

Abstract
White matter injury following ischemic stroke is a major cause of functional disability. Injury to both myelinated axons and oligodendrocytes, the myelin producing cells in the central nervous system, occurs in experimental models of ischemic stroke. Age-related changes in white matter vulnerability to ischemia have been extensively studied and suggest that both the perinatal and the aged periods are times of increased white matter vulnerability. However, sensitivity of white matter following stroke in the juvenile brain has not been evaluated. Interestingly, the late pediatric period is an important developmental stage, as it is the time of maximal myelination. The current study demonstrates that neurons in late pediatric/juvenile striatum are vulnerable to ischemic damage, with neuronal injury being comparable in juvenile and adult mice following ischemia. By contrast, actively myelinating striatal oligodendrocytes in the juvenile brain are resistant to ischemia, whereas adult oligodendrocytes are quite sensitive. As a result, myelin sheaths are remarkably intact and axons survive well in the injured striatum of juvenile mice. In addition to relative resistance of juvenile white matter, other glial responses were very different in juvenile and adult mice following cerebral ischemia, including differences in astrogliosis, fibrosis, NG2-cell reactivity, and vascular integrity. Together, these responses lead to long-term preservation of brain parenchyma in juvenile mice, compared to severe tissue loss and scarring in adult mice. Overall, the current study suggests that equivalent ischemic insults may result in less functional deficit in children compared to adults and an environment more conducive to long-term recovery.
Keywords: Stroke, Oligodendrocyte, Myelin, Astrocyte, Vasculature, Gliosis
Introduction
Ischemic stroke impacts both white and gray matter in the human brain. However, most experimental stroke research has focused on ischemia in gray matter, with less attention on its impact in white matter. White matter damage has local effects at the primary site of damage, as well as distal effects on brain regions with which white matter axons communicate. Age-dependent vulnerability to stroke has been noted. Both early post-natal as well as aging white matter are highly sensitive to ischemia, and unique molecular mechanisms underlie these differences (Back and Rosenberg 2014; Baltan et al. 2008). It is imperative to understand how ischemia affects white matter, and how these effects change during all stages of brain development.
Juvenile arterial ischemic stroke affects up to 1,000 children in the United States each year, with a vast majority of surviving children suffering long-term neurological deficit with varying degrees of disability (Roach et al. 2008b). Initial clinical studies suggest that recovery from stroke is greater in older juvenile patients compared to strokes occurring shortly after birth (Allman and Scott 2013; Baltan et al. 2008; Everts et al. 2008; Pavlovic et al. 2006; Roach et al. 2008a; Westmacott et al. 2010) or in adulthood(Anderson et al. 2011; Ellis et al. 2014). A similar pattern of age-related stroke recovery exists in rodents (Yager et al., 2006; Saucier et al., 2007). In order to understand the mechanisms and responses that may be unique to the juvenile developmental time period, we utilized a recently developed mouse model of juvenile arterial ischemic stroke (Herson et al. 2013). (To prevent confusion with other studies of pediatric hypoxia/ischemia (Vannucci and Vannucci 2005), we refer to this age (P21-P25) as the juvenile period.) The effects of ischemia during this juvenile developmental period have been strikingly understudied. The current studies demonstrate that ischemia in juvenile mice is far less damaging to white matter compared to adults.
The juvenile period is important, as it is the peak of central nervous system (CNS) myelination. Oligodendrocytes are the myelin producing cells in the CNS and a major cellular constituent of white matter, along with myelinated axons and white matter astrocytes. Oligodendrocytes are vulnerable to cerebral ischemia at multiple stages of development. Immature, pre-myelinating oligodendrocytes are highly susceptible to ischemic cell death following neonatal ischemia (Back et al. 2002a; Back et al. 2002b), whereas in the adult, it is the mature oligodendrocytes that are damaged by ischemia, leading to myelin loss, and eventually axonal injury (Dewar et al. 2003). Myelinated axons also display age-dependent sensitivity to ischemic injury, with developing white matter axons having high susceptibility (McCarran and Goldberg 2007).
In active myelination, oligodendrocytes generate massive amounts of membrane, estimated to produce a myelin surface area over 100 times the surface area of the cell (Pfeiffer et al. 1993). At this time, these cells have very high metabolic rates and are sensitive to prolonged deprivation of energy substrates (Rinholm et al. 2011; Yan and Rivkees 2006). Therefore, we would expect that actively myelinating oligodendrocytes would be particularly vulnerable to ischemic injury during this time point. In order to test this hypothesis, we examined the glial responses following experimentally-induced stroke in juvenile mice (20–25 days old).
Here, we show unexpectedly that oligodendrocytes in the juvenile brain are remarkably resistant to ischemic injury. We also show that oligodendrocyte progenitor cells (OPCs, NG2 progenitor cells), astrocytes, pericytes, and blood vessels respond differently to juvenile stroke compared to adult stroke. These differing responses to ischemia could underlie the greater degree of recovery observed in juvenile stroke survivors.
Materials and Methods
Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Colorado School of Medicine, and conformed to the National Institutes of Health guidelines for the care and use of animals in research. Male C57Bl/6 mice were used for most studies (Charles River, Inc.); male PLP-EGFP transgenic mice (Mallon et al. 2002) were also used.
Middle Cerebral Artery Occlusion (MCAo)
MCAo was induced by methods previously published for juvenile male mice at postnatal day (P) 20 to 25(Herson et al. 2013) or adult male mice at 8–12 weeks old (Jia et al. 2011). Briefly, cerebral ischemia was induced for 45 minutes of reversible MCAo via the intraluminal suture method under isoflurane anesthesia. Adequacy of MCAo was confirmed by laser Doppler flowmetry measured over the ipsilateral parietal cortex. The occluding suture was then removed, thereby restoring blood flow to the affected region. Mice were monitored closely following the surgical procedure and were sacrificed at the following time points: 24 hours, 3 days, 7 days, and 30 days.
Infarct Volume Analysis
Twenty-four hours after reperfusion adult and juvenile mice were deeply anesthetized with isoflurane and brains removed for analysis of infarct volume. Each brain was sliced into five 2 mm thick coronal sections and immediately placed in a 1.2% solution of 2,3,5-triphenyltetrazolium chloride (TTC, Sigma) for 30 minutes at 37°C followed by fixation in 10% formalin for 24 hours. Each aspect of all stained coronal slices was photographed using a digital camera; infarction was measured with digital image analysis software (ImageJ) and integrated across all 5 slices. To account for the effect of edema, the infarcted volume was estimated and expressed as a percentage of the contralateral structure.
Immunohistochemistry
After the reperfusion period, mice were anesthetized and tissue was fixed by transcardial perfusion with PBS followed by 4% paraformaldehyde. The brain was dissected, postfixed overnight, and transferred to cryoprotection solution (20% glycerol in 0.1 M Sorenson’s buffer, pH 7.6). The immunohistochemistry protocol was adapted from previously described studies (Trapp et al. 1997). Brains were sectioned at 30 μm on a sliding microtome and sections were stored at 4°C in cryostorage solution (30% ethylene glycol, 30% sucrose, and 1% PVP-40 in 0.1 M Sorenson’s buffer). Free-floating sections were washed in PBS and antigen retrieval was performed with 10 mM sodium citrate (pH 6.0) + 0.05% Tween-20 for 10 minutes at 550 W in a PELCO BioWave Pro tissue processor (Ted Pella). Sections were then washed in PBS, permeabilized with 0.3%–10% Triton X-100 (dependent on specific antibodies), blocked with 5% normal donkey serum, and incubated with primary antibody for 1–3 days at 4°C. The following primary antibodies were used: mouse anti-CC1 (Calbiochem); rabbit anti-fibrinogen (Abcam); chicken anti-GFAP (Abcam); rabbit anti-GSTπ (ENZO); rabbit anti-MOG (Abcam); rabbit anti-NG2 (a gift from Dr. William Stallcup, Sanford-Burnham Institute); rabbit anti-Olig2 (a gift from Dr. Charles Stiles, Harvard University); rabbit anti-platelet-derived growth factor alpha (PDGFRα, Santa Cruz); mouse anti-NeuN (Millipore); rat anti-PLP/DM20 (clone AA3; (Yamamura et al. 1991); mouse anti-SMI32 (Covance); mouse anti-SMI94 (MBP, Covance); goat anti-Sox10 (Santa Cruz); rat anti-CD13 (AbD Serotec); rabbit anti-Glut-1 (Thermo Scientific); rabbit anti-nitrotyrosine (Santa Cruz); mouse anti-HO-1-1 (Abcam). After washing with PBS, sections were incubated with the appropriate secondary antibodies (Jackson ImmunoResearch) for 2 hours at room temperature and mounted on glass slides in Vectashield Mounting Media (Vector Laboratories). TUNEL staining was performed according to manufacturer’s protocol (Roche), followed by antibody co-staining, as described above. Diaminobenzidine (DAB) staining was performed according to manufacturer’s protocol (Avidin-Biotin Complex Kit and DAB Kit, Vector Laboratories). Fluorescent images were acquired using a Leica TCS SP5 II confocal microscope using identical imaging parameters for ipsilateral and contralateral images. DAB images were acquired with a Leica DMR microscope and DFC290HD digital camera.
Cell Counting
Cells were quantified from images acquired in the lateral aspect of the injured (ipsilateral) striatum and corresponding images in the uninjured (contralateral) striatum. At least two images were taken from the ipsilateral and contralateral striatum per stained section, both near the box in Figure 1, and at least two sections were stained per animal. Four animals per group were analyzed. Cell death was quantified by positive TUNEL staining. NeuN positive neurons were quantified. Olig2 or Sox10-positive oligodendrocytes were quantified. Oligodendrocyte precursor cells were identified by co-expression of PDGFRα and Sox10. Mature oligodendrocytes were identified by co-expression of CC1 and Sox10. The Cell Counter plug-in on Fiji software (Schindelin et al. 2012) was used for all cell count analyses. Images were obtained at 40× magnification; all images have size bars defined in the figure legends.
Figure 1. Cell death after MCAo.
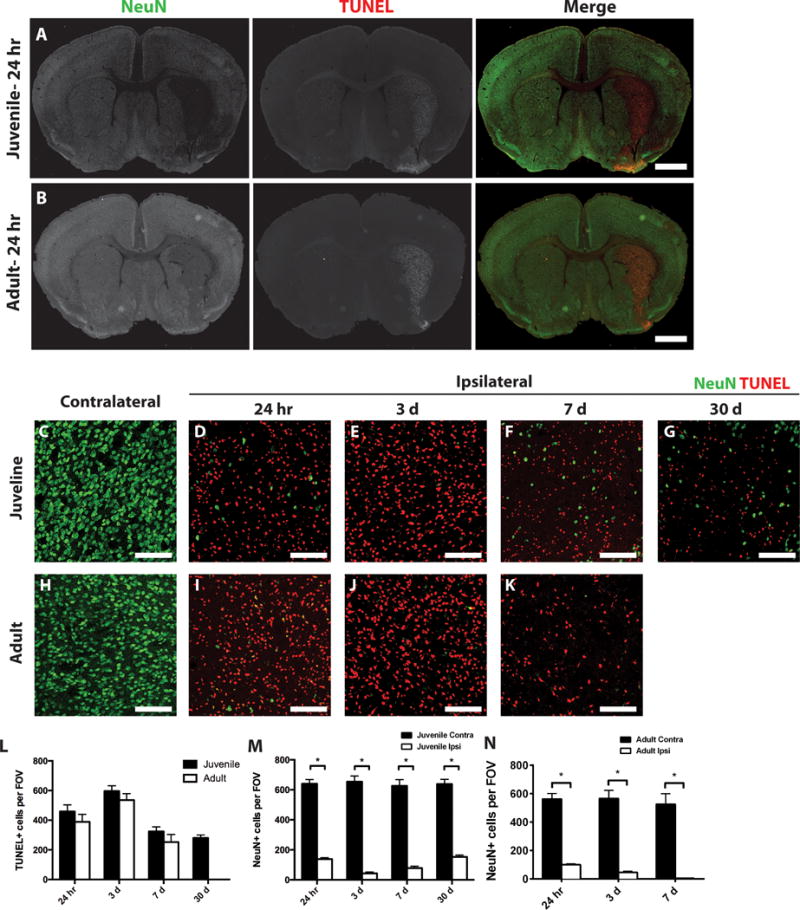
Ischemic (ipsilateral) and control (contralateral) juvenile (A) and adult (B) striatum sections were stained for dying cells (TUNEL, red) and neurons (NeuN, green). Low magnification images identify localized TUNEL-positive cells and reduced neurons in the ipsilateral striatum. Box indicates general area that is imaged for all high magnification images. At higher magnification greater discrimination of the contrast of ipsilateral and contralateral TUNEL-positive cells or neurons is seen in juvenile (C–G) or adult (H–K) tissue at 24 h post-MCAo (D,I), 3 days post-MCAo (E,J) or 7 days post MCAo (F,K). (L) Quantification of dying cells in the ipsilateral striatum from juvenile (dark bar) and adult (open bar) samples. (M,N) Quantification of remaining neurons in ipsilateral (Ipsi, open bar) versus contralateral (Contra, dark bar) striatum from juvenile (L) and adult (M) samples. White scale bars: 1mm (A,B); 100 lm (C–K). *P value < 0.05.
Axon damage analysis
APP fluorescence per unit area was measured for 50 axon bundles per animal using FIJI software. Images were taken under identical imaging setting and thresholded to the same values in order to measure relative fluorescent units (RFU) per unit area for each axon bundle.
Blood vessel analysis
Blood vessels were analyzed in two ways. The lengths of each blood vessel segment were traced using Glut-1 immunostaining and measured using the Measure and Label plug-in on Fiji software. The individual lengths were summed to attain the total vessel length for each image. Second, a MATLAB program was written in-house to measure the number of connected objects (corresponding to the number of individual blood vessel segments) and the area of co-localization between CD13 and Glut-1 (corresponding to the amount of vessel-associated pericytes). The images were adjusted for non-uniform background staining and positive pixels were identified using a set threshold. The area covered by positive pixels was quantified in each channel. Individual blood vessel segments were quantified by counting the number of connected objects with more than 2 pixels. To facilitate the identification of individual blood vessel segments, the staining boundaries were filled in using the “imfill” function, and connected objects were quantified. At least four images were measured per animal, with four animals per group. Blood vessel analyses were done at 25× magnification. All images have size bars defined in the figure legends.
Electron microscopy
Animals were perfused with modified Karnovsky’s fixative (2% paraformaldehyde/2.5% glutaraldehyde) in phosphate buffer, pH 7.4. The brain was dissected and stored in postfix until embedding. Striatal tissue was isolated from 1mm coronal slices between 1.00 and −0.40 of Bregma. Tissue was postfixed in 1% osmium tetroxide, dehydrated in graded acetone, and resin embedded in Embed 812 (Electron Microscopy Services) using a PELCO BioWave Pro tissue processor (Ted Pella). Striatal pieces were oriented such that sections could be cut in a coronal plane to visualize myelinated axon fibers. Ultrathin sections (80 nm) were mounted on copper grids, stained with uranyl acetate and lead citrate, and viewed at 80 kV on a Tecnai G2 transmission electron microscope (FEI).
G-ratio analysis
G-ratios of myelinated fibers were calculated as the ratio of the diameter of the axon to the diameter of the myelinated fiber. Diameters were derived by measuring the respective perimeters using FIJI software. A minimum of 100 axons per condition were quantified, and samples were analyzed from four animals per experimental group.
Statistical analysis
Unpaired, two-tailed Student’s t-test was used to calculate statistical significance between ipsilateral and contralateral regions within each group. For comparison between juvenile and adult axon damage, a two-way ANOVA was used to calculate statistical significance between groups. Statistical significance was defined as a p-value less than 0.05. Graphs represent the mean ± SEM. Graphs were created and statistics were performed using Prism 6 software (www.graphpad.com).
Results
Cell death and neuronal loss after juvenile MCAo
The current studies were undertaken to assess the relative damage induced by MCAo in juvenile mice relative to adult mice. Male adult (2–3 months) and juvenile (P20–25) mice were subjected to 45 minutes of MCAo, and metabolic activity was analyzed 24 hours after reperfusion by TTC stain. Adult and juvenile mice exhibited nearly identical ischemic damage after MCAo, with edema-corrected hemispheric volumes of metabolically-deficient tissue of 46 ± 4.2% and 43 ± 4.5%, respectively. To characterize the cellular responses and extend the observation time beyond the acute stage, adult and juvenile mice were subjected to 45 min MCAo and brains analyzed by immunohistochemistry at 24 hours, 3 or 7 days after reperfusion. Considering the well-established sensitivity of both neurons and oligodendrocytes to hypoxia/ischemia (Back et al. 2002a; Valeriani et al. 2000), we analyzed the effects of MCAo on the survival of neurons and oligodendrocytes. Brain sections from juvenile or adult MCAo mice were stained with NeuN, a marker of neuronal nuclei and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling), to detect DNA fragmentation in apoptotic cells (Figure 1). Increased cell death and neuron loss was seen in particular in the striatum, with comparable damage to both juvenile and adult tissue (Figure 1 A,B). Cell death was not observed in the contralateral striatum of either juvenile or adult mice, where many NeuN-positive cells were detected (Figure 1A,B,C,H). In both juvenile and adult mice, cell death was observed in the ipsilateral striatum as early as 24 hours post-MCAo (Figure 1A,B,D,I,L). Cell death peaked at 3 days post-MCAo (Figure 1E,J,L), but was still occurring at 7 days post-MCAo (Figure 1F,K,L). There were no statistically significant differences in the number of TUNEL-positive cells between juvenile and adult samples at any time point (Figure 1L). Together, these results suggest that acute metabolic damage, as assessed by TTC staining, and cell death, as determined by TUNEL staining, were comparable between juvenile and adult mice after 45 minutes of reversible MCAo.
At the same time that there were dying cells, the number of NeuN-positive neurons dramatically decreased in the injured striatum after MCAo in both juvenile and adult mice. In juvenile mice, the neuronal population in the ipsilateral striatum was severely reduced at 24 hours post-MCAo compared to the contralateral hemisphere (Figure 1A,C,D,M). The neuronal population decreased further after 3 days post-MCAo (Figure 1E,M) and remained low at 7 days post-MCAo (Figure 1F,M). In adult mice, the neuronal population in the ipsilateral striatum was also severely reduced compared to the contralateral hemisphere at 24 hours post-MCAo (Figure 1B,H,I,N). The neuronal population in the adult striatum decreased further at 3 days post-MCAo (Figure 1J,N) and it was almost completely abolished at 7 days post-MCAo (Figure 1K,N). Together, these results suggest that the neuronal loss in the striatum was comparable for juvenile and adult mice after MCAo, and that neuronal loss occurred rapidly. These changes were still noted at 30 day post-MCAo in juvenile animals, where significant TUNEL+ cells were still seen and reduced neurons, although by that time point, some increase in the number of neurons was noted (Figure 1G,L,M). Comparable analysis could not be made for adult tissue, which was severely damaged at that time point.
Oligodendrocyte precursor cell responses in ischemic tissue after MCAo
Quite unexpectedly, when oligodendrocyte lineage cells were quantified after MCAo, very few oligodendrocyte lineage cells in the juvenile striatum were lost, i.e., they appeared highly resistant to ischemic damage. At 24 h post-MCAo, no reduction in the number of proteolipid protein (PLP)-EGFP-positive oligodendrocyte lineage cells was noted in the juvenile ipsilateral striatum compared to the contralateral striatum (Fig. 2A,B,G), while the number of PLP-EGFP-positive oligodendrocyte lineage cells was statistically significantly reduced in adult mice as early as 24 h post MCAo (Fig. 2C,D,H). In this mouse, enhanced green fluorescent protein (EGFP) is expressed under control of the myelin PLP promoter (Mallon et al., 2002), and it allows both quantification of oligodendrocyte lineage cell bodies and assessment of cell morphology. We examined the impact of ischemia on both OPCs and mature oligodendrocytes using stage-specific oligodendrocyte antigenic markers as well. Tissues were immunolabeled with PDGFRa, a marker for OPCs; CC1, a marker for mature oligodendrocytes; and Sox10, a transcription factor expressed by both populations (Fig. 2I–P). In juvenile mice, OPCs were unaffected by ischemia in the ipsilateral striatum at 24 h post-MCAo relative to the contralateral striatum (Fig. 2I,J,Q). OPC numbers were significantly elevated in the injured juvenile striatum at 3 days post-MCAo compared to the corresponding contralateral striatum (Fig. 2I,K,Q), and they remained elevated at 7 days post-MCAo (Fig. 2I,L,Q). OPCs have been shown to respond to injury (Levine, 2015), and it is likely that the increases in OPCs may result from their response to injury.
Figure 2. Oligodendrocytes are preserved in juvenile, but not adult tissue after MCAo.
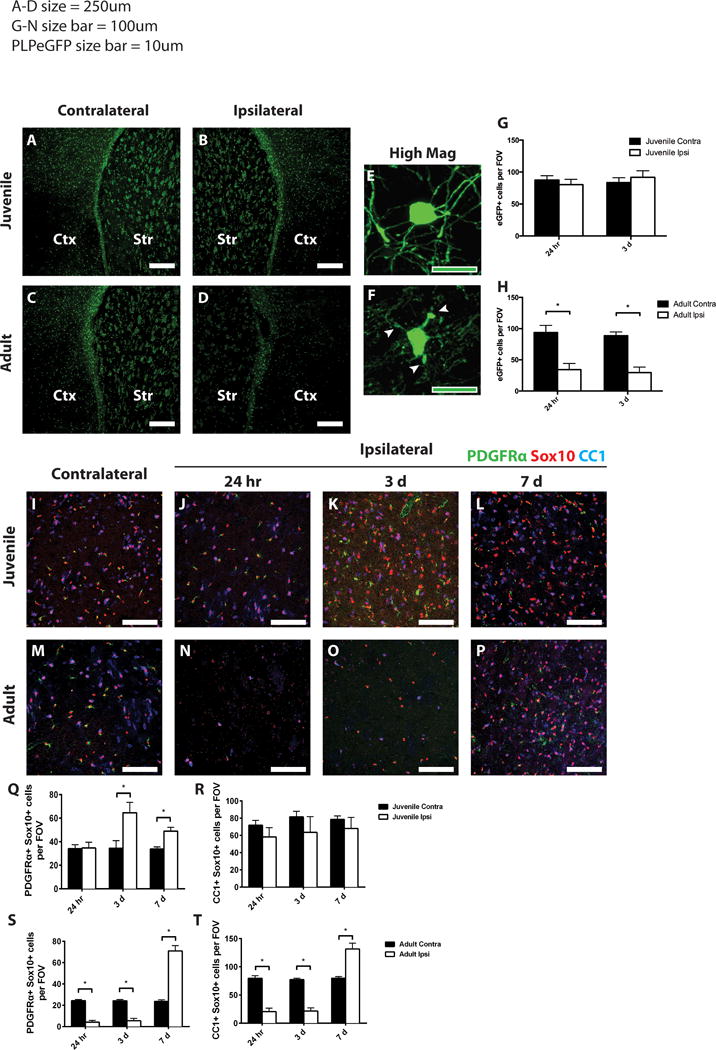
PLP-EGFP mice were analyzed after 45 min MCAo. Control (contralateral, A,C) or ischemic (ipsilateral, B,D) striatum (Str) and lateral cortex (Ctx) from juvenile (A, B) or adult (C, D) mice 24 h after MCAo were imaged. Note presence of PLP-EGFP cells in ipsilateral striatum in juvenile tissue (B) but loss in adult tissue (D). At higher magnification, cell morphology indicates preserved oligodendrocyte morphology and intact myelin processes in juvenile striatum (E), but damaged oligodendrocyte in adult striatum (F). (G–H) Quantification of PLP-EGFP cells in juveline (G) and adult (H) striatum. Ischemic (ipsilateral) and control (contralateral) juvenile (I–L) and adult (M–P) sections were immunostained to detect OPCs (PDGFRa, green), total oligodendrocytes (Sox10, red), and mature oligodendrocytes (CC1, blue). OPCs (Q,S), identified by double staining with PDGFRa and Sox10, were quantified in juvenile (Q) and adult (S) striatum from ischemic (Ipsi, open bars) and control (Contra, dark bars) samples. Mature oligodendrocytes (R,T) were identified by double staining with CC1 and Sox10, and quantified in juvenile (R) and adult (T) striatum from ischemic (Ipsi, open bars) and control (Contra, dark bars) samples. White scale bars: 250 lm (A–D); 100 lm (I–P). Green scale bars: 10 lm (E,F). *P value < 0.05.
By contrast, adult OPCs were severely affected by MCAo during the early period after ischemia. A significant decrease in the PLP-EGFP positive cells was observed in the ipsilateral striatum compared to the contralateral striatum at 24 h post-MCAo (Fig. 2C,D). At 24 h post-MCAo, the number of surviving PDGFRa-positive OPCs was significantly decreased in the ipsilateral striatum relative to contralateral striatum (Fig. 2M,N,S) and remained significantly decreased at 3 days post-MCAo (Fig. 2M,O,S). At 7 days post-MCAo, however, the number of OPCs in the injured striatum was significantly elevated relative to contralateral striatum (Fig. 2M,P,S). Overall, these results suggest that juvenile OPCs are more resistant to ischemic injury than adult OPCs, and they respond earlier to the injury than adult OPCs.
Mature oligodendrocytes are resistant to ischemia in the juvenile brain
To our surprise, as with the OPCs, mature oligodendrocytes (CC1/Sox10-double positive cells) in the juvenile brain were not significantly affected by MCAo at any of the time points examined relative to the contralateral hemisphere (Fig. 2IL, R). In contrast, in the adult brain, mature oligodendrocytes were diminished at 24 h post-MCAo in the ipsilateral striatum compared to the contralateral striatum (Fig. 2M,N,T) and the number of mature oligodendrocytes remained decreased at 3 days post-MCAo (Fig. 2M,O,T). Unexpectedly, between 3 and 7 days post-MCAo, the loss of mature oligodendrocytes, as defined by CC1 expression, was reversed, and by 7 days, the number of CC1-expressing oligodendrocytes was significantly elevated in the adult ipsilateral striatum relative to the contralateral striatum (Fig. 2M,P,T).
To confirm the remarkable resistance of mature oligodendrocytes following juvenile ischemic stroke, the morphological changes in juvenile or adult mice after MCAo were assessed in the PLP-EGFP transgenic mice. At 24 hours post-MCAo in juvenile mice, PLP-EGFP-expressing oligodendrocyte cell bodies were large and seemingly healthy with intact myelin processes (Figure 2E). In adult mice at 24 hours post-MCAo, the PLP-EGFP-expressing cells were shrunken with fragmented and disorganized myelin processes (Figure 2F).
Myelin damage, axon pathology, and long-term tissue loss are spared in juvenile, but not adult, ischemia
Ischemia causes ultrastructural damage to both oligodendrocytes and myelin in adult rodents (Goldenberg-Cohen et al., 2005; Pantoni et al., 1996). Therefore, we examined the myelinated axon bundles that course though the striatum (pencil fibers) for ischemia-induced damage in adult and juvenile tissue. Juvenile myelin remained intact compared to contralateral tissue at all time points examined (Fig. 3A–D). G-ratio analysis demonstrated that the thickness of myelin was unaffected in the ipsilateral fibers compared to the contralateral fibers at all time points examined (Fig. 3I). In contrast, adult tissue showed severe ultrastructural damage, including demyelination and axon swelling, as early as 24 h post-MCAo and continuing through 7 days post-MCAo (Fig. 3E–H). The tissue was then examined for histological signs of axon pathology within the myelinated pencil fibers. SMI32 monoclonal antibody detects non-phosphorylated neurofilament, which in normal tissue is found in cell bodies, dendrites and thick axons. SMI32 also accumulates in damaged axons, and has been used as a measure of axonal pathology (Sherriff et al., 1994). High magnification images from axonal fibers in the juvenile samples 7 days post-MCAo showed scattered signs of axon pathology, including SMI32 immunoreactivity and axonal swellings in the ipsilateral striatum (Fig. 3J,K). By contrast, adult axonal fibers displayed numerous axonal swellings and increased SMI32 staining (Fig. 3J,L).
Figure 3. Myelin damage, axon pathology, and long-term tissue loss after MCAo.
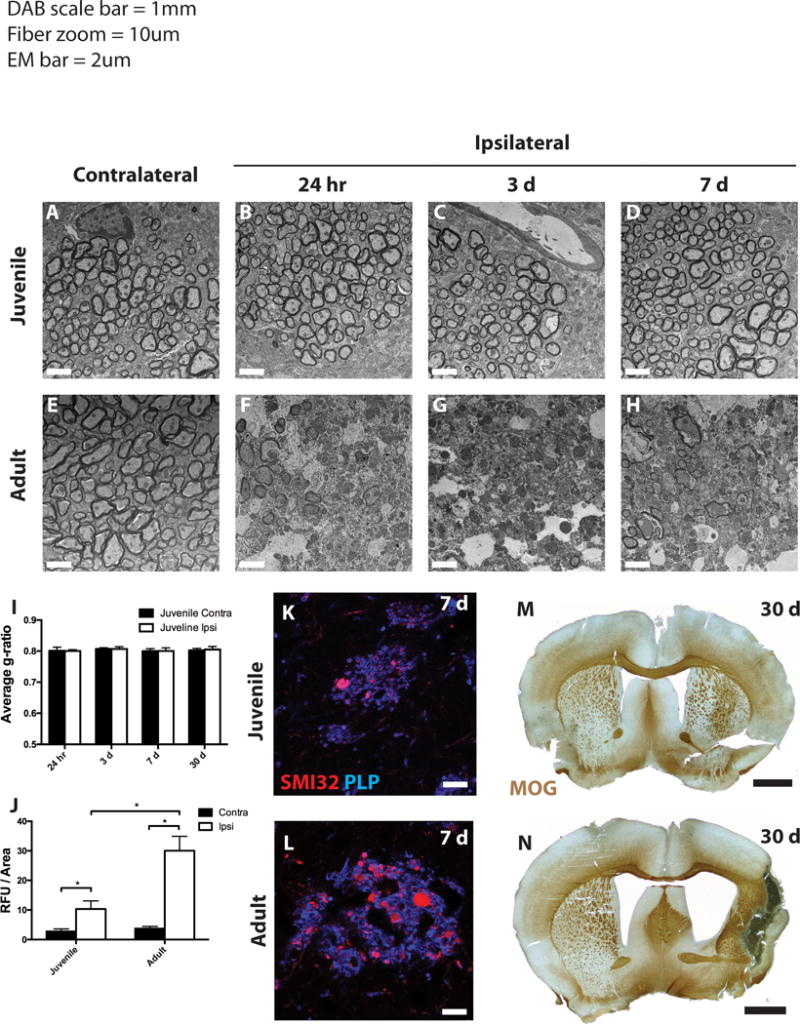
Electron micrographs of myelinated striatal axons from control and ischemic juvenile (A–D) and adult (E–H) mice. (I,J) G ratios and axon damage quantification, respectively, were determined for myelinated axons in the contralateral (dark bars) and ipsilateral (open bars) striatum from juvenile mice (I) and both age groups (J). (K,L) At 7 days post-MCAo, axonal pathology and myelin were determined by SMI32 (red) and PLP (blue) immunoreactivity in juvenile (K) and adult (L) mice. (M,N) At 30 days post-MCAo, tissue was stained for myelin with myelin-oligodendrocyte glycoprotein (MOG) antibody. Scale bars in A–H: 2 lm; K,L: 10 lm; M,N: 1 mm.
To assess the long-term consequences of MCAo on juvenile and adult tissue, we analyzed animals 30 days after MCAo. Tissue was immunostained for myelin oligodendrocyte glycoprotein (MOG) and low magnification images allow analysis of global changes in gross anatomy in these tissues. Juvenile tissue showed overall preservation of all major brain regions, including striatal and cortical tissue, 30 days after MCAo (Fig. 3M). Conversely, adult tissue showed major loss of striatal and cortical tissue in the ipsilateral hemisphere, resulting in cavitation, severe scar formation, and enlargement of the lateral ventricle (Fig. 3N). This tissue loss precluded detailed comparison of longterm consequences of MCAo between samples of these age groups, but in general, immunohistochemical analysis of juvenile tissue indicated little damage (data not shown).
Differential Injury Progression in Juvenile Versus Adult Stroke
Given the dramatic differences in tissue damage between juvenile and adult tissue after ischemia, we next investigated other cellular responses to ischemia that might differ between juvenile and adult stroke. At 24 h post-MCAo, there was diffuse astrogliosis as defined by GFAP immunoreactivity, in both juvenile and adult cortex, corpus callosum, and striatum (Fig. 4A,B,F,G). By 3 days post-MCAo, GFAP-positive astrocytes were evident in the cortex and corpus callosum surrounding the lesion core in both juvenile and adult tissue, with significant GFAP-immunoreactivity in the medial striatum in the juvenile samples (Fig. 4C,H). In juvenile mice starting at 7 days and continuing through 30 days post-MCAo, the GFAP-immunoreactivity was expressed through the entire ipsilateral striatum, including the lesion core (Fig. 4D,E), whereas the astrocytic response remained confined to the periinfarct region in adult mice at 7 days post-MCAo (Fig. 4I).
Figure 4. Differential gliotic responses following MCAo in juvenile versus adult mice.
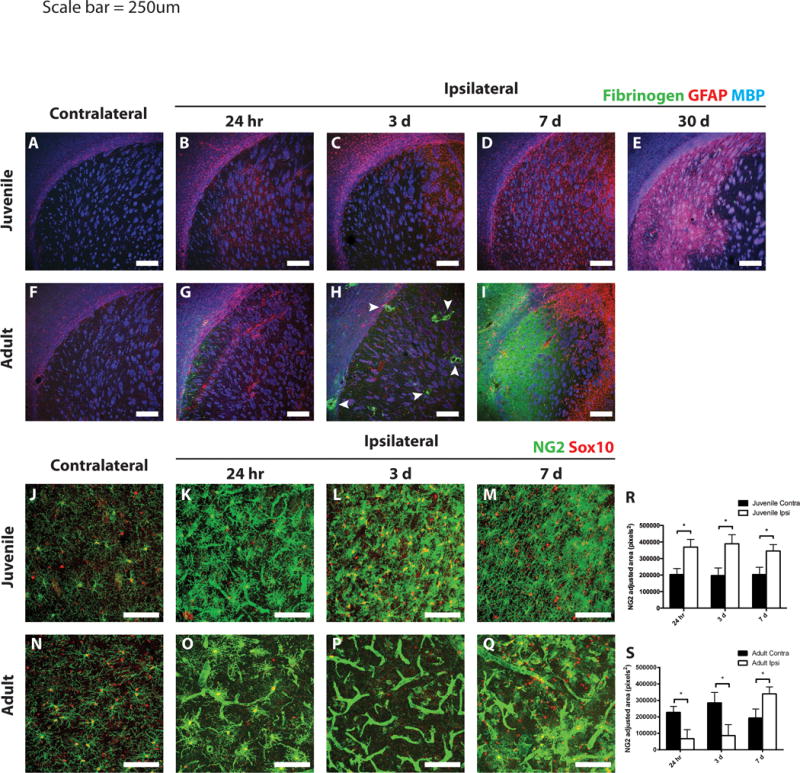
Ischemic and control juvenile (A–E) and adult (F–I) sections were immunostained to detect fibrinogen (green), GFAP (red), and MBP (blue). Scant fibrinogen immunoreactivity was observed in the injured juvenile striatum at any time point examined (A–E). Fibrinogen deposition occurred as early as 24 h after MCAo in the adult striatum (G), with large deposits surrounding blood vessels at 3 days (H, arrowheads). By 7 days, a large sheet of fibrinogen was observed throughout the core of the lesion in adult tissue (I). Astrogliosis was observed surrounding the ischemic core in both juvenile and adult striatum at 3 days post-stroke (C and H, respectively). Peri-infarct astrogliosis was maintained in adults at 7 days post-stroke (I), whereas diffuse astrogliosis throughout the injured striatum was observed in juvenile mice and continued out to 30 days (D,E). Differential NG2-cell response following MCAo in juvenile versus adult mice. Ischemic and control juvenile (J–M) and adult (N–Q) sections were immunostained to detect NG2 (green) and Sox10 (red). Images of juvenile tissue demonstrate hypertrophic NG2-cells (costained with Sox10) and some NG2 immunoreactivity on blood vessels at 24 h post-MCAo (K). Increased density of NG2-cells at 3- and 7-days post-MCAo in juvenile mice (L,M) was seen. Images from adult tissue demonstrate loss of NG2-cells at 24 h (O) and 3 days (P) post-MCAo with high levels of NG2 expression localized to blood vessels. NG2 cells repopulated the adult lesion at 7 days post-MCAo, while blood vessel NG2 immunoreactivity remained prevalent (Q). (R,S) Quantification of non-vascular NG2 immunostaining in the contralateral (dark bars) and ipsilateral (open bars) striatum from juveile (R) and adult (S) mice. White scale bars: 250 lm (A–I), 100 lm (J–Q).
Another major element of damage following ischemia is fibrinogen deposition, which leads to fibrotic scar formation. In juvenile tissue, very little fibrinogen immunoreactivity was noted at any time point examined (Fig. 4A–E). In adult tissue, however, some fibrinogen deposition was observed starting as early as 24 h post-MCAo (Fig. 4G). At 3 days post-MCAo, heavy fibrinogen deposition was observed in the ischemic core, particularly around blood vessels (Fig. 4H, arrowheads). By 7 days, a broad sheet of fibrinogen deposition was observed throughout the ischemic core in adult tissue (Fig. 4I). Thus, astrogliosis was more widespread in the juvenile ischemic core while fibrinogen deposition was limited. The combination of these two factors may have protective effects in juvenile mice, maintaining the injured tissue and preventing long-term tissue loss.
We assessed the effect of ischemia on NG2 proteoglycanexpressing OPCs (NG2-cells). In juvenile mice at 24 h post-MCAo, NG2-cells became hypertrophic with increased NG2 immunoreactivity (Fig. 4K). Similarly, in adult tissue at 24 h post-MCAo, NG2-cells were also hypertrophic; however there were fewer of them (Fig. 4O,S). In addition to altered NG2-cells, there were changes in NG2 expression associated with the vasculature, where it is known to be expressed by pericytes (Ozerdem et al., 2002). Both juvenile and adult tissue at 24 h post-MCAo had increased NG2 immunoreactivity on blood vessels in the lesion core (Fig. 4K,O). In juvenile samples at 3 and 7 days post-MCAo in the ipsilateral striatum, we observed less vascular NG2 immunoreactivity and increased numbers of NG2-cells that still appeared hypertrophic (Fig. 4L,M,R). In adult tissue, on the other hand, there were very few NG2 cells in the lesion core at 3 days post-MCAo. Instead, NG2 immunoreactivity was primarily localized to the vasculature (Fig. 4P,S). At 7 days post-MCAo in adult tissue, vascular NG2 immunoreactivity was present; however NG2-cells were now present in the injured tissue (Fig. 4Q).
Maintained vascular integrity within the ischemic core in juvenile mice
Given the increased NG2 immunoreactivity on blood vessels in the lesion core of adult animals after stroke, we examined these samples for differences in vascular integrity. Tissue was stained for Glut-1, a marker of vascular endothelia, and CD13, a pericyte marker (Fig. 5). Vascular content was quantified either as total vessel length or the number of vessel segments in the tissue. In juvenile tissue, there were no differences in the total vessel length or the number of vascular segments in the ischemic core compared to the corresponding contralateral region, at any time point examined (Fig. 5A– E,J,K). In the first 24 h post-MCAo in the adult, there was little change in total vessel length and vessel count in the ischemic core (Fig. 5G,M,N). However, after 3 days, there was a significant decrease in both total vessel length and number of vessel segments compared to the corresponding contralateral region (Fig. 5H,M,N), which became more severe at 7 days post-MCAo (Fig. 5I,M,N).
Figure 5. Vascular and pericyte responses to MCAo in juvenile versus adult mice.
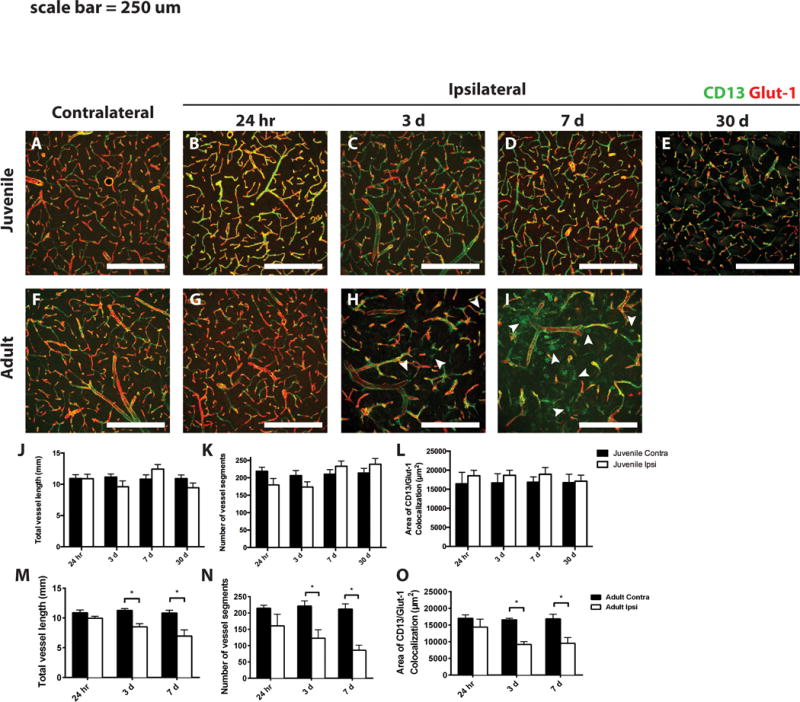
Ischemic and control juvenile (A–E) and adult (F–I) sections were immunostained to detect CD13 (green) and Glut-1 (red). Arrowheads in H and I indicate extravascular CD13 immunostaining. Quantification of total vessel length and total number of vessel segments in juvenile (J and K, respectively) and adult (M and N, respectively) striatum from ischemic (Ipsi, open bars) and control (Contra, dark bars) samples. The area of colocalized CD13 and Glut-1 immunoreactivity was quantified in juvenile (L) and adult (O) samples. White scale bars: 250 lm. *P value < 0.05.
To quantify the effects of MCAo on vascular pericytes, we measured the area of colocalized immunostaining between CD13, a pericyte marker, and Glut-1 at low magnification. The effects of MCAo on vascular pericytes mirrored those of the blood vessels. In juvenile samples, CD13 immunoreactivity was preserved at all time points examined (Fig. 5A–E,L). As with the vasculature, there was little change in CD13 immunoreactivity at 24 h post-MCAo in adult samples (Fig. 5F,G,O). However, after 3 and 7 days, there was a significant decrease in CD13 immunoreactivity that colocalized with Glut-1 (Fig. 5H,I,O). Interestingly, starting at 3 days post-MCAo and continuing at 7 days, we observed extravascular CD13 expression in the striatal parenchyma (Fig. 5H,I arrowheads), an effect not observed in juvenile samples at any time.
Enhanced protection from oxidative damage in juvenile oligodendrocytes
Because juvenile oligodendrocytes exhibited reduced sensitivity to ischemia compared to adult oligodendrocytes, we sought to identify a possible mechanistic explanation for this difference in ischemic susceptibility. Glutathione S-transferase pi (GSTπ) is an enzyme that is upregulated in response to oxidative stress and plays a key role in reducing the byproducts of oxidative stress (Hayes et al. 2005; Huang et al. 2004). In the CNS, GSTπ is expressed specifically in myelinating oligodendrocytes (Tansey and Cammer 1991; Zhang et al. 2014). We observed high basal levels of GSTπ immunoreactivity in oligodendrocytes (Sox10-positive cells) in the control contralateral juvenile striatum (Figure 6A), consistent with the concept that these cells are in a state of active myelination. GSTπ immunoreactivity remained high in striatal oligodendrocytes after juvenile MCAo throughout the reperfusion period (Figure 6B–D). By contrast, in the control contralateral adult striatum, very few Sox10-positive oligodendrocytes expressed GSTπ (Figure 6E). However, in response to ischemia, a marked upregulation of GSTπ in the ipsilateral striatum was seen at all time points examined (Figure 6F–H).
Figure 6. Decreased oxidative damage and increased anti-oxidant capacity after juvenile MCAo compared to adult.
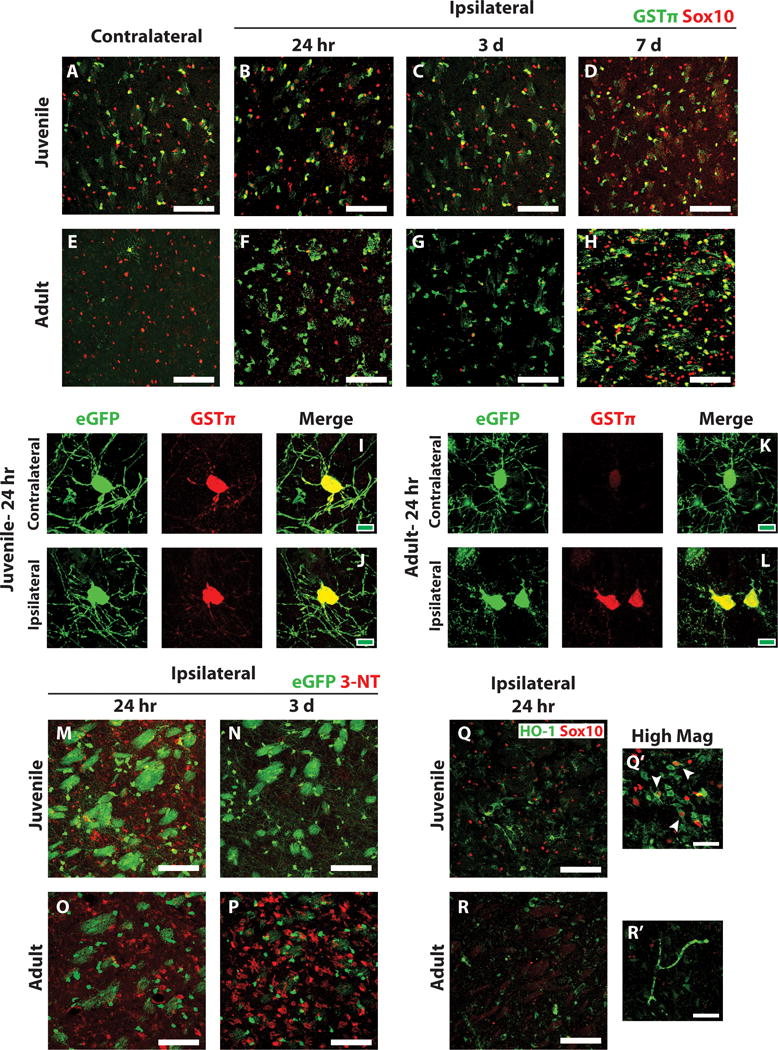
(A–H) Ischemic and control juvenile (A–D) and adult (E–H) sections were immunostained to detect GSTp (green) and Sox10 (red). High basal expression of GSTp was observed in control juvenile striatal oligodendrocytes (A) and low basal expression in control adult striatal oligodendrocytes (E). GSTp expression remained high in juvenile striatal oligodendrocytes after MCAo (B–D) and was upregulated by remaining adult oligodendrocytes after MCAo (F–H). (I–L) High magnification images of PLP-EGFP-expressing oligodendrocytes 24 h after MCAo demonstrate high GSTp expression in EGFP-expressing oligodendrocytes in both the control contralateral (I) and injured ipsilateral (J) striatum of juvenile mice. Low GSTp expression was observed in control contralateral EGFP-expressing oligodendrocytes in adult mice (K) and was upregulated in injured EGFP-expressing oligodendrocytes in the ipsilateral striatum (L). Ischemic PLP-EGFP-expressing juvenile (M, N) and adult (O, P) sections were immunostained to detect nitrotyrosine (3-NT, red). Moderate 3-NT expression was observed in juvenile animals at 24 hr after MCAo (M), which generally resolved 3 days after MCAo (N). High 3-NT expression was observed in adult animals at 24 hr post-MCAo (O) and it remained high after 3 days (P). Sections were also immunostained to detect HO-1 (Q, R, green) and Sox10 (red). High HO-1 was observed in juvenile sections at 24 hr post-MCAo (Q), and often colocalized with Sox10 expressing oligodendrocytes (Q’, arrowheads). Adult expression of HO-1 after MCAo was much lower (R). White scale bars: 100 lm (A–H, M–R), 40 lm (Q’, R’). Green scale bars: 10 lm (I–L).
Morphologically, the GSTp-positive cells in the ipsilateral adult striatum resemble oligodendrocytes; however we were unable to identify them as such because Sox10 immunostaining was lost after MCAo in adult mice (Fig. 6F,G). To confirm that GSTp was upregulated in adult oligodendrocytes after MCAo, we immunostained tissue from PLP-EGFP expressing mice for GSTp after MCAo. In these mice, EGFP was still present in deteriorating oligodendrocytes in the adult striatum (Fig. 6L). As observed in wild type mice, juvenile PLP-EGFP mice expressed high levels of GSTp in oligodendrocytes in both the contralateral and ipsilateral striatum (Fig. 6I,J). In contrast, PLP-EGFP-positive oligodendrocytes in the contralateral adult striatum expressed very low levels of GSTp (Fig. 6K). In response to MCAo, remaining EGFP-positive oligodendrocytes in the ipsilateral striatum upregulated GSTp expression, but were shrunken with fragmented myelin processes (Fig. 6L).
We next examined tissues for evidence of oxidative damage and other anti-oxidant responses. Nitrosylated tyrosine is a marker of oxidative stress (Virág et al. 2003; Warner et al. 2004). Immunostaining for nitrotyrosine (3-NT) showed that in juvenile mice, moderate levels of 3-NT were observed at 24 hours post-MCAo (Figure 6M), with only trace amounts left by 3 days post-MCAo (Figure 6N). In contrast, adult tissue showed high levels of 3-NT at 24 hours post-MCAo, which further increased at 3 days post-MCAo (Figure 6O, P respectively). 3-NT immunostaining was not observed in oligodendrocytes in any of the tissue examined, as co-localization with PLP-EGFP was not observed (Figure 6M–P). Heme oxygenase expression is induced by oxidative stress and increased expression of this enzyme is associated with protective effects from oxidative damage (Wang et al. 2008). Immunostaining against heme oxygenase 1 (HO-1) in juvenile and adult tissue showed higher levels of HO-1 in juvenile striatum than adult (Figure 6Q, R respectively). Furthermore, HO-1 co-localized with many Sox10-positive oligodendrocytes in the juvenile striatum (Figure 6Q′, arrowheads). We examined other anti-oxidant enzymes, including glutamate-cysteine ligase, glutathione reductase, and glutathione peroxidase; however, we were unable to detect significant expression of these enzymes by immunohistochemistry (data not shown). Together, these data suggest that compared to adults, juvenile mice experience less oxidative damage and have an increased anti-oxidant capacity.
Discussion
Juvenile stroke has received very little research attention despite major economic and quality of life considerations. This is in part due to a lack of experimental models with which to study juvenile stroke. Thus, the recent development of a rodent experimental model of juvenile ischemic stroke allows detailed investigation of the pathological events that underlie this condition (Herson et al. 2013), and most importantly, direct comparison with adult stroke using the same model, as shown in this study. The majority of stroke research focuses on gray matter damage, with proportionally less attention paid to white matter and myelinated axons, where damage to axons and glia apparently evolves by different mechanisms (Fern et al., 2014). Ischemic stroke and white matter damage are particularly understudied in juvenile tissue. The current study describes for the first time the glial and white matter response after juvenile MCAo. We made the counter-intuitive observation that juvenile oligodendrocytes were remarkably resistant to ischemic injury, despite their high metabolic demand during this developmental period of active myelin production. Consistent with resistance of juvenile oligodendrocytes to ischemia, myelinated axons were preserved in the juvenile brain compared to adult brain. Thus, the overall progression of injury in the juvenile and adult brains was significantly different, due to differential glial cell responses following cerebral ischemia.
Not surprisingly, both juvenile and adult mice exhibited extensive, and roughly equivalent, ischemic injury and neuronal cell death after MCAo (Figures 1). However, despite widespread neuron loss, relative preservation of oligodendrocytes and myelin was seen in juvenile mice. Oligodendrocytes have been shown previously to be highly sensitive to ischemia in both neonatal and adult tissue (Back and Rosenberg 2014; Dewar et al. 2003; Sozmen et al. 2012). Other studies have reported that adult oligodendrocytes exhibit signs of ultrastructural damage as early as 30 minutes after ischemia (Pantoni et al. 1996). While we cannot rule out the possibility that juvenile oligodendrocytes in our model could undergo some level of damage immediately after ischemia and then recover by 24 hours post-MCAo, adult oligodendrocytes eventually died, whereas the juvenile oligodendrocytes in this study did not. Furthermore, the g ratio of the myelin 24 hrs after MCAo in juvenile mice is completely normal (Figure 3) is very strong evidence that this is not repair.
While the mechanism of resistance in juvenile oligodendrocytes remains unclear, we made the surprising observation of different patterns of antioxidant expression in juvenile and adult striatal oligodendrocytes. Oxidative stress is a major contributing factor to the pathophysiology of ischemic stroke (Allen and Bayraktutan 2009). Oligodendrocytes in the neonatal and adult brains have been shown to be damaged by oxidative damage following hypoxia-ischemia injury (Back and Rosenberg 2014; Dewar et al. 2003; Mifsud et al. 2014). However, a direct comparison between ages using the same experimental model had not been performed prior to this report. We observed that uninjured juvenile oligodendrocytes in the striatum expressed high levels of GSTπ and HO-1 and corresponding adult cells expressed low amounts of these enzymes. In the ischemic core, GSTπ remained elevated in juvenile oligodendrocytes and was upregulated by injured adult oligodendrocytes. Interestingly, our data in adult animals are consistent with observations in adult human patients, where upregulation of blood GSTπ serves as a reliable time indicator of stroke onset (Turck et al. 2012). While little is known regarding the role of GSTπ in healthy or injured oligodendrocytes, extensive evidence indicates that GSTπ is an important protective factor following oxidative damage in other cellular contexts ((Hayes et al. 2005; Huang et al. 2004). Therefore, it is likely that enhanced anti-oxidant capacity in juvenile oligodendrocytes contributes to their relative insensitivity to ischemic injury.
White matter damage is observed after neonatal hypoxia/ischemia, resulting in degeneration and maturation arrest of late oligodendrocyte progenitors, reduced myelination, neuro-axonal degeneration, and long-term cognitive impairment (Back et al. 2002b; Huang et al. 2009; Segovia et al. 2008). Furthermore, stroke in adult mice causes myelin loss, neurodegeneration, sensory/motor deficits, and memory impairment (Blasi et al. 2014; Kalesnykas et al. 2008; Zhou et al. 2013). Similar to the preservation of oligodendrocytes, juvenile myelin was also preserved in the striatum after MCAo with only minimal axon damage. Adult myelin was severely impacted after MCAo and signs of severe axon pathology were also present. This is significant because in adult mice, there is both primary neuron loss as a result of ischemic damage as well as secondary neuron loss as a result of axon degeneration. Since juvenile myelinated axons are maintained after MCAo, secondary injury is presumably also decreased, which could be a significant contributor to the enhanced long-term recovery after stroke in juvenile mice compared to adults.
In investigating other glial responses to MCAo, we observed diffuse astrogliosis throughout the lesioned striatum in juvenile mice without evidence of fibrotic scar formation. In contrast, adult tissue displayed a well-characterized demarcation of the ischemic core by reactive astrocytes, as observed previously (Pekny and Nilsson, 2005;(Fernández-Klett and Priller 2014), which eventually became engulfed by fibrinogen deposition, resulting in permanent tissue loss. The tissue that was lost in adult mice was maintained in juvenile mice. Similarly, we observed a gradual decrease in both vascular integrity and associated pericyte population after MCAo in adult mice, consistent with previous studies demonstrating vascular density correlating to stroke outcome in patients (Krupinski et al. 1994) and a role for pericytes in tissue pathology after ischemia (Fernández-Klett et al. 2013; Hall et al. 2014; Sbarbati et al. 1996). Interestingly, both the blood vessel network and their associated pericytes remained intact in juvenile mice after MCAo. However, the mechanisms responsible for vasculature destruction in adult mice, but maintenance in juvenile mice, are not clear. Fibrinogen deposition around blood vessels occurred in adult mice at 3 days post-MCAo, at the same time that the vasculature was lost. However, whether fibrinogen was deposited around these vessels because they were damaged, or the deposition caused the damage to vessels, is currently unknown. Stromal pericytes have been described to contribute to fibrotic scar formation after stroke (Fernández-Klett et al. 2013), and could be the source of fibrotic scar formation observed in our tissue (Figure 4G, H). The current studies demonstrate that oligodendrocyte and OPC survival in juvenile tissue correlates with blood vessel integrity after MCAo, endothelial cells, and pericytes. These vascular changes could contribute to the long-term maintenance of tissue observed in juvenile tissue compared to adult tissue. The current studies suggest complex interactions within the “oligovascular niche”, in which there is bidirectional cross talk between the cerebral endothelium and oligodendrocytes/OPCs (Arai and Lo 2009; Pham et al. 2012; Yuen et al. 2014). Further studies are warranted to assess whether the relative preservation of oligodendrocytes and OPCs influences the preservation of vessel integrity, or vice versa. Thus, our data indicate that while cell death and neuronal loss reveals comparable acute injury in the adult and juvenile brain, progression of injury during the subacute and chronic stages is different between juvenile and adult mice. This observation has profound implications regarding long-term functional deficits and potential for histological and functional plasticity in the juvenile brain after injury.
Interestingly, we also observed sparing of juvenile oligodendrocyte precursor cells (OPCs) after MCAo. OPCs are the most proliferative cell in the brain and are evenly distributed throughout brain parenchyma (Nishiyama et al. 2002). OPCs have traditionally been viewed with regard to their ability to respond to demyelination and they differentiate into new myelin-forming oligodendrocytes after injury, but it is becoming increasingly obvious that they have much more diverse functions both in healthy and injured brain tissue (Hughes et al. 2013; Xu et al. 2011; Yang et al. 2013). In this light, OPCs are more accurately labeled as NG2-cells (or NG2-glia) based on their expression of the NG2 proteoglycan and their distinct functionality (Nishiyama et al. 2009). In order to understand if this population of cells might mediate some of the differences noted following juvenile MCAo relative to adult MCAo, we examined the NG2-cell response in these samples. We observed a different profile of NG2-cell response in the juvenile and adult mice, with juvenile brain having large numbers of NG2-cells within the ipsilateral striatum during the acute phase (3 days), while the adults did not. This could be indicative of a protective role for NG2-cells in the post-ischemic brain. Indeed, deficiency of NG2-cells in adults results in greater infarct volume after MCAo (Wang et al. 2011), highlighting a potential protective role of NG2-cells after ischemic injury. Interestingly, a role in the repair process is also indicated for NG2-cells via the generation of new, myelin-forming mature oligodendrocytes (Honsa et al. 2012). Consistent with this possible restorative role, we observed recovery of NG2-cells in the adult brain at delayed time points (>7 days).
In conclusion, we have described very different pathological sequelae after MCAo in juvenile and adult mice. Despite similar amounts of striatal cell death and neuronal loss between the two age groups, we observed very different glial responses. Whereas oligodendrocytes and OPCs were lost early after MCAo in adult mice, these cells were spared in juvenile mice, perhaps due to enhanced antioxidant defenses. We suggest a model in which the “oligovascular niche” is maintained in the juvenile brain, culminating in unique cellular events (diffuse gliosis, reduced fibrosis) that maintain the brain parenchyma to a greater extent than that observed in adult tissue. These results suggest that specific glial responses are important mediators of long-term tissue preservation after ischemic injury in juvenile mice and represent important therapeutic targets in order to improve stroke treatment and recovery in all age groups.
Main Points.
Oligodendrocytes/myelinated axons are resistant to ischemia induced by MCAo in juvenile tissue, while local neurons die. Other glial/vascular responses to MCAo at this age are also different from adult ischemia, and tissue survival is much greater.
Acknowledgments
This work was supported by an American Heart Association Bugher Foundation grant (WBM, PSH, and RJT) and National Institutes of Health grant F31 NS081834 (JTA). The authors thank Dorothy Dill for technical assistance with electron microscopy and the entire Macklin lab for helpful discussion.
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–70. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- Allman C, Scott RB. Neuropsychological sequelae following pediatric stroke: a nonlinear model of age at lesion effects. Child neuropsychology: a journal on normal and abnormal development in childhood and adolescence. 2013;19:97–107. doi: 10.1080/09297049.2011.639756. [DOI] [PubMed] [Google Scholar]
- Anderson V, Spencer-Smith M, Wood A. Do children really recover better? Neurobehavioural plasticity after early brain insult. Brain. 2011;134:2197–2221. doi: 10.1093/brain/awr103. [DOI] [PubMed] [Google Scholar]
- Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. Journal of Neuroscience. 2009;29:4351–4355. doi: 10.1523/JNEUROSCI.0035-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. Journal of Neuroscience. 2002a;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Volpe JJ, Kinney HC. Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. JNeuropatholExpNeurol. 2002b;61:197–211. doi: 10.1093/jnen/61.2.197. [DOI] [PubMed] [Google Scholar]
- Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia. 2014;62:1790–1815. doi: 10.1002/glia.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Besancon EF, Mbow B, Ye Z, Hamner MA, Ransom BR. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. Journal of Neuroscience. 2008;28:1479–1489. doi: 10.1523/JNEUROSCI.5137-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi F, Wei Y, Balkaya M, Tikka S, Mandeville JB, Waeber C, Ayata C, Moskowitz MA. Recognition Memory Impairments After Subcortical White Matter Stroke in Mice. Stroke; a journal of cerebral circulation. 2014 doi: 10.1161/STROKEAHA.114.005324. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP. Oligodendrocytes and ischemic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2003;23:263–274. doi: 10.1097/01.WCB.0000053472.41007.F9. [DOI] [PubMed] [Google Scholar]
- Ellis C, McGrattan K, Mauldin P, Ovbiagele B. Costs of pediatric stroke care in the United States: a systematic and contemporary review. Expert review of pharmacoeconomics & outcomes research. 2014;14:643–650. doi: 10.1586/14737167.2014.933672. [DOI] [PubMed] [Google Scholar]
- Everts R, Pavlovic J, Kaufmann F, Uhlenberg B, Seidel U, Nedeltchev K, Perrig W, Steinlin M. Cognitive functioning, behavior, and quality of life after stroke in childhood. Child Neuropsychol. 2008;14:323–38. doi: 10.1080/09297040701792383. [DOI] [PubMed] [Google Scholar]
- Fern RF, Mature C, Stys PK. White matter injury: Ischemic and nonischemic. Glia. 2014;62:1780–89. doi: 10.1002/glia.22722. [DOI] [PubMed] [Google Scholar]
- Fernández-Klett F, Potas JR, Hilpert D, Blazej K, Radke J, Huck J, Engel O, Stenzel W, Genové G, Priller J. Early loss of pericytes and perivascular stromal cell-induced scar formation after stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:428–439. doi: 10.1038/jcbfm.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Klett F, Priller J. The fibrotic scar in neurological disorders. Brain pathology (Zurich, Switzerland) 2014;24:404–413. doi: 10.1111/bpa.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg-Cohen N, Guo Y, Margolis F, Cohen Y, Miller NR, Bernstein SL. Oligodendrocyte dysfunction after induction of experimental anterior optic nerve ischemia. Investigative ophthalmology & visual science. 2005;46:2716–2725. doi: 10.1167/iovs.04-0547. [DOI] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annual review of pharmacology and toxicology. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- Herson PS, Bombardier CG, Parker SM, Shimizu T, Klawitter J, Klawitter J, Quillinan N, Exo JL, Goldenberg NA, Traystman RJ. Experimental pediatric arterial ischemic stroke model reveals sex-specific estrogen signaling. Stroke. 2013;44:759–63. doi: 10.1161/STROKEAHA.112.675124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honsa P, Pivonkova H, Dzamba D, Filipova M, Anderova M. Polydendrocytes display large lineage plasticity following focal cerebral ischemia. PLoS One. 2012;7:e36816. doi: 10.1371/journal.pone.0036816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Tan PH, Tan BKH, Bay BH. GST-pi expression correlates with oxidative stress and apoptosis in breast cancer. Oncology reports. 2004;12:921–925. [PubMed] [Google Scholar]
- Huang Z, Liu J, Cheung PY, Chen C. Long-term cognitive impairment and myelination deficiency in a rat model of perinatal hypoxic-ischemic brain injury. Brain Res. 2009;1301:100–9. doi: 10.1016/j.brainres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci. 2013;16:668–76. doi: 10.1038/nn.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Verma S, Nakayama S, Quillinan N, Grafe MR, Hurn PD, Herson PS. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:2160–2168. doi: 10.1038/jcbfm.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalesnykas G, Tuulos T, Uusitalo H, Jolkkonen J. Neurodegeneration and cellular stress in the retina and optic nerve in rat cerebral ischemia and hypoperfusion models. Neuroscience. 2008;155:937–947. doi: 10.1016/j.neuroscience.2008.06.038. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke; a journal of cerebral circulation. 1994;25:1794–1798. doi: 10.1161/01.str.25.9.1794. [DOI] [PubMed] [Google Scholar]
- Levine J. The reactions and role of NG2 glia in spinal cord injury. Brain Res. 2015 doi: 10.1016/j.brainres.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon BS, Shick HE, Kidd GJ, Macklin WB. Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. JNeurosci. 2002;22:876–885. doi: 10.1523/JNEUROSCI.22-03-00876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarran WJ, Goldberg MP. White matter axon vulnerability to AMPA/kainate receptor-mediated ischemic injury is developmentally regulated. Journal of Neuroscience. 2007;27:4220–4229. doi: 10.1523/JNEUROSCI.5542-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifsud G, Zammit C, Muscat R, Di Giovanni G, Valentino M. Oligodendrocyte pathophysiology and treatment strategies in cerebral ischemia. CNS Neurosci Ther. 2014;20:603–12. doi: 10.1111/cns.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Watanabe M, Yang Z, Bu J. Identity, distribution, and development of polydendrocytes: NG2-expressing glial cells. Journal of neurocytology. 2002;31:437–455. doi: 10.1023/a:1025783412651. [DOI] [PubMed] [Google Scholar]
- Ozerdem U, Monosov E, Stallcup WB. NG2 proteoglycan expression by pericytes in pathological microvasculature. Microvascular research. 2002;63:129–134. doi: 10.1006/mvre.2001.2376. [DOI] [PubMed] [Google Scholar]
- Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke; a journal of cerebral circulation. 1996;27:1641–6. doi: 10.1161/01.str.27.9.1641. discussion 1647. [DOI] [PubMed] [Google Scholar]
- Pavlovic J, Kaufmann F, Boltshauser E, Capone Mori A, Gubser Mercati D, Haenggeli CA, Keller E, Lutschg J, Marcoz JP, Ramelli GP, et al. Neuropsychological problems after paediatric stroke: two year follow-up of Swiss children. Neuropediatrics. 2006;37:13–9. doi: 10.1055/s-2006-923932. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–34. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- Pham L-DD, Hayakawa K, Seo JH, Nguyen M-N, Som AT, Lee BJ, Guo S, Kim K-W, Lo EH, Arai K. Crosstalk between oligodendrocytes and cerebral endothelium contributes to vascular remodeling after white matter injury. Glia. 2012;60:875–881. doi: 10.1002/glia.22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. Regulation of oligodendrocyte development and myelination by glucose and lactate. JNeurosci. 2011;31:538–548. doi: 10.1523/JNEUROSCI.3516-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach ES, Golomb MR, Adams R, Biller J, Daniels S, Deveber G, Ferriero D, Jones BV, Kirkham FJ, Scott RM, et al. Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008a;39:2644–91. doi: 10.1161/STROKEAHA.108.189696. [DOI] [PubMed] [Google Scholar]
- Roach ES, Golomb MR, Adams R, Biller J, Daniels S, Deveber G, Ferriero D, Jones BV, Kirkham FJ, Scott RM, et al. Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. 2008b. pp. 2644–2691. [DOI] [PubMed] [Google Scholar]
- Sbarbati A, Pietra C, Baldassarri AM, Guerrini U, Ziviani L, Reggiani A, Boicelli A, Osculati F. The microvascular system in ischemic cortical lesions. Acta neuropathologica. 1996;92:56–63. doi: 10.1007/s004010050489. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia KN, McClure M, Moravec M, Luo NL, Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS, et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63:520–30. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherriff FE, Bridges LR, Gentleman SM, Sivaloganathan S, Wilson S. Markers of axonal injury in post mortem human brain. Acta neuropathologica. 1994;88:433–439. doi: 10.1007/BF00389495. [DOI] [PubMed] [Google Scholar]
- Sozmen EG, Hinman JD, Carmichael ST. Models that matter: white matter stroke models. Neurotherapeutics. 2012;9:349–58. doi: 10.1007/s13311-012-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey FA, Cammer W. A pi form of glutathione-S-transferase is a myelin- and oligodendrocyte-associated enzyme in mouse brain. JNeurochem. 1991;57:95–102. doi: 10.1111/j.1471-4159.1991.tb02104.x. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Nishiyama A, Cheng D, Macklin WB. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. The Journal of cell biology. 1997;137:459–468. doi: 10.1083/jcb.137.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turck N, Robin X, Walter N, Fouda C, Hainard A, Sztajzel R, Wagner G, Hochstrasser DF, Montaner J, Burkhard PR, et al. Blood glutathione S-transferase-π as a time indicator of stroke onset. PloS one. 2012;7:e43830. doi: 10.1371/journal.pone.0043830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeriani V, Dewar D, McCulloch J. Quantitative assessment of ischemic pathology in axons, oligodendrocytes, and neurons: attenuation of damage after transient ischemia. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2000;20:765–771. doi: 10.1097/00004647-200005000-00002. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev Neurosci. 2005;27:81–6. doi: 10.1159/000085978. [DOI] [PubMed] [Google Scholar]
- Virág L, Szabó E, Gergely P, Szabó C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicology letters. 2003;140–141:113–124. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- Wang P, Tian WW, Song J, Guan YF, Miao CY. Deficiency of NG2+ cells contributes to the susceptibility of stroke-prone spontaneously hypertensive rats. CNS Neurosci Ther. 2011;17:327–32. doi: 10.1111/j.1755-5949.2011.00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xing Z, Vosler PS, Yin H, Li W, Zhang F, Signore AP, Stetler RA, Gao Y, Chen J. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: role of enhanced DNA repair. Stroke. 2008;39:2587–95. doi: 10.1161/STROKEAHA.107.509158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner DS, Sheng H, Batinić-Haberle I. Oxidants, antioxidants and the ischemic brain. The Journal of experimental biology. 2004;207:3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- Westmacott R, Askalan R, MacGregor D, Anderson P, Deveber G. Cognitive outcome following unilateral arterial ischaemic stroke in childhood: effects of age at stroke and lesion location. Dev Med Child Neurol. 2010;52:386–93. doi: 10.1111/j.1469-8749.2009.03403.x. [DOI] [PubMed] [Google Scholar]
- Xu JP, Zhao J, Li S. Roles of NG2 glial cells in diseases of the central nervous system. Neurosci Bull. 2011;27:413–21. doi: 10.1007/s12264-011-1838-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura T, Konola JT, Wekerle H, Lees MB. Monoclonal antibodies against myelin proteolipid protein: identification and characterization of two major determinants. Journal of Neurochemistry. 1991;57:1671–1680. doi: 10.1111/j.1471-4159.1991.tb06367.x. [DOI] [PubMed] [Google Scholar]
- Yan H, Rivkees SA. Hypoglycemia influences oligodendrocyte development and myelin formation. Neuroreport. 2006;17:55–59. doi: 10.1097/01.wnr.0000192733.00535.b6. [DOI] [PubMed] [Google Scholar]
- Yang Q-K, Xiong J-X, Yao Z-X. Neuron-NG2 cell synapses: novel functions for regulating NG2 cell proliferation and differentiation. BioMed Research International. 2013;2013:402843. doi: 10.1155/2013/402843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SPJ, Zahed H, Maltepe E, Rowitch DH. Oligodendrocyte-Encoded HIF Function Couples Postnatal Myelination and White Matter Angiogenesis. Cell. 2014;158:383–396. doi: 10.1016/j.cell.2014.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Zhuang J, Li J, Ooi E, Bloom J, Poon C, Lax D, Rosenbaum DM, Barone FC. Long-term post-stroke changes include myelin loss, specific deficits in sensory and motor behaviors and complex cognitive impairment detected using active place avoidance. PLoS One. 2013;8:e57503. doi: 10.1371/journal.pone.0057503. [DOI] [PMC free article] [PubMed] [Google Scholar]