

Abstract
Neurofibromatosis type 2 (NF2) is a tumor-forming disease of the nervous system caused by deletion or by loss-of-function mutations in NF2, encoding the tumor suppressing protein neurofibromin 2 (also known as schwannomin or merlin). Neurofibromin 2 is a member of the ezrin, radixin, moesin (ERM) family of proteins regulating the cytoskeleton and cell signaling. The correlation of the tumor-suppressive function and conformation (open or closed) of neurofibromin 2 has been subject to much speculation, often based on extrapolation from other ERM proteins, and controversy. Here we show that lipid binding results in the open conformation of neurofibromin 2 and that lipid binding is necessary for inhibiting cell proliferation. Collectively, our results provide a mechanism in which the open conformation is unambiguously correlated with lipid binding and localization to the membrane, which are critical for the tumor-suppressive function of neurofibromin 2, thus finally reconciling the long-standing conformation and function debate.
Neurofibromin 2 (NF2) is a tumour suppressor that inhibits cell growth. Here the authors combine functional, biochemical, and structural studies and show that lipid-bound NF2 adopts an open conformation and that NF2 lipid binding is required for inhibition of cell proliferation.
Introduction
Mutations in the NF2 gene cause Neurofibromatosis type 2 (NF2), a nervous system tumor-forming disease that is characterized by the development of bilateral vestibular schwannomas1. NF2 has an incidence of 0.004% of the population, where it is dominantly inherited. However, more than half of NF2 patients develop the disease from loss-of function due to de novo mutations1,2. NF2 manifests itself in the development of multiple schwannomas on cranial and peripheral nerves as well as of ependymomas and meningiomas that can only be treated surgically where loss-of function of the involved nerve is often an unpreventable consequence. Thus, chemotherapies to slow or eliminate tumor formation are urgently needed, especially since NF2 mutations that result in inactivation of its gene product, neurofibromin 2 (also known as schwannomin or merlin for moesin-ezrin-radixin-like protein), are also found in spontaneous meningiomas and schwannomas and several other types of cancers, such as breast, colorectal, clear cell renal cell carcinoma, hepatic, glioma multiforme, mesothelioma, and prostate. Thus, understanding the neurofibromin 2 activation mechanism will increase our knowledge of the role of NF2 mutations in cancer and might eventually aid prognosis and future chemotherapeutic therapies.
Neurofibromin 2 belongs to the FERM (band 4.1, ezrin, radixin, moesin) gene family characterized by an N-terminal FERM domain (Fig. 1a) that binds to the plasma membrane, a central α-helical domain, and a C-terminal tail domain that binds to the cortical actin cytoskeleton3. In contrast, neurofibromin 2 has a unique cytoskeleton binding site on its N-terminal domains4. By directly or indirectly linking the actin cytoskeleton, neurofibromin 2 is proposed to be involved in organization and maintenance of cytoskeleton architecture beneath plasma membrane. Neurofibromin 2 is a unique tumor suppressor protein that inhibits cell growth both from plasma membrane and in the nucleus upon cell confluency5,6. For example, research carried out by several groups indicated that neurofibromin 2 suppresses cellular growth either inhibiting conversion of Ras/Rac-GDP bound forms to GTP bound form or by hampering Ras recruitment at the plasma membrane7–9. Neurofibromin 2 also attenuates growth factor receptors expression and activity at the plasma membrane in both drosophila and mammals10 and exerts its growth-suppressive function in the nucleus, where it inhibits the DCAF ubiquitin ligase activity11. Neurofibromin 2 is a major regulator of Hippo signaling that is involved in the conserved kinase cascade that inhibits organ overgrowth through cytoplasmic of sequestration of YAP by phosphorylation, and blocking its ability to promote transcriptional enhancer activation domain (TEAD)–dependent transcription of genes involved in proliferation and survival such as CTGF, Cyr61, Axl, BIRC5, and PTGS212–14.
Fig. 1.
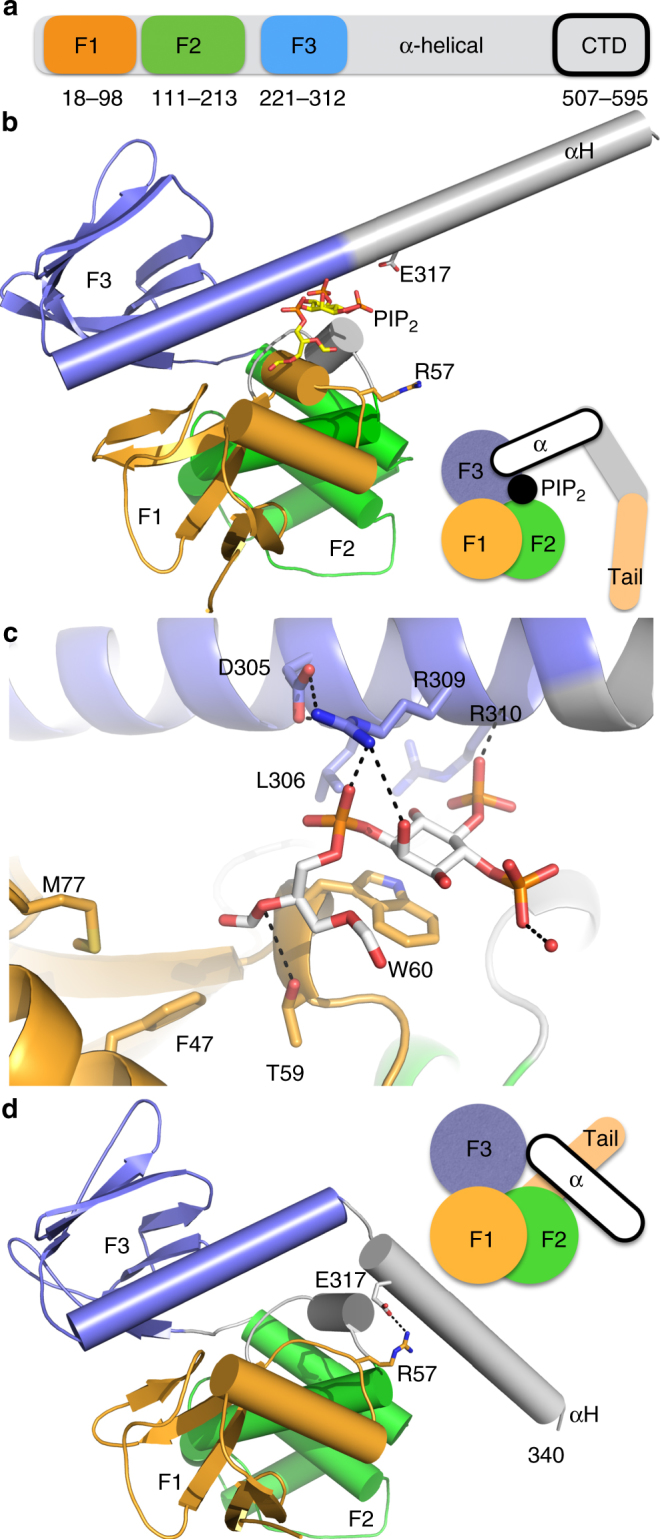
Lipid binding to neurofibromin 2 causes major conformational changes. a Schematic depiction of the domain organization of full-length neurofibromin 2. The F1, F2, and F3 FERM subdomains and the C-terminal domain (CTD) are indicated that are connected by the central α-helical domain. b The 2.61 Å PIP2-bound neurofibromin 2 structure. The F1 subdomain (residues 18–98) is shown in light orange, F2 (residues 111–213) in green, and F3 (residues 221–312) in blue. The α-helix from the central helical domain, αH, is shown in gray (residues 315–339). The cartoon illustrates binding of PIP2 to full-length neurofibromin 2 with its head–tail interaction severed. c Close-up view of the PIP2 binding site. The carbon atoms of PIP2 are shown in gray, of the neurofibromin 2 F1 subdomain in orange, and F3 in blue. Hydrogen bonds are indicated and key binding residues are labeled. d View of the closed FERM neurofibromin 2 structure (PDB entry 1isn34) in the same orientation as our lipid-bound structure shown in panel b. The electrostatic interaction between E317 OE2 and R57 NH2 is indicated (and the distance is 2.9 Å) that is severed in our lipid-bound structure shown in b. The cartoon highlights the distinct conformation of α-helix αH from the central domain
The neurofibromin 2 head–tail interaction is critical for controlling the tumor-suppressive function of neurofibromin 215,16, which seems also to be regulated by phosphorylation. Specifically, protein kinase A and p21-activated kinases phosphorylate S518 residing on the tail domain, thereby inactivating the tumor-suppressive activity of neurofibromin 217–20, while the myosin phosphatase MYPT1-PP1δ activates its tumor suppressor function by dephosphorylating S51821. However, the relationship between neurofibromin 2 activity and conformation is highly controversial6. Originally, by extrapolation from other ERM proteins, neurofibromin 2 was assumed to be phosphorylated at the plasma membrane in its open conformation to promote receptor mediated signaling events that control cell proliferation and survival22,23. However, in its closed conformation, neurofibromin 2 does not seem to bind to growth factor receptors in its supposedly closed, tumor suppressor active, phosphorylated form24,25. More recently, fluorescence resonance energy transfer, mutagenesis, and small-angle neutron scattering studies suggested that unphosphorylated neurofibromin 2 is in its closed conformation that can interact with binding partners including phosphatidylinositol 4,5-bisphosphate (PIP2)26–28.
Here we report the crystal structure of the neurofibromin 2 N-terminal domain in complex with PIP2, which reveals a large conformational change upon lipid binding. Our hydrogen-deuterium exchange experiments with full-length neurofibromin 2 confirm that lipid binding severs the neurofibromin 2 head–tail interaction. Finally, by introducing our structure-based and patient-derived mutagenesis of neurofibromin 2 in live cells, we find that the neurofibromin 2 conformational changes associated with lipid binding and potentially membrane attachment are necessary for neurofibromin 2 inhibition of cell proliferation and that lipid binding and membrane attachment are necessary for neurofibromin 2 to exert its cell growth inhibiting functions and to inhibit YAP activity.
Results
Lipid binding alters the conformation of neurofibromin 2
Neurofibromin 2 localizes to membrane rafts29,30, the hotspot for many signaling events, and its membrane attachment has been shown to be important for its anti-proliferative functions31–33. To gain detailed molecular insight into this important interaction, we determined the 2.71 Å crystal structure of neurofibromin 2 (residues 1–339) in complex with PIP2 (Fig. 1b; Supplementary Tables 1, 2). The crystal structure contains two polypeptide chains, two polyethylene glycols, two phosphates, and two PIP2 molecules in the asymmetric unit (Supplementary Fig. 1, 2). The two polypeptide chains in the asymmetric unit are very similar and can be superimposed with r.m.s.d. of 0.219 Å for 2,309 atoms (Supplementary Fig. 3a). In both polypeptide chains, the PIP2 inositol head group is sandwiched between Trp-60 and Arg-309, the 5′ inositol phosphate interacts with the backbone of Arg-310 and the 1-phosphate group is in hydrogen-bonding distance to Arg-309 (Fig. 1c, d; Supplementary Fig. 3b; Supplementary Table 3). The oxygen atoms of the diacylglycerol interact with the hydroxyl group of Thr-59.
Akin to the known apo structure (PDB entry 1isn34), our PIP2-bound structure comprises the entire FERM domain and the following additional α-helix from the helical domain (αH; residues 315–340) (Fig. 1d). While the lipid-bound FERM domain part of the structure (residues 21–312) resembles the apo FERM structure, the α-helix of the helical domain αH undergoes a large structural change and adopts a distinct conformation. In the apo structure, a short loop (residues 312–315) allows αH to interact with the F1 FERM subdomain (specifically by electrostatic interactions of Arg-57 with Asp-314). Upon PIP2 binding, the acyl group stacks with the side chain of Arg-57 at this interface and severs the FERM and αH interaction further by exposing its 5′ phosphate group into the solvent. This causes the loop (residues 312–315) that keeps αH interacting with F1 in the apo structure, to become helical, and thereby extending the last F3 α-helix (residues 290–312) to form one continuous long α-helix (residues 290–339) instead in the lipid-bound structure by a ~60° relative rotation.
Lipid binding severs the neurofibromin 2 head–tail interaction
To confirm our structural findings in solution, we performed hydrogen-deuterium exchange (HDX) and mass spectrometry experiments on the full-length protein in the presence and absence of PIP2 micelles (Supplementary Fig. 4, Supplementary Data 1). In agreement with our neurofibromin 2/PIP2 and the head/tail crystal structures, the regions that showed most differential deuterium exchange or the highest deprotection profiles in the full-length apo versus PIP2-bound proteins were residues 207–213, where particularly Ile-210 was identified in engaging in hydrophobic interactions with the tail domain35 (Fig. 2a). A further region that showed the most deprotection (residues 122–126) is not involved in the head–tail interface which thus likely presents an interaction interface with the central helical domain that is missing in the crystal structures. Thus, PIP2 releases the tail domain as well as parts of the central helical domain.
Fig. 2.
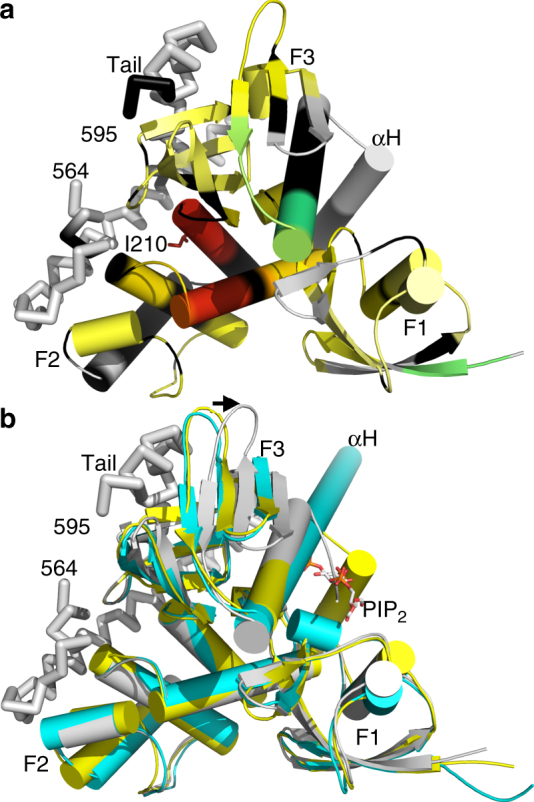
Hydrogen-deuterium exchange (HDX) and mass spectrometry mapping onto the neurofibromin 2 head/tail structure (PDB entry 4zrj35). a Differential deuterium exchange (ΔHDX) between apo and PIP2 bound residues are color coded from red to blue with warm colors representing increased conformational dynamics (red being the relative highest D2O uptake) and cool colors representing decreased conformational dynamics (blue being the lowest D2O uptake); gray, no statistically significant changes between compared conditions; black, regions that have no sequence coverage or include prolines that have no amide hydrogen exchange activity. I210 that binds the tail domain and shows largest degree of deprotection upon binding to PIP2 is shown in sticks and labeled. The tail domain is shown as a Cα-trace. b Superposition of the apo structure (yellow) onto the head–tail (PDB entry 4zrj35; gray), and PIP2-bound structures (cyan). PIP2 is shown as spheres and the tail domain as a Cα-trace. The arrow indicates the movement upon binding of the tail domain of 7.3 Å at the tip of loop (residue K279)
Superposition of the apo (PDB entry 1isn34) and the FERM/tail structures (PDB entry 4zrj35) onto our lipid-bound structure further confirms that our lipid-bound structure is in its open conformer (Fig. 2b). While all three structures are similar, the F3 subdomain is moved over 7 Å in the head/tail structure compared to the lipid-bound and apo structures to allow binding of the tail domain. Importantly, in our lipid-bound structure, the F3 subdomain is in the conformer seen in the apo structure in the space that is occupied by the tail domain in the head/tail structure. Thus, lipid binding is consistent with the unbound, open conformation.
The neurofibromin 2/PIP2 open conformation is the active state
Isothermal calorimetry data for the FERM domain and full-length proteins with PIP2 micelles determined binding constants of 3.3 μM and 0.65 μM, respectively28. To determine the correlation between the open lipid-bound conformation and tumor-suppressive function, we first generated a lipid binding deficient mutant (T59V, W60E, R309Q, R310Q) neurofibromin 2 (Fig. 3a, Supplementary Fig. 5). By lipid co-sedimentation assay, we found that wild-type neurofibromin 2 binds PIP2, while our lipid binding deficient mutant did not. Thus, residues Thr-59, Trp-60, Arg-309, and Arg310 are crucial for neurofibromin 2 attachment to the cell membrane.
Fig. 3.

The conformation of neurofibromin 2 dictates its binding. a Lipid co-sedimentation analysis of the PIP2 binding to wild type and our lipid binding deficient mutant neurofibromin 2. wild-type neurofibromin 2 (residues 1–339) is soluble in the absence of PIP2 and pellets in the presence of PIP2. Mutant (T59V, W60E, R309Q, R310Q) neurofibromin 2 (residues 1–339) remains soluble in the absence and presence of PIP2. S supernatant, P pellet, WT wild type, LBD lipid binding deficient. b Lipid co-sedimentation analysis of the PIP2 binding to wild-type and disease-derived mutant neurofibromin 2. Wild-type neurofibromin 2 (residues 1–339) is soluble in the absence of PIP2 and pellets in the presence of PIP2. Mutant (W60C) neurofibromin 2 (residues 1–339) is soluble in the absence of PIP2, while a small fraction pellets in the presence of PIP2. S supernatant, P pellet, WT wild type. Microscale thermophoresis (MST) measurements show the binding of PIP2 to c wild-type full-length neurofibromin 2 (Kd = 8.02 ± 0.91 μM) or to d our lipid binding deficient (LBD) mutant (T59V, W60E, R309Q, R310Q; Kd = 859.23 ± 184.65 μM). MST measurements show binding of LATS1 (residues 69–100) to e wild type (Kd = 39.31 ± 4.25 μM), f the neurofibromin/PIP2 complex (Kd = 3.77 ± 0.72 μM), or g to our LBD neurofibromin 2 mutant (Kd = 175.54 ± 34.49 μM). No binding was observed for the artificially closed A585W-R588K (AR) mutants to h PIP2, or i LATS1. Error bars represent ±S.D., n = 3 (three independent measurements with the same laser power)
We next sought to understand the effect of the patient-derived W60C neurofibromin 2 mutant onto binding to the plasma membrane given that a key lipid binding residue, Trp-60, was replaced. We found that the W60C mutant bound lipids about four times weaker compared to the wild type as judged by our co-sedimentation assay and gel electrophoresis (Fig. 3b, Supplementary Fig. 5). Unfortunately, we were unable to obtain soluble proteins for other disease relevant mutations near the lipid binding pocket (F62S and L64P) to evaluate their effects on membrane attachment.
To link the conformational effects of lipid binding on neurofibromin 2 to its function, we determined the binding of full-length GST-tagged wild type and mutant neurofibromin 2 to the Hippo pathway kinase LATS1 and PIP2 by microscale thermophoresis (MST). Wild-type neurofibromin 2 bound PIP2 with 8 μM (Fig. 3c), while we obtained a weak affinity (Kd of 0.859 mM) for our lipid binding deficient mutant with respect to PIP2 (Fig. 3d). The LATS1 kinase bound wild-type neurofibromin 2 with 39 μM (Fig. 3e) and this interaction was increased about ten-fold in the presence of PIP2 (Fig. 3f). Our lipid binding deficient mutant bound LATS1 about 4.5 times weaker (0.175 mM; Fig. 3g) compared to wild-type neurofibromin 2. Consistent with the notion that lipid binding induces the open conformer, the constitutively closed neurofibromin 2 A585W/R588K mutant did not bind to PIP2 (Fig. 3h) or LATS1 (Fig. 3i). Thus, PIP2 binding releases the neurofibromin 2 tail domain from its head domain to then allow the open neurofibromin 2 conformer to recruit the downstream interaction partners in the Hippo signaling.
To assess the functional significance of lipid binding to neurofibromin 2, we examined the ability of our lipid binding deficient neurofibromin 2 to inhibit cell growth and compared this to neurofibromin 2 wild type (Fig. 4a–f, Supplementary Fig. 6). NF2-null mouse schwannoma cells (SC4) or immortalized human Schwann cells that stably suppress neurofibromin 2 expression (hSCλ-shNF2), or immortalized human embryonic kidney cells (293 T), were transfected with expression plasmids for wild type and mutant neurofibromin 2 and cell numbers were counted up to 72 h. While expression of wild-type neurofibromin 2 suppressed the growth of these cells, the lipid binding deficient neurofibromin 2 expressing cells grew at rates similar to controls.
Fig. 4.
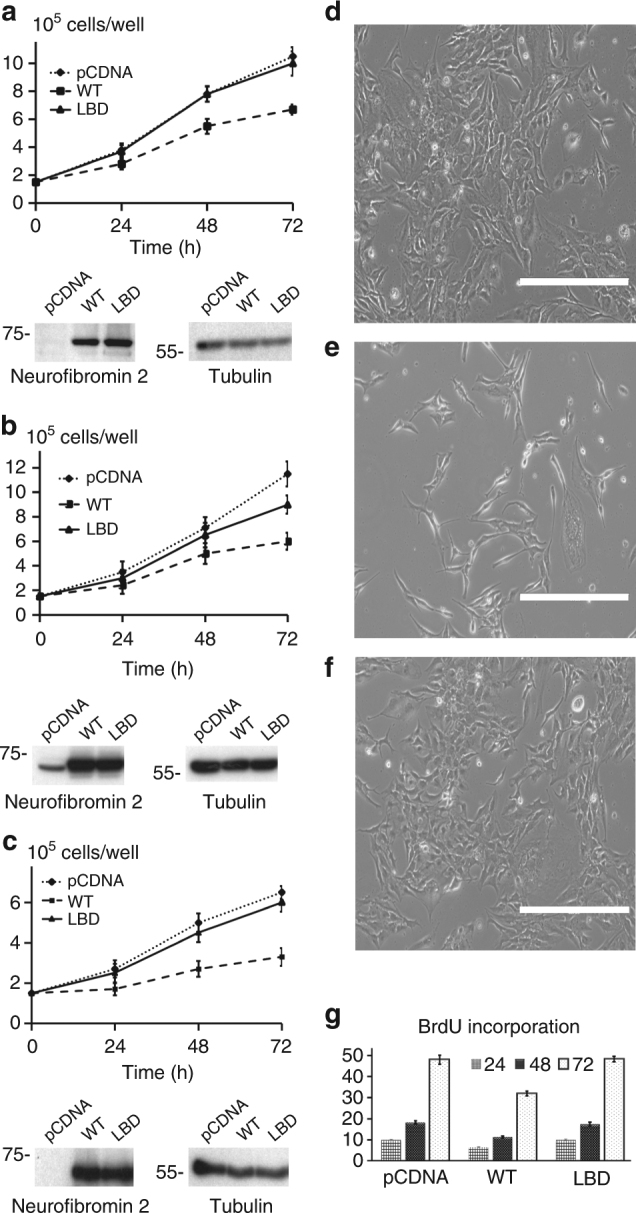
Lipid binding deficient mutants of neurofibromin 2 display impaired inhibition of cell proliferation. a SC4, b HEK293T, or c hSCλ-shNF2 cells were transfected with expression vectors for wild type and lipid binding deficient neurofibromin 2 or empty vector control (pCDNA). Total cell numbers were counted over 72 h. Means of each data point were calculated from three independent biological replicates conducted in triplicate. Error bars represent ± S.D. Immunoblot analysis was used to verify similar expression levels of the indicated neurofibromin 2 alleles. Tubulin was used as a control. The blots shown are representative of three biological replicates. For SC4 cells: difference between pCDNA and lipid binding deficient neurofibromin 2, < 0.7680 (i.e., not significant); pCDNA and wild-type neurofibromin 2, < 0.0001 (i.e., significant); lipid binding deficient and wild-type neurofibromin 2 proteins, < 0.0001 (i.e., significant). For HEK293T cells: difference between pCDNA and lipid binding deficient neurofibromin 2, <0.0013 (i.e., significant); pCDNA and wild-type neurofibromin 2, <0.0001 (i.e., significant); lipid binding deficient and wild-type neurofibromin 2 proteins, <0.0001 (i.e., significant). For hSCλ-shNF2 cells: difference between pCDNA and lipid binding deficient neurofibromin 2, <0.2476 (i.e., not significant); pCDNA and wild-type neurofibromin 2, <0.0001 (i.e., significant); the lipid binding deficient and wild-type neurofibromin 2 proteins, <0.0001 (i.e., significant). Scalebar size is 400 μm. d–f Phase contrast microscopy images, taken at the 72 h time point, of SC4 cells that were used in the BrdU cell proliferation assay. SC4 cells transfected with d pCDNA, e neurofibromin 2, and f the neurofibromin 2 lipid binding deficient mutant. g Cells expressing lipid binding deficient mutants of neurofibromin 2 display impaired inhibition of BrdU incorporation. SC4 cells were transfected with expression vectors for wild type and lipid binding deficient neurofibromin 2 or empty vector control (pCDNA) and BrdU incorporation was assessed over 72 h. Means of each data point were calculated from three independent biological replicates conducted in triplicate. Error bars represent ± S.D. Difference between pCDNA and our lipid binding deficient mutant neurofibromin 2, <1.0000 (i.e., not significant); pCDNA and wild-type neurofibromin 2, <0.000 (i.e., significant); wild type and our lipid binding deficient mutant, <0.0001 (i.e., significant)
To establish whether cells expressing the lipid binding deficient mutant neurofibromin 2 affect cell proliferation rates compared to wild-type neurofibromin 2 transfected cells, BrdU incorporation was assessed over a 72-hour period (Fig. 4g). In agreement with the cell counting experiments, wild-type neurofibromin 2 cells displayed reduced rates of BrdU incorporation compared to cells transfected with expression plasmids for the control and the lipid binding deficient mutant. This suggests that conformational changes associated with lipid binding and potentially membrane attachment are necessary for neurofibromin 2 inhibition of cell proliferation.
We have previously shown that neurofibromin 2 can function as a regulator of small G-proteins from the Rac1/cdc42 family and of the Hippo-YAP pathway9,36. Thus, we first assessed whether expression of the lipid binding deficient mutant can reduce levels of active Rac1 (Rac1-GTP) and compared this to wild-type neurofibromin 2 (Fig. 5a, b Supplementary Figure 7). As expected, transfection of either SC4 or 293 T cells with an expression vector for wild-type neurofibromin 2 resulted in decreased Rac1-GTP levels. However, expression of the neurofibromin 2 lipid binding deficient mutant did not significantly alter the levels of Rac1-GTP in transfected cells. Thus, lipid binding and membrane attachment are necessary for neurofibromin 2 to exert its cell growth inhibiting functions.
Fig. 5.
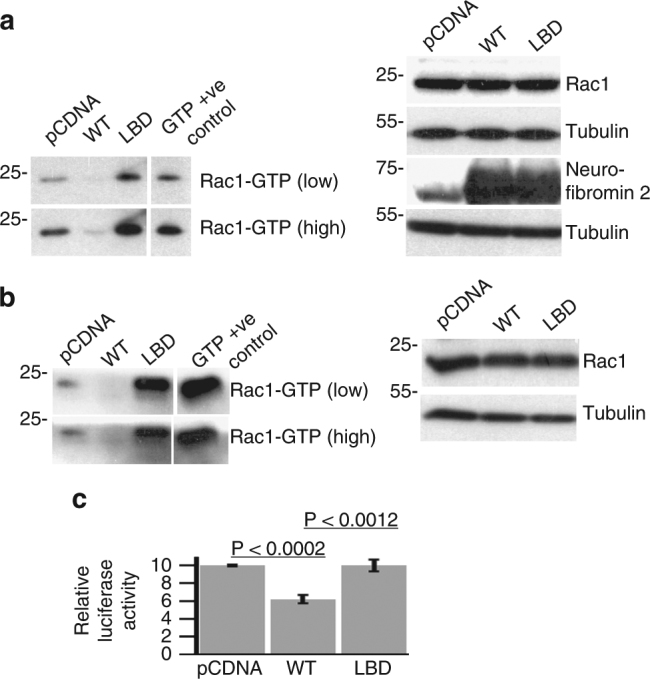
The lipid binding deficient mutant of neurofibromin 2 displays impaired inhibition of Rac1 activation and YAP activity. a 293 T or b SC4 cells were transfected with expression vectors for wild type or lipid binding deficient neurofibromin 2 or empty vector control (pCDNA) and levels of active Rac1 (Rac1-GTP) were assessed after 48 h. Levels of total Rac1, neurofibromin 2, and tubulin were assessed as controls. The blots shown are representative of three biological replicates. c HEK293T cells were transfected with expression vectors for wild type or lipid binding deficient neurofibromin 2 or empty vector control (pCDNA) along with YAP-driven luciferase and Renilla luciferase reporters. Activity of the luciferase reporter was assessed 24 h post transfection. Means of each data point were calculated from three independent biological replicates conducted in triplicate. Error bars represent ± S.D
As mentioned above, neurofibromin 2 is a key component of Hippo signaling and through which it regulates YAP transcriptional activity by inhibiting its translocation into the nucleus14. To assess the impact of the lipid binding deficient mutants against YAP activity, we co-transfected the neurofibromin 2 expression plasmids along a luciferase reporter plasmid for YAP-TEAD transcriptional activity, which served as a reporter for YAP activity (Fig. 5c). As expected, the expression of wild-type neurofibromin 2 resulted in a reduction of the YAP-TEAD reporter activity compared to the control. However, expression of the lipid binding mutant of neurofibromin 2 did not inhibit reporter activity. Thus, as in the case of active Rac1, lipid binding and membrane attachment are necessary for neurofibromin 2 to inhibit YAP activity.
Discussion
Stabilization of neurofibromin 2 at the membrane is an important process that regulates several cellular functions of neurofibromin 2. For example, the direct neurofibromin 2-paxillin interaction has been shown to be involved in the stabilization of neurofibromin 2 at the plasma membrane, which is important for its tumor-suppressive functions37. Despite the mapping of the paxillin-binding site on neurofibromin 2 to residue W60 by biochemical and cell biology studies, it is unclear how the stabilization occurs at the cell membrane38. Significantly, a mutation of this tryptophan residue to a cysteine was found to be present in NF2 patients38. Based on our neurofibromin 2/PIP2complex structure, residue W60 is in fact involved in binding to the lipid, and thereby stabilizing the adhesion complex by anchoring neurofibromin 2 to the plasma membrane. Indeed, the NF2 patient derived W60C mutant binds lipids about four-fold weaker compared to wild type, suggesting the importance of W60 in binding to the plasma membrane. The sequestration to the cell membrane might aid paxillin recruitment to cell adhesion sites where it binds directly to neurofibromin 2 and stabilizes the adhesion complex.
The structural model of auto-inhibition and cycling between closed/resting and open/active conformational states of ERM proteins is often employed to explain the function of neurofibromin 239,40. While the open conformation of ERM proteins represents their active states, the evidence that this is also the case for neurofibromin 2 is debated41,42. Previous studies in mammalian cells have shown that S518 is phosphorylated and that the S518D phosphomimic mutation blocks the intermolecular FERM-tail interactions17,43. However, S518 is located far from the head–tail domain interface in the closed A585W, R588K (“AR”) neurofibromin 2 mutant structure27,28,35. Studies in Drosophila indicate that deletion of the tail domain results in a constitutively active neurofibromin 2 that provides full genetic rescue on a null background44 suggesting that, like ERM proteins, the open form of neurofibromin 2 is active in vivo27.
Our crystal structure of neurofibromin 2 in complex with PIP2 now solve this long-standing debate (Fig. 6) as it reveals the lipid induced severing of the head–tail interaction which is supported by hydrogen-deuterium exchange data of full-length neurofibromin 2. Upon binding, PIP2 rearranges a loop into an α-helix, releases the interaction of the first α-helix αH of the central domain with the FERM domain to instead form one continuous long α-helix that ultimately displaces the tail domain. Our data uncover a mechanism of conformational changes that transition the closed form of neurofibromin 2 to its open conformer and link its dynamic states to previously characterized neurofibromin 2 functions. For example, it was shown previously that binding of Angiomotin to the neurofibromin 2 C-terminal region (residues 451–550) releases the auto-inhibition of the head/tail interaction and exposes the neurofibromin 2 FERM domain to interact with LATS1/235. Although conceptually similar, our data indicate a model where binding of PIP2 to the FERM domain itself releases an auto-inhibitory C-terminal domain lock and might allow neurofibromin 2 to interact with its interacting partners such as LATS1/2 and Angiomotin. A systematic screen for identification of interacting partners of wild-type neurofibromin 2 versus our lipid binding deficient mutant (T59V, W60E, R309Q, R310Q) might reveal additional mechanistic insights into neurofibromin 2 tumor suppressor functions.
Fig. 6.
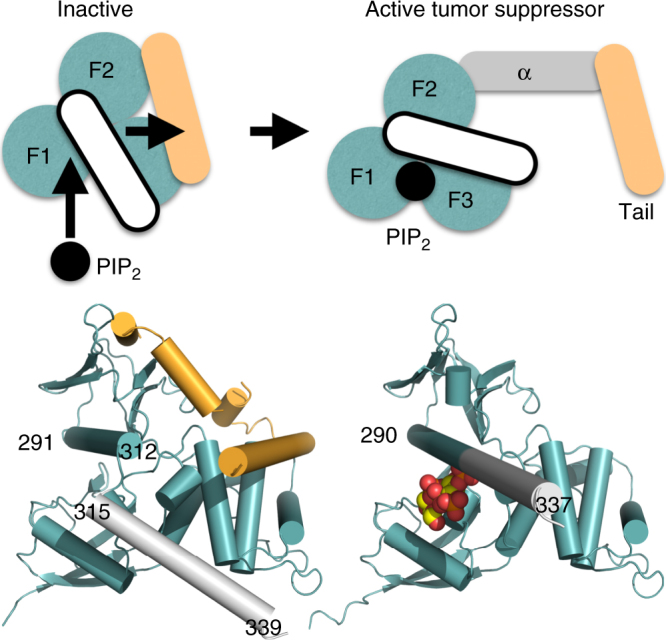
Activation of the neurofibromin 2 tumor-suppressive function. In its inactive state, neurofibromin 2 is in a closed conformation through interactions of the FERM domain (teal; F1, residues 18–98; F2, residues 111–213; F3, residues 220–312) and the tail domain (pale orange). The α-helix C-terminal of F3 (residues 315–339; white) does not interact with the tail domain. PIP2 binds to F1 and the last α-helix (residues 291–312; not depicted) of F3, thereby causing the last F3 α-helix (residues 291–312) and the following α-helix αH (residues 315–339) to rearrange as one long and continuous α-helix (residues 290–337), thereby displacing the tail domain and severing the head–tail interaction which results in active tumor suppressor functions. The central α-helical domain is shown in gray. The head/tail neurofibromin 2 crystal structure (head structure from PDB entry 1isn34; tail structure from PDB entry 4zrj35) is shown below the schematic on the bottom left and our PIP2-bound structure on the bottom right
Our HDX data show that both in the absence and presence of lipid, the neurofibromin 2 tail domain exchanges deuterium, which is consistent with the weak head–tail interaction (Kd of 3 μM) compared to the other ERM proteins where for example the binding affinity of the moesin tail domain for its FERM domain is 16 nM35. Thus, in its unbound state, neurofibromin 2 is in a semi-open conformation which is consistent with structural and biochemical studies16,27,35. Our HDX data clearly show that upon addition of PIP2, the neurofibromin 2 FERM domain is fully exposed with moderate to high deuterium exchanges of FERM domain residues whereby the differential HDX (HDXapo−HDXPIP2) analysis shows that the upon addition of lipid micelles, the neurofibromin 2 FERM and tail domains are completely severed. This observation is in agreement with the low resolution SANS experiments28. In contrast to wild-type neurofibromin 2, our binding data show that the constitutively closed AR mutant does not bind lipids or LATS1 suggesting that neurofibromin 2 is not fully closed as also suggested by our HDX data. Upon binding to PIP2, neurofibromin 2 binds LATS1 ten-fold tighter, implying that PIP2 fully releases the neurofibromin 2 head–tail interaction. Collectively, in its unbound state, neurofibromin 2 exists in a semi-open conformation and phosphoinositides induce the open conformation.
Our functional studies suggest that the lipid binding deficient mutations significantly impair the functions of neurofibromin 2, as these mutations impaired the ability of neurofibromin 2 to inhibit cell proliferation. Neurofibromin 2 has been shown to mediate anti-proliferative effects through several downstream signaling pathways. These include signaling through small G-proteins at regions of cell-cell contacts. Neurofibromin 2 and Rich1 bind to Angiomotin at junctional structures and inhibit Rac1 and downstream signaling into the MAPK pathway9. In addition, neurofibromin 2 has been shown to regulate signaling through YAP on multiple levels13,36,45. Our findings demonstrate that both lipid binding deficient and the open forms of neurofibromin 2 are impaired in their ability to regulate signaling through these pathways. Whether the lipid-bound open conformer impacts the nuclear functions of neurofibromin 211,46 remains to be determined.
Methods
DNA constructs and protein preparation
The neurofibromin 2 constructs, residues 1–312 and 1–339, were cloned into pGEX-2T expression vector (GE Life Sciences) using the full-length human neurofibromin 2 plasmid (addgene plasmid number 11629, Supplementary Table 4). The lipid binding deficient mutant (T59V, W60E, R309Q, R310Q) was generated by site-directed mutagenesis from the wild-type neurofibromin 2 FERM domain by using the QuickChange site-directed mutagenesis kit (Agilent Technologies). For functional studies in mammalian cells, wild type and mutant human full-length neurofibromin 2 were cloned using restriction-free cloning method47 into a modified pCDNA3 vector (Invitrogen) containing an N-terminal flag tag. All plasmid constructs were sequenced and verified.
We cloned the full-length neurofibromin 2 lipid binding deficient and A585W-R588K (AR) mutants into the pGEX-6P-1 plasmid using the restriction-free cloning method and site-directed mutagenesis while we cloned the patient-derived W60C mutant into the PGEX vector.
The neurofibromin 2 proteins (residues 1–339 and 1–595) were expressed in Escherichia coli strain BL21(DE3) Rosetta2 (Novagen) at 25 °C for 20 h and cells were induced for protein expression using either 0.05 mM or 0.2 mM IPTG. These cells were harvested by centrifugation at 5,000× g for 15 min and lysed by sonication in lysis buffer (20 mM Tris, pH 7.5, 400 mM NaCl, and 0.1 mM EDTA) and the lysate was clarified at 100,000× g for 30 min. The supernatant was loaded onto a GST column equilibrated with lysis buffer and the proteins were eluted with 10 mM glutathione in lysis buffer. The proteins were dialyzed overnight in 20 mM Tris (pH 7.5), 400 mM NaCl, 2 mM DTT, and 0.1 mM EDTA and digested with PreScission protease at 4 °C and these proteins were further purified by size exclusion chromatography (SEC) using a 26/60 Superdex 75 column (GE Healthcare) that was pre-equilibrated with 20 mM Tris (pH 7.5), 400 mM NaCl, 1 mM DTT, and 0.1 mM EDTA. For HDX studies, the human full-length GST-tagged neurofibromin 2 supernatant was loaded onto a GST column equilibrated with lysis buffer and the proteins were eluted with 10 mM glutathione in lysis buffer. The proteins were dialyzed overnight in 20 mM Tris (pH 7.5), 500 mM NaCl, 2 mM DTT, 2% glycerol, and 0.1 mM EDTA, and digested with PreScission protease at 4 °C. Proteins were further purified by SEC using a 16/60 Superdex 200 column (GE Healthcare) that was pre-equilibrated with 20 mM Tris (pH 7.5), 500 mM NaCl, 1 mM DTT, 2% glycerol, and 0.1 mM EDTA48. All the proteins were concentrated, and aliquots were frozen in liquid N2 and stored at −80 °C.
Neurofibromin 2/PIP2 co-crystallization
Our initial attempts to obtain crystals of the neurofibromin 2 FERM domain (residues 1–312) in complex with PIP2 were unsuccessful. We used varying concentrations of PIP2diC8 ranging from 20 to 500 μM, but were unable to obtain crystals. Thus, we used a larger neurofibromin 2 construct (residues 1–339) and 100 μM lipid and screened several commercially available crystallization screens (over 1,000 conditions) and obtained one single crystallization hit from the Index screen of Hampton Research within 1 day only at 20 °C. We improved these crystals by incubating 300 μM neurofibromin 2 with 300 μM PIP2diC8 (Avanti Polar Lipids) on ice for 12 h and by removing the precipitate by centrifugation (15,800× g for 30 min at 4 °C). Plate-like crystals grew within 2 days by hanging drop vapor diffusion (1 μl sample plus 1 μl reservoir) from 100 mM HEPES (pH 7.5) and 50 mM MgCl2(H2O)6 and by varying polyethylene glycol (PEG 550 monomethyl ether) concentrations from 25% to 35% at 20 °C that diffracted X-rays at beamline 22ID at the Advanced Photon Source, Argonne National Laboratory to ∼3 Å Bragg spacings. Crystals were directly flash frozen and stored in liquid nitrogen at 100 K.
X-ray data collection and processing
X-ray diffraction data were collected on beamline 22ID at the Advanced Photon Source at Argonne National Laboratory and integrated and scaled using XDS and AIMLESS as implemented in autoPROC49. Isotropic (to 3.09 Å) and anisotropic (to 2.61 Å) data reduction statistics are provided in Supplementary Table 1.
Structure determination and crystallographic refinement
Phases were obtained by molecular replacement using the program MOLREP50 by using the neurofibromin 2 FERM domain structure (PDB)34 as the search model. Molecular replacement solutions were only obtained when we truncated the model to residues 20–309. Two molecules in the asymmetric unit were identified in space group P21 resulting in a calculated volume to mass ratio of 2.68 Å3/Da, corresponding to a solvent content of 0.5455.
For crystallographic refinement, we used anisotropically scaled high-resolution data to 2.61 Å with an ellipsoidal completeness of 0.903. The neurofibromin 2/PIP2 complex structure was refined by performing maximum likelihood as implemented in BUSTER51 by imposing target restraints. The model was improved by non-crystallographic symmetry restraints through local structure similarity restraints52. Iterative cycles of model building were performed using Coot53, and model bias was minimized by building into composite omit maps. The model was further refined and improved by using TLS groups for each polypeptide chain with BUSTER. The missing extended α-helix (residues 310–339) was unambiguously resolved with clear electron density after initial refinement with BUSTER and was built manually using α-helical restraints in Coot. Optimized PIP2 ligand coordinates and ligand restraints were obtained from the Grade web server (grade.globalphasing.org). The electron density maps were sharpened using Coot to ensure the directionality and identity of the α-helices in particular at this moderate resolution. The final model contains two polypeptide chains, two phosphates, and two PIP2 molecules in the asymmetric unit. The quality of the final model was assessed using MolProbity54, which revealed no outliers with >98% of the amino acid residues in the favored region of the Ramachandran plot. Crystallographic refinement statistics are provided in Supplementary Table 2.
Lipid co-sedimentation assay
Lipid binding to wild type and mutant neurofibromin 2 FERM domains was assayed as described previously55. Briefly, lipid vesicles of phosphatidylcholine (PC) and PIP2 were prepared in chloroform to a final composition of 80% chicken egg PC and 20% porcine brain PIP2 (Avanti polar lipids). The lipid mixture was dried under Argon stream and resuspended in 20 mM Tris (pH 7.5), 400 mM NaCl, 0.1 mM EDTA, and 2 mM DTT (resuspension buffer). Small unilamellar vesicles were then produced by sonication after incubating at 37 °C for 60 min. Samples containing 50 μg of total lipid in 15 μl suspension and 50 μM wild type or mutant neurofibromin 2 domain proteins were incubated at 4 °C for 1 h followed by centrifugation at 100,000× g for 30 min. The supernatant and pellet were separated carefully and the pellet was washed twice and resuspended in 15 μl resuspension buffer. The supernatant and pellet samples were analyzed by SDS-PAGE.
Differential HDX-Mass Spectrometry
HDX-MS analysis of the target protein involves two stages, first peptide identification and second on-exchange HDX. For peptide identification, peptic peptides were fragmented using data-dependent MS/MS selecting top five most abundant ions per scan event using an Orbitrap mass spectrometer (Q Exactive, ThermoFisher) coupled to an automated liquid handling system and HPLC56. Peptide identification was performed by submitting raw MS/MS data files to Mascot (Matrix Science). Peptides with a MASCOT score greater than 20 were included in the HDX peptide set and the search was repeated against a decoy (reverse) sequence. Ambiguous identifications were not included in the HDX peptide set. MS/MS spectra for these peptides were verified by manual inspection.
The following procedure was used to perform on-exchange HDX. Neurofibromin 2 (10 μM) was incubated with the pure PIP2 micelles at a 1:10 protein-to-ligand molar ratio for 1 h at room temperature. Next, 5 μl of sample was diluted into 20 μl D2O buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM DTT) and incubated for various time points (0, 10, 60, 300, and 900 s) at RT. The deuterium exchange was then slowed by mixing with 25 μl of ice cold 3 M urea and 1% trifluoroacetic acid. Quenched samples were immediately frozen and remained on dry ice until they were injected into the HDX platform. Upon injection, samples were passed through an immobilized pepsin column (2 mm × 2 cm) at 200 μl min−1 and the digested peptides were captured on a 2 mm × 1 cm C8 trap column (Agilent) and desalted. Peptides were separated using a 2.1 mm × 5 cm C18 column (1.9 μl Hyperkin Gold, Thermo Scientific) with a 5 min linear gradient of 4–40% CH3CN and 0.3% formic acid. Both protein pepsin digestion and peptide separation were conducted at 4 °C to aid in deuterium retention. MS data were acquired using on the Orbitrap described above using a measured resolving power of 65,000 at m/z 400. All on-exchange HDX analyses were performed in triplicate, with single preparations of each protein/ligand complex. The intensity weighted mean m/z centroid value of each peptide envelope was calculated using HDX Workbench57 and these values were converted into percentage of deuterium incorporation. This was accomplished by determining the observed averages of the undeuterated and fully deuterated spectra and using the conventional formula described elsewhere58. Statistical significance for the differential HDX data is determined by an unpaired t-test for each time point57. Back-exchange corrections were made on the basis of an estimated 70% deuterium recovery and accounting for the known 80% deuterium content of the deuterium exchange buffer.
The HDX data from all overlapping peptides were consolidated to individual amino acid values using a residue averaging approach where for each residue, the deuterium incorporation values and peptide lengths from all overlapping peptides were assembled. A weighting function was applied in which shorter peptides were weighted more heavily than longer peptides. Each of the weighted deuterium incorporation values were then averaged to produce a single value per each amino acid. The initial two residues of each peptide, as well as prolines, were omitted from the calculations59.
MST Assay
Specific binding of PIP2diC8 and LATS1 (residues 69 to 100) to N terminally GST-tagged full-length neurofibromin 2 proteins (wild type, AR, and lipid binding deficient) were measured by the MST method. These proteins were either labeled with maleimide conjugated cysteine reactive or NHS conjugated lysine reactive NT-647 red dyes. Malemide labeling dyes were used for PIP2 titrations to avoid interference with PIP2 binding site. Unlabeled PIP2diC8 (10 mM) or LAST1 residues 69–100 (2 mM) was titrated into a fixed concentration of labeled proteins (5 μM). Binding reactions were carried out in a buffer containing 20 mM Tris (pH 7.5), 300 mM NaCl, and 0.01% Tween-20. Before loading the samples, the reactions were incubated for 10 minutes. Samples were loaded into NT.115 standard treated capillaries (Nanotemper Technologies) and data were measured using Monolith NT.115 pico apparatus (Nanotemper Technologies). The data were recorded at room temperature using the red LED at 20% (GREEN filter; excitation 515–525 nm, emission 560–585 nm) and IR-Laser power at either 20% or 40%. Data analyses were performed with NTAnalysis and only the data with same IR-Laser power were averaged and were plotted using “logistic” function in the Origin software.
Cell culture and transfection conditions
HEK293 cells were purchased from ATCC, hSC2λ cells were a gift from Dr. Margaret Wallace (https://www.nature.com/articles/labinvest201688), and SC4 cells were a gift from Dr. Helen Morrison7. The cell lines used in this study were authenticated by short tandem repeat DNA profiling (DDC Medical). In addition, the cells were tested every 3 months for mycoplasma contamination. HEK293, hSC2λ, and SC4 cells were maintained in low glucose Dulbecco’s Modified Eagle’s Medium (Gibco) + 10% fetal bovine serum (Atlas Biologicals) and antibiotics (100 units ml−1 penicillin and 100 μg ml−1 Streptomycin) (Gibco) at 37 °C in a humidified atmosphere of 5% CO2 (v/v). Experiments were executed with cells grown to 70–80% confluence. Transfections were done using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), as previously described60.
Immunoprecipitation and Rac1-GTP pull-down
Cells were transfected with 8 μg of plasmid DNA (empty vector control or different neurofibromin 2 alleles) and Rac1-GTP levels were determined using a pull-down approach employing a fusion protein comprised of the p21-binding domain of PAK1 fused to GST, according to the manufacturer’s instructions (Millipore, #17-441).
BrdU Incorporation Assay
Cells were seeded at of 2 × 105 cells/ well in 6-well dishes. On the following day, cells were transfected with 2 μg of plasmid DNA (empty vector control or different neurofibromin 2 alleles). After 24 h of transfection, the cells were trypsinized and were seeded at 2 × 104 cells/well 96-well tissue culture dishes. At 48 h, 72 h, and 96 h post transfection, the cells were processed and analyzed using a BrdU cell proliferation assay kit following manufacturer’s instructions (Millipore, #2750). Plates were read with a spectrophotometer microplate reader (450 nm) (SpectraMax M5; Molecular Devices).
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 6cds. Other data are available from the corresponding author upon request.
Electronic supplementary material
Supplementary Information (PDF 41078 kb)
Description of Additional Supplementary Files(PDF 169 kb)
Acknowledgements
We are indebted to Erumbi Rangarajan (The Scripps Research Institute) for continuous insightful discussions and help in general, to the staff of the Southeast Regional Collaborative Access Team for synchrotron support, and Dinesh Dhurvas Chandrasekaran (Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences) and Dipak N. Patil (The Scripps Research Institute) for help in generating with some of the expression constructs. We thank Candice Morrow for help with Supplementary Data 1. This is publication #29541 from The Scripps Research Institute. T. I. is supported by grants from the National Institute of Health, the Department of Defense, and by start-up funds provided to The Scripps Research Institute from the State of Florida. K.C. was a Children’s Tumor Foundation fellow. This work was supported in part by NIH grant NS077952 to J. K.
Author contributions
K. C. determined the neurofibromin 2/PIP2 crystal structure and performed the lipid co-sedimentation and MST experiments; A. J. S. and G. B. helped with crystallography; J. Z. performed the HDX experiments; V. M. performed the cell proliferation and Rac1 and YAP activity assays; and T. I. supervised the project and wrote the manuscript with J. K. and P. R. G.
Competing interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-018-03648-4.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J. Rare Dis. 2009;4:16. doi: 10.1186/1750-1172-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asthagiri AR, et al. Neurofibromatosis type 2. Lancet. 2009;373:1974–1986. doi: 10.1016/S0140-6736(09)60259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trofatter JA, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;75:826. doi: 10.1016/0092-8674(93)90501-G. [DOI] [PubMed] [Google Scholar]
- 4.Xu HM, Gutmann DH. Merlin differentially associates with the microtubule and actin cytoskeleton. J. Neurosci. Res. 1998;51:403–415. doi: 10.1002/(SICI)1097-4547(19980201)51:3<403::AID-JNR13>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 5.Cooper J, Giancotti FG. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014;588:2743–2752. doi: 10.1016/j.febslet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrilli AM, Fernandez-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2016;35:537–548. doi: 10.1038/onc.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison H, et al. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007;67:520–527. doi: 10.1158/0008-5472.CAN-06-1608. [DOI] [PubMed] [Google Scholar]
- 8.Okada T, Lopez-Lago M, Giancotti FG. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol. 2005;171:361–371. doi: 10.1083/jcb.200503165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi C, et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell. 2011;19:527–540. doi: 10.1016/j.ccr.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lallemand D, Saint-Amaux AL, Giovannini M. Tumor-suppression functions of merlin are independent of its role as an organizer of the actin cytoskeleton in Schwann cells. J. Cell Sci. 2009;122:4141–4149. doi: 10.1242/jcs.045914. [DOI] [PubMed] [Google Scholar]
- 11.Li W, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140:477–490. doi: 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerrant W, et al. YAP mediates tumorigenesis in neurofibromatosis type 2 by promoting cell survival and proliferation through a COX-2-EGFR signaling axis. Cancer Res. 2016;76:3507–3519. doi: 10.1158/0008-5472.CAN-15-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014;13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gronholm M, et al. Homotypic and heterotypic interaction of the neurofibromatosis 2 tumor suppressor protein merlin and the ERM protein ezrin. J. Cell Sci. 1999;112:895–904. doi: 10.1242/jcs.112.6.895. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen R, Reczek D, Bretscher A. Hierarchy of merlin and ezrin N- and C-terminal domain interactions in homo- and heterotypic associations and their relationship to binding of scaffolding proteins EBP50 and E3KARP. J. Biol. Chem. 2001;276:7621–7629. doi: 10.1074/jbc.M006708200. [DOI] [PubMed] [Google Scholar]
- 17.Shaw RJ, et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev. Cell. 2001;1:63–72. doi: 10.1016/S1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 18.Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J. Biol. Chem. 2002;277:883–886. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- 19.Surace EI, Haipek CA, Gutmann DH. Effect of merlin phosphorylation on neurofibromatosis 2 (NF2) gene function. Oncogene. 2004;23:580–587. doi: 10.1038/sj.onc.1207142. [DOI] [PubMed] [Google Scholar]
- 20.Alfthan K, Heiska L, Gronholm M, Renkema GH, Carpen O. Cyclic AMP-dependent protein kinase phosphorylates merlin at serine 518 independently of p21-activated kinase and promotes merlin-ezrin heterodimerization. J. Biol. Chem. 2004;279:18559–18566. doi: 10.1074/jbc.M313916200. [DOI] [PubMed] [Google Scholar]
- 21.Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature. 2006;442:576–579. doi: 10.1038/nature04856. [DOI] [PubMed] [Google Scholar]
- 22.Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000;101:259–270. doi: 10.1016/S0092-8674(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 23.Scoles DR. The merlin interacting proteins reveal multiple targets for NF2 therapy. Biochim. Biophys. Acta. 1785;32-54:2008. doi: 10.1016/j.bbcan.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 2007;177:893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James MF, Beauchamp RL, Manchanda N, Kazlauskas A, Ramesh V. A NHERF binding site links the betaPDGFR to the cytoskeleton and regulates cell spreading and migration. J. Cell Sci. 2004;117:2951–2961. doi: 10.1242/jcs.01156. [DOI] [PubMed] [Google Scholar]
- 26.Hennigan RF, et al. Fluorescence resonance energy transfer analysis of merlin conformational changes. Mol. Cell. Biol. 2010;30:54–67. doi: 10.1128/MCB.00248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sher I, Hanemann CO, Karplus PA, Bretscher A. The tumor suppressor merlin controls growth in its open state, and phosphorylation converts it to a less-active more-closed state. Dev. Cell. 2012;22:703–705. doi: 10.1016/j.devcel.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ali Khajeh J, et al. Molecular conformation of the full-length tumor suppressor NF2/Merlin—a small-angle neutron scattering study. J. Mol. Biol. 2014;426:2755–2768. doi: 10.1016/j.jmb.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown DA. Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiol. (Bethesda) 2006;21:430–439. doi: 10.1152/physiol.00032.2006. [DOI] [PubMed] [Google Scholar]
- 30.Stickney JT, Bacon WC, Rojas M, Ratner N, Ip W. Activation of the tumor suppressor merlin modulates its interaction with lipid rafts. Cancer Res. 2004;64:2717–2724. doi: 10.1158/0008-5472.CAN-03-3798. [DOI] [PubMed] [Google Scholar]
- 31.Deguen B, et al. Impaired interaction of naturally occurring mutant NF2 protein with actin-based cytoskeleton and membrane. Hum. Mol. Genet. 1998;7:217–226. doi: 10.1093/hmg/7.2.217. [DOI] [PubMed] [Google Scholar]
- 32.Stokowski RP, Cox DR. Functional analysis of the neurofibromatosis type 2 protein by means of disease-causing point mutations. Am. J. Hum. Genet. 2000;66:873–891. doi: 10.1086/302812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mani T, et al. FERM domain phosphoinositide binding targets merlin to the membrane and is essential for its growth-suppressive function. Mol. Cell. Biol. 2011;31:1983–1996. doi: 10.1128/MCB.00609-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimizu T, et al. Structural basis for neurofibromatosis type 2. Crystal structure of the merlin FERM domain. J. Biol. Chem. 2002;277:10332–10336. doi: 10.1074/jbc.M109979200. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, et al. Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Res. 2015;25:801–817. doi: 10.1038/cr.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yi C, et al. The p130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci. Signal. 2013;6:ra77. doi: 10.1126/scisignal.2004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Valle C, et al. Paxillin binds schwannomin and regulates its density-dependent localization and effect on cell morphology. Nat. Genet. 2002;31:354–362. doi: 10.1038/ng930. [DOI] [PubMed] [Google Scholar]
- 38.Manetti ME, et al. Stability of the tumor suppressor merlin depends on its ability to bind paxillin LD3 and associate with beta1 integrin and actin at the plasma membrane. Biol. Open. 2012;1:949–957. doi: 10.1242/bio.20122121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gary R, Bretscher A. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol. Biol. Cell. 1995;6:1061–1075. doi: 10.1091/mbc.6.8.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henry MD, Gonzalez Agosti C, Solomon F. Molecular dissection of radixin: distinct and interdependent functions of the amino- and carboxy-terminal domains. J. Cell Biol. 1995;129:1007–1022. doi: 10.1083/jcb.129.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 42.McClatchey AI. Merlin and ERM proteins: unappreciated roles in cancer development? Nat. Rev. Cancer. 2003;3:877–883. doi: 10.1038/nrc1213. [DOI] [PubMed] [Google Scholar]
- 43.Shaw RJ, McClatchey AI, Jacks T. Regulation of the neurofibromatosis type 2 tumor suppressor protein, merlin, by adhesion and growth arrest stimuli. J. Biol. Chem. 1998;273:7757–7764. doi: 10.1074/jbc.273.13.7757. [DOI] [PubMed] [Google Scholar]
- 44.LaJeunesse DR, McCartney BM, Fehon RG. Structural analysis of Drosophila merlin reveals functional domains important for growth control and subcellular localization. J. Cell Biol. 1998;141:1589–1599. doi: 10.1083/jcb.141.7.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu J, et al. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev. Cell. 2010;18:288–299. doi: 10.1016/j.devcel.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell. 2014;26:48–60. doi: 10.1016/j.ccr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Unger T, Jacobovitch Y, Dantes A, Bernheim R, Peleg Y. Applications of the Restriction Free (RF) cloning procedure for molecular manipulations and protein expression. J. Struct. Biol. 2010;172:34–44. doi: 10.1016/j.jsb.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 48.Chinthalapudi K, Patil D, Rangarajan E, Rader C, Izard T. Lipid-directed vinculin dimerization. Biochemistry. 2015;54:2758–2768. doi: 10.1021/acs.biochem.5b00015. [DOI] [PubMed] [Google Scholar]
- 49.Vonrhein C, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. doi: 10.1107/S0021889897006766. [DOI] [Google Scholar]
- 51.Bricogne G., et al. BUSTER version 2.9. (Global Phasing Ltd., Cambridge, 2011).
- 52.Smart O. S., et al. Refinement with local structure similarity restraints (LSSR) enables exploitation of information from related structures and facilitates use of NCS. Abstr. Annu. Meet Am. Crystallogr. Assoc. abstr. TP139 (2008).
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Davis IW, Murray LW, Richardson JS, Richardson DC. Molprobity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chinthalapudi K, Rangarajan ES, Brown DT, Izard T. Differential lipid binding of vinculin isoforms promotes quasi-equivalent dimerization. Proc. Natl Acad. Sci. USA. 2016;113:9539–9544. doi: 10.1073/pnas.1600702113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chalmers MJ, et al. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- 57.Pascal BD, et al. HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keppel TR, Weis DD. Mapping residual structure in intrinsically disordered proteins at residue resolution using millisecond hydrogen/deuterium exchange and residue averaging. J. Am. Soc. Mass Spectrom. 2015;26:547–554. doi: 10.1007/s13361-014-1033-6. [DOI] [PubMed] [Google Scholar]
- 60.Moleirinho S., et al. Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. Elife6, e23966 (2017). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information (PDF 41078 kb)
Description of Additional Supplementary Files(PDF 169 kb)
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 6cds. Other data are available from the corresponding author upon request.