

The neural basis of reserve capacity in Alzheimer’s disease is yet to be fully determined. Franzmeier et al. show that greater left frontal hub connectivity within the fronto-parietal control network is associated with greater resilience of cognitive performance during the early stages of autosomal dominant and sporadic Alzheimer’s disease.
Keywords: Alzheimer’s disease, cognitive reserve, resting state connectivity, memory, dementia biomarkers
Abstract
Patients with Alzheimer’s disease vary in their ability to sustain cognitive abilities in the presence of brain pathology. A major open question is which brain mechanisms may support higher reserve capacity, i.e. relatively high cognitive performance at a given level of Alzheimer’s pathology. Higher functional MRI-assessed functional connectivity of a hub in the left frontal cortex is a core candidate brain mechanism underlying reserve as it is associated with education (i.e. a protective factor often associated with higher reserve) and attenuated cognitive impairment in prodromal Alzheimer’s disease. However, no study has yet assessed whether such hub connectivity of the left frontal cortex supports reserve throughout the evolution of pathological brain changes in Alzheimer’s disease, including the presymptomatic stage when cognitive decline is subtle. To address this research gap, we obtained cross-sectional resting state functional MRI in 74 participants with autosomal dominant Alzheimer’s disease, 55 controls from the Dominantly Inherited Alzheimer’s Network and 75 amyloid-positive elderly participants, as well as 41 amyloid-negative cognitively normal elderly subjects from the German Center of Neurodegenerative Diseases multicentre study on biomarkers in sporadic Alzheimer’s disease. For each participant, global left frontal cortex connectivity was computed as the average resting state functional connectivity between the left frontal cortex (seed) and each voxel in the grey matter. As a marker of disease stage, we applied estimated years from symptom onset in autosomal dominantly inherited Alzheimer’s disease and cerebrospinal fluid tau levels in sporadic Alzheimer’s disease cases. In both autosomal dominant and sporadic Alzheimer’s disease patients, higher levels of left frontal cortex connectivity were correlated with greater education. For autosomal dominant Alzheimer’s disease, a significant left frontal cortex connectivity × estimated years of onset interaction was found, indicating slower decline of memory and global cognition at higher levels of connectivity. Similarly, in sporadic amyloid-positive elderly subjects, the effect of tau on cognition was attenuated at higher levels of left frontal cortex connectivity. Polynomial regression analysis showed that the trajectory of cognitive decline was shifted towards a later stage of Alzheimer’s disease in patients with higher levels of left frontal cortex connectivity. Together, our findings suggest that higher resilience against the development of cognitive impairment throughout the early stages of Alzheimer’s disease is at least partially attributable to higher left frontal cortex-hub connectivity.
Introduction
Biomarker studies in Alzheimer’s disease have revealed a temporal sequence of the development of brain pathologies and cognitive decline. In autosomal dominantly inherited Alzheimer’s disease, a successive emergence of abnormal CSF concentrations of amyloid-β, tau, microglial activation, grey matter atrophy and cerebral glucose hypometabolism has been reported (Bateman et al., 2012; Benzinger et al., 2013; Suarez-Calvet et al., 2016). Similar findings have been obtained in sporadic late onset Alzheimer’s disease (Jack et al., 2012), providing strong evidence for cascading pathologies that lead to cognitive deficits and ultimately dementia. The extent of cognitive decrease at any level of Alzheimer’s disease pathologies, however, varies considerably between individuals (Vemuri et al., 2011; Mufson et al., 2016). Even in autosomal dominantly inherited Alzheimer’s disease (ADAD), where the course of Alzheimer’s disease development is strongly determined by mutations in genes encoding presinilin-1 (PSEN1), presenilin-2 (PSEN2) or amyloid precursor protein (APP), the cognitive status remains relatively stable in some patients despite advanced levels of amyloid-β and tau pathology (Lim et al., 2016). The theory of reserve posits that such a discrepancy between brain pathology and cognitive impairment is not a prediction error due to incomplete detection of neuronal pathologies, but is due to higher resilience against the effects of pathology (Park and Reuter-Lorenz, 2009; Stern, 2012). In other words, some people may show higher ability to maintain cognitive abilities relatively well at any given level of brain pathology. At the epidemiological level, more years of education, higher general cognitive ability, occupational attainment and other cognitively challenging life experiences (Valenzuela and Sachdev, 2006), or higher physical fitness (Okonkwo et al., 2014; Tolppanen et al., 2015; Duzel et al., 2016) have been associated with higher cognitive abilities in ageing and Alzheimer’s disease. Neuroimaging studies on structural brain differences showed a larger premorbid brain volume to be an additional protective factor in Alzheimer’s disease (Perneczky et al., 2010; Meng and D'Arcy, 2012; Sumowski et al., 2014). However, protective lifestyle factors and brain volume are gross measures that are not informative about protective functional brain mechanisms. Hence, the overall aim of the current study was to test particular functional brain features that support higher resilience of cognitive performance during the course of Alzheimer’s disease.
The fronto-parietal control network has been proposed to play a key role in mental health and reserve in ageing (Cole et al., 2014; Medaglia et al., 2017). The fronto-parietal control network harbours major brain hubs (Cole et al., 2010) and controls the activity of other functional networks across different cognitive domains (Cole et al., 2013). Thus, this network may be fundamental for maintaining efficient functional brain processes that underlie cognitive performance when brain damage occurs. We recently showed that higher global connectivity of a hub in the fronto-parietal control network is associated with higher reserve in amyloid-positive mild cognitively impaired patients who are at increased risk of developing Alzheimer’s disease dementia (Franzmeier et al., 2017a). Specifically, we showed that higher global functional connectivity of the left frontal cortex (gLFC-connectivity, Brodmann area 6/44) was associated with: (i) more years of education (i.e. an epidemiological factor that is associated with lower risk of Alzheimer’s disease); and (ii) an attenuated impact of Alzheimer’s disease-related posterior parietal glucose hypometabolism on memory performance (Franzmeier et al., 2017b). Particularly the global functional connectivity of that hub region of the fronto-parietal control network in the left rather than the right hemisphere has been related to reserve-associated protective factors including higher general cognitive ability (i.e. fluid intelligence) and better cognitive control in young to middle-aged subjects (Cole et al., 2012, 2015). Together, these results suggest that gLFC-connectivity may contribute to reserve at an early stage in Alzheimer’s disease, and motivated us to ask whether gLFC-connectivity is associated with higher reserve during the entire course of the disease from preclinical Alzheimer’s disease to dementia.
Here, we hypothesized that at higher levels of gLFC-connectivity the level of impairment in global cognition (Mini-Mental-State Examination, MMSE) and episodic memory (delayed free recall) are shifted relative to levels of biomarkers of Alzheimer’s disease related neurodegeneration. To assess the specificity of our hypothesis we conducted control analyses for global connectivity of additional regions including: (i) the LFCs’ right homotopic region (i.e. right frontal cortex, RFC); and (ii) two null-regions within the visual and motor cortices for which we did not expect any significant effects. We tested our hypothesis based on resting state functional MRI data, cognitive and biomarker levels obtained in two cohorts including: (i) participants with ADAD recruited within the Dominantly Inherited Alzheimer Network (DIAN); and (ii) elderly subjects with sporadic Alzheimer’s disease recruited within the German Center for Neurodegenerative Diseases longitudinal study on cognition and dementia (DELCODE). The study design of DIAN allowed us to examine the association between gLFC-connectivity and cognitive performance against the dynamic changes of biomarkers of Alzheimer’s disease pathology years before onset of dementia. The inclusion of sporadic Alzheimer’s disease provided the opportunity to test whether any effects of gLFC-connectivity on reserve can be generalized towards the more common age-related form of accumulating Alzheimer’s disease pathology in elderly subjects. Thus, the current cross-sectional study assessed for the first time a core candidate functional brain substrate underlying higher cognition during the course of Alzheimer’s disease from preclinical to dementia stages of Alzheimer’s disease.
Materials and methods
Participants
The DIAN sample of ADAD and controls
Seventy-four mutation carriers exhibiting Alzheimer’s disease causing mutations in genes PSEN1, PSEN2 or APP, and 55 non-mutation carrier siblings were included from the DIAN database (Clinical Trial Identifier NCT00869817) (Moulder et al., 2013). Beyond the inclusion criteria of DIAN, the requirement for inclusion in the current study was the presence of baseline resting state functional MRI, T1 structural MRI, CSF biomarker assessments, partial volume corrected global Pittsburgh compound B-PET (PiB-PET) binding data, and neuropsychological test scores. Because these stricter inclusion criteria resulted in the selection of a subsample from the larger DIAN cohort (n = 177 mutation carriers, and n = 136 non-mutation carriers), we tested for selection bias by comparing baseline characteristics [age, gender, years of education, estimated years from symptom onset (EYO)] between the selected sample (n = 129) and the excluded (n = 184) sample recruited in DIAN, using two-sample t-tests for continuous and chi-squared tests for categorical variables. No significant (i.e. P > 0.05) differences between groups in any of the variables were found, suggesting that the selected sample is representative of the DIAN cohort. For details on the entire DIAN sample versus the selected DIAN sample please see Supplementary Table 1. Distributions of the main biomarkers and cognitive test scores of the selected DIAN sample can be found in Supplementary Fig. 1. The EYO was defined as the difference between a participant’s age and the parental age of symptom onset, as an estimate of the number of years until symptom onset in mutation carriers. The measurement of global PiB-PET and CSF biomarkers of amyloid-β, tau phosphorylated at serine-181 (p-tau181) and total tau have been described previously (Bateman et al., 2012; Benzinger et al., 2013). Local ethical approval and written informed consent had been obtained from each participant by the respective DIAN centres.
The DELCODE sample of sporadic Alzheimer’s disease and controls
A total of 116 elderly participants were included from the DELCODE study encompassing participants with cognitively normal status (n = 49), subjective cognitive decline (n = 40), mild cognitive impairment (MCI, n = 14) and Alzheimer’s disease dementia (n = 13). DELCODE is an ongoing prospective longitudinal study on the development of neuroimaging and biofluid-derived markers for the early detection of late onset sporadic Alzheimer’s disease. The study is conducted at eight University hospitals within the German Center for Neurodegenerative Diseases. Participants were defined as cognitively normal when showing a Clinical Dementia Rating of zero and neither reporting cognitive complaints nor scoring lower than 1.5 standard deviations (SD) of age-, sex-, and education-adjusted norms on all subtests of the German CERAD-NP Plus battery (Berres et al., 2000), which includes the Trail Making Test versions A and B (Reitan, 1958) and letter fluency in addition to the original version of the CERAD battery (Morris et al., 1989). Subjective cognitive decline was defined by criteria including: (i) Clinical Dementia Rating ≤ 0.5; (ii) normal performance on the tests of CERAD-NP Plus (i.e. a criterion of cognitively normal); and (iii) subjective experience of cognitive decline between the last 6 months and 5 years as reported in semi-structured interviews conducted by a physician with the study participant. Mild cognitive impairment was diagnosed at a Clinical Dementia Rating ≤ 0.5 and cognitive performance level being 1.5 SD below the age- and education adjusted norm for the word-list delayed recall trial of the CERAD-NP-Plus battery (Petersen et al., 2014), thus single- and multi-domain amnestic mild cognitive impairment was included. Alzheimer’s disease dementia was diagnosed when fulfilling the clinical NINDCS/ADRDA criteria of probable Alzheimer’s disease (McKhann et al., 2011). All participants included in the current study underwent lumbar puncture to determine CSF levels of amyloid-β42, amyloid-β40, p-tau181, total tau and were a priori dichotomized as showing either normal or abnormal CSF amyloid-β levels (CSF amyloid-β+ versus CSF amyloid-β−), based on the previously established cut-off for CSF amyloid-β42/40 ratio < 0.1 (Janelidze et al., 2016). All participants had to have complete baseline data available including resting state functional MRI, T1 structural MRI, CSF biomarker levels, and neurocognitive test scores. When testing for selection bias by comparing baseline characteristics (age, gender, education) between the selected sample (n = 116) and the excluded sample (n = 250) using two-sample t-tests for continuous and chi-squared tests for categorical variables, no significant (i.e. P > 0.05) differences between groups in any of the variables were found, suggesting that the selected sample is representative of the whole DELCODE cohort. Details of the entire versus the selected DELCODE sample can be found in Supplementary Table 2. Biomarkers and cognitive test score distributions of the selected DELCODE sample are illustrated in Supplementary Fig. 1.
Assessment of general cognitive ability and memory performance
As a measure of general cognitive function, we used the MMSE score (Folstein et al., 1975). For episodic memory, the delayed free recall test included in the Wechsler Memory Scale Logical Memory II test was used (Kent, 2013). These two measures of global cognition and episodic memory were selected from a larger set of psychometric tests since those tests were obtained in both DIAN and DELCODE and thus yielded comparability between both studies.
MRI acquisition
In both DIAN and DELCODE, all MRI scans were acquired on 3 T MRI systems (Siemens) with unified scanning protocols for each study. High resolution structural images were obtained using a T1-MPRAGE sequence (DIAN: repetition time/echo time = 2300 ms/2.95 ms, flip angle = 9°, 1.1 × 1.1 × 1.2 mm voxels; DELCODE: repetition time/echo time = 2500 ms/4.33 ms, flip angle = 7°, 1 mm isotropic voxels) with whole-brain coverage. Resting state functional MRI images were recorded using a T2*-weighted EPI sequence (DIAN: repetition time/echo time = 2230 ms/30 ms, flip angle = 80°, 3.3 mm isotropic voxels, 140 volumes; DELCODE: repetition time/echo time = 2580 ms/30 ms, flip angle = 80°, 3.5 mm isotropic voxels, 180 volumes). In DELCODE, the participants were instructed to keep their eyes closed and not to fall asleep prior to the resting state scan. In DIAN, resting state functional MRI was acquired with eyes open.
MRI preprocessing and connectivity analysis
The same processing pipelines were applied to the MRI scans from the DIAN and DELCODE cohort. Note that all processing steps were conducted separately for each sample, i.e. no data were pooled across DELCODE and DIAN but analysed separately within each study.
Preprocessing of structural MRI and spatial normalization
T1 MRI images were spatially normalized using SPM12 (Wellcome Trust Centre for Neuroimaging, University College London, UK: www.fil.ion.ucl.ac.uk/spm). In a first step, T1-weighted images were segmented into probabilistic tissue maps of grey matter, white matter and CSF compartments (Ashburner and Friston, 2005). Non-linear spatial normalization parameters were estimated using SPM’s DARTEL toolbox (Ashburner, 2007), yielding a sample-specific template that was subsequently registered to a template in MNI standard space in order to estimate affine transformation parameters. In a next step, the non-linear DARTEL and affine transformation parameters were jointly applied to the segmented grey matter maps to obtain participant specific grey matter maps in MNI space. All grey matter maps in MNI space were then averaged and binarized at a voxel value >0.3 to obtain a sample-specific grey matter mask for functional connectivity analyses.
As a surrogate for regional brain atrophy that is highly related to Alzheimer’s disease pathology and to memory impairment (Petersen et al., 2000), we assessed the volumes of the bilateral hippocampi using T1 MRI data, applying a previously described fully automated approach that was validated using manual segmentation (Mak et al., 2011). For details, see Supplementary material.
Preprocessing of resting state functional MRI data
All functional images were visually inspected for artefacts prior to analyses. For each participant, all functional MRI volumes were slice-time corrected, realigned to the first volume, and subsequently registered to the native space high resolution 3D T1-weighted image. The non-linear (i.e. DARTEL) and affine registration transformation matrix were again jointly applied to the registered functional MRI volumes to spatially normalize all images to MNI space followed by a smoothing step with an 8 mm full-width at half-maximum Gaussian kernel. To remove noise from the spatially normalized resting state functional MRI data, we removed the linear trend and applied a band-pass filter with a frequency band of 0.01–0.08 Hz. Additionally, we regressed out the six motion parameters and the functional MRI signal averaged across the white matter and CSF. Since functional connectivity analyses can be potentially confounded by motion even after realignment and motion regression (Power et al., 2012), we performed motion scrubbing following a previously established protocol (Power et al., 2014). To this end, we computed the framewise displacement (i.e. the average spatial displacement between two adjacent volumes) for each volume of the resting state scan. Volumes that exhibited a framewise displacement >0.5 mm were censored together with one preceding and two subsequent volumes. Participants for whom >30% of the resting-state volumes had to be censored after scrubbing were not included in the current study.
Assessment of global connectivity
GLFC-connectivity was assessed via seed-based functional connectivity analysis following a previously described approach (Cole et al., 2012; Franzmeier et al., 2017b). In brief, we created a binary sphere centred around the left frontal cortex (LFC) (MNI: x = −42, y = 6, z = 28; Brodmann area 6/44; Fig. 1) with a radius of 8 mm, which was used as the seed region for connectivity analyses. The LFC seed region of interest was superimposed onto the preprocessed, scrubbed and grey matter-masked resting state functional MRI data to extract the mean functional MRI time series within the LFC region of interest. Next, we calculated Fisher z-transformed Pearson moment correlations between the LFC-region of interest time series and each remaining grey matter voxel. To obtain the gLFC-connectivity score, we averaged all positive correlations across voxels belonging to the grey matter for each participant consistent with our and others previous analyses of gLFC-connectivity (Cole et al., 2012; Franzmeier et al., 2017a, b). Note that negative connections were not included, since positive and negative connectivity values would cancel each other out during averaging. In addition to the LFC, global connectivity was also computed for three control regions of interest: (i) the LFCs’ right hemispheric homotopic region (i.e. RFC, x = 42, y = 6, z = 28) to test left hemispheric specificity; and (ii) two null regions including one in the occipital pole (x = −19, y = −102, z = −3), as well as M1 in the motor cortex (x = −38, y = −22, z = 56). Note that we used group-specific rather than subject-specific regions of interest for the current analyses, since there is to date no reliable gold-standard on how to define subject-specific functional clusters that may serve as individual seed-regions of interest.
Figure 1.
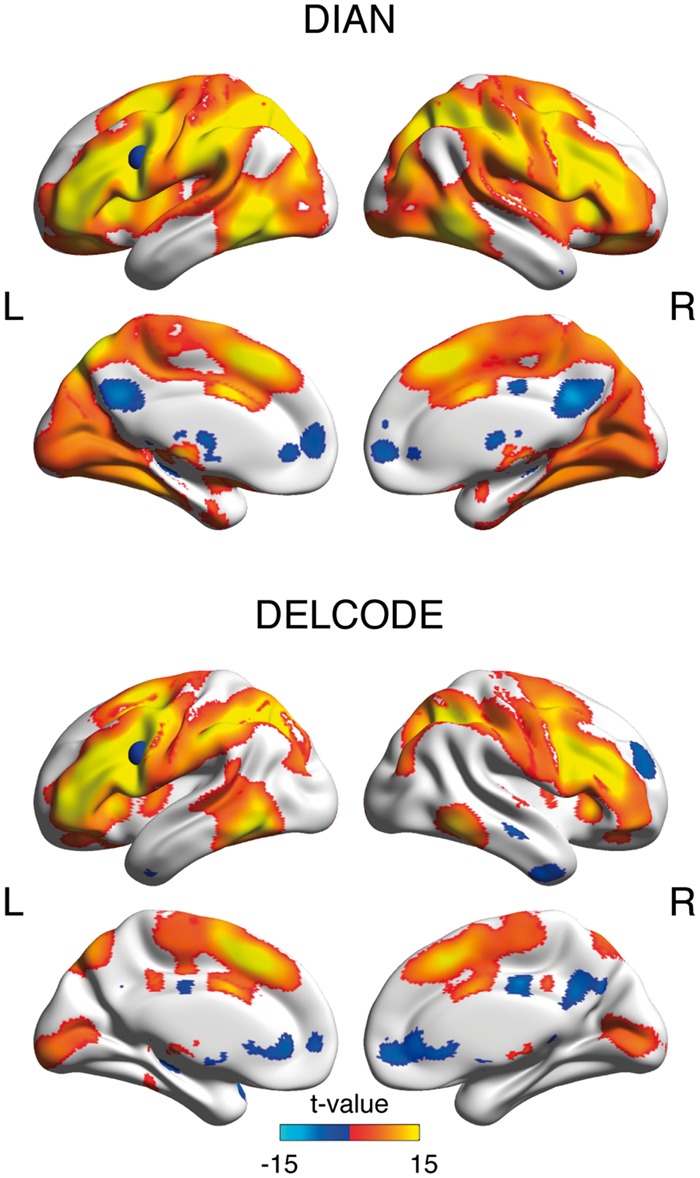
Seed-based LFC connectivity pattern. Surface renderings of significant LFC-connectivity in the DIAN and DELCODE sample at a voxel threshold of α < 0.001, family-wise error corrected at the cluster level at α < 0.05. The LFC-region of interest that was used for seed-based functional connectivity analyses is superimposed as a blue sphere on the left hemisphere.
To address potentially confounding effects of multi-site functional MRI acquisition we included centre as a random effect in our main analyses. In addition, we tested for each study (DIAN and DELCODE) whether global connectivity scores differed between scanner models (Verio, Trio, Prisma in DIAN and Verio, Trio, Skyra in DELCODE) using ANCOVAs controlling for age, gender and education. Here, no significant differences in global connectivity were detected between scanner models for the LFC [DIAN: F(4,124) = 0.176, P = 0.839; DELCODE F(4,111) = 0.674, P = 0.570], the RFC [DIAN: F(4,124) = 0.144, P = 0.867; DELCODE: F(4,111) = 0.361, P = 0.549], the occipital pole [DIAN: F(4,124) = 2.254, P = 0.109; DELCODE: F(4,111) = 0.1.419, P = 0.241] or M1 [DIAN: F(4,124) = 0.219, P = 0.804; DELCODE: F(4,111) = 1.240, P = 0.299].
Statistical analysis
Demographics and cognitive scores were compared between mutation carriers and non-mutation carriers within the DIAN study and between diagnostic groups within the DELCODE study, using ANOVAs (for comparisons between more than two groups), two-sample t-tests (for two groups) and chi-square tests (for categorical variables). GLFC-connectivity, CSF-tau, CSF-p-tau181 and global partial-volume corrected PIB-PET uptake measures were log- and z-transformed, after which there were no deviations from a normal distribution as assessed via Shapiro-Wilk tests. All subsequent analyses were performed using these transformed values. Note that no data were pooled across the DIAN or DELCODE sample for our analyses.
Next, we tested whether the effect of Alzheimer’s disease severity on cognitive ability is attenuated at higher levels of gLFC-connectivity. For DIAN, the primary linear mixed effects models included the interaction term EYO × gLFC-connectivity and main effects of these variables on either MMSE or logical memory delayed free recall. As a secondary measure of disease stage, we used CSF-tau instead of EYO. All linear mixed effects models were tested separately in mutation carrier and non-mutation carrier groups and were adjusted for gender and family affiliation as fixed effects, and centre as a random effect.
To test whether the gLFC-connectivity × EYO interaction term improves the model fit, we compared the full model to the reduced model that did not include the interaction term as a predictor. We compared the Akaike information criterion between the full and the reduced model to assess whether including the interaction term yielded a lower Akaike information criterion, indicating a better model fit. Consistent with the approach in previous DIAN studies (Bateman et al., 2012), age was not included in the models due to collinearity with EYO. However, DIAN participants are relatively young (mean age = 37 years), thus ageing is unlikely to account for any effects of EYO or CSF-tau on cognition in mutation carriers. These analyses were conducted in an equivalent fashion for global connectivity of the RFC, the occipital pole and M1.
Next, we modelled the interaction effect (EYO × gLFC-connectivity) on global cognition (i.e. MMSE). In addition, we estimated the trajectories of biomarker alterations as done previously based on cross-sectional data (Bateman et al., 2012), so that the course of cognitive alterations could be compared to that of biomarkers of Alzheimer’s disease pathological brain differences. In line with a previous approach (Suarez-Calvet et al., 2016), we fitted polynomial mixed effects models for each outcome variable (MMSE, CSF-tau, precuneus FDG-PET, global PiB-PET binding, and hippocampal volume) as a function of EYO, quadratic (EYO2) and cubic (EYO3) terms, gender, and family affiliation as fixed effects and site as random effect (Bateman et al., 2012), stratified for mutation carriers and non-mutation carriers. A first order interaction of EYO × gLFC connectivity was included, in addition, when global cognition was the dependent variable. For each outcome variable, the best model was chosen based on the Akaike information criterion. We then plotted the predicted difference between mutation carriers and non-mutation carriers at each EYO, divided by the standard deviation of the respective variable within the pooled sample. Since we adopted the modelling approach from previous studies on biomarker changes in DIAN, the results for the biomarker trajectories were highly consistent with previous reports (Bateman et al., 2012; Benzinger et al., 2013; Suarez-Calvet et al., 2016). This plot was restricted to an EYO range between −20 and +10 because of low numbers of participants at extreme values of EYO and to prevent identifiability of subjects based on extreme values of EYO (as required by DIAN publication guidelines).
Similarly, for DELCODE, we computed linear mixed effects models to test the interaction between CSF-tau (as a marker of disease severity that closely tracks memory decline) (Brier et al., 2016) and gLFC-connectivity on either MMSE or logical memory delayed recall, adjusting for age and gender as fixed effects and for centre as a random effect. These models were computed separately in the control (CSF amyloid-β−) and Alzheimer’s disease (CSF amyloid-β+) groups. In addition, we computed reduced models without the interaction term as a predictor and assessed differences in the model fit (i.e. Akaike information criterion) between the full and the respective reduced model. Separate models were computed for each dependent variable including global connectivity of the RFC, the occipital pole and M1. In an exploratory analysis, we additionally tested the effect of global connectivity × CSF-tau on the word-list delayed recall subscale of the ADAS-Cog battery, which was only available in the DELCODE sample.
To model the effect of gLFC-connectivity on the trajectories of cognitive changes across increasing disease severity (CSF-tau levels), we applied the same polynomial mixed effects approach as used in the DIAN sample, this time stratified by CSF-amyloid-β status. That is, we computed polynomial mixed effects models including CSF-tau (quadratic and cubic terms), controlled for age, gender (fixed effects) and centre (random effect) to predict biomarkers (i.e. CSF-amyloid-β42/40 ratio, hippocampal volume). A first order interaction of CSF-tau × gLFC connectivity was included, in addition, when MMSE was the dependent variable.
A schematic illustration of these above described main analyses is illustrated in Supplementary Fig. 2.
In additional models, we tested whether education (as a protective factor associated with higher reserve) predicted gLFC-connectivity in both samples, DIAN and DELCODE. These analyses were adjusted for gender and centre (random effect) as well as EYO and family affiliation in DIAN and age in DELCODE.
All analyses were computed using the freely available R statistical software package (http://www.r-project.org) (R Core Team, 2014), standardized beta coefficients were considered significant when meeting an alpha-threshold of α < 0.05. For our main analyses of the interaction gLFC-connectivity times Alzheimer’s disease severity (i.e. EYO or CSF-tau) on global cognition (i.e. MMSE) and episodic memory (i.e. logical memory delayed recall) (four analyses across two samples), we applied a Bonferroni-corrected alpha-threshold of α < 0.0125.
Results
Sample characteristics
Baseline characteristics and group differences are displayed in Table 1 for DIAN and Table 2 for DELCODE. Positive functional connectivity of the LFC seed region to other brain regions covered primarily the fronto-parietal control network in both the DIAN and DELCODE samples (Fig. 1), consistent with our previous work (Franzmeier et al., 2017b).
Table 1.
Baseline characteristics of the DIAN sample of ADAD and controls
| ADAD-MC | ADAD-NC | Cohen’s d | T-value | P-value | |
|---|---|---|---|---|---|
| (n = 74) | (n = 55) | ||||
| Age | 37.49 (10.05) | 37.84 (10.31) | 0.034 | 0.193 | 0.848 |
| Gender (female/male) | 42/32 | 34/21 | 0.563 | ||
| Years of education | 14.47 (3.2) | 15.51 (2.16) | 0.38 | 2.19 | 0.030 |
| EYO | −9.82 (11.00) | −9.61 (11.77) | 0.02 | 0.104 | 0.919 |
| Global PiB-PET | 2.12 (1.25) | 1.04 (0.05) | 1.22 | 6.430 | <0.001 |
| CSF-tau | 110 (89.48) | 55.19 (22.2) | 0.84 | 4.439 | <0.001 |
| CSF-p-tau181 | 60.82 (35.58) | 29.77 (9.41) | 1.19 | 6.305 | <0.001 |
| gLFC-connectivity | 0.27 (0.07) | 0.30 (0.07) | 0.43 | 2.445 | 0.016 |
| Logical memory delayed recall | 10.08 (6.13) | 13.98 (3.71) | 0.77 | 4.168 | <0.001 |
| MMSE | 27.04 (5.1) | 29.45 (1.02) | 0.66 | 3.455 | <0.001 |
MC = mutation carrier; NC = non-mutation carrier. Values are presented as mean (SD).
Table 2.
Baseline characteristics of the DELCODE sample of sporadic Alzheimer’s disease
| Sporadic AD (Aβ+) | CN | SCD | Cohen’s d | MCI | Cohen’s d | ADD | Cohen’s d | F-value | P-value |
|---|---|---|---|---|---|---|---|---|---|
| (n = 75) | (n = 25) | (n = 23) | (SCD versus CN) | (n = 14) | (MCI versus CN) | (n = 13) | (ADD versus CN) | ||
| Age | 67.76 (5.23)b,c | 72.26 (4.16)a | 0.95 | 74.64 (5.34)a | 1.30 | 71.31 (6.18) | 0.62 | 6.160 | <0.001 |
| Gender (female/male) | 16/9 | 10/13 | 5/9 | 9/4 | 0.164 | ||||
| Years of education | 14.64 (2.93) | 14.87 (3.81) | 0.07 | 14.71 (3.58) | 0.02 | 14.00 (3.11) | 0.21 | 0.192 | 0.901 |
| CSF-Aβ42/40 ratio | 0.08 (0.02)c,d | 0.08 (0.02)c,d | 0.00 | 0.06 (0.02)a,b | 1.00 | 0.04 (0.01)a,b | 2.53 | 15.456 | <0.001 |
| CSF- tau | 357.08 (136.91)c,d | 395.16 (178.58)d | 0.24 | 534.97 (172.61)a,d | 1.14 | 818.53 (322.62)a,b,c | 1.86 | 17.616 | <0.001 |
| CSF-p-tau181 | 49.08 (17.06)d | 53.96 (25.89)d | 0.22 | 71.99 (21.37)d | 1.18 | 101.6 (45.58)a,b,c | 1.53 | 12.220 | <0.001 |
| gLFC-connectivity | 0.23 (0.05) | 0.25 (0.05) | 0.40 | 0.23 (0.05) | 0.00 | 0.22 (0.03) | 0.24 | 1.535 | 0.213 |
| LM delayed recall | 14.57 (8.15)d | 12.00 (7.18) | 0.33 | 10.60 (6.47) | 0.54 | 6.69 (7.26)a | 1.02 | 2.905 | 0.041 |
| MMSE | 29.20 (0.96)c,d | 29.39 (0.78)c,d | 0.22 | 27.71 (1.68)a,b,d | 1.09 | 23.85 (2.82)a,b,c | 2.54 | 43.066 | <0.001 |
| Controls (Aβ−) | CN | SCD | Cohen’s d | T-value | P-value | ||||
| (n = 41) | (n = 24) | (n = 17) | (SCD versus CN) | ||||||
| Age | 67.29 (4.6) | 71.06 (5.53) | 0.74 | 2.376 | 0.023 | ||||
| Gender (female/male) | 15/9 | 9/8 | 0.540 | ||||||
| Years of education | 14.54 (2.62) | 15.35 (3.16) | 0.27 | 0.896 | 0.392 | ||||
| CSF-Aβ42/40 ratio | 0.12 (0.02) | 0.12 (0.01) | 0.00 | 0.957 | 0.957 | ||||
| CSF-tau | 320.78 (112.02) | 355.84 (114.32) | 0.31 | 0.979 | 0.334 | ||||
| CSF-p-tau181 | 51.57 (16.28) | 50.35 (21.1) | 0.06 | 0.591 | 0.581 | ||||
| gLFC-connectivity | 0.25 (0.06) | 0.23 (0.04) | 0.39 | 1.067 | 0.293 | ||||
| LM delayed recall | 16.56 (8.26) | 9.06 (2.19) | 1.24 | 1.824 | 0.076 | ||||
| MMSE | 29.67 (0.76) | 29.06 (0.9) | 0.73 | 2.336 | 0.024 | ||||
Post hoc Tukey test significant (P < 0.05) for aversus cognitively normal (CN); bversus subjective cognitive decline (SCD); cversus mild cognitive impairment (MCI); dversus Alzheimer’s disease dementia (ADD). Values are presented as mean (SD).
Aβ = amyloid-β; LM = logical memory.
DIAN
To test our major hypothesis that greater gLFC-connectivity moderates the impact of Alzheimer’s disease pathology on cognition in the mutation carrier group of the DIAN sample, we used linear mixed effects models, where we tested the interaction between gLFC-connectivity and EYO on cognitive measures. We found a significant interaction of gLFC-connectivity × EYO on both MMSE [β/standard error (SE) = 0.269/0.099, P = 0.008; Fig. 2A] and logical memory delayed free recall (β/SE = 0.275/0.106, P = 0.012; Fig. 2B). The full-models including the interaction term showed a better model fit (i.e. Akaike information criterion) as compared to the reduced models for MMSE (186.6 versus 192.1) and logical memory delayed free recall (182.8 versus 186.3). Visual inspection of Fig. 2A and B reveals that at higher levels of gLFC-connectivity, higher EYO (i.e. closer proximity to the estimated age of symptom onset) was associated with relatively less decline in MMSE and logical memory delayed free recall when compared to those with lower levels of gLFC-connectivity within the mutation carrier group. No interaction effect of EYO × gLFC-connectivity on any cognitive measures was found in the non-mutation carrier group. When using CSF-tau instead of EYO as a marker of disease stage (data not shown in Fig. 2), we found a similar result pattern in mutation carriers: at higher levels of gLFC-connectivity, the impact of CSF-tau on cognition was attenuated for MMSE (β/SE = 0.314/0.078, P < 0.001). However, such an effect was not found for logical memory delayed free recall in the mutation carrier group. For the non-mutation carrier group, no significant interaction between CSF-tau and gLFC-connectivity on any cognitive measure was observed. All abovementioned analyses were conducted equivalently for the control regions including the RFC, the occipital pole and M1. For the RFC, we found similar but less strong effects as for the LFC, i.e. a significant interaction gRFC-connectivity × EYO on MMSE (β/SE = 0.169/0.078, P = 0.018) but no interaction on logical memory delayed free recall (β/SE = 0.189/0.112, P = 0.095). These RFC results, however, did not survive Bonferroni correction. The full model including the gRFC-connectivity × EYO interaction term showed a better model fit (i.e. Akaike information criterion) compared to the reduced models for MMSE (172.3 versus 179.1). As expected, no effects were found for the occipital pole or M1. Detailed results of these control analyses are summarized for each control region of interest in Supplementary Tables 3–5.
Figure 2.

Interaction gLFC-connectivity × Alzheimer’s disease severity on cognition. Scatterplots of the interaction gLFC-connectivity × Alzheimer’s disease severity on cognitive performance in the ADAD (DIAN) and sporadic Alzheimer’s disease (DELCODE) sample. For DIAN, the estimated years from symptom onset (EYO) are plotted against the MMSE score (A) and the delayed free recall score of the logical memory scale (B). For DELCODE, CSF-tau levels are plotted against MMSE (C) and logical memory delayed free recall (D). For illustrational purposes, groups of high and low gLFC-connectivity (defined via median split) are plotted separately, statistical interactions were calculated using continuous measures. Dashed lines indicate 95% confidence intervals. P-values of the interaction terms are displayed for each graph. Aβ = amyloid-β.
Figure 3A illustrates the dynamic development of biomarkers and MMSE in mutation carriers versus non-mutation carriers across EYO, as predicted by polynomial mixed models based on cross-sectional data (Bateman et al., 2012). Specifically, the cross-sectionally estimated temporal evolution of impairment in global cognition is shown for mutation carriers with high versus low gLFC-connectivity as defined by median split. The decline in MMSE in individuals with high gLFC-connectivity is shifted to a later time point as compared to individuals with low gLFC-connectivity.
Figure 3.
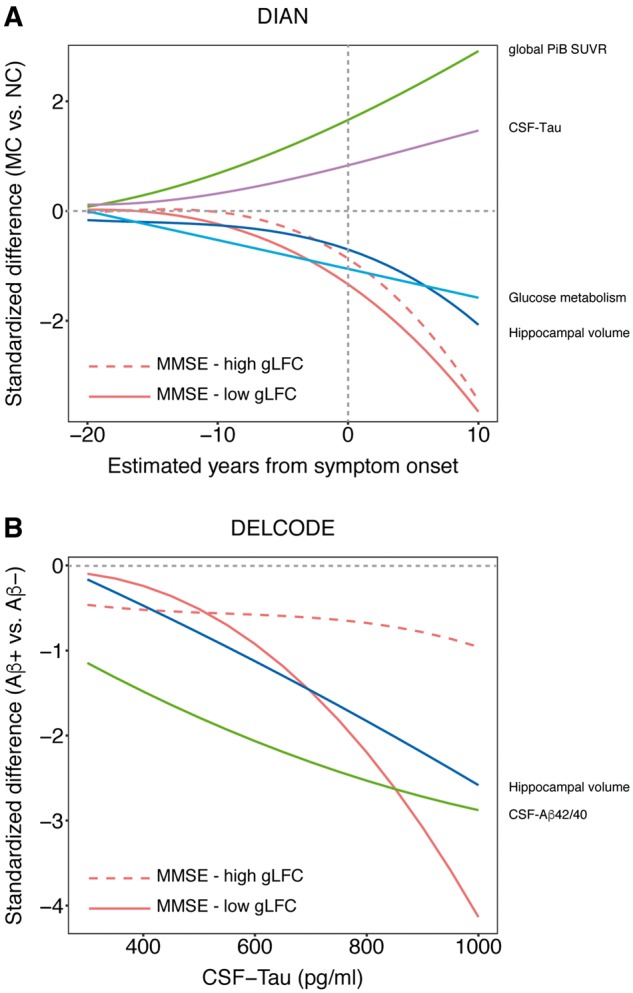
Cognitive and biomarker changes. Cognitive and biomarker changes as a function of Alzheimer’s disease severity. MMSE is plotted separately for individuals with high versus low gLFC-connectivity (as defined via median split). For the DIAN sample (A) we plotted the standardized difference between mutation carriers (MC) and non-mutation carriers (NC) against the EYO based on the polynomial linear mixed models that best fit each marker. The plot suggests that high gLFC-connectivity is associated with a delay of cognitive decline towards a later timepoint with more progressed levels of Alzheimer’s disease pathology. For the DELCODE sample (B) we plotted the standardized difference between CSF-amyloid-β+ and CSF-amyloid-β− subjects against CSF-tau levels. Congruent with the DIAN sample, the plot suggests that cognitive decline is shifted to a later time point in individuals with high levels of gLFC-connectivity.
DELCODE
Next, we aimed to generalize findings on gLFC-connectivity to elderly subjects with evidence for sporadic Alzheimer’s disease pathophysiology (i.e. CSF-amyloid-β+), hence we tested whether greater levels of gLFC-connectivity attenuated the effects of Alzheimer’s disease stage on the same cognitive measures (i.e. MMSE and logical memory delayed recall) in the DELCODE sample. Since EYO is not available in sporadic Alzheimer’s disease subjects, we used CSF total tau levels as a marker of disease severity that is closely linked to neurodegeneration and memory decline (Brier et al., 2016). The interaction gLFC-connectivity × CSF-tau was significant for both global cognition (MMSE: β/SE = 0.285/0.087, P = 0.002; Fig. 2C) and logical memory delayed free recall (β/SE = 0.336/0.126, P = 0.011; Fig. 2D) within the CSF-amyloid-β+ group. The full models including the interaction term showed a better model fit (i.e. Akaike information criterion) as compared to the null-model for MMSE (194.0 versus 199.2) and logical memory delayed free recall (212.1 versus 215.4). The scatterplots of the interaction effect (Fig. 2C and D) show that the effects of CSF-tau on global cognition (MMSE) and logical memory delayed free recall were attenuated at higher levels of gLFC-connectivity. In an exploratory analysis in the DELCODE cohort, we detected an interaction gLFC-connectivity × CSF-tau on ADAS-Cog word-list delayed recall (β/SE = 0.289/0.137, P = 0.039) that was congruent with our main analyses. When tested in the CSF-amyloid-β− control group, all abovementioned interaction models were non-significant. For the RFC, the interaction for gRFC-connectivity × CSF-tau the results were not significant, neither for logical memory delayed free recall (β/SE = 0.215/0.121, P = 0.078) nor MMSE (P = 0.47). As expected, no effects were detected for the occipital pole or M1. Detailed statistics of these control analyses are displayed for each region of interest in Supplementary Tables 3–5.
Table 3.
Summary of linear mixed effects models
| β (SE) | T-value | P-value | Overall R2 | |
|---|---|---|---|---|
| ADAD-MC | ||||
| MMSEa | ||||
| EYO × gLFC-connectivity | 0.269 (0.099) | 2.721 | 0.008c | 0.498 |
| EYO | −0.576 (0.106) | −5.359 | <0.001c | |
| gLFC-connectivity | 0.216 (0.094) | 2.292 | 0.025 | |
| LM delayed recalla | ||||
| EYO × gLFC-connectivity | 0.275 (0.106) | 2.585 | 0.012c | 0.445 |
| EYO | −0.458 (0.109) | −4.208 | <0.001c | |
| gLFC-connectivity | 0.047 (0.095) | 0.494 | 0.623 | |
| Sporadic AD−Aβ+ | ||||
| MMSEb | ||||
| CSF-tau × gLFC-connectivity | 0.285 (0.087) | 3.266 | 0.002c | 0.375 |
| CSF-tau | −0.553 (0.071) | −7.758 | <0.001c | |
| gLFC-connectivity | 0.159 (0.072) | 2.207 | 0.031 | |
| LM delayed recallb | ||||
| CSF-tau × gLFC-connectivity | 0.336 (0.126) | 2.672 | 0.011c | 0.292 |
| CSF-tau | −0.327 (0.111) | −2.925 | 0.005c | |
| gLFC-connectivity | −0.011 (0.112) | −0.099 | 0.922 | |
aModel controlled for gender, family ID (fixed effects) and site (random effect).
bModel controlled for age, gender (fixed effects) and site (random effect).
cP < 0.05, Bonferroni corrected.
Aβ = amyloid-β; AD = Alzheimer’s disease; LM = Logical memory; MC = mutation carrier; NC = non-mutation carrier.
Figure 3B illustrates the change in cognition and biomarkers across the spectrum of CSF-tau levels as predicted by polynomial mixed effects models. With increasing CSF-tau concentrations, there is a decrease in CSF-amyloid-β42/40 ratio and hippocampal volume indicating more progressed Alzheimer’s disease. Similar to the results in DIAN, the cross-sectionally estimated trajectory of the decline in MMSE is shifted to a later time point in individuals with high gLFC-connectivity when compared to individuals with low gLFC-connectivity, reflecting the significant interaction gLFC-connectivity × CSF-tau in the CSF-amyloid-β+ group.
Is global functional connectivity of the left frontal cortex altered in Alzheimer’s disease?
Next, we assessed whether gLFC-connectivity is reduced in subjects with Alzheimer’s disease pathology. For DIAN, an ANCOVA analysis showed that gLFC-connectivity was reduced in mutation carriers compared to non-mutation carriers (P = 0.0164), controlled for age, gender, education, and site. However, markers of disease severity including EYO, CSF-tau global PiB-PET uptake and hippocampal volume were not associated with gLFC-connectivity in mutation carriers, as tested by linear mixed effects analyses. These results suggest that gLFC-connectivity is relatively stable across the course of the disease. No associations between gLFC connectivity and any of the variables including EYO, CSF-tau, global PiB-PET uptake, and hippocampal volume were found in the non-mutation carriers. For DELCODE, no differences in gLFC-connectivity were detected between CSF-amyloid-β+ and CSF-amyloid-β- groups and no associations were found between gLFC-connectivity and age or disease severity as assessed by CSF-tau, CSF-amyloid-β42/40 ratio or hippocampal volume, neither in the CSF-amyloid-β+ nor CSF-amyloid-β– groups.
Associations between global functional connectivity of the left frontal cortex and education
We previously reported that education, a protective factor in ageing and neurodegenerative diseases that is often used as a surrogate marker of reserve, is associated with higher gLFC connectivity in prodromal Alzheimer’s disease (Franzmeier et al., 2017b, c). In general agreement with our previous findings, linear mixed models showed that more years of education were associated with greater gLFC-connectivity in ADAD mutation carriers at trend level significance (β/SE = 0.172/0.117, P = 0.072), and in the CSF-amyloid-β+ of the sporadic Alzheimer’s disease sample (β/SE = 0.248/0.114, P = 0.033).
Discussion
The main finding of the current cross-sectional study is that in participants with higher levels of gLFC-connectivity, the effect of Alzheimer’s disease pathology on cognition was attenuated compared to participants with lower levels of gLFC-connectivity. These results were consistent in patients with ADAD (DIAN) and sporadic Alzheimer’s disease (DELCODE), suggesting that higher gLFC-connectivity allows better maintenance of cognitive abilities during the progression of Alzheimer’s disease.
From the perspective of dynamic biomarker development, higher reserve should translate into a shift in the temporal trajectory of cognitive decrease during the progression of Alzheimer’s disease. Modelling the trajectory of cognitive differences in MMSE and delayed recall against increases in Alzheimer’s disease pathology (CSF-tau, EYO) showed for participants with higher gLFC-connectivity a shift of the cross-sectionally estimated trajectories of cognitive decline to the right, i.e. to more progressed levels of Alzheimer’s disease pathology. A strength of the DIAN study in ADAD is the presence of relatively ‘pure’ Alzheimer’s disease pathology that can be tracked well ahead of dementia onset. Still, the generalizability of findings to the more common sporadic form of Alzheimer’s disease is important. Using CSF-tau as a marker of disease severity that is closely linked to cognitive symptoms in CSF-amyloid-β+ subjects of the DELCODE study, we replicated the findings of a higher preservation of episodic memory and global cognition at more progressed levels of Alzheimer’s disease in participants with higher levels of gLFC-connectivity. This result is also consistent with our previous study in prodromal Alzheimer’s disease, where higher gLFC-connectivity was associated with attenuated impact of Alzheimer’s disease-related parietal glucose hypometabolism on memory (Franzmeier et al., 2017b). Thus, the detection of the association between gLFC-connectivity and reserve is not specific to ADAD.
Since in ADAD the pathological development shows a genetically-driven and stereotypical progression of Alzheimer’s disease pathology, the question of why some participants show higher or lower cognitive performance than expected based on the pathological level is particularly intriguing (Aguirre-Acevedo et al., 2016). In fact, the variability in cognitive decreases at any level of neurodegeneration in ageing and Alzheimer’s disease is not a mere random error of prediction based on pathology, but shows a systematic bias (Reed et al., 2010; Zahodne et al., 2013, 2015). Demographic factors such as education or premorbid verbal cognitive abilities are associated with higher than expected memory performance in ageing and Alzheimer’s disease (Rentz et al., 2010; Ewers et al., 2013; Soldan et al., 2013; Topiwala et al., 2015). However, such protective factors are global in nature and difficult to attribute to structural or functional brain changes (Solé-Padullés et al., 2009; Arenaza-Urquijo et al., 2013; Barulli and Stern, 2013). The current findings thus address the need for an identification of functional brain differences that underlie reserve, i.e. the ability to maintain cognition at a higher level in the presence of pathology.
Why is global connectivity of the LFC hub an important characteristic of brain function and, in particular, reserve? Global connectivity is a graph theoretical measure of the class centrality that captures the connectivity of a node in the whole connectome (Bullmore and Sporns, 2009). The LFC is among the top 5% of globally connected brain regions (Cole et al., 2010). Changes in hubs have particularly strong global impact on the brain compared to connectivity differences in other brain regions and are critical for the resilience of brain networks to targeted attack (Achard et al., 2006; Alstott et al., 2009; Santarnecchi et al., 2015). Reduction of hub connectivity has been associated with different neurodegenerative and psychiatric diseases (Buckner et al., 2009; Crossley et al., 2014), while increased hub connectivity is associated with better cognition (Cole et al., 2012; Liu et al., 2017). These results support the notion that connectivity of hubs is important for sustaining cognition when developing Alzheimer’s disease.
The LFC is a hub closely linked to the fronto-parietal control network. This functional network is particularly important in flexibly regulating the activity of different functional networks (Chen et al., 2013). Such a network regulation by the fronto-parietal control network is critical for successful cognitive task performance (Kragel and Polyn, 2015), particularly when other functional networks become impaired (Cole et al., 2014). Thus, as a hub of the fronto-parietal control network, the LFC may be of critical importance for the regulation of other networks to support successful cognition (Cole et al., 2015).
As shown by our control analyses, we could detect only a single significant interaction effect of RFC × EYO on MMSE, which did not survive Bonferroni correction. No effects for unimodal brain regions including M1 and the visual cortex were observed. This result pattern suggests that the findings are specific to the LFC, where predominantly the LFC rather than the RFC supports reserve. Our findings echo previous reports of resting state functional MRI assessed connectivity in a similar LFC area to be critical for supporting functional network resilience during simulated targeted attack of hub nodes (Santarnecchi et al., 2015). Together, these findings suggest that gLFC-connectivity is a feasible substrate of higher resilience of cognition in the face of neuropathology.
We found no decreases of gLFC-connectivity in sporadic Alzheimer’s disease and only a mild reduction in ADAD, suggesting the gLFC-connectivity is relatively stable in Alzheimer’s disease. Previous studies reported that brain hubs are in general implicated in neurodegenerative and psychiatric diseases (Crossley et al., 2014); however, the posterior and hippocampal hubs are selectivity impaired in Alzheimer’s disease (Yu et al., 2017), potentially due to their overlap with sites of early Alzheimer’s disease pathology (Buckner et al., 2009). This has been consistently shown in sporadic and autosomal dominantly inherited Alzheimer’s disease, where key hubs such as the posterior cingulate, precuneus or hippocampus show disruptions early in the disease course even prior to the onset of cognitive symptoms (Sorg et al., 2007; Mevel et al., 2011; Damoiseaux et al., 2012; Chhatwal et al., 2013). In contrast, we could show that connectivity of the LFC hub region remains relatively unchanged across different Alzheimer’s disease severity levels. Thus, the relatively spared LFC hub may be well posed to subserve the maintenance of cognition during the course of Alzheimer’s disease. At the same time, gLFC-connectivity showed at no stage of Alzheimer’s disease an increase, thus we found no indication of a temporary compensatory increase in response to pathology. Rather, differences in gLFC-connectivity may have existed before disease onset. In line with our previous study (Franzmeier et al., 2017c), we observed in the current study an association between more years of education and higher gLFC-connectivity, suggesting that gLFC-connectivity is possibly shaped by early life experiences, consistent with view of cognitive reserve (Stern, 2012). Thus, higher gLFC-connectivity is most likely a pre-morbid existing trait that enables to better cope with pathological changes during diseases such as Alzheimer’s disease.
For the interpretation of the current results, potential caveats should be taken into consideration. First, as a measure of hubness, we used global connectivity, which in graph theoretical terms is also called weighted degree centrality. It should be noted that alternative measures of centrality have been proposed such as the page-rank centrality and eigenvector centrality (Zuo et al., 2012). However, we chose the current global connectivity measure as it is the most widely used index of centrality applied in human brain connectomics (Buckner et al., 2009; Wang et al., 2010; Cole et al., 2012, 2015; Zuo et al., 2012; Santarnecchi et al., 2015; Franzmeier et al., 2017a, b). Thus, in order to facilitate comparability with previous findings, we focused on the current measure of global connectivity.
Second, the current study included mostly subjects within the range of asymptomatic or mild cognitive impairment of Alzheimer’s disease. Thus, a generalization to more severe dementia stages needs to be tested in future studies. It is conceivable that the influence of the LFC or any substrate of reserve wanes as the disease becomes more severe and network failure becomes more profound (Jones et al., 2016).
Third, in order to facilitate comparability across samples we focused our analyses on the MMSE and logical memory delayed free recall scores. A more comprehensive testing of episodic memory is desirable, but was not possible since the overlap of memory tests between the studies was limited. In DELCODE, the widely used verbal word list learning test as included in the ADAS-Cog battery was also available, for which we found the same result pattern as for the logical memory delayed free recall and MMSE. The current results are also consistent with our previous results on gLFC-connectivity on memory, where more comprehensive memory tests were used (Franzmeier et al., 2017b). Still, future studies may utilize a wider breadth of neuropsychological tests to capture global cognitive and memory abilities in a more comprehensive manner.
Fourth, we included only cross-sectional data rather than longitudinal assessment, thus, individual trajectories of cognitive changes could not be assessed. However, the trajectories assessed across the spectrum of Alzheimer’s disease severity at the cross-sectional level are highly consistent with the longitudinal trajectories of biomarker changes. Thus, especially in ADAD, the cross-sectionally estimated trajectories may be a proxy of the expected trajectories when assessed longitudinally, which awaits further validation once sufficient longitudinal data become available in studies such as DIAN or DELCODE.
Lastly, we assessed only Alzheimer’s disease among other neurodegenerative diseases. Resilience of cognition to pathologies has also been reported in fronto-temporal dementia, vascular dementia or multiple sclerosis (Zieren et al., 2013; Martins Da Silva et al., 2015; Premi et al., 2017). It is an open question whether gLFC-connectivity subserves reserve also in those disorders.
As an outlook, the current identification of the LFC hub may provide a suitable target for therapeutic intervention. Previous studies have shown that connectivity of frontal hubs can be enhanced by cognitive training (Takeuchi et al., 2017) and higher frontal connectivity can be induced by transcranial magnetic stimulation or transcranial direct-current stimulation (Chen et al., 2013; Kim et al., 2016). Enhancement of gLFC-connectivity by stimulation techniques, cognitive training, or physical exercise (Duzel et al., 2016) may provide an attractive non-invasive secondary prevention approach to sustain cognitive resilience in Alzheimer’s disease (Clark and Parasuraman, 2014).
Supplementary Material
Acknowledgements
We would like to thank all the researchers in the DIAN (www.dian-info.org/personnel.htm) and DELCODE study. We acknowledge the altruism of the DIAN and DELCODE participants and their families.
Funding
The study was funded by an ERC career integration grant (PCIG12-GA-2012-334259 to M. E.), LMUexcellent (to M.E.) and Alzheimer Forschung Initiative (to M.E.), and the the National Institute for Health Research University College London Hospitals Biomedical Research Centre and the MRC Dementias Platform UK (MR/L023784/1 and MR/009076/1 to M.R.). Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer’s Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA) and the German Center for Neurodegenerative Diseases (DZNE, to J.L. and M.J.). The DELCODE study was funded by the German Center for Neurodegenerative Diseases (DZNE), Study-ID: BN012DZNE.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- ADAD
autosomal dominantly inherited Alzheimer’s disease
- DELCODE
German Center for Neurodegenerative Diseases longitudinal study on cognition and dementia
- DIAN
Dominantly Inherited Alzheimer Network
- EYO
estimated years from symptom onset
- gLFC-connectivity
global functional connectivity of the left frontal cortex
- LFC
left frontal cortex
- MMSE
Mini-Mental-State Examination
- RFC
right frontal cortex
References
- Achard S, Salvador R, Whitcher B, Suckling J, Bullmore E. A resilient, low-frequency, small-world human brain functional network with highly connected association cortical hubs. J Neurosci 2006; 26: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Acevedo DC, Lopera F, Henao E, Tirado V, Munoz C, Giraldo M, et al. Cognitive decline in a Colombian Kindred with autosomal dominant Alzheimer disease: a retrospective cohort study. JAMA Neurol 2016; 73: 431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alstott J, Breakspear M, Hagmann P, Cammoun L, Sporns O. Modeling the impact of lesions in the human brain. PLoS Comput Biol 2009; 5: e1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenaza-Urquijo EM, Molinuevo JL, Sala-Llonch R, Sole-Padulles C, Balasa M, Bosch B, et al. Cognitive reserve proxies relate to gray matter loss in cognitively healthy elderly with abnormal cerebrospinal fluid amyloid-beta levels. J Alzheimers Dis 2013; 35: 715–26. [DOI] [PubMed] [Google Scholar]
- Ashburner J. A fast diffeomorphic image registration algorithm. NeuroImage 2007; 38: 95–113. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. Unified segmentation. NeuroImage 2005; 26: 839–51. [DOI] [PubMed] [Google Scholar]
- Barulli D, Stern Y. Efficiency, capacity, compensation, maintenance, plasticity: emerging concepts in cognitive reserve. Trends Cogn Sci 2013; 17: 502–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012; 367: 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger TL, Blazey T, Jack CR Jr, Koeppe RA, Su Y, Xiong C, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci USA 2013; 110: E4502–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berres M, Monsch AU, Bernasconi F, Thalmann B, Stahelin HB. Normal ranges of neuropsychological tests for the diagnosis of Alzheimer's disease. Stud Health Technol Inform 2000; 77: 195–9. [PubMed] [Google Scholar]
- Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med 2016; 8: 338ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci 2009; 29: 1860–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullmore E, Sporns O. Complex brain networks: graph theoretical analysis of structural and functional systems. Nat Rev Neurosci 2009; 10: 186–98. [DOI] [PubMed] [Google Scholar]
- Chen AC, Oathes DJ, Chang C, Bradley T, Zhou ZW, Williams LM, et al. Causal interactions between fronto-parietal central executive and default-mode networks in humans. Proc Natl Acad Sci USA 2013; 110: 19944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Schultz AP, Johnson K, Benzinger TL, Jack C Jr, Ances BM, et al. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 2013; 81: 736–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark VP, Parasuraman R. Neuroenhancement: enhancing brain and mind in health and in disease. Neuroimage 2014; 85 (Pt 3): 889–94. [DOI] [PubMed] [Google Scholar]
- Cole MW, Ito T, Braver TS. Lateral prefrontal cortex contributes to fluid intelligence through multinetwork connectivity. Brain Connect 2015; 5: 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MW, Pathak S, Schneider W. Identifying the brain's most globally connected regions. Neuroimage 2010; 49: 3132–48. [DOI] [PubMed] [Google Scholar]
- Cole MW, Repovs G, Anticevic A. The frontoparietal control system: a central role in mental health. Neuroscientist 2014; 20: 652–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MW, Reynolds JR, Power JD, Repovs G, Anticevic A, Braver TS. Multi-task connectivity reveals flexible hubs for adaptive task control. Nat Neurosci 2013; 16: 1348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MW, Yarkoni T, Repovs G, Anticevic A, Braver TS. Global connectivity of prefrontal cortex predicts cognitive control and intelligence. J Neurosci 2012; 32: 8988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley NA, Mechelli A, Scott J, Carletti F, Fox PT, McGuire P, et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain 2014; 137 (Pt 8): 2382–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoiseaux JS, Prater KE, Miller BL, Greicius MD. Functional connectivity tracks clinical deterioration in Alzheimer's disease. Neurobiol Aging 2012; 33: 828.e19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duzel E, van Praag H, Sendtner M. Can physical exercise in old age improve memory and hippocampal function? Brain 2016; 139 (Pt 3): 662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M, Insel PS, Stern Y, Weiner MW; Alzheimer's Disease Neuroimaging Initiative (ADNI). Cognitive reserve associated with FDG-PET in preclinical Alzheimer disease. Neurology 2013; 80: 1194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–98. [DOI] [PubMed] [Google Scholar]
- Franzmeier N, Caballero MÁA, Taylor ANW, Simon-Vermot L, Buerger K, Ertl-Wagner B, et al. Resting-state global functional connectivity as a biomarker of cognitive reserve in mild cognitive impairment. Brain Imaging Behav 2017a; 11: 368–82. [DOI] [PubMed] [Google Scholar]
- Franzmeier N, Duering M, Weiner M, Dichgans M, Ewers M; Alzheimer's Disease Neuroimaging Initiative (ADNI). Left frontal cortex connectivity underlies cognitive reserve in prodromal Alzheimer disease. Neurology 2017b; 88: 1054–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmeier N, Göttler J, Grimmer T, Drzezga A, Áraque-Caballero MA, Simon-Vermot L, et al. Resting-state connectivity of the left frontal cortex to the default mode and dorsal attention network supports reserve in mild cognitive impairment. Front Aging Neurosci 2017c; 9: 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Lowe V, et al. Shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch Neurol 2012; 69: 856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze S, Zetterberg H, Mattsson N, Palmqvist S, Vanderstichele H, Lindberg O, et al. CSF Abeta42/Abeta40 and Abeta42/Abeta38 ratios: better diagnostic markers of Alzheimer disease. Ann Clin Transl Neurol 2016; 3: 154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Knopman DS, Gunter JL, Graff-Radford J, Vemuri P, Boeve BF, et al. Cascading network failure across the Alzheimer's disease spectrum. Brain 2016; 139 (Pt 2): 547–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent P. The evolution of the Wechsler Memory Scale: a selective review. Appl Neuropsychol Adult 2013; 20: 277–91. [DOI] [PubMed] [Google Scholar]
- Kim K, Ekstrom AD, Tandon N. A network approach for modulating memory processes via direct and indirect brain stimulation: toward a causal approach for the neural basis of memory. Neurobiol Learn Mem 2016; 134 (Pt A): 162–77. [DOI] [PubMed] [Google Scholar]
- Kragel JE, Polyn SM. Functional interactions between large-scale networks during memory search. Cereb Cortex 2015; 25: 667–79. [DOI] [PubMed] [Google Scholar]
- Lim YY, Hassenstab J, Cruchaga C, Goate A, Fagan AM, Benzinger TL, et al. BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer's disease. Brain 2016; 139 (Pt 10): 2766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xia M, Dai Z, Wang X, Liao X, Bi Y, et al. Intrinsic brain hub connectivity underlies individual differences in spatial working memory. Cereb Cortex 2017; 27: 5496–508. [DOI] [PubMed] [Google Scholar]
- Mak HK, Zhang Z, Yau KK, Zhang L, Chan Q, Chu LW. Efficacy of voxel-based morphometry with DARTEL and standard registration as imaging biomarkers in Alzheimer's disease patients and cognitively normal older adults at 3.0 Tesla MR imaging. J Alzheimers Dis 2011; 23: 655–64. [DOI] [PubMed] [Google Scholar]
- Martins Da Silva A, Cavaco S, Moreira I, Bettencourt A, Santos E, Pinto C, et al. Cognitive reserve in multiple sclerosis: protective effects of education. Mult Scler 2015; 21: 1312–21. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medaglia JD, Pasqualetti F, Hamilton RH, Thompson-Schill SL, Bassett DS. Brain and cognitive reserve: translation via network control theory. Neurosci Biobehav Rev 2017; 75: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, D'Arcy C. Education and dementia in the context of the cognitive reserve hypothesis: a systematic review with meta-analyses and qualitative analyses. PLoS One 2012; 7: e38268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevel K, Chetelat G, Eustache F, Desgranges B. The default mode network in healthy aging and Alzheimer's disease. Int J Alzheimers Dis 2011; 2011: 535816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology 1989; 39: 1159–65. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Snider BJ, Mills SL, Buckles VD, Santacruz AM, Bateman RJ, et al. Dominantly inherited Alzheimer network: facilitating research and clinical trials. Alzheimers Res Ther 2013; 5: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Malek-Ahmadi M, Snyder N, Ausdemore J, Chen K, Perez SE. Braak stage and trajectory of cognitive decline in noncognitively impaired elders. Neurobiol Aging 2016; 43: 101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo OC, Schultz SA, Oh JM, Larson J, Edwards D, Cook D, et al. Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology 2014; 83: 1753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DC, Reuter-Lorenz P. The adaptive brain: aging and neurocognitive scaffolding. Annu Rev Psychol 2009; 60: 173–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perneczky R, Wagenpfeil S, Lunetta KL, Cupples LA, Green RC, Decarli C, et al. Head circumference, atrophy, and cognition: implications for brain reserve in Alzheimer disease. Neurology 2010; 75: 137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Caracciolo B, Brayne C, Gauthier S, Jelic V, Fratiglioni L. Mild cognitive impairment: a concept in evolution. J Intl Med 2014; 275: 214–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Jack CR Jr, Xu YC, Waring SC, O'Brien PC, Smith GE, et al. Memory and MRI-based hippocampal volumes in aging and AD. Neurology 2000; 54: 581–7. [DOI] [PubMed] [Google Scholar]
- Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. NeuroImage 2012; 59: 2142–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Mitra A, Laumann TO, Snyder AZ, Schlaggar BL, Petersen SE. Methods to detect, characterize, and remove motion artifact in resting state fMRI. NeuroImage 2014; 84: 320–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premi E, Grassi M, van Swieten J, Galimberti D, Graff C, Masellis M, et al. Cognitive reserve and TMEM106B genotype modulate brain damage in presymptomatic frontotemporal dementia: a GENFI study. Brain 2017; 140: 1784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed BR, Mungas D, Farias ST, Harvey D, Beckett L, Widaman K, et al. Measuring cognitive reserve based on the decomposition of episodic memory variance. Brain 2010; 133 (Pt 8): 2196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitan RM. Validity of the Trail Making Test as an Indicator of organic brain damage. Percept Mot Skills 1958; 8: 271–76. [Google Scholar]
- Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 2010; 67: 353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarnecchi E, Rossi S, Rossi A. The smarter, the stronger: intelligence level correlates with brain resilience to systematic insults. Cortex 2015; 64: 293–309. [DOI] [PubMed] [Google Scholar]
- Soldan A, Pettigrew C, Li S, Wang MC, Moghekar A, Selnes OA, et al. Relationship of cognitive reserve and cerebrospinal fluid biomarkers to the emergence of clinical symptoms in preclinical Alzheimer's disease. Neurobiol Aging 2013; 34: 2827–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé-Padullés C, Bartrés-Faz D, Junqué C, Vendrell P, Rami L, Clemente IC, et al. Brain structure and function related to cognitive reserve variables in normal aging, mild cognitive impairment and Alzheimer's disease. Neurobiol Aging 2009; 30: 1114–24. [DOI] [PubMed] [Google Scholar]
- Sorg C, Riedl V, Muhlau M, Calhoun VD, Eichele T, Laer L, et al. Selective changes of resting-state networks in individuals at risk for Alzheimer's disease. Proc Natl Acad Sci USA 2007; 104: 18760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol 2012; 11: 1006–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med 2016; 8: 369ra178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumowski JF, Rocca MA, Leavitt VM, Dackovic J, Mesaros S, Drulovic J, et al. Brain reserve and cognitive reserve protect against cognitive decline over 4.5 years in MS. Neurology 2014; 82: 1776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Taki Y, Nouchi R, Sekiguchi A, Kotozaki Y, Nakagawa S, et al. Neural plasticity in amplitude of low frequency fluctuation, cortical hub construction, regional homogeneity resulting from working memory training. Sci Rep 2017; 7: 1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. Available from: http://www.R-project.org/ (23 January 2018, date last accessed). [Google Scholar]
- Tolppanen AM, Solomon A, Kulmala J, Kareholt I, Ngandu T, Rusanen M, et al. Leisure-time physical activity from mid- to late life, body mass index, and risk of dementia. Alzheimers Dement 2015; 11: 434–43.e6. [DOI] [PubMed] [Google Scholar]
- Topiwala A, Allan CL, Valkanova V, Zsoldos E, Filippini N, Sexton CE, et al. Resilience and MRI correlates of cognitive impairment in community-dwelling elders. Br J Psychiatry 2015; 207: 435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela MJ, Sachdev P. Brain reserve and dementia: a systematic review. Psychol Med 2006; 36: 441–54. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Weigand SD, Przybelski SA, Knopman DS, Smith GE, Trojanowski JQ, et al. Cognitive reserve and Alzheimer's disease biomarkers are independent determinants of cognition. Brain 2011; 134 (Pt 5): 1479–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zuo X, He Y. Graph-based network analysis of resting-state functional MRI. Front Syst Neurosci 2010; 4: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Engels MM, Hillebrand A, van Straaten EC, Gouw AA, Teunissen C, et al. Selective impairment of hippocampus and posterior hub areas in Alzheimer's disease: an MEG-based multiplex network study. Brain 2017; 140: 1466–85. [DOI] [PubMed] [Google Scholar]
- Zahodne LB, Manly JJ, Brickman AM, Narkhede A, Griffith EY, Guzman VA, et al. Is residual memory variance a valid method for quantifying cognitive reserve? A longitudinal application. Neuropsychologia 2015; 77: 260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahodne LB, Manly JJ, Brickman AM, Siedlecki KL, Decarli C, Stern Y. Quantifying cognitive reserve in older adults by decomposing episodic memory variance: replication and extension. J Int Neuropsychol Soc 2013; 19: 854–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieren N, Duering M, Peters N, Reyes S, Jouvent E, Herve D, et al. Education modifies the relation of vascular pathology to cognitive function: cognitive reserve in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Neurobiol Aging 2013; 34: 400–7. [DOI] [PubMed] [Google Scholar]
- Zuo XN, Ehmke R, Mennes M, Imperati D, Castellanos FX, Sporns O, et al. Network centrality in the human functional connectome. Cereb Cortex 2012; 22: 1862–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.