

Abstract
CD4+ T cells play a critical role in the response to chronic viral infections during the acute phase and in the partial containment of infections once chronic infection is established. As infection persists, the virus-specific CD4+ T cell response begins to shift in phenotype. The predominant change described in both mouse and human studies of chronic viral infection is a decrease in detectable Th1 responses. Some Th1 loss is due to decreased proliferative potential and decreased cytokine production in the setting of chronic antigen exposure. However, recent data suggest that Th1 dysfunction is accompanied by a shift in the differentiation pathway of virus-specific CD4+ T cells, with enrichment for cells with a T follicular helper cell (Tfh) phenotype. A Tfh-like program during chronic infection has now been identified in virus-specific CD8+ T cells as well. In this review, we discuss what is known about CD4+ T cell differentiation in chronic viral infections, with a focus on the emergence of the Tfh program and the implications of this shift with respect to Tfh function and the host-pathogen interaction.
VIRAL INFECTIONS AND THE T CELL RESPONSE
Chronic infections are a ubiquitous part of human history and can lead to dysfunction and neoplastic transformation of infected cells and affected organs1. Chronic infection can additionally lead to dysfunction of responding immune cells. However, many of these infections contribute to the shape of the normal immune system. For example, laboratory mice have less mature immune systems than mice experimentally infected with multiple pathogens or co-housed with so-called “dirty” or pet store mice1–3. The balance of persisting infections, immunity, and clinical disease is dependent on the ongoing interplay between host immune responses and the pathogen. Many studies of CD8+ T cells have dissected the mechanisms critical to pathogen containment. However, studies of CD8+ T cell responses have also identified CD8+ T cell dysfunction when a viral infection is chronic. This phenotype has been described as T cell exhaustion, wherein chronic antigen exposure leads to decreased proliferative potential and effector function of antigen-specific cells (reviewed in 4). Mechanistic studies of CD8+ T cell exhaustion in chronic infections has identified clinically relevant molecules such as inhibitory receptors (e.g. programmed death-1 (PD-1) and cytotoxic lymphocyte associated protein-4 (CTLA-4)) that, when targeted with blocking reagents, can improve CD8+ T cell function5,6. Such checkpoint blockade immunotherapies are now mainstays in the treatment of many cancers7, with trials beginning for the study of these immunotherapies in chronic infections8,9 (see clinicaltrials.gov). In addition to CD8+ T cells, CD4+ T cells are also critical in the containment of chronic infections. However, our understanding of the effects of chronic infection on CD4+ T cell differentiation and function remains incomplete.
The roles of CD4+ T cells in the control of infection are broad. CD4+ T cell function is traditionally defined by the provision of help to other effector cells, such as enhancement of the CD8+ T cell response, promotion of CD8+ T cell memory, enhancement of phagocytic or oxidative burst activities of myeloid cells, and B-cell mediated help. These functions are mediated by CD4+ T cells that can be broadly grouped into subsets such as Type 1 helper cells (Th1), Type 2 (Th2), Type 17 (Th17), regulatory T cells (Treg), T follicular helper (Tfh), and cytotoxic CD4+ T cells10. The commitment of CD4+ T cells to one of these lineages is influenced by signals received during the initial priming interaction with antigen presenting cells (APC), including cytokines, costimulatory signals, and signals derived from the quality and duration of the T cell receptor (TCR) binding to the major histocompatibility (MHC) II:peptide complex11. Post-priming signals also influence the continued differentiation of CD4+ T cell helper lineages. The ways in which persisting antigen and chronic infection affect the initial differentiation and continued function of CD4+ T cells are a growing area of research.
The importance of CD4+ T cell responses can depend on the nature and duration of an infection. In mouse models of acute viral infection, CD4+ T cells are dispensable for viral control, although subsequent memory CD8+ T cell responses can be impaired12–14. In contrast, containment of infections with persisting viruses is often dependent on CD4+ T cell help. The importance of CD4+ T cells in chronic viral infection has been demonstrated both in mouse models of chronic viral infection and in human infections with hepatitis B and C viruses 12,15–18. As a result, understanding how the biology of CD4+ T cells shifts as an infection persists is important to the overall understanding of how these infections may be better treated or controlled.
In chronic infections, pathogen-specific CD4+ T cell differentiation and subset distribution can differ from that observed following acutely cleared infections19. For example, during many viral infections in humans and mice, CD4+ T cells differentiate into antiviral IFNγ-producing Th1 cells,10,20 with perhaps a small subset of cytotoxic CD4+ T cells that may be related to Th1 cells10,21. In addition, Tfh cells develop during acute infections or vaccinations, providing essential help to driving B cell responses, germinal center development, antibody affinity maturation, and the development of long-lived plasma cells (Box 1) 19,20,22.
Box 1. Tfh differentiation in acute viral infection.
Tfh are a subset of CD4+ T cells that provide help to B cells within lymphoid tissues for the generation of affinity-matured antibodies, long-lived plasma cells, and memory B cells22. Tfh express CXCR5, a chemokine receptor that allows trafficking into B cell follicles, and their differentiation requires the transcription factor Bcl623–26. Most of our knowledge of Tfh development comes from murine models. In acute microbial infections, Tfh development begins in the environment of initial CD4+ T cell priming by dendritic cells27. Bcl6 transcription in mice then requires IL-6 signaling through Stat1 in the early phases of Tfh differentiation28,29. A high concentration of IL-2 in the environment inhibits Tfh development, whereas IL-10 has been shown to promote Tfh over Th1 responses30,31. Several other transcription factors (e.g. Lef-1, and Tcf-1) and costimulatory molecules (e.g. ICOS) are also important in the initiation of the mouse Tfh program32–36. While these processes are critical in early Tfh differentiation, nascent Tfh cells require functional B cell interactions to complete Tfh commitment37,38. Finally, the T cell receptor (TCR) of the naïve cell may also shape the differentiation process, given that properties of TCR binding and signaling have been demonstrated to drive Tfh differentiation39–44.
The full scope of events that lead to Tfh differentiation remains to be elucidated. This is especially true for human Tfh, where IL-12 takes place of IL-6, and where mechanistic studies remain limited27,45. Approaches to understanding human Tfh responses, such as the recently performed in vitro screen of recombinant proteins in human Tfh differentiation and the evaluation of genotype and phenotype in human primary immunodeficiencies are of particular value to identifying which findings in mice can be potentially translated to humans46,47.
During chronic infections, loss of typical antiviral Th1 responses can occur19,48,49, and there also appears to be a major skewing towards a Tfh-phenotype instead of a more prototypical Th1-like anti-microbial fate19,50,51. More broadly, both virus-specific CD4+ T cell responses and unrelated CD4+ T cell responses display a pattern of transcription factor and differentiation marker expression that suggest there are altered T helper subset lineage decisions during chronic viral infection in mice19,52. Recent work has also demonstrated that these lineage decisions are plastic, and CD4+ T cells can retain the ability to acquire functions of other subsets11,53. To develop therapies and vaccines for chronic infections, it is essential to dissect the functional role of altered CD4+ T cell differentiation patterns during such infections, and to identify common pathways that can be exploited for therapeutic gain.
This review discusses several paradigmatic chronic viral infections to examine the changes in CD4+ T cells that occur during chronic viral infection. In particular, we explore the emerging data on chronic antigen exposure and chronic inflammation and the corresponding shift towards a Tfh phenotype. We review lymphocytic choriomeningitis virus (LCMV) mouse infections, as well as human and nonhuman primate infections with HCV, SIV, and HIV; our aim is to promote research that serves to identify the potential mechanisms for Tfh phenotype shifts during chronic viral infections, as well as the ensuing consequences for both host and pathogen.
CD4+ T RESPONSES IN MOUSE MODELS OF CHRONIC INFECTION
CD4+ T Cell Responses in LCMV Infection
The study of LCMV in the mouse has provided many cellular and mechanistic insights into immune responses to chronic infections. LCMV exists in the form of acutely resolving strains such as Armstrong and chronic strains, such as clone 13 and WE. These viruses differ in a few amino acids that lead to dramatic differences in the ability of the immune system to clear infection54. Following infection of immunocompetent mice with acute strains of LCMV, virus is cleared by days 8–10 post infection. In contrast, chronic strains of LCMV maintain viremia and widespread replication for ~2–3 months post infection, and even after viremia resolves, virus persists for extended periods of time in other tissues, such as the brain, salivary gland, and kidney 55,56. CD4+ T cells are dispensable for the control of acute LCMV infection, but they are critical in the control of chronic infection because CD4+ T cell deficiency, even transiently, causes unresolving life-long viremia upon infection with strains such as LCMV clone 1312.
Several studies have assessed the differentiation and function of CD4+ T cells in chronic LCMV clone 13 compared to acute LCMV Armstrong (Arm) infection. Initially, the number and frequency of effector CD4+ T cells that produce Th1 cytokines IFNγ and TNF is similar between LCMV Arm and clone 13 infections49. However, after the first week of infection, the frequency of Th1-cytokine producing CD4+ T cells decreases in clone 13 infection, and LCMV-specific CD4+ T cells do not efficiently produce IFNγ or TNF following stimulation ex vivo49. The defects in CD4+ T cell cytokine production persist even after viremia is controlled, and the LCMV-specific CD4+ T cells that are present ~3–4 months after clone 13 infection respond poorly and fail to proliferate to in vivo re-challenge, compared to those from Arm infection49. These features of poor functionality in the setting of chronic antigen exposure are emblematic of T cell exhaustion because defects in effector function are one of the defining features of this state of T cell dysfunction. The causes of CD4+ T cell exhaustion or dysfunction in chronic LCMV are not fully understood, but chronic antigen stimulation is unlikely to be the sole cause. Indeed, reduction of signaling by either TGFβ or TNF have been recently shown to preserve CD4+ T cell function during chronic LCMV infection57,58. To understand whether the shift away from Th1 cytokine production reflects exhaustion alone or whether it reflects a broader change in LCMV-specific CD4+ T cells, the transcriptional landscape of all LCMV-specific CD4+ T cells during clone 13 infection have been assessed (19). In addition to losses of Th1 cytokine production, LCMV-specific CD4+ T cells in chronic infection lose the transcriptional programming associated with a Th1 response(19). Whether this reflects reversible transcriptional changes or instead reflects more permanent epigenetic changes remains to be determined.
A shift towards the Tfh phenotype in chronic LCMV infection
The loss of Th1 frequency in chronic LCMV infection is accompanied by a gain in Tfh. One of the first studies to describe an increase in Tfh frequency during chronic viral infection was published ~10 years after Tfh cells were defined as a separate CD4+ T cell lineage, using chemokine receptor CXCR5 surface expression to identify these cells50. Both clone 13 and Arm infections led to high numbers of CXCR5+ LCMV-specific CD4+ T cells in the initial days after infection, comprising 40% of LCMV-specific CD4+ T cells in the spleen during Arm infection and 20% in clone 13 infection50. As clone 13 infection persisted, however, the frequency of CXCR5+ CD4+ T cells increased by 3-fold, reaching approximately 2/3 of the total LCMV-specific cell number by day 3050. In contrast, the frequency of CXCR5+ LCMV-specific cells in the Arm-infected mice remained relatively constant50. As a result, Tfh-like CD4+ T cells were highly overrepresented in chronic LCMV infection compared to resolved, acute LCMV infection50. These Tfh-like CD4+ T cells in chronic LCMV infection provided help to B cells in vitro, suggesting that there was acquisition of some features of true Tfh cells, such as the ability to help coordinate B cell responses and antibody production50.
Confirming this enrichment for Tfh cells more broadly, transcriptional analysis of altered CD4+ T cell differentiation in chronic infection identified a loss in Th1 transcriptional profiles during clone 13 infection in addition of enrichment for Tfh-associated transcripts19. These cells additionally acquired phenotypic (CXCR5) and functional (IL-21 secretion) features of Tfh cells, as observed by other groups19,40. As we work to understand this shift towards Tfh phenotype and function, it is interesting to note that some features of Tfh gained after chronic antigen exposure, including CXCR5 expression, can be partially downregulated with removal of chronic antigen stimulation40,50. Still, other experiments have demonstrated that LCMV-specific Tfh cells could retain transcription factor Bcl6 expression even 2 weeks following their adoptive transfer to uninfected mice50. Together, these studies suggest that chronic infection preferentially drives Tfh cell generation and that part of the Tfh program, such as Bcl6 expression, may be a fixed, rather than a transient response to stimulation or inflammation50. Thus, chronic viral infections may have long-lasting impacts on the overall pattern of CD4+ T cell differentiation, including a skewing towards a Tfh phenotype, even after these infections resolved. Of relevance, there may be implications for viral infections such as chronic HCV, which can now be essentially cured with direct acting antivirals, but nevertheless, where re-exposure/re-activation can occur in high risk patient groups.
Newer themes in follicular programs during chronic LCMV infection
Recent data suggest that Tfh-associated gene networks upregulated during chronic antigen exposure may not be limited to the CD4+ T cell lineage only. A population of CD8+ T cells expressing the germinal center-driving chemokine receptor CXCR5 emerge during chronic LCMV infection (as well as in SIV and HIV in non-human primates and humans, respectively; see below)59–61. This is interesting because only CD4+ T cells are thought to play a role in the germinal center reaction. However, the LCMV-specific CD8+ T cells appear by day 8 post infection and also express other Tfh-associated proteins including ICOS, Bcl6, and PD-159,61,62. The development of CXCR5+ CD8+ T cells in clone 13 infection is dependent on the transcription factor Tcf-1, consistent with its role in CD4+ Tfh differentiation (Box 1)32–34,59,61. The functional role of CXCR5+ CD8+ T cells is incompletely understood, but recent data in LCMV suggest that CXCR5+ LCMV-specific CD8+ T cells control viremia better than their CXCR5-negative counterparts59,62. The ability to study a follicular program in CD8+ T cells occurring mainly during a chronic infection might allow us to dissect certain features of Tfh cells that might be indicative of an acute response, versus those that could represent an adaptation to chronic antigen exposure and inflammation.
One common feature in both CD4+ and CD8+ T cells with a follicular program during chronic infection is an interferon stimulated gene (ISG) signature19,61. ISGs increase after type I interferon (IFN-I) signaling. Early during infection, detectable circulating IFN levels (and corresponding ISGs) are induced, followed by rapid waning to undetectable levels; these kinetics are similar in conditions of acute or chronic LCMV infection63,64. However, in LCMV-specific CD4+ T cells, the ISG signature persists during chronic infection, even after peripherally measureable type I IFN protein levels decline19. Further, as noted in the CD4+ T cell response to chronic LCMV, Tfh-like CD8+ T cells also exhibit an increased ISG signature61. It is unclear why the pathways associated with IFN signaling are maintained in virus-specific T cells despite a decline in detectable IFN-I63,64. Several possibilities exist. First, it is possible that low levels of IFN-I continue to be produced in chronic infection and this residual IFN-I (below the limits of detection) sustains this ISG signature. Newer, more sensitive IFN-I detection assays65 may be able to address this question. Second, other cytokines might sustain the ISG signature initially induced by IFN-I. For example, a critical role for cytokine signaling through the IL-6 receptor pathway in the maintenance of a Tfh phenotype during chronic LCMV infection has been shown66. Moreover, IL-27, another IL-6 family cytokine, plays a key role in CD4+ T cell responses in chronic infection and might also induce ISGs67. How these two cytokines relate to the ISG response in Tfh cells, however, remains to be determined. Finally, it may be that subsets of virus-specific CD4+ T cells generated during chronic viral infection, such as Tfh, have access to locations where local signals (perhaps local IFN-I) sustain an ISG signature. In the future, it will be important to distinguish these possibilities and dissect the importance of inflammatory signals such as IFN-I and chronic TCR signaling in driving Tfh development and maintenance during chronic infections (Figure 1). An accurate definition of the processes driving increased follicular transcriptional programs -- such as persisting ISG signatures -- should help identify settings of potential Tfh cell dysfunction, or perhaps enable the development of predictions for the effects of immunomodulatory drugs during chronic infections.
Figure 1. Tfh cell differentiation during acute and chronic viral infections.
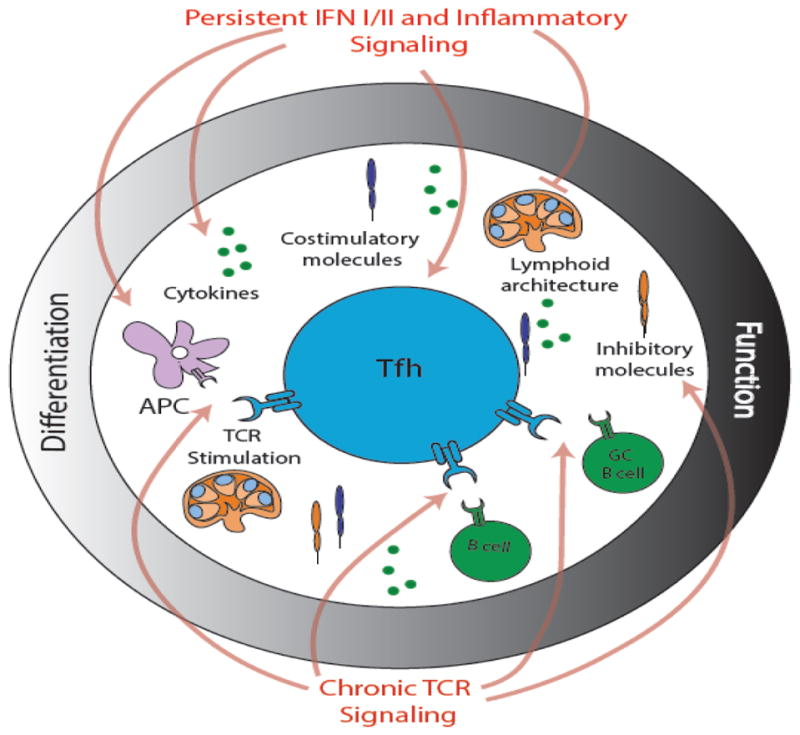
Commitment to the Tfh lineage after an acute viral infection is a multi-step response requiring both priming by antigen presenting cells (APCs) and additional interactions with B cells. Tfh cell differentiation is further informed by a balance of cytokine, costimulatory, and inhibitory signals that allow the Tfh cell program to mature. Following differentiation, the function of Tfh cells is informed by anatomic location and by the tight coordination of Tfh cell interactions with B cells within the follicle and germinal center. These spatial interactions are guided by the lymphoid architecture. Chronic viral infections are associated with increased Tfh frequencies, increased Tfh-like transcriptional programs in CD8+ T cells, as well as by a shift from high quality antibody production, to lower quality, albeit higher quantity of antibody production. The mechanisms that drive this process remain incompletely understood, but continued TCR stimulation and interferon (IFN) signaling due to chronic viral infection presumably affect many components of Tfh cell differentiation and function, including disruption of the lymphoid architecture; changes in the polarization of the APC; direct effects on B cells and germinal center (GC) B cells; alterations in the cytokine milieu; and changes in costimulatory and inhibitory molecule expression as a result of persistent TCR stimulation.
Functional Role of Tfh Cells in Chronic LCMV Infection
Does differentiation towards a Tfh program during chronic infections provide a benefit for the host? Possible mechanisms of benefit include (i) reduction of Th1-driven immunopathology, (ii) support of the CD8+ T cell response, and (iii) a more canonical role for Tfh cell function, namely, providing help to enable a robust antibody response. A role for reduced immunopathology has been supported by vaccination with an LCMV CD4+ T cell epitope driving Th1 responses68. Mice that were immunized with the CD4+ T cell epitope experienced marked mortality after challenge with LCMV clone 13 as compared to unimmunized mice68. This mortality was associated with an extended duration of the Th1 response, while control mice demonstrated the expected shift towards expressing transcripts associated with Tfh, Treg, and exhausted T cells68. A role for Tfh in CD8+ T cell support has also been studied using the canonical Tfh cytokine, IL-2169–71. However, it should be noted that the cellular origin of IL-21 is not necessarily limited to Tfh72. Compared to wild type controls, LCMV-specific CD8+ T cells in IL-21 receptor deficient mice have been shown to be present in lower numbers, and to develop features of more severe exhaustion69. This effect has been partially attributed to direct signaling of IL-21 on CD8+ T cells, observed when IL-21R deficient T cells were adoptively transferred into wild type hosts69. In addition, another study demonstrated that exogenous IL-21 administration improved CD8+ T cell cytokine production and decreased viral titers71. Finally, ablation of the IL-6 family receptor gp130 in CD4+ T cells during chronic LCMV infection led to decreased Tfh and CD8+ T cell frequencies, decreased IL-21 production, reduced LCMV-specific antibody production, and an inability to contain virus73. These data strongly support the notion that differentiation away from a Th1 phenotype, increasing IL-21 production, and sustaining the Tfh response may have benefits to the host. However, whether Tfh cells generated during chronic infection do function optimally is less clear.
A primary function of Tfh is to support class-switched, high-affinity antibody responses from B cells. However, despite increased Tfh frequencies in chronic LCMV infection, the development of neutralizing antibodies does not occur for many months74. Instead, there is a considerable increase in total and non-neutralizing LCMV-specific immunoglobulin74,75, and chronically infected mice can show signs of hypergammaglobulinemia and glomerulonephritis75,76. This high production of poor quality antibodies and delayed production of neutralizing antibodies suggests that there may be dysregulated interactions between Tfh and B cells in chronic LCMV infection, leading to a poor ability of Tfh to help B cells produce optimal antibodies74,75. Supporting the hypothesis of dysregulated Tfh:B cell interactions during chronic LCMV infection, a pair of recent studies have demonstrated a plasmablast response upon deletion of LCMV-specific B cells and a loss of the germinal center B cell response due to early IFN signaling77,78. Together these defects were reported to lead to impaired production of high affinity antibody77,78. While the studies focused on B cell effects of IFN signaling, adoptive transfer experiments demonstrated that the IFN-induced defect was not intrinsic to B cells but exhibited cell-intrinsic effects in monocytes as well as in both CD8+ and CD4+ T cell populations77,78. These advances only begin to define the abnormalities in Tfh:B cell interactions that arise during chronic infection. Moreover, given that the timely development of neutralizing antibodies is central to viral control79, further work is needed to understand which steps of the Tfh:B cell interaction are perturbed by a persisting virus. Consequently, mechanistic studies in chronic LCMV that focus on investigating the changes in B cell dynamics, Tfh quality, and the lymphoid environment will be useful in informing on the characteristics of human immune dysfunction that accompany chronic viral infections.
CD4+ T CELLS IN HUMAN AND NON-HUMAN PRIMATE CHRONIC VIRAL INFECTIONS
A major goal of studying chronic infections in mice is to understand, prevent, and treat human disease. Our ability to translate findings from mice to humans has faced several challenges. First, understanding how the immune response changes as a pathogen transitions from an acute to a chronic infection requires the identification of patients in acute stages of infection. Cohorts developed to track high-risk patients have provided invaluable samples and datasets in recent years80,81. However, the difficulty in establishing these cohorts necessarily limits scientific progress. Second, our understanding of the CD4+ T cell response to chronic infections in humans is limited by the need for HLA class II-matched tetramers that allow pathogen-specific CD4+ T cells to be identified, regardless of whether the cells generate a detectable cytokine or not. Given that a hallmark of immune responses to chronic infection is decreased function, studies of CD4+ T cells that rely on cytokine production are likely to fail to capture dysfunctional cells or those that produce low or no cytokines (e.g. Tfh). Human HLA class II diversity is a tremendous challenge in the development of broadly applicable reagents, but recent efforts to develop suites of HLA class II tetramers for chronic infections such as HIV, HCV and tuberculosis have allowed an increased understanding of human CD4+ T cell differentiation during chronic infections82,83. However, even tetramers can introduce a bias, given that they can only identify responses to a single epitope (Figure 2). A newer technique that we and others have used is the assessment of activation induced markers (AIM)84,85. Upregulation of AIMs such as CD69, CD200, CD71 and CD40L can be used to identify antigen-specific cells that do not produce high levels of cytokines84,85. While AIMs have been used in particular to identify antigen-specific Tfh, it is also possible that clusters of AIMs are specific for some T helper subsets and not others, which would also introduce bias; this warrants further investigation.
Figure 2. Limitations of the most commonly used assays to characterize CD4+ T cell responses in humans.

Virus-specific CD4+ T cells are heterogeneous, and assays that rely on cytokine production alone will inherently miss the breadth of specific cells. This poor detection is especially true in chronic viral infections, where T cells can exhibit impaired cytokine production (Exhausted Th1 cells, Th1-Ex). Use of in vitro stimulation and assessment cytokine production by multiplex immunoassay allows for a diverse capture of secreted cytokines but does not detect cells with minimal cytokine secretion and this approach is unable to identify the sources of expression and co-expression. Flow cytometry after in vitro stimulation allows for resolution of individual cells but is limited to cells that produce robust quantities of cytokines. Recent studies have used activation induced markers to identify activated cells that do not produce readily detectable cytokine upon TCR stimulation. HLA class II tetramers allow for the isolation of all CD4+ T cells with specificity for a given epitope, regardless of whether the cells can proliferate or produce cytokines. Transcriptional and epigenetic analyses of these cells gives a broad understanding of their phenotypes, although the phenotype of identified cells is likely biased by the epitope. Further, bulk transcriptional analyses of even tetramer-sorted cells are still limited by the inability to resolve whether transcriptional programs are expressed in separate cell populations or whether genes are co-expressed in the same cells. Single cell approaches such as flow cytometry, mass cytometry, and single cell sequencing allow for resolution of such true heterogeneity.
To date, studies of CD4+ T cells in human chronic viral infections have shown many similarities with studies and emerging concepts from the mouse model of LCMV infection. In particular, HCV and HIV infections have been associated with i) a loss of Th1-associated cytokine production48,83; ii) a shift towards Tfh differentiation 51,86; and iii) evidence of dysfunctional Tfh:B cell interactions87–89. Nevertheless, distinct features of each infection (e.g. direct infection of CD4+ T cells, including Tfh, by HIV) require detailed interrogation in humans. Of note, most human immunologic studies to date have been performed on blood, while mouse studies of chronic infection are commonly performed using the spleen. Moving forward, it will be critical to explore the immunology of chronic infection within human tissues such as lymph nodes, spleen, and at other sites of viral infection. Further, clinical studies will greatly benefit from an improved understanding of whether and how Tfh responses detectable in the blood relate to those within lymphoid tissues.
CD4+ T Cells in Hepatitis C Virus (HCV) Infection
HCV is a self-limited viral infection in approximately 25% of those who are infected but becomes a chronic infection in the majority of individuals90. Untreated HCV infection is associated with the eventual development of liver fibrosis and hepatocellular carcinoma91. While the difference between acute and chronic LCMV infection in mice is mediated by a change in the viral genome, differences in acute resolution versus progression to chronic HCV infection in humans are likely mediated by differences in host immunity, including virus-specific CD4+ T cell responses92. Chimpanzee studies have shown that CD4+ T cells are required for protection against disease following immunization or prior to infection93. In humans, viral clearance has been associated with increased peripheral blood HCV-specific CD4+ T cell proliferation during acute infection83,92–94. HLA class II tetramers have been used ex vivo to measure HCV-specific CD4+ T cell responses from HCV-infected patients; these studies reported a lack of proliferating HCV-specific CD4+ T cells in individuals who developed chronic HCV infection, mainly due to CD4+ T cell deletion or hyporesponsiveness95,96. Specifically, tetramer-positive HCV-specific CD4+ T cells were present in individuals with chronic infection, but the frequency was considerably lower than in individuals whose infection had been cleared95,96. In addition, a detailed study of HCV-specific CD4+ T cells found that both the initial magnitude of the CD4+ T cell response and breadth of targeted epitopes was similar between individuals who developed chronic infection and those who cleared the infection, given that a similar frequency of CD4+ T cells from these individuals produced effector cytokines when stimulated, and furthermore, the number of individual peptides from the virus recognized by CD4+ T cells was not different83. Together, these data suggest that on the one hand, the initial repertoire of CD4+ T cells probably does not alter the ability of the immune system to clear the virus; yet, on the other hand, both the decreased numbers and proliferative potential of HCV-specific CD4+ T cells may be associated with HCV persistence83,95,96. The reasons why some individuals develop a more robust CD4+ T cell response than others is not well understood and may represent a fruitful area of future investigation.
In addition to the decreased number and proliferation potential, the functional quality of HCV-specific CD4+ effector T cells also seems to correlate with protection from reinfection. For instance, in patients spontaneously clearing a primary HCV infection, those clearing a secondary infection upon challenge are more likely to harbor polyfunctional HCV-specific CD4+ T cells (producing more than one Th1-associated cytokine) than individuals unable to clear the secondary HCV infection; this suggests, perhaps, an important role for CD4+ T cell effector functions in HCV control97. Unfortunately, studies of CD4+ T cells in HCV infection have been largely limited to an assessment of frequency, proliferation, and Th1 cytokine production; other detailed aspects of CD4+ T cell phenotype and differentiation state remain quite limited. Thus, a broader interrogation of the CD4+ T cell functional landscape during HCV infection requires further research. The advent of curative antivirals for HCV infection may also provide a rare opportunity to study whether and how the CD4+ T cell response corrects after the virus is eliminated.
Tfh Responses in HCV Infection
Most of the analyses concerning CD4+ T cell responses in HCV infection have been focused on evaluating Th1-cytokine producing cells, but some data in HCV suggest that this infection behaves similarly to LCMV with respect to Tfh. Early IL-21 production by HCV-specific CD4+ T cells has been correlated with individuals who go on to resolve infection98, although these cells were not otherwise identified as Tfh. In the chronic phase of infection, increased frequencies of HCV-specific Tfh cells have been described, as observed in LCMV50,86. Specifically, HCV-specific Tfh cells were enriched in the liver relative to CD4+ T cells from the livers of patients with non-viral hepatitis or when compared to the overall frequency of Tfh in peripheral blood86. These cells were implicated as Tfh-like because they not only expressed CXCR5 and PD-1 by protein, but they also expressed the Tfh cytokine IL-21 following in vitro expansion, further identifying them as probable Tfh. Thus, we posit that these data support a common model of chronic viral infection that leads to a predominant Tfh phenotype, particularly at the site of infection, although further testing is warranted. This shift towards a Tfh phenotype may have disease relevance if the consequence is a loss of more directly antiviral Th1 cells, or, if the excess of Tfh-like cells leads to a dysregulated antibody response.
HCV infection has also been associated with dysregulated immunoglobulin responses. Indeed, a subset of HCV-infected patients has been reported to experience considerable cryoglobulinemia and antibody accumulation, potentially suggesting that amplified B cell responses and/or T cell help might be induced by chronic HCV87. Despite increased total antibody, HCV-specific antibody responses are typically poorly effective and fail to control viral replication88. These data are consistent with observations in LCMV74,75. In addition, humoral responses against other antigens might be compromised in patients with chronic HCV; specifically, one study noted that antibody responses against hepatitis B vaccination were lower in individuals with chronic HCV than in uninfected individuals99. This observation suggested that Tfh:B cell dysfunctional interactions might be due, at least in part, to a putative bystander effect from surrounding inflammation, although this has not been directly tested. Thus, it will be important to understand whether an increased Tfh program in chronic HCV infection is limited to pathogen-specific cells or whether surrounding inflammation might contribute to dysfunctional Tfh:B cell interactions among non-HCV-specific cells. Distinguishing between direct and bystander effects on Tfh:B cell interactions might help shape therapeutics that could target inflammation versus viral replication directly, aiming to improve the production of quality antibody responses to other challenges, including vaccination and respiratory viruses.
CD4+ T Cells and Human Immunodeficiency Virus (HIV) and Simian Immunodeficiency VIRUS (SIV) infections
Approximately 37 million people are currently infected with HIV-1 worldwide, and the virus continues to infect ~2 million people each year100. Because HIV infection is primarily an infection of CD4+ T cells, considerable efforts have focused on defining the nature of HIV-1-specific CD4+ T cell responses. In acute HIV-1 infection, CD4+ T cells produce the Th1 cytokine IFNγ and are directed against several HIV-1 epitopes48. Other helper subsets have been described in acute disease and linked to disease progression, including Th1-like cells with cytotoxic potential and Th17 cells based on changes in the frequency of these subsets in relation to viral load, overall CD4 counts and/or progression to AIDS101,102,103. Although acute HIV-1 infection generates robust Th1 CD4+ T cell responses, these responses decline within days to weeks, resulting in a decreased frequency of detectable HIV-1-specific IFNγ-producing CD4+ T cells and a reduction in the number of antigens to which the Th1 cells respond48. The HIV-specific Th1 function does not recover after successful initiation of antiretroviral therapy (ART), although transcriptional signatures of inhibitory pathways across all CD4+ T cells do decrease with effective viral control48,58. It remains unclear whether the loss of HIV-specific Th1 CD4+ T cell responses in chronic HIV-1 infection is due to preferential infection of HIV-1-specific CD4+ T cells104 or due to persistent CD4+ T cell dysfunction or exhaustion.
To dissect the relationship between the chronic phase of HIV-1 infection and functionality of CD4+ T cells, several groups have compared cohorts of patients with differing levels of immunologic or ART-induced viral control105,106. Patients able to sustain immunologic control of virus, termed elite controllers, have a higher frequency of HIV-1-specific CD4+ T cells able to produce all three major Th1-associated cytokines (IFNγ, TNF, and IL-2) compared to patients with uncontrolled disease, and on ART, although many of the patients on ART were not shown to be fully suppressed105. There is also some evidence that early initiation of ART can prevent HIV-specific CD4+ T cell dysfunction106. However, an important remaining question is whether ART suppression initiated later in infection allows CD4+ T cells to return to their normal function and subset distribution or whether CD4+ T cells continue to remain altered as a result of the initial insult of chronic infection. To understand this question, approaches that profile HIV-specific CD4 T cells beyond measurement of Th1 cytokine production will be important. To date, analyses of broader transcriptional and differentiation programs in these cells have been limited.
Tfh in HIV Infection
HIV is primarily an infection of lymphoid tissues, and like LCMV and HCV, Tfh have been noted to increase at the site of HIV-1-infection51,107,108. As has been observed in LCMV, CXCR5+ virus-specific CD8+ T cells have also recently been shown to increase in both HIV and in SIV infections62,109, suggesting that the upregulation of a follicular program in both CD4+ and CD8+ T cells is conserved and that mice may be used to model these processes. Animal modeling in HIV has already helped to describe ways in which the increased Tfh response may be altered by chronic infection. Specifically, data from SIV-infection models suggest that Tfh cells exhibit an altered transcriptional signature compared to Tfh cells from SIV-negative lymph nodes, with ISGs being the most differentially expressed trasncripts110. Thus, although, it is not clear whether the mechanisms driving a Tfh-like program in HIV are due to persisting antigen or whether other aspects of the inflammatory environment drive non-HIV specific cells towards a Tfh lineage, there appears to be an alteration in Tfh differentiation and development with HIV-1 infection.
The relationship between HIV and Tfh is further complicated by the fact that Tfh are a major cellular reservoir of replication-competent virus51,111–114. As a result, HIV-1 has been associated with increased frequencies of the very cell type it infects51,111. Moreover, histologic analysis of SIV-infection in non-human primates has suggested that infected Tfh are enriched within the B cell follicles of lymph nodes, which may in turn enable the virus to escape surveillance by cytotoxic CD8+ T cells normally excluded from this location113. Given that the majority of infected Tfh express programmed cell death protein 1 (PD-1), there is additional interest in the potential role of anti-PD1 immunotherapy in modulating the treatment of HIV-1 infected individuals. Indeed, candidate agents targeting PD-1 might be used to deplete this cellular reservoir or provoke viral reactivation from latency (i.e. by activating these CD4+ T cells via PD-1 blockade), thus allowing the virus to be seen by CD8+ T cells and eliminated8,9,115,116. Moreover, there are major efforts underway to develop putative prophylactic and therapeutic antibody-based vaccines for HIV-1 infection, and these may heavily depend on effective Tfh responses117, because production of effective antibodies by vaccination requires functional Tfh:B cell interactions. Tfh cells are therefore an important component of adaptive immunity, and a deeper understanding of the perturbed functional role of these cells from HIV infection may improve our understanding of HIV pathogenesis, prevention, and presumably, future cure of HIV infections118. Nevertheless, interrogation of Tfh in lymph nodes and other lymphoid tissues, as well as the location where Tfh:B cell interactions occur, is necessarily limited by access to tissues, although tissue-based research remains a vital goal. Because access to such lymphoid tissues is challenging, the study of HIV and Tfh has also been focused on Tfh-like cells that can be found in peripheral blood: circulating Tfh (box 2). Hence, an important future goal is to understand the relationship(s) between such blood Tfh-like cells and Tfh in lymphoid tissues, both in health, and during chronic infections.
Box 2. Circulating Tfh Cells.
To investigate the biology of Tfh in HIV-1 infection in clinical trials and across broad patient groups, we and others have studied the more accessible Tfh-like cells in peripheral blood, termed circulating Tfh (cTfh). The precise ontology of these cells remains unclear. For example, whether cTfh arise from cells that were formerly Tfh in the B cell follicle/germinal center remains to be determined. Nevertheless, cTfh changes have been shown to correlate with immune responses to vaccination against influenza and other viruses36,84,119–122. In HIV-1 infections, cTfh have been correlated with the presence of naturally occuring broadly neutralizing antibodies and with response to prophylactic vaccination123,121. Circulating Tfh have also been found to be infected more frequently than other non-Tfh blood CD4+ subsets and to limit HIV-1 specific responses112,121. Of note, cTfh are not all identical in their transcriptional profile or ability to provide B cell help124. Circulating Tfh have been described as being polarized towards Th1, Th2, and Th17 cells, while cTfh that express ICOS and other markers of activation have been shown to contain vaccine-specific responses36,84,120,124. Moving forward, it will be critical to define the ways in which cTfh can reflect the ongoing lymph node biology of HIV infection and immune dysfunction. In so doing, particular attention should be given to the identification of cTfh subsets that will be most informative.
Tfh function in HIV
As in LCMV and HCV infections, HIV-1 infection has been associated not only with increased Tfh frequency at the site of infection but also with abnormal B cell responses. For instance, hypergammaglobulinemia has long been a well-documented consequence of untreated HIV-1 infection75,89. This alteration could be postulated to be due to disturbed Tfh:B cell interactions, wherein Tfh cell help is no longer limiting. Consistent with this notion, increased Tfh frequencies in HIV-infected lymph nodes have also shown that peripheral hypergammaglobulinemia can correlate with Tfh Bcl6 expression51. However, despite this correlation, in vitro studies of lymph node Tfh cells from ART-naïve, HIV-1-infected individuals have indicated that these cells are impaired in their ability to provide B cell help when compared to equal numbers of Tfh from HIV-1-uninfected lymph nodes108. These functional defects may be modifiable, as cTfh from viremic individuals can improve function when cultured with IL-2 blocking antibody125.
Although Tfh are clearly abnormal in ART-naïve HIV-1 infection, less is understood about Tfh differentiation and function following ART control. Of note, lymph node Tfh remain at an elevated frequency, even in patients on ART, although the frequency is lower than that observed in ART-naïve patients51. Whether long-term ART allows Tfh frequencies to return to normal remains unclear. In addition, a continued deficit in Tfh function during ART suppression has been further suggested by several studies demonstrating that specific antibody responses to influenza and pneumococcal vaccination are impaired during ART-controlled HIV-1 infection126–129. Noteworthy, studies in LCMV and Tfh differentiation have focused on a shift towards the Tfh phenotype in virus-specific cells. In contrast, the increased Tfh frequencies and Tfh dysfunction analyses described with HIV-1 infection have included all Tfh populations, regardless of specificity. Therefore, it is not clear which aspects of the Tfh:B cell dysfunction in HIV are due to persisting antigen (as would be expected for virus-specific cells40) or to an inflammatory milieu that affects Tfh:B cell interactions more broadly. Such broad effects might be hypothesized to occur through the direct effects of IFN signaling and cytokine changes during Tfh differentiation, taltered B cell functions, and/or through disruption of normal lymphoid architecture130. Indeed, altered lymphoid architecture may be especially important during HIV-1 infection, as lymph node fibrosis has been reported, despite immunologic and antiretroviral control; indeed, such lymphoid tissue fibrosis might distort the normal structure of the B cell follicle where Tfh:B cell interactions occur131,132. Studies that define the anatomical and molecular causes of altered Tfh and B cell function in HIV infection will thus help provide insights into candidate therapies for immune restoration. Further, these studies may have the potential to reveal mechanisms that allow the field to target or engineer Tfh cells to help reduce viral reservoirs.
CONCLUDING REMARKS
Chronic infections are a normal part of human immune development that can be contained by, but also alter, CD4+ T cell responses1–3. Collectively, LCMV, HCV, SIV and HIV-1 infections indicate a direct relationship between chronic viral infection and Tfh cell biology. The pathways that drive Tfh differentiation are incompletely understood and may include features of both chronic antigen stimulation and of a shifting inflammatory environment. Moreover, while Tfh cells in chronic infection can provide B cell help, it remains unclear how phenotypically and functionally similar Tfh cells generated during chronic infection are to Tfh cells generated during acute infection, even after infection is controlled. Of note, some aspects of these follicular pathways are shared by CD8+ T cells during chronic infection, and many of the genes upregulated by these follicular-like CD8+ and CD4+ T cells are also found to be upregulated by exhausted CD8+ T cells in chronic infection and cancer19. Consequently, Just as studies in CD8+ T cell exhaustion have led to therapeutic advances in cancer, in addition to new trials in chronic infections, understanding CD4+ T cell dysfunction in chronic infection has the potential to inform new approaches to subverting immune dysfunction, improve vaccination strategies, and hopefully, contribute to the cure of chronic viral infections (see Outstanding Questions and Box 3).
Outstanding Questions.
What drives increased Tfh lineage differentiation in CD4+ and CD8+ T cells during chronic viral infections?
Are Tfh cells derived during chronic viral infections distinct from Tfh cells found after acute viral infections or vaccination? What is the functional significance of these differences and similarities?
Are there common pathways of differentiation, inflammation or transcriptional control in CD4+ T cells that negatively impact humoral immunity in chronic viral infections?
Is the skewed distribution of pathogen-specific CD4+ T cells in chronic viral infections reversible? If so, can this characteristic be exploited for therapeutic purposes?
Box 3. Clinician’s corner.
HIV-1 is known to impair immune responses even when CD4+ T cell counts are normal, but other chronic infections also impair T and B cell immunity.
Impaired T cell immunity can be due to loss of immune cells, but it can also be due to a loss or change in cellular function.
Certain T cell changes (such as exhaustion) noted in chronic viral infections may overlap with those noted during chronic antigen exposure in cancers.
Altered humoral immunity and decreased vaccine responses have been observed in chronic viral infections, and these defects may be due, in part, to alterations in the follicular helper subset of CD4+ T cells (Tfh).
Understanding changes in CD4+ (as well as CD8+) T cells during chronic viral infection may inform treatment of these infections and can, by extension, additionally lend insight to further developing cancer immunotherapy strategies.
Trends.
Chronic viral infections can impair CD4+ T cell responses, with a loss of Th1 function.
CD4+ T cells are skewed towards a T follicular helper (Tfh) lineage during chronic viral infections in mice and in humans and away from a potentially more antiviral Th1 state
A Tfh-like program can also occur in some CD8+ T cells perhaps giving these cells access to the B cell follicle where they might interact with viral reservoirs.
Increased Tfh cells may assist CD8+ T cells and prevent immune-mediated damage when a viral infection cannot be cleared.
Tfh cells provide help to B cells for antibody class-switching and affinity maturation, but despite a Tfh bias in chronic viral infections, effective humoral immunity is often impaired.
Acknowledgments
L.A.V. was supported by NIH Grant KL2TR001879. R.S.H. was supported by the NIH grants AI114852 and AG047773. Work in the Wherry lab is supported by the Penn Center for AIDS Research (P30 AI045008) the National Institutes of Health (NIH) grants AI112521, AI115977, AI117950, AI105343, AI082630 as well as the U.S. Broad Agency Announcements Grant NIHAI2010085. E.J.W. is also supported by the Parker Institute for Cancer Immunotherapy.
Glossary
- Activation induced markers
Proteins upregulated on the cell surface following activating signals through the T cell receptor (TCR)
- Affinity maturation
Progressive selection of higher affinity B cell-produced antibodies
- Anti-PD-1 immunotherapy
A type of checkpoint blockade immunotherapy that binds to PD-1 on the T cell surface and blocks PD-1 inhibitory signaling
- Antiretroviral therapy
Combination therapy directed against different stages of the HIV-1 life cycle, usually consisting of two or three medications
- B cell follicle
Collection of B cells within the lymphoid tissue, largely consisting of naïve B cells
- Bcl6
Transcription factor encoded by the gene BCL6 that acts as a transcriptional repressor and as an epigenetic modifier in complex with other proteins. Germinal center cells characteristically express high levels of this transcription factor.
- Broadly neutralizing antibodies
Class of antibodies able to neutralize many different quasi-species of HIV-1 from different individuals
- Bystander effect
Secondary, non-specific effects of a secreted molecule, such as a cytokine on an adjacent cell.
- Checkpoint blockade immunotherapies
Clinical therapy with recombinant antibodies that bind inhibitory receptors (e.g. PD-1, CTLA-4). These antibodies prevent inhibitory molecules from exerting a ‘check’ on the immune response and are used to reinvigorate, for instance, CD8+ T cell responses.
- Circulating Tfh (cTfh)
CD4+ T cells in the peripheral blood that express CXCR5 and PD-1 and are able to provide help to B cells in vitro
- Class-switch
Process by which B cells switch the production of one antibody isotype to another isotype, i.e. IgM to IgG
- Cryoglobulinemia
Medical disorder characterized by the production of immunoglobulins that complex together and clump in cold temperatures, leading to blood vessel inflammation known as vasculitis
- Cytotoxic CD4+ T cells
A subset of CD4+ T cells that express granzyme B and perforin and can exhibit cytotoxic activity.
- Effector function
Cellular production of anti-pathogen, anti-tumor or cell communication molecules such as cytokines, chemokines or granzymes, effecting some change (e.g. in the target cell, other immune cells or tissues
- Elite controllers
HIV-1 infected patients who maintain low or undetectable plasma viral loads without receiving antiretroviral therapy
- Epigenetic
Regulating gene expression without changes in DNA sequence, often used to refer to processes of DNA methylation, post-translational histone modifications and chromatin accessibility.
- Epitope
The sequence of amino acids that is recognized by the TCR when presented in the context of HLA class I or class II molecules or the portion of a molecule that is recognized by the B cell receptor.
- Germinal center
A structure within the B cell follicle of lymphoid tissues in which B cells compete for antigen and T cell help to undergo affinity maturation. The germinal center is the site where B cells commit to become memory B cells or plasma cells, with the latter being the source of circulating antibodies.
- Glomerulonephritis
Medical disorder characterized by inflammation of the glomeruli in the kidney, thereby affecting the ability of the kidney to properly filter blood. One cause of glomerulonephritis is deposition of immunoglobulin complexes.
- HCV infection
HCV is a small, enveloped, positive-sense single-stranded RNA virus responsible for causing chronic infection and long-term liver injury
- Humoral responses
Typically refers to the antibody response to an infection but also includes other immunologically-active secreted proteins including complement and anti-microbial peptides
- Hypergammaglobulinemia
Medical condition referring to elevated levels of circulating IgG antibodies, usually in response to chronic infection or inflammation
- Inhibitory receptors (T cells)
Cell-surface proteins whose expression serves to dampen T cell activation
- Interferon-stimulated gene (ISG) signatures
represent a group of hundreds of genes whose expression is induced by the effect of interferons, that may exert antiviral activity. Some scenarios involve characteristic patterns of expression of ISGs, referred to as signatures.
- Lymphoid architecture
The physical organization and makeup of the lymphoid tissue
- Neoplastic transformation
Transition of a benign tissue lesion into one with invasive, malignant features
- Neutralizing antibodies
Antibodies with particular ability to obstruct, hinder, or otherwise block the biological activity of a toxin or pathogen
- HLA class II-matched tetramers
A biotinyated multimer consisting of four identical Human Leukocyte Antigen (HLA) proteins loaded with peptide. Biotinyated multimers are labeled using streptavidin linked to a fluorochrome or other molecular reporter. Tetramer technology works when the tetramer is composed of an HLA that matches the HLA type of the individual. HLA Class II tetramers “loaded” with a peptide antigen permit identification of the antigen-specific T cells.
- Plasmablast response
Acute increase in circulating frequency of plasmablasts, an early stage of antibody-secreting cells, typically seen following vaccination or infection
- Plasma cells
Late stage of B cell differentiation characterized by the ability to produce large amounts of antibodies
- Programmed cell death protein 1 (PD-1)
A cell surface inhibitory receptor that belongs to the immunoglobulin superfamily and is predominantly expressed on lymphocytes.
- Targeted epitopes
Antigens that are the primary targets of T cell recognition and activity
- T cell exhaustion
A state of cellular dysfunction characterized by poor cytokine expression and poor proliferation in response to a stimulus despite presence of high levels of antigen. Exhaustion is commonly associated with increased surface expression of inhibitory molecules.
- T cell priming
Binding of the T cell receptor complex with its cognate antigen, leading to differentiation of the T cell.
- T helper type 1 cells (Th1)
A subset of CD4+ T cells that is driven by the transcription factor T-bet and typically produces the cytokine IFN-gamma. Th1 cells are typically understood to promote antiviral and cell-mediated immunity.
- T helper type 2 cells (Th2)
A subset of CD4+ T cells that is driven by the transcription factor GATA3 and typically produces the cytokine IL-4. Th2 cells are involved in the response to some extracellular pathogens and have been implicated in the pathogenicity of allergic responses.
- T helper type 17 cells (Th17)
A subset of CD4+ T cells that is driven by the transcription factor RORyT and typically produces the cytokine IL-17. Th17 cells are involved in mucosal immunity and autoimmunity.
- T follicular helper cells (Tfh)
A subset of CD4+ T cells that is driven by the transcription factor Bcl6 and typically produce the cytokine IL-21. Tfh provide B cell help in the germinal center for antibody affinity maturation and plasma cell development.
- Regulatory T cells (Treg)
A subset of T cells that is driven by the transcription factor FoxP3. Regulatory T cells inhibit other T cell responses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Virgin HW, Wherry EJ, Ahmed R. Redefining Chronic Viral Infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 2.Beura LK, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reese TA, et al. Sequential Infection with Common Pathogens Promotes Human-like Immune Gene Expression and Altered Vaccine Response. Cell Host & Microbe. 2016;19:713–719. doi: 10.1016/j.chom.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pauken KE, Wherry EJ. SnapShot: T Cell Exhaustion. Cell. 2015;163:1038–1038.e1. doi: 10.1016/j.cell.2015.10.054. [DOI] [PubMed] [Google Scholar]
- 5.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 6.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 7.Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med. 2016;375:1767–1778. doi: 10.1056/NEJMra1514296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Garff G, et al. Transient HIV-specific T cells increase and inflammation in an HIV-infected patient treated with nivolumab. AIDS. 2017;31:1048–1051. doi: 10.1097/QAD.0000000000001429. [DOI] [PubMed] [Google Scholar]
- 9.Davar D, Wilson M, Pruckner C, Kirkwood JM. Case Report PD-1 Blockade in Advanced Melanoma in Patients with Hepatitis C and/or HIV. Case Reports in Oncological Medicine. 2015:1–5. doi: 10.1155/2015/737389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sallusto F. Heterogeneity of Human CD4 +T Cells Against Microbes. Annu Rev Immunol. 2016;34:317–334. doi: 10.1146/annurev-immunol-032414-112056. [DOI] [PubMed] [Google Scholar]
- 11.O’Shea JJ, Paul WE. Mechanisms Underlying Lineage Commitment and Plasticity of Helper CD4+ T Cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. Journal of Virology. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 15.Zajac AJ, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planz O, et al. A critical role for neutralizing-antibody-producing B cells, CD4(+) T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virus: implications for adoptive immunotherapy of virus carriers. Proceedings of the National Academy of Sciences. 1997;94:6874–6879. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asabe S, et al. The Size of the Viral Inoculum Contributes to the Outcome of Hepatitis B Virus Infection. Journal of Virology. 2009;83:9652–9662. doi: 10.1128/JVI.00867-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shoukry NH, Cawthon AG, Walker CM. Cell-Mediated Immunity and the Outcome of Hepatitis C Virus Infection. Annu Rev Microbiol. 2004;58:391–424. doi: 10.1146/annurev.micro.58.030603.123836. [DOI] [PubMed] [Google Scholar]
- 19.Crawford A, et al. Molecular and Transcriptional Basis of CD4+ T Cell Dysfunction during Chronic Infection. Immunity. 2014;40:289–302. doi: 10.1016/j.immuni.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hale JS, et al. Distinct Memory CD4+ T Cells with Commitment to T Follicular Helper- and T Helper 1-Cell Lineages Are Generated after Acute Viral Infection. Immunity. 2013;38:805–817. doi: 10.1016/j.immuni.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown DM, Lee S, Garcia-Hernandez MDLL, Swain SL. Multifunctional CD4 Cells Expressing Gamma Interferon and Perforin Mediate Protection against Lethal Influenza Virus Infection. Journal of Virology. 2012;86:6792–6803. doi: 10.1128/JVI.07172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol. 2015;15:185–189. doi: 10.1038/nri3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nurieva RI, et al. Bcl6 Mediates the Development of T Follicular Helper Cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu Di, et al. The Transcriptional Repressor Bcl-6 Directs T Follicular Helper Cell Lineage Commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Kroenke MA, et al. Bcl6 and Maf Cooperate To Instruct Human Follicular Helper CD4 T Cell Differentiation. The Journal of Immunology. 2012;188:3734–3744. doi: 10.4049/jimmunol.1103246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitt N, et al. Human Dendritic Cells Induce the Differentiation of Interleukin-21-Producing T Follicular Helper-like Cells through Interleukin-12. Immunity. 2009;31:158–169. doi: 10.1016/j.immuni.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nurieva RI, et al. Generation of T Follicular Helper Cells Is Mediated by Interleukin-21 but Independent of T Helper 1, 2, or 17 Cell Lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting Edge: STAT1 Is Required for IL-6-Mediated Bcl6 Induction for Early Follicular Helper Cell Differentiation. The Journal of Immunology. 2013;190:3049–3053. doi: 10.4049/jimmunol.1203032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballesteros-Tato A, et al. Interleukin-2 Inhibits Germinal Center Formation by Limiting T Follicular Helper Cell Differentiation. Immunity. 2012;36:847–856. doi: 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tian Y, Mollo SB, Harrington LE, Zajac AJ. IL-10 Regulates Memory T Cell Development and the Balance between Th1 and Follicular Th Cell Responses during an Acute Viral Infection. The Journal of Immunology. 2016;197:1308–1321. doi: 10.4049/jimmunol.1502481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu T, et al. TCF1 Is Required for the T Follicular Helper Cell Response to Viral Infection. Cell Reports. 2015;12:2099–2110. doi: 10.1016/j.celrep.2015.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi YS, et al. LEF-1 and TCF-1 orchestrate TFH differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. 2015;16:980–990. doi: 10.1038/ni.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu L, et al. The transcription factor TCF-1 initiates the differentiation of TFH cells during acute viral infection. Nat Immunol. 2015;16:991–999. doi: 10.1038/ni.3229. [DOI] [PubMed] [Google Scholar]
- 35.Choi YS, et al. ICOS Receptor Instructs T Follicular Helper Cell versus Effector Cell Differentiation via Induction of the Transcriptional Repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herati RS, et al. Circulating CXCR5+PD-1+ response predicts influenza vaccine antibody responses in young adults but not elderly adults. J Immunol. 2014;193:3528–3537. doi: 10.4049/jimmunol.1302503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goenka R, et al. Cutting Edge: Dendritic Cell-Restricted Antigen Presentation Initiates the Follicular Helper T Cell Program but Cannot Complete Ultimate Effector Differentiation. The Journal of Immunology. 2011;187:1091–1095. doi: 10.4049/jimmunol.1100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnett LG, et al. B Cell Antigen Presentation in the Initiation of Follicular Helper T Cell and Germinal Center Differentiation. The Journal of Immunology. 2014;192:3607–3617. doi: 10.4049/jimmunol.1301284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tubo NJ, et al. Single Naive CD4+ T Cells from a Diverse Repertoire Produce Different Effector Cell Types during Infection. Cell. 2013;153:785–796. doi: 10.1016/j.cell.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baumjohann D, et al. Persistent Antigen and Germinal Center B Cells Sustain T Follicular Helper Cell Responses and Phenotype. Immunity. 2013;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 41.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keck S, et al. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc Natl Acad Sci USA. 2014;111:14852–14857. doi: 10.1073/pnas.1403271111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb LMC, Linterman MA. Signals that drive T follicular helper cell formation. Immunology. 2017 doi: 10.1111/imm.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knowlden ZAG, Sant AJ. CD4 T cell epitope specificity determines follicular versus non-follicular helper differentiation in the polyclonal response to influenza infection or vaccination. Nature Publishing Group. 2016:1–13. doi: 10.1038/srep28287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma CS, et al. Early commitment of naïve human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol. 2009;87:590–600. doi: 10.1038/icb.2009.64. [DOI] [PubMed] [Google Scholar]
- 46.Locci M, et al. Activin A programs the differentiation of human TFH cells. Nat Immunol. 2016;17:976–984. doi: 10.1038/ni.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma CS, et al. Unique and shared signaling pathways cooperate to regulate the differentiation of human CD4+ T cells into distinct effector subsets. J Exp Med. 2016;213:1589–1608. doi: 10.1084/jem.20151467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oxenius A, et al. Variable fate of virus-specific CD4(+) T cells during primary HIV-1 infection. Eur J Immunol. 2001;31:3782–3788. doi: 10.1002/1521-4141(200112)31:12<3782::aid-immu3782>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 49.Brooks DG, Teyton L, Oldstone MBA, McGavern DB. Intrinsic Functional Dysregulation of CD4 T Cells Occurs Rapidly following Persistent Viral Infection. Journal of Virology. 2005;79:10514–10527. doi: 10.1128/JVI.79.16.10514-10527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fahey LM, et al. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. 2011;208:987–999. doi: 10.1084/jem.20101773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindqvist M, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest. 2012;122:3271–3280. doi: 10.1172/JCI64314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riou C, et al. HIV Skews the Lineage-Defining Transcriptional Profile of Mycobacterium tuberculosis-Specific CD4+ T Cells. The Journal of Immunology. 2016;196:3006–3018. doi: 10.4049/jimmunol.1502094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Current Opinion in Immunology. 2012;24:297–302. doi: 10.1016/j.coi.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matloubian M, Kolhekar SR, Somasundaram T, Ahmed R. Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. Journal of Virology. 1993;67:7340–7349. doi: 10.1128/jvi.67.12.7340-7349.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. Journal of Virology. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beura LK, et al. Lymphocytic choriomeningitis virus persistence promotes effector-like memory differentiation and enhances mucosal T cell distribution. Journal of Leukocyte Biology. 2015;97:217–225. doi: 10.1189/jlb.1HI0314-154R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis GM, Wehrens EJ, Labarta-Bajo L, Streeck H, Zuniga EI. TGF-β receptor maintains CD4 T helper cell identity during chronic viral infections. J Clin Invest. 2016:1–15. doi: 10.1172/JCI87041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beyer M, et al. Tumor-necrosis factor impairs CD4+ T cell–mediated immunological control in chronic viral infection. Nat Immunol. 2016;17:593–603. doi: 10.1038/ni.3399. [DOI] [PubMed] [Google Scholar]
- 59.Leong YA, et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nature Publishing Group. 2016:1–13. doi: 10.1038/ni.3543. [DOI] [PubMed] [Google Scholar]
- 60.He R, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature Publishing Group. 1AD:1–20. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 61.Im SJ, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016:1–20. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He R, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature. 2016;537:412–428. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 63.Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MBA. Persistent Virus Infection Inhibits Type I Interferon Production by Plasmacytoid Dendritic Cells to Facilitate Opportunistic Infections. Cell Host & Microbe. 2008;4:374–386. doi: 10.1016/j.chom.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee LN, Burke S, Montoya M, Borrow P. Multiple Mechanisms Contribute to Impairment of Type 1 Interferon Production during Chronic Lymphocytic Choriomeningitis Virus Infection of Mice. The Journal of Immunology. 2009;182:7178–7189. doi: 10.4049/jimmunol.0802526. [DOI] [PubMed] [Google Scholar]
- 65.Rodero MP, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med. 2017;214:1547–1555. doi: 10.1084/jem.20161451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. 2011;334:825–829. doi: 10.1126/science.1208421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshida H, Hunter CA. The Immunobiology of Interleukin-27. Annu Rev Immunol. 2015;33:417–443. doi: 10.1146/annurev-immunol-032414-112134. [DOI] [PubMed] [Google Scholar]
- 68.Penaloza-MacMaster P, et al. Vaccine-elicited CD4 T cells induce immunopathology after chronic LCMV infection. Science. 2015;347:278–282. doi: 10.1126/science.aaa2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elsaesser H, Sauer K, Brooks DG. IL-21 Is Required to Control Chronic Viral Infection. Science. 2009;324:1569–1572. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frohlich A, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science. 2009;324:1576–1580. doi: 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- 71.Yi JS, Du M, Zajac AJ. A Vital Role for Interleukin-21 in the Control of a Chronic Viral Infection. Science. 2009;324:1576–1576. doi: 10.1126/science.1175194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tian Y, Zajac AJ. IL-21 and T Cell Differentiation: Consider the Context. Trends in Immunology. 2016;37:557–568. doi: 10.1016/j.it.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harker JA, Dolgoter A, Zuniga EI. Cell-Intrinsic IL-27 and gp130 Cytokine Receptor Signaling Regulates Virus-Specific CD4+ T Cell Responses and Viral Control during Chronic Infection. Immunity. 2013;39:548–559. doi: 10.1016/j.immuni.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Battegay M, et al. Impairment and delay of neutralizing antiviral antibody responses by virus-specific cytotoxic T cells. The Journal of Immunology. 1993;151:5408–5415. [PubMed] [Google Scholar]
- 75.Hunziker L, et al. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat Immunol. 2003;4:343–349. doi: 10.1038/ni911. [DOI] [PubMed] [Google Scholar]
- 76.Buchmeier MJ, Oldstone MB. Virus-induced immune complex disease: identification of specific viral antigens and antibodies deposited in complexes during chronic lymphocytic choriomeningitis virus infection. The Journal of Immunology. 1978;120:1297–1304. [PubMed] [Google Scholar]
- 77.Fallet B, et al. Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Science Immunology. 2016;1 doi: 10.1126/sciimmunol.aah6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moseman EA, Wu T, la Torre de JC, Schwartzberg PL, McGavern DB. Type I interferon suppresses virus-specific B cell responses by modulating CD8(+) T cell differentiation. Science Immunology. 2016;1 doi: 10.1126/sciimmunol.aah3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bergthaler A, et al. Impaired Antibody Response Causes Persistence of Prototypic T Cell–Contained Virus. PLoS Biol. 2009;7:e1000080–11. doi: 10.1371/journal.pbio.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grebely J, et al. Hepatitis C virus clearance, reinfection, and persistence, with insights from studies of injecting drug users: towards a vaccine. The Lancet Infectious Diseases. 2012;12:408–414. doi: 10.1016/S1473-3099(12)70010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Loggerenberg F, et al. Establishing a Cohort at High Risk of HIV Infection in South Africa: Challenges and Experiences of the CAPRISA 002 Acute Infection Study. PLoS ONE. 2008;3:e1954–8. doi: 10.1371/journal.pone.0001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laher F, et al. HIV Controllers Exhibit Enhanced Frequencies of Major Histocompatibility Complex Class II Tetramer +Gag-Specific CD4 +T Cells in Chronic Clade C HIV-1 Infection. Journal of Virology. 2017;91:e02477–16–17. doi: 10.1128/JVI.02477-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schulze zur Wiesch J, et al. Broadly directed virus-specific CD4 +T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J Exp Med. 2012;209:61–75. doi: 10.1084/jem.20100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herati RS, et al. Successive annual influenza vaccination induces a recurrent oligoclonotypic memory response in circulating T follicular helper cells. Science Immunology. 2017;2:eaag2152–34. doi: 10.1126/sciimmunol.aag2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dan JM, et al. A Cytokine-Independent Approach To Identify Antigen-Specific Human Germinal Center T Follicular Helper Cells and Rare Antigen-Specific CD4+ T Cells in Blood. J Immunol. 2016;197:983–993. doi: 10.4049/jimmunol.1600318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raziorrouh B, et al. Virus-Specific CD4+ T Cells Have Functional and Phenotypic Characteristics of Follicular T-Helper Cells in Patients With Acute and Chronic HCV Infections. Gastroenterology. 2016;150:696–706.e3. doi: 10.1053/j.gastro.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 87.Dustin LB, Charles ED. Primary, post-primary and non-specific immunoglobulin M responses in HCV infection. Antivir Ther. 2012;17:1449–1452. doi: 10.3851/IMP2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fafi-Kremer S, et al. Neutralizing Antibodies and Pathogenesis of Hepatitis C Virus Infection. Viruses. 2012;4:2016–2030. doi: 10.3390/v4102016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lane HC, et al. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1983;309:453–458. doi: 10.1056/NEJM198308253090803. [DOI] [PubMed] [Google Scholar]
- 90.Abdel-Hakeem MS, Shoukry NH. Protective immunity against hepatitis C: many shades of gray. Front Immunol. 2014;5:274. doi: 10.3389/fimmu.2014.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364:2429–2438. doi: 10.1056/NEJMcp1006613. [DOI] [PubMed] [Google Scholar]
- 92.Takaki A, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med. 2000;6:578–582. doi: 10.1038/75063. [DOI] [PubMed] [Google Scholar]
- 93.Grakoui A, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302:659–662. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 94.Missale G, et al. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J Clin Invest. 1996;98:706–714. doi: 10.1172/JCI118842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lucas M, et al. Tracking Virus-Specific CD4+ T Cells during and after Acute Hepatitis C Virus Infection. PLoS ONE. 2007;2:e649–11. doi: 10.1371/journal.pone.0000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Day CL, et al. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest. 2003;112:831–842. doi: 10.1172/JCI18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abdel-Hakeem MS, B dard N, Murphy D, Bruneau J, Shoukry NH. Signatures of Protective Memory Immune Responses During Hepatitis C Virus Reinfection. Gastroenterology. 2014;147:870–881.e8. doi: 10.1053/j.gastro.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kared H, Fabre T, Bédard N, Bruneau J, Shoukry NH. Galectin-9 and IL-21 Mediate Cross-regulation between Th17 and Treg Cells during Acute Hepatitis C. PLoS Pathog. 2013;9:e1003422–18. doi: 10.1371/journal.ppat.1003422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moorman JP, et al. Impaired hepatitis B vaccine responses during chronic hepatitis C infection: Involvement of the PD-1 pathway in regulating CD4+ T cell responses. Vaccine. 2011;29:3169–3176. doi: 10.1016/j.vaccine.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.World Health Organization. [Accessed: 30 September 2016];1 Global summary of the AIDS epidemic. 2014 Available at. [Google Scholar]
- 101.Soghoian DZ, et al. HIV-Specific Cytolytic CD4 T Cell Responses During Acute HIV Infection Predict Disease Outcome. Science Translational Medicine. 2012;4:123ra25–123ra25. doi: 10.1126/scitranslmed.3003165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Falivene J, et al. Th17 and Th17/Treg ratio at early HIV infection associate with protective HIV-specific CD8+ T-cell responses and disease progression. Nature Publishing Group. 2015:1–14. doi: 10.1038/srep11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brenchley JM, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008;112:2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Douek DC, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 105.Kannanganat S, et al. Human Immunodeficiency Virus Type 1 Controllers but Not Noncontrollers Maintain CD4 T Cells Coexpressing Three Cytokines. Journal of Virology. 2007;81:12071–12076. doi: 10.1128/JVI.01261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cellerai C, et al. Early and Prolonged Antiretroviral Therapy Is Associated with an HIV-1-Specific T-Cell Profile Comparable to That of Long-Term Non-Progressors. PLoS ONE. 2011;6:e18164–15. doi: 10.1371/journal.pone.0018164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pantaleo G, et al. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proceedings of the National Academy of Sciences. 1991;88:9838–9842. doi: 10.1073/pnas.88.21.9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cubas RA, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. 2013;19:494–499. doi: 10.1038/nm.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mylvaganam GH, et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV infection. Proc Natl Acad Sci USA. 2017;114:1976–1981. doi: 10.1073/pnas.1621418114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Petrovas C, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest. 2012;122:3281–3294. doi: 10.1172/JCI63039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perreau M, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. 2013;210:143–156. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pallikkuth S, et al. Peripheral T Follicular Helper Cells Are the Major HIV Reservoir within Central Memory CD4 T Cells in Peripheral Blood from Chronically HIV-Infected Individuals on Combination Antiretroviral Therapy. Journal of Virology. 2016;90:2718–2728. doi: 10.1128/JVI.02883-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fukazawa Y, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. 2015;21:132–139. doi: 10.1038/nm.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Colineau L, et al. HIV-Infected Spleens Present Altered Follicular Helper T Cell (Tfh) Subsets and Skewed B Cell Maturation. PLoS ONE. 2015;10:e0140978–19. doi: 10.1371/journal.pone.0140978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 116.Seung E, et al. PD-1 Blockade in Chronically HIV-1-Infected Humanized Mice Suppresses Viral Loads. PLoS ONE. 2013;8:e77780–10. doi: 10.1371/journal.pone.0077780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Havenar-Daughton C, Lee JH, Crotty S. Tfh cells and HIV bnAbs, an immunodominance model of the HIV neutralizing antibody generation problem. Immunol Rev. 2017;275:49–61. doi: 10.1111/imr.12512. [DOI] [PubMed] [Google Scholar]
- 118.Miles B, Miller SM, Connick E. CD4 T Follicular Helper and Regulatory Cell Dynamics and Function in HIV Infection. Front Immunol. 2016;7:1545–9. doi: 10.3389/fimmu.2016.00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bentebibel SE, et al. Induction of ICOS+CXCR3+CXCR5+ TH Cells Correlates with Antibody Responses to Influenza Vaccination. Science Translational Medicine. 2013;5:176ra32–176ra32. doi: 10.1126/scitranslmed.3005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bentebibel S-E, et al. ICOS+PD-1+CXCR3+ T follicular helper cells contribute to the generation of high-avidity antibodies following influenza vaccination. Nature Publishing Group. 2016:1–8. doi: 10.1038/srep26494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schultz BT, et al. Circulating HIV-Specific Interleukin-21(+)CD4(+) T Cells Represent Peripheral Tfh Cells with Antigen-Dependent Helper Functions. Immunity. 2016;44:167–178. doi: 10.1016/j.immuni.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 122.Li S, et al. Metabolic Phenotypes of Response to Vaccination in Humans. Cell. 2017;169:862–866.e17. doi: 10.1016/j.cell.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Locci M, et al. Human Circulating PD-1+CXCR3 CXCR5+ Memory Tfh Cells Are HighlyFunctional and Correlate with Broadly Neutralizing HIV Antibody Responses. Immunity. 2013;39:758–769. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morita R, et al. Human Blood CXCR5+CD4+ T Cells Are Counterparts of T Follicular Cells and Contain Specific Subsets that Differentially Support Antibody Secretion. Immunity. 2011;34:108–121. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cubas R, et al. Reversible Reprogramming of Circulating Memory T Follicular Helper Cell Function during Chronic HIV Infection. J Immunol. 2015;195:5625–5636. doi: 10.4049/jimmunol.1501524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Parmigiani A, et al. Impaired antibody response to influenza vaccine in HIV-infected and uninfected aging women is associated with immune activation and inflammation. PLoS ONE. 2013;8:e79816. doi: 10.1371/journal.pone.0079816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.George VK, et al. HIV infection Worsens Age-Associated Defects in Antibody Responses to Influenza Vaccine. J Infect Dis. 2015;211:1959–1968. doi: 10.1093/infdis/jiu840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Abudulai LN, et al. Production of IgG antibodies to pneumococcal polysaccharides is associated with expansion of ICOS+ circulating memory T follicular-helper cells which is impaired by HIV infection. PLoS ONE. 2017;12:e0176641–22. doi: 10.1371/journal.pone.0176641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tebas P, et al. Poor immunogenicity of the H1N1 2009 vaccine in well controlled HIV-infected individuals. AIDS. 2010;24:2187–2192. doi: 10.1097/QAD.0b013e32833c6d5c. [DOI] [PubMed] [Google Scholar]
- 130.Wilson EB, et al. Blockade of Chronic Type I Interferon Signaling to Control Persistent LCMV Infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sanchez JL, et al. Lymphoid Fibrosis Occurs in Long-Term Nonprogressors and Persists With Antiretroviral Therapy but May Be Reversible With Curative Interventions. J Infect Dis. 2015;211:1068–1075. doi: 10.1093/infdis/jiu586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zeng M, Haase AT, Schacker TW. Lymphoid tissue structure and HIV-1 infection: life or death for T cells. Trends in Immunology. 2012;33:306–314. doi: 10.1016/j.it.2012.04.002. [DOI] [PubMed] [Google Scholar]