

Abstract
Gram-negative bacteria comprise the majority of microbes that cause infections that are resistant to pre-existing antibiotics. The complex cell wall architecture contributes to their ability to form biofilms, which are often implicated in hospital-acquired infections. Biofilms promote antibiotic resistance by enabling the bacteria to survive hostile environments such as UV radiation, pH shifts, and antibiotics. The outer membrane of Gram-negative bacteria contains lipopolysaccharide (LPS), which plays a role in adhesion to surfaces and formation of biofilms. The main focus of this work was the synthesis of a library of glycolipids designed to be simplified analogues of the Lipid A, the membrane embedded portion component of LPS, to be tested as substrates or inhibitors of Heptosyltransferase I (HepI or WaaC, a glycosyltransferase enzyme involved in the biosynthesis of LPS). Fourteen analogues were synthesized successfully and characterized. While these compounds were designed to function as nucleophilic substrates of HepI, they all demonstrated mild inhibition of HepI. Kinetic characterization of inhibition mechanism identified that the compounds exhibited uncompetitive and mixed inhibition of HepI. Since both uncompetitive and mixed inhibition result in the formation of an Enzyme-Substrate-inhibitor complex, molecular docking studies (using AutoDock Vina) were performed, to identify potential allosteric binding site for these compounds. The inhibitors were shown to bind to a pocket formed after undergoing a conformational change from an open to a closed active site state. Inhibition of HepI via an allosteric site suggest that disruption of protein dynamics might be a viable mechanism for the inhibition of HepI and potentially other enzymes of the GT-B structural class.
Keywords: LPS, Lipopolysaccharide, Heptosyltransferase, Glycosyltransferase, GT-B, Inhibitor Design, AutoDock Vina, Inhibition Kinetics, Structural Rearrangement, Allosterism
Graphical abstract
Highlights
Alkylated monosaccharides inhibit Heptosyltransferase I
Compounds are uncompetitive with sugar acceptor substrate
Compounds have mixed inhibition with sugar donor substrate
Docking revealed putative allosteric inhibition pocket
As the number of drug resistant (and multiple drug resistant) bacteria continue to increase, the need for novel drug targets becomes increasingly pressing.1 The outer membrane (OM) of Gram-negative bacteria provides a crucial layer of protection against many antibiotics.2 Bacterial lipopolysaccharide (LPS), also known as endotoxin, is a major component of the OM. LPS is important for interactions with other cells, as in its interaction with human lipopolysaccharide binding protein to trigger a TLR-4 immune response,3–5 and it maintains structurally similar architecture throughout Gram-negative bacteria (but with variation of the specific sugars incorporated) regardless of the ecological niche or growth conditions of the bacteria.6 LPS consists of three domains – a glycolipid known as Lipid A, which is covalently linked to the core oligosaccharide, which is, in turn, linked to a polysaccharide known as the O-antigen (Figure 1).7, 8 All three components of LPS are required for the virulence of Gram-negative bacteria, but only Lipid A is required for survival of the bacteria. Studies have shown that Lipid A is the minimal LPS structure required for growth of E. coli and other Gram-negative bacteria.9, 10
Figure 1.

Representative structure of LPS from E. coli. (Hep) L-glycero-D-manno-heptose; (Gal) galactose; (Glc) glucose; (KDO) 3-deoxy-D-manno-oct-2-ulosonic acid; (NGa) N-acetyl- galactosamine; (NGc) N-acetyl-glucosamine. Modified from Petsch and Anspach.20
Heptosyltransferase I (HepI, also known as WaaC) is a member of the GT-B structural family and it is involved in the synthesis of the inner core of LPS.11 The enzyme is known to catalyze the addition of the first L-glycero-D-manno-heptose (Hep) to the inner 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) residue of the Kdo2-Lipid A molecule in LPS.9, 11–13 HepI is an inverting glycosyltransferase, which utilizes a direct-displacement reaction mechanism.11, 14–16 HepI has been observed to undergo conformational changes associated with substrate binding and like other GT-B enzymes it is predicted to undergo an open-to-closed conformational transition before catalysis.11, 15–19
Previous studies in E. coli, Campylobacter jejuni and other bacteria have shown that HepI knockouts can produce viable mutant cell lines that synthesize a truncated LPS that lack Heptose residues (and the subsequent sugars) and display a deep-rough phenotype.9, 11–13, 21 Increased sensitivity to hydrophobic antibiotics, detergents and bile salts and decreased virulence for pathogenic Gram-negative bacteria have been associated with this rough phenotype.9, 21, 22 Based on these observations, HepI has been the target for drug design from multiple labs.23–26 No efforts to date have identified potent inhibitors of HepI, leading our group to investigate the overall chemical and dynamical mechanism.17, 27, 28
HepI has been shown to efficiently utilize a number of Kdo2-Lipid A analogue molecules as acceptor substrates (nucleophiles), including a partially deacylated (ODLA) and a fully deacylated (FDLA) Kdo2-Lipid A molecule.28 These findings raised questions on the diversity of substrate nucleophiles that would be tolerated by HepI. In this work, a series of acylated monosaccharide molecules were designed as Lipid A analogues (Figure 2) in the hope of identifying a minimal substrate. The alkyl chains of the analogues were expected to mimic the acyl chains of the Lipid-A molecule, while the sugar residues mimic the Kdo residues of Lipid A. From a stereochemical perspective, galactose analogues were predicted to be the best nucleophiles because they share the stereoconfiguration of Kdo at the C-4 position.
Figure 2.

Structural comparison of E. coli Kdo2-Lipid A (A) and Lipid A analogues (B and C). The sugar residues of the analogues are labeled in red and they are expected to mimic the Kdo residues of Kdo2-Lipid A, also labeled in red. The alkyl chains of the analogues are expected to mimic one of the acyl chains of Kdo2-Lipid A. One of the acyl chains in A is also labeled in blue.
The synthesis of these compounds builds off of the literature on the synthesis of allyl glycosides, with further functionalization of these compounds through metatheses of a series of 1-alkenes of different length tails. Fourteen alkylated sugar compounds were successfully synthesized using glucose and galactose as starting materials (Figure 3 and S1, S2). Each of the syntheses began with per-acetylation of the sugar starting material in reactions of modest yield (47 % and 53 % for Glucose pentaacetate (15) and galactose pentaacetate (16), respectively).29, 30 After acetylation, nucleophilic bromide was used to displace acetate from the C-1 position using two slightly different methods previously published by Rodebaugh and Fraser-Reid, and by Ella-Menye, for the glucose (17) and galactose (18) forms, respectively.30, 31 Using the α-bromo sugar substrates, two different sets of allylation conditions could be used to afford product with either retention or inversion of configuration at the anomeric position. β-allyl glucose tetraacetate (19) and β-allyl galactose tetraacetate (20) were synthesized according to a method by Talley et al. after unsuccessful attempts using methods of Rodebaugh and Fraser-Reid and Yoon et al.31,32,33 The α-allyl glucose tetraacetate (21) and α-allyl galactose tetraacetate (22) were synthesized following the protocol by Lemieux and Morgan,34 which used 17 or 18, 2,4,6-trimethylpyridine, tetrabutylammonium bromide and ethanol to achieve displacement of the anomeric bromine by the allylic alcohol nucleophile.
Figure 3.

Complete list of simplified Lipid A analogues.
Various 1-alkenes (1-decene, 1-octene, 1-heptene and 1-hexene) were used for metathesis reactions to elongate the allyl-containing alkyl chains in both glucose and galactose analogues. The β-metathesis syntheses involved the reaction between compounds 19 – 22 and the corresponding 1-alkene in the presence of Grubbs catalyst using the methods published by Plettenburg et al.35 The 2nd generation Grubbs catalyst was used for reactions with 1-decene and 1-octene whereas the 1st generation Grubbs was used for all metatheses with 1-heptene and 1-hexene. The α-metathesis reactions were performed using the same methods, however, the reactions that were performed using the 2nd generation Grubbs catalyst produced very low yields. Use of the 1st generation Grubbs catalyst significantly improved yields, perhaps due to less steric crowding than with the 2nd generation Grubbs catalyst.
Deacetylation of the α- and β-metathesis products was performed successfully using a 3:3:1 solution of methanol, water, and triethylamine.35 The structures of all compounds were confirmed by 1H, COSY, and 13C NMR. Additionally, compounds 1–12, 23–26 and 31– 34 were also analyzed by electro-spray mass spectrometry (ES-MS). The 1H, COSY, and 13C NMR, as well as the ES-MS spectra are shown in Supplemental Materials.
Once synthesized, all compound were first screened to determine if they could be used as substrates by HepI. Having previously determined that HepI can utilize a tetrasaccharide as its acceptor substrate (nucleophile), we wanted to ascertain if a monosaccharide could also be utilized as a substrate.28 Prior to testing the alkylated monosaccharides, galactose and glucose as well as α- and β-methyl glycosides were first used to see if the deacylated monosaccharides could serve as a substrate. None of these commercial compounds promoted the HepI glycosyltransferase reaction. The compound library, synthesized above, were then examined, and there was also no detectable HepI activity for any of these compounds (data not shown). Retrospectively, this is not surprising because multiple GTs, like many enzymes, had previously demonstrated high substrate utilization fidelity.15, 16, 36 While glucose and galactose are somewhat similar to KDO, these sugars lacks a carboxylic acid as well as C7 and C8 positions. Additionally, glucose and galactose are both hydroxylated at the C2 position while KDO is dehydroxylated at the corresponding ring carbon (the C2 position in glucose or galactose corresponds to C3 in KDO).
The molecules were then screened to determine if they could inhibit HepI catalysis with the substrates, ODLA (the partially deacylated Kdo2-Lipid A) and ADP-Heptose. Each molecule, to some extent, showed inhibition of HepI activity (Table 1). Interestingly, side by side comparison of each glucose compound with its galactose analogue revealed that in every case except for the octene-β-glycoside, the glucose compounds demonstrated a higher level of inhibition than the galactose compounds. Also of interest, the commercially available detergent n-Octyl-β-D-glucopyranoside also showed a small degree of inhibition of HepI. Ki values were determined for all compounds that exhibited inhibition ≥ 20% (Table 1). The smallest Ki values were in the high micromolar range, with the best alkylated monosaccharide hexene-β-D-glucopyranoside (7) giving rise to a Ki of 340 μM. D-glucose and methyl-α-D-glucopyranoside demonstrated similar levels of inhibition. All of these compounds bind with lower affinity than the substrates, which have Km values between 3–10 μM.11, 27, 28 Since compound 7 was the acylated monosaccharide with the lowest Ki, it was used to determine if these compounds are competitive, noncompetitive, or uncompetitive inhibitors with either the donor (ADP-Heptose) or the acceptor (ODLA) substrates of HepI. Inhibition by 7 displayed noncompetitive (mixed-competitive) inhibition against ADP-Heptose as indicated by the decrease in Vmax as inhibitor concentration increases and a characteristic double reciprocal plot (Figure 4A) where lines intersect at a point not on the y-axis. A noncompetitive inhibitor by definition has binding affinity for both free enzyme and enzyme substrate (ES) complex. The equation describing this inhibition, v = vmax[S]ks(1+[I]ki)+[S](1+[I]αki), includes an additional term α used to distinguish if an inhibitor has a preference for either free enzyme (α>1), ternary complex (α<1) or no preference (α=1).37 As inhibitor concentration increases, the changes in Km (increase, decrease or unaffected) can be used to distinguish if α is >1, <1, or =1, respectively. As concentration of 7 increase, Km decreases, thus α<1, suggesting that 7 prefers to bind to the ES complex (Table 2). Unlike ADP-Heptose, inhibition by 7 against ODLA resulted in a double reciprocal plot with parallel lines diagnostic of uncompetitive inhibition; this is also evident by the decrease in both Km and vmax but no change in vmax/Km (Figure 4B and Table 2). The uncompetitive inhibition profile means that 7 binds exclusively to the ES complex when ODLA concentrations are varied. Since inhibition experiments with both substrates suggest a preference for these compounds to bind to the ES complex, this suggests that these compounds are binding to a putative allosteric site in HepI.
Table 1.
Inhibition parameters for modified monosccharide compound library
| Compound | % Inhibition | Ki (μM) | |
|---|---|---|---|
| D-galactose | - | 28.5 | 500 ± 260 |
| methyl-α-D-galactopyranoside | - | 8.8 | |
| methyl-β-D-galactopyranoside | - | 7 | |
| allyl-β-D-galactopyranoside | 14 | 10 | |
| hexene-β-D-galactopyranoside | 8 | 22.6 | 570 ± 240 |
| heptene-β-D-galactopyranoside | 6 | 7.8 | |
| octene-α-D-galactopyranoside | 12 | 8.8 | |
| octene-β-D-galactopyranoside | 4 | 6.3 | |
| decene-α-D-galactopyranoside | 10 | NA | |
| s-β-D-galactopyranoside | 2 | 5 | |
| D-glucose | - | 36 | 280 ± 200 |
| methyl-α-D-glucopyranoside | - | 27.8 | 370 ± 160 |
| methyl-β-D-glucopyranoside | - | 19.5 | 680 ± 370 |
| allyl-β-D-glucopyranoside | 13 | 19.6 | 650 ± 410 |
| n-Octyl-β-D-glucopyranoside | - | 8.3 | |
| hexene-β-D-glucopyranoside | 7 | 29.7 | 340± 150 |
| heptene-β-D-glucopyranoside | 5 | 11.9 | |
| octene-α-D-glucopyranoside | 11 | 16.7 | |
| octene-β-D-glucopyranoside | 3 | 4.2 | |
| decene-α-D-glucopyranoside | 9 | NA | |
| decene-β-D-glucopyranoside | 1 | 23.4 | 520 ± 280 |
Figure 4.

Lineweaver-Burk plots for compound 7 a) against ADP-Heptose and b) against ODLA. Each point represents an average of a triplicate set of data.
Table 2.
Kinetics parameters for compound 7.
| ADP-Heptose | ODLA | |||
|---|---|---|---|---|
| [Inhibitor], μM | KM (μM) | kcat (s−1) | KM (μM) | kcat (s−1) |
| 1000 | 38 ±10 | 0.76 ±0.08 | 65 ±27 | 0.9 ±0.2 |
| 100 | 135 ±19 | 1.6 ±0.1 | 108.3 ±0.6 | 1.3 ±0.004 |
| 10 | 277 ±40 | 2.8 ±0.3 | 114 ±4 | 1.4 ±0.03 |
To gain a better idea as to where the compounds bind on HepI, dockings of these alkylated monosaccharides were conducted using AutoDock Vina v1.1.2. The library of compounds were docked into (1) the “open” apo HepI structure (2GT1.pdb), (2) the “open” HepI•ADP-Heptose complex structure (2H1H.pdb) and (3) a “closed” apo model, previously developed.17 In addition, four additional protein-ligand complexes were evaluated, where each contained one of the substrate molecules (ADP-Heptose bound to the C-terminal domain or FDLA, the fully deacylated Kdo2-Lipid A analogue which by virtue of the removal of the acyl chains is an easier compound to use for docking, was docked into the N-terminal domain) complexed with the apo form of 2H1H and the closed model of HepI. All dockings were performed within four grid boxes: (a) containing the entire protein, (b) the part of the active site contacted by Lipid A (the N-terminal domain), (c) the part contacted by ADP-Heptose, and (d) the entire active site (Table S1). The active site boxes were designed as to include the entire ligand and any residues within five Å of the ligand in question. Dockings to these structures were performed to assess the effect of interactions with the endogenous substrates and observe any differences in binding that might occur when the compounds are physically displaced from either binding site.
The average energies of the alkylated monosaccharides were generally in the range of -4.5 to -6 kcal/mol, with binding energy minimums only slightly lower (Tables S2 and S3). There was a definite trend in energies that shorter-chained alkylated monosaccharides had somewhat weaker binding affinities. It was also notable that the control dockings of substrates showed a significantly higher affinity than any of the inhibitors; ADP-Heptose was the tightest binding ligand (average energy: −8.0 kcal/mol; minimum energy: −8.9 kcal/mol), while FDLA also showed lower affinity than the library compounds (average energy: −7.2 kcal/mol; minimum energy: −8.4 kcal/mol). This observation is unsurprising as all compounds were already shown experimentally to have lower affinity than the Lipid A analogues.
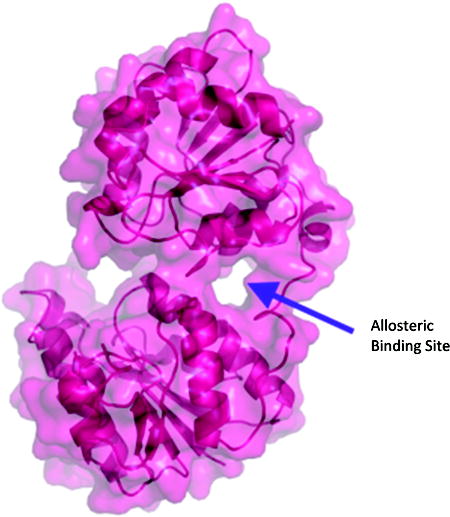
The binding energies and binding sites of the ligands bound are highly conserved regardless of the structure to which they were docked. Figure S1 shows the general patterns for docking to 2H1H. The majority of ligands, when docked, bound to the ADP-Heptose site. Interestingly, this trend is unaffected by whether the starting protein PDB contained one of the substrates. The results were substantially different when the protein used was the closed model of HepI. Perhaps most significantly, a putative allosteric site was identified. This site, no analogue of which exists in 2H1H or 2GT1, is a “tunnel” between the ADP-Heptose binding site and the linker region between the N- and C-terminal domains of HepI, which forms during the simulatated closure of HepI (where upon the two domains coming together to bring the substrates proximal; Figure 5). The site was identified when super imposing images of dockings to this closed structure showed significant clustering of docking poses in this site. It was therefore included in the binding site analyses alongside the ADP-Heptose and FDLA sites.
Figure 5.

Putative Allosteric site in the closed model of HepI. When viewed from the side with the “surface” view activated, the allosteric site appears as a tunnel between the linker region (at the right side) and the spade between the two domains. No such gap exists in the structures of 2H1H or 2GT1.
Generally, longer-chain ligands preferentially bound in the allosteric site and shorter-chain ligands bound more in the ADP-Heptose site, e.g. decene-α-D-glucopyranoside with 95 poses in the ADP-Heptose site, 9 in FDLA site, and 340 in the allosteric site, versus D-galactose with 188 poses in the ADP-Heptose site, 1 in the FDLA site, and 81 in the allosteric site. The substrates, understandably, defied these trends, with nearly 100% of FDLA poses binding in both the ADP-Heptose and FDLA sites, and a negligible number in the allosteric site. ADP-Heptose bound primarily (~75%) in the ADP-Heptose site, with the remainder being split approximately half in each the FDLA and allosteric sites.
Additionally, the putative allosteric site is also significant in understanding the role of conformational change in the dynamics of HepI. Although there is no crystal structure of a ternary HepI complex, the existence of such a conformational change is supported by analogous changes in related GT-B enzymes.38, 39 Previous dynamic studies on HepI, have suggested that a conformational change is induced by the binding of Lipid A. 17, 27 Although the alkylated monosaccharides were designed with the idea of creating a substrate or competitive inhibitor to bind and displace Lipid A, the docking data suggest that they may primarily inhibiting the ES complex in an uncompetitive (e.g. by binding to and somehow altering the closed conformation such that catalysis in impeded) and/or mixed-competitive (both competitive and uncompetitive) manner. These findings are consistent with the kinetics data, since both non-competitive and uncompetitive inhibition was observed. Thus, the putative allosteric site represents a compelling future direction in understanding the inhibition of HepI and other GT-B structural enzymes.
In conclusion, this work demonstrates through experimental and computational studies that the library of compounds herein bind to HepI in a previously undescribed allosteric pocket. For enzymes that undergo conformational changes during catalysis, this analysis highlights that the disruption of protein dynamics can be an important mechanism for the design of inhibitors. While these inhibitors were not particularly potent, it is understandable that inhibitors for this allosteric site wouldn’t necessarily have the same structural features as the substrates. This result is important for consideration when trying to inhibit HepI, GT-B structural enzymes, as well as other enzymes that undergo conformational changed during chemistry.
Supplementary Material
Abbreviations
- GT
glycosyltransferase
- HepI
heptosyltransferase I
- Heptose
L-glycero-D-manno-heptose
- ADP-Heptose
ADP- L-glycero-D-manno-heptose
- LPS
lipopolysaccharide
- ODLA
O-deacylated E. coli Kdo2-LipidA
- FDLA
fully-deacylated E. coli Kdo2-LipidA
Footnotes
This work was supported by NIH grants 1R15AI119907-01.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ventola CL. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharmacy and Therapeutics. 2015;40(4):277–283. [PMC free article] [PubMed] [Google Scholar]
- 2.Delcour AH. Outer membrane permeability and antibiotic resistance. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2009;1794(5):808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen VT, Barlow RS, Fegan N, Turner MS, Dykes GA. Role of Capsular Polysaccharides and Lipooligosaccharides in Campylobacter Surface Properties, Autoagglutination, and Attachment to Abiotic Surfaces. Foodborne Pathogens and Disease. 2013;10(6):506–513. doi: 10.1089/fpd.2012.1365. [DOI] [PubMed] [Google Scholar]
- 4.Schumann Ralf R. Old and new findings on lipopolysaccharide-binding protein: a soluble pattern-recognition molecule. Biochemical Society Transactions. 2011;39(4):989–993. doi: 10.1042/BST0390989. [DOI] [PubMed] [Google Scholar]
- 5.Ding PH, Jin LJ. The role of lipopolysaccharide-binding protein in innate immunity: a revisit and its relevance to oral/periodontal health. Journal of Periodontal Research. 2014;49(1):1–9. doi: 10.1111/jre.12081. [DOI] [PubMed] [Google Scholar]
- 6.Silipo A, De Castro C, Lanzetta R, Parrilli M, Molinaro A. Lipopolysaccharides. In: König H, Claus H, Varma A, editors. Prokaryotic Cell Wall Compounds: Structure and Biochemistry. Berlin, Heidelberg: Springer Berlin Heidelberg; 2010. pp. 133–153. [Google Scholar]
- 7.Gronow S, Brade H. Invited review: Lipopolysaccharide biosynthesis: which steps do bacteria need to survive? Journal of Endotoxin Research. 2001;7(1):3–23. [PubMed] [Google Scholar]
- 8.Raetz CRH, Whitfield C. Lipopolysaccharide Endotoxins. Annual review of biochemistry. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadrmas JL, Crh R. Enzymatic Synthesis of Lipopolysaccharide in Escherichia coli Purification and Propterites of Heptosyltransferase I. The Journal of Biological Chemistry. 1998;273(5):2799–2807. doi: 10.1074/jbc.273.5.2799. [DOI] [PubMed] [Google Scholar]
- 10.Raetz CR. Biochemistry of endotoxins. Annual review of biochemistry. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 11.Grizot S, Salem M, Vongsouthi V, et al. Structure of the Escherichia coli heptosyltransferase WaaC: binary complexes with ADP and ADP-2-deoxy-2-fluoro heptose. Journal of molecular biology. 2006;363(2):383–394. doi: 10.1016/j.jmb.2006.07.057. [DOI] [PubMed] [Google Scholar]
- 12.Klena JD, Gray SA, Konkel ME. Cloning, sequencing, and characterization of the lipopolysaccharide biosynthetic enzyme heptosyltransferase I gene (waaC) from Campylobacter jejuni and Campylobacter coli. Gene. 1998;222(2):177–185. doi: 10.1016/s0378-1119(98)00501-0. [DOI] [PubMed] [Google Scholar]
- 13.Sirisena DM, Brozek KA, MacLachlan PR, Sanderson KE, Raetz CR. The rfaC gene of Salmonella typhimurium. Cloning, sequencing, and enzymatic function in heptose transfer to lipopolysaccharide. Journal of Biological Chemistry. 1992;267(26):18874–18884. [PubMed] [Google Scholar]
- 14.Coutinho PM, Deleury E, Davies GJ, Henrissat B. An Evolving Hierarchical Family Classification for Glycosyltransferases. Journal of Molecular Biology. 2003;328(2):307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- 15.Lairson L, Henrissat B, Davies G, Withers S. Glycosyltransferases: structures, functions, and mechanisms. Annual review of biochemistry. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 16.Albesa-Jové D, Giganti D, Jackson M, Alzari P, Guerin M. Structure-function relationships of membrane-associated GT-B glycosyltransferases. Glycobiology. 2014;24(2):108–124. doi: 10.1093/glycob/cwt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Czyzyk D, Sawant S, Ramirez-Mondragon C, Hingorani M, Taylor E. Escherichia coli Heptosyltransferase I: Investigation of Protein Dynamics of a GT-B Structural Enzyme. Biochemistry. 2013:5158–5160. doi: 10.1021/bi400807r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulichak AM, Losey HC, Lu W, Wawrzak Z, Walsh CT, Garavito RM. Structure of the TDP-epi-Vancosaminyltransferase GtfA from the Chloroeremomycin Biosynthetic Pathway. Proceedings of the National Academy of Sciences. 2003;100(16):9238–9243. doi: 10.1073/pnas.1233577100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vetting MW, Frantom PA, Blanchard JS. Structural and Enzymatic Analysis of MshA from Corynebacterium glutamicum: Substrate-Assisted Catalysis. Journal of Biological Chemistry. 2008;283(23):15834–15844. doi: 10.1074/jbc.M801017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petsch D, Anspach FB. Endotoxin removal from protein solutions. Journal of Biotechnology. 2000;76(2–3):97–119. doi: 10.1016/s0168-1656(99)00185-6. [DOI] [PubMed] [Google Scholar]
- 21.Kanipes MI, Papp-Szabo E, Guerry P, Monteiro MA. Mutation of waaC, Encoding Heptosyltransferase I in Campylobacter jejuni 81–176, Affects the Structure of both Lipooligosaccharide and Capsular Carbohydrate. Journal of Bacteriology. 2006;188(9):3273–3279. doi: 10.1128/JB.188.9.3273-3279.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rietschel ET, Kirikae T, Schade FU, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1994;8(2) doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 23.Durka M, Buffet K, Iehl J, Holler M, Nierengarten J-F, Vincent SP. The Inhibition of Liposaccharide Heptosyltransferase WaaC with Multivalent Glycosylated Fullerenes: A New Mode of Glycosyltransferase Inhibition. Chemistry – A European Journal. 2012;18(2):641–651. doi: 10.1002/chem.201102052. [DOI] [PubMed] [Google Scholar]
- 24.Tikad A, Fu H, Sevrain CM, Laurent S, Nierengarten J-F, Vincent SP. Mechanistic Insight into Heptosyltransferase Inhibition by using Kdo Multivalent Glycoclusters. Chemistry – A European Journal. 2016;22(37):13147–13155. doi: 10.1002/chem.201602190. [DOI] [PubMed] [Google Scholar]
- 25.Ramaraj Sivakumar RVP, Korlakunta, Jayaveera N. Docking Studies of Benzimidazole Derivatives on Peptide Deformylase and Heptosyltransferase Waac As Antibacterial Agent. International Journal of Pharmaceutical Research and Innovation. 2011;4:1–5. [Google Scholar]
- 26.Moreau F, Desroy N, Genevard JM, et al. Discovery of new Gram-negative antivirulence drugs: structure and properties of novel E. coli WaaC inhibitors. Bioorganic & medicinal chemistry letters. 2008;18(14):4022–4026. doi: 10.1016/j.bmcl.2008.05.117. [DOI] [PubMed] [Google Scholar]
- 27.Cote JM, Ramirez-Mondragon CA, Siegel ZS, et al. The Stories Tryptophans Tell: Exploring Protein Dynamics of Heptosyltransferase I from Escherichia coli. Biochemistry. 2017 doi: 10.1021/acs.biochem.6b00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czyzyk D, Liu C, Taylor E. Lipopolysaccharide biosynthesis without the lipids: recognition promiscuity of Escherichia coli heptosyltransferase I. Biochemistry. 2011;50(49):10570–10572. doi: 10.1021/bi201581b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfrom ML, Wood HB. Racemic Glucose. Journal of the American Chemical Society. 1949;71(9):3175–3176. [Google Scholar]
- 30.Ella-Menye JR, Nie X, Wang G. Synthesis of octahydropyrano[3,2-b]pyrrole-2-carboxylic acid derivatives from D-mannose. Carbohydrate research. 2008;343(10–11):1743–1753. doi: 10.1016/j.carres.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 31.Rodebaugh R, Fraser-Reid B. Evidence for Cyclic Bromonium Ion Transfer in Electrophilic Bromination of Alkenes: Reaction of omega-Alkenyl Glycosides with Aqueous N-Bromosuccinimide. Tetrahedron. 1996;52(22):7663–7678. [Google Scholar]
- 32.Yoon S, Kim D, Shin JEN. A Facile Synthesis of p-Nitrophenyl Glycosides. Bull Korean Chem Soc. 1994;15(7):559–563. [Google Scholar]
- 33.Talley EA, Vale MD, Yanovsky E. Allyl Ethers of Carbohydrates .3. Ethers of Glucose and Galactose. Journal of the American Chemical Society. 1945;67(11):2037–2039. [Google Scholar]
- 34.Lemieux RU, Morgan AR. Preparation and Configurations of Tri-O-Acetyl-Alpha-D-Glucopyranose 1,2-(Orthoesters) Can J Chemistry. 1965;43(8):2199. [Google Scholar]
- 35.Plettenburg O, Mui C, Bodmer-Narkevitch V, Wong CH. Rapid preparation of glycolipid libraries by cross metathesis. Adv Synth Catal. 2002;344(6–7):622–626. [Google Scholar]
- 36.Breton C, Šnajdrová L, Jeanneau C, Koča J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2005;16(2):29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- 37.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery : A Guide for Medicinal Chemists and Pharmacologists (1) Hoboken, US: Wiley-Interscience; 2005. [PubMed] [Google Scholar]
- 38.Vetting MW, Frantom PA, Blanchard JS. Structural and Enzymatic Analysis of MshA from Corynebacterium glutamicum: SUBSTRATE-ASSISTED CATALYSIS. Journal of Biological Chemistry. 2008:283. doi: 10.1074/jbc.M801017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulichak AM, Losey HC, Lu W, Wawrzak Z, Walsh CT, Garavito RM. Structure of the TDP-epi-vancosaminyltransferase GtfA from the chloroeremomycin biosynthetic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9238–9243. doi: 10.1073/pnas.1233577100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.