), so to determine the mechanism of lower plasma TG, we injected mice with Poloxamer-407, which prevents LPL-mediated TG-lipolysis, and found identical TG secretion in control and GSI-treated mice (Figure 1E). Next, we found that TG excursion in response to olive oil gavage was attenuate in GSI-treated mice (Figure 1F). Collectively, these results demonstrate that GSIs reduce plasma TG, likely by a mechanism independent of known GSI inhibition of hepatocyte Notch.
Hepatocyte-specific γ-secretase inhibition also lowers plasma TG
These results could be confounded by known GI toxicity of GSIs (van Es et al., 2005), and potential interference with intestinal lipid absorption. To exclude this possibility, we ablated the non-redundant γ-secretase targeting subunit, Nicastrin (Aster and Blacklow, 2012), specifically in hepatocytes (albumin-Cre:Nicastrinflox/flox, henceforth L-Ncst mice). L-Ncst mice showed reduced Ncst expression (Figure 2A) and Nicastrin protein levels (Figure 2B) as compared to Cre- control (Nicastrinflox/flox) mice, leading to lower Psen1 and Psen2 protein levels (Figure 2B) due to inhibition of auto-catalytic γ-secretase enzymatic activity (Brunkan and Goate, 2005). As in GSI-treated mice, despite unchanged body weight, body composition and liver TG (Figure S2A–S2C), L-Ncst mice showed both improved glucose tolerance (Figure 2C) as well as lower plasma TG (Figure 2D) as compared to Cre- control mice. Also similar to GSI-treated mice, fractionation of plasma lipoproteins by density gradient ultracentrifugation or size-exclusion fast protein liquid chromatography showed reduced TG in VLDL and LDL fractions in L-Ncst mice (not shown and Figure 2E), no change in VLDL-TG secretion (Figure 2F) but a marked reduction in appearance of plasma TG after an oral lipid load (Figure 2G).
Figure 2. Hepatocyte-specific γ-secretase inhibition lowers plasma TG.
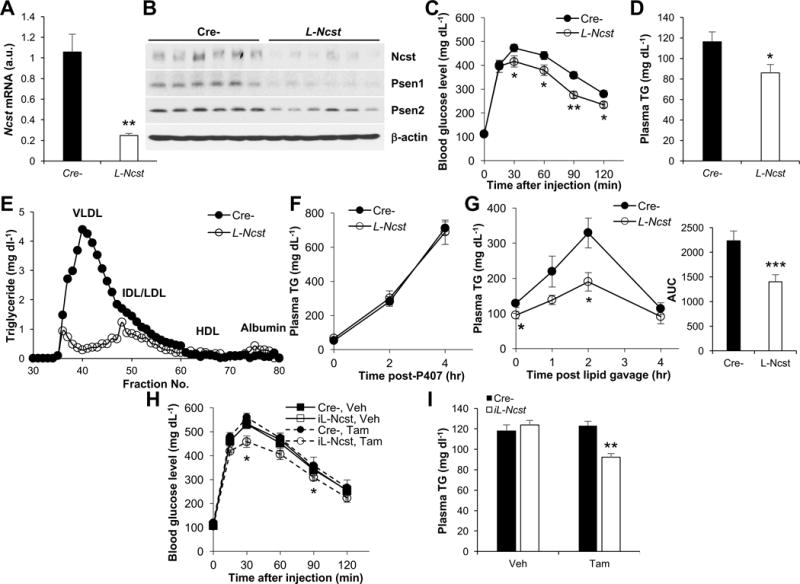
(A and B) Hepatic Ncst mRNA (A) and Western blot for Nicastrin (Ncst), Presenilin 1 (Psen1), and Presenilin 2 (Psen2) (B) in Cre- (Nicastrinflox/flox) or L-Ncst (albumin-Cre;Nicastrinflox/flox) mice (n=6 per group). (C) Intraperitoneal glucose tolerance test (GTT) in Cre- or L-Ncst mice (n=6 per group). (D and E) Plasma TG (D) and fractionated pooled plasma TG by FPLC (E) in 5 h-fasted Cre- or L-Ncst mice (n=6 per group). (F and G) Plasma TG following i.p. injection with poloxamer-407 (1g per kg body weight) (F), or after oral olive-oil gavage (G) in 5 h-fasted Cre- or L-Ncst mice (n=6–7 per group). (H and I) GTT (H) and plasma TG (I) in Cre- or iL-Ncst mice treated with vehicle (corn oil) or tamoxifen to induce recombination (n=6 per group). *P < 0.05, **P < 0.01, ***P < 0.001 as compared to the indicated control by two-way ANOVA. All data are shown as the means ± s.e.m.
These data indicate that hepatocyte γ-secretase affects normal plasma lipid handling, but to eliminate any possible confounding developmental effects of γ-secretase inhibition (de Vera Mudry et al., 2012; Vujovic et al., 2007), we crossed Ncstflox/flox mice with SA-Cre-ERT2 transgenic mice (Schuler et al., 2004) (henceforth, iL-Ncst mice), to allow for both temporal and conditional disruption of hepatocyte γ-secretase. Tamoxifen-treatment of adult iL-Ncst mice reduced Ncst mRNA and protein levels (Figure S2D and S2E). Consistent with GSI-treated and L-Ncst mice, tamoxifen-treated iL-Ncst mice showed normal body weight, adiposity and liver TG (Figure S2F–S2H), but parallel improvements in glucose tolerance (Figure 2H) and plasma TG (Figure 2I) as compared to Cre- mice or vehicle-treated iL-Ncst mice.
Ncst ASO reduces plasma TG
Although GSIs are in advanced clinical trials for cancer (Andersson and Lendahl, 2014), repurposing these drugs to reduce plasma TG is likely precluded by dose-limiting GI toxicity. We theorized that more specific cellular targeting, by use of a liver-selective antisense oligonucleotide (ASO) (Bennett and Swayze, 2010), would avoid GI side-effects while retaining metabolic potency. We assayed 384 ASOs targeting Ncst mRNA for in vitro ability to reduce Ncst mRNA expression, and moved the 4 most potent ASOs (Figure S3A) forward to in vivo testing in C57BL/6 mice. All 4 ASOs, dosed once weekly at 25 mg per kg body weight for 6 weeks, reduced liver Ncst and Psen2 levels as compared to a control ASO composed of the same chemistry and oligonucleotide length (Figure S3B). We picked ASO #806498-3 (henceforth Ncst ASO), based on in vitro predictions of low toxicity and in vivo observations of unchanged body weight and plasma ALT in our pilot experiment (Figure S3C and S3D), coupled with >90% Ncst knockdown efficiency. We administered Ncst ASO to chow- or HFD-fed C57BL/6 wild-type mice (Figure S3E), and found reduced liver Ncst mRNA, Nicastrin protein levels (Figure 3A and 3B and Figure S3F), plasma TG (Figure 3C and Figure S3G), and improved glucose tolerance (Figure 3D and Figure S3H). Ncst ASO treatment appeared to be well tolerated, without intestinal metaplasia as seen with GSI treatment (Real and Ferrando, 2009), and did not affect plasma transaminases, liver TG or glycogen levels (Figure S3I–S3M). Similar to GSIs, however, Ncst ASO reduced plasma TG in both Cre- and L-Rbpj mice, consistent with a Notch-independent effect (Figure S3N). Finally, to test the molecular and tissue specificity of Ncst ASO, we treated Cre- and L-Ncst mice with Ncst ASO. As expected, Ncst ASO reduced plasma TG and the appearance of plasma TG after olive oil gavage in Cre-mice, but not in L-Ncst mice (Figure 3E and 3F). Together, these data indicate that Ncst ASO reduces plasma TG with additional benefits on glucose homeostasis.
Figure 3. Ncst ASO reduces plasma TG in a liver-specific manner.
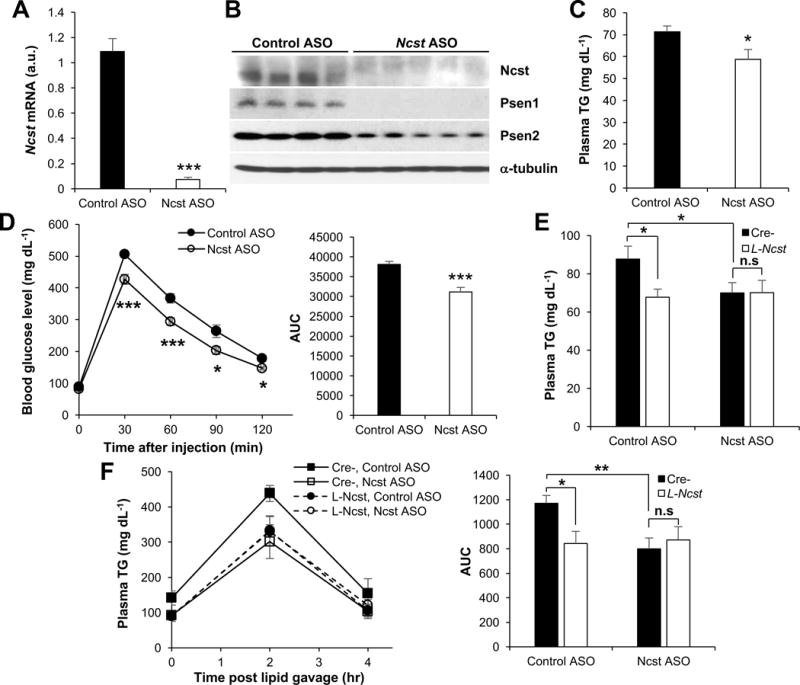
(A and B) Hepatic Ncst mRNA (A) and protein levels (B) in HFD-fed C57BL/6 mice injected with control or Ncst ASO (n=6–7 per group). (C) Plasma TG in mice injected with control of Ncst ASO (n=6–7 per group). (D) GTT or AUC during GTT (right) in HFD-fed mice injected with control of Ncst ASO (n=6–7 per group). (E and F) Plasma TG (E) or plasma TG after oral olive-oil gavage and AUC during olive-oil gavage (F) in 5 h-fasted Cre- or L-Ncst mice injected with either control or Ncst ASO (n=7 per group). *P < 0.05, **P < 0.01, ***P< 0.001 as compared to the indicated control by two-way ANOVA. All data are shown as the means ± s.e.m.
γ-secretase regulates endocytosis of TRLs
We next turned our attention to the mechanism by which hepatocyte γ-secretase regulates plasma TG. As TG secretion was unaffected, we hypothesized that TG lipolysis and/or uptake of TRLs was increased with γ-secretase inhibition, perhaps by transcriptional effects on one of several apolipoproteins secreted by liver that regulate LPL activity. But we observed unchanged Apoc3, Angptl3 and other apolipoprotein genes, or their protein products, in livers from GSI-treated, Ncst ASO, L-Ncst and iL-Ncst mice (not shown and Figure S4A–S4C), leading to unchanged LPL activity in plasma of Cre- and L-Ncst mice (Figure S4D).
Nevertheless, consistent with the known concordance with plasma TG (Schonfeld et al., 1979), we observed reduced plasma ApoC3 in these same mice (Figure 4A and Figure S4E–S4G), independent of the prandial state (Figure S4H). As ApoC3 appearance in plasma was similar in Poloxamer 407-treated Cre- and L-Ncst mice (Figure 4B), these data suggested that ApoC3 disappearance from plasma was accelerated with hepatic γ-secretase inhibition, suggestive of enhanced uptake of VLDL/LDL particles. Indeed, we observed a similar reduction in ApoB and ApoC3 in VLDL fractions isolated from L-Ncst mice (Figure 4C), consistent with lower non-HDL cholesterol in L-Ncst as compared to Cre- control mice (Figure S4I). But to test this hypothesis directly and confirm cell-autonomous effects, we exposed primary hepatocytes derived from L-Ncst (or Cre- control) mice or shNcst (or shControl)-expressing McA-RH7777 rat hepatoma cells (Wang et al., 1999) to purified human VLDL or LDL particles, in the presence of both proteasome (MG-132) and lysosomal (chloroquine) inhibitors to prevent degradation of internalized protein. Using this sensitive and specific uptake assay, we found more intracellular human ApoB and human ApoC3 in L-Ncst hepatocytes (Figure 4D) and shNcst cells (Figure 4E) as compared to the relevant controls. Concordantly, less ApoB and ApoC3 remained in media applied to cells lacking γ-secretase (Figure 4F and 4G). We observed similar findings in L-Ncst hepatocytes or shNcst cells exposed to plasma from Ad-human ApoC3-transduced mice (Figure S4J and S4K) – after 24hr incubation, only 25% of input ApoC3 remained in the medium exposed to shNcst cells, as compared to 60% in shControl cells (Figure S4L). Finally, we injected 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)-labeled VLDL particles into Cre- and L-Ncst mice, and measured remaining plasma label at various time-points post-intravenous injection, to test the kinetics of VLDL extraction in vivo (Altenburg et al., 2008; Li et al., 2014). Plasma Dil-VLDL was more rapidly cleared, with concomitantly greater hepatic label internalization in L-Ncst than Cre- mice (Figure 4H and 4I). These data show that γ-secretase inhibition increases cellular uptake of TRLs, to reduce plasma TG and non-HDL cholesterol.
Figure 4. γ-secretase regulates the uptake of TRLs.
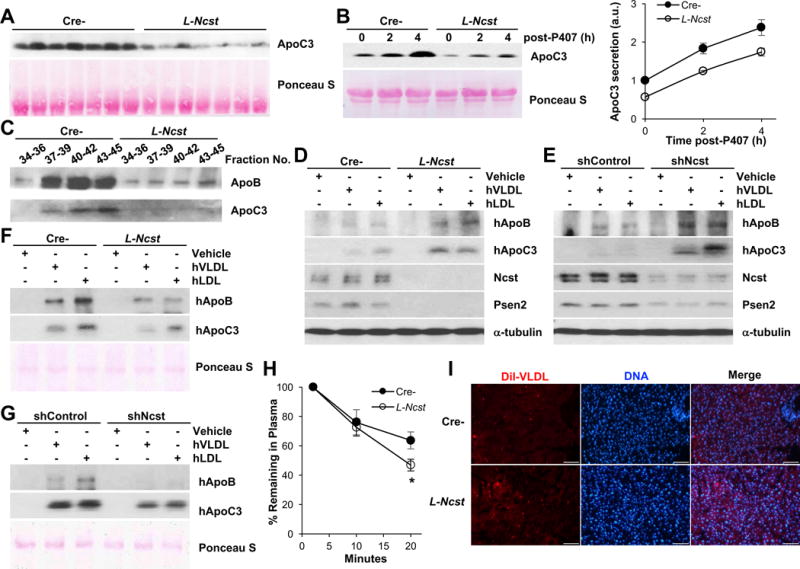
(A) Representative Western blots of plasma ApoC3 in Cre- or L-Ncst. (B) Plasma ApoC3 before and after i.p. Injection of poloxamer-407 in Cre- or L-Ncst mice, with quantitation relative to time 0. (C) Western blot of ApoB and ApoC3 in the VLDL fraction of plasma from Cre- and L-Ncst mice. (D and E) Western blots for intracellular human ApoB and ApoC3 in primary hepatocytes isolated from Cre- or L-Ncst mice (D) and shControl or shNcst-stable McA-RH7777 cells (E) incubated with VLDL or LDL purified from human plasma in the presence of MG-132 and chloroquine. (F and G) Western blot of remaining hApoB and hApoC3 in media of primary hepatocyte from Cre- or L-Ncst mice (F) and shControl or shNcst-stable McA-RH7777 cells (G). (H, I) Dil-labeled VLDL disappearance from plasma (H), and Dil staining in livers (I) of Cre- or L-Ncst mice (n=5–6 per group). The scale bar represents 100 μm. *P < 0.05 as compared to the indicated control by two-way ANOVA. All data are shown as the means ± s.e.m.
γ-secretase regulates LDLR protein stability
TRLs arising from LPL-mediated lipolysis are cleared by multiple hepatic receptors, including LDL receptor (LDLR), LDLR-related protein 1 (LRP1), and syndecan-1 (SDC), the predominant hepatic heparin sulfate proteoglycan (HSPG) (Bishop et al., 2008; Foley et al., 2013; MacArthur et al., 2007; Stanford et al., 2009). As both LDLR and LRP1 have been reported as putative γ-secretase targets (Hemming et al., 2008), we hypothesized that one or more of these mediate the effect of γ-secretase inhibition on TRL clearance, and tested for ApoB and ApoC3 uptake from human VLDL and LDL particles by L-Ncst or shNcst cells in the presence of Pitstop2 (Dutta et al., 2012; von Kleist et al., 2011), an inhibitor of both clathrin-dependent and -independent endocytosis. As predicted, Pitstop2 negated the differences between controls and cells lacking γ-secretase (Figure 5A and 5B and Figure S5A and S5B).
Figure 5. γ-secretase regulates endocytosis of TRLs.
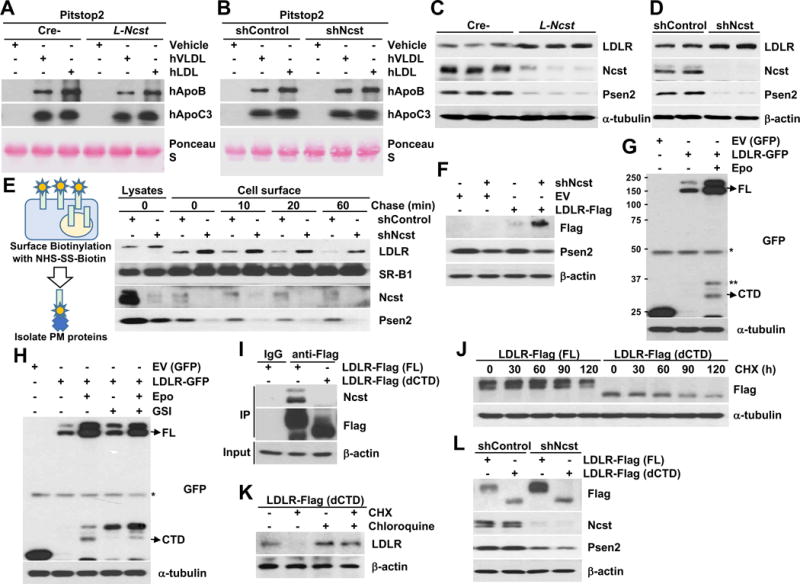
(A and B) Western blot of hApoB and hApoC3 in media of primary hepatocytes from Cre- or L-Ncst mice (A) and shControl or shNcst-stable McA-RH7777 cells (B), with Pitstop 2. (C and D) Western blot of hepatic LDLR in Cre- or L-Ncst (C), or shControl or shNcst cells (D). (E) Cell surface biotinylation analysis in shControl or shNcst-stable McA-RH7777 cells. (F) Western blot of exogenous LDLR with C-terminal Flag tags in shControl or shNcst-stable McA-RH7777 cells. (G, H) Western blots for C-terminal GFP-tagged LDLR (or α-tubulin), in the presence of epoxomicin and/or GSI. FL=full-length LDLR, CTD=LDLR C-terminal domain; *= nonspecific band, **=C-terminal LDLR fragment. (I) Western blots of co-immunoprecipitated Nicastrin, in McA-RH7777 cells transfected with Flag-tagged full-length (FL) or LDLR lacking the C-terminal domain (dCTD). (J) Western blots from McA-RH7777 cells transfected with Flag-tagged FL or dCTD, then treated with 50 μg ml−1 cycloheximide (CHX) for the indicated times, and quantification of Flag normalized to β-actin, relative to time 0. (K) McA-RH7777 cells transfected with LDLR-dCTD-Flag plasmids, pretreated with chloroquine for 1 h before incubation with CHX for 4 hrs. (L) Western blots from shControl or shNcst-stable McA-RH7777 cells, transfected with FL- or LDLR-dCTD-Flag. *P < 0.05 as compared to the indicated control by two-way ANOVA. All data are shown as the means ± s.e.m.
Next, we surveyed the candidate receptors, and found a specific and striking increase in both total and plasma membrane LDLR in L-Ncst livers and shNcst cells, without apparent change in LDLR intracellular trafficking dynamics (Figure 5C–5E) or Ldlr mRNA expression with γ-secretase inhibition (Figure S5C and S5D). Further, we found a robust increase in transfected LDLR (with a non-native promoter) in shNcst cells as compared with control cells (Figure 5F and Figure S5E). These data suggest a post-transcriptional effect of γ-secretase on LDLR protein stability. Known pathways that regulate post-transcriptional levels of LDLR include PCSK9 (pro-protein convertase subtilisin/kexin type 9) and Idol (inducible degrader of the LDLR) (Arsenault et al., 2014; Lakoski et al., 2009; Zelcer et al., 2009), but we observed no difference of hepatic Pcsk9 or Idol expression, and PCSK9 protein levels and secretion with γ-secretase inhibition (Figure S5F–S5K). Thus, we considered that LDLR may be a direct γ-secretase substrate – indeed, in the presence of epoxomicin to inhibit proteasomal degradation of LDLR cleavage products, we observed both the predicted γ-secretase-cleaved LDLR cytoplasmic tail domain (CTD), and another C-terminal fragment oft-seen in γ-secretase substrates (Figure 5G). With GSI treatment, however, LDLR CTD levels were reduced with parallel C-terminal fragment accumulation (Figure 5H), confirming our hypothesis of γ-secretase-dependent LDLR cleavage. In fact, we observed that Nicastrin directly binds full-length (FL)-LDLR, but not a CTD-deficient LDLR mutant (dCTD) (Figure 5I) that mimics the structure of LDLR post-γ-secretase cleavage (Figure S5L). We next predicted that dCTD-LDLR is inherently unstable, and transfected McA-RH7777 cells with dCTD- or FL-LDLR, then treated with cycloheximide to block new protein synthesis. As predicted, dCTD has lower steady state protein levels, likely accounted for by increased degradation (Figure 5J), rescued by chloroquine treatment to block LDLR lysosomal degradation (Figure 5K). We find that LDLR degradation is γ-secretase-dependent, as FL but not dCTD-LDLR accumulated in chloroquine-treated shNcst cells (Figure 5L). In sum, these data support the conclusion that Nicastrin binds LDLR, allowing γ-secretase-mediated intramembrane proteolysis, releasing the short LDLR CTD (which is quickly degraded by the proteasome) and rendering LDLR susceptible to lysosomal degradation.
γ-secretase effects on plasma TG are mediated by LDLR, independent of PCSK9
As PCSK9 reroutes internalized LDLR for intracellular degradation (Lagace, 2014), we considered whether the PCSK9 action may be potentiated (Tveten et al., 2013) by previous or concomitant γ-secretase-mediated LDLR cleavage. To test this, we administered Ncst ASO in combination with alirocumab, a PCSK9 monoclonal antibody in clinical use (Robinson et al., 2015), to HFD-fed wild-type mice (Figure S6A). As expected (Berger et al., 2017), alirocumab reduced plasma cholesterol levels (Figure S6B), but had no effect on plasma TG. Ncst ASO reduced plasma TG in both control and alirocumab-treated mice, but we observed neither synergy nor competition between these inhibitors (Figure S6C).
These data suggest that γ-secretase regulates LDLR turnover, independent of PCSK9. Next, to test LDLR-dependence of γ-secretase effect on plasma TG, we administered Ncst ASO to Ldlr knockout (Ldlr−/−), and in parallel, Ldlr ASO-treated mice (Figure 6A and 6B). Consistent with earlier results, Ncst ASO decreased plasma TG and ApoC3 (Figure 6C, 6D and S6D), as well as plasma TG excursion after olive oil gavage, but not in Ldlr−/− or concomitantly-treated Ldlr ASO mice (Figure 6E and 6F). Collectively, these experiments show that the γ-secretase destabilizes LDLR, and that inhibition of hepatic γ-secretase activity increases hepatic LDLR to facilitate increased TRL uptake and lower plasma TG.
Figure 6. Hepatic γ-secretase inhibition decreases plasma TG in a LDLR-dependent manner.
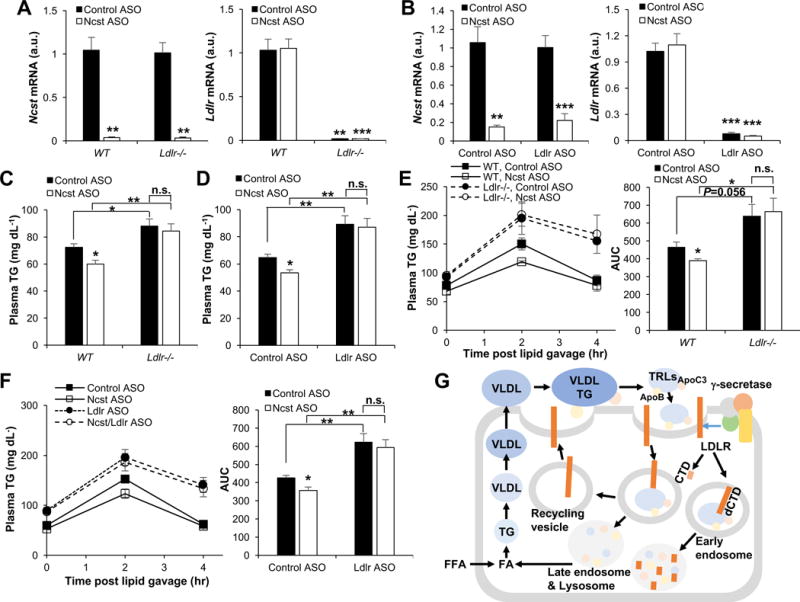
(A and B) Hepatic Ncst and Ldlr expression, (C and D) plasma TG and (E and F) plasma TG before and after oral olive-oil gavage with AUC during olive-oil gavage (right) in Ncst ASO-treated wild-type or Ldlr−/− mice or mice concomitantly injected with control or Ldlr ASO (n=5–7 per group). *P < 0.05, **P < 0.01 as compared to the indicated control by two-way ANOVA. All data are shown as the means ± s.e.m. (G) Model representing the effects of hepatic γ-secretase on LDLR degradation to regulate plasma TG.
DISCUSSION
γ-secretase-mediated proteolysis of integral membrane proteins is required for diverse biological processes (Lal and Caplan, 2011). One γ-secretase target of particular interest is Notch, as recent work has shown Notch activity to be increased in obesity and associated metabolic conditions such as T2D and NAFLD/NASH (Fukuda et al., 2012; Lee et al., 2016; Valenti et al., 2013), perhaps in an ill-fated attempt to regenerate hepatic functional capacity (Geisler and Strazzabosco, 2015). Our data show that treatment with GSIs, or more specific approaches to blocking liver γ-secretase activity (Ncst ASO; L-Ncst or iL-Ncst mice), recapitulates the insulin-sensitizing effects of loss of hepatocyte Notch signaling, but simultaneously reduces plasma TG and non-HDL cholesterol in a Notch-independent manner. These data support the dual role of hepatic γ-secretase to regulate glucose homeostasis and lipid homeostasis by diverse mechanisms, but why this proteolytic complex, functionally conserved from flies and worms to humans (Tomita et al., 2001), has evolved to play such a critical role in metabolic processes disrupted by the obese state is unclear. In addition, further study is required to test whether hepatic γ-secretase activity is constitutive, or may be regulated by increased nutrient availability and/or lipid flux to the liver. For example, Hif-1α, known to regulate glucose metabolism in neoplastic and obese tissue (Denko, 2008; Jiang et al., 2011), has been shown to increase γ-secretase activity in breast cancer (Villa et al., 2014). Our findings suggest that other hormonal and nutrient stimuli may have similar effects.
Our data show that Nicastrin directly targets LDLR for γ-secretase-mediated proteolysis as predicted by a prior proteomics screen (Hemming et al., 2008), which in turn renders LDLR susceptible to lysosomal degradation, and decreases uptake of TG-rich VLDL/LDL particles (Gordts et al., 2016) but we cannot rule out indirect mechanisms that may contribute to TRL clearance. The very short (~50aa) LDLR CTD released is then rapidly degraded by the proteasome, consistent with other γ-secretase substrates (Lal and Caplan, 2011). Of note, we find additional LDLR C-terminal fragments that suggest that LDLR may be shed by other secretases prior to γ-secretase cleavage, but the necessity for this processing for Nicastrin recognition and γ-secretase activity warrants further investigation. Nonetheless, our results show that LDLR lacking its short CTD, which represents the end-product of γ-secretase activity, is inherently unstable and undergoes lysosomal degradation. This experiment unintentionally recapitulates data from Brown and Goldstein from decades earlier, where C-terminal premature stop mutations of LDLR decreased receptor stability (van Driel et al., 1987). We hypothesize that these artificial mutations actually reveal inherent biology, that γ-secretase prevents normal LDLR recycling and hepatic TRL removal (Figure 6F), perhaps to ensure sufficient lipid delivery to other tissues.
Our data further suggest a parallel path for LDLR degradation, above and beyond PCSK9-mediated LDLR lysosomal degradation, with different effects on plasma lipids. PCSK9 loss-of-function or pharmacologic inhibition lowers LDL cholesterol, but effects on plasma TG in treated patients are less robust and reproducible (Everett et al., 2015). Consistent with these data, we found that administration of a PCSK9 mAb, alirocumab, did not further reduce plasma TG in Ncst ASO-treated mice. We postulate that increased “baseline” hepatocyte plasma membrane LDLR, by dint of γ-secretase inhibition, allows rapid binding and uptake of TRLs, which may distinguish this approach from PCSK9 antagonism that preferentially increases LDLR recycling. Alternatively, LDLR PTMs or association with other receptors may determine cargo preference, or it may simply reflect competition for LDLR pools. Notably, simultaneous inhibition of γ-secretase and PCSK9 appeared to lower both TG and cholesterol, but further work is necessary to test if PCSK9 and γ-secretase have synergistic and additive effects on lipoprotein metabolism in animal models with elevated non-HDL cholesterol (Lagace, 2014). Nevertheless, these data indicate that γ-secretase inhibition attenuates multiple metabolic comorbidities of the obese state.
LIMITATIONS OF STUDY
Our work demonstrates the novel role of hepatic γ-secretase to regulate LDLR, which in turn contributes to enhanced clearance of TRLs. In parallel with other metabolic benefits of γ-secretase inhibition, these data may provide rationale for development of liver-specific γ-secretase inhibitors (i.e. Ncst ASO) for simultaneous treatment of multiple cardiovascular risk factors in an at-risk obese and insulin-resistant population. However, several questions remain unanswered, as alluded to above. For instance, although Ncst ASO does not lower plasma TG in the absence of LDLR, these data do not rule-out the possibility that γ-secretase regulates TRL clearance through indirect, but still LDLR-dependent, mechanisms. Additionally, how LDLR is differentially regulated by γ-secretase and PCSK9, to affect uptake of TRLs and cholesterol respectively, is unclear. Finally, as all data presented is derived from rodent models, translation to human biology is unknown – along these lines, it would be of great interest to interrogate the lipid/lipoprotein phenotypes of patients treated with GSIs in clinical trials for Alzheimer’s disease and cancer, although these data may be confounded by these disease processes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Utpal B. Pajvani (up2104@columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Male wild-type C57BL/6 (strain #662) and male leptin-deficient ob/ob (strain #632) mice were purchased from Jackson Labs. We crossed albumin-Cre or SA-Cre-ERT2 (Schuler et al., 2004) and Nicastrinflox/flox mice (Sparling et al., 2016; Tabuchi et al., 2009) on a C57BL/6 background to generate albumin-Cre;Nicastrinflox/flox (L-Ncst) and SA-Cre-ERT2;Nicastrinflox/flox (iL-Ncst) mice, respectively. Male iL-Ncst were treated with i.p. injections of 1 mg tamoxifen dissolved in corn oil (or vehicle) for 3 consecutive days. All mice were housed 3–5 animals per cages, with a 12 h light/dark cycle, in a temperature-controlled environment and were fed normal chow (Purina Mills 5053) or high-fat diet (18.4% calories/carbohydrates, 21.3% calories/protein and 60.3% calories/fat derived from lard; Harlan Laboratories, TD.06414). Male L-Ncst and iL-Ncst mice aged 8–24 weeks were used for most experiments. All mice studied were included in subsequent analysis. All animal experiments were approved by the Columbia University Institutional Animal Care and Utilization Committee.
Cell cultures studies
Primary hepatocytes were isolated from 8 to 10-week-old male mice as previously described (Kim et al., 2016; Kim et al., 2017; Pajvani et al., 2011). Isolated primary hepatocytes were cultured in DMEM supplemented with 23 mM HEPES, 10% FBS and antibiotics at 37°C and 5% CO2. McA-RH7777 cells stably expressing shControl or shNcst were selected with 1 μg ml−1 puromycin and maintained in DMEM supplemented with 10% FBS, 10% horse serum and antibiotics at 37°C and 5% CO2.
METHODS DETAILS
Chemicals
Inhibitor studies were performed as previously described (Pajvani et al., 2011). In brief, we suspended dibenzazepine (Syncom; 2 μmol per kg body weight) in vehicle [(0.5% Methocel E4M (wt/vol, Colorcon) and 0.1% (vol/vol) Tween-80 (Sigma)) and sonicated for 2 min to achieve a homogeneous suspension before daily (for 5 d) intraperitoneal injection (Pajvani et al., 2013). Pitstop2 and Pitstop2 negative control were purchased from Abcam.
Adenovirus studies
GFP, N1-IC, Fc, Notch1 decoy, and ApoC3 adenoviruses have been described (Hernandez et al., 2010; Kim et al., 2016; Kim et al., 2017; Pajvani et al., 2013; Sundaram et al., 2010) For in vivo studies, we injected between 2.5–5 × 108 purified viral particles per gram body weight, performed metabolic analysis on days 3–7 and killed the mice at day 7 or 10 post-injection.
Metabolic analyses
Blood tail vein glucose was measured using a glucose meter (OneTouch). Glucose tolerance tests were performed by intraperitoneal injection of 2 g per kg body weight glucose after a 16 h fast at room temperature. Hepatic lipids were extracted by the Folch method (Folch et al., 1957), and plasma and hepatic triglyceride (Thermo) and Cholesterol E (Wako) were measured using colorimetric assays according to the manufacturer’s protocol.
Hepatic glycogen assay
Hepatic glycogen was measured as previously described (Pajvani et al., 2011). Briefly, 100 mg liver was homogenized in 6% (vol/vol) perchloric acid (PCA), adjusted to pH 6–7 with KOH, then incubated with 1 mg ml-1 amyloglucosidase in 0.2 M acetate (pH 4.8) at 42°C with shaking for 2 h. Glucose released (glycogen breakdown value minus PCA value) was quantified using a glucose assay kit (Sigma).
Hepatic VLDL-TG secretion and lipoprotein profile
Rates of hepatic VLDL-TG secretion rates was measured using poloxamer-407 (Millar et al., 2005), dissolved in saline and injected i.p. at 1 g per kg body weight in mice that were briefly (5 h) fasted before detergent injection. Tail blood samples was collected in capillary tubes, and plasma TG measured as above. The total lipoprotein fraction of pooled plasma (200 μl) was prepared by gel filtration chromatography using Superose 6 column (GE).
Dil-labeled VLDL uptake assay
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)-labeled VLDL uptake assay has been described (Altenburg et al., 2008; Johnson et al., 2011). Briefly, 50 μg Dil-labeled VLDL (Alfa Aesar) was intravenously injected into mice, plasma collected at 2, 10 and 20 minutes after injection, and Dil-labeled VLDL in plasma determined using a fluorescence microplate reader (EnVision, PerkinElmer). Livers were fixed with 4% paraformaldehyde 20 minutes later, incubated in 30% sucrose for cryopreservation, followed by embedding with cryo-embedding media (OCT; Tissue Tek). Slides from frozen liver sections were incubated with Hoechst 33342 to stain DNA and representative pictures taken with a fluorescence microscope coupled to an AxioCam camera.
Immunohistochemistry
Small intestine was fixed in 4% paraformaldehyde, incubated in 30% sucrose, then embedded with OCT. Slides from frozen sections were stained with Periodic acid-Schiff (PAS), and numbers of goblet cells and representative pictures were taken with a light microscope coupled to an AxioCam camera.
Lipoprotein lipase (LPL) and hepatic lipase (HL) activity assay
Post-heparin plasma was collected 5 minutes after an injection of heparin (100 U per kg body weight). Plasma total lipase was determined in triplicate using 10% intralipid/[3H]TG emulsion as a substrate. The contribution of HL was measured by including 1 M NaCl in the assay, and the activity of HL was subtracted from the total lipase activity to estimate the activity of LPL.
Western blotting
Liver and cellular lysates were obtained by centrifugation as previously described (Kim et al., 2016). Immunoblots were conducted on 3–5 samples randomly chosen within each experimental cohort with antibodies against Nicastrin (#5665), Presenilin 1 (#5643), Presenilin 2 (#9979), DYKDDDDK-tag (#14793), and β-actin (#4970) from Cell Signaling; mouse ApoC3 from Ionis; human ApoC3 (33A-R1a) from Academy Biomedical; human ApoB (MAB4124) and PCSK9 (AF3985) from R&D systems; ApoB (ab20737), LDLR (ab30532), Syndecan-1 (ab128936) from Abcam; and GFP (sc-9996) from Santa Cruz.
RNA/quantitative PCR
We isolated RNA with TRIzol (Invitrogen) or NucleoSpin RNA (Clontech), synthesized cDNA with the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) and performed quantitative RT-PCR with a Power SYBR Green PCR master mix (Promega) in a CFX96 Real-Time PCR detection system (Bio-Rad).
VLDL/LDL or human ApoC3-containing particle uptake assay
Purified human VLDLs or LDLs (Lee Biosolutions) or human ApoC3 (hApoC3)-containing plasma lipoproteins which were obtained from wild-type mice transduced with adenovirus encoding hApoC3, were applied to McA-RH7777 cells for 6 h in the presence of MG-132 and chloroquine. Residual hApoB or hApoC3 in the medium, as well as cell lysates were subjected to Western blot using an antibody specific to human ApoB or ApoC3.
Cell-surface biotinylation assay
Cell-surface biotinylation assay was performed according to the method established by Bretscher and Lutter (Bretscher and Lutter, 1988) with some modification. In brief, 90–95% confluent cells were washed with ice-cold PBS and incubated with 0.25 mg ml−1 EZ-link sulfo-NHS-SS-biotin (sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate; Thermo Scientific) at 4°C for 30 min. Cells were washed with ice-cold TBS, then incubated in complete DMEM at 37°C for the indicated times, washed in PBS and lys ed in TNT buffer. Lysates were cleared by centrifugation for 15 min at 14,000 rpm at 4°C. Ali quots were taken of the whole cell extracts, and the remainder of the lysates were incubated with immobilized NeutrAvidin beads slurry (Thermo Scientific). The NeutrAvidin beads were then washed four times with lysis buffer and boiled in SDS sample buffer, and the eluate analyzed by Western blot.
ASO and PCSK9 antibody studies
Control, Ncst and Ldlr ASOs were synthesized by Ionis Pharmaceuticals. Eight-week-old male C57BL/6 mice fed on normal chow or high fat diet were injected i.p. at 5 (Ldlr ASO) or 25 (Ncst ASO) mg per kg body weight once weekly for 6 weeks prior to sacrifice. Alirocumab at 10 mg per kg body weight was injected s.c. once weekly for 3 weeks into HFD-fed control and Ncst ASO-treated mice, prior to sacrifice.
Quantification and statistical analysis
Based on preliminary data from GSI-treated mice, we calculated that 6–7 mice/group would be needed to detect a 25% difference in plasma TG in L-Ncst, iL-Ncst and Ncst ASO-treated mice, with a power of 0.8 and p<0.05. To assess statistical significance, we performed an unpaired two-tailed t test for comparison of 2 groups, or ANOVA followed by unpaired two-tailed t or Tukey tests for studies involving multiple groups. No animals or data was excluded from any analysis. All data shown as mean ± s.e.m. Sample size and statistical details can be found in the figure legends.
Supplementary Material
Highlights.
Nicastrin targets LDLR for γ-secretase-mediated cleavage
Blocking hepatocyte γ-secretase increases liver LDLR stability
γ-secretase inhibitors increase TRL uptake to reduce plasma TG
Systemic or liver-selective γ-secretase inhibitors improve glucose metabolism
Acknowledgments
We thank A. Flete and T. Kolar for excellent technical support, as well as Drs., Jeffrey Esko, Philip Gordts, Jiandie D. Lin, Lale Ozcan, Nobuyo Maeda and Lance Johnson for insightful discussion and various reagents. This work was supported by NIH DK103818 (UBP), HL46095 (IJG), HL92969 (IJG) and HL125649 (RAH), an Edward Mallinckrodt, Jr. Foundation Grant (UBP), a Paul Marks Scholarship (UBP), the Canadian Institutes of Health Research Grant #MOP 123279 (ZY), and an AHA Scientist Development Grant 17SDG33660031 (KK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, K.K. and U.B.P.; Methodology, K.K. and U.B.P.; Investigation, K.K., I.J.G., M.J.G., M.S., E.B., S.L., Z.Y. and U.B.P.; Resources, L.Q., D.M. and P.C.; Writing – Original Draft, K.K. and U.B.P.; Writing – Review & Editing, K.K., I.J.G., R.A.H., H.N.G. and U.B.P.; Supervision, U.B.P.; Funding Acquisition, U.B.P., I.J.G., R.A.H., Z.Y. and K.K.
DECLARATION OF INTERESTS
The authors declare that they have no competing financial interest in the work described, but UBP is an inventor of a patent held by Columbia University related to this work.
References
- Adiels M, Boren J, Caslake MJ, Stewart P, Soro A, Westerbacka J, Wennberg B, Olofsson SO, Packard C, Taskinen MR. Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1697–1703. doi: 10.1161/01.ATV.0000172689.53992.25. [DOI] [PubMed] [Google Scholar]
- Altenburg M, Arbones-Mainar J, Johnson L, Wilder J, Maeda N. Human LDL receptor enhances sequestration of ApoE4 and VLDL remnants on the surface of hepatocytes but not their internalization in mice. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:1104–1110. doi: 10.1161/ATVBAHA.108.164863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson ER, Lendahl U. Therapeutic modulation of Notch signaling–are we there yet? Nature reviews. Drug discovery. 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- Arsenault BJ, Pelletier-Beaumont E, Almeras N, Tremblay A, Poirier P, Bergeron J, Despres JP. PCSK9 levels in abdominally obese men: association with cardiometabolic risk profile and effects of a one-year lifestyle modification program. Atherosclerosis. 2014;236:321–326. doi: 10.1016/j.atherosclerosis.2014.07.010. [DOI] [PubMed] [Google Scholar]
- Aster JC, Blacklow SC. Targeting the Notch pathway: twists and turns on the road to rational therapeutics. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2418–2420. doi: 10.1200/JCO.2012.42.0992. [DOI] [PubMed] [Google Scholar]
- Ayyobi AF, Brunzell JD. Lipoprotein distribution in the metabolic syndrome, type 2 diabetes mellitus, and familial combined hyperlipidemia. The American journal of cardiology. 2003;92:27j–33j. doi: 10.1016/s0002-9149(03)00613-1. [DOI] [PubMed] [Google Scholar]
- Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annual review of pharmacology and toxicology. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- Berger JM, Loza Valdes A, Gromada J, Anderson N, Horton JD. Inhibition of PCSK9 does not improve lipopolysaccharide-induced mortality in mice. Journal of lipid research. 2017;58:1661–1669. doi: 10.1194/jlr.M076844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi P, Kuang S. Notch signaling as a novel regulator of metabolism. Trends in endocrinology and metabolism: TEM. 2015;26:248–255. doi: 10.1016/j.tem.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JR, Stanford KI, Esko JD. Heparan sulfate proteoglycans and triglyceride-rich lipoprotein metabolism. Current opinion in lipidology. 2008;19:307–313. doi: 10.1097/MOL.0b013e3282feec2d. [DOI] [PubMed] [Google Scholar]
- Bretscher MS, Lutter R. A new method for detecting endocytosed proteins. The EMBO journal. 1988;7:4087–4092. doi: 10.1002/j.1460-2075.1988.tb03302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkan AL, Goate AM. Presenilin function and gamma-secretase activity. Journal of neurochemistry. 2005;93:769–792. doi: 10.1111/j.1471-4159.2005.03099.x. [DOI] [PubMed] [Google Scholar]
- Brunzell JD. Clinical practice. Hypertriglyceridemia. The New England journal of medicine. 2007;357:1009–1017. doi: 10.1056/NEJMcp070061. [DOI] [PubMed] [Google Scholar]
- Brunzell JD, Ayyobi AF. Dyslipidemia in the metabolic syndrome and type 2 diabetes mellitus. The American journal of medicine. 2003;115(Suppl 8A):24s–28s. doi: 10.1016/j.amjmed.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends in endocrinology and metabolism: TEM. 2011;22:353–363. doi: 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A, Blatchford PJ, Quackenbush K, Messersmith W. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Investigational new drugs. 2014;32:739–745. doi: 10.1007/s10637-014-0083-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- de Vera Mudry MC, Regenass-Lechner F, Ozmen L, Altmann B, Festag M, Singer T, Muller L, Jacobsen H, Flohr A. Morphologic and functional effects of gamma secretase inhibition on splenic marginal zone B cells. International journal of Alzheimer’s disease. 2012;2012:289412. doi: 10.1155/2012/289412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nature reviews Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. The New England journal of medicine. 2013;369:341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- Dutta D, Williamson CD, Cole NB, Donaldson JG. Pitstop 2 is a potent inhibitor of clathrin-independent endocytosis. PloS one. 2012;7:e45799. doi: 10.1371/journal.pone.0045799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett BM, Smith RJ, Hiatt WR. Reducing LDL with PCSK9 Inhibitors–The Clinical Benefit of Lipid Drugs. The New England journal of medicine. 2015;373:1588–1591. doi: 10.1056/NEJMp1508120. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. The Journal of biological chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- Foley EM, Gordts PL, Stanford KI, Gonzales JC, Lawrence R, Stoddard N, Esko JD. Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:2065–2074. doi: 10.1161/ATVBAHA.113.301637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ, Chudnovskiy A, Yamazaki H, Croce K, Weissleder R, et al. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1868–1877. doi: 10.1073/pnas.1116889109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology (Baltimore, Md) 2015;61:382–392. doi: 10.1002/hep.27268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wang-Iverson P, Norum R, Brown WV. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. The Journal of clinical investigation. 1986;78:1287–1295. doi: 10.1172/JCI112713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. The Journal of clinical investigation. 2016;126:2855–2866. doi: 10.1172/JCI86610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Proteomic profiling of gamma-secretase substrates and mapping of substrate requirements. PLoS biology. 2008;6:e257. doi: 10.1371/journal.pbio.0060257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C, Molusky M, Li Y, Li S, Lin JD. Regulation of hepatic ApoC3 expression by PGC-1beta mediates hypolipidemic effect of nicotinic acid. Cell metabolism. 2010;12:411–419. doi: 10.1016/j.cmet.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Azrolan N, O’Connell A, Walsh A, Breslow JL. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science (New York, NY) 1990;249:790–793. doi: 10.1126/science.2167514. [DOI] [PubMed] [Google Scholar]
- Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, Gonzalez FJ. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes. 2011;60:2484–2495. doi: 10.2337/db11-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Arbones-Mainar JM, Fox RG, Pendse AA, Altenburg MK, Kim HS, Maeda N. Apolipoprotein E4 exaggerates diabetic dyslipidemia and atherosclerosis in mice lacking the LDL receptor. Diabetes. 2011;60:2285–2294. doi: 10.2337/db11-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong MC, Hofker MH, Havekes LM. Role of ApoCs in lipoprotein metabolism: functional differences between ApoC1, ApoC2, and ApoC3. Arteriosclerosis, thrombosis, and vascular biology. 1999;19:472–484. doi: 10.1161/01.atv.19.3.472. [DOI] [PubMed] [Google Scholar]
- Kim K, Qiang L, Hayden MS, Sparling DP, Purcell NH, Pajvani UB. mTORC1-independent Raptor prevents hepatic steatosis by stabilizing PHLPP2. Nature communications. 2016;7:10255. doi: 10.1038/ncomms10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Ryu D, Dongiovanni P, Ozcan L, Nayak S, Ueberheide B, Valenti L, Auwerx J, Pajvani UB. Degradation of PHLPP2 by KCTD17, via a Glucagon-Dependent Pathway, Promotes Hepatic Steatosis. Gastroenterology. 2017;153:1568–1580.e1510. doi: 10.1053/j.gastro.2017.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace TA. PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells. Current opinion in lipidology. 2014;25:387–393. doi: 10.1097/MOL.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH. Genetic and metabolic determinants of plasma PCSK9 levels. The Journal of clinical endocrinology and metabolism. 2009;94:2537–2543. doi: 10.1210/jc.2009-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal M, Caplan M. Regulated intramembrane proteolysis: signaling pathways and biological functions. Physiology (Bethesda, Md) 2011;26:34–44. doi: 10.1152/physiol.00028.2010. [DOI] [PubMed] [Google Scholar]
- Le NA, Gibson JC, Ginsberg HN. Independent regulation of plasma apolipoprotein C-II and C-III concentrations in very low density and high density lipoproteins: implications for the regulation of the catabolism of these lipoproteins. Journal of lipid research. 1988;29:669–677. [PubMed] [Google Scholar]
- Lee YH, Yun MR, Kim HM, Jeon BH, Park BC, Lee BW, Kang ES, Lee HC, Park YW, Cha BS. Exogenous administration of DLK1 ameliorates hepatic steatosis and regulates gluconeogenesis via activation of AMPK. International journal of obesity. 2016;40:356–365. doi: 10.1038/ijo.2015.173. 2005. [DOI] [PubMed] [Google Scholar]
- Li X, Zhu M, Penfold ME, Koenen RR, Thiemann A, Heyll K, Akhtar S, Koyadan S, Wu Z, Gremse F, et al. Activation of CXCR7 limits atherosclerosis and improves hyperlipidemia by increasing cholesterol uptake in adipose tissue. Circulation. 2014;129:1244–1253. doi: 10.1161/CIRCULATIONAHA.113.006840. [DOI] [PubMed] [Google Scholar]
- MacArthur JM, Bishop JR, Stanford KI, Wang L, Bensadoun A, Witztum JL, Esko JD. Liver heparan sulfate proteoglycans mediate clearance of triglyceride-rich lipoproteins independently of LDL receptor family members. The Journal of clinical investigation. 2007;117:153–164. doi: 10.1172/JCI29154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Li H, Lee D, Oliver P, Quarfordt SH, Osada J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. The Journal of biological chemistry. 1994;269:23610–23616. [PubMed] [Google Scholar]
- Mamo JC, Watts GF, Barrett PH, Smith D, James AP, Pal S. Postprandial dyslipidemia in men with visceral obesity: an effect of reduced LDL receptor expression? American journal of physiology. Endocrinology and metabolism. 2001;281:E626–632. doi: 10.1152/ajpendo.2001.281.3.E626. [DOI] [PubMed] [Google Scholar]
- Masucci-Magoulas L, Goldberg IJ, Bisgaier CL, Serajuddin H, Francone OL, Breslow JL, Tall AR. A mouse model with features of familial combined hyperlipidemia. Science (New York, NY) 1997;275:391–394. doi: 10.1126/science.275.5298.391. [DOI] [PubMed] [Google Scholar]
- Millar JS, Cromley DA, McCoy MG, Rader DJ, Billheimer JT. Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR-1339. Journal of lipid research. 2005;46:2023–2028. doi: 10.1194/jlr.D500019-JLR200. [DOI] [PubMed] [Google Scholar]
- Nishina PM, Lowe S, Wang J, Paigen B. Characterization of plasma lipids in genetically obese mice: the mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism: clinical and experimental. 1994;43:549–553. doi: 10.1016/0026-0495(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. Jama. 2007;298:299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- Norum RA, Lakier JB, Goldstein S, Angel A, Goldberg RB, Block WD, Noffze DK, Dolphin PJ, Edelglass J, Bogorad DD, et al. Familial deficiency of apolipoproteins A-I and C-III and precocious coronary-artery disease. The New England journal of medicine. 1982;306:1513–1519. doi: 10.1056/NEJM198206243062503. [DOI] [PubMed] [Google Scholar]
- Pajvani UB, Qiang L, Kangsamaksin T, Kitajewski J, Ginsberg HN, Accili D. Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nature medicine. 2013;19:1054–1060. doi: 10.1038/nm.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J, Accili D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nature medicine. 2011;17:961–967. doi: 10.1038/nm.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, Post W, McLenithan JC, Bielak LF, Peyser PA, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science (New York, NY) 2008;322:1702–1705. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Real PJ, Ferrando AA. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia. 2009;23:1374–1377. doi: 10.1038/leu.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, Sulis ML, Barnes K, Sawai C, Homminga I, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nature medicine. 2009;15:50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. The New England journal of medicine. 2015;372:1489–1499. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- Schonfeld G, George PK, Miller J, Reilly P, Witztum J. Apolipoprotein C-II and C-III levels in hyperlipoproteinemia. Metabolism: clinical and experimental. 1979;28:1001–1010. doi: 10.1016/0026-0495(79)90004-0. [DOI] [PubMed] [Google Scholar]
- Schuler M, Dierich A, Chambon P, Metzger D. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis (New York, NY: 2000) 2004;39:167–172. doi: 10.1002/gene.20039. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiological reviews. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shih Ie M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer research. 2007;67:1879–1882. doi: 10.1158/0008-5472.CAN-06-3958. [DOI] [PubMed] [Google Scholar]
- Sparling DP, Yu J, Kim K, Zhu C, Brachs S, Birkenfeld AL, Pajvani UB. Adipocyte-specific blockade of gamma-secretase, but not inhibition of Notch activity, reduces adipose insulin sensitivity. Molecular metabolism. 2016;5:113–121. doi: 10.1016/j.molmet.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, Witztum JL, Esko JD. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. The Journal of clinical investigation. 2009;119:3236–3245. doi: 10.1172/JCI38251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram M, Zhong S, Bou Khalil M, Links PH, Zhao Y, Iqbal J, Hussain MM, Parks RJ, Wang Y, Yao Z. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. Journal of lipid research. 2010;51:150–161. doi: 10.1194/jlr.M900346-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Chen G, Sudhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:7290–7301. doi: 10.1523/JNEUROSCI.1320-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita T, Watabiki T, Takikawa R, Morohashi Y, Takasugi N, Kopan R, De Strooper B, Iwatsubo T. The first proline of PALP motif at the C terminus of presenilins is obligatory for stabilization, complex formation, and gamma-secretase activities of presenilins. The Journal of biological chemistry. 2001;276:33273–33281. doi: 10.1074/jbc.M011152200. [DOI] [PubMed] [Google Scholar]
- Tveten K, Strom TB, Berge KE, Leren TP. PCSK9-mediated degradation of the LDL receptor generates a 17 kDa C-terminal LDL receptor fragment. Journal of lipid research. 2013;54:1560–1566. doi: 10.1194/jlr.M034371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti L, Mendoza RM, Rametta R, Maggioni M, Kitajewski C, Shawber CJ, Pajvani UB. Hepatic notch signaling correlates with insulin resistance and nonalcoholic fatty liver disease. Diabetes. 2013;62:4052–4062. doi: 10.2337/db13-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Driel IR, Davis CG, Goldstein JL, Brown MS. Self-association of the low density lipoprotein receptor mediated by the cytoplasmic domain. The Journal of biological chemistry. 1987;262:16127–16134. [PubMed] [Google Scholar]
- van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- Villa JC, Chiu D, Brandes AH, Escorcia FE, Villa CH, Maguire WF, Hu CJ, de Stanchina E, Simon MC, Sisodia SS, et al. Nontranscriptional role of Hif-1alpha in activation of gamma-secretase and notch signaling in breast cancer. Cell reports. 2014;8:1077–1092. doi: 10.1016/j.celrep.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Vujovic S, Henderson SR, Flanagan AM, Clements MO. Inhibition of gamma-secretases alters both proliferation and differentiation of mesenchymal stem cells. Cell proliferation. 2007;40:185–195. doi: 10.1111/j.1365-2184.2007.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tran K, Yao Z. The activity of microsomal triglyceride transfer protein is essential for accumulation of triglyceride within microsomes in McA-RH7777 cells. A unified model for the assembly of very low density lipoproteins. The Journal of biological chemistry. 1999;274:27793–27800. doi: 10.1074/jbc.274.39.27793. [DOI] [PubMed] [Google Scholar]
- Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Molecular cancer therapeutics. 2010;9:1618–1628. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. The gamma-secretase complex: membrane-embedded proteolytic ensemble. Biochemistry. 2006;45:7931–7939. doi: 10.1021/bi060799c. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Kopan R. Intramembrane proteolysis: theme and variations. Science (New York, NY) 2004;305:1119–1123. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science (New York, NY) 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C. Updates on apolipoprotein CIII: fulfilling promise as a therapeutic target for hypertriglyceridemia and cardiovascular disease. Current opinion in lipidology. 2014;25:35–39. doi: 10.1097/MOL.0000000000000040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.