

Abstract
Glaucoma is the leading cause of irreversible blindness worldwide. Although most glaucoma patients are elderly, congenital glaucoma and glaucomas of childhood are also important causes of visual disability. Primary congenital glaucoma (PCG) is isolated, non-syndromic glaucoma that occurs in the first three years of life and is a major cause of childhood blindness. Other early-onset glaucomas may arise secondary to developmental abnormalities, such as glaucomas that occur with aniridia or as part of Axenfeld-Rieger syndrome. Congenital and childhood glaucomas have strong genetic bases and disease-causing mutations have been discovered in several genes. Mutations in three genes (CYP1B1, LTBP2, TEK) have been reported in PCG patients. Axenfeld-Rieger syndrome is caused by mutations in PITX2 or FOXC1 and aniridia is caused by PAX6 mutations. This review discusses the roles of these genes in primary congenital glaucoma and glaucomas of childhood.
Introduction
Glaucoma is a heterogeneous group of optic nerve diseases that share two clinical features, a characteristic injury to the optic nerve (cupping) and a corresponding pattern of visual field loss. The anterior segment of the eye is filled with an aqueous fluid that is created in the ciliary body and exits the eye through the trabecular meshwork. The trabecular meshwork is a porous tissue located in the angle created where the cornea intersects the iris - the iridocorneal angle. Abnormalities in the structure or conformation of the iridocorneal angle may limit outflow of aqueous humor and cause a rise in intraocular pressure, which is a strong risk factor for developing glaucoma (1).
While the majority of glaucoma patients are adults, early onset forms of glaucoma are also important causes of visual disability. Glaucomas that occur before three years of age without overt structural defects of the eye are termed primary congenital glaucoma (PCG). Conversely, developmental glaucomas occur secondarily to recognizable malformations of the anterior segment of the eye (iris, iridocorneal angle, etc.). PCG and developmental glaucomas have strong genetic bases and are the subject of this review.
Primary Congenital Glaucoma (OMIM: 231300)
PCG is the most common form of pediatric glaucoma (2) and accounts for up to 18% of childhood blindness (3,4). The incidence of PCG varies geographically, being most common in Saudi Arabia (1/2,500) (5) and among consanguineous populations (1/1,250 in Slovakian Roma) (6), and least common in Western countries (1/10,000) (7). Symptoms are nonspecific and include tearing, light sensitivity, eye rubbing, and irritability. High intraocular pressure leads to enlarged eyes, cloudy corneas, cracks in the cornea (Haab striae), and optic nerve cupping (8,9).
While the majority of PCG cases are sporadic, up to 40% are familial and follow autosomal recessive inheritance patterns with variable penetrance (7). Genetic analyses of affected families have yielded four loci associated with PCG: GLC3A on 2p21 (10), GLC3B on 1p36 (11), GLC3C (12) and GLC3D (13) adjacent to but not overlapping one another on 14q24. PCG-causing mutations have been identified in genes within two of the four loci. Cytochrome P450 1B1 (CYP1B1) mutations were discovered within the GLC3A locus and are the most common known cause of PCG (14). CYP1B1 encodes a metabolizing enzyme of the cytochrome P450 family. Mutations in latent transforming growth factor beta binding protein 2 (LTBP2), located in the GLC3D locus (15), have also been associated with PCG. LTBP2 encodes an extracellular matrix protein involved in cell adhesion and structural maintenance of connective tissues. PCG-causing mutations have not yet been found within the GLC3B or GLC3C loci. Most recently, mutations in a third gene, tunica interna endothelial cell kinase (TEK), have been reported in patients with PCG (16).
Cytochrome P450 1B1 (CYP1B1)
Autosomal recessive mutations in CYP1B1 are the most common known cause of PCG. Over 150 variants have been identified worldwide and the prevalence of CYP1B1 mutations in PCG varies greatly with ethnicity (17). A few specific CYP1B1 mutations account for a majority of PCG cases in select populations (i.e. E387K in Slovakian Roma and G61E in Saudi Arabia) (18,19). One mutation, R390H, is especially common among Chinese, Iranian, Indian, and Pakistani cases of PCG (17,20–22). Pathogenic CYP1B1 alleles have been detected in a smaller fraction of PCG cases in Japan (20%) (23) and the United States (14.9%) (24). Although CYP1B1 mutations are most commonly detected in PCG patients, mutations have also been rarely reported in patients with a range of other phenotypes including aniridia (25), Peters anomaly (26) Axenfeld-Rieger syndrome (27), juvenile open angle glaucoma (28), and primary open angle glaucoma (29).
CYP1B1 is a member of the cytochrome P450 family of membrane-bound oxidase enzymes that have broad roles in metabolism and produce hormones and other metabolic intermediates during development (30). While most other cytochromes are highly expressed in the liver, CYP1B1 is more abundant in extra-hepatic tissues including lung, colon, kidney and eye (31). The precise mechanism by which CYP1B1 mutations cause PCG is unknown. However, CYP1B1 is expressed in the ciliary body and trabecular meshwork, tissues of the eye that regulate intraocular pressure (14,32). Recent studies propose that CYP1B1 may be essential in the development and function of the trabecular meshwork (33,34). Moreover, glaucoma-causing mutations reportedly decrease CYP1B1 enzyme stability, abundance, or catalytic activity (35–41). Together these data suggest that CYP1B1 mutations may alter trabecular meshwork function, cause dysregulation of intraocular pressure, optic nerve damage, and ultimately PCG.
Latent transforming growth factor beta binding protein 2 (LTBP2)
Autosomal recessive LTBP2 variants are a rare cause of PCG, with cases primarily reported in consanguineous families from Pakistan and Iran (15,42). LTBP2 mutations are also responsible for many cases of PCG in Slovakian Roma, with the R299X mutation in LTBP2 accounting for over half of CYP1B1-negative cases (43). However, LTBP2 mutations have not been detected in PCG cohorts from northern India, the United Kingdom, the United States, or China (24,44–46). Recently, mutations in LTBP2 were found to cause congenital glaucoma in Siamese cats, providing additional support for the role of this gene in PCG (47).
LTBP2 encodes an extracellular matrix protein with putative roles in cell adhesion (48,49) and elastin microfibril assembly (50–52). LTBP2 is highly expressed in tissues that are rich in elastic fibers, such as lungs and arteries (53). LTBP2 is also expressed in ocular tissues that are vital to regulation of intraocular pressure and glaucoma biology, including the trabecular meshwork and ciliary body. Moreover, LTBP2 is essential for development of the anterior chamber and ciliary zonules (15,53). Mutations in LTBP2 are, consequently, a plausible cause of congenital abnormalities of ocular structures that may lead to increased intraocular pressure and PCG.
Homozygous LTBP2 mutations have also been reported in rare cases with a range of other ocular abnormalities (megalocornea, microspherophakia, ectopia lentis, primary open angle glaucoma, pseudoexfoliation syndrome, primary angle-closure glaucoma and Weill-Marchesani syndrome) (54–59).
Tunica interna endothelial cell kinase (TEK)
Recently, transgenic "knock-out" mice that are deficient for the angiopoietin 1 and 2 (Angpt1 and Angpt2) genes or the angiopoietin receptor also known as tunica interna endothelial cell kinase (Tek) gene were shown to have signs of PCG including enlarged globes, high intraocular pressure, retinal ganglion cell loss, and optic nerve damage. These phenotypes of the transgenic mice were attributed to an absence of ocular structures required for fluid drainage (i.e. Schlemm canal) and have implicated the angiopoietin signaling pathways in PCG pathogenesis (60). Furthermore, a dose response to Tek deficiency was demonstrated. Mice heterozygous for the Tek "knock-out" mutation develop abnormalities in outflow structures and elevation in intraocular pressure with intermediate severity. Tek haploinsufficiency can compromise aqueous humor outflow in mice (16).
Heterozygous mutations in the human TEK gene were discovered in 10 of 189 unrelated human PCG patients. Seven of these ten PCG cases had no family history of glaucoma, while additional affected family members were identified for three PCG probands. Pedigree analyses reported autosomal dominant inheritance with variable expressivity and incomplete penetrance (16). Several of the TEK mutations detected in human PCG patients were shown to have functional effects in cell culture assays including altered transcription, reduced protein production, altered post-translational modification, and aberrant trafficking. TEK regulates vasculogenesis and is highly expressed in blood vessels and lymphatic endothelia, as well as in the endothelium of Schlemm canal (61,62). Although their specific role in glaucoma pathogenesis is unknown, it is likely that TEK mutations cause alterations in the development of ocular structures necessary for normal aqueous outflow and regulation of intraocular pressure and subsequently predispose development of congenital glaucoma.
Developmental Glaucomas
Axenfeld-Rieger syndrome (OMIM: 180500)
Axenfeld-Rieger syndrome encompasses a heterogeneous collection of disorders with ocular and systemic features. Axenfeld-Rieger syndrome is rare and has a prevalence of 1:200,000 and an autosomal dominant pattern of inheritance with variable expressivity but high penetrance (63). Ocular features are bilateral, congenital, and may be caused by abnormal differentiation and migration of neural crest cells during the formation of the anterior ocular structures (64). Most patients have a white line on the corneal endothelium along its peripheral edge (posterior embryotoxon) and a range of iris abnormalities including a decentered pupil (corectopia), additional holes in the iris (polycoria), and generalized hypoplasia (Fig. 1A and B). Strands or broad bands of iris may project across the iridocorneal angle and up onto the cornea (iris processes). A high iris insertion may restrict aqueous from leaving the eye through the trabecular meshwork and cause elevated intraocular pressure (63–66). Microscopic abnormalities of the trabecular meshwork may also impair fluid drainage from the eye (64). Consequently, elevated intraocular pressure and glaucoma develop in 50% of Axenfeld-Rieger syndrome patients (64,67,68). Non-ocular features of Axenfeld-Rieger syndrome include flat mid-facies (maxillary hypoplasia and a flat, broad nose); a reduction in the number or size of teeth (hypodontia or microdontia, Fig. 1C); and redundant tissue around the umbilicus (63,64,66). Various congenital heart defects may also be present, including atrial septal defects (69,70).
Figure 1.
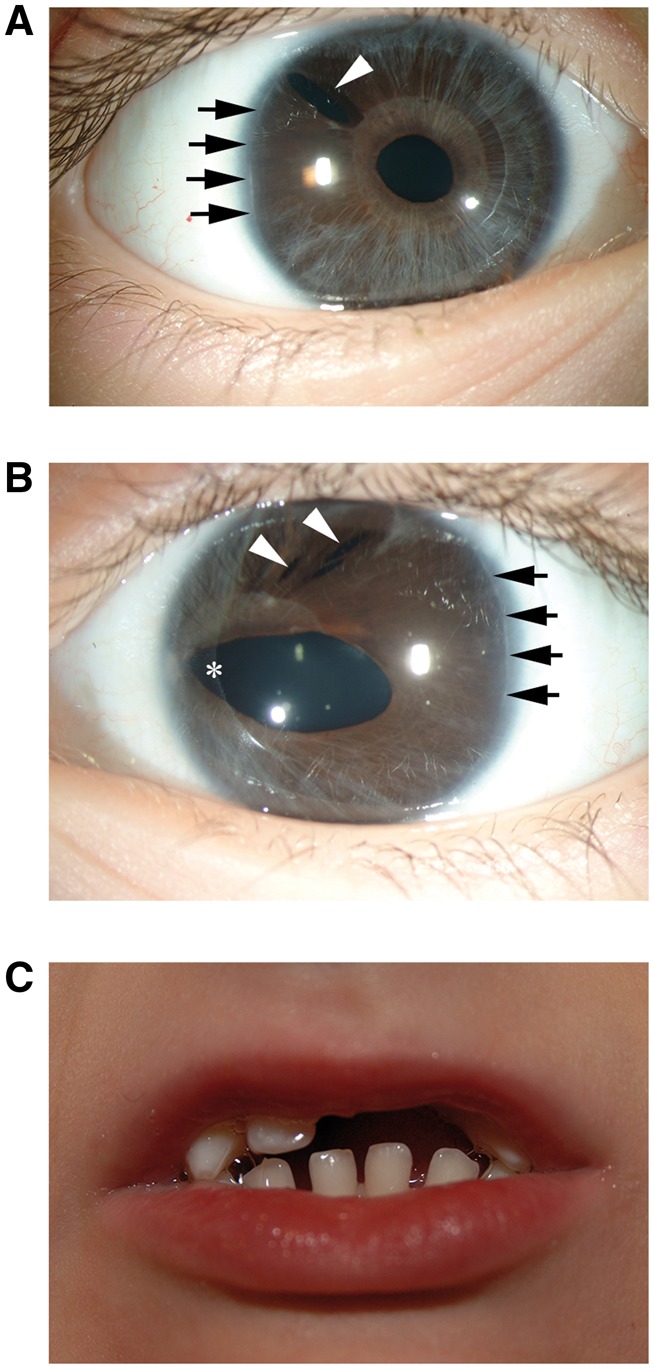
Clinical features of Axenfeld-Rieger Syndrome in a patient with a PITX2 mutation. DNA sequencing of the PITX2 gene in a female with Axenfeld-Rieger syndrome identified a novel heterozygous mutation of a canonical splicing sequence within intron 3, IVS3-1delG. This patient had classic features of Axenfeld-Rieger syndrome including bilateral iris hypoplasia (panel A and B); posterior embryotoxon (panel A and B indicated with black arrows); polycoria (panel A and B indicated with white arrowheads); and corectopia (panel B indicated with an asterisk). This patient also had hypodontia and microdontia (panel C).
Linkage analysis of large pedigrees with dominantly inherited Axenfeld-Rieger syndrome led to the discovery of disease-causing mutations in two genes, paired-like homeodomain 2 (PITX2) (68) and forkhead box C1 (FOXC1) (71).
Paired-like homeodomain transcription factor 2 (PITX2)
A range of PITX2 (originally termed RIEG1) mutations have been associated with Axenfeld-Rieger syndrome including missense mutations, nonsense mutations, splice site mutations (68), and copy number variations (72). PITX2 encodes a member of the bicoid class of homeodomain transcription factors, which play critical roles in embryonic development and tissue morphogenesis. PITX2 encodes several alternatively spliced isoforms, which all contain a partial or complete DNA-binding homeodomain termed solurshin. The majority of Axenfeld-Rieger-causing defects are missense mutations within the homeodomain (73).
The precise mechanisms by which PITX2 mutations cause Axenfeld-Rieger syndrome are not fully known, but relate to PITX2 haploinsufficiency and dominant negative effects (74–76). Some PITX2 mutations detected in Axenfeld-Rieger patients have been shown to impart functional changes in cell-based in vitro assays, including impaired ability to bind DNA and altered transactivation activity in reporter assays (76). In rare cases, hypermorphic alleles of PITX2 were identified in Axenfeld-Rieger patients. These cases suggest that strict regulation of PITX2 dosage is necessary for normal development and function in the eye (76).
PITX2 is expressed in the tissues most affected by Axenfeld-Rieger syndrome (68,77). Moreover, studies using experimental mouse models have demonstrated that PITX2 is required for normal ocular development and have helped to reveal a range of abnormalities arising from PITX2 deficiencies (78,79). Knock-out of Pitx2 reduces the abundance of astrocytes and retinal vasculature in neural crest-derived tissues (78). These deficits suggest that developmental abnormalities of the optic nerve and retina may contribute to glaucoma susceptibility in Axenfeld-Rieger patients. Mice heterozygous for a Pitx2 null mutation also recapitulate the ocular malformations of Axenfeld-Rieger syndrome and associated glaucoma (80). Together, these data confirm a role for PITX2 mutations in the development of Axenfeld-Rieger syndrome.
PITX2 mutations have also been associated with another developmental abnormality characterized by adhesions between the central cornea and lens known as Peters anomaly (81). Mutations in PITX2 have also been discovered in patients diagnosed with iris hypoplasia, iridogoniodysgenesis, mesodermal dysgenesis, and anterior segment cleavage syndrome, which likely represent conditions located on a spectrum of phenotypes best described as Axenfeld-Rieger syndrome (63).
Forkhead box transcription factor C1 (FOXC1)
Missense mutations, deletions, and duplications of FOXC1 (previously termed FKHL5) are another cause of Axenfeld-Rieger syndrome (71,82–84). FOXC1 encodes a member of the forkhead box family of transcription factors, which function as important regulators of embryogenesis, cell migration, differentiation, and fate determination (85). The defining and functional feature of this family of transcription factors is the conserved 110-amino acid forkhead domain, which mediates DNA binding and protein-protein interactions.
Mechanisms by which FOXC1 mutations cause Axenfeld-Rieger syndrome are not precisely known, but may often relate to FOXC1 haploinsufficiency. Mutations that alter the Foxc1 gene dosage in mice confirm a crucial role in the development of the eye and Axenfeld-Rieger syndrome. Haploinsufficiency of Foxc1 is associated with iris hypoplasia, corectopia, and embryotoxon in mice (86).
Many (71,87), but not all (71) FOXC1 mutations in human Axenfeld-Rieger syndrome patients influence the forkhead domain and can impair localization (88,89), DNA-binding, and transactivation (90,91). In rare cases, mutations in the forkhead domain have been identified in Axenfeld-Rieger patients that result in expression of mutant FOXC1 at levels similar to that of wild-type, but with differential phosphorylation. Abnormal phosphorylation of FOXC1 hinders nuclear localization, DNA-binding, and transactivation (92). Each mutation that interferes with FOXC1’s function as a transcription factor has the potential to cause Axenfeld-Rieger syndrome. FOXC1 has recently been identified as a risk factor for primary open angle glaucoma, suggesting a role in adult-onset glaucoma as well (93).
FOXC1 is expressed in the tissues with key roles in the pathophysiology of Axenfeld-Rieger syndrome. FOXC1 is expressed in the neural crest-derived tissues that form the drainage structures of the eye (trabecular meshwork) during development and in the adult iris (94). FOXC1 is also detectable in non-ocular tissues affected by Axenfeld Rieger syndrome including the heart and heart valves (95,96) with alternative transcripts expressed in a tissue-specific manner (94). These expression patterns along with several cases of heart valve and atrial septal defects in patients with Axenfeld-Rieger syndrome suggest that congenital heart disease may be an additional component of this syndrome associated with FOXC1 mutations (95).
PITX2 and FOXC1 both encode transcription factors that interact directly and influence their respective transcriptional activity. PITX2 is a negative regulator of FOXC1. Consequently, mutations in PITX2 may cause Axenfeld-Rieger syndrome, at least in part, by altering the function of FOXC1 (87).
Axenfeld-Rieger syndrome is heterogeneous. Additional Axenfeld-Rieger syndrome loci have been mapped to chromosomes 13q24 and 16q24, however, the specific disease-causing genes in these loci have not yet been discovered (97).
Aniridia (OMIM: 106210)
Aniridia is a rare developmental eye disease characterized by an underdeveloped iris that has a prevalence of 1/64,000 to 1/96,000 and either autosomal dominant inheritance or sporadic occurrence (98). Although the name "aniridia" suggests a complete absence of the iris, all patients have some iris tissue ranging from a tiny vestige that may be difficult to recognize to a normal appearing iris (Fig. 2B and C). These abnormal irides may collapse against the peripheral cornea and obstruct outflow of aqueous humor, leading to increased intraocular pressure, optic nerve damage (Fig. 2D and E) and a secondary glaucoma. Corneal pannus, foveal hypoplasia, and cataract (Fig. 2B and C) are other important features of aniridia (99). Pannus alters the transparency of the cornea and may significantly compromise vision, while foveal hypoplasia is failure of the retina to develop structures required for high-resolution central vision. Aniridia has variable expressivity, with patients showing a range of severity for corneal pannus, foveal hypoplasia, and iris abnormalities (98).
Figure 2.

Clinical features of aniridia in a patient with a PAX6 mutation. Whole genome DNA sequencing of the PAX6 gene in a female with aniridia and secondary glaucoma detected a tandem inversion (hg19:g.[chr11:31763505-40323405inv;chr11:40323414-43536635inv];[=]) on chromosome 11p13 (panel A) including the PAX6 gene. This inversion moved PAX6 away from a vital enhancer (DRR) (105) that is normally downstream of the PAX6 gene (top, wild-type configuration) to an inactive position millions of base pairs away (bottom, mutant configuration). This patient had an absence of visible iris tissue in both eyes (panel B, right eye; panel C, left eye). The edges of the natural lenses (indicated with black arrows) were visible due to the absence of visible iris tissue. There is bilateral cataract indicated by black arrowheads. Finally, the white arrowheads indicate Ahmed drainage valves that were surgically implanted to control intraocular pressure. This patient has severe secondary glaucoma with a near total cupping of the right optic nerve head (panel D) and a large optic cup with a thin rim of neural tissue remaining on the left optic nerve head (panel E). A normal optic nerve head is shown in panel F for comparison.
Paired box 6 (PAX6)
Missense mutations in PAX6 have been associated with autosomal dominant inheritance of aniridia (100), while deletions and rearrangements have been associated with sporadic cases (101). PAX6 encodes a transcription factor that has profound effects on ocular development. PAX6 activates expression of other genes via its DNA-binding domains (a paired and a paired-type homeodomain) and a proline, serine, threonine-rich transactivating domain. Mutations that cause aniridia may alter PAX6 functional domains (i.e. missense mutations), cause a truncation of the encoded protein, or disrupt enhancer sequences, such as a distant downstream regulatory region (DRR) element (102–105). A common feature of disease-causing mutations is that they alter the dose of functional PAX6 transcription factor, which leads to dysregulation of downstream transcription factors and gene expression patterns, resulting in congenital malformations that may promote glaucoma (106).
Phenotypes similar to aniridia have been detected in animal models with mutations in PAX6 orthologues. Small eye (Sey) mice do not develop eyes or noses and die soon after birth (107). This developmental abnormality in mice was shown to be due to a Pax6 mutation in parallel with discoveries of PAX6 mutations in human patients with aniridia (100,102). A similar mutation of the Drosophila Pax6 gene is associated with an eyeless (ey) phenotype (108). PAX6 has a highly conserved and central role in ocular development and aniridia (109).
Sporadic cases of aniridia may be caused by a large deletion of chromosome 11p13 spanning the PAX6 gene, which has been associated with a contiguous gene syndrome, WAGR (Wilms tumor, Aniridia, Genitourinary abnormalities, and mental Retardation). In addition to deletion of the PAX6 gene, sporadic aniridia patients may also have a deletion of the neighboring tumor suppressor gene, WT1, and increased risk for Wilms tumor. All children with sporadic cases of aniridia should be investigated for a chromosome 11p13 deletion and the associated risk for a potentially lethal Wilms tumor (110).
Conclusion
Glaucomas of infancy and childhood have important genetic components to their pathogenesis. Over the last two decades, many disease-causing mutations have been identified. These include mutations in three genes (CYP1B1, LTBP2, and TEK) that have been associated with primary congenital glaucoma (PCG). Mutations in a different set of genes have been detected in patients with secondary glaucomas of childhood. Specifically, PITX2 and FOXC1 mutations have been associated with Axenfeld-Rieger syndrome, while mutations in PAX6 have been associated with aniridia. Patients with Axenfeld-Rieger syndrome or with aniridia are at high risk for developing secondary glaucoma. Notably, the genes that have been associated with these secondary glaucomas (PITX2, FOXC1, and PAX6) are all transcription factors that control ocular development. In each of these forms of glaucoma, disease-causing mutations lead to microscopic and/or visible developmental malformations of ocular structures that regulate drainage of fluid from the eye and ultimately cause elevated intraocular pressure and optic nerve damage. Despite the significant discoveries of many glaucoma-causing mutations in several genes, a large proportion of PCG, Axenfeld-Rieger syndrome, and aniridia cases do not have known molecular genetic causes. Continuing research is needed both to identify the additional glaucoma-causing mutations in novel genes that are responsible for disease in these patients and to translate this knowledge into new treatments.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health [R01EY023512 and R01EY017673] and Leonard and Marlene Hadley.
References
- 1. Kwon Y.H., Fingert J.H., Kuehn M.H., Alward W.L.M. (2009) Primary open-angle glaucoma. N. Engl. J. Med., 360, 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taylor R.H., Ainsworth J.R., Evans A.R., Levin A.V. (1999) The epidemiology of pediatric glaucoma: the Toronto experience. J. AAPOS, 3, 308–315. [DOI] [PubMed] [Google Scholar]
- 3. Gilbert C.E., Canovas R., Kocksch d., Canovas R., Foster A. (1994) Causes of blindness and severe visual impairment in children in Chile. Dev. Med. Child Neurol., 36, 326–333. [DOI] [PubMed] [Google Scholar]
- 4. Tabbara K.F., Badr I.A. (1985) Changing pattern of childhood blindness in Saudi Arabia. Br. J. Ophthalmol., 69, 312–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bejjani B.A., Stockton D.W., Lewis R.A., Tomey K.F., Dueker D.K., Jabak M., Astle W.F., Lupski J.R. (2000) Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Hum. Mol. Genet., 9, 367–374. [DOI] [PubMed] [Google Scholar]
- 6. Genĉík A. (1989) Epidemiology and genetics of primary congenital glaucoma in Slovakia. Description of a form of primary congenital glaucoma in gypsies with autosomal-recessive inheritance and complete penetrance. Dev. Ophthalmol., 16, 76–115. [PubMed] [Google Scholar]
- 7. Ho C.L., Walton D.S. (2004) Primary congenital glaucoma: 2004 update. J. Pediatr. Ophthalmol. Strabismus, 41, 271–288. [DOI] [PubMed] [Google Scholar]
- 8. deLuise V.P., Anderson D.R. (1983) Primary infantile glaucoma (congenital glaucoma). Surv. Ophthalmol., 28, 1–19. [DOI] [PubMed] [Google Scholar]
- 9. Ko F., Papadopoulos M., Khaw P.T. (2015) Primary congenital glaucoma. Prog. Brain. Res., 221, 177–189. [DOI] [PubMed] [Google Scholar]
- 10. Sarfarazi M., Akarsu A.N., Hossain A., Turacli M.E., Aktan S.G., Barsoum-Homsy M., Chevrette L., Sayli B.S. (1995) Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics, 30, 171–177. [DOI] [PubMed] [Google Scholar]
- 11. Akarsu A.N., Turacli M.E., Aktan S.G., Barsoum-Homsy M., Chevrette L., Sayli B.S., Sarfarazi M. (1996) A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum. Mol. Genet., 5, 1199–1203. [DOI] [PubMed] [Google Scholar]
- 12. Stoilov I.R., Sarfarazi M. (2002) The third genetic locus (GLC3C) for primary congenital glaucoma (PCG) maps to chromosome 14q24.3. Invest. Ophthalmol. Vis. Sci., 43, 3015.. [PubMed] [Google Scholar]
- 13. Firasat S., Riazuddin S.A., Hejtmancik J.F., Riazuddin S. (2008) Primary congenital glaucoma localizes to chromosome 14q24.2-24.3 in two consanguineous Pakistani families. Mol. Vis., 14, 1659–1665. [PMC free article] [PubMed] [Google Scholar]
- 14. Stoilov I., Akarsu A.N., Sarfarazi M. (1997) Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet., 6, 641–647. [DOI] [PubMed] [Google Scholar]
- 15. Ali M., McKibbin M., Booth A., Parry D.A., Jain P., Riazuddin S.A., Hejtmancik J.F., Khan S.N., Firasat S., Shires M.. et al. (2009) Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet., 84, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Souma T., Tompson S.W., Thomson B.R., Siggs O.M., Kizhatil K., Yamaguchi S., Feng L., Limviphuvadh V., Whisenhunt K.N., Maurer-Stroh S.. et al. (2016) Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J. Clin. Invest., 126, 2575–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li N., Zhou Y., Du L., Wei M., Chen X. (2011) Overview of Cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Exp. Eye Res., 93, 572–579. [DOI] [PubMed] [Google Scholar]
- 18. Plásilová M., Stoilov I., Sarfarazi M., Kádasi L., Feráková E., Ferák V. (1999) Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary congenital glaucoma. J. Med. Genet., 36, 290–294. [PMC free article] [PubMed] [Google Scholar]
- 19. Bejjani B.A., Lewis R.A., Tomey K.F., Anderson K.L., Dueker D.K., Jabak M., Astle W.F., Otterud B., Leppert M., Lupski J.R. (1998) Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital glaucoma in Saudi Arabia. Am. J. Hum. Genet., 62, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rauf B., Irum B., Kabir F., Firasat S., Naeem M.A., Khan S.N., Husnain T., Riazuddin S., Akram J., Riazuddin S.A. (2016) A spectrum of CYP1B1 mutations associated with primary congenital glaucoma in families of Pakistani descent. Hum. Genome Var., 3, 16021.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Y., Sorenson C.M., Sheibani N. (2015) Cytochrome P450 1B1 and primary congenital glaucoma. J. Ophthalmic Vis. Res., 10, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chitsazian F., Tusi B.K., Elahi E., Saroei H.A., Sanati M.H., Yazdani S., Pakravan M., Nilforooshan N., Eslami Y., Mehrjerdi M.A.Z.. et al. (2007) CYP1B1 mutation profile of Iranian primary congenital glaucoma patients and associated haplotypes. J. Mol. Diagn., 9, 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mashima Y., Suzuki Y., Sergeev Y., Ohtake Y., Tanino T., Kimura I., Miyata H., Aihara M., Tanihara H., Inatani M.. et al. (2001) Novel cytochrome P4501B1 (CYP1B1) gene mutations in Japanese patients with primary congenital glaucoma. Invest. Ophthalmol. Vis. Sci., 42, 2211–2216. [PubMed] [Google Scholar]
- 24. Lim S.H., Tran-Viet K.N., Yanovitch T.L., Freedman S.F., Klemm T., Call W., Powell C., Ravichandran A., Metlapally R., Nading E.B.. et al. (2013) CYP1B1, MYOC, and LTBP2 mutations in primary congenital glaucoma patients in the United States. Am. J. Ophthalmol., 155, 508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alzuhairy S., Abu-Amero K.K., Al-Shahwan S., Edward D.P. (2015) A novel CYP1B1 mutation with congenital glaucoma and total aniridia. Ophthalmic Genet., 36, 89–91. [DOI] [PubMed] [Google Scholar]
- 26. Vincent A., Billingsley G., Priston M., Glaser T., Oliver E., Walter M., Ritch R., Levin A., Héon E. (2006) Further support of the role of CYP1B1 in patients with Peters anomaly. Mol. Vis., 12, 506–510. [PubMed] [Google Scholar]
- 27. Chavarria-Soley G., Michels-Rautenstrauss K., Caliebe A., Kautza M., Mardin C., Rautenstrauss B. (2006) Novel CYP1B1 and known PAX6 mutations in anterior segment dysgenesis (ASD). J. Glaucoma, 15, 499–504. [DOI] [PubMed] [Google Scholar]
- 28. Acharya M., Mookherjee S., Bhattacharjee A., Bandyopadhyay A.K., Daulat Thakur S.K., Bhaduri G., Sen A., Ray K. (2006) Primary role of CYP1B1 in Indian juvenile-onset POAG patients. Mol. Vis., 12, 399–404. [PubMed] [Google Scholar]
- 29. Pasutto F., Chavarria-Soley G., Mardin C.Y., Michels-Rautenstrauss K., Ingelman-Sundberg M., Fernández-Martínez L., Weber B.H.F., Rautenstrauss B., Reis A. (2010) Heterozygous loss-of-function variants in CYP1B1 predispose to primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci., 51, 249–254. [DOI] [PubMed] [Google Scholar]
- 30. Stoilov I., Jansson I., Sarfarazi M., Schenkman J.B. (2001) Roles of cytochrome p450 in development. Drug Metabol. Drug Interact., 18, 33–55. [DOI] [PubMed] [Google Scholar]
- 31. Li F., Zhu W., Gonzalez F.J. (2017) Potential role of CYP1B1 in the development and treatment of metabolic diseases. Pharmacol Ther., doi: 10.1016/j.pharmthera.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stoilov I., Akarsu A.N., Alozie I., Child A., Barsoum-Homsy M., Turacli M.E., Or M., Lewis R.A., Ozdemir N., Brice G.. et al. (1998) Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am. J. Hum. Genet., 62, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao Y., Wang S., Sorenson C.M., Teixeira L., Dubielzig R.R., Peters D.M., Conway S.J., Jefcoate C.R., Sheibani N. (2013) Cyp1b1 mediates periostin regulation of trabecular meshwork development by suppression of oxidative stress. Mol. Cell Biol., 33, 4225–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mookherjee S., Acharya M., Banerjee D., Bhattacharjee A., Ray K. (2012) Molecular basis for involvement of CYP1B1 in MYOC upregulation and its potential implication in glaucoma pathogenesis. PLoS ONE, 7, e45077.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. López-Garrido M.P., Blanco-Marchite C., Sánchez-Sánchez F., López-Sánchez E., Chaqués-Alepuz V., Campos-Mollo E., Salinas-Sánchez A.S., Escribano J. (2010) Functional analysis of CYP1B1 mutations and association of heterozygous hypomorphic alleles with primary open-angle glaucoma. Clin. Genet., 77, 70–78. [DOI] [PubMed] [Google Scholar]
- 36. López-Garrido M.P., Medina-Trillo C., Morales-Fernandez L., Garcia-Feijoo J., Martínez-de-la-Casa J.M., García-Antón M., Escribano J. (2013) Null CYP1B1 genotypes in primary congenital and nondominant juvenile glaucoma. Ophthalmology, 120, 716–723. [DOI] [PubMed] [Google Scholar]
- 37. Chavarria-Soley G., Sticht H., Aklillu E., Ingelman-Sundberg M., Pasutto F., Reis A., Rautenstrauss B. (2008) Mutations in CYP1B1 cause primary congenital glaucoma by reduction of either activity or abundance of the enzyme. Hum. Mutat., 29, 1147–1153. [DOI] [PubMed] [Google Scholar]
- 38. Campos-Mollo E., López-Garrido M.P., Blanco-Marchite C., Garcia-Feijoo J., Peralta J., Belmonte-Martínez J., Ayuso C., Escribano J. (2009) CYP1B1 mutations in Spanish patients with primary congenital glaucoma: phenotypic and functional variability. Mol. Vis., 15, 417–431. [PMC free article] [PubMed] [Google Scholar]
- 39. Medina-Trillo C., Ferre-Fernández J.J., Aroca-Aguilar J.D., Bonet-Fernández J.M., Escribano J. (2016) Functional characterization of eight rare missense CYP1B1 variants involved in congenital glaucoma and their association with null genotypes. Acta Ophthalmol., 94, e555–e560. [DOI] [PubMed] [Google Scholar]
- 40. Jansson I., Stoilov I., Sarfarazi M., Schenkman J.B. (2001) Effect of two mutations of human CYP1B1, G61E and R469W, on stability and endogenous steroid substrate metabolism. Pharmacogenetics, 11, 793–801. [DOI] [PubMed] [Google Scholar]
- 41. Choudhary D., Jansson I., Sarfarazi M., Schenkman J.B. (2008) Characterization of the biochemical and structural phenotypes of four CYP1B1 mutations observed in individuals with primary congenital glaucoma. Pharmacogenet. Genomics, 18, 665–676. [DOI] [PubMed] [Google Scholar]
- 42. Narooie-Nejad M., Paylakhi S.H., Shojaee S., Fazlali Z., Rezaei Kanavi M., Nilforushan N., Yazdani S., Babrzadeh F., Suri F., Ronaghi M.. et al. (2009) Loss of function mutations in the gene encoding latent transforming growth factor beta binding protein 2, LTBP2, cause primary congenital glaucoma. Hum. Mol. Genet., 18, 3969–3977. [DOI] [PubMed] [Google Scholar]
- 43. Azmanov D.N., Dimitrova S., Florez L., Cherninkova S., Draganov D., Morar B., Saat R., Juan M., Arostegui J.I., Ganguly S.. et al. (2011) LTBP2 and CYP1B1 mutations and associated ocular phenotypes in the Roma/Gypsy founder population. Eur. J. Hum. Genet., 19, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mohanty K., Tanwar M., Dada R., Dada T. (2013) Screening of the LTBP2 gene in a north Indian population with primary congenital glaucoma. Mol. Vis., 19, 78–84. [PMC free article] [PubMed] [Google Scholar]
- 45. Sharafieh R., Child A.H., Khaw P.T., Fleck B., Sarfarazi M. (2013) LTBP2 gene analysis in the GLC3C-linked family and 94 CYP1B1-negative cases with primary congenital glaucoma. Ophthalmic Genet., 34, 14–20. [DOI] [PubMed] [Google Scholar]
- 46. Chen X., Chen Y., Fan B.J., Xia M., Wang L., Sun X. (2016) Screening of the LTBP2 gene in 214 Chinese sporadic CYP1B1-negative patients with primary congenital glaucoma. Mol. Vis., 22, 528–535. [PMC free article] [PubMed] [Google Scholar]
- 47. Kuehn M.H., Lipsett K.A., Menotti-Raymond M., Whitmore S.S., Scheetz T.E., David V.A., O'Brien S.J., Zhao Z., Jens J.K., Snella E.M.. et al. (2016) A Mutation in LTBP2 Causes Congenital Glaucoma in Domestic Cats (Felis catus). PLoS ONE, 11, e0154412.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vehviläinen P., Hyytiäinen M., Keski-Oja J. (2003) Latent transforming growth factor-beta-binding protein 2 is an adhesion protein for melanoma cells. J. Biol. Chem., 278, 24705–24713. [DOI] [PubMed] [Google Scholar]
- 49. Hyytiäinen M., Keski-Oja J. (2003) Latent TGF-beta binding protein LTBP-2 decreases fibroblast adhesion to fibronectin. J. Cell. Biol., 163, 1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hirai M., Ohbayashi T., Horiguchi M., Okawa K., Hagiwara A., Chien K.R., Kita T., Nakamura T. (2007) Fibulin-5/DANCE has an elastogenic organizer activity that is abrogated by proteolytic cleavage in vivo. J. Cell. Biol., 176, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fujikawa Y., Yoshida H., Inoue T., Ohbayashi T., Noda K., von Melchner H., Iwasaka T., Shiojima I., Akama T.O., Nakamura T. (2017) Latent TGF-β binding protein 2 and 4 have essential overlapping functions in microfibril development. Sci. Rep., 7, 43714.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hirani R., Hanssen E., Gibson M.A. (2007) LTBP-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with LTBP-1 for binding to this microfibrillar protein. Matrix Biol., 26, 213–223. [DOI] [PubMed] [Google Scholar]
- 53. Inoue T., Ohbayashi T., Fujikawa Y., Yoshida H., Akama T.O., Noda K., Horiguchi M., Kameyama K., Hata Y., Takahashi K.. et al. (2014) Latent TGF-β binding protein-2 is essential for the development of ciliary zonule microfibrils. Hum. Mol. Genet., 23, 5672–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khan A.O., Aldahmesh M.A., Alkuraya F.S. (2011) Congenital megalocornea with zonular weakness and childhood lens-related secondary glaucoma - a distinct phenotype caused by recessive LTBP2 mutations. Mol. Vis., 17, 2570–2579. [PMC free article] [PubMed] [Google Scholar]
- 55. Kumar A., Duvvari M.R., Prabhakaran V.C., Shetty J.S., Murthy G.J., Blanton S.H. (2010) A homozygous mutation in LTBP2 causes isolated microspherophakia. Hum. Genet., 128, 365–371. [DOI] [PubMed] [Google Scholar]
- 56. Desir J., Sznajer Y., Depasse F., Roulez F., Schrooyen M., Meire F., Abramowicz M. (2010) LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur. J. Hum. Genet., 18, 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jelodari-Mamaghani S., Haji-Seyed-Javadi R., Suri F., Nilforushan N., Yazdani S., Kamyab K., Elahi E. (2013) Contribution of the latent transforming growth factor-β binding protein 2 gene to etiology of primary open angle glaucoma and pseudoexfoliation syndrome. Mol. Vis., 19, 333–347. [PMC free article] [PubMed] [Google Scholar]
- 58. Safari I., Akbarian S., Yazdani S., Elahi E. (2015) A possible role for LTBP2 in the etiology of primary angle closure glaucoma. J. Ophthalmic Vis. Res., 10, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Haji-Seyed-Javadi R., Jelodari-Mamaghani S., Paylakhi S.H., Yazdani S., Nilforushan N., Fan J.B., Klotzle B., Mahmoudi M.J., Ebrahimian M.J., Chelich N.. et al. (2012) LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum. Mutat., 33, 1182–1187. [DOI] [PubMed] [Google Scholar]
- 60. Thomson B.R., Heinen S., Jeansson M., Ghosh A.K., Fatima A., Sung H.K., Onay T., Chen H., Yamaguchi S., Economides A.N.. et al. (2014) A lymphatic defect causes ocular hypertension and glaucoma in mice. J. Clin. Invest., 124, 4320–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kizhatil K., Ryan M., Marchant J.K., Henrich S., John S.W.M. (2014) Schlemm's canal is a unique vessel with a combination of blood vascular and lymphatic phenotypes that forms by a novel developmental process. PLoS Biol., 12, e1001912.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Park D.Y., Lee J., Park I., Choi D., Lee S., Song S., Hwang Y., Hong K.Y., Nakaoka Y., Makinen T.. et al. (2014) Lymphatic regulator PROX1 determines Schlemm's canal integrity and identity. J. Clin. Invest., 124, 3960–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alward W.L. (2000) Axenfeld-Rieger syndrome in the age of molecular genetics. Am. J. Ophthalmol., 130, 107–115. [DOI] [PubMed] [Google Scholar]
- 64. Shields M.B. (1983) Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans. Am. Ophthalmol. Soc., 81, 736–784. [PMC free article] [PubMed] [Google Scholar]
- 65. Shields M.B., Buckley E., Klintworth G.K., Thresher R. (1985) Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Surv. Ophthalmol., 29, 387–409. [DOI] [PubMed] [Google Scholar]
- 66. Shields M.B. (2001) Axenfeld-Rieger and iridocorneal endothelial syndromes: two spectra of disease with striking similarities and differences. J. Glaucoma, 10, S36–S38. [DOI] [PubMed] [Google Scholar]
- 67. Fitch N., Kaback M. (1978) The Axenfeld syndrome and the Rieger syndrome. J. Med. Genet., 15, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Semina E.V., Reiter R., Leysens N.J., Alward W.L., Small K.W., Datson N.A., Siegel-Bartelt J., Bierke-Nelson D., Bitoun P., Zabel B.U.. et al. (1996) Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet., 14, 392–399. [DOI] [PubMed] [Google Scholar]
- 69. Du R.F., Huang H., Fan L.L., Li X.P., Xia K., Xiang R. (2016) A Novel mutation of FOXC1 (R127L) in an Axenfeld-Rieger syndrome family with glaucoma and multiple congenital heart diseases. Ophthalmic Genet., 37, 111–115. [DOI] [PubMed] [Google Scholar]
- 70. Gripp K.W., Hopkins E., Jenny K., Thacker D., Salvin J. (2013) Cardiac anomalies in Axenfeld-Rieger syndrome due to a novel FOXC1 mutation. Am. J. Med. Genet., 161A, 114–119. [DOI] [PubMed] [Google Scholar]
- 71. Nishimura D.Y., Swiderski R.E., Alward W.L., Searby C.C., Patil S.R., Bennet S.R., Kanis A.B., Gastier J.M., Stone E.M., Sheffield V.C. (1998) The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat. Genet., 19, 140–147. [DOI] [PubMed] [Google Scholar]
- 72. Reis L.M., Tyler R.C., Volkmann Kloss B.A., Schilter K.F., Levin A.V., Lowry R.B., Zwijnenburg P.J.G., Stroh E., Broeckel U., Murray J.C.. et al. (2012) PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur. J. Hum. Genet., 20, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hjalt T.A., Semina E.V. (2005) Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev. Mol. Med., 7, 1–17. [DOI] [PubMed] [Google Scholar]
- 74. Flomen R.H., Vatcheva R., Gorman P.A., Baptista P.R., Groet J., Barisić I., Ligutic I., Nizetić D. (1998) Construction and analysis of a sequence-ready map in 4q25: Rieger syndrome can be caused by haploinsufficiency of RIEG, but also by chromosome breaks approximately 90 kb upstream of this gene. Genomics, 47, 409–413. [DOI] [PubMed] [Google Scholar]
- 75. Saadi I., Semina E.V., Amendt B.A., Harris D.J., Murphy K.P., Murray J.C., Russo A.F. (2001) Identification of a dominant negative homeodomain mutation in Rieger syndrome. J. Biol. Chem., 276, 23034–23041. [DOI] [PubMed] [Google Scholar]
- 76. Priston M., Kozlowski K., Gill D., Letwin K., Buys Y., Levin A.V., Walter M.A., Héon E. (2001) Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Hum. Mol. Genet., 10, 1631–1638. [DOI] [PubMed] [Google Scholar]
- 77. Walter M.A. (2003) PITs and FOXes in ocular genetics: the Cogan lecture. Invest. Ophthalmol. Vis. Sci., 44, 1402–1405. [DOI] [PubMed] [Google Scholar]
- 78. Evans A.L., Gage P.J. (2005) Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum. Mol. Genet., 14, 3347–3359. [DOI] [PubMed] [Google Scholar]
- 79. Gage P.J., Suh H., Camper S.A. (1999) Dosage requirement of Pitx2 for development of multiple organs. Development, 126, 4643–4651. [DOI] [PubMed] [Google Scholar]
- 80. Chen L., Gage P.J. (2016) Heterozygous Pitx2 null mice accurately recapitulate the ocular features of Axenfeld-Rieger syndrome and congenital glaucoma. Invest. Ophthalmol. Vis. Sci., 57, 5023–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Doward W., Perveen R., Lloyd I.C., Ridgway A.E., Wilson L., Black G.C. (1999) A mutation in the RIEG1 gene associated with Peters' anomaly. J. Med. Genet., 36, 152–155. [PMC free article] [PubMed] [Google Scholar]
- 82. Lehmann O.J., Ebenezer N.D., Jordan T., Fox M., Ocaka L., Payne A., Leroy B.P., Clark B.J., Hitchings R.A., Povey S.. et al. (2000) Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. Am. J. Hum. Genet., 67, 1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lehmann O.J., Ebenezer N.D., Ekong R., Ocaka L., Mungall A.J., Fraser S., McGill J.I., Hitchings R.A., Khaw P.T., Sowden J.C.. et al. (2002) Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletions. Invest. Ophthalmol. Vis. Sci., 43, 1843–1849. [PubMed] [Google Scholar]
- 84. Nishimura D.Y., Searby C.C., Alward W.L., Walton D., Craig J.E., Mackey D.A., Kawase K., Kanis A.B., Patil S.R., Stone E.M.. et al. (2001) A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am. J. Hum. Genet., 68, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lehmann O.J., Sowden J.C., Carlsson P., Jordan T., Bhattacharya S.S. (2003) Fox's in development and disease. Trends Genet., 19, 339–344. [DOI] [PubMed] [Google Scholar]
- 86. Smith R.S., Zabaleta A., Kume T., Savinova O.V., Kidson S.H., Martin J.E., Nishimura D.Y., Alward W.L., Hogan B.L., John S.W. (2000) Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum. Mol. Genet., 9, 1021–1032. [DOI] [PubMed] [Google Scholar]
- 87. Honkanen R.A., Nishimura D.Y., Swiderski R.E., Bennett S.R., Hong S., Kwon Y.H., Stone E.M., Sheffield V.C., Alward W.L.M. (2003) A family with Axenfeld-Rieger syndrome and Peters Anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene. Am. J. Ophthalmol., 135, 368–375. [DOI] [PubMed] [Google Scholar]
- 88. Berry F.B., Lines M.A., Oas J.M., Footz T., Underhill D.A., Gage P.J., Walter M.A. (2006) Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum. Mol. Genet., 15, 905–919. [DOI] [PubMed] [Google Scholar]
- 89. Saleem R.A., Banerjee-Basu S., Berry F.B., Baxevanis A.D., Walter M.A. (2003) Structural and functional analyses of disease-causing missense mutations in the forkhead domain of FOXC1. Hum. Mol. Genet., 12, 2993–3005. [DOI] [PubMed] [Google Scholar]
- 90. Murphy T.C., Saleem R.A., Footz T., Ritch R., McGillivray B., Walter M.A. (2004) The wing 2 region of the FOXC1 forkhead domain is necessary for normal DNA-binding and transactivation functions. Invest. Ophthalmol. Vis. Sci., 45, 2531–2538. [DOI] [PubMed] [Google Scholar]
- 91. Weisschuh N., Dressler P., Schuettauf F., Wolf C., Wissinger B., Gramer E. (2006) Novel mutations of FOXC1 and PITX2 in patients with Axenfeld-Rieger malformations. Invest. Ophthalmol. Vis. Sci., 47, 3846–3852. [DOI] [PubMed] [Google Scholar]
- 92. Ito Y.A., Footz T.K., Murphy T.C., Courtens W., Walter M.A. (2007) Analyses of a novel L130F missense mutation in FOXC1. Arch. Ophthalmol., 125, 128–135. [DOI] [PubMed] [Google Scholar]
- 93. Bailey J.N.C., Loomis S.J., Kang J.H., Allingham R.R., Gharahkhani P., Khor C.C., Burdon K.P., Aschard H., Chasman D.I., Igo R.P.. et al. (2016) Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat. Genet., 48, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mears A.J., Jordan T., Mirzayans F., Dubois S., Kume T., Parlee M., Ritch R., Koop B., Kuo W.L., Collins C.. et al. (1998) Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am. J. Hum. Genet., 63, 1316–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Swiderski R.E., Reiter R.S., Nishimura D.Y., Alward W.L., Kalenak J.W., Searby C.S., Stone E.M., Sheffield V.C., Lin J.J. (1999) Expression of the Mf1 gene in developing mouse hearts: implication in the development of human congenital heart defects. Dev. Dyn., 216, 16–27. [DOI] [PubMed] [Google Scholar]
- 96. Pierrou S., Hellqvist M., Samuelsson L., Enerbäck S., Carlsson P. (1994) Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J., 13, 5002–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Phillips J.C., del Bono E.A., Haines J.L., Pralea A.M., Cohen J.S., Greff L.J., Wiggs J.L. (1996) A second locus for Rieger syndrome maps to chromosome 13q14. Am. J. Hum. Genet., 59, 613–619. [PMC free article] [PubMed] [Google Scholar]
- 98. Brauner S.C., Walton D.S., Chen T.C. (2008) Aniridia. Int. Ophthalmol. Clin., 48, 79–85. [DOI] [PubMed] [Google Scholar]
- 99. Hingorani M., Hanson I., van Heyningen V. (2012) Aniridia. Eur. J. Hum. Genet., 20, 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jordan T., Hanson I., Zaletayev D., Hodgson S., Prosser J., Seawright A., Hastie N., van Heyningen V. (1992) The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet., 1, 328–332. [DOI] [PubMed] [Google Scholar]
- 101. Grønskov K., Olsen J.H., Sand A., Pedersen W., Carlsen N., Bak Jylling A.M., Lyngbye T., Brøndum-Nielsen K., Rosenberg T. (2001) Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum. Genet., 109, 11–18. [DOI] [PubMed] [Google Scholar]
- 102. Glaser T., Walton D.S., Maas R.L. (1992) Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat. Genet., 2, 232–239. [DOI] [PubMed] [Google Scholar]
- 103. Hanson I., Churchill A., Love J., Axton R., Moore T., Clarke M., Meire F., van Heyningen V. (1999) Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum. Mol. Genet., 8, 165–172. [DOI] [PubMed] [Google Scholar]
- 104. Bhatia S., Bengani H., Fish M., Brown A., Divizia M.T., de Marco R., Damante G., Grainger R., van Heyningen V., Kleinjan D.A. (2013) Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet., 93, 1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kleinjan D.A., Seawright A., Mella S., Carr C.B., Tyas D.A., Simpson T.I., Mason J.O., Price D.J., van Heyningen V. (2006) Long-range downstream enhancers are essential for Pax6 expression. Dev. Biol., 299, 563–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Davis-Silberman N., Kalich T., Oron-Karni V., Marquardt T., Kroeber M., Tamm E.R., Ashery-Padan R. (2005) Genetic dissection of Pax6 dosage requirements in the developing mouse eye. Hum. Mol. Genet., 14, 2265–2276. [DOI] [PubMed] [Google Scholar]
- 107. Hogan B.L., Horsburgh G., Cohen J., Hetherington C.M., Fisher G., Lyon M.F. (1986) Small eyes (Sey): a homozygous lethal mutation on chromosome 2 which affects the differentiation of both lens and nasal placodes in the mouse. J. Embryol. Exp. Morphol., 97, 95–110. [PubMed] [Google Scholar]
- 108. Quiring R., Walldorf U., Kloter U., Gehring W.J. (1994) Homology of the eyeless gene of Drosophila to the Small eye gene in mice and Aniridia in humans. Science, 265, 785–789. [DOI] [PubMed] [Google Scholar]
- 109. Cvekl A., Callaerts P. (2017) PAX6: 25th anniversary and more to learn. Exp. Eye Res., 156, 10–21. [DOI] [PubMed] [Google Scholar]
- 110. Nelson L.B., Spaeth G.L., Nowinski T.S., Margo C.E., Jackson L. (1984) Aniridia. A review. Surv. Ophthalmol., 28, 621–642. [DOI] [PubMed] [Google Scholar]