

Abstract
Tissue infiltration by circulating monocytes is a critical step in the initiation and augmentation of type 2 inflammatory responses in the lungs. Our studies demonstrate that IL-33−/− mice have a defect in monocyte extravasation from the vasculature to the lung interstitium during induction of type 2 inflammatory responses. This result suggests that monocyte migration to the lungs is IL-33 dependent, and we found that administration of exogenous recombinant IL-33 is sufficient to restore monocyte localization to the lung interstitium. Further investigation of the effect of early administration of recombinant IL-33 on the lungs identified upregulation of multiple chemokines including the monocyte chemoattractants CCL2, CCL7, and CCL22. Importantly, blockade of G-protein coupled receptor–dependent signaling, and thereby chemokine receptor activity, inhibited IL-33–driven monocyte recruitment. CCR2 deficiency prevented recruitment of monocytes to the lung extravascular space during allergic sensitization, and resulted in reduced eosinophilia after allergen challenge. Thus, IL-33 plays a critical role in the initiation of type 2 inflammatory responses by inducing upregulation of chemokines that promote monocyte recruitment to the lung interstitium.
INTRODUCTION
Asthma is a chronic disease of the airways that is most commonly characterized by a type 2 inflammatory response (1–3). Multiple immune cell populations contribute to this response, but many unanswered questions remain about the role of APCs during allergic airway responses. In particular, there has been greater interest in understanding the role monocytes play during allergic airway inflammation. Studies in which circulating monocytes were depleted demonstrated decreased allergic lung inflammation, suggesting that monocytes are important for initiating allergic lung responses (4–6).
Murine monocytes predominantly circulate through the vasculature at steady state and can be traditionally classified into two groups based on Ly6C expression. Ly6C− monocytes have been associated with patrolling blood vessel walls and playing an anti-inflammatory role during tissue injury (7, 8). In contrast, Ly6C+ monocytes are thought to rapidly infiltrate inflamed tissues where they can then differentiate into CD11b+ monocyte-derived dendritic cells (moDCs) or monocyte-derived macrophages (9). Other studies have also shown that Ly6Chi monocytes can act as precursors to Ly6Clo monocytes (10). Several investigations have elucidated key chemokines that promote monocyte trafficking. Chemokine receptors are differentially expressed between Ly6C− monocytes and Ly6C+ monocytes (11). Ly6C− monocytes express high levels of CX3C-chemokine receptor 1 (CX3CR1), which binds to CX3CL1 (12). This ligand was found to be upregulated in human endothelial cells through a CD40-TNF receptor associated factor pathway (13). In contrast, Ly6C+ monocytes have low expression of CX3CR1 but high expression of CCR2. CCL2 and CCL7 have been shown to bind to CCR2 and promote monocyte recruitment (14). Upregulation of these ligands can be found in various murine models including bacterial infections, peritonitis, autoimmune encephalitis, atherosclerosis, and asthma (15, 16). Although it has been shown that monocytes accumulate in response to upregulation of these chemokines, it is still unclear how early signals in allergic sensitization upregulate these monocyte chemoattractants in the lungs.
Early signals implicated in initiating type 2 lung inflammation include IL-33, which acts upon a variety of cell populations including both non-hematopoietic cells and hematopoietic cells. In response to IL-33, innate lymphoid cells type 2, mast cells, basophils, and type 2 Th cells promote type 2 inflammation (17). We previously demonstrated that immune complex (IC)–mediated and house dust mite–mediated type 2 lung inflammation was dependent on the IL-33/ST2 pathway (18, 19). Thus, our objective in this study was to investigate mechanisms by which IL-33 produced early after sensitization promotes allergic lung inflammation.
MATERIALS AND METHODS
Mice
C57BL/6 wild-type (WT) mice were purchased from Harlan Laboratories. FcγRIII−/− mice and CCR2−/− mice were purchased from the Jackson Laboratory. IL-33−/− mice were a gift from S. Nakae (University of Tokyo, Tokyo, Japan) (20). Animals were housed in a specific pathogen-free facility maintained by the University of Chicago Animal Resources Center. The studies conformed to the principles set forth by the Animal Welfare Act and the National Institutes of Health guidelines for the care and use of animals in biomedical research.
Allergic sensitization and challenge in a mouse model
Grade V chicken egg OVA (A5503; Sigma-Aldrich), rabbit anti–chicken egg OVA IgG (α-OVA) (C6534; Sigma-Aldrich), recombinant mouse IL-33 (BioLegend), and pertussis toxin (PTX) (516560; EMD Millipore) were used in murine experiments. Mice were administered α-OVA serum i.v. on day 0, followed by a fluorescently labeled OVA (OVA-FITC or OVA-AF647; Invitrogen) sensitization intratracheally (i.t.) 24 h later. At the time of sacrifice, the mice received an i.v. injection of anti-CD45 (30-F11; BioLegend). After 5 min, the lungs were perfused and harvested to allow for identification of cell localization in the vasculature (i.v. CD45+) or lung interstitium (i.v. CD45−). Lungs were processed into single-cell suspensions as previously described for staining and analysis by flow cytometry (18, 19). For some experiments, mice were further challenged on days 8, 9, and 10, sacrificed on day 11, and bronchoalveolar lavage (BAL) was analyzed for cellular composition.
Flow cytometric analysis
For flow cytometric analysis, 1 × 106 cells were resuspended in 100 μl of flow cytometric buffer (PBS containing 0.1% sodium azide and 1% BSA). Cells were blocked with 20 μl of anti-CD16/32 (2.4G2) supernatant and stained with fluorescently conjugated Abs specific for mouse CD11b (M1/70), CD11c (N418), CD103 (2E7), F4/80 (BM8), I-A/I-E(M5/114.15.2), Ly-6C(HK1.4)(BioLegend); orSiglec-F (E50-2440) (BD Pharmingen). Ly6C− monocytes were gated as SSCloCD11clo/−MHC class II (MHC II)lo/−CD11b+F4/80+Ly6C−; Ly6C+ monocytes were SSCloCD11clo/−MHC IIlo/−CD11b+F4/80+Ly6C+; CD11b+ moDCs were CD11c+MHC IIhiCD103−CD11b+Ly6C+; CD11b+ conventional dendritic cells were CD11c+MHC IIhiCD103−CD11b+Ly6C−; and CD103+ conventional dendritic cells were CD11c+MHC IIhiCD103+CD11b−. Flow cytometric analysis was performed on a BD LSR Fortessa (BD Biosciences), and the data were analyzed with FlowJo software (Tree Star).
Quantitative PCR array and analysis
IL-33−/− mice were instilled i.t. with PBS (n = 4) or 2.5 μg of recombinant IL-33 (rIL-33) (n = 5) in a total of 50 μl PBS. The lungs were harvested 6 h later, minced, and homogenized in RLT buffer using a Qiagen TissueRuptor. RNA was purified from each mouse lung homogenate (Qiagen RNeasy Plus Mini Kit) and cDNA was prepared for each sample (Qiagen RT2 First Strand Kit). RT2 Profiler PCR Array Mouse Chemokines and Receptors kit (Qiagen) was used on pooled samples for quantitative PCR (qPCR) array, which was performed on a CFX96 Real Time System (Bio-Rad). Array data were analyzed using the data analysis web portal at http://www.qiagen.com/geneglobe. Cycle threshold (CT) values were normalized to the housekeeping genes Gusb and Hsp90ab1, after which ΔΔCT calculations were performed and transcript fold change over PBS controls was determined using the 2−ΔΔCT formula. Results for Ccl2, Ccl7, and Ccl22 were confirmed by individual qPCRs performed on cDNA from individual mice. PCR primers were as follows: HPRT, forward 5′-TGATCAGT-CAACGGGGGACA-3′, reverse 5′-TTCGAGAGGTCCTTTTCA-CCA-3′; CCL2, forward 5′-GGCCTGCTGTTCACAGTTGC-3′, reverse 5′-CCTGCTGCTGGTGATCCTCT-3′; CCL7, forward 5′-TGTGCCTGCTGCTCATAGCC-3′, reverse 5′-ACATAGCAGCATGTGGATGCATTG-3′; CCL22, forward 5′-CGCAAGCCTG-GCGTTGTTT-3′, reverse 5′-CCTCCCTGGACCACACCAGA-3′.
Statistical analysis
GraphPad Prism software was used to perform statistical analyses, and a p value < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, #p < 10−10; ns, not significant). An unpaired Student two-tailed t test was used to analyze experiments with two groups. Error bars represent the SEM.
Study approval
The University of Chicago Animal Resources Center approved all animal procedures.
RESULTS
Localization of monocytes to the lung interstitium after allergen challenge is IL-33 dependent
Monocytes are primarily present in the vasculature but are known to extravasate into tissues at the earliest stages of inflammation (21). Our previous study had found that IC formation in vivo promoted type 2 inflammatory responses in the lung by up-regulating IL-33 as early as 3 h after administering allergen (18, 19). To examine the influence of IL-33 in the early stages of this model, we have addressed whether OVA-IC–induced IL-33 affects the early migration of APCs into the lung tissue. To induce IC formation in vivo, mice are injected with OVA-specific serum on day 0, followed by an OVA i.t. sensitization on day 1 (19). Utilizing fluorescently labeled OVA and intravascular staining of CD45 (22), we identify APCs that take up Ag and whether they migrate into the extravascular space.
As IL-33 upregulation is seen as early as 3 h after administering allergen (18, 19), we investigated the early effects OVA-ICs had on monocytes at 30 min, 1 h, and 3 h postsensitization. As expected, the Ly6C− and Ly6C+ monocytes were primarily localized in the vasculature (Fig. 1). Although monocytes took up minimal amounts of OVA alone, Ag uptake was significantly enhanced in the presence of OVA-IC (Fig. 1, Supplemental Fig. 1). Furthermore, OVA-ICs drove Ly6C+ monocytes to localize to the lung interstitium as early as 3 h after sensitization (Fig. 1B) and continued through to 18 h (Fig. 2A). These findings demonstrate that OVA-IC enhanced Ag uptake by Ly6C− and Ly6C+ monocytes, and promoted Ly6C+ monocyte localization into the lung interstitium.
FIGURE 1. OVA-ICs enhance monocyte uptake of Ag and promote Ly6C+ monocyte accumulation in the lung extravascular space within 3 h.

WT mice received α-OVA (OVA-IC) or IgG-depleted serum (OVA) i.v. followed by a fluorescently labeled OVA (OVA-FITC) sensitization i.t. 24 h later. Mice were sacrificed at 30 min, 1 h, and 3 h after sensitization. Representative flow plots of Ag uptake (OVA-FITC) versus localization (i.v. CD45) for (A) Ly6C− monocytes and (B) Ly6C+ monocytes. The data are from two independent experiments with at least six mice analyzed per group.
FIGURE 2. Monocyte migration from the vasculature to the interstitium is reduced in IL-33−/− mice.
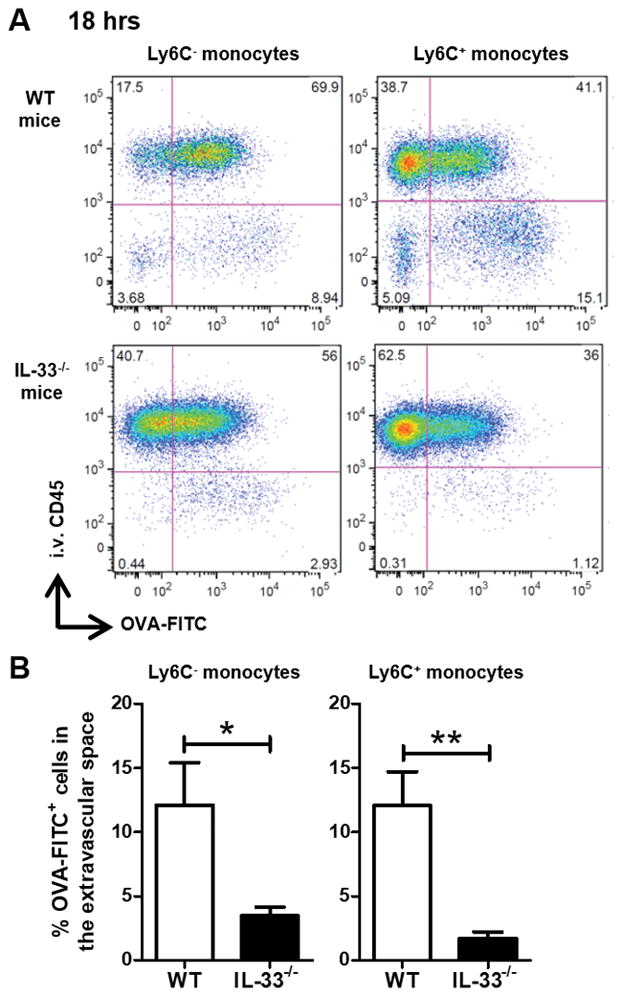
WT and IL-33−/− mice received α-OVA serum i.v. followed by a fluorescently labeled OVA (OVA-FITC) sensitization i.t. 24 h later. The mice were sacrificed 18 h after sensitization. Minutes prior to sacrifice, the mice received an i.v. injection of anti-CD45 to allow for identification of cell localization in the vasculature (i.v. CD45+) or extravascular space (i.v. CD45−). (A) Representative flow plots of Ag uptake (OVA-FITC) versus localization (i.v. CD45) for Ly6C− and Ly6C+ monocytes. (B) Percentage of OVA-FITC+ cells in the extravascular space is plotted. Data represent the mean ± SEM, and the combined data from at least two independent experiments with a total of at least six mice analyzed per group. *p < 0.05, **p < 0.01.
To determine if there is a role for IL-33 in the monocyte migration in the OVA-IC model, we sensitized WT and IL-33−/− mice. No significant difference was noted in the frequency of Ly6C− and Ly6C+ monocytes at baseline between WT and IL-33−/− mice (data not shown). However, after challenge, IL-33−/− mice displayed a significant decrease in the percentage of Ag-positive Ly6C− and Ly6C+ monocytes localized to the lung interstitium (Fig. 2). Given that our previous work found IL-33 upregulation to be downstream of FcγRIII, we analyzed whether a similar defect was present in FcγRIII−/− mice. Compared to WT mice, FcγRIII−/− mice had a significant decrease in the presence of Ag-positive monocytes in the lung interstitium (Supplemental Fig. 2). These results demonstrated that during early sensitization, recruitment and localization of monocytes to the lung interstitium was IL-33 and FcγRIII dependent. Taken together with our previous investigations (18, 19), these data suggest that augmentation of type 2 inflammatory responses through IL-33 upregulation may be driven by the early accumulation of monocytes.
CCR2-deficient mice have reduced eosinophilia after allergen sensitization and challenge
CCR2 is an important chemokine receptor for monocyte egress from the bone marrow (11). Thus, CCR2−/− mice have a severe reduction in the number of circulating monocytes and are widely used as monocyte-deficient mice. We hypothesized that monocyte accumulation in the lung extravascular space during sensitization was required for inflammation during allergen challenge. To evaluate monocyte responses to allergen sensitization in these mice, WT or CCR2−/− mice were administered OVA-specific serum on day 0, followed by an OVA i.t. sensitization on day 1. As above, Ag uptake and localization of the monocytes were evaluated 18 h after OVA sensitization. We were surprised to find that an almost significant proportion of Ly6C+ monocytes took up Ag and were recruited to the lung extravascular space even in the absence of CCR2 (Fig. 3A). However, as expected, the total number of Ag-positive Ly6C+ monocytes accumulated in the lung interstitium in the CCR2-deficient mice compared with in WT mice was greatly reduced (Fig. 3A).
FIGURE 3. CCR2 is partially responsible for regulating Ly6C+ monocyte migration from the vasculature to the interstitium after OVA-IC sensitization and lung eosinophilia after allergen challenge.

α-OVA serum (OVA-IC) was administered i.v. to naive mice, whereas control mice received either (A) PBS or (B) α-OVAdepl. The next day the mice were challenged i.t. with (A) OVA-FITC or (B) OVA. (A) After 18 h the mice were sacrificed. Minutes prior to sacrifice, the mice received an i.v. injection of anti-CD45 to allow for discrimination between cells in the vasculature and interstitium. Percentage and number of monocytes in the extravascular space are plotted. (B) On days 1, 8, 9, and 10 the mice were challenged i.t. with OVA. On day 11 the mice were sacrificed. Airway inflammation was assessed by determining the number of eosinophils and CD4+ T cells in the BAL by flow cytometry. The data represent the combined data from at least two independent experiments with a total of at least three mice analyzed per group. Data represent the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns, not significant.
To address whether the reduction in monocyte recruitment in the CCR2−/− mice affected IC-induced allergic inflammation, mice were sensitized as described above but were additionally challenged with OVA i.t. on days 8, 9, and 10. On day 11, type 2 airway inflammation was assessed by measuring the numbers of eosinophils and CD4+ T cells in the BAL. Although CCR2-sufficient and CCR2-deficient mice developed equal degrees of BAL CD4+ T cell infiltration, CCR2-deficient mice had reduced eosinophilia compared with their CCR2-sufficient counterparts (Fig. 3B). Importantly, others have reported the same defect in eosinophilia in a house dust mite model of allergic inflammation (23). Thus, monocyte accumulation in the lung interstitium after sensitization is required for optimal eosinophilia after challenge.
Administration of exogenous rIL-33 restores Ly6C+ monocyte localization to the lung interstitium
We had previously demonstrated that administration of exogenous rIL-33 during sensitization was sufficient to restore allergic lung inflammation in FcRγ−/− mice (18). Because both monocyte recruitment to the lungs and IL-33 upregulation were early events in the development of type 2 inflammatory responses in the lungs, we sought to determine if exogenous IL-33 was sufficient to restore lung interstitial localization of Ag-positive monocytes. Mice were sensitized with OVA-specific serum on day 0, and on day 1 mice received OVA-FITC with or without rIL-33. Notably, administration of rIL-33 to the IL-33−/− mice restored lung interstitial localization of Ag-positive Ly6C+ monocytes (Fig. 4A). There was a trending increase in the Ly6C− monocytes, but it was not quite significant (Fig. 4A).
FIGURE 4. Administration of exogenous rIL-33 restores Ly6C+ monocyte localization to the extravascular space in response to OVA-IC and is sufficient to promote monocyte extravasation without Ag.

(A) IL-33−/− mice received α-OVA i.v. followed by a fluorescently labeled OVA (OVA-FITC) sensitization with or without rIL-33 24 h later. Percentage of OVA-FITC+ monocytes in the extravascular space is plotted. (B) In the absence of any Ag or antisera, IL-33−/− mice received i.t. PBS or rIL-33. The mice were sacrificed 18 h after challenge. Example flow plots of F4/80+ monocytes (SSCloCD11clo/−MHC IIlo/−CD11b+). Percentage of monocytes in the extravascular space is plotted. Data represent the mean ± SEM combined from at least two independent experiments with a total of at least six mice analyzed per group. *p < 0.05, **p < 0.01.
To determine whether IL-33 alone is sufficient to drive monocytes into the lung interstitium in the absence of Ag, PBS or rIL-33 alone was i.t. administered to IL-33−/− mice. The proportion of Ly6C+ monocytes in the lung interstitium was significantly higher in the mice that had received rIL-33 compared with the PBS-only controls (Fig. 4B). This effect was limited to Ly6C+ monocytes as the localization of Ly6C− monocytes was unaltered by rIL-33 (Fig. 4B). Thus, exogenous rIL-33 was sufficient to promote Ly6C+ monocyte accumulation independent of allergen.
IL-33 promotes the expression of monocyte chemoattractants in the lungs
IL-33 has previously been found to induce production of cytokines and chemokines in a variety of disease states, including asthma (24, 25). As monocyte recruitment is sensitive to many chemokines, and because we showed that some monocyte recruitment occurs even in the absence of CCR2, we hypothesized that IL-33 promoted the upregulation of additional monocyte chemoattractants in the lungs. To determine whether IL-33 modulates chemokine expression, PBS or rIL-33 was administered to IL-33−/− mice, and the lungs were harvested 6 h later for cDNA preparation. A qPCR array was performed to analyze the differential expression of 84 chemokines and their receptors. Notably, the mice that had received rIL-33 displayed increased expression of multiple monocyte chemoattractants such as CCL2, CCL4, CCL7, CCL8, CCL22, and CXCL10 (Fig. 5, Supplemental Table I). In particular, CCR2 is well known for its role in promoting recruitment of circulating monocytes to the lung (11). In addition, IL-33 promoted expression of several other chemokines including CCL11 and CCL24, which attract eosinophils expressing CCR3; CCL17, which recruits CCR4-expressing cells such as T cells; and CCL19, which chemo-attracts dendritic cells, B cells, and T cells expressing the receptor CCR7. Thus, the introduction of IL-33 into the lungs altered the chemokine milieu to promote the recruitment of several inflammatory populations including monocytes.
FIGURE 5. IL-33 upregulates chemokines in the lungs, including monocyte chemoattractants.
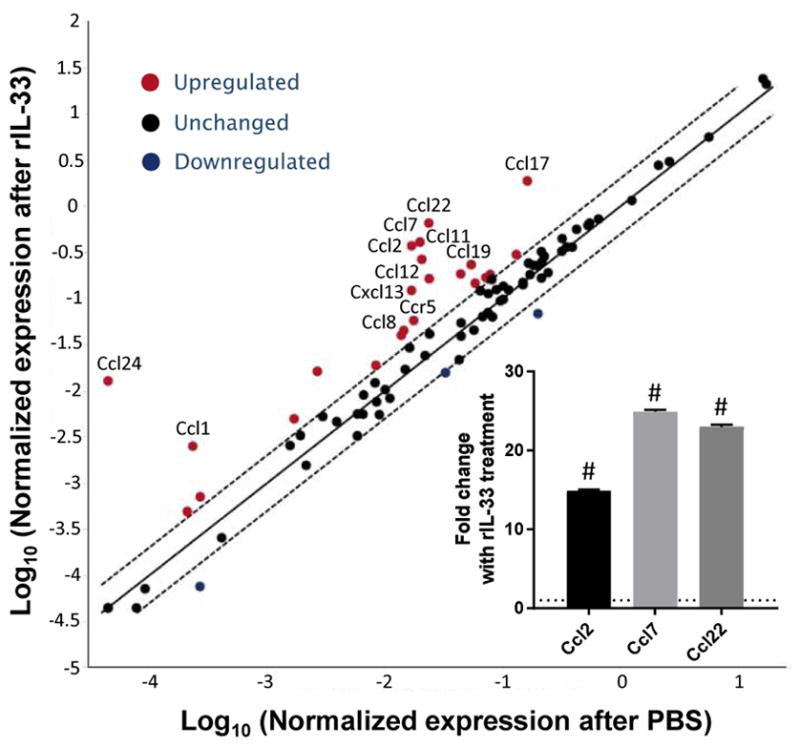
IL-33−/− mice received i.t. rIL-33 (n = 5) or PBS (n = 4). The mice were sacrificed 6 h later and RNA was purified from total lung homogenate. cDNA was prepared and pooled for each group. A qPCR array was used to assess transcript abundance of genes encoding chemokines and chemokine receptors. Relative expression was calculated using the ΔΔCT method as compared with the housekeeping genes Gusb and Hsp90ab1. Inset: qPCR for Ccl2, Ccl7, and Ccl22 was performed on individual mouse cDNA samples generated from mouse lung homogenates. Chemokine transcripts were normalized to the housekeeping gene Hprt. Dotted line represents expression levels of PBS-treated mice. rIL-33–treated mice (n = 5) were compared with PBS-treated mice (n = 4). #p < 10−10.
Monocyte accumulation in the lung interstitium is G-protein coupled receptor dependent
As we had demonstrated that IL-33 upregulated chemokines in the lungs, we questioned whether monocyte accumulation in response to OVA-ICs was chemokine dependent. PTX has previously been shown to inhibit signaling of G-protein coupled receptors (GPCRs), including chemokine receptors (26). Thus, treatment with PTX allowed us to inhibit all chemokine receptor activity simultaneously, as opposed to targeting a particular chemokine receptor. Mice were treated with PTX (or PBS control) on day 0 at the same time as administration of αOVA serum, followed by an OVA i.t. sensitization the next day. Notably, treatment with PTX inhibited extravascular accumulation of Ly6C+ monocytes after OVA-IC treatment with a trending decrease in Ly6C− monocytes (Fig. 6). These results demonstrated that the accumulation of monocytes in the lung interstitium after OVA-IC treatment occurred as a result of chemokine-dependent migration and is not a result of direct chemotactic effects of IL-33.
FIGURE 6. PTX treatment abolishes IL-33–dependent monocyte migration to the lung extravascular space in response to OVA-IC.

IL-33−/− mice received α-OVA i.v. and either PBS or PTX i.p. OVA-AF647 sensitization with rIL-33 was delivered i.t. 24 h later. The mice were sacrificed 18 h after challenge. (A) Representative flow plots of Ag uptake (OVA-AF647) versus localization (i.v. CD45). (B) Percentage of OVA-AF647+ cells in the extravascular space. Data represent the mean ±SEM combined from three independent experiments with a total of at least 10 mice analyzed per group. ****p < 0.0001. ns, not significant.
DISCUSSION
Using a murine model of asthma, we demonstrated that IL-33 was critical for the recruitment of Ly6C+ monocytes from the vasculature to the lung parenchyma by upregulating monocyte chemoattractants including CCL2 and CCL7 (14). Blocking GPCR signaling inhibited Ly6C+ monocyte localization to the extravascular space, demonstrating a critical role for IL-33–induced chemokines in monocyte migration. Monocyte infiltration has been shown to be important in promoting the development of type 2 inflammatory responses, either through direct effector functions or differentiation into CD11b+ moDCs (6, 23). Indeed, we found that CCR2−/− mice, in which monocyte accumulation is attenuated, had reduced eosinophilia after challenge. Studies have found that CD11b+ moDCs can produce chemokines as well as migrate to draining lymph nodes to prime and activate naive CD4+ T cells (23). Thus, IL-33–induced monocyte recruitment during allergic sensitization may be critical to augmenting type 2 inflammatory responses in the lungs through multiple mechanisms.
As IL-33 was found to upregulate multiple chemokines, pharmacologic targeting of IL-33 may be able to broadly reduce type 2 inflammatory responses. Attempts to block chemokines and GPCR signaling in patients with asthma has been difficult to achieve as there is a diverse number of chemokines upregulated during allergic airway inflammation and there is much redundancy among chemokines and their receptors (27, 28). Pharmacologic approaches to blockone particular mediator (i.e., CXCR2, IL-8/CXCL8, eotaxin/CCR3, and others) have met limited success and fail to improve or modify lung function (29). Identifying novel pathways to halt the disease early on is critical because repeated exposure to allergens leads to cumulative tissue damage and pathological changes that affect respiratory function (30). Our findings suggest that a more comprehensive strategy for inhibiting multiple downstream effectors may be achieved by targeting IL-33 to inhibit the development of type 2 inflammatory responses in the lungs.
Supplementary Material
Antigen uptake in monocytes is enhanced by OVA-IC. WT mice received α-OVAdepl serum (OVA) or α-OVA serum (OVA-IC) i.v. followed by a fluorescently labeled OVA (OVA-FITC) challenge i.t. 24 hours later. 18 hours after challenge, the mice were sacrificed and FITC expression on monocytes was assessed by flow cytometry. (Black – unchallenged, Green – OVA, Purple – OVA-IC.) The data are representative plots with at least six mice analyzed per group.
Monocyte migration from the vasculature to the interstitium is dependent on FcγRIII. WT and FcγRIII−/− mice received α-OVA (OVA-IC) i.v. followed by a fluorescently labeled OVA (OVA-FITC) challenge 24 hours later. 18 hours after challenge, the mice were sacrificed. (A) Representative flow plots of antigen uptake (OVA-FITC) versus localization (i.v. CD45) for Ly6C− and Ly6C+ monocytes. (B) Percentage of OVA-FITC+ cells in the extravascular space is plotted. Data represent the mean ±SEM. The data are from two independent experiments with at least six mice analyzed per group.
IL-33 alters chemokine expression in the lungs. IL-33−/− mice received recombinant IL-33 (rIL-33) or PBS i.t. The mice were sacrificed six hours later and RNA was purified from total lung homogenate. cDNA was prepared and a qPCR array was used to assess transcript abundance of genes encoding chemokines and chemokine receptors, which were calculated using the delta-delta CT method.
Acknowledgments
This work was supported by National Institutes of Health Grants R21AI094408, R01HL118758, and R01AI125644 (to A.I.S.), R01HL122489 (to H.R.T.), R01HL118758-03S1, T32GM007281, and F30HL137309 (to D.F.C.); and a Naomi Ragins-Goldsmith Fellowship, University of Chicago (to M.Y.T.). The University of Chicago Cancer Center Core Facilities utilized are supported by National Institutes of Health Grant P30CA14599.
We thank K. Blaine and D. Decker for technical support, as well as C. Hrusch for reading and insightful comments on the manuscript.
Abbreviations used in this article
- BAL
bronchoalveolar lavage
- CT
cycle threshold
- IC
immune complex
- i.t
intratracheal, intratracheally
- MHC II
MHC class II
- moDC
monocyte-derived dendritic cell
- PTX
pertussis toxin
- qPCR
quantitative PCR
- rIL-33
recombinant IL-33
- WT
wild type
Footnotes
DISCLOSURES
The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Locksley RM. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16:45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 3.Aleman F, Lim HF, Nair P. Eosinophilic endotype of asthma. Immunol Allergy Clin North Am. 2016;36:559–568. doi: 10.1016/j.iac.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Zasłona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, Peters-Golden M. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 2014;193:4245–4253. doi: 10.4049/jimmunol.1400580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tashiro H, Takahashi K, Hayashi S, Kato G, Kurata K, Kimura S, Sueoka-Aragane N. Interleukin-33 from monocytes recruited to the lung contributes to house dust mite-induced airway inflammation in a mouse model. PLoS One. 2016;11:e0157571. doi: 10.1371/journal.pone.0157571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349–362. doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
- 7.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 8.Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, Hedrick CC. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 10.Sunderkötter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 11.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 13.Greene JA, Portillo JA, Lopez Corcino Y, Subauste CS. CD40-TRAF signaling upregulates CX3CL1 and TNF-α in human aortic endothelial cells but not in retinal endothelial cells. PLoS One. 2015;10:e0144133. doi: 10.1371/journal.pone.0144133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy RM, Wüthrich M, Klein BS. Chitin elicits CCL2 from airway epithelial cells and induces CCR2-dependent innate allergic inflammation in the lung. J Immunol. 2012;189:2545–2552. doi: 10.4049/jimmunol.1200689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith DE. IL-33: a tissue derived cytokine pathway involved in allergic inflammation and asthma. Clin Exp Allergy. 2010;40:200–208. doi: 10.1111/j.1365-2222.2009.03384.x. [DOI] [PubMed] [Google Scholar]
- 18.Tjota MY, Hrusch CL, Blaine KM, Williams JW, Barrett NA, Sperling AI. Signaling through FcRγ-associated receptors on dendritic cells drives IL-33-dependent TH2-type responses. J Allergy Clin Immunol. 2014;134:706–713.e8. doi: 10.1016/j.jaci.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tjota MY, Williams JW, Lu T, Clay BS, Byrd T, Hrusch CL, Decker DC, de Araujo CA, Bryce PJ, Sperling AI. IL-33-dependent induction of allergic lung inflammation by FcγRIII signaling. J Clin Invest. 2013;123:2287–2297. doi: 10.1172/JCI63802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci USA. 2010;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingersoll MA, Platt AM, Potteaux S, Randolph GJ. Monocyte trafficking in acute and chronic inflammation. Trends Immunol. 2011;32:470–477. doi: 10.1016/j.it.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, Masopust D. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9:209–222. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. 2013;38:322–335. doi: 10.1016/j.immuni.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 24.Yagami A, Orihara K, Morita H, Futamura K, Hashimoto N, Matsumoto K, Saito H, Matsuda A. IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol. 2010;185:5743–5750. doi: 10.4049/jimmunol.0903818. [DOI] [PubMed] [Google Scholar]
- 25.Nabe T, Wakamori H, Yano C, Nishiguchi A, Yuasa R, Kido H, Tomiyama Y, Tomoda A, Kida H, Takiguchi A, et al. Production of interleukin (IL)-33 in the lungs during multiple antigen challenge-induced airway inflammation in mice, and its modulation by a glucocorticoid. Eur J Pharmacol. 2015;757:34–41. doi: 10.1016/j.ejphar.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Sun L, Ye RD. Role of G protein-coupled receptors in inflammation. Acta Pharmacol Sin. 2012;33:342–350. doi: 10.1038/aps.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukacs NW, Miller AL, Hogaboam CM. Chemokine receptors in asthma: searching for the correct immune targets. J Immunol. 2003;171:11–15. doi: 10.4049/jimmunol.171.1.11. [DOI] [PubMed] [Google Scholar]
- 28.Adcock IM, Caramori G, Chung KF. New targets for drug development in asthma. Lancet. 2008;372:1073–1087. doi: 10.1016/S0140-6736(08)61449-X. [DOI] [PubMed] [Google Scholar]
- 29.Durham AL, Caramori G, Chung KF, Adcock IM. Targeted anti-inflammatory therapeutics in asthma and chronic obstructive lung disease. Transl Res. 2016;167:192–203. doi: 10.1016/j.trsl.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holt PG, Sly PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat Med. 2012;18:726–735. doi: 10.1038/nm.2768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Antigen uptake in monocytes is enhanced by OVA-IC. WT mice received α-OVAdepl serum (OVA) or α-OVA serum (OVA-IC) i.v. followed by a fluorescently labeled OVA (OVA-FITC) challenge i.t. 24 hours later. 18 hours after challenge, the mice were sacrificed and FITC expression on monocytes was assessed by flow cytometry. (Black – unchallenged, Green – OVA, Purple – OVA-IC.) The data are representative plots with at least six mice analyzed per group.
Monocyte migration from the vasculature to the interstitium is dependent on FcγRIII. WT and FcγRIII−/− mice received α-OVA (OVA-IC) i.v. followed by a fluorescently labeled OVA (OVA-FITC) challenge 24 hours later. 18 hours after challenge, the mice were sacrificed. (A) Representative flow plots of antigen uptake (OVA-FITC) versus localization (i.v. CD45) for Ly6C− and Ly6C+ monocytes. (B) Percentage of OVA-FITC+ cells in the extravascular space is plotted. Data represent the mean ±SEM. The data are from two independent experiments with at least six mice analyzed per group.
IL-33 alters chemokine expression in the lungs. IL-33−/− mice received recombinant IL-33 (rIL-33) or PBS i.t. The mice were sacrificed six hours later and RNA was purified from total lung homogenate. cDNA was prepared and a qPCR array was used to assess transcript abundance of genes encoding chemokines and chemokine receptors, which were calculated using the delta-delta CT method.