

Summary
BRAF and MEK inhibitors have improved clinical outcomes in advanced, BRAFV600-mutated melanomas. Acquired resistance occurs in most patients, with numerous and diverse drivers. We obtained pre-treatment and progression biopsies from a patient who progressed on dabrafenib and trametinib. In addition to a preserved BRAFV600E mutation, an internal deletion (rearrangement) of BRAF was observed in the progression sample. This deletion involved exons 2-8, which includes the Ras-binding domain, and is analogous to previously documented BRAF fusions and splice variants known to reactivate RAS-RAF-MEK-ERK signaling. In a large cohort of melanomas, 10 additional internal deletions were identified (0.4% of all melanomas; 9 of which had concurrent BRAF mutations), as well as sporadically in other tumor types. Thus, we describe a novel mechanism of resistance to BRAF and MEK inhibition.
Keywords: BRAF, internal deletion, resistance, rearrangement, dabrafenib, trametinib, vemurafenib
The combination of BRAF and MEK inhibitors produce potent responses and improved overall survival in patients with advanced melanoma harboring BRAFV600 mutations (Long et al., 2016). Acquired resistance limits the clinical benefit of these agents in most patients, and arises at a median of 9-11 months on therapy (Flaherty et al., 2012; Robert et al., 2014). Mechanisms of acquired resistance are numerous and diverse, and include alterations activating the RAS-RAF-MEK-ERK pathway, and those promoting alternative signaling networks (Rizos et al., 2014; Shi et al., 2014; Van Allen et al., 2014). Genomic and transcriptomic alterations driving resistance that affect BRAF specifically include gene amplification and alternate splicing. The latter alteration removes the inhibitory RAS-binding domain (RBD), thus promoting constitutive signaling (Poulikakos et al., 2011).
Other genomic alterations in BRAF have been described across the spectrum of human tumors. These include atypical (non-V600) BRAF mutations, BRAF fusions, and BRAF kinase domain duplications, that each promote RAS-RAF-MEK-ERK signaling, and are variably sensitive to RAF or MEK inhibitors (Botton et al., 2013; Dahlman et al., 2012; Hutchinson et al., 2013; Klempner et al., 2016). Short, oncogenic internal deletions in BRAF have also been described in pancreatic cancer, which confer resistance to BRAF inhibitors (Chen et al., 2016).
Large internal deletions (also termed rearrangements) in BRAF that delete the RBD would be predicted to confer similar functional consequences as BRAF fusions or alternatively-spliced BRAF. These alterations, however, have not been described in human cancers to our knowledge. Here, we describe a large BRAF internal deletion in a melanoma sample with acquired resistance to BRAF and MEK inhibitors, and evaluate the incidence of BRAF internal deletions across human cancer types.
A woman in her 30s with advanced melanoma with axillary lymph node, liver, thoracic spine, and subcutaneous disease involvement presented to our clinic. She was highly symptomatic from an extensive disease burden with pain and fatigue. Laboratory testing showed elevated lactate dehydrogenase; molecular testing revealed a BRAFV600E mutation. She was initiated on dabrafenib and trametinib and experienced rapid clinical improvement (Figure 1). A CT scan performed two months following start of therapy demonstrated partial response in nearly all visible tumors (-32% decrease in measurable tumor diameters). Four months following treatment initiation, she experienced increasing size of a subcutaneous back lesion, and rise in LDH. She was continued on dabrafenib and ipilimumab was added in hopes of inducing a durable response; trametinib was discontinued for safety reasons (given prior reports of colon perforation with the triple combination) (Minor et al., 2015). She experienced further progressive disease in numerous lesions approximately 2 months later. Based on the rapid painful tumor growth, she underwent palliative resection of the subcutaneous lesion on her back. Thereafter, she was treated with pembrolizumab without response and ultimately died of progressive disease.
Figure 1. Timecourse of treatment and biopsies.
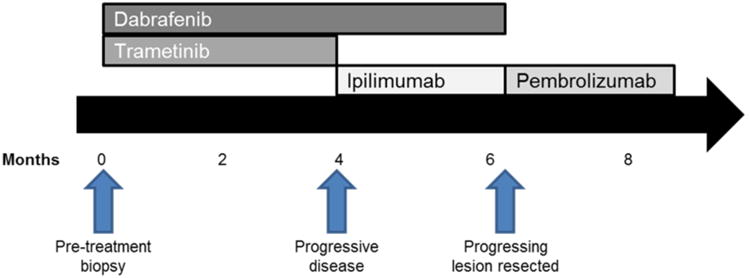
The subcutaneous tumor on the patient's back was biopsied one week prior to initiation of dabrafenib and trametinib, and the same lesion was resected following disease progression (Figure 1). Both samples were evaluated by an extensively validated hybrid-capture based next generation sequencing platform that sequences exons from 315 genes and introns from 28 genes (Frampton et al., 2013). Sequencing from both the pre-treatment and disease-progression samples identified BRAFV600E, PTENG129E, and TERT promoter mutations, as well as CDKN2A/B loss. Minimal change in BRAFV600E mutant allele frequency was noted between pre-treatment (42%) and progression (41%) tumors. The sample obtained at disease progression was also found to have a BRAF internal deletion (rearrangement). Breakpoints in introns 1 and 8 were identified, thus resulting in the loss of exons 2-8, which includes the inhibitory RAS-binding domain (Figure 2A). The breakpoints resulted in an internal, in-frame deletion rather than a fusion product, which was confirmed by RNA sequencing (Figure 2B; Supplemental Figure 2). IGF1R amplification was also identified in the progression sample only, consistent with prior data that resistance to BRAF + MEK combination therapy is usually mediated by multiple resistance mechanisms functioning in concert (Long et al., 2014; Moriceau et al., 2015; Wagle et al., 2014).
Figure 2.
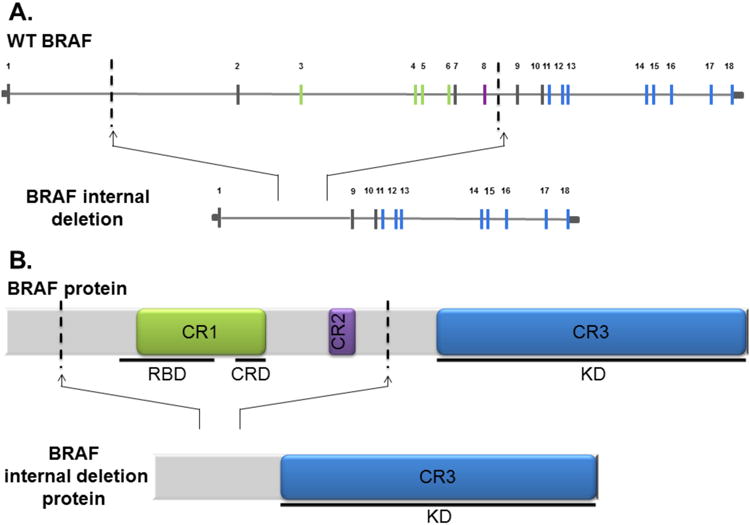
(A) Scaled genomic representation of BRAF (top) and BRAF internal deletion observed in our case (bottom). Exons are numerically labeled. Exons encoding highly conserved protein domains are depicted in green (CR1), purple (CR2), and blue (CR3). Dashed lines indicate the breakpoints found in the patient case report that resulted in the B-Raf internal deletion variant depicting the loss of exons 2-8. (B) Protein schematic of full length B-Raf (top). Highly conserved protein domains are depicted in green, purple, and blue: CR1 including the inhibitory Ras binding domain (RBD) as well as the cysteine rich domain (CRD) required for Ras binding; CR2; and CR3 which contains the entire kinase domain. Dashed lines indicate genomic breakpoint locations in our observed case.
We then surveyed the spectrum of tumors sequenced by Foundation Medicine, to assess for additional BRAF internal deletions that remove the RBD, while sparing the kinase domain. Among 2439 melanomas, 10 additional BRAF internal deletions were identified (0.4% of all melanomas). Of these 10 samples, 9 had concurrent BRAF mutations, including BRAFV600 mutations in 6 and atypical, non-V600 BRAF mutations in 3 (Table 1). No NRAS or KRAS mutations were observed in these samples. BRAF internal deletions involving the RBD and sparing the kinase domain were also identified in a diverse set of 16 other samples, including in multiple myeloma, glioblastoma, breast cancer, non-small cell lung cancer, and others (Table 1; Supplemental Figure 1). A surprisingly high number of these (7 of 16; 44%) were found to have concurrent BRAF mutations.
Table 1. Genomic and pathologic details of tumors harboring BRAF internal deletions.
| Disease Ontology | N-Ras Mutations | K-Ras Mutations | B-Raf Mutations | BRAF Exons Deleted |
|---|---|---|---|---|
| unknown primary melanoma | ND | ND | V600 K601>E | 2-8 |
| unknown primary melanoma | ND | ND | V600E | 4-8 |
| unknown primary melanoma | ND | ND | V600K | 2-10 |
| unknown primary melanoma | ND | ND | ND | 2-10 |
| unknown primary melanoma | ND | ND | V600E | 3-10 |
| skin melanoma | ND | ND | V600E | 2-8 |
| skin melanoma | ND | ND | T599I | 3-8 |
| skin melanoma | ND | ND | V600E | 2-8 |
| head and neck melanoma | ND | ND | L597S | 2-8 |
| vulva melanoma | ND | ND | D594G | 3-8 |
| bladder urothelial (transitional cell) carcinoma | ND | ND | D587H | 3-8 |
| bone marrow multiple myeloma | ND | ND | V600E | 2-8 |
| bone marrow multiple myeloma | ND | ND | V600E | 2-8 |
| bone marrow multiple myeloma | ND | ND | ND | 2-10 |
| brain glioblastoma | ND | ND | ND | 3-8 |
| breast carcinoma (nos) | ND | ND | ND | 4-7 |
| breast invasive ductal carcinoma | ND | ND | ND | 5-9 |
| lung adenocarcinoma | ND | G12C | ND | 2-9 |
| lung non-small cell lung carcinoma | ND | ND | D594G | 4-8 |
| lung non-small cell lung carcinoma | ND | ND | G464A | 4-8 |
| nasopharynx and paranasal sinuses carcinoma | ND | ND | L485F | 3-8 |
| pancreas ductal adenocarcinoma | ND | ND | S467L | 2-10 |
| prostate acinar adenocarcinoma | ND | ND | ND | 4-8 |
| thymus thymoma | ND | ND | ND | 3-10 |
| unknown primary adenocarcinoma | ND | ND | ND | 2-7 |
| unknown primary germ cell tumor | ND | amplification | ND | 3-8 |
ND: Not detected. NOS: Not otherwise specified
Comprehensive genomic profiling of paired pre-treatment and disease progression biopsies, has uncovered numerous mechanisms of resistance to targeted therapies in diverse tumor types. Using this approach, we identified a novel BRAF internal deletion mimicking previously reported alternative splicing events that confers resistance to BRAF and MEK inhibition. While other alterations in BRAF arise in the context of BRAF inhibitor therapy (amplifications and alternative splicing) or in BRAFV600 wild type samples (fusions, kinase duplications, and short internal deletions), this is the first report of a large BRAF internal deletion involving exons encoding the RAS-binding domain. Interestingly, in melanoma, all but one of these tumors harbored concurrent BRAF mutations. Taken with the results of our paired biopsies, we would hypothesize that these internal deletions may serve as weak oncogenes on their own, but enhance mutant BRAF signaling. An unexpectedly high number of patients with other cancers also harbored concurrent BRAF point mutations, providing further evidence for this hypothesis.
Our case suggests that BRAF internal deletions likely contribute to acquired resistance to BRAF +/- MEK inhibition in concert with other resistance mechanisms. One could also speculate that these alterations drive primary resistance to BRAF inhibitors if present in the pre-treatment setting. We elected not to model these alterations in vitro, since our groups and others have extensively shown that removal of the RAS-binding domain through fusions or alternate splicing activates RAS-RAF-MEK-ERK signaling and mediates resistance to BRAF inhibitors (Hutchinson et al., 2013; Poulikakos et al., 2010; Shi et al., 2014). Characterizing effects of particular BRAF internal deletions is an important future direction. Further, evaluating the effect of tumor heterogeneity and outgrowth of resistant clones is another critical question (Shi et al., 2014). It also remains unclear how the addition of a MEK inhibitor contributes to the development of BRAF internal deletions, as compared with patients treated with BRAF inhibitor monotherapy.
Other potential resistance mechanisms were identified in this patient as well, including PTEN mutations. Pre-existing PTEN alterations decrease the duration of, but do not preclude therapeutic responses, whereas acquired mutations/loss appear to mediate (or contribute to) therapeutic resistance (Van Allen et al., 2014). Thus, PTEN mutations may have contributed to the short duration of response in this case (4 months). IGF1R genomic amplification may have also played a role, as overexpression at the protein level has been described as a mechanism of acquired resistance to BRAF inhibitors (Nazarian et al., 2010). These intrinsic and acquired genomic alterations likely mediated resistance to BRAF/MEK inhibition synergistically. Several elegant studies have demonstrated that resistance to combination therapy is driven by multiple mechanisms functioning in concert, supporting this notion (Long et al., 2014; Moriceau et al., 2015; Villanueva et al., 2013; Wagle et al., 2014).
We and others have also demonstrated that MEK or unselective RAF inhibitors (e.g. sorafenib) may be effective in patients with BRAF fusions (Botton et al., 2013; Hutchinson et al., 2013; Menzies et al., 2015). In the context of BRAF or BRAF + MEK inhibitor resistant melanoma, these agents are unlikely to provide a significant clinical benefit as monotherapy. However, in tumors harboring BRAF internal deletions without concurrent BRAFV600 mutations (including those with concurrent non-V600 mutations), MEK or RAF inhibition should be investigated. In conclusion, this is the first report of a BRAF internal deletion to our knowledge and defines an uncommon but recurrent genetic subset of melanomas and other cancers.
Supplementary Material
Supplemental Figure 1 Schematic of additional B-Raf internal deletion variants identified in Foundation Medicine's database of all interrogated cancers.
Supplemental Figure 2: Chromosomal breakpoints in BRAF gene are shown in pre- (A,C) and post- (B,D) treatment specimens. The 400 bp genomic region chr7:140,490,823-140,491,222 is shown (A,B) and the 400bp genomic region chr7:140,611,466-140,611,865 is shown (C,D). 49 basepair sequencing reads are shown, with bases matching the reference sequence in gray and mismatching bases indicted with colors. Reads belonging to pairs that map to different chromosomes, greater than 2,000 basepairs apart on the same chromosome, and with improper relative mapping direction are shown in color and in a separate track (A,B).
Significance.
This is the first report of a BRAF internal deletion affecting the RAS-binding domain in a similar fashion to fusions or splice variants in BRAF. BRAF internal deletions were identified in melanomas and other tumor types, and thus comprise a recurrent, albeit uncommon genetic subset of cancer. Finally, the identification of this alteration in a progression sample (but not pre-treatment) suggests BRAF internal deletions mediate resistance to BRAF/MEK inhibition.
Acknowledgments
Funding: DBJ is supported by K23 CA204726 and by the James C. Bradford, Jr. Fund in Melanoma Cancer Research.
Footnotes
Conflicts of Interest: DBJ is on advisory boards for BMS, Genoptix, Incyte, and Merck. ZC, SA, DF, SB, JSR, VAM, and PJS are employees of Foundation Medicine. CML has served as a consultant for Pfizer, Novartis, Genoptix, Sequenom, Clovis, and Ariad and has been an invited speaker for Abbott and Qiagen. JAS has served as a consultant and Advisory Boards for Novartis, Genentech, Array, and Merck.
References
- Botton T, Yeh I, Nelson T, Vemula SS, Sparatta A, Garrido MC, Allegra M, Rocchi S, Bahadoran P, Mccalmont TH, et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment cell & melanoma research. 2013 doi: 10.1111/pcmr.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Zhang Y, Van Horn RD, Yin T, Buchanan S, Yadav V, Mochalkin I, Wong SS, Yue YG, Huber L, et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer discovery. 2016;6:300–15. doi: 10.1158/2159-8290.CD-15-0896. [DOI] [PubMed] [Google Scholar]
- Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, Atefi M, Su Z, Branch S, Lyle P, et al. BRAF L597 mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer discovery. 2012 doi: 10.1158/2159-8290.CD-12-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. The New England journal of medicine. 2012 doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson KE, Lipson D, Stephens PJ, Otto G, Lehmann BD, Lyle PL, Vnencak-Jones CL, Ross JS, Pietenpol JA, Sosman JA, et al. BRAF Fusions Define a Distinct Molecular Subset of Melanomas with Potential Sensitivity to MEK Inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:6696–702. doi: 10.1158/1078-0432.CCR-13-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempner SJ, Bordoni R, Gowen K, Kaplan H, Stephens PJ, Ou SH, Ali SM. Identification of BRAF Kinase Domain Duplications Across Multiple Tumor Types and Response to RAF Inhibitor Therapy. JAMA oncology. 2016;2:272–4. doi: 10.1001/jamaoncol.2015.4437. [DOI] [PubMed] [Google Scholar]
- Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nature communications. 2014;5:5694. doi: 10.1038/ncomms6694. [DOI] [PubMed] [Google Scholar]
- Long GV, Weber JS, Infante JR, Kim KB, Daud A, Gonzalez R, Sosman JA, Hamid O, Schuchter L, Cebon J, et al. Overall Survival and Durable Responses in Patients With BRAF V600-Mutant Metastatic Melanoma Receiving Dabrafenib Combined With Trametinib. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016;34:871–8. doi: 10.1200/JCO.2015.62.9345. [DOI] [PubMed] [Google Scholar]
- Menzies AM, Yeh I, Botton T, Bastian BC, Scolyer RA, Long GV. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment cell & melanoma research. 2015 doi: 10.1111/pcmr.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor DR, Puzanov I, Callahan MK, Hug BA, Hoos A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment cell & melanoma research. 2015;28:611–2. doi: 10.1111/pcmr.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J, et al. Tunable-Combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Cotargeting but Result in Melanoma Drug Addiction. Cancer cell. 2015;27:240–56. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:1965–77. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. The New England journal of medicine. 2014 doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer discovery. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer discovery. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell reports. 2013;4:1090–9. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer discovery. 2014;4:61–8. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 Schematic of additional B-Raf internal deletion variants identified in Foundation Medicine's database of all interrogated cancers.
Supplemental Figure 2: Chromosomal breakpoints in BRAF gene are shown in pre- (A,C) and post- (B,D) treatment specimens. The 400 bp genomic region chr7:140,490,823-140,491,222 is shown (A,B) and the 400bp genomic region chr7:140,611,466-140,611,865 is shown (C,D). 49 basepair sequencing reads are shown, with bases matching the reference sequence in gray and mismatching bases indicted with colors. Reads belonging to pairs that map to different chromosomes, greater than 2,000 basepairs apart on the same chromosome, and with improper relative mapping direction are shown in color and in a separate track (A,B).