

Abstract
Glomerular mesangial cell hypertrophy contributes to the complications of diabetic nephropathy. The mechanism by which high glucose induces mesangial cell hypertrophy is poorly understood. Here we explored the role of the platelet-derived growth factor receptor-β (PDGFRβ) tyrosine kinase in driving the high glucose-induced mesangial cell hypertrophy. We show that high glucose stimulates the association of the PDGFRβ with PI 3 kinase leading to tyrosine phosphorylation of the latter. High glucose-induced Akt kinase activation was also dependent upon PDGFRβ and its tyrosine phosphorylation at 740/751 residues. Inhibition of PDGFRβ activity, its downregulation and expression of its phospho-deficient (Y740/751F) mutant inhibited mesangial cell hypertrophy by high glucose. Interestingly, expression of constitutively active Akt reversed this inhibition, indicating a role of Akt kinase downstream of PDGFRβ phosphorylation in this process. The transcription factor Hif1α is a target of Akt kinase. siRNAs against Hif1α inhibited the high glucose-induced mesangial cell hypertrophy. In contrast, increased expression of Hif1α induced hypertrophy similar to high glucose. We found that inhibition of PDGFRβ and expression of PDGFRβ Y740/751F mutant significantly inhibited the high glucose-induced expression of Hif1α. Importantly, expression of Hif1α countered the inhibition of mesangial cell hypertrophy induced by siPDGFRβ or PDGFRβ Y740/751F mutant. Finally, we show that high glucose-stimulated PDGFRβ tyrosine phosphorylation at 740/751 residues and the tyrosine kinase activity of the receptor regulate the transforming growth factor-β (TGFβ) expression by Hif1α. Thus we define the cell surface PDGFRβ as a major link between high glucose and its effectors Hif1α and TGFβ for induction of diabetic mesangial cell hypertrophy.
Keywords: Diabetic nephropathy, Renal hypertrophy, Receptor tyrosine kinase
1. Introduction
The pathology of diabetic kidney disease consists of changes in the glomeruli with altered hemodynamics and matrix protein expansion [1,2]. The initial structural changes include renal hypertrophy especially glomerular hypertrophy, which leads to hyperfiltration and microalbuminuria. This is followed by fibrosis [3]. In fact, mesangial matrix expansion correlates well with the development of diabetic nephropathy. One third of the glomerular cell types consists of mesangial cells, which are considered as vascular pericytes of the glomerulus. These cells respond to hyperglycemia and contribute to the whole glomerular hypertrophy [4–6]. Hyperglycemia increases the production of hormones and cytokines such as angiotensin II and TGFβ, which contribute to the pathology of mesangial cells [2].
Importantly, the prominent growth factors the mesangial cells respond to are the PDGFs [7]. Mammalian genome codes for four different isotypes of this growth factor: A, B, C and D [8]. By homo or heterodimerization, these proteins form five isoforms. These isoforms bind to two distinct receptors, PDGFRα and PDGFRβ, with variable affinity and specificity. Although PDGF AB and BB bind to both receptors, the PDGF DD interacts with PDGFRββ with high affinity and to a lesser extent to the heterodimeric PDGFRαβ. On the other hand PDGF AA and CC bind to PDGFRα with high affinity; however, CC can interact with PDGFRαβ at a lower affinity [9]. Mesangial cells express all five dimeric PDGFs and their three trimeric receptors [10]. PDGF BB, CC and DD are mitogenic for mesangial cells and play a prominent role in mesangioproliferative glomerulonephritis [7,11,12,13]. On the other hand PDGF AA is not mitogenic for mesangial cells [13]. In fact PDGF BB, which functions predominantly through binding to the PDGFRββ, is the most potent mitogen and activator of various downstream signaling pathways [13,14]. Furthermore, mice deficient in either PDGF-B chain or PDGFRβ lack development of mesangial cells [15]. Although proliferation of glomerular mesangial cells is not a predominant feature of diabetic nephropathy, glomerular expression of PDGF-A is increased in patients with diabetic nephropathy [16]. Augmented urinary excretion of PDGF BB has been reported in the type 2 diabetes patients [17]. Similarly, PDGF B and its receptor beta are expressed in the diabetic mesangial cells [16,18]. Since mesangial cell hypertrophy represents an early event in the pathology of diabetic nephropathy, whether PDGFRβ signal transduction contributes to this phenomenon has not been investigated. In the present study, we show that high glucose initiates the PI 3 kinase/Akt signaling cascade to drive the expression of Hif1α via activation of PDGFRβ. The PDGFRβ autophosphorylation sites Tyr-740 and Tyr-751 are necessary for the expression of Hif1α. Furthermore, we show that tyrosine phosphorylation of these sites in PDGFRβ contributes to the mesangial cell hypertrophy via Hif1α and TGFβ expression.
2. Materials and methods
2.1. Reagents
The following reagents were purchased from Sigma: D-glucose, D-mannitol, protease inhibitor cocktail, phenylmethylsulfonyl fluoride, Na3VO4, NP-40, JNJ-10198409 (JNJ) and actin antibody. Tissue culture reagents were obtained from Life Technologies. Antibodies for phospho-p85 (Tyr-458), p85, phospho-PDGFRβ (Tyr-857), phospho-PDGFRβ (Tyr-740), phospho-PDGFRβ (Tyr-751), PDGFRβ, phospho-Akt (Ser-473), phospho-Akt (Thr-308), Akt, phospho-GSK3β (Ser-9) and GSK3β were obtained from Cell Signaling. TGFβ was purchased from R & D. TGFβ antibody was obtained from Abcam. Hif1α antibody, scramble RNA and pooled siRNAs against PDGFRβ were purchased from Santa Cruz. Anti-HA antibody was obtained from Covance. FuGENE transfection reagent and the OPTIMEM transfection medium were purchased from Promega and Life Technology, respectively. MK 2206 was obtained from Selleck Chemicals. 35S-methionine was purchased from PerkinElmer. The PDGFRβ (Y740/751F) mutant plasmid was a gift from Dr. Carl Heldin (Ludwig Institute for Cancer Research, Uppsala University, Sweden). The plasmids expressing HA-tagged Hif1α and HA-tagged Akt K179M were described previously [19].
2.2. Cell culture
Human glomerular mesangial cells were grown in DMEM with 5 mM glucose in the presence of 10% fetal bovine serum as described previously [20,21]. For experiments, cells were grown to confluency and incubated with serum free medium for 24 h. Serum-starved cells were then treated with 25 mM glucose for 24 h or for indicated periods of time. 5 mM glucose plus 20 mM mannitol was used as osmotic control.
2.3. Cell lysis, immunoblotting and immunoprecipitation
After incubation with high glucose, the mesangial cell monolayer was lysed in RIPA buffer (20 mM Tris-HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% NP-40, 1 mM Na3VO4, 1 mM PMSF and 0.1% protease inhibitor cocktail) at 4 °C for 30 min. The lysed cells were scraped along with the debris into Eppendorf tube. The cell extracts were centrifuged at 10,000 ×g for 20 min at 4 °C. The supernatant was collected. After determining the protein concentration, equal amounts of proteins were separated by SDS polyacrylamide gel electrophoresis. The separated proteins were transferred to PVDF membrane. The membrane containing the proteins was immunoblotted with indicated antibodies. The protein bands were developed with HRP-conjugated secondary antibody using ECL reagent as described previously [4,22,23]. For immunoprecipitation, equal amounts of proteins were incubated with the indicated antibody on ice for 30 min. Protein G agarose conjugated beads were then added. The mixture was rotated at 4 °C for overnight. The immune beads were washed with RIPA buffer and resuspended in SDS sample buffer [21]. The denatured proteins were separated by electrophoresis. The separated proteins were then immunoblotted with the indicated antibody as described above.
2.4. Transfection
The mesangial cells were seeded at 80% confluency. Next day, the cells were transfected with the plasmid vectors or siRNAs against PDGFRβ using FuGENE HD according to vendor’s protocol as described previously [4,22]. After 24 h of transfection, the cells were starved in serum free medium and treated with high glucose as described above.
2.5. Protein synthesis
After incubation with high glucose, the mesangial cells were incubated with 35S-methionine and protein synthesis was determined as [35S]-methionine incorporation as described previously [4,5,22].
2.6. Measurement of cellular hypertrophy
At the end of the incubation period, the mesangial cells were trypsinized. The cells were counted in a hemocytometer. After counting, the cells were centrifuged at 4000 ×g at 4 °C. The cell pellet was washed with PBS and lysed in RIPA buffer as described above. The protein content in the cells was determined. Hypertrophy was expressed as an increase in the ratio of total cellular protein content to the cell number as described previously [4,22].
2.7. Statistics
The mean ± SE of indicated measurements is shown. The significance of the results was determined using the Graph Pad Prism software. Analysis of variance followed by Students-Newman-Keuls analysis was used as described previously [24,25]. A p value of < 0.05 was considered significant.
3. Results
3.1. Tyrosine phosphorylation of PDGFRβ is necessary for high glucose-induced PI 3 kinase phosphorylation
Recent work demonstrated that the regulatory subunit of PI 3 kinase, the p85 protein, is tyrosine phosphorylated when the PI 3 kinase is activated [26]. We examined the tyrosine phosphorylation of p85 by high glucose. Incubation of mesangial cells with high glucose increased the phosphorylation of p85 at Tyr-458 in a time-dependent manner (Fig. 1A). Since PI 3 kinase is activated by growth factor receptors and recent report showed increased expression of PDGFRβ in diabetic renal glomeruli [18], we tested the status of tyrosine phosphorylation of this receptor in mesangial cells. High glucose time-dependently enhanced the autophosphorylation of PDGFRβ at Tyr-857 (Fig. 1B). To determine the requirement of the tyrosine kinase activity of PDGFRβ for phosphorylation of p85 subunit of PI 3 kinase, we used JNJ-10198409 (JNJ), a PDGFRβ inhibitor [27]. JNJ inhibited the tyrosine phosphorylation of p85 concomitant with attenuation of the autophosphorylation of the PDGFRβ (Fig. 1C). Furthermore, the requirement of PDGFRβ for p85 phosphorylation was also confirmed by siRNAs against this tyrosine kinase (Fig. 1D).
Fig. 1.
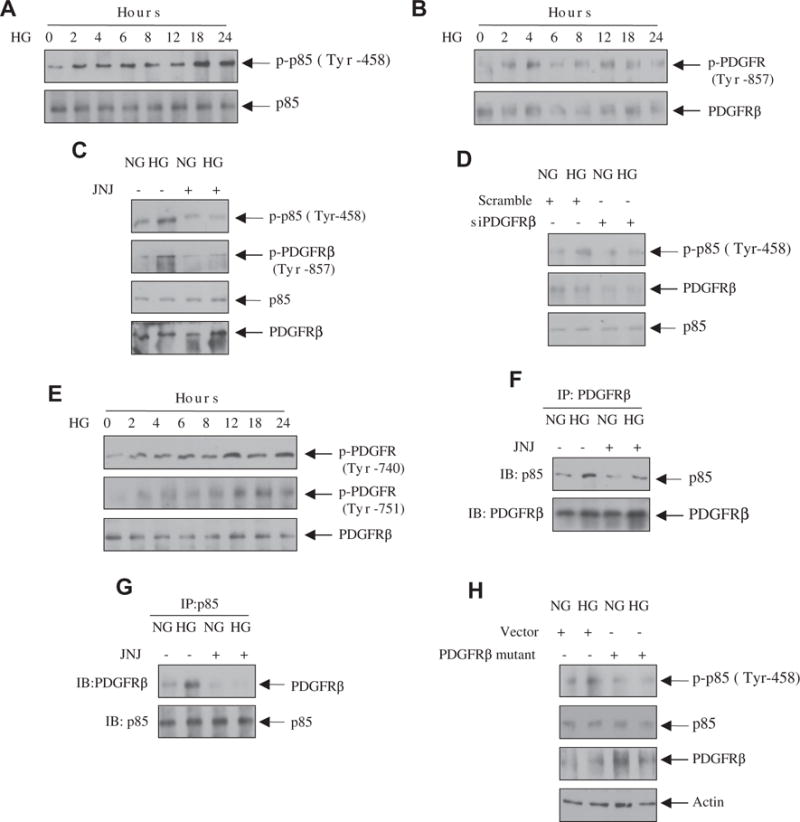
High glucose increases the association of PI 3 kinase with the PDGFRβ leading to its phosphorylation. (A, B and E) Mesangial cells were incubated with high glucose (HG, 25 mM glucose) for the indicated time periods. As control (0 h), 20 mM mannitol plus 5 mM glucose was used as described in the Materials and methods section. Equal amounts of cell lysates were immunoblotted with phospho-p85 (Tyr-458), p85, phospho-PDGFRβ (Tyr-857), phospho-PDGFRβ (Tyr-740), phospho-PDGFRβ (Tyr-751) and PDGFRβ antibodies as indicated. (C, F and G) Mesangial cells were treated with 0.1 μM JNJ prior to incubation with high glucose (HG) and normal glucose (NG) for 24 h. In panel C, the cell lysates were immunoblotted with the indicated antibodies. In panels F and G, the cell lysates were immunoprecipitated with PDGFRβ (panel F) or with p85 PI 3 kinase subunit (panel G) antibodies. The immunoprecipitates were immunoblotted with p85 (panel F) or PDGFRβ (panel G) antibodies, respectively. (D and H) Mesangial cells were transfected with scramble siRNA or siRNAs against PDGFRβ (panel D) or with vector or PDGFRβ (Y740/Y751F) mutant as described in the Materials and methods section. The transfected cells were incubated with high glucose (HG) or normal glucose (NG) as described above. The cell lysates were immunoblotted with the indicated antibodies.
PDGFRβ-mediated activation of PI 3 kinase requires its association with the receptor through phosphorylated tyrosine residues at 740 and 751 [28,29]. Therefore, we examined the phosphorylation of these residues in PDGFRβ. High glucose time-dependently increased the phosphorylation of Tyr-740 and Tyr-751 in the PDGFRβ (Fig. 1E). To determine the association between PI 3 kinase and PDGFRβ, we performed co-immunoprecipitation experiment. PDGFRβ immunoprecipitates were immunoblotted with p85 antibody. The results in Fig. 1F show association of p85 with the PDGFRβ. Importantly, JNJ, which blocked the tyrosine phosphorylation of PDGFRβ at Tyr-740 and 751 (Supplemental Fig. S1), inhibited this association (Fig. 1F). Reciprocal co-immunoprecipitation experiment confirmed this observation (Fig. 1G). Finally, we examined the requirement of phosphorylated PDGFRβ at Tyr-740 and 751 in p85 phosphorylation by using a phosphorylation deficient mutant of the receptor (Y740F/Y751F) [30]. Expression of the mutant PDGFRβ inhibited the phosphorylation of p85 in response to high glucose (Fig. 1H). These data demonstrate that high glucose increases phosphorylation of PI 3 kinase via enhanced phosphorylation of the PDGFRβ at Tyr-740 and Tyr-751.
3.2. High glucose-induced PDGFRβ controls Akt kinase
Activation of PI 3 kinase promotes Akt kinase activity by inducing phosphorylation of this kinase at the catalytic loop Thr-308 and hydrophobic motif site Ser-473 [31,32]. Incubation of mesangial cells with high glucose increased the phosphorylation of Akt at both these sites in a time dependent manner (Supplementary Fig. S2). Since PDGFRβ was involved in activation of PI 3 kinase, we examined the effect of JNJ on Akt phosphorylation. As shown in Fig. 2A, JNJ inhibited the phosphorylation of Akt at Thr-308 and Ser-473 by high glucose. Since Akt phosphorylation increases its kinase activity, high glucose increased the phosphorylation of the Akt substrate GSK3β, which was inhibited by JNJ (Fig. 2B). Similarly, siRNA-mediated downregulation of PDGFRβ blocked the Akt and GSK3β phosphorylation in response to high glucose (Fig. 2C and D). To determine the role of the PI 3 kinase binding site of the receptor in Akt activation, we transfected the phospho-deficient mutant PDGFRβ (Y740/Y751F) into mesangial cells. Expression of the mutant receptor significantly abrogated the phosphorylation of Akt and GSK3β (Fig. 2E and F). These results indicate that high glucose-stimulated PDGFRβ phosphorylation at Tyr-740 and -751 is required for the Akt activity.
Fig. 2.
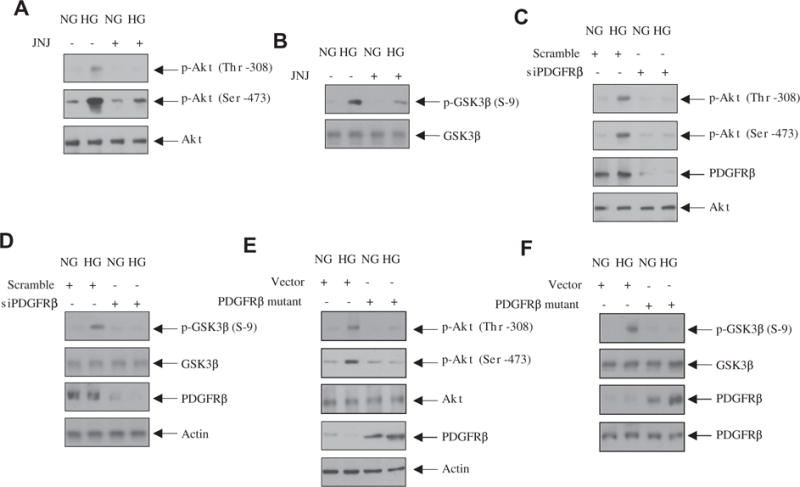
PDGFRβ regulates high glucose-stimulated Akt kinase activity. (A and B) Mesangial cells were treated with 0.1 μM JNJ for 1 h prior to incubation with high glucose (HG) or normal glucose (NG) for 24 h. Equal amounts of cell lysates were immunoblotted with the indicated antibodies. (C–F) Mesangial cells were transfected with siPDGFRβ (panels C and D) or with PDGFRβ Y740/751F (E and F) mutant. The cells were then incubated with high glucose (HG) or normal glucose (NG) for 24 h. The cell lysates were immunoblotted with indicated antibodies.
3.3. PDGFRβ regulates high glucose-induced mesangial cell hypertrophy
We have previously shown a role of Akt kinase in rat mesangial cell hypertrophy [5]. We used a recently developed Akt kinase inhibitor MK2206 in human mesangial cells. MK2206 inhibited the high glucose-stimulated Akt phosphorylation, resulting in inhibition of protein synthesis and mesangial cell hypertrophy (Supplementary Fig. S3A–C). Since Akt kinase activity is regulated by high glucose-stimulated PDGFRβ (Fig. 2), we examined the role of this receptor tyrosine kinase in mesangial cell hypertrophy. Inhibition of PDGFRβ by JNJ significantly inhibited high glucose-induced protein synthesis and hypertrophy of mesangial cells (Fig. 3A and B). Similarly, downregulation of PDGFRβ markedly attenuated the protein synthesis and hypertrophy of mesangial cells by high glucose (Fig. 3C and D). Additionally, the expression of phospho-deficient mutant of the PDGFRβ (Y740/Y751F) significantly suppressed the high glucose-induced mesangial cell protein synthesis and hypertrophy (Fig. 3E and F). To confirm the role of Akt in this process, we used constitutively active myristoylated Akt. Expression of constitutively active Myr Akt in the presence of high glucose reversed the siPDGFRβ- and PDGFRβ (Y740/Y751F)-mediated inhibition of protein synthesis and hypertrophy (Fig. 4A–D). Interestingly, Myr Akt alone was sufficient to induce significantly protein synthesis and hypertrophy similar to high glucose (Fig. 4A–D). These results demonstrate that tyrosine phosphorylation of PDGFRβ at 740 and 751 residues that regulate Akt kinase activity by high glucose controls the mesangial cell hypertrophy.
Fig. 3.
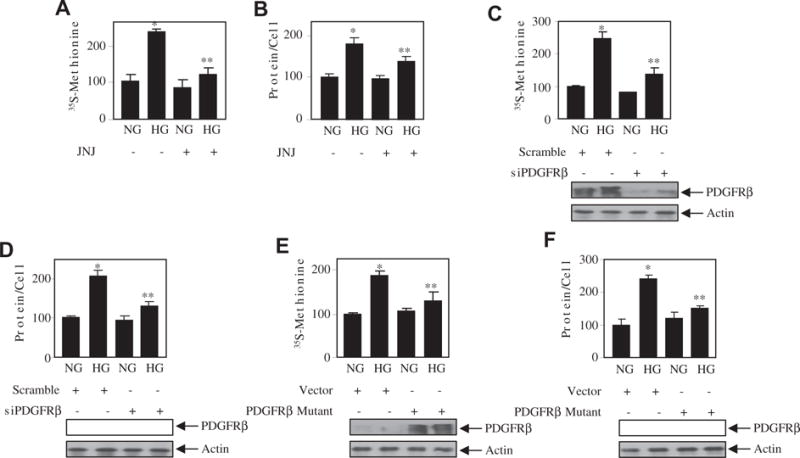
PDGFRβ controls mesangial cell hypertrophy by high glucose. (A and B) Mesangial cells were treated 0.1 μM JNJ for an hour prior to incubation with high glucose (HG) for 24 h. Protein synthesis was measured by 35S-methionine incorporation as described in the Materials and methods section (panel A). Mean ± SE of triplicate measurements is shown. *p < 0.01 vs NG; **p < 0.01 vs HG. Hypertrophy was determined as the ratio of total amount of protein to cell number as described. Mean ± SE of 6 measurements is shown. *p < 0.001 vs NG; **p < 0.05 vs HG [22,25]. (C–F) Mesangial cells were transfected with siPDGFRβ or scramble (panels C and D) or PDGFRβ mutant (Y740/751F) or vector (panels E and F). The transfected cells were incubated with normal glucose (NG) or high glucose (HG) for 24 h. The protein synthesis and hypertrophy were determined as described above. Mean ± SE of 3–6 measurements is shown. *p < 0.01 vs HG. For panels B and E, **p < 0.05 vs HG; for panels C, D and F, **p < 0.01 vs HG. The bottom panels show the expression of PDGFRβ and actin.
Fig. 4.
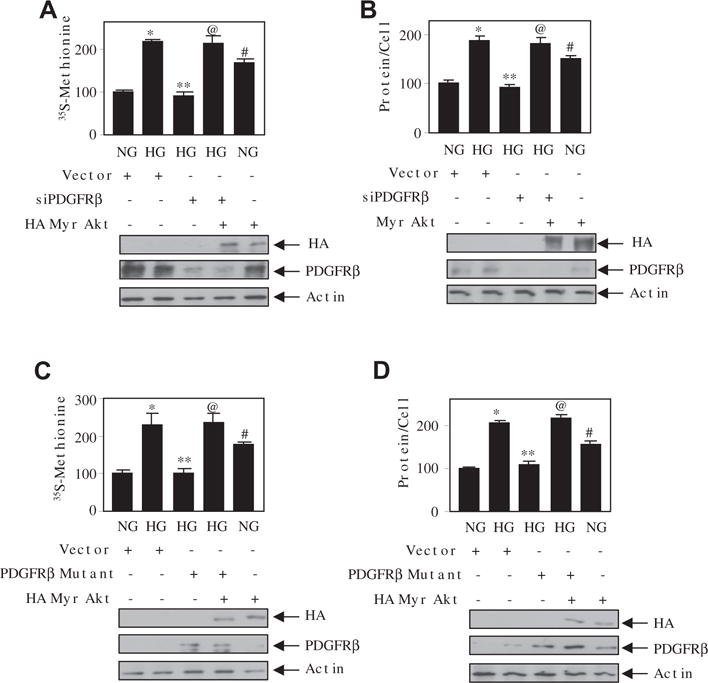
Akt kinase regulates high glucose-induced PDGFRβ-mediated hypertrophy of mesangial cells. Mesangial cells were transfected with siPDGFRβ and Myr Akt (panels A and B) or PDGFRβ mutant (Y740/751F) and HA Myr Akt (panels C and D) as indicated. The transfected cells were incubated with normal glucose (NG) or high glucose (HG). The protein synthesis and hypertrophy were determined as described in the Materials and methods section [22,25]. Mean ± SE of 4 measurements is shown. *p < 0.01 vs NG; **p < 0.01 vs HG alone; @p < 0.01 vs HG + siPDGFRβ or PDGFRβ mutant; #p < 0.01 or 0.05 vs NG. The bottom panels show the expression of HA Myr Akt, PDGFRβ and actin.
3.4. PDGFRβ-regulated Hif1α controls mesangial cell hypertrophy
A role of Hif1α is reported in renal injury [33]. High glucose increased the expression of Hif1α in mesangial cells in a time-dependent manner (Supplementary Fig. S4). PI 3 kinase has been shown to regulate Hif1α in various cells types [34]. In mesangial cells, MK 2206 and dominant negative Akt (K179M) significantly inhibited the expression of Hif1α in response to high glucose (Fig. 5A and B). Since Akt regulates mesangial cell hypertrophy, we determined the involvement of Hif1α in this process. Downregulation of Hif1α significantly inhibited the high glucose-induced protein synthesis and hypertrophy of mesangial cells (Fig. 5C and D). Conversely, overexpression of Hif1α induced protein synthesis and hypertrophy similar to incubation with high glucose (Fig. 5E and F) As we have shown above that high glucose increases the PI 3 kinase/Akt pathway through PDGFRβ, we investigated the role of this receptor tyrosine kinase in Hif1α expression. Incubation of mesangial cells with the PDGFRβ inhibitor JNJ and siPDGFRβ significantly inhibited the expression of Hif1α by high glucose (Fig. 5G and H). Finally, the mutant PDGFRβ deficient in phosphorylation at 740 and 751 tyrosines suppressed the high glucose-stimulated expression of Hif1α (Fig. 5I).
Fig. 5.
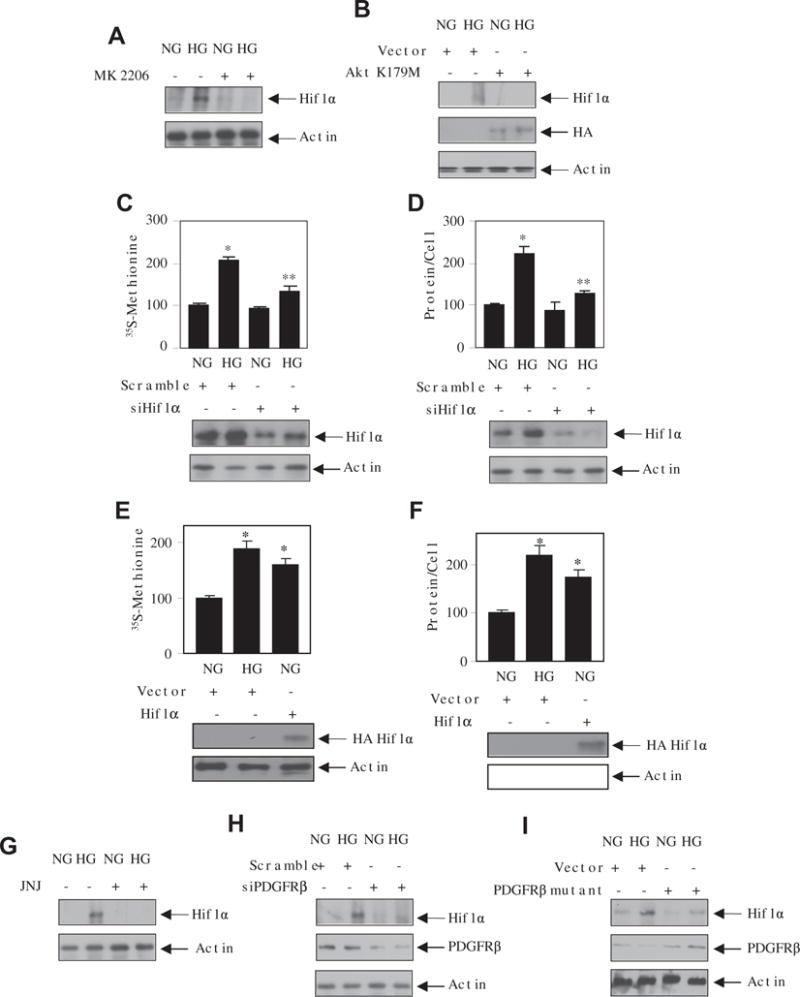
Akt-dependent Hif1α regulates mesangial cell hypertrophy. (A and G) Mesangial cells were treated with 0.1 μM JNJ (panel A) or 1 μM MK 2206 (panel G) for 1 h followed by incubation with normal glucose (NG) or high glucose (HG) for 24 h. The cell lysates were immunoblotted with Hif1α and actin antibodies. (B, H and I) Mesangial cells were transfected with dominant negative HA Akt K179M (panel B) or siPDGFRβ (panel H) or PDGFRβ mutant (Y740/751F) (panel I). The transfected cells were incubated with normal glucose or high glucose. The cell lysates were immunoblotted Hif1α, HA, PDGFRβ or actin antibodies as indicated. (C–F) Mesangial cells were transfected with siHif1α or scramble (panels C and D) or HA Hif1α expression vector (panels E and F). The protein synthesis and hypertrophy were determined as described in the Materials and methods section [22,25]. Mean ± SE of 3–6 measurements is shown. *p < 0.01 or 0.001 vs NG; **p < 0.01 vs HG. The bottom panels show the expression of Hif1α.
Since we have shown that PDGFRβ and Hif1α independently regulate the mesangial cell hypertrophy (Figs. 3 and 5), we examined the interaction between this receptor and the transcription factor in this process. As expected siPDGFRβ significantly inhibited the high glucose-induced protein synthesis and hypertrophy. Co-expression of Hif1α with siPDGFRβ prevented this inhibition (Fig. 6A and B). As the PDGFRβ phospho-deficient mutant inhibits the hypertrophy (Fig. 3F), we tested the effect of Hif1α. Overexpression of Hif1α significantly reversed the PDGFRβ mutant-mediated inhibition of protein synthesis and hypertrophy (Fig. 6C and D). These results demonstrate that tyrosine phosphorylation sites 740/751 in PDGFRβ regulates Hif1α to promote mesangial cell hypertrophy in response to high glucose.
Fig. 6.
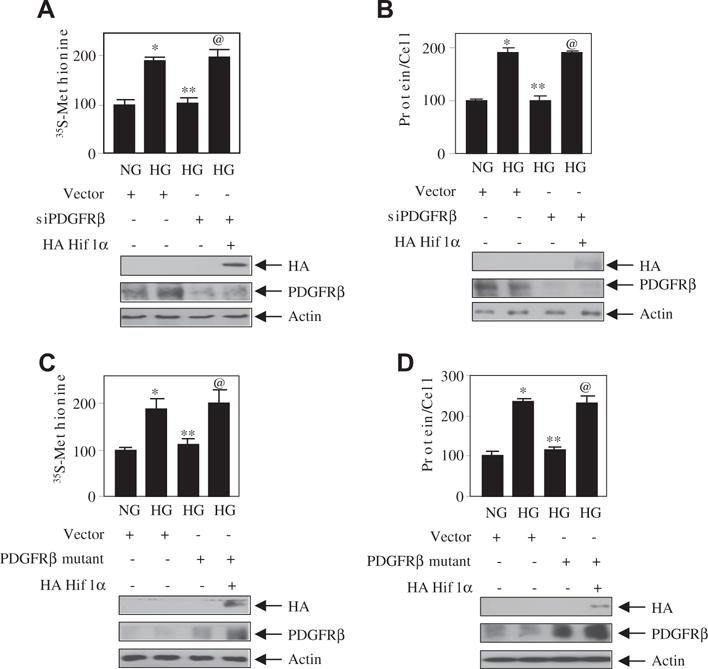
Hif1α downstream of PDGFRβ controls high glucose-induced mesangial cell hypertrophy. Mesangial cells were transfected with siPDGFRβ or PDGFRβ mutant (Y740/751F) along with HA Hif1α. The transfected cells were incubated with normal glucose or high glucose. The protein synthesis and hypertrophy were determined as described in the Materials and methods section [22,25]. The bottom panels show the expression of HA Hif1α, PDGFRβ and actin. Mean ± SE of 3–6 measurement is shown. *p < 0. 01, 0.05 or 0.001 vs NG; **p < 0. 01, 0.05 or 0.001 vs HG; @p < 0.05, 0.01 or 0.001.
3.5. PDGFRβ regulates high glucose-stimulated TGFβ expression by Hif1α
We have previously reported that high glucose-induced mesangial cell hypertrophy is mediated by TGFβ [5]. Since we have shown above a role of PDGFRβ and Hif1α in mesangial cell hypertrophy, we examined the involvement of these proteins in TGFβ expression. Inhibition of PDGFRβ blocked the expression of TGFβ in mesangial cells (Fig. 7A). Similarly, siRNAs against PDGFRβ and the phospho-deficient mutant of PDGFRβ (Y740/751F) significantly inhibited the high glucose-stimulated TGFβ expression (Fig. 7B and C). siRNAs against Hif1α also abrogated the expression of TGFβ in response to high glucose (Fig. 7D). In contrast, overexpression of Hif1α in control cells increased the TGFβ expression similar to high glucose (Fig. 7E). Furthermore, expression of Hif1α reversed the siPDGFRβ- and PDGFRβ mutant (Y740/751F)-mediated inhibition of TGFβ expression (Fig. 7F and G). These results for the first time demonstrate a role of PDGFRβ-mediated Hif1α in the high glucose-induced TGFβ expression.
Fig. 7.
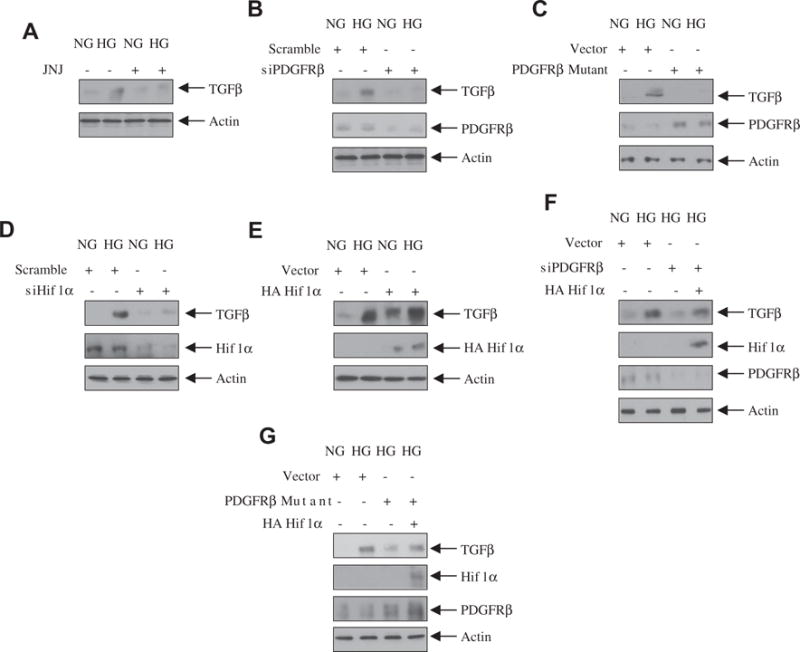
PDGFRβ-stimulated Hif1α regulates high glucose-induced TGFβ expression. (A) Mesangial cells were treated with 0.1 μM JNJ for 1 h prior to incubation with high glucose (HG) or normal glucose (NG) for 24 h. The cell lysates were immunoblotted with TGFβ or actin antibodies. (B–G) Mesangial cells were transfected with siPDGFRβ (panel B) or PDGFRβ mutant (Y7f40/751F) (panel C) or siHIf1α (panel D) or HA Hif1α (panel E) or siPDGFRβ plus HA Hif1α (panel F) or PDGFRβ mutant (Y740/751F) plus HA Hif1α (panel G). The cell lysates were immunoblotted with TGFβ, PDGFRβ, HA and actin antibodies as indicated.
4. Discussion
Here we show that PDGFRβ downstream of high glucose increases PI 3 kinase/Akt signal transduction to induce mesangial cell hypertrophy. Specifically, we show that tyrosine phosphorylation at sites 740/751 on the PDGFRβ is necessary for PI 3 kinase/Akt activation leading to high glucose-induced hypertrophy. Furthermore, the PDGFRβ tyrosine phosphorylation at 740/751 regulates the expression of the transcription factor Hif1α. Finally, we provide the first evidence that Hif1α regulates high glucose-induced TGFβ expression. Thus our findings disclose a new mechanism for high glucose-induced mesangial cell hypertrophy.
Chronic hyperglycemia significantly increases the risk of nephropathy in both type 1 and type 2 diabetes [35]. Several factors and multiple cell types in the kidney contribute to the pathology of diabetic nephropathy [36]. The early changes in the kidney during the progression of diabetic nephropathy includes glomerular hypertrophy. Although the signaling mechanism of cell hypertrophy is poorly understood, a role of the PI 3 kinase product PIP3 has been shown previously to play important role [37]. For example, in Drosophila, overexpression of the PI 3 kinase in the eye and wing increased the organ size [38]. Similarly, overexpression of the constitutively active catalytic subunit of the PI 3 kinase, p110, increased the heart size in mouse. Conversely, dominant negative p110 resulted in reduced heart size [39]. In conjunction with these results, we have shown that PI 3 kinase contributes to renal hypertrophy including glomerular mesangial cell hypertrophy in response to high glucose [5,22]. However, the mechanism of PI 3 kinase activation by high glucose has not been investigated in detail. It is established that activation of PI 3 kinase requires upstream tyrosine kinase activity [40]. We considered the involvement of PDGFRβ as the expression of this receptor tyrosine kinase is increased in the glomerular mesangial area of the kidneys of patients and in the rodent model with diabetic nephropathy [18,41]. Also, we have shown that mesangial cells abundantly express PDGFRβ and its activation stimulates PI 3 kinase activity [12,13,42]. In the present study, we show that activation of PI 3 kinase by high glucose correlates with activating phosphorylation of PDGFRβ in mesangial cells (Fig. 1A and B). PI 3 kinase contains an 85 kDa regulatory subunit, which binds to the tyrosine phosphorylated receptors through the SH2 domains [43]. This association facilitates the tyrosine phosphorylation of the PI 3 kinase [26]. We show that inhibition of PDGFRβ blocks the association of PI 3 kinase with the receptor and tyrosine phosphorylation of PI 3 kinase (Fig. 1C, D, F and G). It was previously described that the tyrosine residues 740 and 751 of the PDGFRβ serve as the docking sites for PI 3 kinase [29]. We demonstrate that high glucose uses these residues of PDGFRβ to phosphorylate the PI 3 kinase (Fig. 1H). These results are the first demonstration of the involvement of the PI 3 kinase binding sites of the PDGFRβ in high glucose-induced signal transduction.
In the PDGF receptor signaling, one of the downstream targets of PI 3 kinase is the Akt kinase [44]. The PI 3 kinase product PIP3 upon binding to the PH domain of Akt increases its initial activating phosphorylation at the Thr-308 residue by the PDK-1 [31,45,46]. Finally, PI 3 kinase-dependent mTORC2 phosphorylates the Ser-473 residue of the Akt [32,47]. In fact, phosphorylation of this residue stabilizes Thr-308 phosphorylation and full activation of Akt [46]. Previously, we have shown increased Akt phosphorylation and activity following exposure of renal cells to high glucose and in the kidneys of mice with diabetes [4,5,22,48]. Our present studies show that high glucose-induced Akt phosphorylation and activation are dependent upon activated PDGFRβ (Fig. 2A–D). In fact, phosphorylation of tyrosines 740/751 of the PDGFRβ is required for high glucose-induced Akt phosphorylation and activation (Fig. 2E and F). Although PI 3 kinase-dependent and independent mechanisms have been reported for full activation of Akt [32,49], our results demonstrating the dependence of the PI 3 kinase on the phosphorylation of PDGFRβ tyrosines 740/751 confirm the lipid kinase dependence of the Akt activation by high glucose in mesangial cells.
As described above that glomerular mesangial cell hypertrophy constitutes a major pathologic feature in the diabetic nephropathy [6]. A role for PI 3 kinase-dependent Akt kinase has been shown in cell size control. For example in Drosophila, expression of Akt in the imaginal discs increased the cell size [50]. Similarly, Akt promoted cardiomyocyte and cardiac hypertrophy when activated in vivo [51]. Also, this action of Akt in cardiac hypertrophy was inhibited by the action of PTEN, which dephosphorylates the PI 3 kinase product and inhibits Akt activity [37]. We have previously shown that PI 3 kinase-dependent Akt kinase contributes to glomerular mesangial cell hypertrophy [23,25]. Activation of various receptor tyrosine kinases including PDGFRβ has been shown to participate in the pathogenesis of diabetic nephropathy [52]. However, specific involvement of receptor tyrosine kinase in high glucose-induced renal cell hypertrophy has not been identified. Our results demonstrate that the PDGFRβ, which activates Akt in mesangial cells, regulates high glucose-induced hypertrophy of these cells (Fig. 3A–D). In fact, the tyrosine phosphorylation of PDGFRβ at the residues 740/751 contributes to mesangial cell hypertrophy (Fig. 3E and F). Importantly, our data provide the evidence that Akt kinase downstream of PDGFRβ contributes to high glucose-induced hypertrophy of mesangial cells (Fig. 4).
Oncogenes including receptor tyrosine kinases and loss of tumor suppressor genes have been shown to increase the levels of Hif1α in the absence of hypoxia [53]. Hif1α is regulated by its de novo expression and E3 ubiquitin-mediated degradation. It dimerizes with the constitutive Hif1β to act as a transcription factor to increase expression of genes involved in cell proliferation, apoptosis, angiogenesis, inflammation, pH homeostasis and metabolic switch to glycolysis [33,54]. Homozygous deletion of Hif1α in mice is embryonically lethal although tissue-specific knockout has shown its role in hematopoiesis, osteogenesis, chondrogenesis, innate immunity, adipogenesis and T cell development [55]. During renal development, Hif1α is highly expressed in the glomerular cells; however in the adult kidney it is expressed in the most cells [33,56]. Renal fibrosis is an end point in chronic kidney disease. Interestingly, pro- as well as anti-fibrotic role of Hif1α has been reported in the literature [33]. For example, induction of Hif1α by cobalt chloride reduced the proteinuria and tubulointerstitial damage in rat model of diabetes [57]. On the other hand, administration of a Hif inhibitor attenuated the mesangial matrix expansion, albuminuria and Nox4 expression in a mouse model of type 1 diabetes [58]. Several reports demonstrate a potent fibrotic role for increased Hif1α in the kidney [59,60]. In fact increased glomerular expression of Hif1α has been shown in the kidneys of patients with moderate to severe diabetic nephropathy [59]. One mechanism by which the abundance of the Hif1α can be maintained at least in certain cancers has recently been worked out. For example, deficiency in the TCA cycle enzymes succinate dehydrogenase and fumarate hydratase in pheochromocytomas and leiomyomata can increase the accumulation of succinate and fumarate to induce a pseudohypoxic state that stabilizes the Hif1α [53]. Recently, using Akita mouse model of diabetic nephropathy, Sharma and coworkers have shown that Nox4-derived hydrogen peroxide inhibits the expression of fumarate hydratase to increase the levels of fumarate in the kidney and in the urine; increased fumarate caused enhanced expression of Hif1α [61]. In agreement with these studies, in vitro we show that high glucose increases the expression of Hif1α in mesangial cells (Supplementary Fig. S4). In the present study, we demonstrate the involvement of PDGFRβ and specifically its 740/751 tyrosine phosphorylation-mediated Akt kinase in high glucose-induced expression of Hif1α (Fig. 5). Furthermore, we provide the first evidence that high glucose-induced Hif1α downstream of PDGFRβ regulates the mesangial cell hypertrophy (Figs. 5 and 6).
A role of TGFβ has been conclusively established in the development of complications of diabetic nephropathy with early glomerular hypertrophy, including mesangial cell hypertrophy [1,6,62]. Administration of anti-TGFβ antibody to streptozotocin-induced type 1 diabetic mice attenuated the glomerular hypertrophy [63]. Similarly, anti-TGFβ antibody prevented the pathologic features of nephropathy in the db/db mouse model of type 2 diabetes [64]. Further evidence for the involvement of TGFβ signaling in glomerular hypertrophy came from the studies where TGFβ receptor II heterozygous mice with streptozotocin-induced diabetes exhibited significantly reduced glomerular hypertrophy [65]. Also, renal cells isolated from TGFβ knock out mice showed impaired response to high glucose-induced hypertrophy [66]. We have shown previously that in cultured mesangial cells, the hypertrophic effect of high glucose is mediated by TGFβ [5]. However, the mechanism by which high glucose increases the expression of TGFβ to induce hypertrophy is poorly understood. We show that activation of PDGFRβ by high glucose increases the expression of TGFβ and mesangial cell hypertrophy (Figs. 3, 7A and B). Overexpression of constitutively active TGFβ receptor I reversed the siPDGFRβ-mediated inhibition of high glucose-induced mesangial cell hypertrophy (data not shown). These results indicate that TGFβ downstream of PDGFRβ contributes to hypertrophy of mesangial cells in response to high glucose. In fact, the tyrosine phosphorylation of PDGFRβ at 740/751 residues is necessary for the high glucose-dependent expression of TGFβ (Fig. 7C and G). Furthermore, we show that high glucose-stimulated Hif1α increased the expression of TGFβ (7D, 7E and 7F). These results are in congruence with a recent in vivo study where the authors observed the expression of Hif1α and TGFβ in the glomeruli of diabetic mice [61]. Recently, we have shown that TGFβ regulates the expression of Hif1α in renal cells [67]. Intriguingly, we demonstrate for the first time that Hif1α regulates the expression of TGFβ and mesangial cell hypertrophy in response to high glucose. Thus Hif1α can act as both an upstream regulator of TGFβ and a downstream effector. Importantly, our data provide significant insight into a potential signal transduction mechanism which involves PDGFRβ Y740/751 phosphorylation, Akt activation, expression of Hif1α and TGFβ for diabetic mesangial hypertrophy.
Supplementary Material
Acknowledgments
This work was supported by the Merit Review Award 2 I01 BX000926 from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service and by the NIH RO1 DK 50190 to GGC. GGC is recipient of Senior Research Career Scientist Award IK6BX003611 from the Department of Veterans Affairs Biomedical Laboratory Research and Development Service. BSK is supported by the VA Merit Review grant I01 BX 001340. NGC is recipient of a research grant from the San Antonio Area Foundation.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cellsig.2017.09.017.
References
- 1.Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med (Maywood) 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 3.Satriano J. Kidney growth, hypertrophy and the unifying mechanism of diabetic complications. Amino Acids. 2007;33:331–339. doi: 10.1007/s00726-007-0529-9. [DOI] [PubMed] [Google Scholar]
- 4.Dey N, Das F, Mariappan MM, Mandal CC, Ghosh-Choudhury N, Kasinath BS, Choudhury GG. MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J Biol Chem. 2011;286:25586–25603. doi: 10.1074/jbc.M110.208066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahimainathan L, Das F, Venkatesan B, Choudhury GG. Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes. 2006;55:2115–2125. doi: 10.2337/db05-1326. [DOI] [PubMed] [Google Scholar]
- 6.Wolf G. Molecular mechanisms of diabetic mesangial cell hypertrophy: a proliferation of novel factors. J Am Soc Nephrol. 2002;13:2611–2613. doi: 10.1681/ASN.V13102611. [DOI] [PubMed] [Google Scholar]
- 7.Abboud HE. Mesangial cell biology. Exp Cell Res. 2012;318:979–985. doi: 10.1016/j.yexcr.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 8.Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Heldin CH. Autocrine PDGF stimulation in malignancies. Ups J Med Sci. 2012;117:83–91. doi: 10.3109/03009734.2012.658119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boor P, Ostendorf T, Floege J. PDGF and the progression of renal disease. Nephrol Dial Transplant. 2014;29(Suppl. 1):i45–i54. doi: 10.1093/ndt/gft273. [DOI] [PubMed] [Google Scholar]
- 11.(a) van Roeyen CR, Ostendorf T, Denecke B, Bokemeyer D, Behrmann I, Strutz F, Lichenstein HS, LaRochelle WJ, Pena CE, Chaudhuri A, Floege J. Biological responses to PDGF-BB versus PDGF-DD in human mesangial cells. Kidney Int. 2006;69:1393–1402. doi: 10.1038/sj.ki.5000332. [DOI] [PubMed] [Google Scholar]; (b) Eitner F, Ostendorf T, Van Roeyen C, Kitahara M, Li X, Aase K, Grone HJ, Eriksson U, Floege J. Expression of a novel PDGF isoform, PDGF-C, in normal and diseased rat kidney. J Am Soc Nephrol. 2002;13:910–917. doi: 10.1681/ASN.V134910. [DOI] [PubMed] [Google Scholar]; (c) Ostendorf T, van Roeyen CR, Peterson JD, Kunter U, Eitner F, Hamad AJ, Chan G, Jia XC, Macaluso J, Gazit-Bornstein G, Keyt BA, Lichenstein HS, LaRochelle WJ, Floege J. A fully human monoclonal antibody (CR002) identifies PDGF-D as a novel mediator of mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 2003;14:2237–2247. doi: 10.1097/01.asn.0000083393.00959.02. [DOI] [PubMed] [Google Scholar]; (d) Jefferson JA, Johnson RJ. Experimental mesangial proliferative glomerulonephritis (the anti-Thy-1.1 model) J Nephrol. 1999;12:297–307. [PubMed] [Google Scholar]
- 12.(a) Choudhury GG, Biswas P, Grandaliano G, Fouqueray B, Harvey SA, Abboud HE. PDGF-mediated activation of phosphatidylinositol 3 kinase in human mesangial cells. Kidney Int. 1994;46:37–47. doi: 10.1038/ki.1994.242. [DOI] [PubMed] [Google Scholar]; (b) Choudhury GG, Karamitsos C, Hernandez J, Gentilini A, Bardgette J, Abboud HE. PI-3-kinase and MAPK regulate mesangial cell proliferation and migration in response to PDGF. Am J Phys. 1997;273:F931–8. doi: 10.1152/ajprenal.1997.273.6.F931. [DOI] [PubMed] [Google Scholar]
- 13.Choudhury GG, Grandaliano G, Jin DC, Katz MS, Abboud HE. Activation of PLC and PI 3 kinase by PDGF receptor alpha is not sufficient for mitogenesis and migration in mesangial cells. Kidney Int. 2000;57:908–917. doi: 10.1046/j.1523-1755.2000.00907.x. [DOI] [PubMed] [Google Scholar]
- 14.van Roeyen CR, Ostendorf T, Floege J. The platelet-derived growth factor system in renal disease: an emerging role of endogenous inhibitors. Eur J Cell Biol. 2012;91:542–551. doi: 10.1016/j.ejcb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 15.(a) Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]; (b) Lindahl P, Hellstrom M, Kalen M, Karlsson L, Pekny M, Pekna M, Soriano P, Betsholtz C. Paracrine PDGF-B/PDGF-Rbeta signaling controls mesangial cell development in kidney glomeruli. Development. 1998;125:3313–3322. doi: 10.1242/dev.125.17.3313. [DOI] [PubMed] [Google Scholar]
- 16.Langham RG, Kelly DJ, Maguire J, Dowling JP, Gilbert RE, Thomson NM. Over-expression of platelet-derived growth factor in human diabetic nephropathy. Nephrol Dial Transplant. 2003;18:1392–1396. doi: 10.1093/ndt/gfg177. [DOI] [PubMed] [Google Scholar]
- 17.Bessa SS, Hussein TA, Morad MA, Amer AM. Urinary platelet-derived growth factor-BB as an early marker of nephropathy in patients with type 2 diabetes: an Egyptian study. Ren Fail. 2012;34:670–675. doi: 10.3109/0886022X.2012.674438. [DOI] [PubMed] [Google Scholar]
- 18.Uehara G, Suzuki D, Toyoda M, Umezono T, Sakai H. Glomerular expression of platelet-derived growth factor (PDGF)-A, -B chain and PDGF receptor-alpha, -beta in human diabetic nephropathy. Clin Exp Nephrol. 2004;8:36–42. doi: 10.1007/s10157-003-0265-8. [DOI] [PubMed] [Google Scholar]
- 19.(a) Mahimainathan L, Ghosh-Choudhury N, Venkatesan B, Das F, Mandal CC, Dey N, Habib SL, Kasinath BS, Abboud HE, Ghosh Choudhury G. TSC2 deficiency increases PTEN via HIF1alpha. J Biol Chem. 2009;284:27790–27798. doi: 10.1074/jbc.M109.028860. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh Choudhury G, Lenin M, Calhaun C, Zhang JH, Abboud HE. PDGF inactivates forkhead family transcription factor by activation of Akt in glomerular mesangial cells. Cell Signal. 2003;15:161–170. doi: 10.1016/s0898-6568(02)00057-8. [DOI] [PubMed] [Google Scholar]
- 20.Choudhury GG, Ghosh-Choudhury N, Abboud HE. Association and direct activation of signal transducer and activator of transcription1alpha by platelet-derived growth factor receptor. J Clin Invest. 1998;101:2751–2760. doi: 10.1172/JCI1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das F, Ghosh-Choudhury N, Bera A, Dey N, Abboud HE, Kasinath BS, Choudhury GG. Transforming growth factor beta integrates Smad 3 to mechanistic target of rapamycin complexes to arrest deptor abundance for glomerular mesangial cell hypertrophy. J Biol Chem. 2013;288:7756–7768. doi: 10.1074/jbc.M113.455782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dey N, Ghosh-Choudhury N, Das F, Li X, Venkatesan B, Barnes JL, Kasinath BS, Choudhury GG. PRAS40 acts as a nodal regulator of high glucose-induced TORC1 activation in glomerular mesangial cell hypertrophy. J Cell Physiol. 2010;225:27–41. doi: 10.1002/jcp.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das F, Ghosh-Choudhury N, Dey N, Bera A, Mariappan MM, Kasinath BS, Ghosh Choudhury G. High glucose forces a positive feedback loop connecting Akt kinase and FoxO1 transcription factor to activate mTORC1 kinase for mesangial cell hypertrophy and matrix protein expression. J Biol Chem. 2014;289:32703–32716. doi: 10.1074/jbc.M114.605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das F, Dey N, Bera A, Kasinath BS, Ghosh-Choudhury N, Choudhury GG. MicroRNA-214 reduces insulin-like growth factor-1 (IGF-1) receptor expression and downstream mTORC1 signaling in renal carcinoma cells. J Biol Chem. 2016;291:14662–14676. doi: 10.1074/jbc.M115.694331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das F, Ghosh-Choudhury N, Mariappan MM, Kasinath BS, Choudhury GG. Hydrophobic motif site-phosphorylated protein kinase CbetaII between mTORC2 and Akt regulates high glucose-induced mesangial cell hypertrophy. Am J Phys Cell Phys. 2016;310:C583–96. doi: 10.1152/ajpcell.00266.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Gu M, Roy S, Raina K, Agarwal C, Agarwal R. Inositol hexaphosphate suppresses growth and induces apoptosis in prostate carcinoma cells in culture and nude mouse xenograft: PI3K-Akt pathway as potential target. Cancer Res. 2009;69:9465–9472. doi: 10.1158/0008-5472.CAN-09-2805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shen X, Kan S, Hu J, Li M, Lu G, Zhang M, Zhang S, Hou Y, Chen Y, Bai Y. EMC6/TMEM93 suppresses glioblastoma proliferation by modulating autophagy. Cell Death Dis. 2016;7:e2043. doi: 10.1038/cddis.2015.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiram-Bab S, Katz LS, Shapira H, Sandbank J, Gershengorn MC, Oron Y. Platelet-derived growth factor BB mimics serum-induced dispersal of pancreatic epithelial cell clusters. J Cell Physiol. 2014;229:743–751. doi: 10.1002/jcp.24493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heldin CH, Ostman A, Ronnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta. 1998;1378:F79–113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 29.Valius M, Kazlauskas A. Phospholipase C-gamma 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor’s mitogenic signal. Cell. 1993;73:321–334. doi: 10.1016/0092-8674(93)90232-f. [DOI] [PubMed] [Google Scholar]
- 30.Wennstrom S, Siegbahn A, Yokote K, Arvidsson AK, Heldin CH, Mori S, Claesson-Welsh L. Membrane ruffling and chemotaxis transduced by the PDGF beta-receptor require the binding site for phosphatidylinositol 3′ kinase. Oncogene. 1994;9:651–660. [PubMed] [Google Scholar]
- 31.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 32.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Wei Q, Guo C, Dong G, Liu Y, Tang C, Dong Z. Hypoxia, HIF, and associated signaling networks in chronic kidney disease. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18050950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Joshi S, Singh AR, Durden DL. MDM2 regulates hypoxic hypoxia-inducible factor 1alpha stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J Biol Chem. 2014;289:22785–22797. doi: 10.1074/jbc.M114.587493. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joshi S, Singh AR, Zulcic M, Durden DL. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1alpha and HIF2alpha stability and tumor growth, angiogenesis, and metastasis. Mol Cancer Res. 2014;12:1520–1531. doi: 10.1158/1541-7786.MCR-13-0682. [DOI] [PubMed] [Google Scholar]
- 35.Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;13:1–14. doi: 10.2215/CJN.11491116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhattacharjee N, Barma S, Konwar N, Dewanjee S, Manna P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: an update. Eur J Pharmacol. 2016;791:8–24. doi: 10.1016/j.ejphar.2016.08.022. [DOI] [PubMed] [Google Scholar]
- 37.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 38.(a) Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 1996;15:6584–6594. [PMC free article] [PubMed] [Google Scholar]; (b) Weinkove D, Neufeld TP, Twardzik T, Waterfield MD, Leevers SJ. Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr Biol. 1999;9:1019–1029. doi: 10.1016/s0960-9822(99)80450-3. [DOI] [PubMed] [Google Scholar]
- 39.Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 2000;19:2537–2548. doi: 10.1093/emboj/19.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013;3:1345–1354. doi: 10.1158/2159-8290.CD-13-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa H, Sasahara M, Haneda M, Koya D, Hazama F, Kikkawa R. Immunohistochemical characterization of glomerular PDGF B-chain and PDGF beta-receptor expression in diabetic rats. Diabetes Res Clin Pract. 2000;48:87–98. doi: 10.1016/s0168-8227(99)00144-8. [DOI] [PubMed] [Google Scholar]
- 42.Choudhury GG, Mahimainathan L, Das F, Venkatesan B, Ghosh-Choudhury N. c-Src couples PI 3 kinase/Akt and MAPK signaling to PDGF-induced DNA synthesis in mesangial cells. Cell Signal. 2006;18:1854–1864. doi: 10.1016/j.cellsig.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 43.McGlade CJ, Ellis C, Reedijk M, Anderson D, Mbamalu G, Reith AD, Panayotou G, End P, Bernstein A, Kazlauskas A, et al. SH2 domains of the p85 alpha subunit of phosphatidylinositol 3-kinase regulate binding to growth factor receptors. Mol Cell Biol. 1992;12:991–997. doi: 10.1128/mcb.12.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.(a) Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]; (b) Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 45.James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, Alessi DR. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J. 1996;315(Pt 3):709–713. doi: 10.1042/bj3150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.(a) Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]; (b) Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, Wang B, Blenis J, Cantley LC, Toker A, Su B, Wei W. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 2015;5:1194–1209. doi: 10.1158/2159-8290.CD-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.(a) Dey N, Bera A, Das F, Ghosh-Choudhury N, Kasinath BS, Choudhury GG. High glucose enhances microRNA-26a to activate mTORC1 for mesangial cell hypertrophy and matrix protein expression. Cell Signal. 2015;27:1276–1285. doi: 10.1016/j.cellsig.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dey N, Das F, Ghosh-Choudhury N, Mandal CC, Parekh DJ, Block K, Kasinath BS, Abboud HE, Choudhury GG. microRNA-21 governs TORC1 activation in renal cancer cell proliferation and invasion. PLoS One. 2012;7:e37366. doi: 10.1371/journal.pone.0037366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ebner M, Lucic I, Leonard TA, Yudushkin I. PI(3,4,5)P3 engagement restricts Akt activity to cellular membranes. Mol Cell. 2017;65:416–431 (e6). doi: 10.1016/j.molcel.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 50.Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ. Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol. 1999;1:500–506. doi: 10.1038/70293. [DOI] [PubMed] [Google Scholar]
- 51.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.(a) Harskamp LR, Gansevoort RT, van Goor H, Meijer E. The epidermal growth factor receptor pathway in chronic kidney diseases. Nat Rev Nephrol. 2016;12:496–506. doi: 10.1038/nrneph.2016.91. [DOI] [PubMed] [Google Scholar]; (b) Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Ren Physiol. 2015;308:F287–97. doi: 10.1152/ajprenal.00533.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 54.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bernhardt WM, Schmitt R, Rosenberger C, Munchenhagen PM, Grone HJ, Frei U, Warnecke C, Bachmann S, Wiesener MS, Willam C, Eckardt KU. Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int. 2006;69:114–122. doi: 10.1038/sj.ki.5000062. [DOI] [PubMed] [Google Scholar]
- 57.Nordquist L, Friederich-Persson M, Fasching A, Liss P, Shoji K, Nangaku M, Hansell P, Palm F. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J Am Soc Nephrol. 2015;26:328–338. doi: 10.1681/ASN.2013090990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nayak BK, Shanmugasundaram K, Friedrichs WE, Cavaglierii RC, Patel M, Barnes J, Block K. HIF-1 mediates renal fibrosis in OVE26 type 1 diabetic mice. Diabetes. 2016;65:1387–1397. doi: 10.2337/db15-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.(a) Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Ren Physiol. 2008;295:F1023–9. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brukamp K, Jim B, Moeller MJ, Haase VH. Hypoxia and podocyte-specific Vhlh deletion confer risk of glomerular disease. Am J Physiol Ren Physiol. 2007;293:F1397–407. doi: 10.1152/ajprenal.00133.2007. [DOI] [PubMed] [Google Scholar]
- 61.You YH, Quach T, Saito R, Pham J, Sharma K. Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol. 2016;27:466–481. doi: 10.1681/ASN.2015030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.(a) Ziyadeh FN. Mediators of diabetic renal disease: the case for TGF-beta as the major mediator. J Am Soc Nephrol. 2004;15(Suppl. 1):S55–7. doi: 10.1097/01.asn.0000093460.24823.5b. [DOI] [PubMed] [Google Scholar]; (b) Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes. 1995;44:1139–1146. doi: 10.2337/diab.44.10.1139. [DOI] [PubMed] [Google Scholar]
- 63.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 64.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim HW, Kim BC, Song CY, Kim JH, Hong HK, Lee HS. Heterozygous mice for TGF-betaIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int. 2004;66:1859–1865. doi: 10.1111/j.1523-1755.2004.00959.x. [DOI] [PubMed] [Google Scholar]
- 66.Chen S, Hoffman BB, Lee JS, Kasama Y, Jim B, Kopp JB, Ziyadeh FN. Cultured tubule cells from TGF-beta1 null mice exhibit impaired hypertrophy and fibronectin expression in high glucose. Kidney Int. 2004;65:1191–1204. doi: 10.1111/j.1523-1755.2004.00492.x. [DOI] [PubMed] [Google Scholar]
- 67.Das F, Bera A, Ghosh-Choudhury N, Abboud HE, Kasinath BS, Choudhury GG. TGFbeta-induced deptor suppression recruits mTORC1 and not mTORC2 to enhance collagen I (alpha2) gene expression. PLoS One. 2014;9:e109608. doi: 10.1371/journal.pone.0109608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.