

Abstract
Cardiac and skeletal striated muscles are intricately designed machines responsible for muscle contraction. Coordination of the basic contractile unit, the sarcomere, and the complex cytoskeletal networks are critical for contractile activity. The sarcomere is comprised of precisely organized individual filament systems that include thin (actin), thick (myosin), titin, and nebulin. Connecting the sarcomere to other organelles (e.g., mitochondria and nucleus) and serving as the scaffold to maintain cellular integrity are the intermediate filaments. The costamere, on the other hand, tethers the sarcomere to the cell membrane. Unique structures like the intercalated disc in cardiac muscle and the myotendinous junction in skeletal muscle help synchronize and transmit force. Intense investigation has been done on many of the proteins that make up these cytoskeletal assemblies. Yet the details of their function and how they interconnect have just started to be elucidated. A vast number of human myopathies are contributed to mutations in muscle proteins; thus understanding their basic function provides a mechanistic understanding of muscle disorders. In this review, we highlight the components of striated muscle with respect to their interactions, signaling pathways, functions, and connections to disease.
Introduction
The cardiac and skeletal striated muscle cytoskeleton is complex, yet intricately organized to coordinate muscle contraction. Numerous cytoskeletal assemblies are present within each muscle cell. For example, in striated muscle, the basic unit of contraction is the sarcomere, comprised of a plethora of structural and regulatory proteins. Intermediate filaments serve as a scaffold that connects the sarcomere to other organelles (such as mitochondria or the nucleus) to maintain cellular integrity and to contribute to mechanotransduction. The sarcomere is tethered to the sarcolemma, the membrane surrounding the myofibril by another cytoskeletal assembly—the costamere. Costameres link the sarcomere to the sarcolemma via the Z-disc and M-band. Individual heart cells are connected by intercalated discs, which synchronize muscle contraction. Skeletal muscle has a specialized structure to transmit force from the sarcomere to the connective tissue of the tendon, referred to as the myotendinous junction. Coordinated action of all the cytoskeletal assemblies is crucial to produce proper contractile function; thereby, disruption in the integrity of any component can often result in cardiac or skeletal myopathies.
A critical key to the pathogenesis of cardiomyopathies was described with the first direct, causal link between mutations in cytoskeletal sarcomeric genes and the development of hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) as well as restrictive cardiomyopathy (RCM), left ventricular noncompaction, and arrhythmogenic right ventricular cardiomyopathy (ARVC). Since then, thousands of mutations have been identified with state-of-the-art genetic testing to be potentially associated with HCM and DCM [for recent reviews see (268,397)]. In fact, mutations are identified in 50% of cardiomyopathy patients but few are verified as direct, bona-fide disease-causing mutations [reviewed in (268)].
Skeletal muscle myopathies are also directly linked to mutations in cytoskeletal components. Muscular dystrophies are a group of over 30 genetic diseases whose clinical features include progressive muscle degeneration and muscle weakness. The most common is Duchenne muscular dystrophy (DMD), which is an X-linked recessive disease that affects 1 in 3500 to 5000 males [reviewed in (438)]. Another example is nemaline myopathy, which affects 1 in 50,000 births [reviewed in (499)]. Advances in next generation sequencing have identified numerous mutations in humans. Together, studies combining in vivo genetic models of human disease (e.g., transgenic mice), isolated cell culture studies including myocytes differentiated from patient induced pluripotent stem cells, and in vitro mechanistic studies have been invaluable in understanding the etiology of skeletal and cardiac myopathies and, currently, in the design of potential personalized therapies.
In this review, we will focus on major striated muscle cytoskeletal assemblies, their components, functions, and how they interact with each other to coordinate muscle contraction. In addition, we will present how altered expression or mutations in these proteins can result in disease.
Sarcomere—The Basic Contractile Unit of Striated Muscle
The sarcomere is the smallest contractile unit of striated muscle (Fig. 1). The lateral boundaries of a sarcomere are defined by protein-dense Z-discs. The I-band is the region on either side of the Z-disc that is devoid of the myosin-containing thick filaments. The A-band comprises the region extending the entire length of the thick filaments, and the M-band resides at the center of the A-band. The sarcomere has three major filament systems that include actin-thin filaments, myosin-thick filaments and the giant protein titin. The force of muscle contraction occurs when the myosin motor protein attaches to the actin filament and pulls the Z-discs toward the M-band. The sarcomere is not a static structure. Although once considered to be solely a stable scaffold for regulatory and structural proteins, it is now known to undergo remarkable, rapid protein exchange, and respond to alterations in muscle load and injury.
Figure 1.
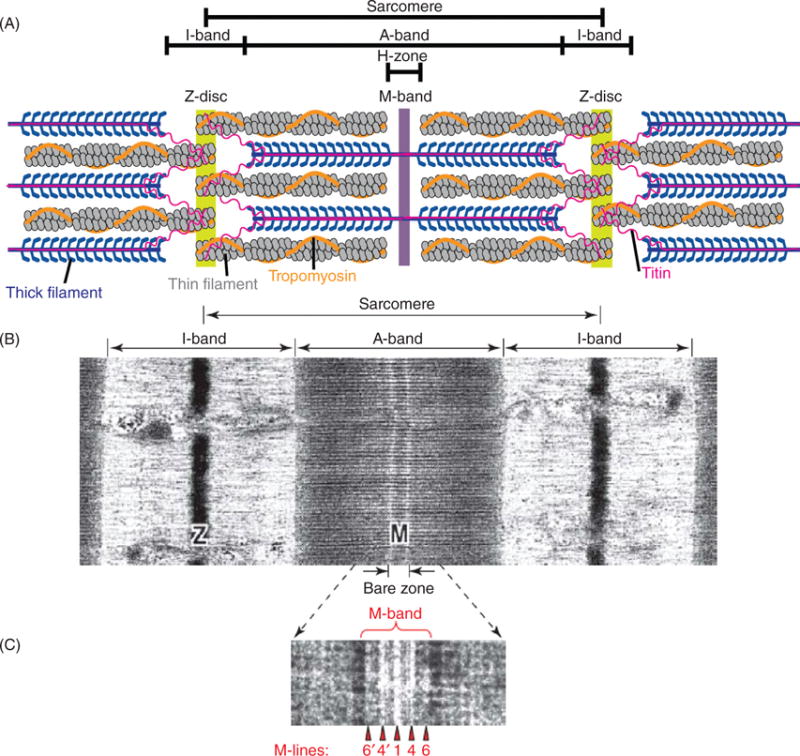
(A) Schematic representation of a cardiac sarcomere (lacking nebulin) illustrating the three major filament systems: actin-based thin filaments (gray), myosin-based thick filaments (blue), and titin (pink). The lateral boundaries of the sarcomere are the Z-discs. The I-bands surrounds the Z-disc and is a region where thin filaments are not superimposed by thick filaments. The A-band region contains thin filaments and thick filaments. The M-band falls within the H-zone, where thick filaments do not interdigitate with thick filaments. (B) Electron micrograph of skeletal muscle sarcomere. (C) Enlarged view of the M-band region. The M-band is composed of a series of three to five electron-dense M-lines: M6′, M4′, M1, M4, and M6. [Part A modified, with permission, from (255); Parts B and C modified, with permission, from (9).]
Z-discs: Borders of contractile units with ever-growing functions and networks of proteins
Z-discs define the lateral borders of striated muscle sarcomeres and cross-link the barbed ends of actin-based thin filaments from adjacent sarcomeres via α-actinin. The Z-disc also serves as an anchor site for the N-terminus of titin and nebulin/nebulette filament systems, making it indispensable for transmission of contractile force. Even though α-actinin was the most well characterized Z-disc protein in the 1990s, there is a plethora of gene products reported to be associated with this region of the sarcomere.
The role of the Z-disc has long transcended from being solely a passive structure providing anchorage to the thin filaments (including nebulin) and titin (Fig. 1). Z-discs and their associated protein networks have been shown to participate in numerous cellular processes including signal transduction and protein turnover in both cardiac and skeletal muscles. Furthermore, mutations in Z-disc-associated proteins are linked with numerous cardiomyopathies and skeletal muscle dystrophies [reviewed in (40,174,175,317)].
Proteins that cross-link actin and anchor thin filaments to the Z-disc
α-ACTININ
α-Actinin is a member of the spectrin superfamily and was originally described to function as an actin filament cross-linker (420). There are four vertebrate α-actinin genes with overlapping functions: ACTA1 and ACTA4 are non-muscle isoforms, ACTA2 and ACTA3 are skeletal muscle isoforms, while only ACTA2 is found in cardiac muscle (51,272). The actin-binding domain at the N-terminus is linked through an α-helical neck to a domain containing four spectrin-like repeats, while the C-terminus is composed of a calmodulin-like domain with two pairs of calcium-binding EF hand motifs (EF 1–2 and 3–4). One α-actinin-2 homodimer cross-links two antiparallel actin filaments of adjacent sarcomeres forming a flexible tetragonal lattice (588). This lattice is key for the rigidity the Z-disc needs to serve as a structural anchor site, while still allowing for the flexibility needed to conform to contractile forces.
As one of the integral Z-disc proteins, α-actinin has a myriad of binding partners with each interaction serving a distinct role in the production of concerted contractile action. Some major Z-disc proteins that interact with ACTA2 are ALP (actinin-associated LIM protein), MLP (muscle LIM protein), N-terminus of titin, myotilin, CapZ, cypher/oracle/ZASP, FATZ (filamin, α-actinin, and telethonin-binding protein at the Z-disc), myopalladin, and myopodin [reviewed in (316,405,639) (Figs. 2 and 3). ACTA2 has also been demonstrated to bind phosphorylase-b, an important metabolic enzyme in the Z-disc (116).
Figure 2.
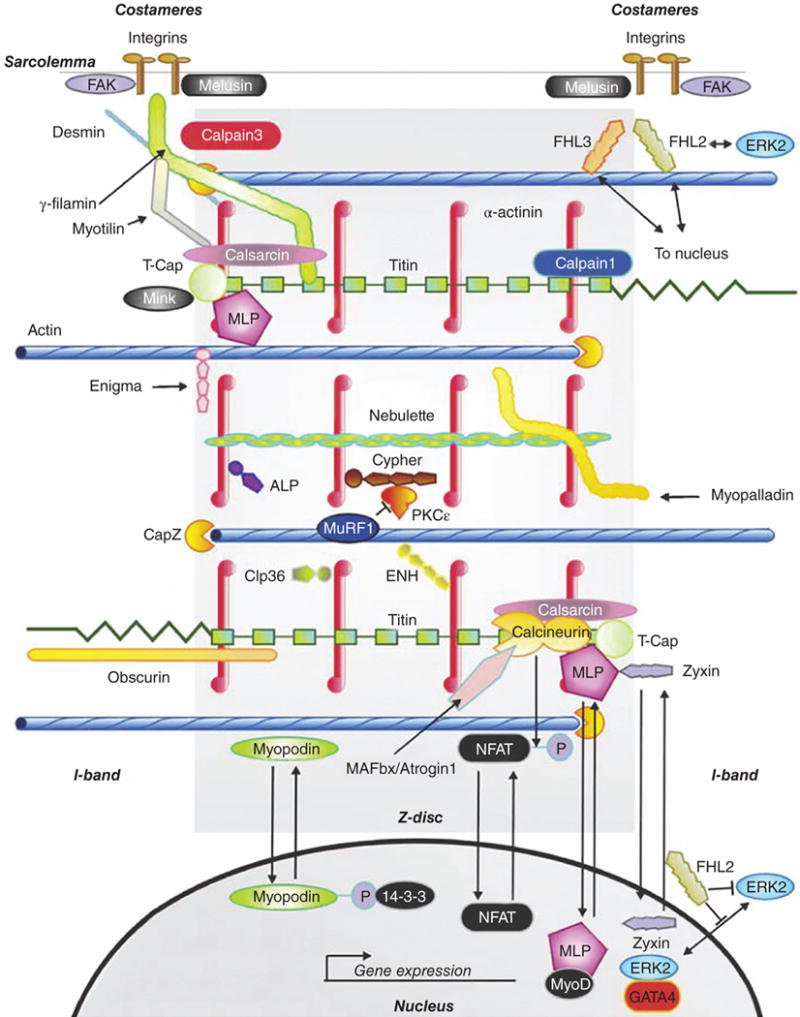
Z-discs define the lateral edge of the sarcomere, and also participate in numerous cellular processes including signal transduction and protein turnover. Abbreviations: FAK, focal adhesion kinase; γ-filamin, also known as Filamin C; FHL, four-and-a-half LIM domains protein; ERK, extracellular signal-regulated kinase; MLP, muscle LIM protein; ALP, actin-associated LIM protein; PKCε, protein kinase C epsilon; MuRF, muscle-ring-finger protein; ENH, enigma-homolog protein; NFAT, nuclear factor of activated T-cells; MAFbx, muscle atrophy F-box (striated muscle-specific E3 ubiquitin ligase) protein; GATA4, GATA sequence-binding zinc-finger transcription factor 4. [Fig. modified, with permission, from (175).]
Figure 3.
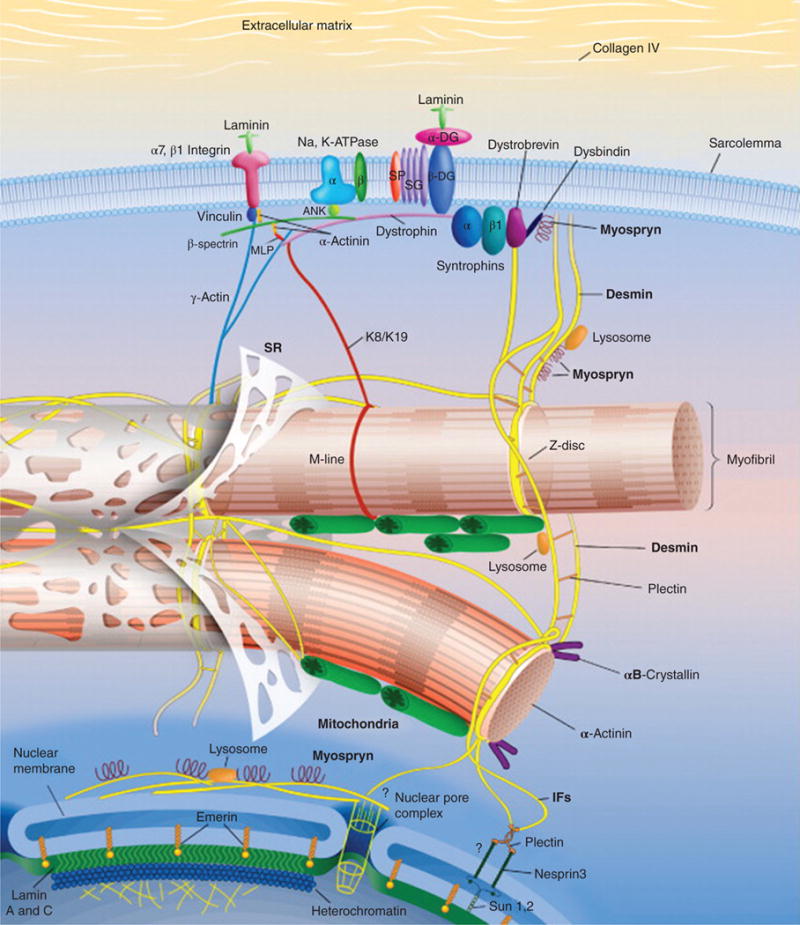
Schematic representation of the intermediate filament (IF) scaffold in striated muscle. The IF scaffold, predominantly composed of desmin (yellow), links the entire contractile apparatus to the sarcolemma and other organelles, such as the nucleus, mitochondria, lysosomes, and potentially the sarcoplasmic reticulum (SR). Desmin interacts with many other proteins including synemin, paranemin, syncoilin, and myospryn. Keratins (K8/K19) link the contractile apparatus to the sarcolemma and interact with the dystrophin-dystroglycan (DG) complex. Overall, the IF scaffold helps maintain the integrity of muscle cytoarchitecture and provide mechanical strength to the cell. Abbreviations: MLP, striated muscle-specific LIM protein; SG, sarcoglycan. [Fig. modified, with permission, from (88).]
Independent studies reported that human mutations in the ACTA2 gene are associated with DCM, HCM, idiopathic ventricular fibrillation, left ventricular noncompaction, and atrial arrhythmias [reviewed in (474)]. ACTA3 is expressed exclusively in type II fast glycolytic skeletal muscles, which are specialized for fast contractions over a short duration (51). Remarkably, 16% of the human population is homozygous for a nonsense mutation in ACTA3, which results in no expression of this protein (368). However, the ACTA3 mutation does not result in any disease symptoms classifying it as a nonessential gene as ACTA2 may be able to compensate (498). In accordance with the lack of observable symptoms in humans, the ACTA3 knockout mouse does not develop any muscle dysfunction, but there is a switch from anaerobic metabolism to more efficient aerobic metabolism (409). Loss of ACTA3 expression in humans has been linked to increased endurance and is associated with world-class athletes, suggesting its positive selection in the human population (624).
FILAMIN-C
Similar to spectrin super-family members, filamin protein family members also bind and cross-link actin. There are three filamin proteins: filamin-A (α isoform), filamin-B (β isoform), and striated muscle-specific filamin-C (γ isoform). The N-terminal actin-binding domain is followed by a central rod domain with 4-24 immunoglobulin-repeats and a C-terminal dimerization domain [reviewed in (706)]. A unique insertion of 81 amino acids in filamin C’s twentieth immunoglobulin-repeat is a Z-disc localization motif (708).
Filamin-C (γ-filamin) is one of the major proteins that serves as a link between the costamere and Z-disc and is involved in signal transduction with integrins (Fig. 2). Filamin-C functions through interactions with sarcolemmal (striated muscle cell membrane) proteins, such as γ-and δ-sarcoglycans of the dystrophin glycoprotein complex (681), β1A-subunit of the integrin receptor complex (214), as well as Z-disc proteins such as myotilin (708) and FATZ (161,214,671) [see section “The Costamere: Protects against mechanical stress and is an important signaling hub” for more information on sarcoglycans and the dystrophin glycoprotein complex]. The calcium-dependent proteolytic enzymes, calpains-1 and -3, regulate filamin-C activity by cleaving it to produce fragments that disrupt its interaction with sarcoglycans (234, 776) (Fig. 2). Filamin-C dimerizes via a C-terminal immunoglobulin domain, which allows it to cross-link and bundle actin filaments (264). An autosomal dominant nonsense mutation, W2710, in the last exon of the human filamin-C gene interferes with its dimerization process, and causes filamin-C to aggregate within skeletal muscle fibers; this phenomenon eventually leads to the disease myofibrillar myopathy (315,722).
MYOTILIN
Myotilin is a vertebrate striated muscle-specific protein also involved in stabilizing and anchoring thin filaments in the Z-disc (602) (Fig. 2). Dimers of myotilin cross-link and stabilize actin filaments in the Z-disc, as well as prevent actin filament depolymerization (603). The domain structure of myotilin includes an N-terminal serine-rich region followed by two immunoglobulin-like domains that are important for dimerization, and a C-terminal PDZ-binding motif (602, 603). Myotilin binds to α-actinin (602), filamin-C (708), and FATZ-1 and -2 (214). Mutations in the myotilin gene, especially in the N-terminal serine-rich region, are linked with several inherited skeletal muscle dystrophies such as limb girdle muscular dystrophy type 1A, myofibrillar myopathy, and spheroid body myopathy, which are collectively referred to as “myotilinopathies” [reviewed in (526,527,620)].
LIM proteins: A scaffold for protein-protein interactions
MUSCLE LIM PROTEIN (MLP)
MLP belongs to the cysteine-rich protein (CRP) family. CSRP1 is found in smooth muscle, CSRP2 in arteries and fibroblasts, and CSRP3 encodes the striated muscle protein MLP (25, 398). MLP is composed of two LIM domains that are surrounded by glycine-rich repeats; LIM domains serve as protein-protein binding sites (738). MLP is a positive regulator of myogenesis and overexpression of MLP increases muscle differentiation in C2C12 skeletal muscle cell culture (25). MLP helps to stabilize the Z-disc through its interactions with α-actinin (398) and anchors the titin-binding protein T-Cap to the Z-disc (318) (Fig. 2). MLP also has diverse roles such as acting as a stretch sensor and signaling protein [see section “Z-disc is an important signaling node in the sarcomere”] (Fig. 4).
Figure 4.
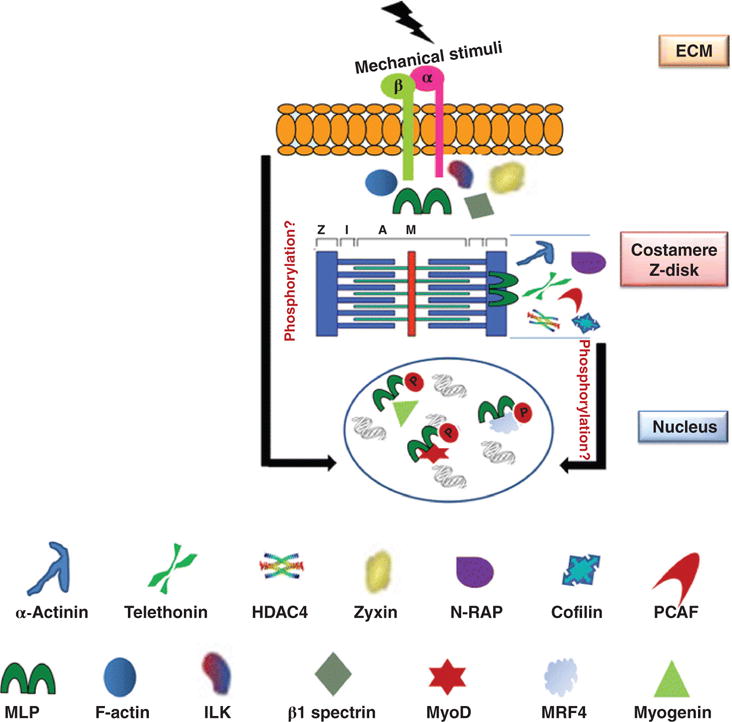
MLP (muscle LIM protein) is a functionally diverse, multicompartment protein. MLP interacts with β1 spectrin, zyxin, and integrin-linked kinase (ILK) in costameres and plays a role in force transmission. MLP also binds to α-actinin to help stabilize the Z-disc. At the intercalated discs, MLP binds to N-RAP. MLP acetylated by HDAC4 [histone acetyltransferases (HATs) and deacetylases] and PCAF (P300/CBP-associated factor) enhance calcium sensitivity and increase contractile function. In addition, MLP and cofilin form a complex and regulate actin dynamics. MLP is an important stretch sensor. The MLP/titin/telethonin (T-Cap) complex plays a key role in stretch-induced signaling. MLP translocates to the nucleus and interacts with transcription factors, which regulate myogenesis [e.g., MyoD, myogenin and MRF4 (muscle-specific regulatory factor 4)]. [Fig. reprinted, with permission, from (84).]
MLP-b is an MLP isoform, resulting from alternative splicing of CSRP3 exons 3 and 4. MLP-b, similar to full length MLP, localizes to the Z-disc and interacts with α-actinin and T-Cap. This isoform also forms oligomers with full-length MLP. Intriguingly, MLP-b has the opposite effect on myogenesis compared to MLP; MLP-b decreases differentiation indicating a distinct role from MLP (701).
While mainly considered a Z-disc protein, MLP has been shown to localize to other regions in myocytes (Fig. 4). MLP interacts with β-spectrin (168), zyxin (600), and integrin-linked kinase (ILK) in costameres (566), as well as nebulin-related anchoring protein (N-RAP) (153) in intercalated discs. MLP also localizes to the M-band, with no binding partners yet identified in this location (319). Consistent with its many subcellular localizations, MLP plays a wide variety of roles in the striated muscle cytoskeletal assemblies. For example, MLP plays a role in force transmission via interactions with β-spectrin (168) and zyxin (600) in the costamere. MLP also binds to histone-deacetylase 4 (HDAC4) in the Z-disc; MLP acetylated by HDAC4 and PCAF (P300/CBP-associated factor) enhances calcium sensitivity and increases contractile function (230). MLP can also form a complex with cofilin-2 and alter actin dynamics (540)
Alterations in MLP levels have been noted in skeletal myopathies. In a nemaline myopathy mouse model generated via a knock-in of the human mutation of α-tropomyosin (M9R), MLP protein levels are significantly increased (605). MLP protein levels are also increased in skeletal muscle from the mouse dysferin knockout model of muscular dystrophy and from humans with facioscapulohumeral muscular dystrophy (719,746). The increase in MLP levels in skeletal muscle myopathies are the opposite of what is seen in heart failure patients (e.g., they present with decreased MLP levels), indicating MLP may have differential roles in skeletal and cardiac muscle during disease. As a positive regulator of myogenesis, the upregulation of MLP may contribute to an attempted repair mechanism, but direct involvement of MLP in these myopathies is yet unknown.
PDZ-LIM FAMILY OF PROTEINS
All PDZ-LIM family members have at least one PDZ domain and at least one LIM domain. Both the PDZ and LIM domains act as a scaffold for protein interactions [reviewed in (215)]. Four PDZ-LIM subfamilies have been identified including: (i) α-actinin-associated LIM protein (ALP), (ii) ENIGMA (Enigma, enigma-homologue, and CYPHER/ZASP), (iii) LMO7, and (iv) LIM-KINASE (338).
ALP SUBFAMILY: All members of the ALP subfamily contain one PDZ domain and one LIM domain, and have multiple splice variants (771). There are four proteins in the ALP subfamily: ALP (PDLIM3), CLP36 (PDLIM1; also known as elfin and CLIM1), RIL (PDLIM4), and mystique (PDLIM2; also known as SLIM). ALP is found in both skeletal and heart muscle (752), while CLP36 is only found in the heart (335) (Fig. 2). ALP binds to α-actinin in both skeletal and cardiac muscle and enhances actin filament cross-linking by α-actinin (547). Mice deficient in ALP develop right ventricular DCM, indicating ALP may be critical for right ventricle function (547). A role for CLP36 has not yet been determined in the heart, but it may be important in myofibrillogenesis (336).
-
ENIGMA SUBFAMILY: Enigma and enigma-homolog protein (ENH) are found in skeletal and heart muscle, and contain one PDZ domain and three LIM domains. Enigma is anchored to the Z-disc via α-actinin-2 and can bind to protein kinase C through its LIM domains, indicating it may be involved in signaling pathways (349). Enigma binds to skeletal muscle-specific tropomyosin (formerly known as β-tropomyosin), possibly serving as an adapter protein for F-actin to recruit signaling proteins (233). ENH and its homologue cypher/oracle/ZASP are important for Z-disc integrity in cardiac and skeletal muscle. ENH forms a complex with short cypher (CypherS) isoform and calsarcin-1, which may help stabilize the Z-disc (110) (Fig. 2).
Global and cardiac-specific ENH knockout mice develop DCM from a loss of cypherS/ENH/calsarcin-1 complex resulting in Z-disc instability (110). Global knockout of the ENH homolog cypher results in development of congenital myopathy and postnatal lethality (775). However, cardiac-specific cypher knockout mice survive to six months old and develop DCM (772). Double knockout of ENH and cypherL (long isoform) is embryonic lethal due to aborted heart development, while ENH and cypherS double knockout mice survive to adulthood. Results from single and double knockout of ENH and cypher indicate that ENH and cypherL are functionally redundant in cardiac development, while ENH and cypherS play a role in Z-disc stability in the adult heart (470). Further highlighting the role of cypher/ZASP in Z-disc stability, mutations in ZASP have been identified in patients with skeletal distal myopathy and DCM (226,656).
LIM-ONLY PROTEIN 7 (LMO7): LMO7 is found at the Z-disc in developing cardiac and adult skeletal muscle. It has one LIM domain, one PDZ domain, and a calponin-homology domain (576). Like all PDZ-family proteins, LMO7 binds to α-actinin (522). Mice with a deletion of Lmo7Δ800 (which removes an 800 Kb region that contains the Lmo7 gene) develop severe muscle degeneration and growth retardation (622). In chicken, mice, and zebrafish, Lmo7 is found in the secondary heart field, outflow tract, inflow tract, and the proepicardium. Knockdown of Lmo7 in zebrafish results in defects in heart and conduction system development, indicating Lmo7 may play a key role in heart development (529).
LIM KINASE: LIM kinases (LIMK1 and LIMK2) have two LIM domains, one PDZ domain, and a protein kinase C domain (514). LIMK1 and LIMK2 are most abundant in neural tissue, but are also present in numerous tissues including in the Z-discs of the developing heart (384, 501). LIMK1 phosphorylates cofilin-1 at serine 3, which inhibits cofilin-1’s function as an actin-severing protein (24). Knockout of LIMK1 and LIMK2 results in severe neuronal effects (437), but little is known about their role in the heart.
FOUR-AND-A-HALF LIM (FHL) FAMILY
The FHL proteins 1, 2, and 3 are expressed primarily in striated muscles (465). FHL1 (also known as SLIM1, KyoT) isoforms have diverse localizations (M-band, I-band, nucleus and cytoplasm) and interact with more than a dozen proteins including human cardiac titin and cardiac myosin-binding protein C (cMyBP-C), as well as proteins of the MAPK signaling pathway [reviewed in (632)]. Both FHL1 and FHL2 bind to titin’s N2B spring region (Fig. 5) and activate downstream signaling pathways, thus serving as an important mechanosensor that triggers hypertrophy in response to strain (223).
Figure 5.
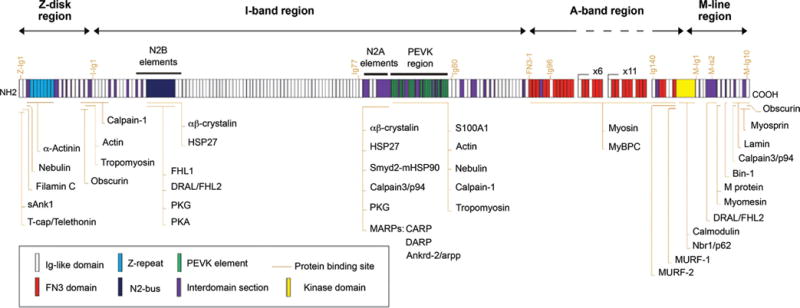
Schematic representation of the titin domain structure and localization of its binding partners in striated muscle. Titin is a huge protein that spans half a sarcomere from the Z-disc to the M-line region. The N-terminal region of titin inserts into the Z-disc, and many of the interaction in this region contribute to mechanosensing, structural integrity, and force transmission. I-band titin contains elastic elements, which play a critical role in passive tension. The A-band region binds to myosin and MyBPc, linking the myosin-based thick filaments to titin. M-band titin is important to both structural support and signaling. Abbreviations: sAnk1, small-ankyrin-1 isoform; FHL1 and 2, four-and-a-half-LIM-domain protein-1 and -2; PKG and PKA, protein kinase-G and -A; MARPs, muscle ankyrin repeat proteins; CARP, cardiac ankyrin repeat protein; ankrd-2/Arpp, ankyrin repeat domain 2; DARP, diabetes-related ankyrin repeat protein; S100A1, S100 calcium-binding protein A1; MyBPC, myosin-binding protein-C; MURF-1 and MURF-2, muscle-specific RING-finger protein-1 and -2; FN3, fibronectin type 3 like domain; Ig-like, immunoglobulin-like domain; N2-bus, N2-B-unique sequence; PEVK, titin region rich in proline (P), glutamate (E), valine (V), and lysine (K). The following titin-binding proteins were not discussed in this review: HSP27, heat shock protein-27; Smyd2, SET and MYND domain-containing protein-2; mHSP90, methylated heat shock protein-90; Nbr1, neighbor of BRCA1 gene-1; Bin-1, bridging integrator-1 [see (105) for discussion of these proteins]. [Fig. reprinted, with permission, from (105).]
Importantly, FHL1 binds to the prohypertrophic transcription factor NFATc1 (nuclear factor of activated T cells, cytoplasmic, calcineurin-dependent 1), enhancing NFAT activity and hypertrophy (127); knockout of prohypertrophic FHL1 in mice results in a blunted response to hypertrophy in the heart and skeletal muscle myopathy (147,634). Dysregulation of FHL1 due to mutations in this protein are causative for several forms of human X-linked skeletal muscle dystrophies: Emery-Dreifuss muscular dystrophy, reducing body myopathy, rigid spine syndrome, scapuloperoneal myopathy, and X-linked myopathy with postural muscle atrophy [reviewed in (126)]. FHL1 upregulation is potentially therapeutic for skeletal myopathies as upregulation can rescue a DMD mouse model (mdx model) by increasing muscle hypertrophy through the NFAT pathway (136). Furthermore, upregulation of FHL1 is detected at early stages of DCM progression in multiple mouse models, making it a promising candidate as a marker of early detection of DCM (195). Human patients with DCM, HCM, and other cardiac pathologies also have increased levels of FHL1 [reviewed in (126,632)].
FHL2 (also known as SLIM 3 and DRAL) is expressed most abundantly in cardiac muscles (100,202). In the sarcomere, FHL2 binds to two distinct regions of titin (N2B and IS2) and localizes to both the Z-disc and M-band, respectively (Figs. 2 and 5). FHL2 docks important metabolic enzymes such as phosphofructokinase, creatine kinase, and adenylate kinase, all of which are key to cross-bridge cycling (356) [see section “Metabolism of ATP: Maintaining a sufficient pool of ATP for proper muscle contraction” for more information on metabolic enzymes] (Fig. 8). Interestingly, FHL2 has the opposite effect on hypertrophy than FHL1. While FHL1 enhances hypertrophic NFAT signaling, FHL2 interacts with calcineurin (which activates NFAT) and suppresses calcineurin activity (270,328). In human heart failure, FHL2 expression is reduced and is no longer localized to the sar-comere. At the M-band, FHL2 binds to metabolic enzymes. In heart failure patients, loss of FHL2 localization also results in decreased activity of creatine kinase, phosphofructokinase, and adenylate kinase, which could contribute to the progression of heart failure (77,294).
Figure 8.
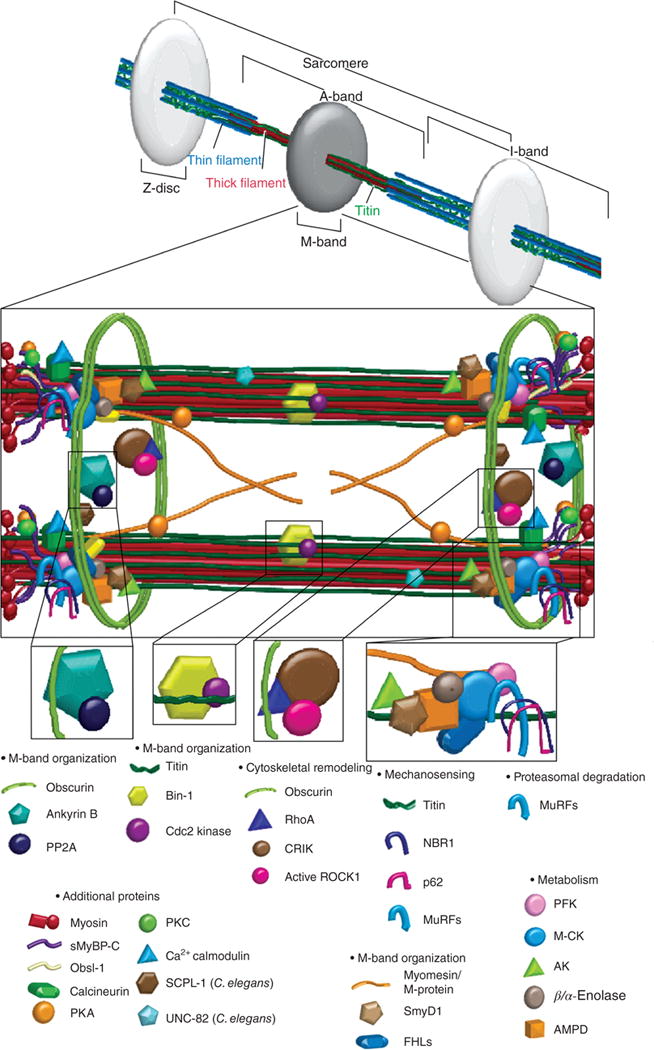
The sarcomeric M-band contains components important for mechanosensing, proteosomal degradation, actin dynamics, metabolism, and signal transduction. Myomesin is a key structural protein of the M-band. MURFs (muscle-specific ring finger protein) are multifunctional proteins that ubiquitinate certain myofibrillar proteins, play a key role in muscle atrophy and regulate hypertrophic signaling. Obscurin interacts with ankyrin and anchors the sarcomere to the sarcoplasmic reticulum; ankyrin and obscurin also sequester PP2A (protein phosphatase 2A) to the M-band. FHLs (four-and-a-half LIM proteins) bind to titin’s N2B spring region and activate downstream signaling pathways, thus serving as an important mechanosensor that triggers hypertrophy in response to strain. FHL2 also docks important metabolic enzymes such as the metabolic enzymes muscle-specific M-CK (creatine kinase), AK (adenylate kinase), and PFK (phosphofructokinase). M-CK anchors the glycolytic enzyme β/α-enolase to the M-band. The muscle isoform of AMPD (adenosine monophosphate deaminase) works with M-CK and AK to monitor local ATP levels. Other proteins identified at the M-band, but not discussed in this review include SmyD1, SCPL-1 (Caenorhabditis elegans), UNC-82 (C. elegans), p62, rhoA, CRIK, and active ROCK1. [Fig. reprinted, with permission, from (277).]
FHL3 (SLIM 2) is predominantly expressed in skeletal muscles and localizes to the Z-disc of mature myofibrils (464) (Fig. 2). FHL3 directly binds to actin, inhibits actin cross-linking activity of α-actinin in vitro, and directly interacts with MyoD to negatively regulate the MyoD-dependent myogenic differentiation process (121,125).
Z-disc is an important signaling node in the sarcomere
MLP
MLP is an important component of the cardiac stretch sensor machinery (Fig. 4). Muscle cells need to respond rapidly to increased mechanical stretch by activating downstream pathways of hypertrophy or myocyte survival [reviewed in (84)]. The MLP/titin/T-Cap complex plays an important role in stretch-induced signaling (Fig. 2). In MLP knockout mice, T-Cap mislocalizes from the titin spring region and the mice develop DCM likely due to their inability to sense passive stretch (318). Following stretch of isolated cardiomyocytes (to simulate contraction), MLP translocates to the nucleus (65), and interacts with transcription factors that regulate myogenesis (e.g., MyoD, myogenin, and MRF4) (327) (Figs. 2 and 4). MLP shuttles between cytoskeletal assemblies in order for the sarcomere to respond to changes in cardiac load.
MLP plays a key role in cardiac disease. MLP anchors the prohypertrophic Ca2+/calmodulin-dependent phosphatase calcineurin to the Z-disc; calcineurin dephosphorylates NFAT (nuclear factor of activated T-cells) and activates the hypertrophic response (Fig. 2). Following myocardial infarction in heterozygous MLP mice (∼50% reduction of MLP), there is a blunted response of the calcineurin/NFAT hypertrophy pathway indicating that MLP plays a pivotal role in stress-induced hypertrophy (252). The connection between MLP levels and calcineurin/NFAT signaling may provide insight into possible therapeutic targets for heart failure patients (778). The MLP knockout mouse was the first genetic mouse model of DCM (26). In humans, mutations in MLP, clustered in the N-terminal region, have been identified that result in both DCM and HCM [reviewed in (702)]. A knock-in mouse model was developed of a MLP mutation, W4R; these mice develop HCM indicating that this mutation in MLP is directly responsible for the development of disease (319).
MUSCLE ANKYRIN REPEAT PROTEINS (MARPs)
The MARP family of proteins has three members: CARP1/Ankrd1 (cardiac ankyrin repeat protein/ankyrin repeat domain 1), CARP2/Ankrd2/Arpp (ankyrin repeat protein with PEST and proline rich region), and CARP3/DARP/Ankrd23 (diabetes-related ankyrin repeat protein). All members of the MARP family contain four ankyrin repeat domains and localize to both titin’s elastic N2A region and the nucleus (446) (Fig. 5), while the N-terminal domain of MARPs is necessary for dimerization (750). While the MARP family is important stress responsive proteins, none of the proteins are required for cardiac development as single, double, and triple knockout of MARP family members results in no detectable cardiac phenotype (43). DARP is the least studied protein in the MARP family. It is expressed in heart and skeletal muscle, and is upregulated following insulin challenge in Type II diabetic mice suggesting a role in metabolism (286).
CARP1 (encoded by the gene ANKRD1) is expressed in cardiomyocytes, and to a lesser extent in skeletal muscle, with expression seen in the earliest stages of heart development (780). CARP1 gene expression is part of the fetal gene program, which increases following the induction of pathologic hypertrophy (15, 348). CARP1 interacts with a wide-variety of proteins such as calsequestrin, desmin, FHL2 myopalladin, and talin (44, 468, 687, 750). In the nucleus, CARP1 binds to numerous transcription factors involved in cell proliferation and differentiation, apoptosis, immune response, and hematopoiesis (325). Overexpression of CARP1 decreases expression of the NF-κB inflammatory pathway possibly to protect muscle from stress and excessive hypertrophy (360). CARP1 is upregulated in patients with DCM, HCM, and ischemic cardiomyopathy (477, 736, 779), and a mutation in the ankrd1 gene is causative for HCM and DCM (27,468).
CARP2/Ankrd2/Arrp is found in type I (slow twitch) skeletal muscle fibers and its levels increase following stretch (311,537). There are very low or undetectable levels of ankrd2 in the heart; however, ankrd2 is upregulated in patients with DCM (289, 467, 477). Ankrd2 is highly responsive to stress and increases following exercise, stretch, and muscle injury (46, 256, 367, 696, 697). The mechanism by which ankrd2 responds to stress is by shuttling to the nucleus and repressing the NF-κB inflammatory pathway; repression of this pathway is dependent on phosphorylation of ankrd2 (50,402). As a key factor in muscle stress responses, it is no surprise that Ankrd2 levels are altered in many myopathies (478,480,537).
MYOPODIN
Myopodin, of the synaptopodin gene family, is another protein that can shuttle between the sarcomere and nucleus. Its cellular localization is dependent on the developmental stage, phosphorylation status, or cellular stress of the myocyte (160, 737). Myopodin directly binds to filamentous actin (F-actin), and colocalizes with α-actinin and filamin-C in Z-discs of nascent and mature myofibrils and plays an important role in organizing early Z-discs (390, 737). Myopodin also has the ability to bundle F-actin in a manner similar to α-actinin during myofibril assembly (389). Myopodin’s binding to 14-3-3 protein and subsequent localization to the nucleus is positively regulated by protein kinase A (PKA) and calcium/calmodulin-dependent kinase II (CaMKII), and is negatively regulated by calcineurin (160) (Fig. 2).
MYOPALLADIN
Myopalladin is another sarcomeric protein containing immunoglobulin-like domains: two in the N-terminal half and three in the C-terminal half. Myopalladin is found both at the Z-disc and in the nucleus (44,446). At the Z-disc, myopalladin directly binds to α-actinin, nebulette, and CARP (407,443) (Fig. 2). Myopalladin mutation Y20C leads to the development of HCM and DCM. The Y20C mutation decreases the nuclear shuttling of myopalladin affecting its binding to CARP1 thus decreasing CARP1 function resulting in upreguation of hypertrophic genes (574). Another myopal-ladin mutation, Q529X, results in loss of α-actinin and neb-ulette binding and leads to the development of RCM (574). Analysis of Q529X knock-in mice indicate that mutant Q529X myopalladin can translocate to the nucleus, although CARP1 activity levels are decreased leading to an increase in fibrotic genes and resulting in progressive RCM (280).
ZYXIN
Zyxin is a phosphoprotein found in focal adhesion sites that also translocates from the Z-disc and nucleus (Fig. 4). The N-terminal half of zyxin contains a conserved leucine-rich region that not only regulates its subcellular distribution, but also serves as a nuclear export signal. Zyxin possesses three LIM domains in its C-terminal half, and its N-terminal proline-rich region interacts with α-actinin and SH3 domain-containing proteins (131, 495). Responding to mechanical stress, zyxin translocates to the nucleus and activates genes responsible for cell survival (303) (Fig. 2).
Other Z-disc proteins
T-CAP: T-Cap (titin-cap or telethonin) is a striated muscle-specific protein that provides a strong tie between the N-termini of two anti-parallel titin molecules in the Z-disc (Fig. 5). Two immunoglobulin-like domains (Z1 and Z2) of titin are bolted together by two unique β-sheets of T-Cap (225,561,704). T-Cap interacts with FATZ (filamin, α-actinin, and telethonin-binding protein of the Z-disc) family members (182, 183), E3 ubiquitin ligases (684), and MinK – aβ-subunit of the delayed rectifier potassium channel (189) (Fig. 2). Human T-Cap mutations are associated with skeletal muscle-related diseases, such as limb-girdle muscular dystrophy type 2G and congenital muscular dystrophy, as well as cardiac muscle-related diseases such as DCM and HCM (163, 250, 318, 463). Knockout of T-Cap in mice results in a mild dystrophic phenotype indicating that a simple loss of or reduction of T-Cap may not explain its role in disease development (414). An alternative hypothesis is that T-Cap phosphorylation plays a key role in its function. T-Cap is constitutively phosphorylated, and T-Cap phosphorylation regulates calcium transients and disrupts T-tubule organization in cardiomyocytes (87).
FATZ FAMILY (FILAMIN, α-ACTININ, AND TELETHONIN-BINDING PROTEIN OF THE Z-DISC)
The FATZ family of proteins interacts with an array of major Z-disc proteins, as its name suggests. FATZ-1 (also known as calsarcin-2 or myozenin-1) and FATZ-3 (calsarcin-3 or myozenin-3) are highly expressed in fast-twitch skeletal muscles, and FATZ-2 (carsarcin-1 or myozenin-2) is found in cardiac and slow-twitch muscles (161, 182, 183, 671). FATZ family members are small proteins with α-helical N- and C-terminal regions flanking a central glycine-rich domain (161, 671). FATZ proteins also bind to other Z-disc proteins, such as myotilin (214) and cypher/oracle/ZASP (175) (Fig. 2). Initially identified as a binding partner of calcineurin, a calcium- and calmodulin-dependent serine/threonine protein phosphatase, it is proposed that FATZ-2 negatively regulates calcineurin signaling activity; FATZ-2 knockout mice have increased levels of calcineurin, which leads to accelerated development of HCM (175, 181, 183). In humans, mutations in FATZ-2 have been linked to development of HCM; however, disease progression may not be linked to altered calcineurin activity (525,595).
TITIN: The largest protein in the human genome
TITIN
Titin, or connectin, is a huge (often called “giant”) protein that spans half of the sarcomere, acts as a molecular spring and is key to the passive mechanical properties of the myofilaments. The human titin gene is made up of more than 38,000 amino acids and contains 363 exons (44). Titin is important for sarcomere stability as it spans the length of a half sarcomere. Numerous functionally diverse partners have been identified to interact along the length of titin (Fig. 5). Identification of these partners has contributed to the identification of new (and often surprising) roles for titin—these will be discussed below.
Z-DISC TITIN
The N-terminal region of titin is anchored in the Z-disc. The first 200 residues contain multiple immunoglobulin repeats and varying numbers (between two and seven) of Z-repeats (modules located within the Z-disc that bind α-actinin and determine Z-disc width); the number of Z-repeats varies depending on the tissue type and developmental stage (198). Regions of N-terminal titin bind proteins such as actin (386), α-actinin (765), T-Cap (225, 472), small ankyrin 1 (331), filamin-C (353), and nebulin/nebulette (747) (Figs. 5 and 6). At the junction of the Z-disc and I-band, the proteins tropomyosin (386), obscurin (764), and calpain (582) bind titin (Fig. 5). These interactions support structural integrity, force transduction, and mechanosensing at the Z-disc (Fig. 7).
Figure 6.
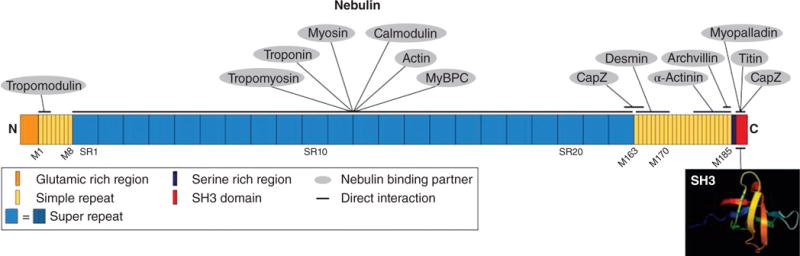
Schematic representation of nebulin domain structure and localization of binding partners in striated muscle. Nebulin is a large protein that interacts with a multitude of sarcomeric proteins including: capZ, titin, myopalladin, α-actinin, and desmin at its C-terminus in the Z-disc; tropomyosin, troponins, myosin, calmodulin, actin, and myosin-binding protein C (MyBPC) along its 22 seven-module super-repeats (blue); and tropomodulin at its N-terminus, though this interaction is likely transient. These protein interactions have given rise to two similar yet distinct functional models—as a molecular ruler and as an actin stabilizer. Archvillin is not discussed in this review. [Fig. modified, with permission, from (330).]
Figure 7.
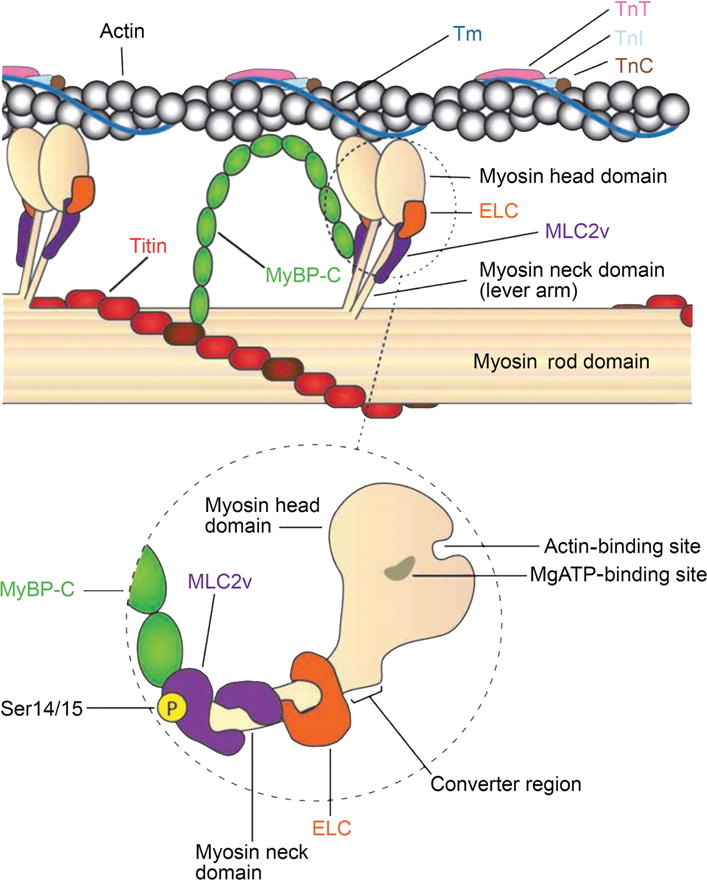
Schematic drawing of thin and thick filament interactions in striated muscle highlighting the major myosin regulatory proteins. Muscle contraction is dependent on the interactions between myosin-based thick filament via the head domain and actin-based thin filament. Thick filament regulatory proteins—myosin essential light chain (ELC), myosin regulatory light chain-2 (MLC2v), and myosin-binding protein C (MyBP-C)—control muscle contraction. MyBP-C interactions with actin, the myosin rod domain, MLC2v, and titin are depicted. The dashed circle is a magnified view highlighting (i) MyBP-C interaction with MLC2v located in the neck domain of myosin, (ii) the actin and MgATP-binding sites located within the myosin head domain, and (iii) MLC2v phosphorylation (Ser14/15) site important in promoting actin-myosin interactions. Abbreviations: Tm, tropomyosin; TnT, Troponin T; TnI, Troponin I; TnC, Troponin C. [Fig. modified, with permission, from (633).]
I-BAND TITIN
The elastic I-band region of titin consists of immunoglobulin sequences with intermittent unique regions. The PEVK region (named because it consists of approximately 70% proline (P), glutamic acid (E), valine (V), and lysine (K) residues) is primarily responsible for titin’s elastic properties (352). The N2A and N2B regions also contain immunoglobulin domains; the N2A region is found in all titin isoforms while the N2B region is only found in cardiac titin (179). The N2B isoform lacks the N2A region and part of the PEVK region. Numerous proteins interact with I-band titin (Fig. 5). In particular, the PEVK domain interacts with calpain-1 (249) and nebulin (407), as well as actin (386) and tropomyosin (581).
Titin’s I-band region (along with collagen) is key to passive tension, in which the muscle lengthens without contractile force. Passive tension can be fine-tuned in response to changes in mechanical demand or exercise. Alternative splicing of titin allows for changes in passive tension. In the heart, the shorter N2B isoform has more passive stiffness (fewer extensible spring regions) compared with the longer N2BA isoform (which has the N2A, N2B and PEVK regions) (691). Interestingly, the expression ratios of these isoforms are altered in hearts of patients suffering from chronic ischemia. Instead of the normal 30:70 N2BA:N2B ratio, patients exhibit an expression ratio closer to 50:50, leading to a decrease in passive muscle stiffness (484). Phosphorylation of the N2B region of titin can also alter its function, reducing passive tension in the heart (185, 757). Titin is a vital adjustable spring that is necessary to respond to changes in mechanical force.
Titin’s I-band region also participates in mechanosensing and the hypertrophy response. Interactions between the skeletal muscle N2A region and calpain 3/p94 suppress calpain’s autolytic activity and therefore protect titin from proteolysis (249) (Fig. 5). The immunoglobulin domains in the N2A or N2B linker regions unravel at low forces, while the PEVK region unravels at high forces (222, 373, 387, 388). With such a response to contractile stress, the I-band region of titin exhibits mechanosensory properties. The N2A region also interacts with muscle-ankyrin repeat proteins (MARPs) to elicit a mechanosensory response (446) (Fig. 5). Four-and-a-half-LIM-domain proteins (FHLs) bind to the N2B region, which in response to biomechanical stress activates hypertrophy pathways (634) [see section “Z-disc is an important signaling node in the sarcomere” for more information on MARP and FHL proteins] (Fig. 5).
A-BAND TITIN
In the A-band, titin interacts with myosin-binding protein C (MyBP-C) and the myosin tail domains, thus linking titin to the thick filaments (275, 648) (Fig. 5). The inextensible A-band region of titin is composed of super repeats of seven fibronectin III domains and four immunoglobulin domains (275). Eleven of these correspond to the C-zone thick filament repeats and may define the number and position of myosin and MyBP-C (178) (Fig. 15B). It has been suggested that the fibronectin III domains position myosin heads adjacent to the thick filament backbone and slightly change the orientation of the heads under stress that may contribute to active force development (287,473).
Figure 15.
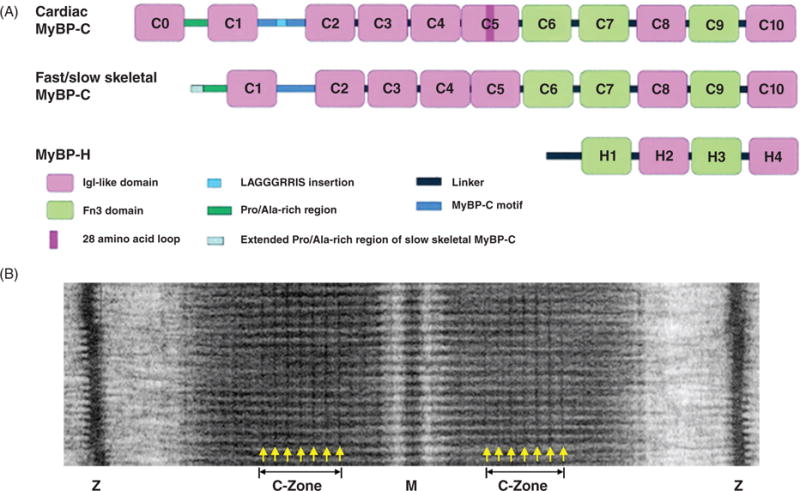
Myosin-binding proteins (MyBP). (A) Schematic drawing of MyBP domain organization. MyBPs are composed of a series of immunoglobulin (Igl-like in pink) and fibronectin type III (Fn3 in green) repeat domains. Domains termed C1 through C10 and a 105-residue linker between C1 and C2 termed the MyBP-C motif (in blue) make up the core structure of MyBP-C isoforms. Cardiac MyBP-C has the addition of an eight IgI-like domain termed C0, a unique amino acid sequence—LAGGGRRIS—insertion (in light blue) in the MyBP-C motif, and a 28 amino acid insertion (in dark pink) in the C5 domain. Slow skeletal MyBP-C differs from the fast isoform with an extended Pro/Ala-rich region at the N-terminus. MyBP-H is the smallest isoform with four domains similar to C7 through C10 of MyBP-C and a unique Pro/Ala-rich linker (in black) region. (B) Example electron micrograph of frog skeletal muscle showing MyBP-C transverse stripes located in the C-Zone. [Part A modified, with permission, from (167); Part B modified, with permission, from (406).]
M-BAND TITIN
Several protein interactions at the titin kinase (TK) domain and nearby regions implicate that M-band titin is important in both signaling and structural support [reviewed in (341)] (Fig. 5). For example, muscle-RING-finger-proteins (MURF) 1 and 2 may mark titin for proteo-somal degradation through the ubiquitin pathway (744, 749) (Fig. 8). The extreme C-terminus of titin binds FHL2 and p94, similar to I-band titin, as well as myospryn (scaffolding protein that regulates calcineurin signaling) and obscurin that serve both structural and signaling roles [reviewed in (196, 385)]. Through interactions with myomesin, M-band titin further stabilizes the thick filament (506) (Fig. 9).
Figure 9.
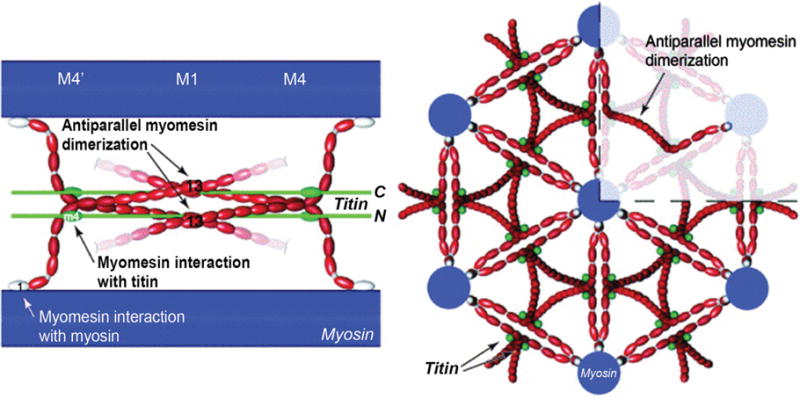
(Right) Longitudinal view of myosin (blue), myomesin (red) and titin (green). The M-band is composed of a series of electron-dense M-lines: M4, M1, and M4′ (see Fig. 1C for an electron micrograph of M-lines). Myomesin family members form antiparallel homodimers through interactions called M-bridges between the C-terminal immunoglobulin domain (labeled 13), and bind to myosin at the N-terminal domain. (Left) Cross-sectional view highlighting myomesin forming an antiparallel dimer. Myomesin acts as a thick filament cross-linking protein. [Fig. reprinted, with permission, from (9).]
Mutations in titin have emerged as a major cause of disease, resulting in both skeletal and cardiac myopathies. Mutations that truncate titin are the most common genetic cause for DCM. In particular, it was found that approximately 25% of idiopathic DCM patients have titin truncation mutations (predominantly in the A-band region), while mutations in titin are rarely seen in HCM patients (257). In addition, mutations and post-translational modifications in titin have been identified in patients with numerous cardiomyopathies including DCM, HCM, RCM, and arrhythmogenic right ventricular myopathy [reviewed in (370,485)].
Titin mutations have been identified in numerous skeletal myopathies, as well [reviewed in (104)]. Examples of muscle myopathies include tibial muscular dystrophy (TMD), hereditary myopathy with early respiratory failure (HMERF) and centronuclear myopathy (CNM). Interestingly, these skeletal myopathies do not present concurrently with cardiac phenotypes. Mutations in skeletal muscle titin are not localized to any one region of the titin molecule. For example, TMD is the result of a mutation in M-band titin; this disease is characterized by atrophy and weakening of the tibialis muscle (235). Titin’s A-band is a hotspot for mutations leading to HMERF; this disease is characterized by weakness of the extremities and diaphragm (555). CNM has been associated with mutations in titin that lead to truncations (97). To date, there are nine additional titin mutations that result in both skeletal and cardiac myopathies [reviewed in (104). Inevitably, more titin mutations will likely be described in the future, highlighting the critical role titin plays in the sarcomere.
ACTIN: The most abundant protein in eukaryotic cells
Actin is the primary component of the sarcomeric thin filament and makes up 20% of the mass of striated muscle. Globular in solution (G-actin), individual actin molecules interact to form tightly regulated, yet highly dynamic, filamentous polymers (F-actin) (Fig. 10). Actin filaments are polar in structure with their barbed ends inserted in the Z-disc and the pointed ends extending into the M-band (Fig. 1). The designation of barbed and pointed end comes from the arrowhead appearance of F-actin bound to heavy meromyosin (HMM) in vitro (283).
Figure 10.
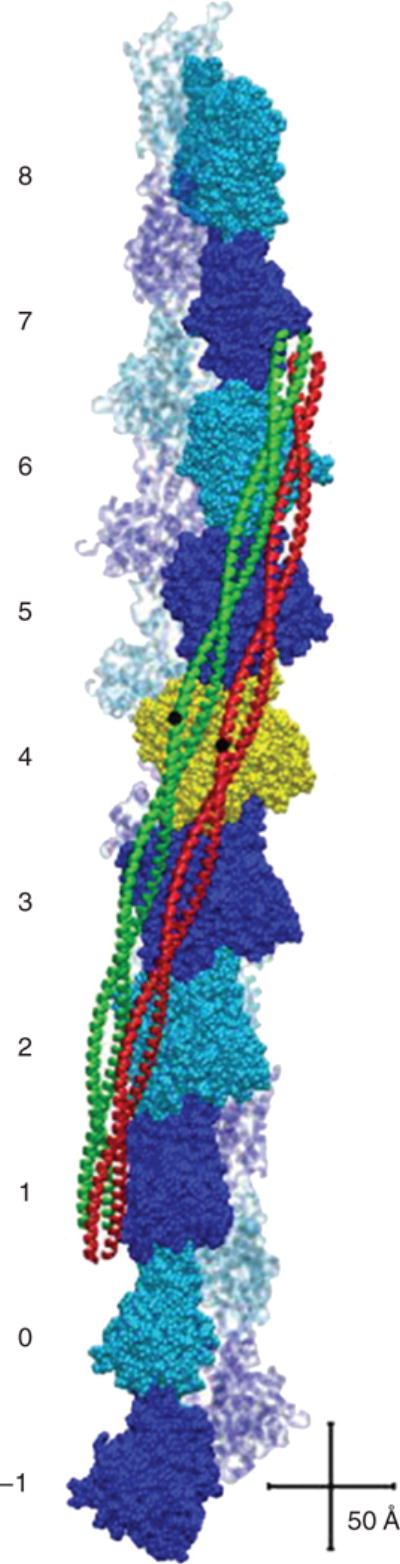
Tropomyosin positions on the surface of F-actin in the presence (green) and absence (red) of myosin. Ten actin-pairs (alternately colored blue and cyan) are shown with the pointed end facing up. Two tropomyosin α-helical chains form coiled-coils that interact with the positively charged groove of actin filaments and form dimers that span seven actin monomers. Tropomyosin regulates interactions between actin-based thin filaments and myosin-based thick filaments to control cross-bridge cycling. Depicted in ribbon representation are tropomyosin coiled-coils in either in the troponin and myosin-free (red), or the myosin head (S1)-decorated (green). Tropomyosin residue 125 is shown in black as a reference point, highlighting the relative sliding between the positions. Scale equals 50Å Actin is numbered -1 to 8. [Fig. reprinted, with permission, from (523).]
The structure of G-actin comprises two domains connected together via two “hinge” strands, with each domain further divided into two subdomains (297, 534) (Fig. 11). Two coiled strands of actin polymers intertwine to make up a single polarized F-actin filament (152)]. Six genes in the human genome encode different actin isoforms that share more than 87% sequence identity: α-skeletal-, α-cardiac-, α-smooth-, β-cytoplasmic-, γ-smooth-, and γ-cytoplasmic-actin [reviewed in (229)]. Though they share high sequence homology, the different isoforms display tissue, as well as cytoskeletal assembly-specific, expression patterns. α-Skeletal- and α-cardiac-actin are the primary isoforms which make up the sarcomeric thin filaments in their respective striated muscle cells (57), while γ-cytoplasmic-actin is found in costameres (598). Non-striated muscle isoforms, β-cytoplasmic- and γ-cytoplasmic-actin, are ubiquitously expressed, while α-smooth-actin is found predominantly in smooth muscle.
Figure 11.
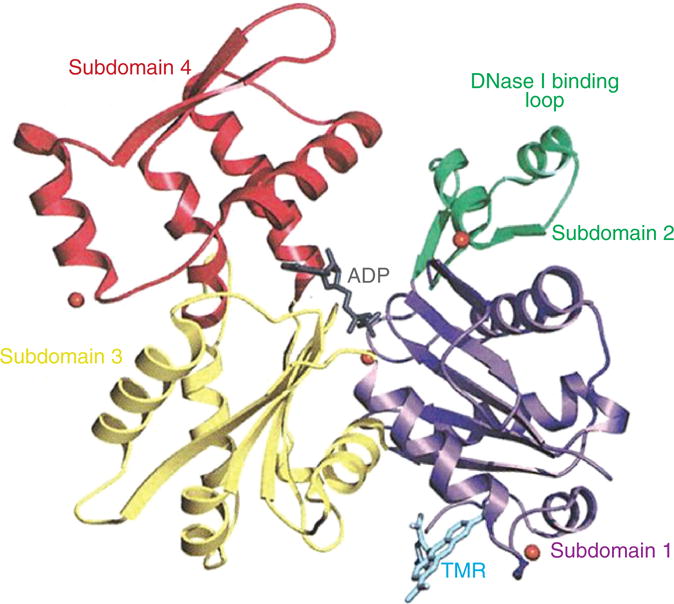
Ribbon structure of globular actin in the ADP-bound state. Actin is an asymmetrical protein composed of four subdomains (subdomain 1 shown in purple, subdomain 2 shown in green, subdomain 3 shown in red, and subdomain 4 shown in yellow) connected via two “hinge” strands. The representation is oriented with the pointed (minus end) at the top and the barbed (plus end) at the bottom. ADP is shown in stick representation bound in the cleft. Shown in cyan in stick representation is tetramethylrhodamine-5-maleimide (TMR), a fluorescent probe that inhibits actin polymerization. [Fig. reprinted, with permission, from (534).]
Mouse models have been instrumental in determining the specialized roles of the different actin isoforms and whether isoforms can compensate for each other. Both cardiac and skeletal actins are necessary for proper sarcomere organization and function. α-Cardiac-actin knockout mice die either before or shortly after birth due to a loss of thin filaments in the sarcomere leading to cardiac failure (345). α-Skeletal-actin knockout mice have normal thin filaments, due to a compensatory upregulation of cardiac actin, but show decreased muscle strength; the knockout mice die within 10 days of birth due to malnutrition (132). Costameres are only mildly disrupted in γ-cytoplasmic-actin knockout mice, however progressive necrosis develops in their skeletal muscle as a result of this disruption (645). In short, analysis of actin isoform-specific knockout mice have revealed that, while different isoforms of actin are often observed to partially compensate for each other, α-skeletal- and α-cardiac-actin are essential for maintaining the integrity of the sarcomere.
As a primary component of the sarcomere, it comes as no surprise that several mutations in striated muscle actin isoforms result in different muscle myopathies. More than 200 missense mutations in ACTA1 (which encodes α-skeletal-actin) are associated with actin myopathy (congenital myopathy with excessive actin filaments) and nemaline myopathy [reviewed in (500)]. Patients suffering from these myopathies experience severe muscle weakness and diminished muscle tone, with nemaline myopathy symptoms typically affecting face, neck, and limb muscles most severely. Approximately one in five patients suffering from nemaline myopathy has mutations in ACTA1; the mutations are, interestingly, not localized to any one region of actin (355). Actin mutations also result in intranuclear rod myopa-thy and congenital fiber type disproportion (354, 615). In the heart, mutations in ACTC (that encodes α-cardiac-actin) are linked to cases of HCM and DCM. HCM-related mutations likely interfere with myosin binding and, therefore, force generation, whereas DCM-related mutations occur elsewhere in the molecule and may disrupt force transmission to adjacent sarcomeres (452, 516, 517). The alterations in force generation and transmission observed as a result of HCM and DCM mutations likely lead to the heart remodeling seen in these myopathies.
Actin-thin filament length regulation: Proper striated muscle function requires precise thin filament lengths
CAPZ
CapZ (or β-actinin) is a highly conserved, barbed-end capping protein (93, 94). There are four genes that encode the different α- and β-isoforms, which interact to form heterodimers: CAPZA1, A2, A3, and CAPZB (244, 609). Three α-subunit isoforms bind differentially to actin and are each encoded by a separate gene, while the three β-subunit isoforms are all alternatively spliced from a single gene. The α1β1 heterodimer is the primary sarcomeric Z-disc isoform in striated muscle (thus called “CapZ”), binding α-actinin, and anchoring the thin filament in the Z-disc (94, 539) (Fig. 2). Overexpression of the β2 isoform (which normally localizes to the intercalated disc) in the heart causes displacement of the β1 isoform from its normal localization at the Z-disc and leads to the development of HCM (243). This study highlights the importance of CapZ; the presence of thin filaments not properly anchored into the Z-disc leads to disease development.
CapZ regulates actin polymerization and depolymerization (731). The C-terminal portion of CapZ α- and β-subunits each bind one terminal actin molecule; the β-subunit remains attached while the α-subunit is dynamically associated with the filament to allow actin monomers to come off and on (381, 759) (Fig. 12). Regulation of actin dynamics at the barbed end may also play a key role in both skeletal and cardiac hypertrophy. During hypertrophic remodeling, new sarcomeres are added in parallel to existing sarcomeres. When hypertrophy is induced in isolated cardiomyocytes by treatment with phenylephrine or endothelin-1, CapZ dynamics increase resulting in destabilization of the Z-disc to insert new sarcomeres (245). Further, when hypertrophy is induced by mechanical strain, actin dynamics increase at the barbed end, which is conducive for the addition of new sarcomeres (191, 380,382) (Fig. 12). Actin dynamics at the barbed end are vital for the sarcomere to respond to changing stress and strain.
Figure 12.
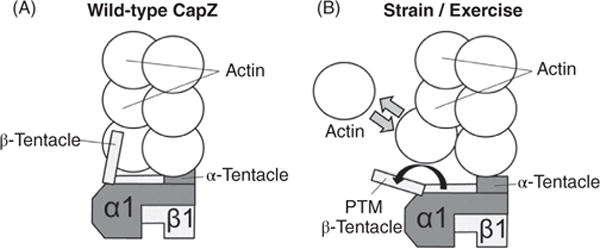
CapZ dynamics at the barbed end of F-actin. (A) CapZ has two subunits: α1 and β1 each with a tentacle that binds one terminal actin. Tightly capped F-actin has a low actin off rate. (B) Following mechanical stimulation (to simulate exercise), the β tentacle undergoes a structural change via post-translation modification (PTM) including phosphorylation on serine-204 and acetylation on lysine-199. The β tentacle shifts off the terminal actin, which increases actin monomer exchange. Regulation of actin dynamics at the barbed end may also play a key role in both skeletal and cardiac hypertrophy. [Fig. modified, with permission, from (381).]
TROPOMODULIN
Tropomodulin (Tmod) is a capping protein at the pointed ends of the thin filaments which blocks elongation and prevents actin depolymerization (732). There are four Tmod isoforms, with two expressed in vertebrate striated muscle: erythrocyte (E-Tmod or Tmod1) and skeletal (Sk-Tmod or Tmod4) (19, 128, 173). Tmod consists of a disordered N-terminus containing two tropomyosin-binding sites, an actin-binding site, and a leucine-rich repeat C-terminal region with a second actin-binding site [reviewed in (209,334,578,758)]. Tmod1 and 4 bind to the N-terminus of nebulin in vitro, but spatial separation in vivo reveals that this interaction is likely transient (95, 211, 427) (Fig. 6). Tmod1 is essential for proper cardiac development and myofibril-logenesis. Mice lacking Tmod1 exhibit perturbed cardiac development and subsequent embryonic lethality (117, 184), a phenotype that can be rescued by cardiac-specific overexpression of Tmod1, indicating that Tmod1 is essential in heart development (430).
Tmod4 is more highly abundant in skeletal muscle when compared to Tmod1 (212). Tmod4 knockout mice are viable and exhibit no detectable skeletal myopathies, however most intriguing was that thin filament lengths were not altered as expected. Tmod1 is able to compensate for the loss of Tmod4 in skeletal muscle to maintain proper thin filament lengths indicating that Tmod1 directly controls skeletal muscle thin filament lengths (212). Finally, Tmod3 caps γ-cytoplasmic actin filament pointed ends in the sarcoplasmic reticulum (SR), contributing to the stabilization of the SR complex. Following loss of Tmod1 in skeletal muscle, Tmod3 leaves the SR to cap the pointed end of the thin filament (209). Therefore, Tmod1 is vital to sarcomeric stability, while loss of Tmod3 and Tmod4 can be stabilized when Tmod1 compensates.
LEIOMODIN
Another less-investigated member of the tropomodulin family is leiomodin (Lmod). Three Lmod isoforms have been identified with each encoded by a separate gene: smooth muscle (Lmod1), cardiac/skeletal (Lmod2), and skeletal (Lmod3) (122). Lmod shares approximately 40% sequence identity with Tmod with some important differences; Lmod lacks the second tropomyosin-binding site found in Tmod and has a C-terminal extension that contains a third actin-binding, Wiskott-Aldrich homology 2 (WH2) domain (122). With three actin-binding sites, Lmod2 has been shown to be a potent actin nucleator in vitro (111). The unique WH2 domain is necessary for Lmod2s function, as removal of this domain makes Lmod2 function as a pointed-end capping protein [e.g., like Tmod1 (695)].
Due to the similar structural domains found in Lmod and Tmod family members, it is likely that they perform similar functions. Interestingly in Xenopus laevis skeletal muscle, knockdown of Lmod3 disrupts sarcomere assembly and overexpression of Tmod4 rescues the phenotype (and vice versa Lmod3 can rescue loss of Tmod4). This study indicates that these closely related family members could functionally compensate for each other in sarcomeric assembly (502). Clinically, mutations in Lmod3 are associated with nemaline myopathy, where 90% of patients with Lmod3 mutations suffer from severe cases of this disease (766). In accordance, Lmod3 knockout mice develop nemaline myopathy (683).
The thin filament pointed end is highly dynamic and its regulation is key in maintaining proper thin filament length (391). Altering levels of both Lmod2 and Tmod1 result in thin filament length changes. Overexpression of Lmod2 in isolated cardiomyocytes results in elongated thin filaments (695), while Lmod2 knockout mice have significantly shorter lengths and a DCM phenotype (545). In contrast, knockdown of Tmod1 in isolated cardiomyocytes results in elongated thin filament lengths, while overexpression of Tmod1 in isolated myocytes and mouse heart [Tmod1 overexpression mouse (TOT)] results in shorter lengths and a DCM phenotype (661, 662). Both the TOT and Lmod2 knockout mice highlight the importance of thin filament length regulation in proper contractile function.
There are a few proposed models for how Tmod1 and Lmod2 may regulate thin filament length. One is that Lmod2 and Tmod1 compete for pointed end binding and thereby fine-tune thin filament lengths (695). Alternatively, an Lmod2-independent mechanism was proposed based on X-ray crystallography. In this model, Tmod1 binds to actin and tropomyosin with multiple low affinity interactions, allowing part of Tmod1 to detach from the pointed end and actin monomer exchange (578). A similar mechanism was proposed for Lmod2 based on X-ray crystallography of Lmod2 bound to actin. This model suggests that Lmod2 binds to tropomyosin and two terminal actins at the pointed end, while the third WH2 actin-binding site recruits an actin monomer and makes it available to elongate the thin filament at the pointed end (109).
FORMIN
Formin proteins regulate assembly of unbranched actin filaments of the eukaryotic cytoskeletal system through their association with the barbed ends of actin filaments, making them ideal candidates as striated muscle actin nucleation factors (570, 572). The actin nucleation factor for initial thin filament assembly has not been identified. However, several independent reports have identified formin as a potential candidate (290,442,455,594).
Formins are a highly conserved family of dimeric proteins that are present in almost all eukaryotes (98,262). There are 15 mammalian formin homologues identified, and 13 are expressed in various sarcomeric localizations in developing mouse cardiomyocytes (594). The GTPase-binding domain, diaphanous (DIA) inhibitory domain and DIA autoregulatory domain of formins contribute to self-regulatory activity (216). In general, actin barbed-end binding proteins inhibit barbed end polymerization. Uniquely, formins not only allow actin monomer addition but also prevent barbed-end capping proteins, such as CapZ, from interfering with thin filament elongation (528).
Amongst seven known Z-disc-associated formins, FHOD3 (formin homology-2 domain containing protein 3) is the best studied. FHOD3 knockout mice are embryonic lethal mainly due to arrested myocardial development at embryonic day 10.5, indicating that FHOD3 plays a key role in regulating actin dynamics during myofibrillogenesis and is vital for heart development (299). Two different coding variants (V1151I and Y1249N) of human FHOD3 gene are associated with increased incidences of familial HCM and DCM, respectively (29,751). Taken together with data from investigations of Lmod2, Tmod1, and CapZ, control of actin dynamics at thin filament ends is critical to maintaining thin filament integrity and proper contractile function [see “The Nebulin family of proteins: a giant regulator of thin filament function” for nebulin’s role in thin filament length regulation].
The Nebulin family of proteins: A giant regulator of thin filament function
NEBULIN
Nebulin is part of a family of proteins including N-RAP, nebulette and LASP-2 (LIM and SH3 protein) [reviewed in (543)]. Nebulin is a huge protein that is highly conserved in vertebrates: 154 central modules (M9-M162) are divided into 22 seven-module super-repeats that associate with actin thin filaments (258). A single nebulin module interacts with one actin monomer, and each super-repeat associates with one thin filament regulatory complex (7 actin monomers:1 tropomyosin:1 troponin complex) (351) (Fig. 6). Nebulin is highly abundant in skeletal muscle, with very low amounts detected in the heart (42,307). Cardiac-specific nebulin knockout mice do not have altered thin filament length indicating that nebulin has a distinct, yet undefined, role in the heart (326).
Nebulin plays many roles in skeletal muscle. It functions in contraction by regulating actin-myosin interactions (41) and regulating calcium uptake to the sarcoplasmic reticulum (532). In addition, nebulin has been shown to regulate Z-disc alignment (686). In the Z-disc, nebulin’s C-terminus contains a serine-rich domain and an SH3 domain that interacts with CapZ, N-WASP, titin, and myopalladin (407, 542, 672) (Fig. 6). Nebulin and N-WASP form a complex at the Z-disc that can nucleate IGF-1-induced actin branches from the barbed end, indicating that nebulin may also play a role in the addition of new sarcomeres following the induction of hypertrophy (672). However, the function of nebulin that is most well studied is its role in thin filament length regulation.
A very popular model of nebulin’s function in thin filament length regulation is that of a “molecular ruler.” Data consistent with this role include the observation that nebulin assembles before actin filaments reach mature length early in myofibrillogenesis (511). The molecular layout of nebulin also suggests a molecular ruler (template) role since its C-terminus lies in the Z-disc with its N-terminus (regions M1-M3) extending to thin filament pointed ends (427) (Fig. 6). Further observations consistent with nebulin having a role as a ruler came from a study showing that the sizes of alternatively spliced nebulin isoforms correlate with thin filament lengths in different muscle fiber types and at different developmental stages (342).
However, inconsistent with nebulin having a strict ruler function, the pointed end of the thin filament extends past the N-terminus of nebulin (95). As such, an alternative model of nebulin’s role in thin filament regulation is that nebulin functions not as a strict molecular ruler but as an actin stabilizer. In support of this model, a truncated nebulin protein can stabilize thin filaments that extend far past its end (544). Consistent with this, a two-segment model has been proposed where it is predicted that nebulin stabilizes a core actin filament length, and actin filaments that extend beyond nebulin are regulated by Tmod1 [reviewed in (210)].
Approximately 50% of nemaline myopathy patients have mutations in nebulin, and further insight into the role of this diverse protein is necessary [reviewed in (593)]. Over 140 recessive variants have been identified in nebulin (499). Due to the heterogeneity of nemaline myopathy pathology and the large size of nebulin, it has been difficult to study the role of this protein in disease development. However, patients typically have reduced levels of nebulin leading to decreased thin filament lengths, decreased contractile force and muscle weakness highlighting that thin filament length regulation is necessary for proper contractile function (361,531,533).
NEBULETTE
Nebulette is the smaller, cardiac-specific nebulin relative (457). Unlike full-length nebulin, nebulette does not span the length of the thin filament. Its nebulin-repeat domain lies in the I-band region and its C-terminus interacts with several Z-disc-associated proteins (157, 271). Knockdown of nebulette decreases thin filament lengths and impairs beating of cultured cardiomyocytes, suggesting that, like nebulin, nebulette may also play a role in stabilizing the thin filament (458). Nebulette knockout mice exhibit no overt cardiac phenotype; however, cardiac stress genes are upregulated and Z-discs are wider indicating nebulette plays a role in Z-disc stability (421). Several nebulette mutations in humans have been associated with DCM, endocardial fibroelastosis, and cardiac failure, highlighting the need to learn more about the roles of this protein (575). Transgenic mice expressing the above mutations reveal that contractile function and calcium homeostasis are compromised leading to the development of DCM (410).
NEBULIN-RELATED ANCHORING PROTEIN (N-RAP)
N-RAP is exclusively expressed in striated muscle and is integral in promoting myofibrillar assembly (91,142,404,411). N-RAP is composed of a LIM domain at its N-terminus followed by 11 nebulin-like single domains and five super-repeats; N-RAP isoforms are alternatively spliced from a single gene (454). The skeletal muscle isoform is termed N-RAP-s, while exon 12 is absent in the cardiac iso-form, N-RAP-c. In the adult myofibril, N-RAP-c is found in the intercalated disc, and N-RAP-s is found at the myotendinous junction (404). N-RAP binds to actin, talin, vinculin, and MLP (153,403) (Fig. 4). At the intercalated disc, N-RAP likely binds to actin filaments and is thought to maintain the stability of F-actin at the intercalated disc junction. N-RAP has also been shown to increase in the early stages of DCM, indicating it may be an early marker in the development of disease (153).
LIM-NEBULETTE (LASP-2)
LASP-1 (LIM and Src homology 3 (SH3) Protein-1) and LASP-2 (LIM and SH3 Protein-2/LIM-Nebulette) are splice variants of nebulette that contains four unique exons from the shared nebulette gene (371, 538, 777). LASP-1 is expressed in non-muscle cells and is found at sites of actin dynamics such focal adhesions, lamellipodia, and filopodia [reviewed in (112)]. LASP-2 is expressed in low levels in skeletal and cardiac muscle and is found at the Z-disc where it interacts with α-actinin and bundles F-actin (777). In Drosophila, knockout of LASP results in decreased thin filament length. LASP has dual localizations at the A-band and I-band and controls proper thin filament spacing via interactions with both actin and myosin (162).
XIN-REPEAT PROTEINS
Xin is part of the Xin-repeat protein family of actin-binding proteins and is alternatively spliced into three isoforms in humans (A-C) (707). Xin is enriched at sites of actin filament anchorage to the plasma membrane such as myotendinous junctions (636). In skeletal muscle, Xin levels increase following muscle damage, and the degree of increase correlates with the degree of damage (494). Knockout of Xin in skeletal muscle results in myopathy with impaired contractile function and decreased satellite cell function. Loss of Xin also results in cytoskeletal instability, leading to apoptosis of satellite cells and an inability of the muscle to regenerate (18). Global Xin (A-C isoforms) knockout mice develop mild cardiomyopathy with perturbed intercalated discs suggesting that Xin is also an integral protein in the intercalated disc (232,530). Xin expression is being investigated as a marker of skeletal muscle damage, as well as of cardiomyopathies.
Tropomyosin and troponins: Key regulators of cross-bridge cycling
TROPOMYOSIN
Along with actin, tropomyosin contributes to proper sarcomeric function as both a structural support and as a regulatory protein. Two α-helical chains form coiled-coils that interact with the positively charged groove of actin filaments (718) and form dimers that span seven actin monomers (266, 734). These dimers interact head-to-tail to span the length of the thin filament (Fig. 1 and 10). Actin filament stiffness increases upon tropomyosin binding and this interaction inhibits thin filament depolymerization (735). Similarly, actin binding to tropomyosin decreases overall tropomyosin dynamics, stabilizing the actin-tropomyosin interaction even further (579).
The human tropomyosin family contains four genes – TPM1 (α-TPM), TPM2 (β-TPM), TPM3 (γ-TPM) and TPM4 (δ-TPM) that encode more than 40 alternatively spliced isoforms [reviewed in (199)]. α- and β-Tropomyosin are approximately 87% identical, and expression ratios vary based on the fiber type and developmental stage. α-Tropomyosin is the primary isoform expressed in cardiac and skeletal muscle, while β-tropomyosin is largely found in slow-twitch muscle fibers (476). α-Tropomyosin is essential for life as knockout of α-tropomyosin in mice is embryonic lethal (587), while overexpression of β-tropomyosin in the mouse heart leads to diastolic dysfunction (475). To our knowledge, no β-tropomyosin knockout mouse has been published.
As part of a complex with the troponins, tropomyosin regulates interactions between actin-based thin filaments and myosin-based thick filaments to control cross-bridge cycling (129,546). Each tropomyosin molecule is associated with one troponin complex [TnI (inhibitory-blocks myosin binding to actin), TnC (binds calcium) and TnT (binds tropomyosin)] and seven actin monomers (Fig. 10). A study using cryoelectron microscopy confirmed many calculated structural predictions and highlighted interactions between a positively charged groove on the outer surface of the actin filament and the largely negative tropomyosin (718). Under conditions of low calcium, tropomyosin covers the outer domain of actin and blocks the myosin-binding site. In the presence of calcium, TnC binds calcium and mediates dissociation of TnI and a conformational shift in tropomyosin, which then exposes myosin-binding sites on actin (101, 365, 714) (Figs. 13 and 14). As actin binds to the myosin heads, it activates myosin ATPase activity, inducing a conformational change in the myosin protein. This movement is referred to as the “power stroke,” sliding the thin filament toward the M-band and leading to contraction of the muscle [reviewed in (663)]. Tropomyosin acts as the “gatekeeper” for force generation as its position on actin directly determines which of the three states actin and myosin are in (677). The three states include the “blocked” state where myosin cannot bind actin, the “closed” state where myosin weakly binds to actin, and the “open” state where myosin strongly binds to actin (431,562). It is of note that there are other models of contraction proposed such as the “fly-casting” model and a four-state model [reviewed in (304)].
Figure 13.
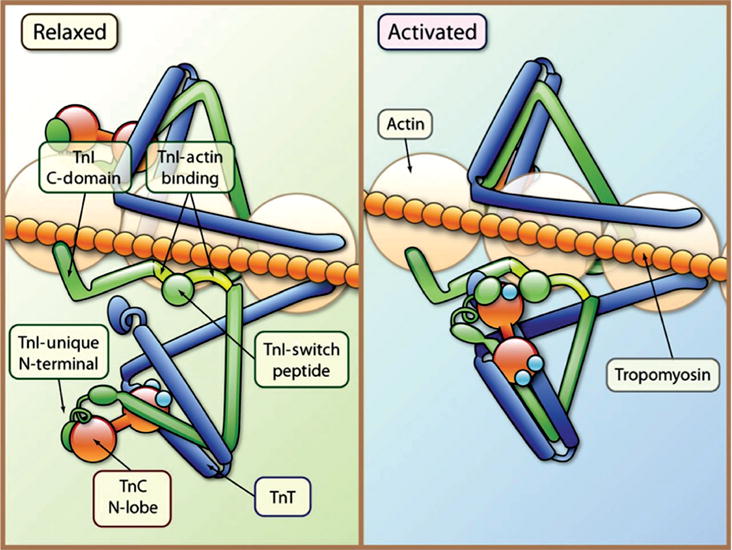
Tropomyosin and the troponin complex regulate striated muscle contraction. Each tropomyosin (orange chain) molecule is associated with one troponin complex [TnI (inhibitory-blocks myosin binding to actin; green), TnC (binds calcium; red barbells), and TnT (binds tropomyosin; blue)] and seven actin monomers. In the relaxed state tropomyosin blocks the myosin-binding site on actin. TnC is weakly bound to TnI; TnI binds to actin (TnI-actin binding) and inhibits myosin from binding to actin. Following the release of calcium (Ca2+), calcium binds to TnC and a patch of residues in the N-terminal domain of TnC is exposed and the interaction of TnC with TnI is enhanced. TnI then dissociates its inhibitory region from actin, and forms a complex with TnT and tropomyosin. Following the conformational change in the troponin complex, tropomyosin shifts and the myosin head binds to actin. [Fig. reprinted, with permission, from (642).]
Figure 14.
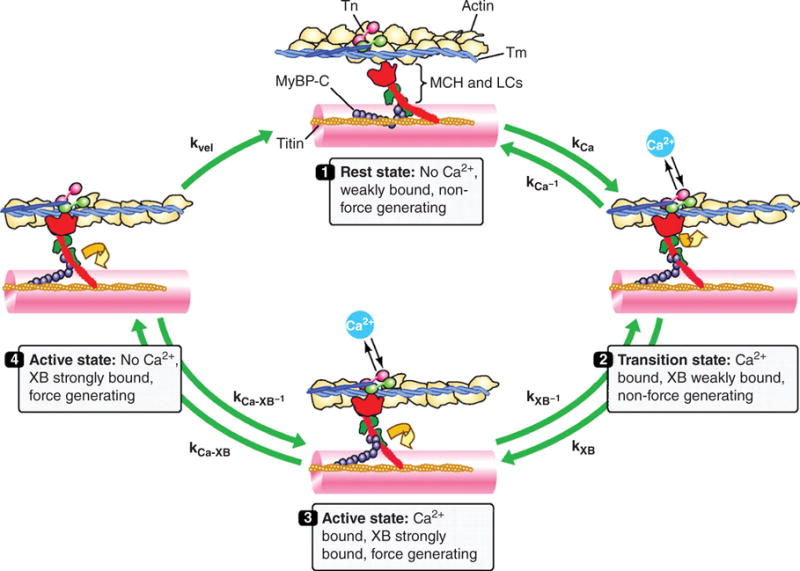
Schematic drawing of the cardiac cross-bridge cycle. Thin-filaments are shown with actin, tropomyosin (Tm) and the troponin (Tn) complex with the Ca2+-binding unit (cTnC) in pink, the Tm-binding unit (cTnT) in blue, and the inhibitory unit (cTnI) in light green. Thick-filament cross-bridges (XB) are shown with myosin heavy chain (MHC; figure illustrating one MHC) in red, myosin light chains (LC) in green, along with myosin-binding protein C (MyBP-C) in purple and titin in orange. Cross-bridges are initially in a rest state (1) where they are weakly bound and do not generate force. Cross-bridges enter a transition state (2) determined by the on (kCa) and off rates (kCa-1) for Ca2+ exchange with cTnC. During this transition state, cross-bridges are weakly bound (kXB-1) and do not generate force. In the active state (3), the cTnT-dependent shift of Tm from its blocking position on actin filaments allows strong cross-bridge binding (kXB) and induces cooperative activation of the thin filament (e.g., increase Ca2+ affinity of cTnC; kCa-XB-1). In the active state (4) with loss of bound Ca2+, the cooperative mechanisms allow a population of cross-bridges to remain active and force generating (kCa-XB). Mechanical feedback termed shortening-induced deactivation (kvel) will transition active cross-bridges back to the resting state. [Fig. modified, with permission, from (265).]
Tropomyosin plays a key role in coordinated activation of the thin filament and contractile function [reviewed in (396)], therefore it is not surprising that mutations in tropomyosin can have severe effects on striated muscle function. In humans, numerous mutations in TPM1 have been linked to familial HCM and DCM [reviewed in (676)]. Further, mutations in TPM2 and TPM3 have been observed in cases of nemaline myopathy (149). Tropomyosin mutations are found at numerous locations along the protein, and different mutations have different effects on the development and severity of disease. Interestingly, mutations in one part of the tropomyosin protein can have effects at a distance in other parts of the protein. For instance, a mutation at position 137 alters tropomyosin flexibility and this alteration is propagated along the protein (762). Discoveries like this add an additional layer of complexity in determining the causative nature of tropomyosin mutations.
TROPONIN COMPLEX
The troponin regulatory complex interacts with the actin-based thin filament and tropomyosin to modulate the actomyosin cross-bridge formation [reviewed in (304)] (Fig. 14). Troponin C (TnC) is the best studied of the troponin subunits, and is expressed in three isoforms: fast skeletal, slow skeletal and cardiac. The slow skeletal and cardiac isoforms are alternatively spliced from TNNC1 (644), and fast skeletal TnC is encoded by TNNC2 (190). TnC is largely composed of calcium-binding EF-hand motifs. Binding of calcium to the EF-motif leads to conformational changes allowing the N-terminal domain of TnC to interact with TnI (428) (Fig. 13).
Troponin I (TnI) is the inhibitory subunit of the troponin complex and is able to block actomyosin ATPase activity in vitro in the presence of other troponins and tropomyosin (363). When TnC interacts with TnI, it dissociates from actin, shifting tropomyosin to allow for weak binding of myosin to actin (Fig. 13). Three different isoforms of TnI are encoded by separate genes: fast skeletal, slow skeletal, and cardiac (59, 685, 725). Structurally, TnI consists of an IT-arm, inhibitory domain, regulatory domain, C-terminal mobile domain, and a cardiac isoform-specific N-terminal domain [reviewed in (304)]. The calcium state of TnI changes its affinity for binding partners based on the binding of calcium to TnC and its subsequent conformational shift (366).
The third player in the troponin regulatory complex is troponin T (TnT). The role of TnT is to anchor the troponin complex to tropomyosin (177, 320, 674) (Fig. 13). Like TnI, TnT has fast and slow skeletal and cardiac isoforms. TnT is composed of an IT-arm region, an inhibitory domain, a switch region, and a C-terminal mobile domain (21, 48, 643, 689). The function of TnT is somewhat controversial. It is thought to organize the regulatory complex as a whole by anchoring TnC and TnI to the thin filament, however TnT may also have roles in muscle contraction through regulation of actomyosin ATPase activity, calcium sensitivity, and force generation in the sarcomere (567,745).
Hundreds of mutations in all three members of the troponin family have been identified to date. Interestingly, troponin and tropomyosin mutations occur in multiple locations along the protein and can lead to DCM, HCM, or restricted cardiomyopathy with varying degrees of severity [reviewed in (401,451,676)].
MYOSIN: The motor driving muscle contraction
MYOSIN
Interacting with the thin filaments, myosin acts as the molecular motor driving muscle contraction and is the main component of the thick filament. Myosin’s make up a diverse group of motor proteins with structural and functional variations seen across 18 distinct classes—11 of which are found in humans (171). Each myosin is composed of one or two heavy chains and several light chain molecules. Myosin heavy chains (MHC) consist of three regions: N-terminal head and neck with a C-terminal tail region (Fig. 7). The conserved head region contains a motor domain that binds filamentous actin and drives actin-based movement (e.g., muscle contraction) via ATP hydrolysis. The neck region transduces force through its lever arm and the highly variable tail region mediates myosin interactions. Myosin light chains (MLC) regulate MHC motor function by binding to the neck region. Together, MHC and MLC coil together to form a unique macromolecular complex capable of driving movement in the cell.
In muscle, the conventional myosin II is the predominant class and is commonly termed “muscle myosin” because it is responsible for powering muscle contraction. Muscle myosin is a hetero-multimer composed of two MHCs and two pairs of non-identical MLCs [reviewed in (597,621)]. The N-terminal region of MHC interacts with two light chains to form the myosin head that binds actin filaments (Fig. 7). The C-terminal regions of the two MHCs interact in a coiled-coil double helix that forms the elongated tail [reviewed in (621)]. The tails of many myosin molecules pack together to form the bulk of the thick filament. Muscle contraction is generated by ATPase activity in the myosin head that is initiated upon binding to F-actin (580) (Fig. 14). The hydrolysis of ATP induces a conformational change in the myosin II molecule, referred to as the “powerstroke.” The effect of the myosin powerstroke is to pull the thin filaments toward the middle of the sarcomere resulting in contraction [reviewed in (663)]. ADP then dissociates and the myosin molecule returns to the relaxed state (281,282) (Fig. 14). To achieve efficient muscle contraction, the interaction between thin and thick filaments must go unhindered.
To accommodate the difference in energy requirements between muscle groups and during development, multiple striated muscle MHC and MLC isoforms exist [reviewed in (584, 728, 740)]. In cardiac muscle of small mammals (e.g., mice), MHC-β (MYH7) is the predominant isoform during development and is replaced by MHC-α (MYH6) in the adult (394). In large mammals such as humans, MHC-α is expressed during development while MHC-β is the predominant isoform in the adult heart [reviewed in (740)]. In failing hearts (mouse and human), MHC can shift to the fetal isoform as a maladaptive response to severe cardiovascular stress (340, 400). This fetal gene program is triggered by stress signaling pathways during cardiac remodeling and is believed to play a role in disease progression [reviewed in (144)].
Like in cardiac muscle, expression of different MHC isoforms differ during developmental and also in a fiber-type specific pattern in skeletal muscle. The MHC-perinatal (MYH8), MHC-embryonic (MYH3), and MHC-1 (MYH7, same as MHC-β seen in cardiac) proteins are present early in development. Once skeletal muscle fibers are restricted to a fiber type, MHC-1 is expressed in slow-twitch, while fast-twitch fibers express MHC-IId/x (MYH1), MHC-IIa (MYH2), and/or MHC-IIb (MYH4). In human fast-twitch muscle, MHC-IIb is present only in a very small subset of specialized muscle, but is observed to re-activate in limb muscle undergoing regeneration after profound degeneration (242). As seen in cardiac muscle, re-expression of fetal myosins occurs in regenerating muscle and during disease (e.g., hypothyroidism) [reviewed in (610)].
Myosin light chains are categorized as either “essential light chains” or “regulatory light chains.” The regulatory light chains are highly regulated by phosphorylation (Fig. 7). Myosin light chain kinase 2 (MYLK2) is expressed in both skeletal and cardiac muscle. Considered as the skeletal-myosin light chain kinase (skMLCK), it is expressed higher in fast twitch muscle fibers (137). Sk-MLCK phosphorylates the regulatory MLC playing a critical role in the potentiation of contraction in fast-twitch muscle fibers (773). Interestingly the skMLCK-deficient mice did not display cardiac abnormalities, although mutations in skMLCK are linked to HCM (23, 137, 563, 773). Myosin light chain kinase 3 (MYLK3) is cardiac-specific (618); cardiac-MLCK3 phosphorylates MLC2, which plays a role in sarcomere organization and cardiomyocyte contraction (99).
It comes as no surprise that mutations in myosin carry functional consequences and lead to myopathy. Mutations in myosin typically result in myosin protein aggregates that accumulate under the sarcolemma, muscle fiber degeneration, and/or impaired myosin motor function [reviewed in (669)]. The first myosin isoform associated with human disease was a mutation in MYH7 causing familial HCM (201). Since its identification, dominant mutations in MYH7 are associated with both hypertrophic and DCM, as well as a myosin storage myopathy called Laing distal myopathy (89,439,670). In skeletal muscle, the first identified skeletal myopathy was a mutation in MYH2 resulting in autosomal dominant myopathy (419). Mutations in skeletal MYH3 and MYH8 result in distal arthrogryposis syndromes (e.g., Freeman-Sheldon and Sheldon-Hall syndromes) (668,690).
Other myosins from different classes are expressed in muscle and are associated with disease. Myosin VI is expressed in both skeletal and cardiac muscle (32,300). As a unique, unconventional myosin motor that travels toward the pointed end (−) end of actin, myosin VI is found mainly at the sarcoplasmic reticulum (300, 742). A point mutation, at H236R within the myosin VI gene is associated with HCM (453). Myosin XIIIb, another unconventional myosin, is expressed in both skeletal and cardiac muscle where it local-izes to the Z-disc. Lack of myosin XIIIb results in embryonic lethality in mice; hearts from these embryos display enlargement of the right atrium and disruption of myofibrillar structures at E10.5, characteristics commonly seen in DCM (16).
Myosin-binding proteins: Linking the thin and thick filaments
MYOSIN BINDING PROTEINS
Another component of the thick filament is the myosin-binding protein family (MyBP). This family consists of myosin-binding protein C (MyBP-C, also known as C-protein) and myosin-binding protein H (MyBP-H, also known as H-protein) (Fig. 15A). Both MyBPs are located in the C zone (A-band region containing cross-bridges) and are restricted to transverse stripes spaced at 43nm intervals (55,130,140,207) (Fig. 15B).
MyBP-C is expressed in three different isoforms: a slow skeletal isoform (human gene MYBPC1), a fast skeletal isoform (human gene MYBPC2), and a cardiac isoform (human gene MYBPC3) (188,197,733). Slow skeletal MyBP-C, originally termed MyBP-X, encompasses four alternatively spliced variants that differ from one another via three novel insertions (2). While the cardiac isoform is tissue-specific, the slow and fast skeletal muscle isoforms are co-expressed in some skeletal muscle types (141,197,585). The slow skeletal isoform is also found in the right atrium and inter-atrial septum in adult mammalian hearts (2). There is only one isoform of MyBP-H (human gene MYBPH) expressed in the Purkinje fibers and fast twitch skeletal muscle fibers (20,35,651,713).
MyBPs are composed of a series of immunoglobulin and fibronectin type III repeat domains (Fig. 15A). MyBP-C isoforms share a core domain structure consisting of seven immunoglobulin and three fibronectin type III domains termed C1 through C10, a 105-residue linker between C1 and C2 called the MyBP-C motif (M-motif), and a proline- and alanine-rich (PA-rich) region near the N-terminus (155, 513) (Fig. 15A). Slow skeletal MyBP-C splice variants have three novel insertions encoded by the retention or exclusion of select exons within C7 and the N- and C-terminus (2). Cardiac MyBP-C (cMyBP-C) isoform differs the most with the addition of an eighth immunoglobulin domain at the N-terminus called C0 and a 28-amino acid insertion in C5 domain making it the largest MyBP (197,763) (Fig. 15A). cMyBP-C also contains a unique amino acid sequence—LAGGGRRIS—within its M-motif that is functionally important (197) (Fig. 15). MyBP-H is the smallest isoform and contains only four domains: two immunoglobulin and two fibronectin type III domains similar to C7 through C10 of MyBP-C with a unique N-terminal PA-rich region (35,713) (Fig. 15A).
MyBPs interact with the thick, thin, and titin filament systems (Figs. 5 and 7). The highly conserved C-terminal C10 domain of both MyBP-C and MyBP-H allows for interaction with myosin tails contributing to the maintenance and stability of the thick filament (298, 462, 469, 513). Additionally, the C-terminus binds titin and is necessary to localize MyBPs to the A-band (188, 205, 206, 350) (Fig. 5). The N-terminus of MyBP-C binds actin filaments, potentially regulating contraction by altering the actin-activated myosin ATPase activity (246,344,406,461,508,650), while MyBP-C’s N-terminal M-motif interacts with the S2 region of myosin and mediates contractile regulation (228,260,652). Interaction with the S2 region of myosin is altered when cMyBP-C is phosphorylated, affecting force generation in the heart (347,599,760).
Although the precise function is still being elucidated, MyBPs are involved in filament assembly and in regulation of contraction (228,298,469,513,627,741,756). The function of MyBP-C is highly regulated by phosphorylation particularly in the N-terminal PA-rich and M-motif regions (1,379). MyBP-C is thought to link the thick and thin filament systems and further regulate cross-bridge cycling by displacing tropomyosin and competing with myosin for actin binding [reviewed in (3,709)]. Additionally, interaction with titin allows MyBP-C to possibly work in concert with all of the filament systems during contraction to impact force development, transmission, sensing, and signaling [reviewed in (709)]. Although MyBP-H has not been shown to bind actin, evidence suggests it is involved in the regulation of contraction since it inhibits actin-activated skeletal muscle myosin ATPase in vitro (756).
As the link to the three filament systems, it is not surprising that mutations in MyBPs greatly affect muscle function and are often linked to striated muscle disease. In skeletal muscle, mutations in the slow-twitch MyBP-C isoform (sMyBP-C) are linked to cases of distal arthrogryposis type I (DA1), a disease characterized by congenital contractures of the distal limbs (231). A reduction in phosphorylation levels of sMyBP-C is associated with aging and diseases like DA1 and DMD (4). Knockdown of fast-twitch MyBP-C in zebrafish result in skeletal myopathy with elevated apoptosis, structural changes (e.g., narrower sarcomeres and shorter thin filaments), and muscle weakness (e.g., decrease in muscle size and force production) (375). Interestingly, MyBP-H is increased in the skeletal muscle from patients with amy-otrophic lateral sclerosis (ALS) patients (123). It is postulated that the uncharacteristically high levels of MyBP-H causes dysregulation in actin-myosin interaction and potentially can serve as a biomarker of ALS (123). Together, regulation of MyBPs expression and phosphorylation in skeletal muscle are critical for proper function.
In the heart, cMyBP-C is considered a key HCM disease gene (67, 730). Mutations in cMyBP-C result in ∼20–25% of cases of human familial HCM (491), with more than 350 identified mutations associated with HCM [reviewed in (90)]. Many mutations in cMyBP-C are missense and cause truncation of the protein (67, 730). Knockout of cMyBP-C in mice (lacking exons 3-10) are fertile and survive to adulthood but exhibit significant cardiac hypertrophy, supporting cMyBP-Cs causative role in HCM (241). While the mechanism for how cMyBP-C causes disease is not known, evidence points to haploinsufficiency of cMyBP-C (417). Increasing evidence exists demonstrating that cMyBP-C plays an important role in cardiac function from the sarcomeric level (e.g., filament assembly and stabilization) to the functional level (e.g., contraction and force production).
M-band: Cross-links Myosin Filaments and Acts as a Hub for Mechanosensing and Metabolism
The M-band is located at the center of the A-band. Electron microscopy reveals that the M-band is composed of a series of three to five electron-dense M-lines: M6, M4, M1, M4′, and M6′ (Fig. 1C). The number and composition of lines vary by fiber type (640). Myosin-based thick filaments are cross-linked in the M-band via “M-bridges” composed of myomesin. The M-band serves an important role in stabilizing the thick filaments during contraction, but it also contains components important for mechanosensing, proteosomal degradation, actin dynamics, metabolism, and signal transduction [reviewed in (277,694)] (Fig. 8).
Structural proteins: M-bridges stabilize myosin in the center of the sarcomere
MYOMESIN FAMILY OF PROTEINS
The myomesin protein family has three members: myomesin-1 (MYOM1), M-protein (myomesin-2, MYOM2), and myomesin-3 (MYOM3). M-protein is restricted to mature fast type II muscle fibers and the adult heart (227), while myomesin-1 is found in all striated muscle fiber types (505). Myomesin-3 is found only in skeletal muscle intermediate twitch fibers (613).
All three myomesin family members have similar domain structures. Each has a unique N-terminal domain followed by identical immunoglobulin- and fibronectin type III-domain repeats (613). To date, myomesin-1 is the only isoform known to form splice variants. Embryonic heart (EH)-myomesin is a splice variant with a 100 amino acid insertion between exons 6 and 7. It is the major myomesin isoform in the developing heart until birth (7). The 100 amino acid insert in EH-myomesin has a coiled conformation that functions like a molecular spring similar to the PEVK region in titin (611). In human DCM expression of EH-myomesin is upregulated, which results in increased M-band elasticity and further progression of DCM. It is unclear whether the upregulation in EH-myomesin is causal or if it is secondary to increased strain that eventually becomes maladaptive (612).
All myomesin family members form antiparallel homodimers through interactions called M-bridges between the C-terminal immunoglobulin domain (357). During contraction and relaxation, the repeating myomesin domains are able to stretch to 2.5 times their original length and act as an elastic band to stabilize the M-band (694). Myomesin, like titin, acts as a sarcomeric stretch sensor [reviewed in (8, 9)]. Importantly, N-terminal domain 1 of myomesin binds to the α-helical domain of myosin, and myomesin domains 4-6 binds to titin’s C-terminus (506). Linking the thick filaments to titin, myomesin-1 serves as the major thick filament cross-linking protein, much like α-actinin in the Z-disc (10) (Fig. 9).
To date, there are no reported knockout or transgenic mice for any of the myomesin family members. However, data from human cardiac and skeletal myopathies suggests that destabilization of the M-band leads to muscle dysfunction. A myomesin missense mutation V1490I was identified in a family with HCM; this mutation interferes with myomesin dimerization (635). In addition, an aberrant splice variant of myomesin has been identified in myotonic dystrophy type 1 (321). RNA-binding motif 20 (RBM20), an RNA-binding protein that regulates alternative splicing and is associated with DCM, has also been shown to cause alternative splicing in myomesin-1 (e.g., EH-myomesin), though the implication of the alternative splicing is unknown (408). Due to the vital importance of myomesin-1 in M-band stability, it is likely that more disease causing mutations or splice variants of myomesin-1 will be discovered in the future.
Proteasomal degradation: Regulation of protein turnover is vital to maintain sarcomeric integrity
MUSCLE-SPECIFIC RING FINGER PROTEIN (MURFs)
MURFs make up a family of proteins implicated in protein turnover and regulation of contractility (Fig. 8). MURFS are E3 ubiquitin ligases that share common structural features: an N-terminal zinc-finger RING domain, a MURF family conserved (MFC) domain, a B-box region, leucine-rich coiled-coil domains, and an acidic C-terminal tail (96, 649). MURF1 is found in all striated muscle (96). MURF2 is found in the heart and at low levels in skeletal muscle (8), while MURF3 is associated with microtubules (649). Via their coil-coiled domain, all of the MURFs can either homo- or heteroligamerize (96,426). MURF1 and MURF2 bind to numerous myofibrillar proteins including titin (Fig. 5), nebulin, N-RAP, troponin-T, cardiac troponin C, myotilin, and T-Cap (749). MURF1 has been shown to ubiquitinate troponin-I and myosin (164,308). While protein turnover is clearly necessary to maintain the integrity of the many sarcomeric components, MURF1 has been identified primarily for its role in muscle atrophy.
MURF1 and MURF2 play key roles in atrophy. Analysis of MURF1 knockout mice reveals that MURF1 confers resistance to skeletal muscle atrophy by targeting proteins for degradation (66, 119). Both MURF1 and MURF2 are necessary for maintenance of soleus type II fibers as double knockout of MURF1/MURF2 mice show atrophy in this muscle group (466). MURF3, while associated with microtubules, may also contribute to muscle atrophy. Simultaneous knockout of MURF1/MURF3 in mice leads to decreased muscle function and resembles myosin storage myopathy characterized by accumulation of myosin aggregates, muscle weakness, and HCM (164, 744). While the MURF family exhibits differential localization, they likely work in conjunction with each other to regulate skeletal muscle atrophy.
In the heart, MURF1 also regulates hypertrophic signaling by inhibiting protein kinase C-ε, a key kinase involved in activating hypertrophy (30) (Fig. 2). Further evidence of MURF1’s role in hypertrophy was demonstrated when MURF1 knockout mice underwent cardiac hypertrophy via transaortic constriction (TAC); the hypertrophic response was exaggerated indicating that MURF1 plays a role in blunting the hypertrophic response. In comparison, MURF2 knockout mice showed no increase in hypertrophy following TAC, indicating MURF2 has a distinct role from MURF1 (744). In another study, double knockout of MURF1/MURF2 in mice resulted in severe cardiac and mild skeletal hypertrophy, which was not seen in the single knockout of either MURF isoform. Therefore, MURF1 and MURF2 are functionally redundant as only one MURF1 or MURF2 functional allele is necessary for normal cardiac function (748). Further evidence that MURF1 and MURF2 regulate the hypertrophic response is that mutations in both proteins have been identified in human patients with HCM (658). The MURF family could be an important target for treatment of maladaptive hypertrophic response or atrophy.
Signal transduction: Maintaining a proper stress response in muscle
OBSCURIN
Obscurin is a giant sarcomeric protein that plays an integral role in myofibrillogenesis for A-band formation and myosin incorporation (332). Like titin, obscurin is composed of predominantly immunoglobulin and fibronectin domains, a calmodulin-binding site, and a Rho guanine nucleotide exchange factor domain (involved in G-protein coupled signaling) (764). Obscurin localizes to the Z-disc via its interaction with Z-disc titin (Figs. 2 and 5), while M-band localization of obscurin occurs through its interaction with myomesin and M-band titin (Fig. 8). Myomesin is necessary for obscurin localization as knockdown of myomesin by siRNA in neonatal mouse cardiomyocytes results in disrupted M-band obscurin (186). However, obscurin knockout mice do not show any disruption of the M-band, indicating it may not be essential in M-band stability (358). Obscurin also appears to play an important role in anchoring the sarcomere to the sarcoplasmic reticulum via interaction with ankyrin, as knockout of obscurin disrupts SR structure (358).
In cardiac muscle, obscurin localizes ankyrin B and protein phosphatase 2A (PP2A) to the M-band (Fig. 8). PP2A levels increase during heart failure, therefore PP2A sequestered at the M-band by obscurin and ankyrin-B may have potential therapeutic value (135). In humans, the obscurin missense mutation R4344Q, which disrupts its binding to titin, has been linked to HCM (28). Obscurin mutations were also found in a high percentage of human familial DCM samples and highlights the need to better understand obscurin’s function in the sarcomere (418).
MUSCLE-SPECIFIC CALPAIN-3/P94
Muscle-specific calpain-3 (CAPN3), also known as p94, is a calcium-dependent intracellular cysteine protease that exhibits autolytic activity and is expressed primarily in skeletal muscle (520, 647), while CAPN1 and CAPN2 are ubiquitously expressed (646). Structurally, CAPN3 is composed of three regions: a proline-rich N-terminal sequence domain followed by unique insert sequence (IS) 1 and 2 regions, with IS2 containing a nuclear localization sequence and a titin-binding site (646, 647). The IS2 region binds within the titin N2A and M-band regions (Fig. 5), and the presence of a nuclear localization sequence provides a signaling link between the sarcomere and nucleus (309, 521, 680). The interaction of CAPN3 with titin suppresses its autolytic activity (521).
CAPN3 is also involved in the sarcomere stretch response. As sarcomeres are overstretched CAPN3’s binding site at titin’s N2A region (near the Z-line) is exposed and CAPN3 translocates from the M-band to the N2A region. CAPN3 binding to the N2A region dissociates MARP2 from titin allowing MARP2 to translocate to the nucleus and increase transcription of stress-response genes (510, 512). Calpain is an important protein in propagating appropriate stress responses.
CAPN3 mutations are causative for limb girdle muscular dystrophy type 2A (LGMD2A) (247, 589). Numerous mutations have been identified with different effects on calpain activity [reviewed in (519)]. For example, in humans, homozygous mutations near the CAPN3-titin-binding site, which lead to decreased CAPN3 protein levels, result in LGMD-like pathology (235, 240). CAPN3 is a high value target to treat LGMD2A and possibly other muscular dystrophies.
Metabolism of ATP: Maintaining a sufficient pool of ATP for proper muscle contraction
MUSCLE-SPECIFIC CREATINE KINASE (M-CK)
The M-band is located near the A-band (where cross-bridge cycling occurs), and is a critical location for localized monitoring and production of ATP. The vast majority of M-CK is part of a soluble pool, but approximately 5% to 10% is bound to the sarcomere (698). M-CK forms a dimer and interacts with myomesin, M-protein and FHL2 (274, 356) (Fig. 8). The primary role of M-CK is to locally replenish ATP for cross-bridge cycling at both the M-band and the sarcoplas-mic reticulum calcium ATPase pump (SERCA) (729). M-CK levels decrease during heart failure (296). In a DCM mouse model (serum response factor knockout mice), insufficient levels of M-CK lead to depolymerization of actin filaments and, therefore, disruption of F-actin:G-actin ratios, suggesting thin filament destabilization is dependent on the contractile state (143). Surprisingly, M-CK knockout mice do not develop cardiomyopathy, likely because of compensation by mitochondrial or cytosolic creatine kinase (607).
OTHER METABOLIC ENZYMES AT THE M-BAND
In addition to M-CK, other critical enzymes in ATP production localize to the M-band (Fig. 8). Adenylate kinase is a phosphotransferase that generates ATP and ADP (146). In addition, the muscle isoform of adenosine monophosphate deaminase (AMPD) works with M-CK and adenylate kinase to monitor local ATP levels (238).
Enolase is a glycolytic enzyme that forms homo- or heterodimers with three subunits: α-, β- and γ-enolase. In cardiac and skeletal muscle, α-enolase localizes to the M-band while β-enolase is diffuse (310) (Fig. 8). M-CK has also been shown to bind β-enolase, thereby M-CK may serve as an anchor for glycolytic complexes on the sarcomere (172). In addition, phosphofructokinase (PFK) is another glycolytic enzyme at the M-band. PFK knockout mice have decreased ATP concentrations and develop HCM (194). Mutations in PFK are causative for type VII glycogen storage disease, which results in a loss of PFK activity in skeletal muscle and accumulation of glycogen (679). Further, the metabolic enzymes M-CK, adenylate kinase, and phosphofructokinase interact with FHL2, which binds to the N2B region of titin (Fig. 5), thus anchoring key metabolic enzymes to the M-band (Fig. 8).
Actin dynamics: Maintaining a soluble pool of G-actin to regulate actin dynamics at the pointed end
CYCLASE-ASSOCIATED PROTEINS (CAPS)
The main function of CAP proteins is to sequester G-actin. CAP1 expression is ubiquitous, while CAP2 is localized to striated muscle, brain, and skin. In mature myofibrils CAP2 localizes primarily to the M-band (549). Knockout of CAP2 in mice leads to the development of DCM and loss of a clear M-band as observed by electron microscopy (550), therefore CAP2 may also play a role in the stability of the M-band.
COFILIN
Cofilin contributes to actin dynamics by severing F-actin to make more G-actin available for polymerization. Both CAP2 and cofilin-2 are localized near the highly dynamic thin filament pointed ends where thin filament lengths are primarily regulated. There are three isoforms: cofilin-1 (nonmuscle), cofilin-2 (striated muscle), and ADF (actin-depolymerizing factor) [reviewed in (711). Cofilin-2 binds to ADP-bound F-actin and twists the filament to sever it [reviewed in (509)]. Cofilin-2 knockout mice display rod-shaped accumulations called nemaline bodies, which consist of thin filament and Z-disc proteins (12). Mutations in cofilin-2 have been identified in patients with nemaline myopathy, similar to the cofilin-2 knockout mouse, resulting in nemaline bodies and progressive muscle weakness (11, 507, 518). Knockdown of cofilin-2 in neonatal rat cardiomyocytes results in an elongation of the thin filaments, indicating a role in actin filament dynamics. Loss of the actin severing protein cofilin-2 leads to accumulation of F-actin resulting in longer thin filament lengths (339).
The Costamere: Protects against Mechanical Stress and Is an Important Signaling Hub
A focal adhesion is a large macromolecular complex at the interface between a cell and the extracellular matrix. In striated muscle, the costamere serves an analogous function to the focal adhesion, sharing a similar protein composition [reviewed in (504)]. The costamere bidirectionally and mechanically links the cytoskeleton to the extracellular matrix; it transmits forces from the sarcomere to the extracellular matrix (“inside-out”) and conversely transmits forces on the extracellular matrix to the myocytes (“outside-in”). The main components of the costamere are the vinculin-talin-integrin system and the dystrophin glycoprotein complex (Figs. 3 and 16). Intermediate filaments align the costamere with the M-band and Z-line [see section “Intermediate filaments: The scaffold that links the entire contractile apparatus to the sarcolemma and other organelles”]
Figure 16.
Schematic representation of costameric proteins, which bidirectionally link the extracellular matrix to the sarcomere. There are two major components of the costamere: the vinculin-talin-integrin complex and the dystrophin glycoprotein complex (DGC). The DGC includes dystrophin, sarcoglycans, α/β dystroglycans, dystrobrevin and syntrophin. Additional integrin-associated proteins include melusin, FAK (focal adhesion kinase), ILK (integrin-linked kinase, PINCH (particularly interesting new cysteine-histidine-rich protein), and kindlin. [Fig. reprinted, with permission, from (292).]
The vinculin-talin-integrin macromolecular complex
VINCULIN
Vinculin is expressed in all cell types. It is localized to both the costamere (cell-matrix junction) and the cardiac intercalated discs (cell-cell junction) [see section “Intercalated discs: Specialized cardiac structures that coordinate contraction”]. In the costamere, vinculin is on the cytosolic side of the sarcolemma where it links actin filaments to the sarcolemma; the head region of vinculin binds α-actinin and talin (83, 723) (Figs. 3 and 16). Vinculin has a N-terminal globular head, and proline-rich linker region connecting it to a rod-shaped tail (36,72,288,444).
Global vinculin KO is embryonic lethal in mice (755); vinculin heterozygous (HET) mice develop normally and are used to evaluate vinculin function in the adult heart (768). Vinculin HET mice progressively develop minor disruption of intercalated disc and costamere structure; however, there is no abnormal cardiac phenotype until hypertrophy is induced with TAC surgery (768). Cardiac-specific knockout (cKO) of vinculin leads to severely disrupted intercalated discs. Only 50% of mice survive past three months, although the survivors develop DCM by 4 months old (767). In depth ultra-structural analysis of vinculin cKO mice before the onset of cardiac dysfunction revealed that loss of vinculin leads to increased myofilament lattice spacing. This, in turn, leads to increased strain and the eventual development of DCM (673). These results also indicate that vinculin maintains tension at the extracellular matrix and loss of vinculin decreases costamere tension, leading to Z-disc expansion and increased lattice spacing.
Proper vinculin localization and regulation is necessary for normal heart function. Vinculin upregulation in cardiomyocytes was seen in multiple aging models, including rhesus monkeys, rats, and Drosophila, indicating it may be a key regulator of cardiac function. Overexpression of vinculin in Drosophila increased lifespan by 150%, suggesting potential therapeutic value in aging human hearts (306). Increased vinculin may be beneficial by reinforcing the cytoskeleton. In addition, vinculin upregulation is also observed following increased mechanical load in cardiomyocytes (e.g., mechanical stretch) and localization is disrupted upon unloading, implicating vinculin as a component of mechanotransduction (631).
TALIN
Talin binds to vinculin (binds α-actinin), and also binds integrins, thus indirectly connecting integrins to the sarcomere (Fig. 16). Talin is a dimeric protein that has a globular head containing a FERM domain (4.1/ezrin/radixin/moesin family) and a flexible rod domain (208, 456). Vertebrates have two talin genes that are 74% identical at the protein level—Tln1 and Tln2 (623); despite similar homology Tln1 and Tln2 have distinct roles. Tln1 is ubiquitously expressed, and in skeletal muscle localizes to costameres and myotendinous junctions. Tln2 is restricted to skeletal muscle, heart, and brain (180, 459). It should be noted that many studies do not distinguish between the two isoforms. Global knockout of Tln1 in mice results in embryonic lethality due to aborted gastrulation, indicating that Tln2 does not compensate for loss of Tln1 (460). On the other hand, global knockout of Tln2 results in mice that are viable and fertile (108). Skeletal muscle-specific Tln1 deletion revealed that Tln1 is not required for the assembly of the myotendinous junction and costamere, but it is crucial for the maintenance of the mechanical integrity of the myotendinous junction as loss of Tln1 lead to a progressive myopathy due to myotendinous junction defects. While Tln2 is also abundant in skeletal muscle, it does not compensate for the loss of Tln1 indicating it has a distinct role (124). Talin also plays a role in mechanosensing; increased load in skeletal muscle results in an increased localization of talin at the myotendinous junction (180).
In cardiac muscle, Tln1 and Tln2 have distinct roles and are developmentally regulated (413). During development, both Tln1 and Tln2 are expressed. In the adult heart, Tln2 is the main isoform and localizes to the costamere. Compared with Tln1, Tln2 has a higher affinity for the predominant integrin isoform in muscle, β1D, and results in the strongest connection between integrins and talin in actively contracting muscles (22). Upon induction of hypertrophy (via phenylephrine or pressure overload) Tln1 is unregulated in the costamere, possibly as a compensatory response. Tln1 is also unregulated in human failing heart biopsies (413). Cardiac specific knockout (cKO) Tln1 mice have no alterations in costamere structure and normal heart function. However, following the induction of hypertrophy, Tln1 cKO mice have a blunted hypertrophy response indicating that reduction of Tln1 levels and translocation to the costamere, despite its low basal expression, may have therapeutic potential (413).
INTEGRINS
Vinculin and talin are tethered to the costamere via their interaction with integrins and serve as adaptor proteins (Fig. 16). Integrins are transmembrane glycoproteins that do not have kinase activity, but are bidirectional signaling receptors that communicate signals from outside the cell in and vice versa. The extracellular domains of integrins are embedded in the extracellular matrix, while the intracellular domain binds talin and vinculin to indirectly connect integrins to α-actinin in the sarcomere (Figs. 3 and 16). Talin binds to the β-cytoplasmic tail of integrins to activate them (85), and binding of talin to integrin is the final step in activation (667). Integrins are αβ heterodimeric proteins made from 18 α- and 8 β-subunits that dimerize to form 24 different receptors, and there are splice variants of the α- and β-subunits [reviewed in (284)]. Different integrin receptor heterodimers vary temporally and during disease. The α5β1A subunit is prevalent in embryonic and neonatal cardiomyocytes, while the α7Bβ1D is the dominant isoform in postnatal and adult cardiomyocytes and strongly stabilizes the costamere (52,78,705). In disease states, the heart reverts to fetal α- and β-subunits (483, 660). In skeletal muscle, integrin isoforms vary developmentally with the α7Bβ1D isoform as the primary isoform in adult costameres; levels and isoforms are altered with exercise and in myopathies [reviewed in (145,660)].
Components of the ECM, such as collagen, laminin, or fibronectin, bind to integrins (Fig. 3) and activate signaling pathways, such as the IPP (ILK-PINCH-Parvin), resulting in expression of key genes that enable the muscle to respond to changes in stretch or load (Fig. 16). Integrin-linked kinase (ILK) is a critical component of the mechanical stretch sensor. PINCH (particularly interesting new cysteine-histidine-rich protein), parvin, and ILK form a complex and bind to the β1-subunit of integrins (107,273) (Fig. 16). The IPP complex then activates signaling pathways involved in hypertrophy and resistance to apoptosis [reviewed in (292)].
OTHER INTEGRIN ASSOCIATED PROTEINS
Melusin (also known as ITBP1BP2) is a chaperone protein that activates compensatory hypertrophy responses in striated muscle. Little is known about the role of melusin in skeletal muscle, but its role in the heart is well documented. Following binding to integrins, melusin activates cardioprotective pathways such as AKT, ERK1/2, and atrial natriuretic protein [reviewed in (678)] (Figs. 2 and 16). In human heart failure patients and animal models of hypertrophy melusin levels are downregulated (81, 138, 148), and overexpression of melusin in mice is cardioprotective (699). Due to melusin’s cardioprotective effects, it is being pursued as a possible therapeutic for heart failure.
Focal adhesion kinase (FAK) is another important mechanosensor in both cardiac and skeletal muscle (Figs. 2 and 16). FAK is a tyrosine kinase that binds to the β1 integrin tail and activates hypertrophy and anti-apoptotic pathways in response to changes in stretch and load, as well as during muscle development [extensively reviewed in (219, 604)]. Kindlin-2 is a protein that binds to the β-cytoplasmic tail and activates integrins (Fig. 16). Cardiac-specific knockout of kindlin-2 results in embryonic lethality, however postnatal cardiac-specific deletion led to fibrosis and heart failure indicating kindlin-2 is essential for integrin function (770).
The dystrophin glycoprotein complex
The dystrophin glycoprotein complex is the second major component of the costamere. It is a large, heteromultimeric protein complex that connects the intracellular cytoskeleton to the extracellular matrix in striated muscle (Figs. 3 and 17). Perturbation of the dystrophin glycoprotein complex and many of its components leads to muscle myopathies. More than 30 mouse models with various muscular dystrophy phenotypes have been generated by inactivating or mutating dystrophin glycoprotein complex components [reviewed in (743)].
Figure 17.
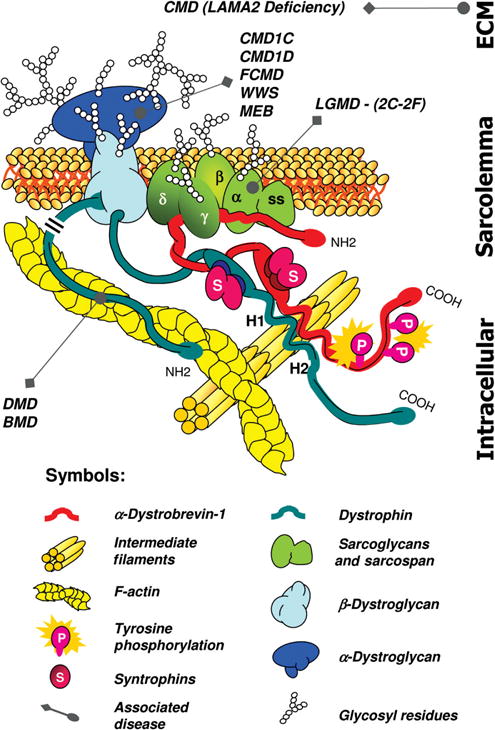
Schematic representation of the dystrophin associated protein complex in muscle. The three subcomplexes are shown: the dystroglycan subcomplex (blue), the dystrobrevin:syntrophin subcomplex (red) and the sarcoglycan:sarcospan subcomplex (green). Also indicated are the muscular dystrophies caused due to defects or deficiencies of proteins within the dystrophin associated protein complex. Abbreviations: BMD, Becker muscular dystrophy; CMD1C-1D, congenital muscular dystrophy type 1C-1D; DMD, Duchenne muscular dystrophy; FCMD, Fukuyama; CMD, LGMD2C-2F, limb-girdle muscular dystrophy type 2C-2F; LAMA2, laminin alpha 2 chain or merosin-deficient muscular dystrophy; MEB, muscle-eye-brain disease; WWS, Walker–Warburg syndrome. [Fig. reprinted, with permission, from (583).]
DYSTROPHIN
Dystrophin is the primary component of the dystrophin glycoprotein complex, which connects the sarcolemma to the extracellular matrix. Dystrophin is made up of two calponin homology (CH) domains followed by a large region of 24 spectrin-like repeats, a cysteine-rich region containing a WW domain, two EF-hand motifs, and a ZZ domain (73,323,324,564). Utrophin (ubiquitously expressed dystrophin) is highly homologous to dystrophin and is down-regulated in adult muscle where dystrophin is expressed, except in myotendinous and neuromuscular junctions (63,422). Through interactions with actin, microtubules, and intermediate filament proteins such as syntrophin, dystrophin forms a link to the extracellular matrix at the costamere [(323, 324, 781), reviewed in (292)] (Figs. 3 and 16). Clinically, dystrophin mutations lead to DMD and Becker muscular dystrophy, a less severe skeletal muscle myopathy (269, 322, 536). Complete deletion of the dystrophin protein perturbs the assembly of the dystrophin glycoprotein complex and leads to the development of DMD, whereas partial deletion of the protein causes Becker muscular dystrophy.
DMD has been studied extensively through one model system in particular: the mdx mouse [reviewed in (412)]. The mdx mouse arose from a spontaneous mutation in the C57BL/10 strain and has a phenotype comparable to human DMD (82). Mice exhibit pseudo-hypertrophy (muscle is enlarged because it is replaced with connective tissue and fat) in several skeletal muscle types (39,479,548). The fibrosis and force reduction in the thoracic diaphragm relates closely to the human disease, though less severe (49,200,218,278,653). In both humans and mice utrophin is upregulated in dystrophin-deficient muscle but, unlike in humans, this upregulation partially rescues the dystrophic phenotype in mdx mice (207). The cardiac phenotype of mdx mice also translates to human DMD; ike in humans, mouse cardiac muscle deterioration worsens with age leading to fibrosis, decreased efficiency and cardiac dysfunction (312,415,490).
SARCOGLYCANS
Other components of the dystrophin glycoprotein complex are the sarcoglycans – a family of single-pass transmembrane proteins [reviewed in (688)]. Sarcoglycans help facilitate interactions with the cytoskeleton and the extracellular matrix, and also stabilize sarcospan [reviewed in (535)] (Fig. 16). In skeletal muscle, the α, β, γ, and δ family members interact to form the sarcoglycan subcomplex of the dystrophin glycoprotein complex; ε-sarcoglycan is homologous to α-sarcoglycan and is expressed in striated muscle early in development, but expression is localized primarily to non-striated muscle tissues in adults (158, 392, 434, 657) (Fig. 17). The extracellular domains of each isoform contain one to three sites for N-glycosylation. N-glycosylation is important for cell-cell and cell-extracellular matrix interactions and is present in several cytoskeletal proteins, including laminins, integrins, and cadherins [(416, 720), reviewed in (118)]. The majority of disease-causing sarcoglycan mutations lie within the extracellular domains (70, 71, 378, 433, 435, 556, 592). These mutations lead to different forms of Limb-Girdle muscular dystrophy, with the severity dependent on the affected isoforms [reviewed in (688)].
SARCOSPAN
Sarcospan is another protein interacting with the dystrophin glycoprotein complex (Fig. 16). It is composed of four transmembrane domains with a large extracellular loop located between domains three and four (133). Able to form homooligomers, sarcospan interacts tightly with the sarcoglycan subcomplex and may keep other dystrophin glycoprotein complex proteins clustered in close proximity (134,445) (Fig. 17). Sarcospan-deficient mice do not develop any obvious signs of striated muscle myopathy (364). However, sarcospan overexpression yields differing phenotypes in a dose-dependent manner. At low levels, sarcospan overexpression alleviates some of the muscular dystrophy manifestations such as muscle degeneration and necrosis seen in mdx mice, but severe dystrophy is observed in sarcospan transgenic mice where overexpression is approximately tenfold (551, 552). These findings indicate that while the levels of sarcospan are important for it to function properly, sarcospan may be, to a degree, functionally redundant to dystroglycan, as it can rescue some of the symptoms of muscular dystrophy in the mdx mouse.
DYSTROPHIN-ASSOCIATED GLYCOPROTEIN
Dystrophin-associated glycoprotein, or dystroglycan, is a heterodimeric protein that associates with the dystrophin glycoprotein complex (Fig. 17). Both the α- and β-dystroglycan subunits are translated and cleaved from the same peptide (285). The C-terminus of the transmembrane β-dystroglycan subunit lies in the cytosol and binds dystrophin in striated muscle or utrophin in other tissue types (295, 423). The α-dystroglycan subunit interacts with the β-subunit on the extracellular surface and serves as a receptor for proteins of the extracellular matrix, such as laminin (156) (Fig. 3).
Several diseases termed “dystroglyconopathies” are believed to be caused by abnormalities of the dystrophin-associated glycoproteins. Fukuyama congenital muscular dystrophy is a disease characterized by progressive muscle weakness and atrophy, central nervous system, and visual effects that affects the protein fukutin, which is thought to play a role in glycosylation of α-dystroglycan. Reduced expression of dystrophin-associated glycoprotein complex proteins has been linked to muscle-eye-brain disease, characterized by diffuse muscle weakness, structural brain and eye abnormalities, and scoliosis with hyperextension of the head. Walker-Warburg Syndrome arises from mutations in protein O-mannosyltransferase and is characterized by severe congenital muscular dystrophy with severe abnormalities in the brain and eyes [reviewed in (165)].
DYSTROBREVIN
Both muscle and nonmuscle dystrobrevins are encoded by two genes—α- and β-DB (62,393,554, 601). α-Dystrobrevin-1 and -2 are known to participate in the skeletal muscle dystrophin glycoprotein complex (Fig. 3). Structurally, α-dystrobrevin contains four major domains: two EF-hand motifs, a ZZ domain, an α-helical coiled-coil dystrophin-binding domain, and a tyrosine kinase substrate domain (62,601). α -Dystrobrevin, like other members of the complex, links other cytoskeletal proteins, intermediate filament proteins, and possibly nitric oxide signaling through interactions with syntrophin to the dystrophin glycoprotein complex (56,64,450,489) (Figs. 3, 16, and 17).
SYNTROPHINS
Syntrophins are scaffold proteins that interact with other members of the dystrophin glycoprotein complex (Figs. 16 and 17). Structurally, syntrophins are composed of an N-terminal PH (Plekstrin Homology) domain, a PDZ domain, followed by a second PH domain, and a unique C-terminus (6,14). Syntrophin is expressed in five known isoforms: α1, β1, β2, γ1, and γ2. Of the five syntrophin isoforms, α1-syntrophin is the primary isoform expressed in skeletal and cardiac muscle (5, 13, 14, 560) (Fig. 3). α1-Syntrophin interacts with dystrophin, dystrobrevin, and neuronal nitric oxide synthase (nNOS), connecting the dystrophin glycoprotein complex to nitric oxide signaling (263,488,726).
Intercalated discs: Specialized cardiac structures that coordinate contraction
Intercalated discs are the main sites of interconnection between adjacent cardiomyocytes. Intercalated discs employ three traditional junctional contacts: adherens-, desmosomal-, and gap junctions [reviewed in (170)] (Fig. 18). During the last decade, however, the term “area composita” has been coined for larger adhering complexes; these are found in higher vertebrates and are comprised of traditional adherens- and desmosomal junctions (76, 176, 557, 558). Each junctional component in a structure as complex as intercalated discs is stringently orchestrated to achieve the coordinated cardiac contractions through properly conducted electrochemical and mechanical signals. Arrhythmogenic or degenerative cardiomyopathies are observed when electrical and mechanical couplings in intercalated discs are defective [reviewed in (496)].
Figure 18.
Structural organization and molecular components of the intercalated disc (ICD). Low-magnification transmission electron micrograph (A) and schematic drawing of cardiac myocardium (B) exhibit characteristic step-like structures of intercalated discs (A, arrowheads) formed through syncytial interconnection of rod-shaped cardiomyocytes. (C and D) Higher magnification view of areas enclosed in A and B, respectively, show three specialized substructures of intercalated discs—fascia adherens (adherens junction), desmosome (desmosomal junction, arrowhead), and gap junction. The ends of gap junction here connect to two adherens junctions (arrows). (D) Molecular components of ICD substructures not only serve as mechanical and electrochemical coupling platforms between adjacent cardiomyocytes, but also interact with major cytoskeletal filament systems (e.g., actin and microtubule cytoskeletons). The following proteins are not discussed in texts: p120, p150, EB1, and protein 4.3). [Parts A and C reprinted, with permission, from (626); B and D reprinted, with permission, from (38).]
Adherens junctions (fasciae adherentes)
Adherens junctions serve as the anchor sites for myofibrillar actin filaments, as well as the mechanical coupling platforms for adjacent cardiomyocytes (170). Adherens junctions bridge the opposing plasma membranes across ∼20 nm intercellular distances and transmit contractile force from one cardiomyocyte to another [reviewed in (436, 496)]. The classical adherens junctions contains two components: a transmembrane component composed of cadherin proteins (e.g., N-cadherin) which links neighboring cardiomyocytes together, and a cytoplasmic plaque component composed of catenin proteins (e.g., α- and β-catenins) through which adherens junctions connect to the actin cytoskeleton (436) (Fig. 18).
N-CADHERIN
The classical vertebrate cadherins are single-pass transmembrane proteins that mediate calcium-dependent cell-cell adhesion at adherens junctions. These proteins contain a prodomain, five extracellular cadherin (EC) repeats with calcium-binding motifs, a transmembrane domain, and a cytoplasmic domain that interacts with catenins [reviewed in (630)]. Even though there are about 20 classical cadherins, N-cadherin is the prominent cadherin expressed in myocardium (436, 630, 716). Activated by calcium, the EC repeats of N-cadherins from opposing membranes join together over a distance of up to 0.5 μ in a homophilic manner (436, 496). Cardiac-specific knockout of N-cadherin (CDH2) results in mice with loss of intercalated disc structures, impaired cardiac function with modest DCM, and rapid onset ventricular tachyarrhythmia followed by sudden cardiac death (333).
CATENIN PROTEINS
Catenin proteins fall under two categories: armadillo (arm) domain-containing catenins (e.g., β-catenin) and vinculin homology domain-containing catenins (e.g., α-catenin). The highly conserved β-catenin protein found in adherens junctions contain a central domain comprised of 12-13 helical arm repeats that interact with N-cadherin. The N-terminal region has an α-catenin-binding site, while the C-terminal region contributes to transcriptional activity in the canonical Wnt signaling pathway (630, 716). Global knockout of β-catenin is embryonic lethal due to its requirement for ectodermal cell layer formation during mammalian gastrulation (236). In cardiac-specific β-catenin KO mice, γ-catenin (plakoglobin protein primarily found in desmosomal junctions, see later) expression is upregulated to achieve normal cardiac development and function (774). Cardiac-specific loss of both β- and γ-catenin proteins results in gap junction remodeling, intercalated disc disassembly, and subsequent ischemic cardiomyopathy followed by sudden cardiac death (664). These results suggest functional redundancy within β- and γ-catenin proteins.
There are three mammalian α-catenin proteins: ubiquitously expressed αE-catenin (which is essential for early embryonic development), cardiac-specific αT-catenin, and neural tissue-specific αN-catenin. Cardiac-specific αE-catenin KO mice, as well as αE-catenin heterozygous-null mice, exhibit significantly higher susceptibility of cardiac rupture after ischemia [reviewed in (715)]. Homozygous αT-catenin-null mice are viable and fertile; however, they are highly susceptible to progressive DCM and ischemic cardiomyopathy. Furthermore, reduced expressions of plakophilin-2, a desmosomal protein with a unique αT-catenin-binding site, and connexin-43, the most abundant gap junction protein, are observed in intercalated discs of αT-catenin-null mice, revealing the cooperative roles of junctional proteins in maintaining the structure and function of intercalated discs (374).
Desmosomal junctions (Desmosomes; Maculae adherentes)
As with adherens junctions and actin cytoskeleton, interactions of desmosomal junctions with the intermediate filaments confer mechanical strength to cardiomyocytes. Desmosomal junctions, like adherens junctions, are composed of a transmembrane component (e.g., desmosomal cadherins) that provides connection across 20 to 35 nm intercellular space, and a cytoplasmic plaque component (e.g., armadillo proteins) that links to the intermediate filament system through intracellular connections [reviewed in (139,496)] (Fig. 18). Mutations in genes encoding desmosomal proteins are associated with inherited autosomal dominant diseases, such as arrhythmogenic right ventricular cardiomyopathy [reviewed in (86,248)].
DESMOSOMAL CADHERINS
Desmosomal cadherins (desmogleins and desmocollins) are single-pass transmembrane proteins that interact in a heterophillic manner to connect two half-desmosomes from adjacent cells. Structurally, desmogleins and desmocollins are composed of five highly conserved EC repeats that contain calcium-binding sites, a cell adhesion recognition site, intracellular and extracellular anchor regions, and an intracellular cadherin-like sequence (630). Desmogleins also contain a short leucine-rich region and a repeat unit domain at the C-terminal end (493). Desmocollins are alternatively spliced into longer ‘a’ isoforms and shorter ‘b’ isoforms that lack the intracellular cadherin-like sequence [reviewed in (139)].
Seven human desmosomal cadherins are classified into four desmogleins (DSG 1-4) and three desmocollins (DSC 1-3) [reviewed in (224)]. A variety of human heart and skin diseases are observed when desmosomal cadherins are disrupted (590). Mutations in human DSG2 and DSC2 genes result in either ARVC or Naxos disease characterized by ARVC, palmoplantar keratoderma, and woolly hair (33,60,261,559,590,665).
ARMADILLO PROTEINS
Plakoglobin (γ-catenin), and plakophilin are both members of the armadillo (arm) protein family, and serve as the linkage between the transmembrane and cytosolic components of desmosomal complex. Plakoglobin is composed of 12-13 central arm repeats flanked by largely unstructured N- and C-terminal tails (279, 727). Plakoglobin is primarily localized to desmosomes due to its higher affinity for desmosomal cadherins than for classical cadherins (113). Without plakoglobin, desmosomal structure is disrupted leading to death by cardiac rupture (596). Loss of the C-terminal tail of plakoglobin leads to Naxos disease (432,497).
Plakophilin is a sickle-shaped protein composed of nine super-helical armadillo repeats, each composed of three α-helices, with a flexible bend between the fifth and sixth repeats (114). There are three human plakophilin isoforms; plakophilin-2 (PKP2) is the only isoform detected in the myocardium (176,441). Among all desmosomal genes, mutations associated with defective PKP2 are the most common associated with ARVC patients (203,710).
DESMOPLAKIN
Desmoplakin is another integral component of the desmosomal complex, linking the cytosolic desmosomal plaque to intermediate filament proteins (e.g., desmin) (301, 302). Desmoplakin is made up of a central coiled-coil rod separating the globular N- and C-terminal head domains. Each head domain is made up of two pairs of spectrin repeats separated by a Srchomology domain (293). The N-terminal domain binds the desmosomal plaque (75, 625, 641, 693). The C-terminal tail contains three plakin repeat regions that may bind intermediate filaments (115).
Gap junctions (Maculae communicantes; Nexuses)
While adherens- and desmosomal junctions enable mechanical coupling between adjacent cardiomyocytes, gap junctions allow conductance of electrochemical signals via passive diffusion. Gap junctions are low-resistance intercellular channels that allow the direct passage of ions and small molecules (up to 1 kDa) between adjacent cells in the myocardium, enabling incredibly fast propagation of electrical impulses through the cytoplasm. Irregular gap junction organization and subsequent improperly regulated electrochemical signals often lead to cardiac arrhythmias [reviewed in (169,217,496)].
CONNEXINS
Twenty-one members of the connexin family of proteins provide the structural basis for gap junctions. Six connexin subunits, of either homotypic or heterotypic nature, make one connexon hemi-channel, and two connexons from apposing membranes adjoin to form a channel within the gap junction structure of up to 3.5 nm in diameter (217, 496) (Fig. 18). Three connexin (Cx) proteins are expressed in mammalian hearts: Cx40, Cx43, and Cx45.
Connexin 40 and Cx45 exhibit overlapping expression patterns, especially in the atrioventricular node and His bundle branches, and appear to contribute in cardiac morphogenesis and conduction of electrical signals (343, 626). Cx43 is the best studied and the most abundantly expressed cardiac connexin protein, especially in adult ventricular myocardium. Therefore, it is not surprising that abnormal Cx43 expression (often followed by intercalated disc remodeling) is detected in various types of progressive cardiomyopathy [reviewed in (169)]. However, altered connexin expressions, not limited to that of Cx43 alone, at both transcript and protein levels are detected in congestive heart failure patients – upregulated Cx43 in HCM, downregulated Cx43 in DCM, and upregulated Cx40 and downregulated Cx43 and Cx45 in ischemic cardiomyopathy (150, 169). Cx43 has many well-established interacting partners, such as cadherins, catenins, and Srckinases. Cx43 is also shown to associate with the actin and microtubule cytoskeletons via its respective direct interactions with zona occludin and tubulin proteins [reviewed in (204)] (Fig. 18).
Myotendinous Junction: A Specialized Skeletal Muscle Complex That Is Important for Transmission of Force
In addition to the costamere, skeletal muscle also has the myotendinous junction to transmit mechanical force. Both the costamere and myotendinous junction share similar protein components. The myotendinous junction lies at the interface between skeletal muscle and the tendon; it is important for force transmission and subsequent locomotion. Ultrastructurally, thin filaments from the terminal Z-lines are bundled through protein interactions and interface with the muscle membrane via the sarcolemma [reviewed in (102)]. The sarcolemma interacts with collagen IV and laminins that are enriched in the basement membrane (31) (Fig. 3). At the myotendinous junction, finger-like interdigitations and a highly invaginated membrane greatly increase the contact area and therefore the structural integrity of the junction under stress and tension (17,481,482,692).
There are two major structural linkages that connect the sarcomere to the myotendinous junction. The α7β1 integrin complex is enriched at the myotendinous junction (45, 449). Loss of the α7 protein leads to a congenital myopathy in mice and humans affecting the myotendinous junction (251, 424). The second involves the dystrophin glycoprotein complex. Laminin 211 (also known as merosin) is a common component between the two linkage systems; it is the primary isoform in the adult skeletal muscle basement membrane (154, 606). Mutations in the LAMA2 gene, which encodes laminin 211, cause merosin-deficient muscular dystrophy (237,253,753).
Intermediate Filaments: The Scaffold That Links the Entire Contractile Apparatus to the Sarcolemma and Other Organelles
Intermediate filaments maintain the integrity of muscle cytoarchitecture and provide mechanical strength to the cell (Fig. 3). These filaments are composed of proteins that are grouped into six types (types I-VI) based on similarities between their amino acid sequences. Proteins from each of the six types are expressed in muscle with desmin being the best-studied major muscle-specific intermediate filament protein. Each intermediate filament protein consists of an N-terminal head and a C-terminal tail, with a central rod domain that coils together to form a dimer between similarly grouped proteins. These dimers associate in either parallel or antiparallel homo-or hetero-oligomers that are highly regulated both spatially and temporally. The detailed molecular interactions between intermediate filaments and other cytoskeletal proteins are still being elucidated, as are their emerging roles in striated muscle diseases.
DESMIN
Desmin is a type III intermediate filament protein found more abundantly in heart muscle (2% of total protein) than in skeletal muscle (0.35% of total protein) (568). Desmin is expressed early in myofibrillogenesis where it aids in establishing the sarcomere (187, 259, 305, 608). Desmin is found at the periphery of Z-discs where it forms a network made from either homopolymers (e.g., desmin only) or heteropolymers (e.g., desmin with vimentin, nestin, synemin, or paranemin) (54,220,362,524,616,761). The desmin network links the Z-discs of adjacent myofibrils to each other and to the sarcolemma, as well as to the mitochondria and nucleus (53,54,440,447,586) (Figs. 2 and 3). Desmin is also found at the M-band where it laterally aligns myofibrils (569). Overall, desmin and its network provide structural stability essential for proper cellular function.
Desmin interacts with many cytoskeletal proteins [see section “Other intermediate filament proteins/desmin-binding partners”]. As previously mentioned, heteropolymers between desmin and other intermediate filament proteins form the network that provides structural stability throughout the cell (54, 220, 362, 524, 616, 761) The desmin network indirectly links to the membrane adhesion complexes via syncolin and synemin (254, 425, 450, 616). Desmin also directly links to the membrane adhesion complexes via interactions with spectrin and ankyrin (359) (Fig. 3). In addition to its Z-disc localization, interaction with skelemin (a splice variant of myomesin) localizes desmin to the M-band, while interactions with plectin link desmin to the costamere (329,569) (Fig. 3). Desmin’s ability to interact with all of these proteins is ablated during differentiation and apoptosis by the proteolytic activity of caspase-6, calpain, and cathepsin B (47,106,487).
While many of desmin’s interactions contribute to its structural role in the cell, desmin also plays a role in cellular signaling [reviewed in (267)]. The dystrophin-associated protein complex involvement in mechanotransduction suggests desmin’s interaction with syncoilin and α-dystrobrevin allows for localized mechanical stress signaling through the sarcolemma (346, 450, 565) (Fig. 3). Myospryn (also known as CMYA5) interacts with desmin (Fig. 3), influencing vesicle trafficking, organelle biogenesis and/or positioning, and phosphorylation in response to changes in muscle activity (313, 337). Through calcium-binding proteins, S100A1 and S100B, desmin can influence calcium homeostasis (193, 571, 717). Lastly, desmin may play a role in regulating gene expression through MyoD and MLH1 (MutL Homolog 1) (79,372). Therefore, in addition to desmin’s network forming the scaffold that stabilizes the cell, desmin also regulates cell homeostasis and survival.
Interestingly, with desmin as one of the earliest muscle-specific proteins detected during myofibrillogenesis, desmin knockout mice are fertile and undergo muscle differentiation, cell fusion, and muscle maturation normally (376, 448). However, after birth, mice devoid of desmin develop myofibril misalignment, costamere disruption, loss of nuclear shape and positioning, abnormal mitochondria, and impairment of force generation (37, 376, 383, 448, 503, 629, 682, 739). Therefore, although desmin is important for proper sarcomeric structure and function, it does not appear to be fundamentally necessary during muscle development in vivo. The lack of detectable abnormalities early on could be attributed to additional proteins that also aid in establishing the sarcomere (e.g., titin), as well as the contribution of keratins, which stabilize the costamere even in the absence of desmin (187, 259, 305, 503, 608). While desmin’s function in maintaining the structural integrity of the cytoskeleton has been established, study of desmin’s role in both skeletal and heart disease, known as desminopathies, continues to grow.
Desminopathies are the result of disorganization of desmin filament networks, largely seen as insoluble desmin-containing aggregates that accumulate in the subsarcolemmal space [reviewed in (120)]. Typically, mutations in desmin that interfere with binding to its partners, particularly the desmin chaperone protein αB-crystalline (Fig. 3) or intermediate filaments synemin and syncoilin, result in desmin-containing aggregates (213, 276, 515, 614). Onset of desminopathies range from childhood to late adulthood and present with mild-to-severe skeletal and cardiac myopathy [reviewed in (34)]. While many studies have provided insight into the complex pathophysiology of this disease, treatment options remain limited and further research is still needed.
Other intermediate filament proteins/desmin-binding partners
VIMENTIN and NESTIN
Desmin interacts and co-localizes at the Z-disc with an assortment of other intermediate filament proteins. During development, desmin’s interaction with vimentin (a type III intermediate filament protein) and nestin (type IV) are important for establishing the cytoskeletal network that stabilizes the cell (220, 369, 619, 638). In mature myocytes, vimentin and nestin are expressed at very low levels, though their expressions are elevated in regenerating skeletal muscle fibers in response to both injury and disease (e.g., dystrophic and inflammatory myopathies) (58,74,192,637,703).
PARANEMIN, SYNCOILIN, AND SYNEMIN (DESMUSLIN)
Later in development, the intermediate filament proteins paranemin (type IV), syncoilin (type III), and synemin (type IV) are expressed, and function to stabilizing the desmin intermediate filament network of mature myocytes, especially during stress (221). Paranemin functions in the organization of desmin intermediate filament networks, whereas syncoilin and synemin link desmin and its network to the dystrophin protein complex via interaction with α-dystrobrevin (254, 425, 450, 616) (Fig. 3). Synemin also plays a role in regulating hypertrophic signaling via interaction with protein kinase A (377). While the interaction of desmin with other intermediate filament proteins are necessary to maintain proper function within the cell, further research needs to be done to better elucidate the molecular role of these interactions in disease.
KERATINS
Keratins, originally termed cytokeratins, were renamed in 2006 to incorporate novel genes uncovered in the sequencing of the human genome (617). Keratins are composed of two of the six types of intermediate filament proteins: type I (acidic) and type II (basic/neutral). In striated muscle, keratins 8 (type II), 18 (type I), and 19 (type I) are the best characterized. Filaments composed of keratin 8 and 19 contribute to linking the contractile apparatus at both the peripheral Z-discs and M-bands to the sarcolemma (503) (Fig. 3). This link involves interactions between the costameric dystrophin-dystroglycan complex (via binding of dystrophin) to keratin 19 (654, 700). Knockout of keratin 19 in mice results in disruption of costameres and an increase in spacing between the sarcolemma and the contractile apparatus. This causes an accumulation of mitochondria, a decrease in contractile force, and a mild skeletal myopathy (399, 628, 655). Therefore, keratin 19 is considered important in costameric organization and regulating the distribution of mitochondria in myocytes. Loss of keratin 19 results in symptoms similar to those observed in facioscapulohumeral muscular dystrophy and, via its binding to dystrophin, may play a role in muscular dystrophy (399, 628, 655). In addition to keratin 8/19 filaments, keratin 8/18 filaments are present in striated muscle. Upregulation of tumor necrosis factor-alpha (TNFα), a proinflammatory cytokine, leads to ectopic expression of keratin 8/18 filaments at the intercalated disc in hearts after tissue injury; this expression results in a cardioprotective effect by maintaining the intercalated disc structure and mitochondrial integrity in cardiomyocytes (541). Taken together, keratin filaments play an important role in maintaining myocyte integrity, mechanotransduction, and in diseases (such as heart failure and muscular dystrophy).
NUCLEAR LAMINS
Lamin is a type V intermediate filament protein that makes up the inner nuclear membrane that create a meshwork essential for nuclear architecture. Additionally, lamins provide attachment sites for intranuclear proteins (e.g., emerin, nesprin-1, nesprin-2), as well as influence chromatin organization and nuclear metabolism (61,239,769) (Figs. 3 and 19). Lamins are ubiquitously expressed and grouped into two types: A-type lamins consists of lamin A and C, which are developmentally regulated splice variants of the LMNA gene, and B-type lamins consist of B1 and B2 expressed from the LMNB1 and LMNB2 genes, respectively (166, 429, 553, 591, 721). With the growing number of muscle diseases associated with the nuclear lamina, known as “laminopathies,” a plethora of research is focused on the role of lamins and lamin-binding proteins in disease.
Figure 19.
Nuclear lamins. (A) Schematic drawing of nuclear lamins and their nearby protein interactions. Nuclear lamins localizes underneath the inner nuclear membrane where they directly bind lamina-associated proteins (e.g., emerin, nesprin-1, and nesprin-2). Nesprin-1 and emerin both interact with nuclear actin and mediate the cortical actin cytoskeleton assembly at the nuclear envelope. The integral membrane protein MAN1 allows lamins to associate with transcription factors (e.g., SMAD), while SUN1/2 allows lamins to associate with microtubules and anchors nesprin-2 to the nuclear envelope. Interactions with barrier-to-autointegration factor (BAF) and lamin B receptor (LBR), as well as directly to chromatin, allows lamins to influence chromatin organization and gene expression. (B) Electron micrograph of the nuclear lamina composed of lamin intermediate filaments and associated proteins that extend between the nuclear pore complexes (NPCs). [Parts A and B modified, with permission, from (80).]
The first identified laminopathy was a mutation in emerin, a lamin A-interacting protein, causing X-linked Emery-Dreifuss muscular dystrophy (EDMD) (61, 712). Since then, the majority of laminopathies identified are associated with mutations in A-type lamins (e.g., the LMNA gene), including Emery-Dreifuss muscular dystrophy, DCM type 1A, limb-girdle muscular dystrophy type 1B, and congenital muscular dystrophy (68, 159, 471, 577). A variety of mouse models has been created to examine the role of A-type lamins. For example, A-type lamin knockout mice, which express a C-terminally truncated lamin A product, exhibit reduced growth rate and present with skeletal muscle dystrophy and DCM before dying around eight weeks after birth (291, 492, 659). The disease progression in this model is consistent with skeletal muscle wasting and cardiomyopathy seen in human Emery-Dreifuss muscular dystrophy patients [reviewed in (69)]. In addition to emerin and lamins, nesprin-1 and -2 are also associated with Emery-Dreifuss muscular dystrophy, as well as other muscle diseases, but little is known about their function [reviewed in (92)].
Overall, it is clear that lamins (especially A-type lamins) and lamin-binding proteins are important for proper nuclear function. Although the pathophysiology remains unclear, two leading hypotheses have emerged: the mechanical stress hypothesis and the gene expression hypothesis [reviewed in (103)]. In the mechanical stress hypothesis, evidence suggests that disrupting the interaction of lamin with desmin leads to a loss of cytoskeletal tension and therefore, defective force transmission throughout the myocytes (492). Alternatively, in the gene expression hypothesis, lamin and its binding partners are required for nuclear assembly and regulate gene expression via interactions with DNA, chromatin, histones, and transcription factors [reviewed in (573)]. While the molecular mechanisms contributing to disease progression in laminopathies remains unclear, the data supporting these two hypotheses provide insight and may not be mutually exclusive in how lamins contribute to the development of disease.
Conclusion
The intricately designed striated muscle is composed of diverse cytoskeletal assemblies. Coordination between the sarcomere, the basic contractile unit, and the complex cytoskeletal network is essential for muscle contraction. In this review, we have discussed the cytoskeletal assemblies composed of a plethora of structural and regulatory molecules that work together to produce contractile activity. With tremendous advances in technology, especially with respect to mass spectroscopy and proteomics, it is inevitable that numerous other important muscle proteins will be identified. Many sarcomeric cytoskeletal proteins have functional mutations that lead to disease, which emphasizes their importance in normal muscle function. Therefore, a basic understanding of their function provides insight into the mechanisms of disease. Identifying disease-causing mutations in cytoskeletal genes is imperative, and focus is now being placed on the power of genetics. Genome-wide screens and genetic testing of patients at risk of developing disease allow earlier intervention and slowing of disease progression as well as personalized regimens for treatment [for discussion see: (675)].
New therapeutics are being developed in an attempt to attenuate the expression of mutant alleles to stop or slow down the progression of myopathies. For example, an exciting approach for treating myopathies is genome editing. The CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) system is a method to potentially remove disease-causing mutations, termed “myoediting” [reviewed in (724)]. Preliminary testing in the DMD mdx mouse model shows that myoediting increased dystrophin expression and resulted in increased muscle strength (395,486,666,754).
It is becoming clear that an oversimplified view of myopathies is that a single gene mutation leads to disease. Additionally, effect(s) of mutations may not occur at the site of mutation. For instance, the helical nature of tropomyosin results in structural effects of mutations seen “at a distance” [reviewed in (676)]. More research is also needed into possible environmental factor or epigenetic factors that affect the penetrance of mutations. One such epigenetic factor that has shown promise is microRNAs (miRNA)—small noncoding RNAs that modulate gene expression. miRNAs have been identified to play a key role in heart failure and may be a potentially useful prognostic marker in cardiovascular disease, as well as in skeletal myopathies [reviewed in (151,314)].
Research continues to demonstrate that striated muscle is a uniquely balanced machine dependent on the interconnection of multiple cytoskeletal assemblies for proper function. With the emergence of new technologies, discovery of new integral components and dissection of the causes of myopathies at the molecular level—it is exciting to envision what the next discoveries in the cytoskeletal muscle field will be. Further investigation of the basic properties of striated muscle proteins is still essential to provide a better understanding of the physiological functional properties of individual muscle proteins and how they contribute to unique cytoskeletal assemblies in health and disease.
Didactic Synopsis.
The major cytoskeletal assemblies discussed are:
-
–
Sarcomere—basic contractile unit of striated muscle
-
–
Costamere—connects sarcomere to cell membrane and functions to protect against mechanical stress
-
–
Interacalated disc—specialized junction between cardiomyocytes that functions to coordinate contraction
-
–
Myotendinous junction—interface between skeletal muscle and tendon. Has role in force transmission
-
–
Intermediate filaments—scaffold that links the contractile apparatus to the sarcolemma and other organelles
Each section introduces a cytoskeletal assembly, discusses the protein components and interactions, signaling pathways, functions, and connections to disease.
Acknowledgments
This work was funded by National Institutes of Health R01HL108625 and R01HL123078 (C. C. Gregorio), the University of Arizona Sarver Heart Center Finley and Florence Brown Endowed Research Award (C. A. Henderson), the University of Arizona Undergraduate Biology Research Program (C. G. Gregorio), National Institutes of Health Predoctoral Fellowship 1F31HL117520 (S. M. Novak), and an American Heart Association Western State Affiliates Postdoctoral Fellowship 16POST31350016 (L. Mi-Mi).
References
- 1.Ackermann MA, Kontrogianni-Konstantopoulos A. Myosin binding protein-C slow is a novel substrate for protein kinase A (PKA) and C (PKC) in skeletal muscle. J Proteome Res. 2011;10:4547–4555. doi: 10.1021/pr200355w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackermann MA, Kontrogianni-Konstantopoulos A. Myosin binding protein-C slow: An intricate subfamily of proteins. J Biomed Biotechnol. 2010;2010:1–10. doi: 10.1155/2010/652065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ackermann MA, Kontrogianni-Konstantopoulos A. Myosin binding protein-C: A regulator of actomyosin interaction in striated muscle. Biomed Res Int. 2011;2011:1–9. doi: 10.1155/2011/636403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ackermann MA, Ward CW, Gurnett C, Kontrogianni-Konstantopoulos A. Myosin binding protein-c slow phosphorylation is altered in Duchenne dystrophy and arthrogryposis myopathy in fast-twitch skeletal muscles. Sci Rep. 2015;19:13235–13250. doi: 10.1038/srep13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. 1993;11:531–540. doi: 10.1016/0896-6273(93)90157-M. [DOI] [PubMed] [Google Scholar]
- 6.Adams ME, Dwyer TM, Dowler LL, White RA, Froehner SC. Mouse α1- and β2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J Biol Chem. 1995;270:25859–25865. doi: 10.1074/jbc.270.43.25859. [DOI] [PubMed] [Google Scholar]
- 7.Agarkova I, Auerbach D, Ehler E, Perriard JC. A novel marker for vertebrate embryonic heart, the EH-myomesin isoform. J Biol Chem. 2000;275:10256–10264. doi: 10.1074/jbc.275.14.10256. [DOI] [PubMed] [Google Scholar]
- 8.Agarkova I, Ehler E, Lange S, Schoenauer R, Perriard JC. M-band: A safeguard for sarcomere stability? J Muscle Res Cell Motil. 2003;24:191–203. doi: 10.1023/A:1026094924677. [DOI] [PubMed] [Google Scholar]
- 9.Agarkova I, Perriard JC. The M-band: An elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol. 2005;15:477–485. doi: 10.1016/j.tcb.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Agarkova I, Schoenauer R, Ehler E, Carlsson L, Carlsson E, Thornell LE, Perriard JC. The molecular composition of the sarcomeric M-band correlates with muscle fiber type. Eur J Cell Biol. 2004;83:193–204. doi: 10.1078/0171-9335-00383. [DOI] [PubMed] [Google Scholar]
- 11.Agrawal PB, Greenleaf RS, Tomczak KK, Lehtokari VL, Wallgren-Pettersson C, Wallefeld W, Laing NG, Darras BT, Maciver SK, Dormitzer PR, Beggs AH. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet. 2007;80:162–167. doi: 10.1086/510402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agrawal PB, Joshi M, Savic T, Chen Z, Beggs AH. Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum Mol Genet. 2012;21:2341–2356. doi: 10.1093/hmg/dds053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahn AH, Freener CA, Gussoni E, Yoshida M, Ozawa E, Kunkel LM. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J Biol Chem. 1996;271:2724–2730. doi: 10.1074/jbc.271.5.2724. [DOI] [PubMed] [Google Scholar]
- 14.Ahn AH, Yoshida M, Anderson MS, Feener CA, Selig S, Hagiwara Y, Ozawa E, Kunkel LM. Cloning of human basic A1, a distinct 59-kDa dystrophin-associated protein encoded on chromosome 8q23-24. Proc Natl Acad Sci U S A. 1994;91:4446–4450. doi: 10.1073/pnas.91.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aihara Y, Kurabayashi M, Saito Y, Ohyama Y, Tanaka T, Takeda S, Tomaru K, Sekiguchi K, Arai M, Nakamura T, Nagai R. Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: Role of M-CAT element within the promoter. Hypertension. 2000;36:48–53. doi: 10.1161/01.HYP.36.1.48. [DOI] [PubMed] [Google Scholar]
- 16.Ajima R, Akazawa H, Kodama M, Takeshita F, Otsuka A, Kohno T, Komuro I, Ochiya T, Yokota J. Deficiency of Myo18B in mice results in embryonic lethality with cardiac myofibrillar aberrations. Genes Cells. 2008;13:987–999. doi: 10.1111/j1365-2443.2008.01226.x. [DOI] [PubMed] [Google Scholar]
- 17.Ajiri T, Kimura T, Ito R, Inokuchi S. Microfibrils in the myotendon junctions. Acta Anat (Basel) 1978;102:433–439. doi: 10.1159/000145668. [DOI] [PubMed] [Google Scholar]
- 18.Al-Sajee D, Nissar AA, Coleman SK, Rebalka IA, Chiang A, Wathra R, van der Ven PF, Orfanos Z, Hawke TJ. Xin-deficient mice display myopathy, impaired contractility, attenuated muscle repair and altered satellite cell functionality. Acta Physiol (Oxf) 2015;214:248–260. doi: 10.1111/apha.12455. [DOI] [PubMed] [Google Scholar]
- 19.Almenar-Queralt A, Lee A, Conley CA, Ribas de Pouplana L, Fowler VM. Identification of a novel tropomodulin isoform, skeletal tropo-modulin, that caps actin filament pointed ends in fast skeletal muscle. J Biol Chem. 1999;274:28466–28475. doi: 10.1074/jbc.274.40.28466. [DOI] [PubMed] [Google Scholar]
- 20.Alyonycheva T, Cohen-Gould L, Siewert C, Fischman DA, Mikawa T. Skeletal muscle-specific myosin binding protein-H is expressed in Purkinje fibers of the cardiac conduction system. Circ Res. 1997;80:665–672. doi: 10.1161/01.res.80.5.665. [DOI] [PubMed] [Google Scholar]
- 21.Anderson PA, Malouf NN, Oakeley AE, Pagani ED, Allen PD. Troponin T isoform expression in humans. A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ Res. 1991;69:1226–1233. doi: 10.1161/01.RES.69.5.1226. [DOI] [PubMed] [Google Scholar]
- 22.Anthis NJ, Wegener KL, Ye F, Kim C, Goult BT, Lowe ED, Vakonakis I, Bate N, Critchley DR, Ginsberg MH, Campbell ID. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. Embo j. 2009;28:3623–3632. doi: 10.1038/emboj.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arad M, Penas-Lado M, Monserrat L, Maron BJ, Sherrid M, Ho CY, Barr S, Karim A, Olson TM, Kamisago M, Seidman JG, Seidman CE. Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005;112:2805–2811. doi: 10.1161/circulationaha.105.547448. [DOI] [PubMed] [Google Scholar]
- 24.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 25.Arber S, Halder G, Caroni P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell. 1994;79:221–231. doi: 10.1016/0092-8674(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 26.Arber S, Hunter JJ, Ross J, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/S0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 27.Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A. Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:334–342. doi: 10.1016/j.jacc.2008.12.082. [DOI] [PubMed] [Google Scholar]
- 28.Arimura T, Matsumoto Y, Okazaki O, Hayashi T, Takahashi M, Inagaki N, Hinohara K, Ashizawa N, Yano K, Kimura A. Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2007;362:281–287. doi: 10.1016/j.bbrc.2007.07.183. [DOI] [PubMed] [Google Scholar]
- 29.Arimura T, Takeya R, Ishikawa T, Yamano T, Matsuo A, Tatsumi T, Nomura T, Sumimoto H, Kimura A. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ J. 2013;77:2990–2996. doi: 10.1253/circj.CJ-13-0255. [DOI] [PubMed] [Google Scholar]
- 30.Arya R, Kedar V, Hwang JR, McDonough H, Li HH, Taylor J, Patterson C. Muscle ring finger protein-1 inhibits PKCepsilon activation and prevents cardiomyocyte hypertrophy. J Cell Biol. 2004;167:1147–1159. doi: 10.1083/jcb.2004.02033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aumailley M, Smyth N. The role of laminins in basement membrane function. J Anat. 1998;193:1–21. doi: 10.1046/j.1469-7580.1998.19310001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avraham KB, Hasson T, Sobe T, Balsara B, Testa JR, Skvorak AB, Morton CC, Copeland NG, Jenkins NA. Characterization of unconventional MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice. Hum Mol Genet. 1997;6:1225–1231. doi: 10.1093/hmg/6.8.1225. [DOI] [PubMed] [Google Scholar]
- 33.Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, Tucker A, Russell SD, Bluemke DA, Dietz HC, Calkins H, Judge DP. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet. 2006;79:136–142. doi: 10.1086/504393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Azzimato V, Genneback N, Tabish AM, Buyandelger B, Knoll R. Desmin, desminopathy and the complexity of genetics. J Mol Cell Cardiol. 2016;92:93–95. doi: 10.1016/j.yjmcc.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 35.Bahler M, Eppenberger HM, Wallimann T. Novel thick filament protein of chicken pectoralis muscle: The 86 kd protein. I. Purification and characterization. J Mol Biol. 1985;186:381–391. doi: 10.1016/0022-2836(85)90112-3. [DOI] [PubMed] [Google Scholar]
- 36.Bakolitsa C, Cohen DM, Bankston LA, Bobkov AA, Cadwell GW, Jennings L, Critchley DR, Craig SW, Liddington RC. Structural basis for vinculin activation at sites of cell adhesion. Nature. 2004;430:583–586. doi: 10.1038/nature02610. [DOI] [PubMed] [Google Scholar]
- 37.Balogh J, Merisckay M, Li Z, Paulin D, Arner A. Hearts from mice lacking desmin have a myopathy with impaired active force generation and unaltered wall compliance. Cardiovasc Res. 2002;53:439–450. doi: 10.1016/s0008-6363(01)00500-4. [DOI] [PubMed] [Google Scholar]
- 38.Balse E, Steele DF, Abriel H, Coulombe A, Fedida D, Hatem SN. Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiol Rev. 2012;92:1317–1358. doi: 10.1152/physrev.00041.2011. [DOI] [PubMed] [Google Scholar]
- 39.Baltgalvis KA, Call JA, Nikas JB, Lowe DA. Effects of prednisolone on skeletal muscle contractility in mdx mice. Muscle Nerve. 2009;40:443–454. doi: 10.1002/mus.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bang ML. Animal models of congenital cardiomyopathies associated with mutations in Z-line proteins. J Cell Physiol. 2016;9999:1–15. doi: 10.1002/jcp.25424. [DOI] [PubMed] [Google Scholar]
- 41.Bang ML, Caremani M, Brunello E, Littlefield R, Lieber RL, Chen J, Lombardi V, Linari M. Nebulin plays a direct role in promoting strong actin-myosin interactions. FASEB J. 2009;23:4117–4125. doi: 10.1096/fj09-137729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bang ML, Chen J. Roles of nebulin family members in the heart. Circ J. 2015;79:2081–2087. doi: 10.1253/circj.CJ-15-0854. [DOI] [PubMed] [Google Scholar]
- 43.Bang ML, Gu Y, Dalton ND, Peterson KL, Chien KR, Chen J. The muscle ankyrin repeat proteins CARP, Ankrd2, and DARP are not essential for normal cardiac development and function at basal conditions and in response to pressure overload. PLoS One. 2014;9:e93638. doi: 10.1371/journal.pone.0093638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bang ML, Mudry RE, McElhinny AS, Trombitas K, Geach AJ, Yamasaki R, Sorimachi H, Granzier H, Gregorio CC, Labeit S. Myopal-ladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001;153:413–427. doi: 10.1083/jcb.153.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bao ZZ, Lakonishok M, Kaufman S, Horwitz AF. α7β1 integrin is a component of the myotendinous junction on skeletal muscle. J Cell Sci. 1993;106:579–589. doi: 10.1242/jcs.106.2.579. [DOI] [PubMed] [Google Scholar]
- 46.Barash IA, Mathew L, Ryan AF, Chen J, Lieber RL. Rapid muscle-specific gene expression changes after a single bout of eccentric contractions in the mouse. Am J Physiol Cell Physiol. 2004;286:C355–C364. doi: 10.1152/ajpcell.00211.2003. [DOI] [PubMed] [Google Scholar]
- 47.Baron CP, Jacobsen S, Purslow PP. Cleavage of desmin by cysteine proteases: Calpains and cathepsin B. Meat Sci. 2004;68:447–456. doi: 10.1016/j.meatsci.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 48.Baryshnikova OK, Li MX, Sykes BD. Modulation of cardiac troponin C function by the cardiac-specific N-terminus of troponin I: Influence of PKA phosphorylation and involvement in cardiomyopathies. J Mol Biol. 2008;375:735–751. doi: 10.1016/j.jmb.2007.10.062. [DOI] [PubMed] [Google Scholar]
- 49.Bates G, Sigurdardottir S, Kachmar L, Zitouni NB, Benedetti A, Petrof BJ, Rassier D, Lauzon AM. Molecular, cellular, and muscle strip mechanics of the mdx mouse diaphragm. Am J Physiol Cell Physiol. 2013;304:C873–C880. doi: 10.1152/ajpcell.00220.2012. [DOI] [PubMed] [Google Scholar]
- 50.Bean C, Verma NK, Yamamoto DL, Chemello F, Cenni V, Filomena MC, Chen J, Bang ML, Lanfranchi G. Ankrd2 is a modulator of NF-κB-mediated inflammatory responses during muscle differentiation. Cell Death Dis. 2014;5:1–13. doi: 10.1038/cddis.2013.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beggs AH, Byers TJ, Knoll JH, Boyce FM, Bruns GA, Kunkel LM. Cloning and characterization of two human skeletal muscle α-actinin genes located on chromosomes 1 and 11. J Biol Chem. 1992;267:9281–9288. [PubMed] [Google Scholar]
- 52.Belkin AM, Zhidkova NI, Balzac F, Altruda F, Tomatis D, Maier A, Tarone G, Koteliansky VE, Burridge K. β1D integrin displaces the β1A isoform in striated muscles: localization at junctional structures and signaling potential in nonmuscle cells. J Cell Biol. 1996;132:211–226. doi: 10.1083/jcb.132.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bellin RM, Huiatt TW, Critchley DR, Robson RM. Synemin may function to directly link muscle cell intermediate filaments to both myofib-rillar Z-lines and costameres. J Biol Chem. 2001;276:32330–32337. doi: 10.1074/jbc.M104005200. [DOI] [PubMed] [Google Scholar]
- 54.Bellin RM, Sernett SW, Becker B, Ip W, Huiatt TW, Robson RM. Molecular characteristics and interactions of the intermediate filament protein synemin Interactions with α-actinin may anchor synemin-containing heterofilaments. J Biol Chem. 1999;274:29493–29499. doi: 10.1074/jbc.274.41.29493. [DOI] [PubMed] [Google Scholar]
- 55.Bennett P, Craig R, Starr R, Offer G. The ultrastructural location of C-protein, X-protein and H-protein in rabbit muscle. J Muscle Res Cell Motil. 1986;7:550–567. doi: 10.1007/bf01753571. [DOI] [PubMed] [Google Scholar]
- 56.Benson MA, Newey SE, Martin-Rendon E, Hawkes R, Blake DJ. Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J Biol Chem. 2001;276:24232–24241. doi: 10.1074/jbc.M010418200. [DOI] [PubMed] [Google Scholar]
- 57.Bergen HR, 3rd, Ajtai K, Burghardt TP, Nepomuceno AI, Muddiman DC. Mass spectral determination of skeletal/cardiac actin isoform ratios in cardiac muscle. Rapid Commun Mass Spectrom. 2003;17:1467–1471. doi: 10.1002/rcm.1075. [DOI] [PubMed] [Google Scholar]
- 58.Berry SE, Andruszkiewicz P, Chun JL, Hong J. Nestin expression in end-stage disease in dystrophin-deficient heart: Implications for regeneration from endogenous cardiac stem cells. Stem Cells Transl Med. 2013;2:848–861. doi: 10.5966/sctm2012-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhavsar PK, Brand NJ, Yacoub MH, Barton PJR. Isolation and characterization of the human cardiac troponin I gene (TNNI3) Genomics. 1996;35:11–23. doi: 10.1006/geno.1996.0317. [DOI] [PubMed] [Google Scholar]
- 60.Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, Schouten M, Jongbloed R, Cox MG, van Wolferen M, Rodriguez LM, van Gelder IC, Bikker H, Suurmeijer AJ, van den Berg MP, Mannens MM, Hauer RN, Wilde AA, van Tintelen JP. Desmoglein-2 and desmocollin-2 mutations in dutch arrhythmogenic right ventricular dysplasia/cardiomypathy patients: Results from a multicenter study. Circ Cardiovasc Genet. 2009;2:418–427. doi: 10.1161/circgenetics.108.839829. [DOI] [PubMed] [Google Scholar]
- 61.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 62.Blake DJ, Nawrotzki R, Peters MF, Froehner SC, Davies KE. Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J Biol Chem. 1996;271:7802–7810. doi: 10.1074/jbc.271.13.7802. [DOI] [PubMed] [Google Scholar]
- 63.Blake DJ, Tinsley JM, Davies KE. Utrophin: A structural and functional comparison to dystrophin. Brain Pathol. 1996;6:37–47. doi: 10.1111/j1750-3639.1996.tb00781.x. [DOI] [PubMed] [Google Scholar]
- 64.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 65.Boateng SY, Senyo SE, Qi L, Goldspink PH, Russell B. Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J Mol Cell Cardiol. 2009;47:426–435. doi: 10.1016/j.yjmcc.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 67.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- 68.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 69.Bonne G, Quijano-Roy S. Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol. 2013;113:1367–1376. doi: 10.1016/b978-0-444-59565-2.00007-1. [DOI] [PubMed] [Google Scholar]
- 70.Bönnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally EM, Duggan DJ, Angelini C, Hoffman EP, Ozawa E, Kunkel LM. β-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995;11:266–273. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- 71.Bönnemann CG, Wong J, Hamida CB, Hamida MB, Hentati F, Kunkel LM. LGMD 2E in Tunisia is caused by a homozygous missense mutation in β-sarcoglycan exon 3. Neuromuscul Disord. 1998;8:193–197. doi: 10.1016/S0960-8966(98)00014-5. [DOI] [PubMed] [Google Scholar]
- 72.Borgon RA, Vonrhein C, Bricogne G, Bois PR, Izard T. Crystal structure of human vinculin. Structure. 2004;12:1189–1197. doi: 10.1016/j.str.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 73.Bork P, Sudol M. The WW domain: A signalling site in dystrophin? Trends Biochem Sci. 1994;19:531–533. doi: 10.1016/0968-0004(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 74.Bornemann A, Schmalbruch H. Anti-vimentin staining in muscle pathology. Neuropathol Appl Neurobiol. 1993;19:414–419. doi: 10.1111/j.1365-2990.1993.tb00463.x. [DOI] [PubMed] [Google Scholar]
- 75.Bornslaeger EA, Godsel LM, Corcoran CM, Park JK, Hatzfeld M, Kowalczyk AP, Green KJ. Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci. 2001;114:727–738. doi: 10.1242/jcs.114.4.727. [DOI] [PubMed] [Google Scholar]
- 76.Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur J Cell Biol. 2006;85:469–485. doi: 10.1016/j.ejcb.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 77.Bovill E, Westaby S, Crisp A, Jacobs S, Shaw T. Reduction of four-and-a-half LIM-protein 2 expression occurs in human left ventricular failure and leads to altered localization and reduced activity of metabolic enzymes. J Thorac Cardiovasc Surg. 2009;137:853–861. doi: 10.1016/j.jtcvs.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 78.Brancaccio M, Cabodi S, Belkin AM, Collo G, Koteliansky VE, Tomatis D, Altruda F, Silengo L, Tarone G. Differential onset of expression of α7 and β1D integrins during mouse heart and skeletal muscle development. Cell Adhes Commun. 1998;5:193–205. doi: 10.3109/15419069809040291. [DOI] [PubMed] [Google Scholar]
- 79.Brieger A, Adryan B, Wolpert F, Passmann S, Zeuzem S, Trojan J. Cytoskeletal scaffolding proteins interact with Lynch-Syndrome associated mismatch repair protein MLH1. Proteomics. 2010;10:3343–3355. doi: 10.1002/pmic.2009.00672. [DOI] [PubMed] [Google Scholar]
- 80.Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol Rev. 2006;86:967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 81.Brokat S, Thomas J, Herda LR, Knosalla C, Pregla R, Brancaccio M, Accornero F, Tarone G, Hetzer R, Regitz-Zagrosek V. Altered melusin expression in the hearts of aortic stenosis patients. Eur J Heart Fail. 2007;9:568–573. doi: 10.1016/j.ejheart.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 82.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burridge K, Mangeat P. An interaction between vinculin and talin. Nature. 1984;308:744–746. doi: 10.1038/308744a0. [DOI] [PubMed] [Google Scholar]
- 84.Buyandelger B, Ng KE, Miocic S, Piotrowska I, Gunkel S, Ku CH, Knoll R. MLP (muscle LIM protein) as a stress sensor in the heart. Pflugers Arch. 2011;462:135–142. doi: 10.1007/s00.424-011-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The Talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274:28071–28074. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- 86.Calkins H. Arrhythmogenic right ventricular dysplasia/cardiomyopathy-three decades of progress. Circ J. 2015;79:901–913. doi: 10.1253/circj.CJ-15-0288. [DOI] [PubMed] [Google Scholar]
- 87.Candasamy AJ, Haworth RS, Cuello F, Ibrahim M, Aravamudhan S, Kruger M, Holt MR, Terracciano CM, Mayr M, Gautel M, Avkiran M. Phosphoregulation of the titin-cap protein telethonin in cardiac myocytes. J Biol Chem. 2014;289:1282–1293. doi: 10.1074/jbc.M113.479030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res. 2007;313:2063–2076. doi: 10.1016/j.yexcr.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 89.Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, Graw SL, Feiger J, Zhu XZ, Dao D, Ferguson DA, Bristow MR, Mestroni L. α-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112:54–59. doi: 10.1161/circulation-aha.104.507699. [DOI] [PubMed] [Google Scholar]
- 90.Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 2015;573:188–197. doi: 10.1016/j.gene.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carroll S, Lu S, Herrera AH, Horowits R. N-RAP scaffolds I-Z-I assembly during myofibrillogenesis in cultured chick cardiomyocytes. J Cell Sci. 2004;117:105–114. doi: 10.1242/jcs.00847. [DOI] [PubMed] [Google Scholar]
- 92.Cartwright S, Karakesisoglou I. Nesprins in health and disease. Semin Cell Dev Biol. 2014;29:169–179. doi: 10.1016/j.semcdb.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 93.Casella JF, Craig SW, Maack DJ, Brown AE. Cap Z(36/32), a barbed end actin-capping protein, is a component of the Z-line of skeletal muscle. J Cell Biol. 1987;105:371–379. doi: 10.1083/jcb.105.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Casella JF, Maack DJ, Lin S. Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. J Biol Chem. 1986;261:10915–10921. [PubMed] [Google Scholar]
- 95.Castillo A, Nowak R, Littlefield KP, Fowler VM, Littlefield RS. A nebulin ruler does not dictate thin filament lengths. Biophys J. 2009;96:1856–1865. doi: 10.1016/j.bpj.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol. 2001;306:717–726. doi: 10.1006/jmbi.2001.4448. [DOI] [PubMed] [Google Scholar]
- 97.Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, Swanson LC, Soemedi R, Vasli N, Iannaccone ST, Shieh PB, Shur N, Dennison JM, Lawlor MW, Laporte J, Markianos K, Fairbrother WG, Granzier H, Beggs AH. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. 2013;81:1205–1214. doi: 10.1212/WNL.0b013e3182a6ca62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chalkia D, Nikolaidis N, Makalowski W, Klein J, Nei M. Origins and evolution of the formin multigene family that is involved in the formation of actin filaments. Mol Biol Evol. 2008;25:2717–2733. doi: 10.1093/molbev/msn215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki H, Sato N, Chien KR, Kasahara H. Identification of cardiac-specific myosin light chain kinase. Circ Res. 2008;102:571–580. doi: 10.1161/circresaha.107.161687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chan KK, Tsui SK, Lee SM, Luk SC, Liew CC, Fung KP, Waye MM, Lee CY. Molecular cloning and characterization of FHL2, a novel LIM domain protein preferentially expressed in human heart. Gene. 1998;210:345–350. doi: 10.1016/S0378-1119(97)00644-6. [DOI] [PubMed] [Google Scholar]
- 101.Chandy IK, Lo JC, Ludescher RD. Differential mobility of skeletal and cardiac tropomyosin on the surface of F-actin. Biochemistry. 1999;38:9286–9294. doi: 10.1021/bi983073s. [DOI] [PubMed] [Google Scholar]
- 102.Charvet B, Ruggiero F, Le Guellec D. The development of the myotendinous junction. A review. Muscles Ligaments Tendons J. 2012;2:53–63. [PMC free article] [PubMed] [Google Scholar]
- 103.Chatzifrangkeskou M, Bonne G, Muchir A. Nuclear envelope and striated muscle diseases. Curr Opin Cell Biol. 2015;32:1–6. doi: 10.1016/j.ceb.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 104.Chauveau C, Bonnemann CG, Julien C, Kho AL, Marks H, Talim B, Maury P, Arne-Bes MC, Uro-Coste E, Alexandrovich A, Vihola A, Schafer S, Kaufmann B, Medne L, Hübner N, Foley AR, Santi M, Udd B, Topaloglu H, Moore SA, Gotthardt M, Samuels ME, Gautel M, Ferreiro A. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23:980–991. doi: 10.1093/hmg/ddt494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chauveau C, Rowell J, Ferreiro A. A rising titan: TTN review and mutation update. Hum Mutat. 2014;35:1046–1059. doi: 10.1002/humu.22611. [DOI] [PubMed] [Google Scholar]
- 106.Chen F, Chang R, Trivedi M, Capetanaki Y, Cryns VL. Caspase proteolysis of desmin produces a dominant-negative inhibitor of intermediate filaments and promotes apoptosis. J Biol Chem. 2003;278:6848–6853. doi: 10.1074/jbc.M212021200. [DOI] [PubMed] [Google Scholar]
- 107.Chen H, Huang XN, Yan W, Chen K, Guo L, Tummalapali L, Dedhar S, St-Arnaud R, Wu C, Sepulveda JL. Role of the integrin-linked kinase/PINCH1/α-parvin complex in cardiac myocyte hypertrophy. Lab Invest. 2005;85:1342–1356. doi: 10.1038/labinvest.3700345. [DOI] [PubMed] [Google Scholar]
- 108.Chen NT, Lo SH. The N-terminal half of talin2 is sufficient for mouse development and survival. Biochem Biophys Res Commun. 2005;337:670–676. doi: 10.1016/j.bbrc.2005.09.100. [DOI] [PubMed] [Google Scholar]
- 109.Chen X, Ni F, Kondrashkina E, Ma J, Wang Q. Mechanisms of leiomodin 2-mediated regulation of actin filament in muscle cells. Proc Natl Acad Sci U S A. 2015;112:12687–12692. doi: 10.1073/pnas.1512464112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cheng H, Kimura K, Peter AK, Cui L, Ouyang K, Shen T, Liu Y, Gu Y, Dalton ND, Evans SM, Knowlton KU, Peterson KL, Chen J. Loss of enigma homolog protein results in dilated cardiomyopathy. Circ Res. 2010;107:348–356. doi: 10.1161/circresaha.110.218735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chereau D, Boczkowska M, Skwarek-Maruszewska A, Fujiwara I, Hayes DB, Rebowski G, Lappalainen P, Pollard TD, Dominguez R. Leiomodin is an actin filament nucleator in muscle cells. Science. 2008;320:239–243. doi: 10.1126/science.1155313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chew CS, Chen X, Parente JA, Jr, Tarrer S, Okamoto C, Qin HY. Lasp-1 binds to non-muscle F-actin in vitro and is localized within multiple sites of dynamic actin assembly in vivo. J Cell Sci. 2002;115:4787–4799. doi: 10.1242/jcs.00174. [DOI] [PubMed] [Google Scholar]
- 113.Chitaev NA, Leube RE, Troyanovsky RB, Eshkind LG, Franke WW, Troyanovsky SM. The binding of plakoglobin to desmosomal cadherins: Patterns of binding sites and topogenic potential. J Cell Biol. 1996;133:359–369. doi: 10.1083/jcb.133.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Choi H-J, Weis WI. Structure of the armadillo repeat domain of plakophilin 1. J Mol Biol. 2005;346:367–376. doi: 10.1016/j.jmb.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 115.Choi HJ, Park-Snyder S, Pascoe LT, Green KJ, Weis WI. Structures of two intermediate filament-binding fragments of desmoplakin reveal a unique repeat motif structure. Nat Struct Biol. 2002;9:612–620. doi: 10.1038/nsb818. [DOI] [PubMed] [Google Scholar]
- 116.Chowrashi P, Mittal B, Sanger JM, Sanger JW. Amorphin is phosphorylase; phosphorylase is an α-actinin-binding protein. Cell Motil Cytoskeleton. 2002;53:125–135. doi: 10.1002/cm.10059. [DOI] [PubMed] [Google Scholar]
- 117.Chu X, Chen J, Reedy MC, Vera C, Sung KLP, Sung LA. E-Tmod capping of actin filaments at the slow-growing end is required to establish mouse embryonic circulation. Am J Physiol Heart Circ Physiol. 2003;284:1827–1838. doi: 10.1152/ajpheart.00947.2002. [DOI] [PubMed] [Google Scholar]
- 118.Clark E, Brugge J. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 119.Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6:376–385. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 120.Clemen CS, Herrmann H, Strelkov SV, Schroder R. Desminopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:47–75. doi: 10.1007/s00401-012-1057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Coghill ID, Brown S, Cottle DL, McGrath MJ, Robinson PA, Nandurkar HH, Dyson JM, Mitchell CA. FHL3 is an actin-binding protein that regulates α-actinin-mediated actin bundling: FHL3 localizes to actin stress fibers and enhances cell spreading and stress fiber disassembly. J Biol Chem. 2003;278:24139–24152. doi: 10.1074/jbc.M213259200. [DOI] [PubMed] [Google Scholar]
- 122.Conley CA, Fritz-Six KL, Almenar-Queralt A, Fowler VM. Leiomodins: larger members of the tropomodulin (Tmod) gene family. Genomics. 2001;73:127–139. doi: 10.1006/geno.2000.6501. [DOI] [PubMed] [Google Scholar]
- 123.Conti A, Riva N, Pesca M, Iannaccone S, Cannistraci CV, Corbo M, Previtali SC, Quattrini A, Alessio M. Increased expression of myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim Biophys Acta. 2014;1842:99–106. doi: 10.1016/j.bbadis.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 124.Conti FJ, Felder A, Monkley S, Schwander M, Wood MR, Lieber R, Critchley D, Muller U. Progressive myopathy and defects in the maintenance of myotendinous junctions in mice that lack talin 1 in skeletal muscle. Development. 2008;135:2043–2053. doi: 10.1242/dev.015818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cottle DL, McGrath MJ, Cowling BS, Coghill ID, Brown S, Mitchell CA. FHL3 binds MyoD and negatively regulates myotube formation. J Cell Sci. 2007;120:1423–1435. doi: 10.1242/jcs.004739. [DOI] [PubMed] [Google Scholar]
- 126.Cowling BS, Cottle DL, Wilding BR, D’Arcy CE, Mitchell CA, McGrath MJ. Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: A comprehensive review of the clinical, histological and pathological features. Neuromuscul Disord. 2011;21:237–251. doi: 10.1016/j.nmd.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 127.Cowling BS, McGrath MJ, Nguyen MA, Cottle DL, Kee AJ, Brown S, Schessl J, Zou Y, Joya J, Bonnemann CG, Hardeman EC, Mitchell CA. Identification of FHL1 as a regulator of skeletal muscle mass: Implications for human myopathy. J Cell Biol. 2008;183:1033–1048. doi: 10.1083/jcb.2008.04077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cox PR, Zoghbi HY. Sequencing, expression analysis, and mapping of three unique human tropomodulin genes and their mouse orthologs. Genomics. 2000;63:97–107. doi: 10.1006/geno.1999.6061. [DOI] [PubMed] [Google Scholar]
- 129.Craig R, Lehman W. Crossbridge and tropomyosin positions observed in native, interacting thick and thin filaments. J Mol Biol. 2001;311:1027–1036. doi: 10.1006/jmbi.2001.4897. [DOI] [PubMed] [Google Scholar]
- 130.Craig R, Offer G. The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond B Biol Sci. 1976;192:451–461. doi: 10.1098/rspb..1976.0023. [DOI] [PubMed] [Google Scholar]
- 131.Crawford AW, Michelsen JW, Beckerle MC. An interaction between zyxin and α-actinin. J Cell Biol. 1992;116:1381–1393. doi: 10.1083/jcb.116.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Crawford K, Flick R, Close L, Shelly D, Paul R, Bove K, Kumar A, Lessard J. Mice lacking skeletal muscle actin show reduced muscle strength and growth deficits and die during the neonatal period. Mol Cell Biol. 2002;22:5887–5896. doi: 10.1128/MCB.22(16)5887-5896.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Crosbie RH, Heighway J, Venzke DP, Lee JC, Campbell KP. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J Biol Chem. 1997;272:31221–31224. doi: 10.1074/jbc.272.50.31221. [DOI] [PubMed] [Google Scholar]
- 134.Crosbie RH, Lebakken CS, Holt KH, Venzke DP, Straub V, Lee JC, Grady RM, Chamberlain JS, Sanes JR, Campbell KP. Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J Cell Biol. 1999;145:153–165. doi: 10.1083/jcb.145.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cunha SR, Mohler PJ. Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J Biol Chem. 2008;283:31968–31980. doi: 10.1074/jbc.M806050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.D’Arcy CE, Feeney SJ, McLean CA, Gehrig SM, Lynch GS, Smith JE, Cowling BS, Mitchell CA, McGrath MJ. Identification of FHL1 as a therapeutic target for Duchenne muscular dystrophy. Hum Mol Genet. 2014;23:618–636. doi: 10.1093/hmg/ddt449. [DOI] [PubMed] [Google Scholar]
- 137.Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, Aletras AH, Wen H, Epstein ND. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–641. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 138.De Acetis M, Notte A, Accornero F, Selvetella G, Brancaccio M, Vecchione C, Sbroggio M, Collino F, Pacchioni B, Lanfranchi G, Aretini A, Ferretti R, Maffei A, Altruda F, Silengo L, Tarone G, Lembo G. Cardiac overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circ Res. 2005;96:1087–1094. doi: 10.1161/01.RES.0000168028.36081.e0. [DOI] [PubMed] [Google Scholar]
- 139.Delva E, Tucker DK, Kowalczyk AP. The desmosome. Cold Spring Harb Perspect Biol. 2009;1:1–17. doi: 10.1101/cshperspect.a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dennis JE, Shimizu T, Reinach FC, Fischman DA. Localization of C-protein isoforms in chicken skeletal muscle: Ultrastructural detection using monoclonal antibodies. J Cell Biol. 1984;98:1514–1522. doi: 10.1083/jcb.98.4.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dhoot GK, Hales MC, Grail BM, Perry SV. The isoforms of C protein and their distribution in mammalian skeletal muscle. J Muscle Res Cell Motil. 1985;6:487–505. doi: 10.1007/bf00712585. [DOI] [PubMed] [Google Scholar]
- 142.Dhume A, Lu S, Horowits R. Targeted disruption of N-RAP gene function by RNA interference: A role for N-RAP in myofibril organization. Cell Motil Cytoskeleton. 2006;63:493–511. doi: 10.1002/cm.2014.1. [DOI] [PubMed] [Google Scholar]
- 143.Diguet N, Mallat Y, Ladouce R, Clodic G, Prola A, Tritsch E, Blanc J, Larcher JC, Delcayre C, Samuel JL, Friguet B, Bolbach G, Li Z, Mericskay M. Muscle creatine kinase deficiency triggers both actin depolymerization and desmin disorganization by advanced glycation end products in dilated cardiomyopathy. J Biol Chem. 2011;286:35007–35019. doi: 10.1074/jbc.M111.252395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta. 2013;1832:2414–2424. doi: 10.1016/j.bbadis.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 145.Docheva D, Popov C, Alberton P, Aszodi A. Integrin signaling in skeletal development and function. Birth Defects Res C Embryo Today. 2014;102:13–36. doi: 10.1002/bdrc.21059. [DOI] [PubMed] [Google Scholar]
- 146.Dolken G, Leisner E, Pette D. Immunofluorescent localization of glycogenolytic and glycolytic enzyme proteins and of malate dehydrogenase isozymes in cross-striated skeletal muscle and heart of the rabbit. Histochemistry. 1975;43:113–121. doi: 10.1007/BF00492440. [DOI] [PubMed] [Google Scholar]
- 147.Domenighetti AA, Chu PH, Wu T, Sheikh F, Gokhin DS, Guo LT, Cui Z, Peter AK, Christodoulou DC, Parfenov MG, Gorham JM, Li DY, Banerjee I, Lai X, Witzmann FA, Seidman CE, Seidman JG, Gomes AV, Shelton GD, Lieber RL, Chen J. Loss of FHL1 induces an age-dependent skeletal muscle myopathy associated with myofibrillar and intermyofibrillar disorganization in mice. Hum Mol Genet. 2014;23:209–225. doi: 10.1093/hmg/ddt412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Donker DW, Maessen JG, Verheyen F, Ramaekers FC, Spatjens RL, Kuijpers H, Ramakers C, Schiffers PM, Vos MA, Crijns HJ, Volders PG. Impact of acute and enduring volume overload on mechanotransduction and cytoskeletal integrity of canine left ventricular myocardium. Am J Physiol Heart Circ Physiol. 2007;292:H2324–H2332. doi: 10.1152/ajpheart.00392.2006. [DOI] [PubMed] [Google Scholar]
- 149.Donner K, Ollikainen M, Ridanpaa M, Christen HJ, Goebel HH, de Visser M, Pelin K, Wallgren-Pettersson C. Mutations in the β-tropomyosin (TPM2) gene—a rare cause of nemaline myopathy. Neuromuscul Disord. 2002;12:151–158. doi: 10.1016/S0960-8966(01)00252-8. [DOI] [PubMed] [Google Scholar]
- 150.Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–371. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 151.Duygu B, de Windt LJ, da Costa Martins PA. Targeting microRNAs in heart failure. Trends Cardiovasc Med. 2015;26:99–110. doi: 10.1016/j.tcm.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 152.Egelman EH. The structure of F-actin. J Muscle Res Cell Motil. 1985;6:129–151. doi: 10.1007/BF00713056. [DOI] [PubMed] [Google Scholar]
- 153.Ehler E, Horowits R, Zuppinger C, Price RL, Perriard E, Leu M, Caroni P, Sussman M, Eppenberger HM, Perriard JC. Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol. 2001;153:763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ehrig K, Leivo I, Argraves WS, Ruoslahti E, Engvall E. Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc Natl Acad Sci U S A. 1990;87:3264–3268. doi: 10.1073/pnas.87.9.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Einheber S, Fischman DA. Isolation and characterization of a cDNA clone encoding avian skeletal muscle C-protein: an intracellular member of the immunoglobulin superfamily. Proc Natl Acad Sci U S A. 1990;87:2157–2161. doi: 10.1073/pnas.87.6.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ervasti J, Campbell K. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Esham M, Bryan K, Milnes J, Holmes WB, Moncman CL. Expression of nebulette during early cardiac development. Cell Motil Cytoskeleton. 2007;64:258–273. doi: 10.1002/cm.2018.0. [DOI] [PubMed] [Google Scholar]
- 158.Ettinger AJ, Feng G, Sanes JR. ε-Sarcoglycan, a broadly expressed homologue of the gene mutated in Limb-Girdle Muscular Dystrophy 2D. J Biol Chem. 1997;272:32534–32538. doi: 10.1074/jbc.272.51.32534. [DOI] [PubMed] [Google Scholar]
- 159.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/nejm199912023412302. [DOI] [PubMed] [Google Scholar]
- 160.Faul C, Dhume A, Schecter AD, Mundel P. Protein kinase A, Ca2+/calmodulin-dependent kinase II, and calcineurin regulate the intracellular trafficking of myopodin between the Z-disc and the nucleus of cardiac myocytes. Mol Cell Biol. 2007;27:8215–8227. doi: 10.1128/mcb00950-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Faulkner G, Pallavicini A, Comelli A, Salamon M, Bortoletto G, Ievolella C, Trevisan S, Kojic S, Dalla Vecchia F, Laveder P, Valle G, Lanfranchi G. FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J Biol Chem. 2000;275:41234–41242. doi: 10.1074/jbc.M007493200. [DOI] [PubMed] [Google Scholar]
- 162.Fernandes I, Schock F. The nebulin repeat protein Lasp regulates I-band architecture and filament spacing in myofibrils. J Cell Biol. 2014;206:559–572. doi: 10.1083/jcb.2014.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ferreiro A, Mezmezian M, Olive M, Herlicoviez D, Fardeau M, Richard P, Romero NB. Telethonin-deficiency initially presenting as a congenital muscular dystrophy. Neuromuscul Disord. 2011;21:433–438. doi: 10.1016/j.nmd.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 164.Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, Richardson JA, Bassel-Duby R, Olson EN. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1-3. J Clin Invest. 2007;117:2486–2495. doi: 10.1172/jci32827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Finsterer J, Ramaciotti C, Wang CH, Wahbi K, Rosenthal D, Duboc D, Melacini P. Cardiac findings in congenital muscular dystrophies. Pediatrics. 2010;126:538–545. doi: 10.1542/peds2010-0208. [DOI] [PubMed] [Google Scholar]
- 166.Fisher DZ, Chaudhary N, Blobel G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A. 1986;83:6450–6454. doi: 10.1073/pnas.83.17.6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: Its role in physiology and disease. Circ Res. 2004;94:1279–1289. doi: 10.1161/01.res.0000127175.2.1818.c2. [DOI] [PubMed] [Google Scholar]
- 168.Flick MJ, Konieczny SF. The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of βI-spectrin. J Cell Sci. 2000;113:1553–1564. doi: 10.1242/jcs.113.9.1553. [DOI] [PubMed] [Google Scholar]
- 169.Fontes MS, van Veen TA, de Bakker JM, van Rijen HV. Functional consequences of abnormal Cx43 expression in the heart. Biochim Biophys Acta. 2012;1818:2020–2029. doi: 10.1016/j.bbamem.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 170.Forbes MS, Sperelakis N. Intercalated discs of mammalian heart: A review of structure and function. Tissue Cell. 1985;17:605–648. doi: 10.1016/0040-8166(85)90001-1. [DOI] [PubMed] [Google Scholar]
- 171.Foth BJ, Goedecke MC, Soldati D. New insights into myosin evolution and classification. Proc Natl Acad Sci U S A. 2006;103:3681–3686. doi: 10.1073/pnas.0506307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Foucault G, Vacher M, Merkulova T, Keller A, Arrio-Dupont M. Presence of enolase in the M-band of skeletal muscle and possible indirect interaction with the cytosolic muscle isoform of creatine kinase. Biochem J. 1999;338:115–121. doi: 10.1042/bj3380115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fowler VM. Identification and purification of a novel Mr 43,000 tropomyosin-binding protein from human erythrocyte membranes. J Biol Chem. 1987;262:12792–12800. [PubMed] [Google Scholar]
- 174.Frank D, Frey N. Cardiac Z-disc signaling network. J Biol Chem. 2011;286:9897–9904. doi: 10.1074/jbc.R110.174268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Frank D, Kuhn C, Katus HA, Frey N. The sarcomeric Z-disc: A nodal point in signalling and disease. J Mol Med (Berl) 2006;84:446–468. doi: 10.1007/s00109-005-0033-1. [DOI] [PubMed] [Google Scholar]
- 176.Franke WW, Borrmann CM, Grund C, Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006;85:69–82. doi: 10.1016/j.ejcb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 177.Franklin AJ, Baxley T, Kobayashi T, Chalovich JM. The C-terminus of troponin T is essential for maintaining the inactive state of regulated actin. Biophys J. 2012;102:2536–2544. doi: 10.1016/j.bpj.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Freiburg A, Gautel M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem. 1996;235:317–323. doi: 10.1111/j1432-1033.1996.00317.x. [DOI] [PubMed] [Google Scholar]
- 179.Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, Granzier H, Labeit S. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res. 2000;86:1114–1121. doi: 10.1161/01.RES.86.11.1114. [DOI] [PubMed] [Google Scholar]
- 180.Frenette J, Tidball JG. Mechanical loading regulates expression of talin and its mRNA, which are concentrated at myotendinous junctions. Am J Physiol. 1998;275:C818–C825. doi: 10.1242/jcs.00303. [DOI] [PubMed] [Google Scholar]
- 181.Frey N, Barrientos T, Shelton JM, Frank D, Rutten H, Gehring D, Kuhn C, Lutz M, Rothermel B, Bassel-Duby R, Richardson JA, Katus HA, Hill JA, Olson EN. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med. 2004;10:1336–1343. doi: 10.1038/nm1132. [DOI] [PubMed] [Google Scholar]
- 182.Frey N, Olson EN. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J Biol Chem. 2002;277:13998–14004. doi: 10.1074/jbc.M200712200. [DOI] [PubMed] [Google Scholar]
- 183.Frey N, Richardson JA, Olson EN. Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc Natl Acad Sci U S A. 2000;97:14632–14637. doi: 10.1073/pnas.260501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fritz-Six KL, Cox PR, Fischer RS, Xu B, Gregorio CC, Zoghbi HY, Fowler VM. Aberrant myofibril assembly in tropomodulin1 null mice leads to aborted heart development and embryonic lethality. J Cell Biol. 2003;163:1033–1044. doi: 10.1083/jcb.2003.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fukuda N, Wu Y, Nair P, Granzier HL. Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform-dependent manner. J Gen Physiol. 2005;125:257–271. doi: 10.1085/jgp.2004.09177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fukuzawa A, Lange S, Holt M, Vihola A, Carmignac V, Ferreiro A, Udd B, Gautel M. Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band–implications for hereditary myopathies. J Cell Sci. 2008;121:1841–1851. doi: 10.1242/jcs.028019. [DOI] [PubMed] [Google Scholar]
- 187.Furst DO, Osborn M, Weber K. Myogenesis in the mouse embryo: Differential onset of expression of myogenic proteins and the involvement of titin in myofibril assembly. J Cell Biol. 1989;109:517–527. doi: 10.1083/jcb.109.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Furst DO, Vinkemeier U, Weber K. Mammalian skeletal muscle C-protein: Purification from bovine muscle, binding to titin and the characterization of a full-length human cDNA. J Cell Sci. 1992;102:769–778. doi: 10.1242/jcs.102.4.769. [DOI] [PubMed] [Google Scholar]
- 189.Furukawa T, Ono Y, Tsuchiya H, Katayama Y, Bang ML, Labeit D, Labeit S, Inagaki N, Gregorio CC. Specific interaction of the potassium channel β-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol. 2001;313:775–784. doi: 10.1006/jmbi.2001.5053. [DOI] [PubMed] [Google Scholar]
- 190.Gahlmann R, Kedes L. Cloning, structural analysis, and expression of the human fast twitch skeletal muscle troponin C gene. J Biol Chem. 1990;265:12520–12528. doi: 10.1016/j.egypro.2011.10.601. [DOI] [PubMed] [Google Scholar]
- 191.Gaikis L, Stewart D, Johnson R, Pyle WG. Identifying a role of the actin capping protein CapZ in β-adrenergic receptor signalling. Acta Physiol (Oxf) 2013;207:173–182. doi: 10.1111/j1748-1716.2012.02470.x. [DOI] [PubMed] [Google Scholar]
- 192.Gallanti A, Prelle A, Moggio M, Ciscato P, Checcarelli N, Sciacco M, Comini A, Scarlato G. Desmin and vimentin as markers of regeneration in muscle diseases. Acta Neuropathol. 1992;85:88–92. doi: 10.1007/bf00304637. [DOI] [PubMed] [Google Scholar]
- 193.Garbuglia M, Verzini M, Sorci G, Bianchi R, Giambanco I, Agneletti AL, Donato R. The calcium-modulated proteins, S100A1 and S100B, as potential regulators of the dynamics of type III intermediate filaments. Braz J Med Biol Res. 1999;32:1177–1185. doi: 10.1590/S0100-879X.1999.001000001. [DOI] [PubMed] [Google Scholar]
- 194.Garcia M, Pujol A, Ruzo A, Riu E, Ruberte J, Arbos A, Serafin A, Albella B, Feliu JE, Bosch F. Phosphofructo-1-kinase deficiency leads to a severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis. PLoS Genet. 2009;5:e1000615. doi: 10.1371/journal.pgen.1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gaussin V, Tomlinson JE, Depre C, Engelhardt S, Antos CL, Takagi G, Hein L, Topper JN, Liggett SB, Olson EN, Lohse MJ, Vatner SF, Vatner DE. Common genomic response in different mouse models of β-adrenergic-induced cardiomyopathy. Circulation. 2003;108:2926–2933. doi: 10.1161/01.cir.000010.1922.18151.7b. [DOI] [PubMed] [Google Scholar]
- 196.Gautel M. The sarcomeric cytoskeleton: Who picks up the strain? Curr Opin Cell Biol. 2011;23:39–46. doi: 10.1016/j.ceb.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 197.Gautel M, Furst DO, Cocco A, Schiaffino S. Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: lack of isoform transcomplementation in cardiac muscle. Circ Res. 1998;82:124–129. doi: 10.1161/01.res.82.1.124. [DOI] [PubMed] [Google Scholar]
- 198.Gautel M, Goulding D, Bullard B, Weber K, Furst DO. The central Z-disk region of titin is assembled from a novel repeat in variable copy numbers. J Cell Sci. 1996;109:2747–2754. doi: 10.1242/jcs.109.11.2747. [DOI] [PubMed] [Google Scholar]
- 199.Geeves MA, Hitchcock-DeGregori SE, Gunning PW. A systematic nomenclature for mammalian tropomyosin isoforms. J Muscle Res Cell Motil. 2015;36:147–153. doi: 10.1007/s10974-014-9389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gehrig SM, Ryall JG, Schertzer JD, Lynch GS. Insulin-like growth factor-I analogue protects muscles of dystrophic mdx mice from contraction-mediated damage. Exp Physiol. 2008;93:1190–1198. doi: 10.1113/expphysiol.2008.042838. [DOI] [PubMed] [Google Scholar]
- 201.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: A β cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-I. [DOI] [PubMed] [Google Scholar]
- 202.Genini M, Schwalbe P, Scholl FA, Remppis A, Mattei MG, Schafer BW. Subtractive cloning and characterization of DRAL, a novel LIM-domain protein down-regulated in rhabdomyosarcoma. DNA Cell Biol. 1997;16:433–442. doi: 10.1089/dna.1997.16.433. [DOI] [PubMed] [Google Scholar]
- 203.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Gross-mann KS, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 204.Giepmans BN. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233–245. doi: 10.1016/j.cardiores.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 205.Gilbert R, Cohen JA, Pardo S, Basu A, Fischman DA. Identification of the A-band localization domain of myosin binding proteins C and H (MyBP-C, MyBP-H) in skeletal muscle. J Cell Sci. 1999;112:69–79. doi: 10.1242/jcs.112.1.69. [DOI] [PubMed] [Google Scholar]
- 206.Gilbert R, Kelly MG, Mikawa T, Fischman DA. The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. J Cell Sci. 1996;109:101–111. doi: 10.1242/jcs.109.1.101. [DOI] [PubMed] [Google Scholar]
- 207.Gilbert R, Nalbantoglu J, Petrof BJ, Ebihara S, Guibinga GH, Tinsley JM, Kamen A, Massie B, Davies KE, Karpati G. Adenovirus-mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum Gene Ther. 1999;10:1299–1310. doi: 10.1089/10430349950017987. [DOI] [PubMed] [Google Scholar]
- 208.Gingras AR, Bate N, Goult BT, Hazelwood L, Canestrelli I, Grossmann JG, Liu H, Putz NS, Roberts GC, Volkmann N, Hanein D, Barsukov IL, Critchley DR. The structure of the C-terminal actin-binding domain of talin. Embo j. 2008;27:458–469. doi: 10.1038/sj.emboj.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gokhin DS, Fowler VM. Cytoplasmic γ-actin and tropomodulin isoforms link to the sarcoplasmic reticulum in skeletal muscle fibers. J Cell Biol. 2011;194:105–120. doi: 10.1083/jcb.2010.11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gokhin DS, Fowler VM. A two-segment model for thin filament architecture in skeletal muscle. Nat Rev Mol Cell Biol. 2013;14:113–119. doi: 10.1038/nrm3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gokhin DS, Lewis RA, McKeown CR, Nowak RB, Kim NE, Littlefield RS, Lieber RL, Fowler VM. Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. J Cell Biol. 2010;189:95–109. doi: 10.1083/jcb.2010.01125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gokhin DS, Ochala J, Domenighetti AA, Fowler VM. Tropomod-ulin1 directly controls thin filament length in both wild-type and tropomodulin4-deficient skeletal muscle. Development. 2015;142:4351–4362. doi: 10.1242/dev.129171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino-Mora C, Sivakumar K, Dalakas MC. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–403. doi: 10.1038/1300. [DOI] [PubMed] [Google Scholar]
- 214.Gontier Y, Taivainen A, Fontao L, Sonnenberg A, van der Flier A, Carpen O, Faulkner G, Borradori L. The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sar-colemma via muscle-specific filamins. J Cell Sci. 2005;118:3739–3749. doi: 10.1242/jcs.02484. [DOI] [PubMed] [Google Scholar]
- 215.Good MC, Zalatan JG, Lim WA. Scaffold proteins: Hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- 217.Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol. 2009;1:1–19. doi: 10.1101/cshperspect.a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gosselin LE, Williams JE. Pentoxifylline fails to attenuate fibrosis in dystrophic (mdx) diaphragm muscle. Muscle Nerve. 2006;33:820–823. doi: 10.1002/mus.20523. [DOI] [PubMed] [Google Scholar]
- 219.Graham ZA, Gallagher PM, Cardozo CP. Focal adhesion kinase and its role in skeletal muscle. J Muscle Res Cell Motil. 2015;36:305–315. doi: 10.1007/s10974-015-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Granger BL, Lazarides E. Desmin and vimentin coexist at the periphery of the myofibril Z disc. Cell. 1979;18:1053–1063. doi: 10.1016/0092-8674(79)90218-6. [DOI] [PubMed] [Google Scholar]
- 221.Granger BL, Lazarides E. Synemin: a new high molecular weight protein associated with desmin and vimentin filaments in muscle. Cell. 1980;22:727–738. doi: 10.1016/0092-8674(80)90549-8. [DOI] [PubMed] [Google Scholar]
- 222.Granzier H, Kellermayer M, Helmes M, Trombitás K. Titin elasticity and mechanism of passive force development in rat cardiac myocytes probed by thin-filament extraction. Biophys J. 1997;73:2043–2053. doi: 10.1016/S0006-3495(97)78234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschope C, McNabb M, Larson DF, Labeit S, Gotthardt M. Truncation of titin’s elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ Res. 2009;105:557–564. doi: 10.1161/circresaha.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Green KJ, Simpson CL. Desmosomes: New perspectives on a classic. J Invest Dermatol. 2007;127:2499–2515. doi: 10.1038/sj.jid.5701015. [DOI] [PubMed] [Google Scholar]
- 225.Gregorio CC, Trombitas K, Centner T, Kolmerer B, Stier G, Kunke K, Suzuki K, Obermayr F, Herrmann B, Granzier H, Sorimachi H, Labeit S. The NH2 terminus of titin spans the Z-disc: Its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J Cell Biol. 1998;143:1013–1027. doi: 10.1083/jcb.143.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Griggs R, Vihola A, Hackman P, Talvinen K, Haravuori H, Faulkner G, Eymard B, Richard I, Selcen D, Engel A, Carpen O, Udd B. Zaspopathy in a large classic late-onset distal myopathy family. Brain. 2007;130:1477–1484. doi: 10.1093/brain/awm006. [DOI] [PubMed] [Google Scholar]
- 227.Grove BK, Cerny L, Perriard JC, Eppenberger HM, Thornell LE. Fiber type-specific distribution of M-band proteins in chicken muscle. J Histochem Cytochem. 1989;37:447–454. doi: 10.1177/37.4.2926123. [DOI] [PubMed] [Google Scholar]
- 228.Gruen M, Gautel M. Mutations in β-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286:933–949. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- 229.Gunning PW, Ghoshdastider U, Whitaker S, Popp D, Robinson RC. The evolution of compositionally and functionally distinct actin filaments. J Cell Sci. 2015;128:2009–2019. doi: 10.1242/jcs.165563. [DOI] [PubMed] [Google Scholar]
- 230.Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem. 2008;283:10135–10146. doi: 10.1074/jbc.M710277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Gurnett CA, Desruisseau DM, McCall K, Choi R, Meyer ZI, Talerico M, Miller SE, Ju JS, Pestronk A, Connolly AM, Druley TE, Weihl CC, Dobbs MB. Myosin binding protein C1: A novel gene for autosomal dominant distal arthrogryposis type 1. Hum Mol Genet. 2010;19:1165–1173. doi: 10.1093/hmg/ddp587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, Williamson RA, Chen J, Lin CI, Lin JJ. Loss of mXinα, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol. 2007;293:H2680–H2692. doi: 10.1152/ajpheart.00806.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Guy PM, Kenny DA, Gill GN. The PDZ domain of the LIM protein enigma binds to β-tropomyosin. Mol Biol Cell. 1999;10:1973–1984. doi: 10.1091/mbc.10.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Guyon JR, Kudryashova E, Potts A, Dalkilic I, Brosius MA, Thompson TG, Beckmann JS, Kunkel LM, Spencer MJ. Calpain 3 cleaves filamin C and regulates its ability to interact with γ- and δ-sarcoglycans. Muscle Nerve. 2003;28:472–483. doi: 10.1002/mus.10465. [DOI] [PubMed] [Google Scholar]
- 235.Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De Seze J, Labeit S, Witt C, Peltonen L, Richard I, Udd B. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492–500. doi: 10.1086/342380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of β-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 237.Hall TE, Bryson-Richardson RJ, Berger S, Jacoby AS, Cole NJ, Hollway GE, Berger J, Currie PD. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin α2-deficient congenital muscular dystrophy. Proc Natl Acad Sci U S A. 2007;104:7092–7097. doi: 10.1073/pnas.0700942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hancock CR, Brault JJ, Terjung RL. Protecting the cellular energy state during contractions: Role of AMP deaminase. J Physiol Pharmacol. 2006;57:17–29. [PubMed] [Google Scholar]
- 239.Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, Trembath RC, Shackleton S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol. 2006;26:3738–3751. doi: 10.1128/mcb.26(10)3738-3751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Haravuori H, Vihola A, Straub V, Auranen M, Richard I, Marchand S, Voit T, Labeit S, Somer H, Peltonen L, Beckmann JS, Udd B. Secondary calpain3 deficiency in 2q-linked muscular dystrophy: Titin is the candidate gene. Neurology. 2001;56:869–877. doi: 10.1212/WNL.56.7.869. [DOI] [PubMed] [Google Scholar]
- 241.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 242.Harrison BC, Allen DL, Leinwand LA. IIb or not IIb? Regulation of myosin heavy chain gene expression in mice and men. Skelet Muscle. 2011;1:1–9. doi: 10.1186/2044-5040-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Hart MC, Cooper JA. Vertebrate isoforms of actin capping protein β have distinct functions in vivo. J Cell Biol. 1999;147:1287–1298. doi: 10.1083/jcb.147.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hart MC, Korshunova YO, Cooper JA. Vertebrates have conserved capping protein α isoforms with specific expression patterns. Cell Motil Cytoskeleton. 1997;38:120–132. doi: 10.1002/(sici)1097-0169.199738:2<120::aid-cm2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 245.Hartman TJ, Martin JL, Solaro RJ, Samarel AM, Russell B. CapZ dynamics are altered by endothelin-1 and phenylephrine via PIP2-and PKC-dependent mechanisms. Am J Physiol Cell Physiol. 2009;296:C1034–C1039. doi: 10.1152/ajpcell.00544.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Hartzell HC. Effects of phosphorylated and unphosphorylated C-protein on cardiac actomyosin ATPase. J Mol Biol. 1985;186:185–195. doi: 10.1016/0022-2836(85)90268-2. [DOI] [PubMed] [Google Scholar]
- 247.Hauerslev S, Sveen M-L, Duno M, Angelini C, Vissing J, Krag TO. Calpain 3 is important for muscle regeneration: Evidence from patients with limb girdle muscular dystrophies. BMC Musculoskelet Disord. 2012;13:1–11. doi: 10.1186/1471-2474-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Haugaa KH, Haland TF, Leren IS, Saberniak J, Edvardsen T. Arrhythmogenic right ventricular cardiomyopathy, clinical manifestations, and diagnosis. Europace. 2015;18:965–972. doi: 10.1093/europace/euv340. [DOI] [PubMed] [Google Scholar]
- 249.Hayashi C, Ono Y, Doi N, Kitamura F, Tagami M, Mineki R, Arai T, Taguchi H, Yanagida M, Hirner S, Labeit D, Labeit S, Sorimachi H. Multiple molecular interactions implicate the connectin/titin N2A region as a modulating scaffold for p94/calpain 3 activity in skeletal muscle. J Biol Chem. 2008;283:14801–14814. doi: 10.1074/jbc.M708262200. [DOI] [PubMed] [Google Scholar]
- 250.Hayashi T, Arimura T, Itoh-Satoh M, Ueda K, Hohda S, Inagaki N, Takahashi M, Hori H, Yasunami M, Nishi H, Koga Y, Nakamura H, Matsuzaki M, Choi BY, Bae SW, You CW, Han KH, Park JE, Knoll R, Hoshijima M, Chien KR, Kimura A. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2192–2201. doi: 10.1016/j.jacc.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 251.Hayashi YK, Chou FL, Engvall E, Ogawa M, Matsuda C, Hirabayashi S, Yokochi K, Ziober BL, Kramer RH, Kaufman SJ, Ozawa E, Goto Y, Nonaka I, Tsukahara T, Wang JZ, Hoffman EP, Arahata K. Mutations in the integrin α7 gene cause congenital myopathy. Nat Genet. 1998;19:94–97. doi: 10.1038/ng0598-94. [DOI] [PubMed] [Google Scholar]
- 252.Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci U S A. 2005;102:1655–1660. doi: 10.1073/pnas.0405488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tome FM, Schwartz K, Fardeau M, Tryggvason K, et al. Mutations in the laminin α 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- 254.Hemken PM, Bellin RM, Sernett SW, Becker B, Huiatt TW, Robson RM. Molecular characteristics of the novel intermediate filament protein paranemin. Sequence reveals EAP-300 and IFAPa-400 are highly homologous to paranemin. J Biol Chem. 1997;272:32489–32499. doi: 10.1074/jbc.272.51.32489. [DOI] [PubMed] [Google Scholar]
- 255.Henderson CA, Gregorio CC. Dynamics of actin in the heart: defining thin filament length. In: Ehler E, editor. Cardiac Cytoarchitecture. Springer:Springer; 2015. pp. 71–88. [Google Scholar]
- 256.Hentzen ER, Lahey M, Peters D, Mathew L, Barash IA, Friden J, Lieber RL. Stress-dependent and -independent expression of the myogenic regulatory factors and the MARP genes after eccentric contractions in rats. J Physiol. 2006;570:157–167. doi: 10.1113/jphysiol.2005.093005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Herrera AH, Elzey B, Law DJ, Horowits R. Terminal regions of mouse nebulin: sequence analysis and complementary localization with N-RAP. Cell Motil Cytoskeleton. 2000;45:211–222. doi: 10.1002/(sici)1097-0169(200003)45:3<211::aid-cm4>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 259.Herrmann H, Fouquet B, Franke WW. Expression of intermediate filament proteins during development of Xenopus laevis. II. Identification and molecular characterization of desmin. Development. 1989;105:299–307. doi: 10.1242/dev.105.2.299. [DOI] [PubMed] [Google Scholar]
- 260.Herron TJ, Rostkova E, Kunst G, Chaturvedi R, Gautel M, Kentish JC. Activation of myocardial contraction by the N-terminal domains of myosin binding protein-C. Circ Res. 2006;98:1290–1298. doi: 10.1161/01.RES.0000222059.54917.ef. [DOI] [PubMed] [Google Scholar]
- 261.Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, MacRae CA, Gerull B. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79:1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Higgs HN, Peterson KJ. Phylogenetic analysis of the formin homology 2 domain. Mol Biol Cell. 2005;16:1–13. doi: 10.1091/mbcE04-07.-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Hillier BJ, Christopherson KS, Prehoda KE, Bredt DS, Lim WA. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science. 1999;284:812–815. doi: 10.2210/pdb1qau/pdb. [DOI] [PubMed] [Google Scholar]
- 264.Himmel M, Van Der Ven PF, Stocklein W, Furst DO. The limits of promiscuity: Isoform-specific dimerization of filamins. Biochemistry. 2003;42:430–439. doi: 10.1021/bi026501+. [DOI] [PubMed] [Google Scholar]
- 265.Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda) 2007;22:73–80. doi: 10.1152/physiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- 266.Hitchcock-DeGregori SE. Tropomyosin: Function follows structure. Adv Exp Med Biol. 2008;644:60–72. doi: 10.1007/978-0-387-85766-4_5. [DOI] [PubMed] [Google Scholar]
- 267.Hnia K, Ramspacher C, Vermot J, Laporte J. Desmin in muscle and associated diseases: Beyond the structural function. Cell Tissue Res. 2015;360:591–608. doi: 10.1007/s00441-014-2016-4. [DOI] [PubMed] [Google Scholar]
- 268.Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: State of the art. Cardiovasc Res. 2015;105:397–408. doi: 10.1093/cvr/cvv025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 270.Hojayev B, Rothermel BA, Gillette TG, Hill JA. FHL2 binds calcineurin and represses pathological cardiac growth. Mol Cell Biol. 2012;32:4025–4034. doi: 10.1128/mcb05948-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Holmes WB, Moncman CL. Nebulette interacts with filamin C. Cell Motil Cytoskeleton. 2008;65:130–142. doi: 10.1002/cm.20249. [DOI] [PubMed] [Google Scholar]
- 272.Honda K, Yamada T, Endo R, Ino Y, Gotoh M, Tsuda H, Yamada Y, Chiba H, Hirohashi S. Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J Cell Biol. 1998;140:1383–1393. doi: 10.1083/jcb.140.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Honda S, Shirotani-Ikejima H, Tadokoro S, Tomiyama Y, Miyata T. The integrin-linked kinase-PINCH-parvin complex supports integrin αIIbβ3 activation. PLoS One. 2013;8:e85498. doi: 10.1371/journal.pone.0085498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hornemann T, Kempa S, Himmel M, Hayess K, Furst DO, Wallimann T. Muscle-type creatine kinase interacts with central domains of the M-band proteins myomesin and M-protein. J Mol Biol. 2003;332:877–887. doi: 10.1016/S0022-2836(03)00921-5. [DOI] [PubMed] [Google Scholar]
- 275.Houmeida A, Holt J, Tskhovrebova L, Trinick J. Studies of the interaction between titin and myosin. J Cell Biol. 1995;131:1471–1481. doi: 10.1083/jcb.131.6.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Howman EV, Sullivan N, Poon EP, Britton JE, Hilton-Jones D, Davies KE. Syncoilin accumulation in two patients with desmin-related myopathy. Neuromuscul Disord. 2003;13:42–48. doi: 10.1016/S0960-8966(02)00181-5. [DOI] [PubMed] [Google Scholar]
- 277.Hu L-YR, Ackermann MA, Kontrogianni-Konstantopoulos A. The sarcomeric M-Region: A molecular command center for diverse cellular processes. Biomed Res Int. 2015;2015:1–25. doi: 10.1155/2015/714197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Huang P, Zhao XS, Fields M, Ransohoff RM, Zhou L. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J. 2009;23:2539–2548. doi: 10.1096/fj09-129833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of β-catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 280.Huby AC, Mendsaikhan U, Takagi K, Martherus R, Wansapura J, Gong N, Osinska H, James JF, Kramer K, Saito K, Robbins J, Khuchua Z, Towbin JA, Purevjav E. Disturbance in Z-disk mechanosensitive proteins induced by a persistent mutant myopalladin causes familial restrictive cardiomyopathy. J Am Coll Cardiol. 2014;64:2765–2776. doi: 10.1016/j.jacc.2014.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Huxley H. The crossbridge mechanism of muscular contraction and its implications. J Exp Biol. 1985;115:17–30. doi: 10.1242/jeb.115.1.17. [DOI] [PubMed] [Google Scholar]
- 282.Huxley H, Hanson J. Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature. 1954;173:973–976. doi: 10.1038/173973a0. [DOI] [PubMed] [Google Scholar]
- 283.Huxley HE. Electron microscope studies on the structure of natural and synthetic protein filaments from striated muscle. J Mol Biol. 1963;7:281–308. doi: 10.1016/s0022-2836(63)80008-x. [DOI] [PubMed] [Google Scholar]
- 284.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 285.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 286.Ikeda K, Emoto N, Matsuo M, Yokoyama M. Molecular identification and characterization of a novel nuclear protein whose expression is up-regulated in insulin-resistant animals. J Biol Chem. 2003;278:3514–3520. doi: 10.1074/jbc.M204563200. [DOI] [PubMed] [Google Scholar]
- 287.Irving T, Wu Y, Bekyarova T, Farman GP, Fukuda N, Granzier H. Thick-filament strain and interfilament spacing in passive muscle: Effect of titin-based passive tension. Biophys J. 2011;100:1499–1508. doi: 10.1016/j.bpj.2011.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Isenberg G, Leonard K, Jockusch BM. Structural aspects of vinculin-actin interactions. J Mol Biol. 1982;158:231–249. doi: 10.1016/0022-2836(82)90431-4. [DOI] [PubMed] [Google Scholar]
- 289.Ishiguro N, Baba T, Ishida T, Takeuchi K, Osaki M, Araki N, Okada E, Takahashi S, Saito M, Watanabe M, Nakada C, Tsukamoto Y, Sato K, Ito K, Fukayama M, Mori S, Ito H, Moriyama M. Carp, a cardiac ankyrin-repeated protein, and its new homologue, Arpp, are differentially expressed in heart, skeletal muscle, and rhabdomyosarcomas. Am J Pathol. 2002;160:1767–1778. doi: 10.1016/s0002-9440(10)61123-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Iskratsch T, Lange S, Dwyer J, Kho AL, dos Remedios C, Ehler E. Formin follows function: A muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J Cell Biol. 2010;191:1159–1172. doi: 10.1083/jcb.2010.05060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Jahn D, Schramm S, Schnolzer M, Heilmann CJ, de Koster CG, Schutz W, Benavente R, Alsheimer M. A truncated lamin A in the Lmna−/− mouse line: Implications for the understanding of laminopathies. Nucleus. 2012;3:463–474. doi: 10.4161/nucl.21676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Jaka O, Casas-Fraile L, Lopez de Munain A, Saenz A. Costamere proteins and their involvement in myopathic processes. Expert Rev Mol Med. 2015;17:e12–e23. doi: 10.1017/erm.2015.9. [DOI] [PubMed] [Google Scholar]
- 293.Jefferson JJ, Ciatto C, Shapiro L, Liem RK. Structural analysis of the plakin domain of bullous pemphigoid antigen1 (BPAG1) suggests that plakins are members of the spectrin superfamily. J Mol Biol. 2007;366:244–257. doi: 10.1016/j.jmb.2006.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Johannessen M, Moller S, Hansen T, Moens U, Van Ghelue M. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell Mol Life Sci. 2006;63:268–284. doi: 10.1007/s00018-005-5438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on β-dystroglycan. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- 296.Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: Architectural integration of energy production and utilization. Circ Res. 2001;89:153–159. doi: 10.1161/hh1401.093440. [DOI] [PubMed] [Google Scholar]
- 297.Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin: DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 298.Kampourakis T, Yan Z, Gautel M, Sun YB, Irving M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc Natl Acad Sci U S A. 2014;111:18763–18768. doi: 10.1073/pnas.1413922112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Kan OM, Takeya R, Abe T, Kitajima N, Nishida M, Tominaga R, Kurose H, Sumimoto H. Mammalian formin Fhod3 plays an essential role in cardiogenesis by organizing myofibrillogenesis. Biol Open. 2012;1:889–896. doi: 10.1242/bio.2012.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Karolczak J, Sobczak M, Majewski L, Yeghiazaryan M, Jakubiec-Puka A, Ehler E, Slawinska U, Wilczynski GM, Redowicz MJ. Myosin VI in skeletal muscle: its localization in the sarcoplasmic reticulum, neuromuscular junction and muscle nuclei. Histochem Cell Biol. 2013;139:873–885. doi: 10.1007/s00418-012-1070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Kartenbeck J, Franke WW, Moser JG, Stoffels U. Specific attachment of desmin filaments to desmosomal plaques in cardiac myocytes. Emboj. 1983;2:735–742. doi: 10.1002/j.1460-2075.1983.tb01493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Kartenbeck J, Schwechheimer K, Moll R, Franke WW. Attachment of vimentin filaments to desmosomal plaques in human meningiomal cells and arachnoidal tissue. J Cell Biol. 1984;98:1072–1081. doi: 10.1083/jcb.98.3.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Kato T, Muraski J, Chen Y, Tsujita Y, Wall J, Glembotski CC, Schaefer E, Beckerle M, Sussman MA. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J Clin Invest. 2005;115:2716–2730. doi: 10.1172/jci24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Katrukha IA. Human cardiac troponin complex. Structure and functions. Biochemistry (Mosc) 2013;78:1447–1465. doi: 10.1134/s0006297913130063. [DOI] [PubMed] [Google Scholar]
- 305.Kaufman SJ, Foster RF. Replicating myoblasts express a muscle-specific phenotype. Proc Natl Acad Sci U S A. 1988;85:9606–9610. doi: 10.1073/pnas.85.24.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Kaushik G, Spenlehauer A, Sessions AO, Trujillo AS, Fuhrmann A, Fu Z, Venkatraman V, Pohl D, Tuler J, Wang M, Lakatta EG, Ocorr K, Bodmer R, Bernstein SI, Van Eyk JE, Cammarato A, Engler AJ. Vinculin network-mediated cytoskeletal remodeling regulates contractile function in the aging heart. Sci Transl Med. 2015;7:292ra299. doi: 10.1126/scitranslmed.aaa5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kazmierski ST, Antin PB, Witt CC, Huebner N, McElhinny AS, Labeit S, Gregorio CC. The complete mouse nebulin gene sequence and the identification of cardiac nebulin. J Mol Biol. 2003;328:835–846. doi: 10.1016/S0022-2836(03)00348-6. [DOI] [PubMed] [Google Scholar]
- 308.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci U S A. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Keira Y, Noguchi S, Minami N, Hayashi YK, Nishino I. Localization of calpain 3 in human skeletal muscle and its alteration in limb-girdle muscular dystrophy 2A muscle. J Biochem. 2003;133:659–664. doi: 10.1093/jb/mvg084. [DOI] [PubMed] [Google Scholar]
- 310.Keller A, Demeurie J, Merkulova T, Geraud G, Cywiner-Golenzer C, Lucas M, Chatelet FP. Fibre-type distribution and subcellular localisation of α and β enolase in mouse striated muscle. Biol Cell. 2000;92:527–535. doi: 10.1016/S0248-4900(00)01103-5. [DOI] [PubMed] [Google Scholar]
- 311.Kemp TJ, Sadusky TJ, Saltisi F, Carey N, Moss J, Yang SY, Sassoon DA, Goldspink G, Coulton GR. Identification of Ankrd2, a novel skeletal muscle gene coding for a stretch-responsive ankyrin-repeat protein. Genomics. 2000;66:229–241. doi: 10.1006/geno.2000.6213. [DOI] [PubMed] [Google Scholar]
- 312.Khairallah M, Khairallah R, Young ME, Dyck JR, Petrof BJ, Des Rosiers C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J Mol Cell Cardiol. 2007;43:119–129. doi: 10.1016/j.yjmcc.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 313.Kielbasa OM, Reynolds JG, Wu CL, Snyder CM, Cho MY, Weiler H, Kandarian S, Naya FJ. Myospryn is a calcineurin-interacting protein that negatively modulates slow-fiber-type transformation and skeletal muscle regeneration. FASEB J. 2011;25:2276–2286. doi: 10.1096/fj10-169219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Kirby TJ, Chaillou T, McCarthy JJ. The role of microRNAs in skeletal muscle health and disease. Front Biosci (Landmark Ed) 2015;20:37–77. doi: 10.2741/4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kley RA, Hellenbroich Y, van der Ven PF, Furst DO, Huebner A, Bruchertseifer V, Peters SA, Heyer CM, Kirschner J, Schroder R, Fischer D, Muller K, Tolksdorf K, Eger K, Germing A, Brodherr T, Reum C, Walter MC, Lochmuller H, Ketelsen UP, Vorgerd M. Clinical and morphological phenotype of the filamin myopathy: A study of 31 German patients. Brain. 2007;130:3250–3264. doi: 10.1093/brain/awm271. [DOI] [PubMed] [Google Scholar]
- 316.Knoll R, Buyandelger B. Z-disc transcriptional coupling, sarcom-eroptosis and mechanoptosis. Cell Biochem Biophys. 2013;66:65–71. doi: 10.1007/s12013-012-9430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Knoll R, Buyandelger B, Lab M. The sarcomeric Z-disc and Z-discopathies. J Biomed Biotechnol. 2011;2011:1–12. doi: 10.1155/2011/569628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/S0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 319.Knoll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kotter S, Babicz K, Sohns M, Miocic S, Didie M, Knoll G, Zimmermann WH, Thelen P, Bickeboller H, Maier LS, Schaper W, Schaper J, Kraft T, Tschope C, Linke WA, Chien KR. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. 2010;106:695–704. doi: 10.1161/circresaha.109.206243. [DOI] [PubMed] [Google Scholar]
- 320.Kobayashi T, Jin L, de Tombe PP. Cardiac thin filament regulation. Pflugers Arch. 2008;457:37–46. doi: 10.1007/s00424-008-0511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Koebis M, Ohsawa N, Kino Y, Sasagawa N, Nishino I, Ishiura S. Alternative splicing of myomesin 1 gene is aberrantly regulated in myotonic dystrophy type 1. Genes Cells. 2011;16:961–972. doi: 10.1111/j1365-2443.2011.01542.x. [DOI] [PubMed] [Google Scholar]
- 322.Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, Meng G, Müller CR, Lindlöf M, Kaariainen H, de la Chapelle A, Kiuru A, Savontaus ML, Gilgenkrantz H, Récan D, Chelly J, Kaplan JC, Covone AE, Archidiacono N, Romeo G, Liechti-Gallati S, Schneider V, Braga S, Moser H, Darras BT, Murphy P, Francke U, Chen JD, Morgan G, Denton M, Greenberg CR, Wrogemann K, Blonden LAJ, van Paassen HMB, van Ommen GJB, Kunkel LM. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am J Hum Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 323.Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- 324.Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 325.Kojic S, Nestorovic A, Rakicevic L, Belgrano A, Stankovic M, Divac A, Faulkner G. A novel role for cardiac ankyrin repeat protein Ankrd1/CARP as a co-activator of the p53 tumor suppressor protein. Arch Biochem Biophys. 2010;502:60–67. doi: 10.1016/j.abb.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 326.Kolb J, Li F, Methawasin M, Adler M, Escobar YN, Nedrud J, Pappas CT, Harris SP, Granzier H. Thin filament length in the cardiac sarcomere varies with sarcomere length but is independent of titin and nebulin. J Mol Cell Cardiol. 2016;30:286–294. doi: 10.1016/j.yjmcc.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Kong Y, Flick MJ, Kudla AJ, Konieczny SF. Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Mol Cell Biol. 1997;17:4750–4760. doi: 10.1128/mcb.17.8.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Kong Y, Shelton JM, Rothermel B, Li X, Richardson JA, Bassel-Duby R, Williams RS. Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to β-adrenergic stimulation. Circulation. 2001;103:2731–2738. doi: 10.1161/01.CIR.103.22.2731. [DOI] [PubMed] [Google Scholar]
- 329.Konieczny P, Fuchs P, Reipert S, Kunz WS, Zeold A, Fischer I, Paulin D, Schroder R, Wiche G. Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J Cell Biol. 2008;181:667–681. doi: 10.1083/jcb.2007.11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ. Muscle giants: Molecular scaffolds in sarcomerogenesis. Physiol Rev. 2009;89:1217–1267. doi: 10.1152/physrev.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Kontrogianni-Konstantopoulos A, Bloch RJ. The hydrophilic domain of small ankyrin-1 interacts with the two N-terminal immunoglobulin domains of titin. J Biol Chem. 2003;278:3985–3991. doi: 10.1074/jbc.M209012200. [DOI] [PubMed] [Google Scholar]
- 332.Kontrogianni-Konstantopoulos A, Catino DH, Strong JC, Sutter S, Borisov AB, Pumplin DW, Russell MW, Bloch RJ. Obscurin modulates the assembly and organization of sarcomeres and the sarcoplasmic reticulum. FASEB J. 2006;20:2102–2111. doi: 10.1096/fj06-5761c.om. [DOI] [PubMed] [Google Scholar]
- 333.Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005;96:346–354. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- 334.Kostyukova AS. Tropomodulins and tropomodulin/tropomyosin interactions. Cell Mol Life Sci. 2008;65:563–569. doi: 10.1007/s00018-007-7347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kotaka M, Kostin S, Ngai S, Chan K, Lau Y, Lee SM, Li H, Ng EK, Schaper J, Tsui SK, Fung K, Lee C, Waye MM. Interaction of hCLIM1, an enigma family protein, with α-actinin 2. J Cell Biochem. 2000;78:558–565. doi: 10.1002/1097-4644(20000915)78:4<558::AID-JCB5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 336.Kotaka M, Lau YM, Cheung KK, Lee SM, Li HY, Chan WY, Fung KP, Lee CY, Waye MM, Tsui SK. Elfin is expressed during early heart development. J Cell Biochem. 2001;83:463–472. doi: 10.1002/jcb.1244. [DOI] [PubMed] [Google Scholar]
- 337.Kouloumenta A, Mavroidis M, Capetanaki Y. Proper perinuclear localization of the TRIM-like protein myospryn requires its binding partner desmin. J Biol Chem. 2007;282:35211–35221. doi: 10.1074/jbc.M704733200. [DOI] [PubMed] [Google Scholar]
- 338.Krcmery J, Camarata T, Kulisz A, Simon HG. Nucleocytoplasmic functions of the PDZ-LIM protein family: New insights into organ development. Bioessays. 2010;32:100–108. doi: 10.1002/bies.2009.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Kremneva E, Makkonen MH, Skwarek-Maruszewska A, Gateva G, Michelot A, Dominguez R, Lappalainen P. Cofilin-2 controls actin filament length in muscle sarcomeres. Dev Cell. 2014;31:215–226. doi: 10.1016/j.devcel.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Krenz M, Robbins J. Impact of β-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. 2004;44:2390–2397. doi: 10.1016/j.jacc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 341.Kruger M, Kotter S. Titin, a central mediator for hypertrophic signaling, exercise-induced mechanosignaling and skeletal muscle remodeling. Front Physiol. 2016;7:1–8. doi: 10.3389/fphys.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Kruger M, Wright J, Wang K. Nebulin as a length regulator of thin filaments of vertebrate skeletal muscles: Correlation of thin filament length, nebulin size, and epitope profile. J Cell Biol. 1991;115:97–107. doi: 10.1083/jcb.115.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Kruger O, Maxeiner S, Kim JS, van Rijen HV, de Bakker JM, Eckardt D, Tiemann K, Lewalter T, Ghanem A, Luderitz B, Willecke K. Cardiac morphogenetic defects and conduction abnormalities in mice homozygously deficient for connexin40 and heterozygously deficient for connexin45. J Mol Cell Cardiol. 2006;41:787–797. doi: 10.1016/j.yjmcc.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 344.Kulikovskaya I, McClellan G, Flavigny J, Carrier L, Winegrad S. Effect of MyBP-C binding to actin on contractility in heart muscle. J Gen Physiol. 2003;122:761–774. doi: 10.1085/jgp.2003.08941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Kumar A, Crawford K, Close L, Madison M, Lorenz J, Doetschman T, Pawlowski S, Duffy J, Neumann J, Robbins J, Boivin GP, O’Toole BA, Lessard JL. Rescue of cardiac α-actin-deficient mice by enteric smooth muscle γ-actin. Proc Natl Acad Sci U S A. 1997;94:4406–4411. doi: 10.1073/pnas.94.9.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Kumar A, Khandelwal N, Malya R, Reid MB, Boriek AM. Loss of dystrophin causes aberrant mechanotransduction in skeletal muscle fibers. FASEB J. 2004;18:102–113. doi: 10.1096/fj03-0453c.om. [DOI] [PubMed] [Google Scholar]
- 347.Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res. 2000;86:51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- 348.Kuo H, Chen J, Ruiz-Lozano P, Zou Y, Nemer M, Chien KR. Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development. 1999;126:4223–4234. doi: 10.1242/dev.126.19.4223. [DOI] [PubMed] [Google Scholar]
- 349.Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Gill GN, Kikkawa U. Protein-protein interaction of zinc finger LIM domains with protein kinase C. J Biol Chem. 1996;271:31029–31032. doi: 10.1074/jbc.271.49.31029. [DOI] [PubMed] [Google Scholar]
- 350.Labeit S, Gautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. Embo j. 1992;11:1711–1716. doi: 10.1002/j.1460-2075.1992.tb05222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Labeit S, Kolmerer B. The complete primary structure of human nebulin and its correlation to muscle structure. J Mol Biol. 1995;248:308–315. doi: 10.1016/S0022-2836(95)80052-2. [DOI] [PubMed] [Google Scholar]
- 352.Labeit S, Kolmerer B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 353.Labeit S, Lahmers S, Burkart C, Fong C, McNabb M, Witt S, Witt C, Labeit D, Granzier H. Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. J Mol Biol. 2006;362:664–681. doi: 10.1016/j.jmb.2006.07.077. [DOI] [PubMed] [Google Scholar]
- 354.Laing NG, Clarke NF, Dye DE, Liyanage K, Walker KR, Kobayashi Y, Shimakawa S, Hagiwara T, Ouvrier R, Sparrow JC, Nishino I, North KN, Nonaka I. Actin mutations are one cause of congenital fibre type disproportion. Ann Neurol. 2004;56:689–694. doi: 10.1002/ana.20260. [DOI] [PubMed] [Google Scholar]
- 355.Laing NG, Nowak KJ. When contractile proteins go bad: the sarcomere and skeletal muscle disease. Bioessays. 2005;27:809–822. doi: 10.1002/bies.20269. [DOI] [PubMed] [Google Scholar]
- 356.Lange S, Auerbach D, McLoughlin P, Perriard E, Schafer BW, Perriard JC, Ehler E. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. J Cell Sci. 2002;115:4925–4936. doi: 10.1242/jcs.00181. [DOI] [PubMed] [Google Scholar]
- 357.Lange S, Himmel M, Auerbach D, Agarkova I, Hayess K, Fürst DO, Perriard J-C, Ehler E. Dimerisation of myomesin: Implications for the structure of the sarcomeric M-band. J Mol Biol. 2005;345:289–298. doi: 10.1016/j.jmb.2004.10.040. [DOI] [PubMed] [Google Scholar]
- 358.Lange S, Ouyang K, Meyer G, Cui L, Cheng H, Lieber RL, Chen J. Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J Cell Sci. 2009;122:2640–2650. doi: 10.1242/jcs.046193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Larsen TH, Dalen H, Sommer JR, Boyle R, Lieberman M. Membrane skeleton in cultured chick cardiac myocytes revealed by high resolution immunocytochemistry. Histochem Cell Biol. 1999;112:307–316. doi: 10.1007/s004180050452. [DOI] [PubMed] [Google Scholar]
- 360.Laure L, Daniele N, Suel L, Marchand S, Aubert S, Bourg N, Roudaut C, Duguez S, Bartoli M, Richard I. A new pathway encompassing calpain 3 and its newly identified substrate cardiac ankyrin repeat protein is involved in the regulation of the nuclear factor-kappaB pathway in skeletal muscle. Febs j. 2010;277:4322–4337. doi: 10.1111/j1742-4658.2010.07820.x. [DOI] [PubMed] [Google Scholar]
- 361.Lawlor MW, Ottenheijm CA, Lehtokari V-L, Cho K, Pelin K, Wallgren-Pettersson C, Granzier H, Beggs AH. Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet Muscle. 2011;1:1–12. doi: 10.1186/2044-5040-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Lazarides E. The distribution of desmin (100 Å) filaments in primary cultures of embryonic chick cardiac cells. Exp Cell Res. 1978;112:265–273. doi: 10.1016/0014-4827(78)90209-4. [DOI] [PubMed] [Google Scholar]
- 363.Leavis PC, Gergely J. Thin filament proteins and thin filament-linked regulation of vertebrate muscle contraction. CRC Crit Rev Biochem. 1984;16:235–305. doi: 10.3109/10409238409108717. [DOI] [PubMed] [Google Scholar]
- 364.Lebakken CS, Venzke DP, Hrstka RF, Consolino CM, Faulkner JA, Williamson RA, Campbell KP. Sarcospan-deficient mice maintain normal muscle function. Mol Cell Biol. 2000;20:1669–1677. doi: 10.1128/mcb.20(5)1669-1677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, Geeves MA, Van Eyk JE, Tobacman LS, Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J Mol Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 366.Lehman W, Rosol M, Tobacman LS, Craig R. Troponin organization on relaxed and activated thin filaments revealed by electron microscopy and three-dimensional reconstruction. J Mol Biol. 2001;307:739–744. doi: 10.1006/jmbi.2001.4514. [DOI] [PubMed] [Google Scholar]
- 367.Lehti M, Kivela R, Komi P, Komulainen J, Kainulainen H, Kyrolainen H. Effects of fatiguing jumping exercise on mRNA expression of titin-complex proteins and calpains. J Appl Physiol 1985. 2009;106:1419–1424. doi: 10.1152/japplphysiol.90660.2008. [DOI] [PubMed] [Google Scholar]
- 368.Lek M, Quinlan KG, North KN. The evolution of skeletal muscle performance: gene duplication and divergence of human sarcomeric α-actinins. Bioessays. 2010;32:17–25. doi: 10.1002/bies.2009.00110. [DOI] [PubMed] [Google Scholar]
- 369.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 370.LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation. 2013;127:938–944. doi: 10.1161/circulationaha.112.139717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Li B, Zhuang L, Trueb B. Zyxin interacts with the SH3 domains of the cytoskeletal proteins LIM-nebulette and Lasp-1. J Biol Chem. 2004;279:20401–20410. doi: 10.1074/jbc.M310304200. [DOI] [PubMed] [Google Scholar]
- 372.Li H, Choudhary SK, Milner DJ, Munir MI, Kuisk IR, Capetanaki Y. Inhibition of desmin expression blocks myoblast fusion and interferes with the myogenic regulators MyoD and myogenin. J Cell Biol. 1994;124:827–841. doi: 10.1083/jcb.124.5.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Li H, Linke WA, Oberhauser AF, Carrion-Vazquez M, Kerkvliet JG, Lu H. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 374.Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, Shang X, De Rycke R, van Roy F, Radice GL. Loss of αT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J Cell Sci. 2012;125:1058–1067. doi: 10.1242/jcs.098640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Li M, Andersson-Lendahl M, Sejersen T, Arner A. Knockdown of fast skeletal myosin-binding protein C in zebrafish results in a severe skeletal myopathy. J Gen Physiol. 2016;147:309–322. doi: 10.1085/jgp.2015.11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Li Z, Colucci-Guyon E, Pincon-Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol. 1996;175:362–366. doi: 10.1006/dbio.1996.0122. [DOI] [PubMed] [Google Scholar]
- 377.Li Z, Parlakian A, Coletti D, Alonso-Martin S, Hourde C, Joanne P, Gao-Li J, Blanc J, Ferry A, Paulin D, Xue Z, Agbulut O. Synemin acts as a regulator of signalling molecules during skeletal muscle hypertrophy. J Cell Sci. 2014;127:4589–4601. doi: 10.1242/jcs.143164. [DOI] [PubMed] [Google Scholar]
- 378.Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, Tome FMS, Fardeau M, Jackson CE, Beckmann JS, Campbell KP. β-sarcoglycan: Characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11:257–265. doi: 10.1016/0960-8966(96)88965-6. [DOI] [PubMed] [Google Scholar]
- 379.Lim MS, Sutherland C, Walsh MP. Phosphorylation of bovine cardiac C-protein by protein kinase C. Biochem Biophys Res Commun. 1985;132:1187–1195. doi: 10.1016/0006-291x(85)91932-1. [DOI] [PubMed] [Google Scholar]
- 380.Lin Y-H, Warren CM, Li J, McKinsey TA, Russell B. Myofibril growth during cardiac hypertrophy is regulated through dual phosphorylation and acetylation of the actin capping protein CapZ. Cell Signal. 2016;28:1015–1024. doi: 10.1016/j.cellsig.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Lin YH, Li J, Swanson ER, Russell B. CapZ and actin capping dynamics increase in myocytes after a bout of exercise and abates in hours after stimulation ends. J Appl Physiol 1985. 2013;114:1603–1609. doi: 10.1152/japplphysiol.01283.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Lin YH, Swanson ER, Li J, Mkrtschjan MA, Russell B. Cyclic mechanical strain of myocytes modifies CapZβ1 post translationally via PKCε; J Muscle Res Cell Motil. 2015;36:329–337. doi: 10.1007/s10974-015-9420-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Linden M, Li Z, Paulin D, Gotow T, Leterrier JF. Effects of desmin gene knockout on mice heart mitochondria. J Bioenerg Biomembr. 2001;33:333–341. doi: 10.1023/A:1010611408007. [DOI] [PubMed] [Google Scholar]
- 384.Lindstrom NO, Neves C, McIntosh R, Miedzybrodzka Z, Vargesson N, Collinson JM. Tissue specific characterisation of Lim-kinase 1 expression during mouse embryogenesis. Gene Expr Patterns. 2011;11:221–232. doi: 10.1016/j.gep.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Res. 2014;114:1052–1068. doi: 10.1161/circresaha.114.301286. [DOI] [PubMed] [Google Scholar]
- 386.Linke WA, Ivemeyer M, Labeit S, Hinssen H, Rüegg JC, Gautel M. Actin-titin interaction in cardiac myofibrils: Probing a physiological role. Biophys J. 1997;73:905–919. doi: 10.1016/S0006-3495(97)78123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Linke WA, Ivemeyer M, Olivieri N, Kolmerer B, Rüegg CJ, Labeit S. Towards a molecular understanding of the elasticity of titin. J Mol Biol. 1996;261:62–71. doi: 10.1006/jmbi.1996.0441. [DOI] [PubMed] [Google Scholar]
- 388.Linke WA, Rudy DE, Centner T, Gautel M, Witt C, Labeit S, Gregorio CC. I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J Cell Biol. 1999;146:631–644. doi: 10.1083/jcb.146.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Linnemann A, Vakeel P, Bezerra E, Orfanos Z, Djinovic-Carugo K, van der Ven PF, Kirfel G, Furst DO. Myopodin is an F-actin bundling protein with multiple independent actin-binding regions. J Muscle Res Cell Motil. 2013;34:61–69. doi: 10.1007/s10974-012-9334-5. [DOI] [PubMed] [Google Scholar]
- 390.Linnemann A, van der Ven PF, Vakeel P, Albinus B, Simonis D, Bendas G, Schenk JA, Micheel B, Kley RA, Furst DO. The sarcomeric Z-disc component myopodin is a multiadapter protein that interacts with filamin and α-actinin. Eur J Cell Biol. 2010;89:681–692. doi: 10.1016/j.ejcb.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 391.Littlefield R, Almenar-Queralt A, Fowler VM. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat Cell Biol. 2001;3:544–551. doi: 10.1038/35078517. [DOI] [PubMed] [Google Scholar]
- 392.Liu LA, Engvall E. Sarcoglycan isoforms in skeletal muscle. J Biol Chem. 1999;274:38171–38176. doi: 10.1074/jbc.274.53.38171. [DOI] [PubMed] [Google Scholar]
- 393.Loh NY, Ambrose HJ, Guay-Woodford LM, DasGupta S, Nawrotzki RA, Blake DJ, Davies KE. Genomic organization and refined mapping of the mouse β-dystrobrevin gene. Mamm Genome. 1998;9:857–862. doi: 10.1007/s003359900883. [DOI] [PubMed] [Google Scholar]
- 394.Lompre AM, Nadal-Ginard B, Mahdavi V. Expression of the cardiac ventricular α- and β-myosin heavy chain genes is developmentally and hormonally regulated. J Biol Chem. 1984;259:6437–6446. [PubMed] [Google Scholar]
- 395.Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Loong CK, Badr MA, Chase PB. Tropomyosin flexural rigidity and single ca(2+) regulatory unit dynamics: Implications for cooperative regulation of cardiac muscle contraction and cardiomyocyte hypertrophy. Front Physiol. 2012;3:1–10. doi: 10.3389/fphys.2012.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Lopes LR, Elliott PM. A straightforward guide to the sarcomeric basis of cardiomyopathies. Heart. 2014;100:1916–1923. doi: 10.1136/heartjnl-2014-305645. [DOI] [PubMed] [Google Scholar]
- 398.Louis HA, Pino JD, Schmeichel KL, Pomies P, Beckerle MC. Comparison of three members of the cysteine-rich protein family reveals functional conservation and divergent patterns of gene expression. J Biol Chem. 1997;272:27484–27491. doi: 10.1074/jbc.272.43.27484. [DOI] [PubMed] [Google Scholar]
- 399.Lovering RM, O’Neill A, Muriel JM, Prosser BL, Strong J, Bloch RJ. Physiology, structure, and susceptibility to injury of skeletal muscle in mice lacking keratin 19-based and desmin-based intermediate filaments. Am J Physiol Cell Physiol. 2011;300:C803–C813. doi: 10.1152/ajpcell.00394.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, Bristow MR. Changes in gene expression in the intact human heart. Downregulation of α-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997;100:2315–2324. doi: 10.1172/jci119770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Lu QW, Wu XY, Morimoto S. Inherited cardiomyopathies caused by troponin mutations. J Geriatr Cardiol. 2013;10:91–101. doi: 10.3724/SP.J.1263.2012.07131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Lun AS, Chen J, Lange S. Probing muscle ankyrin-repeat protein (MARP) structure and function. Anat Rec (Hoboken) 2014;297:1615–1629. doi: 10.1002/ar.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Luo G, Herrera AH, Horowits R. Molecular interactions of N-RAP, a nebulin-related protein of striated muscle myotendon junctions and intercalated disks. Biochemistry. 1999;38:6135–6143. doi: 10.1021/bi982395t. [DOI] [PubMed] [Google Scholar]
- 404.Luo G, Zhang JQ, Nguyen TP, Herrera AH, Paterson B, Horowits R. Complete cDNA sequence and tissue localization of N-RAP, a novel nebulin-related protein of striated muscle. Cell Motil Cytoskeleton. 1997;38:75–90. doi: 10.1002/(sici)1097-0169.199738:1<75::aid-cm7>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 405.Luther PK. The vertebrate muscle Z-disc: Sarcomere anchor for structure and signalling. J Muscle Res Cell Motil. 2009;30:171–185. doi: 10.1007/s10974-009-9189-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Luther PK, Craig R. Modulation of striated muscle contraction by binding of myosin binding protein C to actin. Bioarchitecture. 2011;1:277–283. doi: 10.4161/bioa.1.6.19341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Ma K, Wang K. Interaction of nebulin SH3 domain with titin PEVK and myopalladin: implications for the signaling and assembly role of titin and nebulin. FEBS Lett. 2002;532:273–278. doi: 10.1016/S0014-5793(02)03655-4. [DOI] [PubMed] [Google Scholar]
- 408.Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH, Selbach M, Barton PJ, Cook SA, Rajewsky N, Gotthardt M, Landthaler M, Hubner N. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Invest. 2014;124:3419–3430. doi: 10.1172/jci74523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.MacArthur DG, Seto JT, Raftery JM, Quinlan KG, Huttley GA, Hook JW, Lemckert FA, Kee AJ, Edwards MR, Berman Y, Hardeman EC, Gunning PW, Easteal S, Yang N, North KN. Loss of ACTN3 gene function alters mouse muscle metabolism and shows evidence of positive selection in humans. Nat Genet. 2007;39:1261–1265. doi: 10.1038/ng2122. [DOI] [PubMed] [Google Scholar]
- 410.Maiellaro-Rafferty K, Wansapura JP, Mendsaikhan U, Osinska H, James JF, Taylor MD, Robbins J, Kranias EG, Towbin JA, Purevjav E. Altered regional cardiac wall mechanics are associated with differential cardiomyocyte calcium handling due to nebulette mutations in preclinical inherited dilated cardiomyopathy. J Mol Cell Cardiol. 2013;60:151–160. doi: 10.1016/j.yjmcc.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Manisastry SM, Zaal KJ, Horowits R. Myofibril assembly visualized by imaging N-RAP, α-actinin, and actin in living cardiomyocytes. Exp Cell Res. 2009;315:2126–2139. doi: 10.1016/j.yexcr.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Manning J, O’Malley D. What has the mdx mouse model of Duchenne muscular dystrophy contributed to our understanding of this disease? J Muscle Res Cell Motil. 2015;36:155–167. doi: 10.1007/s10974-015-9406-4. [DOI] [PubMed] [Google Scholar]
- 413.Manso AM, Li R, Monkley SJ, Cruz NM, Ong S, Lao DH, Koshman YE, Gu Y, Peterson KL, Chen J, Abel ED, Samarel AM, Critchley DR, Ross RS. Talin1 has unique expression versus talin 2 in the heart and modifies the hypertrophic response to pressure overload. J Biol Chem. 2013;288:4252–4264. doi: 10.1074/jbc.M112.427484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Markert CD, Meaney MP, Voelker KA, Grange RW, Dalley HW, Cann JK, Ahmed M, Bishwokarma B, Walker SJ, Yu SX, Brown M, Lawlor MW, Beggs AH, Childers MK. Functional muscle analysis of the Tcap knockout mouse. Hum Mol Genet. 2010;19:2268–2283. doi: 10.1093/hmg/ddq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Marques MJ, Oggiam DS, Barbin IC, Ferretti R, Santo Neto H. Long-term therapy with deflazacort decreases myocardial fibrosis in mdx mice. Muscle Nerve. 2009;40:466–468. doi: 10.1002/mus.21341. [DOI] [PubMed] [Google Scholar]
- 416.Marrs JA, Andersson-Fisone C, Jeong MC, Cohen-Gould L, Zurzolo C, Nabi IR, Rodriguez-Boulan E, Nelson WJ. Plasticity in epithelial cell phenotype: Modulation by expression of different cadherin cell adhesion molecules. J Cell Biol. 1995;129:507–519. doi: 10.1083/jcb.129.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/circresaha.109.202440. [DOI] [PubMed] [Google Scholar]
- 418.Marston S, Montgiraud C, Munster AB, Copeland O, Choi O, Dos Remedios C, Messer AE, Ehler E, Knoll R. OBSCN mutations associated with dilated cardiomyopathy and haploinsufficiency. PLoS One. 2015;10:e0138568. doi: 10.1371/journal.pone.0138568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Martinsson T, Oldfors A, Darin N, Berg K, Tajsharghi H, Kyllerman M, Wahlstrom J. Autosomal dominant myopathy: Missense mutation (Glu-706 –> Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci U S A. 2000;97:14614–14619. doi: 10.1073/pnas.250289597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Maruyama K, Ebashi S. α-Actinin, a new structural protein from striated muscle. II. Action on actin. J Biochem. 1965;58:13–19. doi: 10.1093/oxfordjournals.jbchem.a128158. [DOI] [PubMed] [Google Scholar]
- 421.Mastrototaro G, Liang X, Li X, Carullo P, Piroddi N, Tesi C, Gu Y, Dalton ND, Peterson KL, Poggesi C, Sheikh F, Chen J, Bang ML. Nebulette knockout mice have normal cardiac function, but show Z-line widening and up-regulation of cardiac stress markers. Cardiovasc Res. 2015;107:216–225. doi: 10.1093/cvr/cvv156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Matsumura CY, Taniguti AP, Pertille A, Santo Neto H, Marques MJ. Stretch-activated calcium channel protein TRPC1 is correlated with the different degrees of the dystrophic phenotype in mdx mice. Am J Physiol Cell Physiol. 2011;301:C1344–C1350. doi: 10.1152/ajpcell.00056.2011. [DOI] [PubMed] [Google Scholar]
- 423.Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 424.Mayer U, Saher G, Fassler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Poschl E, von der Mark K. Absence of integrin α7 causes a novel form of muscular dystrophy. Nat Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- 425.McCullagh KJ, Edwards B, Poon E, Lovering RM, Paulin D, Davies KE. Intermediate filament-like protein syncoilin in normal and myopathic striated muscle. Neuromuscul Disord. 2007;17:970–979. doi: 10.1016/j.nmd.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 426.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002;157:125–136. doi: 10.1083/jcb.2001.08089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.McElhinny AS, Kolmerer B, Fowler VM, Labeit S, Gregorio CC. The N-terminal end of nebulin interacts with tropomodulin at the pointed ends of the thin filaments. J Biol Chem. 2001;276:583–592. doi: 10.1074/jbc.M005693200. [DOI] [PubMed] [Google Scholar]
- 428.McKay RT, Tripet BP, Hodges RS, Sykes BD. Interaction of the second binding region of troponin I with the regulatory domain of skeletal muscle troponin C as determined by NMR spectroscopy. J Biol Chem. 1997;272:28494–28500. doi: 10.1074/jbc.272.45.28494. [DOI] [PubMed] [Google Scholar]
- 429.McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature. 1986;319:463–468. doi: 10.1038/319463a0. [DOI] [PubMed] [Google Scholar]
- 430.McKeown CR, Nowak RB, Moyer J, Sussman MA, Fowler VM. Tropomodulin1 is required in the heart but not the yolk sac for mouse embryonic development. Circ Res. 2008;103:1241–1248. doi: 10.1161/circresaha.108.178749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/s0006-3495(93)81110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/s0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 433.McNally EM, Duggan D, Rafael Gorospe J, Bönnemann CG, Fanin M, Pegoraro E, Lidov HGW, Noguchi S, Ozawa E, Finkel RS, Cruse RP, Angelini C, Kunkel LM, Hoffman EP. Mutations that disrupt the carboxyl-terminus of γ-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5:1841–1847. doi: 10.1093/hmg/.5.11.1841. [DOI] [PubMed] [Google Scholar]
- 434.McNally EM, Ly CT, Kunkel LM. Human ε-sarcoglycan is highly related to α-sarcoglycan (adhalin), the limb girdle muscular dystrophy 2D gene1. FEBS Lett. 1998;422:27–32. doi: 10.1016/S0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- 435.McNally EM, Yoshida M, Mizuno Y, Ozawa E, Kunkel LM. Human adhalin is alternatively spliced and the gene is located on chromosome 17q21. Proc Natl Acad Sci U S A. 1994;91:9690–9694. doi: 10.1073/pnas.91.21.9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Meng W, Takeichi M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1:1–13. doi: 10.1101/cshperspect.a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/S0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 438.Mercuri E, Muntoni F. Muscular dystrophy: New challenges and review of the current clinical trials. Curr Opin Pediatr. 2013;25:701–707. doi: 10.1097/MOP.0b013e328365ace5. [DOI] [PubMed] [Google Scholar]
- 439.Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, Duff RM, Beckman K, de Visser M, van der Graaff MM, Hedera P, Fink JK, Petty EM, Lamont P, Fabian V, Bridges L, Voit T, Mastaglia FL, Laing NG. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1) Am J Hum Genet. 2004;75:703–708. doi: 10.1086/424760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Mermelstein CS, Andrade LR, Portilho DM, Costa ML. Desmin filaments are stably associated with the outer nuclear surface in chick myoblasts. Cell Tissue Res. 2006;323:351–357. doi: 10.1007/s00441-005-0063-6. [DOI] [PubMed] [Google Scholar]
- 441.Mertens C, Kuhn C, Franke WW. Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol. 1996;135:1009–1025. doi: 10.1083/jcb.135.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Mi-Mi L, Votra S, Kemphues K, Bretscher A, Pruyne D. Z-line formins promote contractile lattice growth and maintenance in striated muscles of C. elegans. J Cell Biol. 2012;198:87–102. doi: 10.1083/jcb.2012.02053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Mikhailov AT, Torrado M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. Int J Dev Biol. 2008;52:811–821. doi: 10.1387/ijdb.082655am. [DOI] [PubMed] [Google Scholar]
- 444.Milam LM. Electron microscopy of rotary shadowed vinculin and vinculin complexes. J Mol Biol. 1985;184:543–545. doi: 10.1016/0022-2836(85)90301-8. [DOI] [PubMed] [Google Scholar]
- 445.Miller G, Wang EL, Nassar KL, Peter AK, Crosbie RH. Structural and functional analysis of the sarcoglycan-sarcospan subcomplex. Exp Cell Res. 2007;313:639–651. doi: 10.1016/j.yexcr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Miller MK, Bang M-L, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol. 2003;333:951–964. doi: 10.1016/j.jmb.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 447.Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–1298. doi: 10.1083/jcb.150.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996;134:1255–1270. doi: 10.1083/jcb.134.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Miosge N, Klenczar C, Herken R, Willem M, Mayer U. Organization of the myotendinous junction is dependent on the presence of α7β1 integrin. Lab Invest. 1999;79:1591–1599. [PubMed] [Google Scholar]
- 450.Mizuno Y, Thompson TG, Guyon JR, Lidov HG, Brosius M, Imamura M, Ozawa E, Watkins SC, Kunkel LM. Desmuslin, an intermediate filament protein that interacts with α-dystrobrevin and desmin. Proc Natl Acad Sci U S A. 2001;98:6156–6161. doi: 10.1073/pnas.111153298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Mogensen J, Hey T, Lambrecht S. A systematic review of phenotypic features associated with cardiac troponin i mutations in hereditary cardiomyopathies. Can J Cardiol. 2015;31:1377–1385. doi: 10.1016/j.cjca..2015.06.015. [DOI] [PubMed] [Google Scholar]
- 452.Mogensen J, Perrot A, Andersen PS, Havndrup O, Klausen IC, Chris-tiansen M, Bross P, Egeblad H, Bundgaard H, Osterziel KJ, Haltern G, Lapp H, Reinecke P, Gregersen N, Borglum AD. Clinical and genetic characteristics of α cardiac actin gene mutations in hypertrophic cardiomyopathy. J Med Genet. 2004;41:1–5. doi: 10.1136/jmg.2003.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Mohiddin SA, Ahmed ZM, Griffith AJ, Tripodi D, Friedman TB, Fananapazir L, Morell RJ. Novel association of hypertrophic cardiomyopathy, sensorineural deafness, and a mutation in unconventional myosin VI (MYO6) J Med Genet. 2004;41:309–314. doi: 10.1136/jmg.2003.01.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Mohiddin SA, Lu S, Cardoso JP, Carroll S, Jha S, Horowits R, Fananapazir L. Genomic organization, alternative splicing, and expression of human and mouse N-RAP, a nebulin-related LIM protein of striated muscle. Cell Motil Cytoskeleton. 2003;55:200–212. doi: 10.1002/cm.10123. [DOI] [PubMed] [Google Scholar]
- 455.Molnar I, Migh E, Szikora S, Kalmar T, Vegh AG, Deak F, Barko S, Bugyi B, Orfanos Z, Kovacs J, Juhasz G, Varo G, Nyitrai M, Sparrow J, Mihaly J. DAAM is required for thin filament formation and Sarcom-erogenesis during muscle development in Drosophila. PLoS Genet. 2014;10:e1004166. doi: 10.1371/journal.pgen.1004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Molony L, McCaslin D, Abernethy J, Paschal B, Burridge K. Properties of talin from chicken gizzard smooth muscle. J Biol Chem. 1987;262:7790–7795. [PubMed] [Google Scholar]
- 457.Moncman CL, Wang K. Nebulette: A 107 kD nebulin-like protein in cardiac muscle. Cell Motil Cytoskeleton. 1995;32:205–225. doi: 10.1002/cm.970320305. [DOI] [PubMed] [Google Scholar]
- 458.Moncman CL, Wang K. Targeted disruption of nebulette protein expression alters cardiac myofibril assembly and function. Exp Cell Res. 2002;273:204–218. doi: 10.1006/excr.2001.5423. [DOI] [PubMed] [Google Scholar]
- 459.Monkley SJ, Pritchard CA, Critchley DR. Analysis of the mammalian talin2 gene TLN2. Biochem Biophys Res Commun. 2001;286:880–885. doi: 10.1006/bbrc.2001.5497. [DOI] [PubMed] [Google Scholar]
- 460.Monkley SJ, Zhou XH, Kinston SJ, Giblett SM, Hemmings L, Priddle H, Brown JE, Pritchard CA, Critchley DR, Fassler R. Disruption of the talin gene arrests mouse development at the gastrulation stage. Dev Dyn. 2000;219:560–574. doi: 10.1002/1097-0177.2000.9999:9999<::aiddvdy1079>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 461.Moos C, Mason CM, Besterman JM, Feng IN, Dubin JH. The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1. J Mol Biol. 1978;124:571–586. doi: 10.1016/0022-2836(78)90172-9. [DOI] [PubMed] [Google Scholar]
- 462.Moos C, Offer G, Starr R, Bennett P. Interaction of C-protein with myosin, myosin rod and light meromyosin. J Mol Biol. 1975;97:1–9. doi: 10.1016/s0022-2836(75)80017-9. [DOI] [PubMed] [Google Scholar]
- 463.Moreira ES, Wiltshire TJ, Faulkner G, Nilforoushan A, Vainzof M, Suzuki OT, Valle G, Reeves R, Zatz M, Passos-Bueno MR, Jenne DE. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet. 2000;24:163–166. doi: 10.1038/72822. [DOI] [PubMed] [Google Scholar]
- 464.Morgan MJ, Madgwick AJ. The LIM proteins FHL1 and FHL3 are expressed differently in skeletal muscle. Biochem Biophys Res Commun. 1999;255:245–250. doi: 10.1006/bbrc.1999.0179. [DOI] [PubMed] [Google Scholar]
- 465.Morgan MJ, Madgwick AJ. Slim defines a novel family of LIM-proteins expressed in skeletal muscle. Biochem Biophys Res Commun. 1996;225:632–638. doi: 10.1006/bbrc.1996.1222. [DOI] [PubMed] [Google Scholar]
- 466.Moriscot AS, Baptista IL, Bogomolovas J, Witt C, Hirner S, Granzier H, Labeit S. MuRF1 is a muscle fiber-type II associated factor and together with MuRF2 regulates type-II fiber trophicity and maintenance. J Struct Biol. 2010;170:344–353. doi: 10.1016/j.jsb.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Moriyama M, Tsukamoto Y, Fujiwara M, Kondo G, Nakada C, Baba T, Ishiguro N, Miyazaki A, Nakamura K, Hori N, Sato K, Shomori K, Takeuchi K, Satoh H, Mori S, Ito H. Identification of a novel human ankyrin-repeated protein homologous to CARP. Biochem Biophys Res Commun. 2001;285:715–723. doi: 10.1006/bbrc.2001.5216. [DOI] [PubMed] [Google Scholar]
- 468.Moulik M, Vatta M, Witt SH, Arola AM, Murphy RT, McKenna WJ, Boriek AM, Oka K, Labeit S, Bowles NE, Arimura T, Kimura A, Towbin JA. ANKRD1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. J Am Coll Cardiol. 2009;54:325–333. doi: 10.1016/j.jacc.2009.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Mouton J, Loos B, Moolman-Smook JC, Kinnear CJ. Ascribing novel functions to the sarcomeric protein, myosin binding protein H (MyBPH) in cardiac sarcomere contraction. Exp Cell Res. 2015;331:338–351. doi: 10.1016/j.yexcr.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 470.Mu Y, Jing R, Peter AK, Lange S, Lin L, Zhang J, Ouyang K, Fang X, Veevers J, Zhou X, Evans SM, Cheng H, Chen J. Cypher and Enigma homolog protein are essential for cardiac development and embryonic survival. J Am Heart Assoc. 2015;4:e001950–e001961. doi: 10.1161/jaha.115.00.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 472.Mues A, van der Ven PF, Young P, Furst DO, Gautel M. Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Lett. 1998;428:111–114. doi: 10.1016/S0014-5793(98)00501-8. [DOI] [PubMed] [Google Scholar]
- 473.Muhle-Goll C, Habeck M, Cazorla O, Nilges M, Labeit S, Granzier H. Structural and functional studies of titin’s fn3 modules reveal conserved surface patterns and binding to myosin S1—a possible role in the Frank-Starling mechanism of the heart. J Mol Biol. 2001;313:431–447. doi: 10.1006/jmbi.2001.5017. [DOI] [PubMed] [Google Scholar]
- 474.Murphy AC, Young PW. The actinin family of actin cross-linking proteins—a genetic perspective. Cell Biosci. 2015;5:1–9. doi: 10.1186/s13.578-015-0029-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Muthuchamy M, Grupp IL, Grupp G, O’Toole BA, Kier AB, Boivin GP, Neumann J, Wieczorek DF. Molecular and physiological effects of overexpressing striated muscle β-tropomyosin in the adult murine heart. J Biol Chem. 1995;270:30593–30603. doi: 10.1074/jbc.270.51.30593. [DOI] [PubMed] [Google Scholar]
- 476.Muthuchamy M, Pajak L, Howles P, Doetschman T, Wieczorek DF. Developmental analysis of tropomyosin gene expression in embryonic stem cells and mouse embryos. Mol Cell Biol. 1993;13:3311–3323. doi: 10.1128/MCB.13.6.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155–162. doi: 10.1161/01.cir.0000135591.37759.af. [DOI] [PubMed] [Google Scholar]
- 478.Nakada C, Tsukamoto Y, Oka A, Nonaka I, Sato K, Mori S, Ito H, Moriyama M. Altered expression of ARPP protein in skeletal muscles of patients with muscular dystrophy, congenital myopathy and spinal muscular atrophy. Pathobiology. 2004;71:43–51. doi: 10.1159/000072961. [DOI] [PubMed] [Google Scholar]
- 479.Nakae Y, Hirasaka K, Goto J, Nikawa T, Shono M, Yoshida M, Stoward PJ. Subcutaneous injection, from birth, of epigallocatechin-3-gallate, a component of green tea, limits the onset of muscular dystrophy in mdx mice: A quantitative histological, immunohistochemical and electrophysiological study. Histochem Cell Biol. 2008;129:489–501. doi: 10.1007/s00418-008-0390-2. [DOI] [PubMed] [Google Scholar]
- 480.Nakamura K, Nakada C, Takeuchi K, Osaki M, Shomori K, Kato S, Ohama E, Sato K, Fukayama M, Mori S, Ito H, Moriyama M. Altered expression of cardiac ankyrin repeat protein and its homologue, ankyrin repeat protein with PEST and proline-rich region, in atrophic muscles in amyotrophic lateral sclerosis. Pathobiology. 2002;70:197–203. doi: 10.1159/000069329. [DOI] [PubMed] [Google Scholar]
- 481.Nakao T. Fine structure of the myotendinous junction and “terminal coupling” in the skeletal muscle of the lamprey, Lampetra japonica. Anat Rec. 1975;182:321–337. doi: 10.1002/ar.1091820306. [DOI] [PubMed] [Google Scholar]
- 482.Nakao T. Some observations on the fine structure of the myotendinous junction in myotomal muscle of the tadpole tail. Cell Tissue Res. 1976;166:241–254. doi: 10.1007/bf00227045. [DOI] [PubMed] [Google Scholar]
- 483.Nawata J, Ohno I, Isoyama S, Suzuki J, Miura S, Ikeda J, Shirato K. Differential expression of α1, α3 and α5 integrin subunits in acute and chronic stages of myocardial infarction in rats. Cardiovasc Res. 1999;43:371–381. doi: 10.1016/s0008-6363(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 484.Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333–1341. doi: 10.1161/01.CIR.0000029803.93022.93. [DOI] [PubMed] [Google Scholar]
- 485.Neiva-Sousa M, Almeida-Coelho J, Falcao-Pires I, Leite-Moreira AF. Titin mutations: The fall of Goliath. Heart Fail Rev. 2015;20:579–588. doi: 10.1007/s10741-015-9495-6. [DOI] [PubMed] [Google Scholar]
- 486.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Nelson WJ, Traub P. Proteolysis of vimentin and desmin by the Ca2+-activated proteinase specific for these intermediate filament proteins. Mol Cell Biol. 1983;3:1146–1156. doi: 10.1128/mcb.3.6.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Newey SE, Benson MA, Ponting CP, Davies KE, Blake DJ. Alternative splicing of dystrobrevin regulates the stoichiometry of syntrophin binding to the dystrophin protein complex. Curr Biol. 2000;10:1295–1298. doi: 10.1016/s0960-9822(00)00760-0. [DOI] [PubMed] [Google Scholar]
- 489.Newey SE, Howman EV, Ponting CP, Benson MA, Nawrotzki R, Loh NY, Davies KE, Blake DJ. Syncoilin, a novel member of the intermediate filament superfamily that interacts with α-dystrobrevin in skeletal muscle. J Biol Chem. 2001;276:6645–6655. doi: 10.1074/jbc.M008305200. [DOI] [PubMed] [Google Scholar]
- 490.Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-g. [DOI] [PubMed] [Google Scholar]
- 491.Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG, Seidman CE. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257. doi: 10.1056/nejm19980430338.1802. [DOI] [PubMed] [Google Scholar]
- 492.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–369. doi: 10.1172/jci19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Nilles LA, Parry DA, Powers EE, Angst BD, Wagner RM, Green KJ. Structural analysis and expression of human desmoglein: A cadherin-like component of the desmosome. J Cell Sci. 1991;99:809–821. doi: 10.1242/jcs.99.4.809. [DOI] [PubMed] [Google Scholar]
- 494.Nilsson MI, Nissar AA, Al-Sajee D, Tarnopolsky MA, Parise G, Lach B, Furst DO, van der Ven PF, Kley RA, Hawke TJ. Xin is a marker of skeletal muscle damage severity in myopathies. Am J Pathol. 2013;183:1703–1709. doi: 10.1016/j.ajpath.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 495.Nix DA, Beckerle MC. Nuclear-cytoplasmic shuttling of the focal contact protein, zyxin: A potential mechanism for communication between sites of cell adhesion and the nucleus. J Cell Biol. 1997;138:1139–1147. doi: 10.1083/jcb.138.5.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM, van Rijen HV. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23–31. doi: 10.1016/j.yjmcc.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 497.Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 498.North KN, Yang N, Wattanasirichaigoon D, Mills M, Easteal S, Beggs AH. A common nonsense mutation results in α-actinin-3 deficiency in the general population. Nat Genet. 1999;21:353–354. doi: 10.1038/7675. [DOI] [PubMed] [Google Scholar]
- 499.Nowak KJ, Davis MR, Wallgren-Pettersson C, Lamont PJ, Laing NG. Clinical utility gene card for: Nemaline myopathy - update 2015. Eur J Hum Genet. 2015;23:e1–e5. doi: 10.1038/ejhg.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Nowak KJ, Ravenscroft G, Laing NG. Skeletal muscle α-actin diseases (actinopathies): Pathology and mechanisms. Acta Neuropathol. 2013;125:19–32. doi: 10.1007/s00401-012-1019-z. [DOI] [PubMed] [Google Scholar]
- 501.Nunoue K, Ohashi K, Okano I, Mizuno K. LIMK-1 and LIMK-2, two members of a LIM motif-containing protein kinase family. Oncogene. 1995;11:701–710. [PubMed] [Google Scholar]
- 502.Nworu CU, Kraft R, Schnurr DC, Gregorio CC, Krieg PA. Leiomodin 3 and tropomodulin 4 have overlapping functions during skeletal myofib-rillogenesis. J Cell Sci. 2015;128:239–250. doi: 10.1242/jcs.152702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.O’Neill A, Williams MW, Resneck WG, Milner DJ, Capetanaki Y, Bloch RJ. Sarcolemmal organization in skeletal muscle lacking desmin: Evidence for cytokeratins associated with the membrane skeleton at costameres. Mol Biol Cell. 2002;13:2347–2359. doi: 10.1091/mbc01-12.-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Oakes PW, Gardel ML. Stressing the limits of focal adhesion mechanosensitivity. Curr Opin Cell Biol. 2014;30:68–73. doi: 10.1016/j.ceb.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Obermann W, Gautel M, Steiner F, Van der Ven P, Weber K, Fürst DO. The structure of the sarcomeric M band: Localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. J Cell Biol. 1996;134:1441–1453. doi: 10.1083/jcb.134.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Obermann WM, Plessmann U, Weber K, Furst DO. Purification and biochemical characterization of myomesin, a myosin-binding and titin-binding protein, from bovine skeletal muscle. Eur J Biochem. 1995;233:110–115. doi: 10.1111/j1432-1033.1995.110_1.x. [DOI] [PubMed] [Google Scholar]
- 507.Ockeloen CW, Gilhuis HJ, Pfundt R, Kamsteeg EJ, Agrawal PB, Beggs AH, Dara Hama-Amin A, Diekstra A, Knoers NV, Lammens M, van Alfen N. Congenital myopathy caused by a novel missense mutation in the CFL2 gene. Neuromuscul Disord. 2012;22:632–639. doi: 10.1016/j.nmd.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Offer G, Moos C, Starr R. A new protein of the thick filaments of vertebrate skeletal myofibrils: Extraction, purification and characterization. J Mol Biol. 1973;74:653–676. doi: 10.1016/0022-2836(73)90055-7. [DOI] [PubMed] [Google Scholar]
- 509.Ohashi K. Roles of cofilin in development and its mechanisms of regulation. Dev Growth Differ. 2015;57:275–290. doi: 10.1111/dgd.12213. [DOI] [PubMed] [Google Scholar]
- 510.Ojima K, Kawabata Y, Nakao H, Nakao K, Doi N, Kitamura F, Ono Y, Hata S, Suzuki H, Kawahara H. Dynamic distribution of muscle-specific calpain in mice has a key role in physical-stress adaptation and is impaired in muscular dystrophy. J Clin Invest. 2010;120:2672–2683. doi: 10.1172/jci40658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Ojima K, Lin ZX, Zhang ZQ, Hijikata T, Holtzer S, Labeit S, Sweeney HL, Holtzer H. Initiation and maturation of I-Z-I bodies in the growth tips of transfected myotubes. J Cell Sci. 1999;112:4101–4112. doi: 10.1242/jcs.112.22.4101. [DOI] [PubMed] [Google Scholar]
- 512.Ojima K, Ono Y, Doi N, Yoshioka K, Kawabata Y, Labeit S, Sorimachi H. Myogenic stage, sarcomere length, and protease activity modulate localization of muscle-specific calpain. J Biol Chem. 2007;282:14493–14504. doi: 10.1074/jbc.M610806200. [DOI] [PubMed] [Google Scholar]
- 513.Okagaki T, Weber FE, Fischman DA, Vaughan KT, Mikawa T, Reinach FC. The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J Cell Biol. 1993;123:619–626. doi: 10.1083/jcb.123.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Okano I, Hiraoka J, Otera H, Nunoue K, Ohashi K, Iwashita S, Hirai M, Mizuno K. Identification and characterization of a novel family of serine/threonine kinases containing two N-terminal LIM motifs. J Biol Chem. 1995;270:31321–31330. doi: 10.1074/jbc.270.52.31321. [DOI] [PubMed] [Google Scholar]
- 515.Olive M, Goldfarb L, Dagvadorj A, Sambuughin N, Paulin D, Li Z, Goudeau B, Vicart P, Ferrer I. Expression of the intermediate filament protein synemin in myofibrillar myopathies and other muscle diseases. Acta Neuropathol. 2003;106:1–7. doi: 10.1007/s00401-003-0695-0. [DOI] [PubMed] [Google Scholar]
- 516.Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, Fananapazir L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:1687–1694. doi: 10.1006/jmcc.2000.1204. [DOI] [PubMed] [Google Scholar]
- 517.Olsson MC, Palmer BM, Stauffer BL, Leinwand LA, Moore RL. Morphological and functional alterations in ventricular myocytes from male transgenic mice with hypertrophic cardiomyopathy. Circ Res. 2004;94:201–207. doi: 10.1161/01.res.0000111521.40760.18. [DOI] [PubMed] [Google Scholar]
- 518.Ong RW, AlSaman A, Selcen D, Arabshahi A, Yau KS, Ravenscroft G, Duff RM, Atkinson V, Allcock RJ, Laing NG. Novel cofilin-2 (CFL2) four base pair deletion causing nemaline myopathy. J Neurol Neurosurg Psychiatry. 2014;85:1058–1060. doi: 10.1136/jnnp-2014-307608. [DOI] [PubMed] [Google Scholar]
- 519.Ono Y, Ojima K, Shinkai-Ouchi F, Hata S, Sorimachi H. An eccentric calpain, CAPN3/p94/calpain-3. Biochimie. 2016;122:169–187. doi: 10.1016/j.biochi.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 520.Ono Y, Ojima K, Torii F, Takaya E, Doi N, Nakagawa K, Hata S, Abe K, Sorimachi H. Skeletal muscle-specific calpain is an intracellular Na+-dependent protease. J Biol Chem. 2010;285:22986–22998. doi: 10.1074/jbc.M110.126946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Ono Y, Torii F, Ojima K, Doi N, Yoshioka K, Kawabata Y, Labeit D, Labeit S, Suzuki K, Abe K, Maeda T, Sorimachi H. Suppressed disassembly of autolyzing p94/CAPN3 by N2A connectin/titin in a genetic reporter system. J Biol Chem. 2006;281:18519–18531. doi: 10.1074/jbc.M601029200. [DOI] [PubMed] [Google Scholar]
- 522.Ooshio T, Irie K, Morimoto K, Fukuhara A, Imai T, Takai Y. Involvement of LMO7 in the association of two cell-cell adhesion molecules, nectin and E-cadherin, through afadin and α-actinin in epithelial cells. J Biol Chem. 2004;279:31365–31373. doi: 10.1074/jbc.M401957200. [DOI] [PubMed] [Google Scholar]
- 523.Orzechowski M, Moore JR, Fischer S, Lehman W. Tropomyosin movement on F-actin during muscle activation explained by energy landscapes. Arch Biochem Biophys. 2014;545:63–68. doi: 10.1016/j.abb..2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Osinska HE, Lemanski LF. Immunofluorescent localization of desmin and vimentin in developing cardiac muscle of Syrian hamster. Anat Rec. 1989;223:406–413. doi: 10.1002/ar.1092230409. [DOI] [PubMed] [Google Scholar]
- 525.Osio A, Tan L, Chen SN, Lombardi R, Nagueh SF, Shete S, Roberts R, Willerson JT, Marian AJ. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2007;100:766–768. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Otey CA, Dixon R, Stack C, Goicoechea SM. Cytoplasmic Ig-domain proteins: Cytoskeletal regulators with a role in human disease. Cell Motil Cytoskeleton. 2009;66:618–634. doi: 10.1002/cm.20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Otey CA, Rachlin A, Moza M, Arneman D, Carpen O. The palladin/myotilin/myopalladin family of actin-associated scaffolds. Int Rev Cytol. 2005;246:31–58. doi: 10.1016/s0074-7696(05)46002-7. [DOI] [PubMed] [Google Scholar]
- 528.Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK. Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature. 2005;433:488–494. doi: 10.1038/nature03251. [DOI] [PubMed] [Google Scholar]
- 529.Ott EB, van den Akker NM, Sakalis PA, Gittenberger-de Groot AC, Te Velthuis AJ, Bagowski CP. The lim domain only protein 7 is important in zebrafish heart development. Dev Dyn. 2008;237:3940–3952. doi: 10.1002/dvdy.2.1807. [DOI] [PubMed] [Google Scholar]
- 530.Otten J, van der Ven PF, Vakeel P, Eulitz S, Kirfel G, Brandau O, Boesl M, Schrickel JW, Linhart M, Hayess K, Naya FJ, Milting H, Meyer R, Furst DO. Complete loss of murine Xin results in a mild cardiac phenotype with altered distribution of intercalated discs. Cardiovasc Res. 2010;85:739–750. doi: 10.1093/cvr/cvp345. [DOI] [PubMed] [Google Scholar]
- 531.Ottenheijm CA, Buck D, de Winter JM, Ferrara C, Piroddi N, Tesi C, Jasper JR, Malik FI, Meng H, Stienen GJ. Deleting exon 55 from the nebulin gene induces severe muscle weakness in a mouse model for nemaline myopathy. Brain. 2013;136:1718–1731. doi: 10.1093/brain/awt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Ottenheijm CA, Fong C, Vangheluwe P, Wuytack F, Babu GJ, Periasamy M, Witt CC, Labeit S, Granzier H. Sarcoplasmic reticulum calcium uptake and speed of relaxation are depressed in nebulin-free skeletal muscle. FASEB J. 2008;22:2912–2919. doi: 10.1096/fj07-104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Ottenheijm CA, Witt CC, Stienen GJ, Labeit S, Beggs AH, Granzier H. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum Mol Genet. 2009;18:2359–2369. doi: 10.1093/hmg/ddp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Otterbein LR, Graceffa P, Dominguez R. The crystal structure of uncomplexed actin in the ADP state. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 535.Ozawa E, Mizuno Y, Hagiwara Y, Sasaoka T, Yoshida M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve. 2005;32:563–576. doi: 10.1002/mus.20349. [DOI] [PubMed] [Google Scholar]
- 536.Ozawa E, Noguchi S, Mizuno Y, Hagiwara Y, Yoshida M. From dystrophinopathy to sarcoglycanopathy: Evolution of a concept of muscular dystrophy. Muscle Nerve. 1998;21:421–438. doi: 10.1002/(sici)1097-4598(199804)21:4<421::aid-mus1>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 537.Pallavicini A, Kojic S, Bean C, Vainzof M, Salamon M, Ievolella C, Bortoletto G, Pacchioni B, Zatz M, Lanfranchi G, Faulkner G, Valle G. Characterization of human skeletal muscle Ankrd2. Biochem Biophys Res Commun. 2001;285:378–386. doi: 10.1006/bbrc.2001.5131. [DOI] [PubMed] [Google Scholar]
- 538.Panaviene Z, Moncman CL. Linker region of nebulin family members plays an important role in targeting these molecules to cellular structures. Cell Tissue Res. 2007;327:353–369. doi: 10.1007/s00441-006-0305-2. [DOI] [PubMed] [Google Scholar]
- 539.Papa I, Astier C, Kwiatek O, Raynaud F, Bonnal C, Lebart MC, Roustan C, Benyamin Y. α-actinin-CapZ, an anchoring complex for thin filaments in Z-line. J Muscle Res Cell Motil. 1999;20:187–197. doi: 10.1023/A:1005489319058. [DOI] [PubMed] [Google Scholar]
- 540.Papalouka V, Arvanitis DA, Vafiadaki E, Mavroidis M, Papadodima SA, Spiliopoulou CA, Kremastinos DT, Kranias EG, Sanoudou D. Muscle LIM protein interacts with cofilin 2 and regulates F-actin dynamics in cardiac and skeletal muscle. Mol Cell Biol. 2009;29:6046–6058. doi: 10.1128/mcb00654-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Papathanasiou S, Rickelt S, Soriano ME, Schips TG, Maier HJ, Davos CH, Varela A, Kaklamanis L, Mann DL, Capetanaki Y. Tumor necrosis factor-α confers cardioprotection through ectopic expression of keratins K8 and K18. Nat Med. 2015;21:1076–1084. doi: 10.1038/nm.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Pappas CT, Bhattacharya N, Cooper JA, Gregorio CC. Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Mol Biol Cell. 2008;19:1837–1847. doi: 10.1091/mbcE07-07.-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Pappas CT, Bliss KT, Zieseniss A, Gregorio CC. The Nebulin family: An actin support group. Trends Cell Biol. 2011;21:29–37. doi: 10.1016/j.tcb.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Pappas CT, Krieg PA, Gregorio CC. Nebulin regulates actin filament lengths by a stabilization mechanism. J Cell Biol. 2010;189:859–870. doi: 10.1083/jcb.2010.01043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Pappas CT, Mayfield RM, Henderson C, Jamilpour N, Cover C, Hernandez Z, Hutchinson KR, Chu M, Nam KH, Valdez JM, Wong PK, Granzier HL, Gregorio CC. Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc Natl Acad Sci U S A. 2015;112:3573–3578. doi: 10.1073/pnas.1508273112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Parry DA, Squire JM. Structural role of tropomyosin in muscle regulation: Analysis of the x-ray diffraction patterns from relaxed and contracting muscles. J Mol Biol. 1973;75:33–55. doi: 10.1016/0022-2836(73)90527-5. [DOI] [PubMed] [Google Scholar]
- 547.Pashmforoush M, Pomies P, Peterson KL, Kubalak S, Ross J, Jr, Hefti A, Aebi U, Beckerle MC, Chien KR. Adult mice deficient in actinin-associated LIM-domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nat Med. 2001;7:591–597. doi: 10.1038/87920. [DOI] [PubMed] [Google Scholar]
- 548.Payne ET, Yasuda N, Bourgeois JM, Devries MC, Rodriguez MC, Yousuf J, Tarnopolsky MA. Nutritional therapy improves function and complements corticosteroid intervention in mdx mice. Muscle Nerve. 2006;33:66–77. doi: 10.1002/mus.20436. [DOI] [PubMed] [Google Scholar]
- 549.Peche V, Shekar S, Leichter M, Korte H, Schröder R, Schleicher M, Holak T, Clemen C, Ramanath-Y B, Pfitzer G. CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell Mol Life Sci. 2007;64:2702–2715. doi: 10.1007/s00018-007-7316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Peche VS, Holak TA, Burgute BD, Kosmas K, Kale SP, Wunderlich FT, Elhamine F, Stehle R, Pfitzer G, Nohroudi K. Ablation of cyclase-associated protein 2 (CAP2) leads to cardiomyopathy. Cell Mol Life Sci. 2013;70:527–543. doi: 10.1007/s00018-012-1142-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Peter AK, Marshall JL, Crosbie RH. Sarcospan reduces dystrophic pathology: Stabilization of the utrophin-glycoprotein complex. J Cell Biol. 2008;183:419–427. doi: 10.1083/jcb.2008.08027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Peter AK, Miller G, Crosbie RH. Disrupted mechanical stability of the dystrophin-glycoprotein complex causes severe muscular dystrophy in sarcospan transgenic mice. J Cell Sci. 2007;120:996–1008. doi: 10.1242/jcs.03360. [DOI] [PubMed] [Google Scholar]
- 553.Peter M, Kitten GT, Lehner CF, Vorburger K, Bailer SM, Maridor G, Nigg EA. Cloning and sequencing of cDNA clones encoding chicken lamins A and B1 and comparison of the primary structures of vertebrate A- and B-type lamins. J Mol Biol. 1989;208:393–404. doi: 10.1016/0022-2836(89)90504-4. [DOI] [PubMed] [Google Scholar]
- 554.Peters MF, O’Brien KF, Sadoulet-Puccio HM, Kunkel LM, Adams ME, Froehner SC. β-Dystrobrevin, a new member of the dystrophin family: Identification, cloning, and protein associations. J Biol Chem. 1997;272:31561–31569. doi: 10.1074/jbc.272.50.31561. [DOI] [PubMed] [Google Scholar]
- 555.Pfeffer G, Barresi R, Wilson IJ, Hardy SA, Griffin H, Hudson J, Elliott HR, Ramesh AV, Radunovic A, Winer JB, Vaidya S, Raman A, Busby M, Farrugia ME, Ming A, Everett C, Emsley HCA, Horvath R, Straub V, Bushby K, Lochmüller H, Chinnery PF, Sarkozy A. Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J Neurol Neurosurg Psychiatry. 2014;85:331–338. doi: 10.1136/jnnp-2012-304728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Piccolo F, Roberds SL, Jeanpierre M, Leturcq F, Azibi K, Beldjord C, Carrie A, Recan D, Chaouch M, Reghis A, El Kerch F, Sefiani A, Voit T, Merlini L, Collin H, Eymard B, Beckmann JS, Romero NB, Tome FMS, Fardeau M, Campbell KP, Kaplan JC. Primary adhalinopathy: A common cause of autosomal recessive muscular dystrophy of variable severity. Nat Genet. 1995;10:243–245. doi: 10.1038/ng0695-243. [DOI] [PubMed] [Google Scholar]
- 557.Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. IV: Coalescence and amalgamation of desmosomal and adhaerens junction components—late processes in mammalian heart development. Eur J Cell Biol. 2007;86:377–391. doi: 10.1016/j.ejcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 558.Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. VI. Different precursor structures in non-mammalian species. Eur J Cell Biol. 2008;87:413–430. doi: 10.1016/j.ejcb.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 559.Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/circulationaha.105.583674. [DOI] [PubMed] [Google Scholar]
- 560.Piluso G, Mirabella M, Ricci E, Belsito A, Abbondanza C, Servidei S, Puca AA, Tonali P, Puca GA, Nigro V. γ1- and γ2-Syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J Biol Chem. 2000;275:15851–15860. doi: 10.1074/jbc.M000439200. [DOI] [PubMed] [Google Scholar]
- 561.Pinotsis N, Petoukhov M, Lange S, Svergun D, Zou P, Gautel M, Wilmanns M. Evidence for a dimeric assembly of two titin/telethonin complexes induced by the telethonin C-terminus. J Struct Biol. 2006;155:239–250. doi: 10.1016/j.jsb.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 562.Pirani A, Vinogradova MV, Curmi PM, King WA, Fletterick RJ, Craig R, Tobacman LS, Xu C, Hatch V, Lehman W. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J Mol Biol. 2006;357:707–717. doi: 10.1016/j.jmb.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 563.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- 564.Ponting CP, Blake DJ, Davies KE, Kendrick-Jones J, Winder SJ. ZZ and TAZ: New putative zinc fingers in dystrophin and other proteins. Trends Biochem Sci. 1996;21:11–13. doi: 10.1016/0968-0004(96)80878-4. [DOI] [PubMed] [Google Scholar]
- 565.Poon E, Howman EV, Newey SE, Davies KE. Association of syncoilin and desmin: Linking intermediate filament proteins to the dystrophin-associated protein complex. J Biol Chem. 2002;277:3433–3439. doi: 10.1074/jbc.M105273200. [DOI] [PubMed] [Google Scholar]
- 566.Postel R, Vakeel P, Topczewski J, Knoll R, Bakkers J. Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin-ECM adhesion complex. Dev Biol. 2008;318:92–101. doi: 10.1016/j.ydbio.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 567.Potter JD, Sheng Z, Pan BS, Zhao J. A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. J Biol Chem. 1995;270:2557–2562. doi: 10.1074/jbc.270.6.2557. [DOI] [PubMed] [Google Scholar]
- 568.Price MG. Molecular analysis of intermediate filament cytoskeleton—a putative load-bearing structure. Am J Physiol. 1984;246:H566–H572. doi: 10.1152/ajpheart.1984.246.4.H566. [DOI] [PubMed] [Google Scholar]
- 569.Price MG. Skelemins: Cytoskeletal proteins located at the periphery of M-discs in mammalian striated muscle. J Cell Biol. 1987;104:1325–1336. doi: 10.1083/jcb.104.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Pring M, Evangelista M, Boone C, Yang C, Zigmond SH. Mechanism of formin-induced nucleation of actin filaments. Biochemistry. 2003;42:486–496. doi: 10.1021/bi026520j. [DOI] [PubMed] [Google Scholar]
- 571.Prosser BL, Wright NT, Hernandez-Ochoa EO, Varney KM, Liu Y, Olojo RO, Zimmer DB, Weber DJ, Schneider MF. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J Biol Chem. 2008;283:5046–5057. doi: 10.1074/jbc.M709231200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C. Role of formins in actin assembly: Nucleation and barbed-end association. Science. 2002;297:612–615. doi: 10.1126/science.1072309. [DOI] [PubMed] [Google Scholar]
- 573.Puckelwartz MJ, Depreux FF, McNally EM. Gene expression, chromosome position and lamin A/C mutations. Nucleus. 2011;2:162–167. doi: 10.4161/nucl.2.3.16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Purevjav E, Arimura T, Augustin S, Huby AC, Takagi K, Nunoda S, Kearney DL, Taylor MD, Terasaki F, Bos JM, Ommen SR, Shibata H, Takahashi M, Itoh-Satoh M, McKenna WJ, Murphy RT, Labeit S, Yamanaka Y, Machida N, Park JE, Alexander PM, Weintraub RG, Kitaura Y, Ackerman MJ, Kimura A, Towbin JA. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum Mol Genet. 2012;21:2039–2053. doi: 10.1093/hmg/dds022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Purevjav E, Varela J, Morgado M, Kearney DL, Li H, Taylor MD, Arimura T, Moncman CL, McKenna W, Murphy RT, Labeit S, Vatta M, Bowles NE, Kimura A, Boriek AM, Towbin JA. Nebulette mutations are associated with dilated cardiomyopathy and endocardial fibroelastosis. J Am Coll Cardiol. 2010;56:1493–1502. doi: 10.1016/j.jacc.2010.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Putilina T, Jaworski C, Gentleman S, McDonald B, Kadiri M, Wong P. Analysis of a human cDNA containing a tissue-specific alternatively spliced LIM domain. Biochem Biophys Res Commun. 1998;252:433–439. doi: 10.1006/bbrc.1998.9656. [DOI] [PubMed] [Google Scholar]
- 577.Quijano-Roy S, Mbieleu B, Bonnemann CG, Jeannet PY, Colomer J, Clarke NF, Cuisset JM, Roper H, De Meirleir L, D’Amico A, Ben Yaou R, Nascimento A, Barois A, Demay L, Bertini E, Ferreiro A, Sewry CA, Romero NB, Ryan M, Muntoni F, Guicheney P, Richard P, Bonne G, Estournet B. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. 2008;64:177–186. doi: 10.1002/ana.21417. [DOI] [PubMed] [Google Scholar]
- 578.Rao JN, Madasu Y, Dominguez R. Mechanism of actin filament pointed-end capping by tropomodulin. Science. 2014;345:463–467. doi: 10.1126/science.1256159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Rayes RF, Kalai T, Hideg K, Geeves MA, Fajer PG. Dynamics of tropomyosin in muscle fibers as monitored by saturation transfer EPR of bi-functional probe. PLoS One. 2011;6:e21277. doi: 10.1371/journal.pone.0021277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 581.Raynaud F, Astier C, Benyamin Y. Evidence for a direct but sequential binding of titin to tropomyosin and actin filaments. Biochim Biophys Acta. 2004;1700:171–178. doi: 10.1016/j.bbapap.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 582.Raynaud F, Fernandez E, Coulis G, Aubry L, Vignon X, Bleimling N, Gautel M, Benyamin Y, Ouali A. Calpain 1-titin interactions concentrate calpain 1 in the Z-band edges and in the N2-line region within the skeletal myofibril. Febs j. 2005;272:2578–2590. doi: 10.1111/j1742-4658.2005.04683.x. [DOI] [PubMed] [Google Scholar]
- 583.Rees MLJ, Lien C-F, Górecki DC. Dystrobrevins in muscle and non-muscle tissues. Neuromuscul Disord. 2007;17:123–134. doi: 10.1016/j.nmd.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 584.Reggiani C, Bottinelli R, Stienen GJ. Sarcomeric myosin isoforms: Fine tuning of a molecular motor. News Physiol Sci. 2000;15:26–33. doi: 10.1152/physiologyonline.2000.15.1.26. [DOI] [PubMed] [Google Scholar]
- 585.Reinach FC, Masaki T, Shafiq S, Obinata T, Fischman DA. Isoforms of C-protein in adult chicken skeletal muscle: Detection with monoclonal antibodies. J Cell Biol. 1982;95:78–84. doi: 10.1083/jcb.95.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Reipert S, Steinbock F, Fischer I, Bittner RE, Zeold A, Wiche G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res. 1999;252:479–491. doi: 10.1006/excr.1999.4626. [DOI] [PubMed] [Google Scholar]
- 587.Rethinasamy P, Muthuchamy M, Hewett T, Boivin G, Wolska BM, Evans C, Solaro RJ, Wieczorek DF. Molecular and physiological effects of α-tropomyosin ablation in the mouse. Circ Res. 1998;82:116–123. doi: 10.1161/01.RES.82.1.116. [DOI] [PubMed] [Google Scholar]
- 588.Ribeiro Ede A, Jr, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjoblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, Holt MR, Aachmann FL, Zagrovic B, Bordignon E, Pirker KF, Sver-gun DI, Gautel M, Djinovic-Carugo K. The structure and regulation of human muscle α-actinin. Cell. 2014;159:1447–1460. doi: 10.1016/j.cell.2014.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 590.Rickelt S, Pieperhoff S. Mutations with pathogenic potential in proteins located in or at the composite junctions of the intercalated disk connecting mammalian cardiomyocytes: A reference thesaurus for arrhythmogenic cardiomyopathies and for Naxos and Carvajal diseases. Cell Tissue Res. 2012;348:325–333. doi: 10.1007/s00441-012-1365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: A developmental study. Development. 1989;105:365–378. doi: 10.1242/dev.105.2.365. [DOI] [PubMed] [Google Scholar]
- 592.Roberds SL, Anderson RD, Ibraghimov-Beskrovnaya O, Campbell KP. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin) J Biol Chem. 1993;268:23739–23742. [PubMed] [Google Scholar]
- 593.Romero NB, Sandaradura SA, Clarke NF. Recent advances in nemaline myopathy. Curr Opin Neurol. 2013;26:519–526. doi: 10.1097/WCO.0b013e328364d681. [DOI] [PubMed] [Google Scholar]
- 594.Rosado M, Barber CF, Berciu C, Feldman S, Birren SJ, Nicastro D, Goode BL. Critical roles for multiple formins during cardiac myofibril development and repair. Mol Biol Cell. 2014;25:811–827. doi: 10.1091/mbcE13-08.-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Ruggiero A, Chen SN, Lombardi R, Rodriguez G, Marian AJ. Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovasc Res. 2013;97:44–54. doi: 10.1093/cvr/cvs294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW, Birchmeier W. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 1996;135:215–225. doi: 10.1083/jcb.135.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Ruppel KM, Spudich JA. Structure-function analysis of the motor domain of myosin. Annu Rev Cell Dev Biol. 1996;12:543–573. doi: 10.1146/annurev.cellbio.12.1.543. [DOI] [PubMed] [Google Scholar]
- 598.Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000;150:1209–1214. doi: 10.1083/jcb.150.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Sadler I, Crawford AW, Michelsen JW, Beckerle MC. Zyxin and cCRP: Two interactive LIM domain proteins associated with the cytoskeleton. J Cell Biol. 1992;119:1573–1587. doi: 10.1083/jcb.119.6.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Sadoulet-Puccio HM, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: An interaction through coiled-coil motifs. Proc Natl Acad Sci U S A. 1997;94:12413–12418. doi: 10.1073/pnas.94.23.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Salmikangas P, Mykkanen OM, Gronholm M, Heiska L, Kere J, Carpen O. Myotilin, a novel sarcomeric protein with two Ig-like domains, is encoded by a candidate gene for limb-girdle muscular dystrophy. Hum Mol Genet. 1999;8:1329–1336. doi: 10.1093/hmg/8.7.1329. [DOI] [PubMed] [Google Scholar]
- 603.Salmikangas P, van der Ven PF, Lalowski M, Taivainen A, Zhao F, Suila H, Schroder R, Lappalainen P, Furst DO, Carpen O. Myotilin, the limb-girdle muscular dystrophy 1A (LGMD1A) protein, cross-links actin filaments and controls sarcomere assembly. Hum Mol Genet. 2003;12:189–203. doi: 10.1093/hmg/ddg020. [DOI] [PubMed] [Google Scholar]
- 604.Samarel AM. Focal adhesion signaling in heart failure. Pflugers Arch. 2014;466:1101–1111. doi: 10.1007/s00424-014-1456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Sanoudou D, Corbett MA, Han M, Ghoddusi M, Nguyen MA, Vlahovich N, Hardeman EC, Beggs AH. Skeletal muscle repair in a mouse model of nemaline myopathy. Hum Mol Genet. 2006;15:2603–2612. doi: 10.1093/hmg/ddl186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Sasaki T, Giltay R, Talts U, Timpl R, Talts JF. Expression and distribution of laminin α1 and α2 chains in embryonic and adult mouse tissues: An immunochemical approach. Exp Cell Res. 2002;275:185–199. doi: 10.1006/excr.2002.5499. [DOI] [PubMed] [Google Scholar]
- 607.Saupe KW, Spindler M, Tian R, Ingwall JS. Impaired cardiac energetics in mice lacking muscle-specific isoenzymes of creatine kinase. Circ Res. 1998;82:898–907. doi: 10.1161/01.res.82.8.898. [DOI] [PubMed] [Google Scholar]
- 608.Schaart G, Viebahn C, Langmann W, Ramaekers F. Desmin and titin expression in early postimplantation mouse embryos. Development. 1989;107:585–596. doi: 10.1242/dev.107.3.585. [DOI] [PubMed] [Google Scholar]
- 609.Schafer DA, Korshunova YO, Schroer TA, Cooper JA. Differential localization and sequence analysis of capping protein β-subunit isoforms of vertebrates. J Cell Biol. 1994;127:453–465. doi: 10.1083/jcb.127.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C. Developmental myosins: Expression patterns and functional significance. Skelet Muscle. 2015;5:1–14. doi: 10.1186/s13395-015-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Schoenauer R, Bertoncini P, Machaidze G, Aebi U, Perriard J-C, Hegner M, Agarkova I. Myomesin is a molecular spring with adaptable elasticity. J Mol Biol. 2005;349:367–379. doi: 10.1016/j.jmb.2005.03.055. [DOI] [PubMed] [Google Scholar]
- 612.Schoenauer R, Emmert MY, Felley A, Ehler E, Brokopp C, Weber B, Nemir M, Faggian GG, Pedrazzini T, Falk V, Hoerstrup SP, Agarkova I. EH-myomesin splice isoform is a novel marker for dilated cardiomyopathy. Basic Res Cardiol. 2011;106:233–247. doi: 10.1007/s00395-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Schoenauer R, Lange S, Hirschy A, Ehler E, Perriard J-C, Agarkova I. Myomesin 3, a novel structural component of the M-band in striated muscle. J Mol Biol. 2008;376:338–351. doi: 10.1016/j.jmb.2007.11.048. [DOI] [PubMed] [Google Scholar]
- 614.Schrickel JW, Stockigt F, Krzyzak W, Paulin D, Li Z, Lubkemeier I, Fleischmann B, Sasse P, Linhart M, Lewalter T, Nickenig G, Lickfett L, Schroder R, Clemen CS. Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J Interv Card Electrophysiol. 2010;28:71–80. doi: 10.1007/s10840-010-9482-8. [DOI] [PubMed] [Google Scholar]
- 615.Schroder JM, Durling H, Laing N. Actin myopathy with nemaline bodies, intranuclear rods, and a heterozygous mutation in ACTA1 (Asp154Asn) Acta Neuropathol. 2004;108:250–256. doi: 10.1007/s00401-004-0888-1. [DOI] [PubMed] [Google Scholar]
- 616.Schweitzer SC, Klymkowsky MW, Bellin RM, Robson RM, Capetanaki Y, Evans RM. Paranemin and the organization of desmin filament networks. J Cell Sci. 2001;114:1079–1089. doi: 10.1242/jcs.114.6.1079. [DOI] [PubMed] [Google Scholar]
- 617.Schweizer J, Bowden PE, Coulombe PA, Langbein L, Lane EB, Magin TM, Maltais L, Omary MB, Parry DA, Rogers MA, Wright MW. New consensus nomenclature for mammalian keratins. J Cell Biol. 2006;174:169–174. doi: 10.1083/jcb.2006.03161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Seguchi O, Takashima S, Yamazaki S, Asakura M, Asano Y, Shintani Y, Wakeno M, Minamino T, Kondo H, Furukawa H, Nakamaru K, Naito A, Takahashi T, Ohtsuka T, Kawakami K, Isomura T, Kitamura S, Tomoike H, Mochizuki N, Kitakaze M. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J Clin Invest. 2007;117:2812–2824. doi: 10.1172/jci30804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 619.Sejersen T, Lendahl U. Transient expression of the intermediate filament nestin during skeletal muscle development. J Cell Sci. 1993;106:1291–1300. doi: 10.1242/jcs.106.4.1291. [DOI] [PubMed] [Google Scholar]
- 620.Selcen D. Myofibrillar myopathies. Neuromuscul Disord. 2011;21:161–171. doi: 10.1016/j.nmd.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Sellers JR. Myosins: A diverse superfamily. Biochim Biophys Acta. 2000;1496:3–22. doi: 10.1016/s0167-4889(00)00005-7. [DOI] [PubMed] [Google Scholar]
- 622.Semenova E, Wang X, Jablonski MM, Levorse J, Tilghman SM. An engineered 800 kilobase deletion of Uchl3 and Lmo7 on mouse chromosome 14 causes defects in viability, postnatal growth and degeneration of muscle and retina. Hum Mol Genet. 2003;12:1301–1312. doi: 10.1093/hmg/ddg140. [DOI] [PubMed] [Google Scholar]
- 623.Senetar MA, McCann RO. Gene duplication and functional divergence during evolution of the cytoskeletal linker protein talin. Gene. 2005;362:141–152. doi: 10.1016/j.gene.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 624.Seto JT, Quinlan KG, Lek M, Zheng XF, Garton F, MacArthur DG, Hogarth MW, Houweling PJ, Gregorevic P, Turner N, Cooney GJ, Yang N, North KN. ACTN3 genotype influences muscle performance through the regulation of calcineurin signaling. J Clin Invest. 2013;123:4255–4263. doi: 10.1172/jci67691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Setzer SV, Calkins CC, Garner J, Summers S, Green KJ, Kowalczyk AP. Comparative analysis of armadillo family proteins in the regulation of a431 epithelial cell junction assembly, adhesion and migration. J Invest Dermatol. 2004;123:426–433. doi: 10.1111/j0022-202X.2004.23319.x. [DOI] [PubMed] [Google Scholar]
- 626.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Shaffer JF, Kensler RW, Harris SP. The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem. 2009;284:12318–12327. doi: 10.1074/jbc.M808850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Shah SB, Love JM, O’Neill A, Lovering RM, Bloch RJ. Influences of desmin and keratin 19 on passive biomechanical properties of mouse skeletal muscle. J Biomed Biotechnol. 2012;2012:1–12. doi: 10.1155/2012/704061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Shah SB, Su FC, Jordan K, Milner DJ, Friden J, Capetanaki Y, Lieber RL. Evidence for increased myofibrillar mobility in desmin-null mouse skeletal muscle. J Exp Biol. 2002;205:321–325. doi: 10.1242/jeb.205.3.321. [DOI] [PubMed] [Google Scholar]
- 630.Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol. 2009;1:1–21. doi: 10.1101/cshperspect.a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Sharp WW, Simpson DG, Borg TK, Samarel AM, Terracio L. Mechanical forces regulate focal adhesion and costamere assembly in cardiac myocytes. Am J Physiol. 1997;273:H546–H556. doi: 10.1152/ajpheart.1997.273.2.H546. [DOI] [PubMed] [Google Scholar]
- 632.Shathasivam T, Kislinger T, Gramolini AO. Genes, proteins and complexes: the multifaceted nature of FHL family proteins in diverse tissues. J Cell Mol Med. 2010;14:2702–2720. doi: 10.1111/j1582-4934.2010.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Sheikh F, Lyon RC, Chen J. Getting the skinny on thick filament regulation in cardiac muscle biology and disease. Trends Cardiovasc Med. 2014;24:133–141. doi: 10.1016/j.tcm.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Sheikh F, Raskin A, Chu P-H, Lange S, Domenighetti AA, Zheng M, Liang X, Zhang T, Yajima T, Gu Y. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biome-chanical stress responses in mice. J Clin Invest. 2008;118:3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Siegert R, Perrot A, Keller S, Behlke J, Michalewska-Wludarczyk A, Wycisk A, Tendera M, Morano I, Ozcelik C. A myomesin mutation associated with hypertrophic cardiomyopathy deteriorates dimerisation properties. Biochem Biophys Res Commun. 2011;405:473–479. doi: 10.1016/j.bbrc.2011.01.056. [DOI] [PubMed] [Google Scholar]
- 636.Sinn HW, Balsamo J, Lilien J, Lin JJ. Localization of the novel Xin protein to the adherens junction complex in cardiac and skeletal muscle during development. Dev Dyn. 2002;225:1–13. doi: 10.1002/dvdy.10131. [DOI] [PubMed] [Google Scholar]
- 637.Sjoberg G, Edstrom L, Lendahl U, Sejersen T. Myofibers from Duchenne/Becker muscular dystrophy and myositis express the intermediate filament nestin. J Neuropathol Exp Neurol. 1994;53:416–423. doi: 10.1097/00005072-199407000.-00014. [DOI] [PubMed] [Google Scholar]
- 638.Sjoberg G, Jiang WQ, Ringertz NR, Lendahl U, Sejersen T. Colocalization of nestin and vimentin/desmin in skeletal muscle cells demonstrated by three-dimensional fluorescence digital imaging microscopy. Exp Cell Res. 1994;214:447–458. doi: 10.1006/excr.1994.1281. [DOI] [PubMed] [Google Scholar]
- 639.Sjoblöm B, Salmazo A, Djinović-Carugo K. α-Actinin structure and regulation. Cell Mol Life Sci. 2008;65:2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Sjöström M, Squire JM. Fine structure of the A-band in cryosections: The structure of the A-band of human skeletal muscle fibres from ultrathin cryosections negatively stained. J Mol Biol. 1977;109:49–68. doi: 10.1016/0889-1605(88)90054-7. [DOI] [PubMed] [Google Scholar]
- 641.Smith EA, Fuchs E. Defining the interactions between intermediate filaments and desmosomes. J Cell Biol. 1998;141:1229–1241. doi: 10.1083/jcb.141.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112:355–366. doi: 10.1161/circresaha.112.268672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem Biophys Res Commun. 2008;369:82–87. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Song WJ, Van Keuren ML, Drabkin HA, Cypser JR, Gemmill RM, Kurnit DM. Assignment of the human slow twitch skeletal muscle/cardiac troponin C gene (TNNC1) to human chromosome 3p21.3–>3p143 using somatic cell hybrids. Cytogenet Cell Genet. 1996;75:36–37. doi: 10.1159/000134453. [DOI] [PubMed] [Google Scholar]
- 645.Sonnemann KJ, Fitzsimons DP, Patel JR, Liu Y, Schneider Martin F, Moss RL, Ervasti James M. Cytoplasmic γ-actin is not required for skeletal muscle development but its absence leads to a progressive myopathy. Dev Cell. 2006;11:387–397. doi: 10.1016/j.devcel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 646.Sorimachi H, Imajoh-Ohmi S, Emori Y, Kawasaki H, Ohno S, Minami Y, Suzuki K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mutypes. Specific expression of the mRNA in skeletal muscle. J Biol Chem. 1989;264:20106–20111. [PubMed] [Google Scholar]
- 647.Sorimachi H, Tsukahara T, Okada-Ban M, Sugita H, Ishiura S, Suzuki K. Identification of a third ubiquitous calpain species–chicken muscle expresses four distinct calpains. Biochim Biophys Acta. 1995;1261:381–393. doi: 10.1016/0167-4781(95)00027-e. [DOI] [PubMed] [Google Scholar]
- 648.Soteriou A, Gamage M, Trinick J. A survey of interactions made by the giant protein titin. J Cell Sci. 1993;104:119–123. doi: 10.1242/jcs.104.1.119. [DOI] [PubMed] [Google Scholar]
- 649.Spencer JA, Eliazer S, Ilaria RL, Jr, Richardson JA, Olson EN. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J Cell Biol. 2000;150:771–784. doi: 10.1083/jcb.150.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Squire JM, Luther PK, Knupp C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J Mol Biol. 2003;331:713–724. doi: 10.1016/s0022-2836(03)00781-2. [DOI] [PubMed] [Google Scholar]
- 651.Starr R, Offer G. H-protein and X-protein. Two new components of the thick filaments of vertebrate skeletal muscle. J Mol Biol. 1983;170:675–698. doi: 10.1016/S0022-2836(83)80127-2. [DOI] [PubMed] [Google Scholar]
- 652.Starr R, Offer G. The interaction of C-protein with heavy meromyosin and subfragment-2. Biochem J. 1978;171:813–816. doi: 10.1042/bj1710813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 654.Stone MR, O’Neill A, Catino D, Bloch RJ. Specific interaction of the actin-binding domain of dystrophin with intermediate filaments containing keratin 19. Mol Biol Cell. 2005;16:4280–4293. doi: 10.1091/mbcE05-02.-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Stone MR, O’Neill A, Lovering RM, Strong J, Resneck WG, Reed PW, Toivola DM, Ursitti JA, Omary MB, Bloch RJ. Absence of keratin 19 in mice causes skeletal myopathy with mitochondrial and sarcolemmal reorganization. J Cell Sci. 2007;120:3999–4008. doi: 10.1242/jcs.009241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Strach K, Reimann J, Thomas D, Naehle CP, Kress W, Kornblum C. ZASPopathy with childhood-onset distal myopathy. J Neurol. 2012;259:1494–1496. doi: 10.1007/s00415-012-6543-1. [DOI] [PubMed] [Google Scholar]
- 657.Straub V, Ettinger AJ, Durbeej M, Venzke DP, Cutshall S, Sanes JR, Campbell KP. ε-Sarcoglycan replaces α-sarcoglycan in smooth muscle to form a unique dystrophin-glycoprotein complex. J Biol Chem. 1999;274:27989–27996. doi: 10.1074/jbc.274.39.27989. [DOI] [PubMed] [Google Scholar]
- 658.Su M, Wang J, Kang L, Wang Y, Zou Y, Feng X, Wang D, Ahmad F, Zhou X, Hui R, Song L. Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathy. Int J Mol Sci. 2014;15:9302–9313. doi: 10.3390/ijms15069302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Sun M, Opavsky MA, Stewart DJ, Rabinovitch M, Dawood F, Wen WH, Liu PP. Temporal response and localization of integrins β1 and β3 in the heart after myocardial infarction: regulation by cytokines. Circulation. 2003;107:1046–1052. doi: 10.1161/01.cir.0000051363.86009.3c. [DOI] [PubMed] [Google Scholar]
- 661.Sussman MA, Baque S, Uhm C-S, Daniels MP, Price RL, Simpson D, Terracio L, Kedes L. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ Res. 1998;82:94–105. doi: 10.1161/01.RES.82.1.94. [DOI] [PubMed] [Google Scholar]
- 662.Sussman MA, Welch S, Cambon N, Klevitsky R, Hewett TE, Price R, Witt SA, Kimball TR. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. J Clin Invest. 1998;101:51–61. doi: 10.1172/JCI1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Sweeney HL, Houdusse A. Structural and functional insights into the Myosin motor mechanism. Annu Rev Biophys. 2010;39:539–557. doi: 10.1146/annurev.biophys.050708.133751. [DOI] [PubMed] [Google Scholar]
- 664.Swope D, Cheng L, Gao E, Li J, Radice GL. Loss of cadherin-binding proteins β-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol Cell Biol. 2012;32:1056–1067. doi: 10.1128/mcb06188-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.Syrris P, Ward D, Asimaki A, Sen-Chowdhry S, Ebrahim HY, Evans A, Hitomi N, Norman M, Pantazis A, Shaw AL, Elliott PM, McKenna WJ. Clinical expression of plakophilin-2 mutations in familial arrhyth-mogenic right ventricular cardiomyopathy. Circulation. 2006;113:356–364. doi: 10.1161/circulationaha.105.561654. [DOI] [PubMed] [Google Scholar]
- 666.Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vanden-berghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin β tails: A final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 668.Tajsharghi H, Kimber E, Kroksmark AK, Jerre R, Tulinius M, Oldfors A. Embryonic myosin heavy-chain mutations cause distal arthrogryposis and developmental myosin myopathy that persists postnatally. Arch Neurol. 2008;65:1083–1090. doi: 10.1001/archneur.65.8.1083. [DOI] [PubMed] [Google Scholar]
- 669.Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:3–18. doi: 10.1007/s00401-012-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol. 2003;54:494–500. doi: 10.1002/ana.10693. [DOI] [PubMed] [Google Scholar]
- 671.Takada F, Vander Woude DL, Tong HQ, Thompson TG, Watkins SC, Kunkel LM, Beggs AH. Myozenin: An α-actinin- and γ-filamin-binding protein of skeletal muscle Z lines. Proc Natl Acad Sci U S A. 2001;98:1595–1600. doi: 10.1073/pnas.041609698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Takano K, Watanabe-Takano H, Suetsugu S, Kurita S, Tsujita K, Kimura S, Karatsu T, Takenawa T, Endo T. Nebulin and N-WASP cooperate to cause IGF-1-induced sarcomeric actin filament formation. Science. 2010;330:1536–1540. doi: 10.1126/science.1197767. [DOI] [PubMed] [Google Scholar]
- 673.Tangney JR, Chuang JS, Janssen MS, Krishnamurthy A, Liao P, Hoshijima M, Wu X, Meininger GA, Muthuchamy M, Zemljic-Harpf A, Ross RS, Frank LR, McCulloch AD, Omens JH. Novel role for vinculin in ventricular myocyte mechanics and dysfunction. Biophys J. 2013;104:1623–1633. doi: 10.1016/j.bpj.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Tanokura M, Tawada Y, Ono A, Ohtsuki I. Chymotryptic subfragments of troponin T from rabbit skeletal muscle. Interaction with tropomyosin, troponin I and troponin C. J Biochem. 1983;93:331–337. doi: 10.1093/oxfordjournals.jbchem.a134185. [DOI] [PubMed] [Google Scholar]
- 675.Tardiff JC. It’s never too early to look subclinical disease in sarcomeric dilated cardiomyopathy. Circ Cardiovasc Genet. 2012;5:483–486. doi: 10.1161/circgenetics.112.964817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRE-SAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Tardiff JC. Tropomyosin and dilated cardiomyopathy: revenge of the actinomyosin “gatekeeper”. J Am Coll Cardiol. 2010;55:330–332. doi: 10.1016/j.jacc.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 678.Tarone G, Brancaccio M. The muscle-specific chaperone protein melusin is a potent cardioprotective agent. Basic Res Cardiol. 2015;110:1–10. doi: 10.1007/s00.395-015-0466-9. [DOI] [PubMed] [Google Scholar]
- 679.Tarui S, Okuno G, Ikura Y, Tanaka T, Suda M, Nishikawa M. Phosphofructokinase deficiency in skeletal muscle. A new type of glycogenosis. Biochem Biophys Res Commun. 1965;19:517–523. doi: 10.1016/0006-291x(65)90156-7. [DOI] [PubMed] [Google Scholar]
- 680.Taveau M, Bourg N, Sillon G, Roudaut C, Bartoli M, Richard I. Calpain 3 is activated through autolysis within the active site and lyses sarcomeric and sarcolemmal components. Mol Cell Biol. 2003;23:9127–9135. doi: 10.1128/mcb.23(24)9127-9135.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681.Thompson TG, Chan YM, Hack AA, Brosius M, Rajala M, Lidov HG, McNally EM, Watkins S, Kunkel LM. Filamin 2 (FLN2): A muscle-specific sarcoglycan interacting protein. J Cell Biol. 2000;148:115–126. doi: 10.1083/jcb.148.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D. Null mutation in the desmin gene gives rise to a cardiomyopathy. J Mol Cell Cardiol. 1997;29:2107–2124. doi: 10.1006/jmcc.1997.0446. [DOI] [PubMed] [Google Scholar]
- 683.Tian L, Ding S, You Y, Li TR, Liu Y, Wu X, Sun L, Xu T. Leiomodin-3-deficient mice display nemaline myopathy with fast-myofiber atrophy. Dis Model Mech. 2015;8:635–641. doi: 10.1242/dmm.019430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.Tian LF, Li HY, Jin BF, Pan X, Man JH, Zhang PJ, Li WH, Liang B, Liu H, Zhao J, Gong WL, Zhou T, Zhang XM. MDM2 interacts with and downregulates a sarcomeric protein, TCAP. Biochem Biophys Res Commun. 2006;345:355–361. doi: 10.1016/j.bbrc.2006.04.108. [DOI] [PubMed] [Google Scholar]
- 685.Tiso N, Rampoldi L, Pallavicini A, Zimbello R, Pandolfo D, Valle G, Lanfranchi G, Danieli GA. Fine mapping of five human skeletal muscle genes: α-Tropomyosin, β-tropomyosin, troponin-I slow-twitch, troponin-I fast-twitch, and troponin-C fast. Biochem Biophys Res Com-mun. 1997;230:347–350. doi: 10.1006/bbrc.1996.5958. [DOI] [PubMed] [Google Scholar]
- 686.Tonino P, Pappas CT, Hudson BD, Labeit S, Gregorio CC, Granzier H. Reduced myofibrillar connectivity and increased Z-disk width in nebulin-deficient skeletal muscle. J Cell Sci. 2010;123:384–391. doi: 10.1242/jcs.042234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Torrado M, Nespereira B, Lopez E, Centeno A, Castro-Beiras A, Mikhailov AT. ANKRD1 specifically binds CASQ2 in heart extracts and both proteins are co-enriched in piglet cardiac Purkinje cells. J Mol Cell Cardiol. 2005;38:353–365. doi: 10.1016/j.yjmcc.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 688.Townsend D. Finding the sweet spot: assembly and glycosylation of the dystrophin-associated glycoprotein complex. Anat Rec (Hoboken) 2014;297:1694–1705. doi: 10.1002/ar.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.Townsend PJ, Barton PJ, Yacoub MH, Farza H. Molecular cloning of human cardiac troponin T isoforms: Expression in developing and failing heart. J Mol Cell Cardiol. 1995;27:2223–2236. doi: 10.1016/S0022-2828(95)91587-7. [DOI] [PubMed] [Google Scholar]
- 690.Toydemir RM, Rutherford A, Whitby FG, Jorde LB, Carey JC, Bamshad MJ. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet. 2006;38:561–565. doi: 10.1038/ng1775. [DOI] [PubMed] [Google Scholar]
- 691.Trombitas K, Redkar A, Centner T, Wu Y, Labeit S, Granzier H. Extensibility of isoforms of cardiac titin: Variation in contour length of molecular subsegments provides a basis for cellular passive stiffness diversity. Biophys J. 2000;79:3226–3234. doi: 10.1016/s0006-3495(00)76555-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Trotter JA, Samora A, Baca J. Three-dimensional structure of the murine muscle-tendon junction. Anat Rec. 1985;213:16–25. doi: 10.1002/ar.1092130104. [DOI] [PubMed] [Google Scholar]
- 693.Troyanovsky SM, Troyanovsky RB, Eshkind LG, Leube RE, Franke WW. Identification of amino acid sequence motifs in desmocollin, a desmosomal glycoprotein, that are required for plakoglobin binding and plaque formation. Proc Natl Acad Sci U S A. 1994;91:10790–10794. doi: 10.1073/pnas.91.23.10790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Tskhovrebova L, Trinick J. Making muscle elastic: the structural basis of myomesin stretching. PLoS-Biology. 2012;10:e1001264–e1001267. doi: 10.1371/journal.pbio.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 695.Tsukada T, Pappas CT, Moroz N, Antin PB, Kostyukova AS, Gregorio CC. Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. J Cell Sci. 2010;123:3136–3145. doi: 10.1242/jcs.07.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Tsukamoto Y, Hijiya N, Yano S, Yokoyama S, Nakada C, Uchida T, Matsuura K, Moriyama M. Arpp/Ankrd2, a member of the muscle ankyrin repeat proteins (MARPs), translocates from the I-band to the nucleus after muscle injury. Histochem Cell Biol. 2008;129:55–64. doi: 10.1007/s00418-007-0348-9. [DOI] [PubMed] [Google Scholar]
- 697.Tsukamoto Y, Senda T, Nakano T, Nakada C, Hida T, Ishiguro N, Kondo G, Baba T, Sato K, Osaki M, Mori S, Ito H, Moriyama M. Arpp, a new homolog of carp, is preferentially expressed in type 1 skeletal muscle fibers and is markedly induced by denervation. Lab Invest. 2002;82:645–655. doi: 10.1038/labinvest.3780459. [DOI] [PubMed] [Google Scholar]
- 698.Turner DC, Wallimann T, Eppenberger HM. A protein that binds specifically to the M-line of skeletal muscle is identified as the muscle form of creatine kinase. Proc Natl Acad Sci U S A. 1973;70:702–705. doi: 10.1073/pnas.70.3.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Unsold B, Kaul A, Sbroggio M, Schubert C, Regitz-Zagrosek V, Bran-caccio M, Damilano F, Hirsch E, Van Bilsen M, Munts C, Sipido K, Bito V, Detre E, Wagner NM, Schafer K, Seidler T, Vogt J, Neef S, Bleckmann A, Maier LS, Balligand JL, Bouzin C, Ventura-Clapier R, Garnier A, Eschenhagen T, El-Armouche A, Knoll R, Tarone G, Hasen-fuss G. Melusin protects from cardiac rupture and improves functional remodelling after myocardial infarction. Cardiovasc Res. 2014;101:97–107. doi: 10.1093/cvr/cvt235. [DOI] [PubMed] [Google Scholar]
- 700.Ursitti JA, Lee PC, Resneck WG, McNally MM, Bowman AL, O’Neill A, Stone MR, Bloch RJ. Cloning and characterization of cytokeratins 8 and 19 in adult rat striated muscle. Interaction with the dystrophin glycoprotein complex. J Biol Chem. 2004;279:41830–41838. doi: 10.1074/jbc.M400128200. [DOI] [PubMed] [Google Scholar]
- 701.Vafiadaki E, Arvanitis DA, Papalouka V, Terzis G, Roumeliotis TI, Spengos K, Garbis SD, Manta P, Kranias EG, Sanoudou D. Muscle LIM protein isoform negatively regulates striated muscle actin dynamics and differentiation. Febs j. 2014;281:3261–3279. doi: 10.1111/febs.12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702.Vafiadaki E, Arvanitis DA, Sanoudou D. Muscle LIM protein: Master regulator of cardiac and skeletal muscle functions. Gene. 2015;566:1–7. doi: 10.1016/j.gene.2015.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Vaittinen S, Lukka R, Sahlgren C, Hurme T, Rantanen J, Lendahl U, Eriksson JE, Kalimo H. The expression of intermediate filament protein nestin as related to vimentin and desmin in regenerating skeletal muscle. J Neuropathol Exp Neurol. 2001;60:588–597. doi: 10.1093/jnen/60.6.588. [DOI] [PubMed] [Google Scholar]
- 704.Valle G, Faulkner G, De Antoni A, Pacchioni B, Pallavicini A, Pandolfo D, Tiso N, Toppo S, Trevisan S, Lanfranchi G. Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett. 1997;415:163–168. doi: 10.1016/S0014-5793(97)01108-3. [DOI] [PubMed] [Google Scholar]
- 705.van der Flier A, Gaspar AC, Thorsteinsdottir S, Baudoin C, Groeneveld E, Mummery CL, Sonnenberg A. Spatial and temporal expression of the β1D integrin during mouse development. Dev Dyn. 1997;210:472–486. doi: 10.1002/(sici)1097-0177(199712)210:4<472::aid-aja10>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 706.van der Flier A, Sonnenberg A. Structural and functional aspects of filamins. Biochim Biophys Acta. 2001;1538:99–117. doi: 10.1016/S0167-4889(01)00072-6. [DOI] [PubMed] [Google Scholar]
- 707.van der Ven PF, Ehler E, Vakeel P, Eulitz S, Schenk JA, Milting H, Micheel B, Furst DO. Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mena/VASP. Exp Cell Res. 2006;312:2154–2167. doi: 10.1016/j.yexcr.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 708.van der Ven PF, Wiesner S, Salmikangas P, Auerbach D, Himmel M, Kempa S, Hayess K, Pacholsky D, Taivainen A, Schroder R, Carpen O, Furst DO. Indications for a novel muscular dystrophy pathway. γ-Filamin, the muscle-specific filamin isoform, interacts with myotilin. J Cell Biol. 2000;151:235–248. doi: 10.1083/jcb.151.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.van Dijk SJ, Bezold KL, Harris SP. Earning stripes: myosin binding protein-C interactions with actin. Pflugers Arch. 2014;466:445–450. doi: 10.1007/s00424-013-1432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.van Tintelen JP, Entius MM, Bhuiyan ZA, Jongbloed R, Wiesfeld AC, Wilde AA, van der Smagt J, Boven LG, Mannens MM, van Langen IM, Hofstra RM, Otterspoor LC, Doevendans PA, Rodriguez LM, van Gelder IC, Hauer RN. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113:1650–1658. doi: 10.1161/circu-lationaha.105.609719. [DOI] [PubMed] [Google Scholar]
- 711.Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008;87:649–667. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 712.Vaughan A, Alvarez-Reyes M, Bridger JM, Broers JL, Ramaekers FC, Wehnert M, Morris GE, Whitfield WGF, Hutchison CJ. Both emerin and lamin C depend on lamin A for localization at the nuclear envelope. J Cell Sci. 2001;114:2577–2590. doi: 10.1242/jcs.114.14.2577. [DOI] [PubMed] [Google Scholar]
- 713.Vaughan KT, Weber FE, Ried T, Ward DC, Reinach FC, Fischman DA. Human myosin-binding protein H (MyBP-H): Complete primary sequence, genomic organization, and chromosomal localization. Genomics. 1993;16:34–40. doi: 10.1006/geno.1993.1136. [DOI] [PubMed] [Google Scholar]
- 714.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 715.Vite A, Li J, Radice GL. New functions for α-catenins in health and disease: From cancer to heart regeneration. Cell Tissue Res. 2015;360:773–783. doi: 10.1007/s00441-015-2123-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716.Vite A, Radice GL. N-cadherin/catenin complex as a master regulator of intercalated disc function. Cell Commun Adhes. 2014;21:169–179. doi: 10.3109/15419061.2014.908853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Volkers M, Rohde D, Goodman C, Most P. S100A1: a regulator of striated muscle sarcoplasmic reticulum Ca2+ handling, sarcomeric, and mitochondrial function. J Biomed Biotechnol. 2010;2010:1–10. doi: 10.1155/2010/178614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 718.von der Ecken J, Muller M, Lehman W, Manstein DJ, Penczek PA, Raunser S. Structure of the F-actin-tropomyosin complex. Nature. 2015;519:114–117. doi: 10.1038/nature14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 719.von der Hagen M, Laval SH, Cree LM, Haldane F, Pocock M, Wappler I, Peters H, Reitsamer HA, Hoger H, Wiedner M, Oberndorfer F, Anderson LV, Straub V, Bittner RE, Bushby KM. The differential gene expression profiles of proximal and distal muscle groups are altered in pre-pathological dysferlin-deficient mice. Neuromuscul Disord. 2005;15:863–877. doi: 10.1016/j.nmd.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 720.von Lampe B, Stallmach A, Riecken EO. Altered glycosylation of integrin adhesion molecules in colorectal cancer cells and decreased adhesion to the extracellular matrix. Gut. 1993;34:829–836. doi: 10.1136/gut.34.6.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Vorburger K, Lehner CF, Kitten GT, Eppenberger HM, Nigg EA. A second higher vertebrate B-type lamin. cDNA sequence determination and in vitro processing of chicken lamin B2. J Mol Biol. 1989;208:405–415. doi: 10.1016/0022-2836(89)90505-6. [DOI] [PubMed] [Google Scholar]
- 722.Vorgerd M, van der Ven PF, Bruchertseifer V, Lowe T, Kley RA, Schroder R, Lochmuller H, Himmel M, Koehler K, Furst DO, Huebner A. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77:297–304. doi: 10.1086/43.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Wachsstock DH, Wilkins JA, Lin S. Specific interaction of vinculin with α-actinin. Biochem Biophys Res Commun. 1987;146:554–560. doi: 10.1016/0006-291x(87)90564-x. [DOI] [PubMed] [Google Scholar]
- 724.Wade M. High-throughput silencing using the CRISPR-Cas9 System: A review of the benefits and challenges. J Biomol Screen. 2015;20:1027–1039. doi: 10.1177/1087057115587916. [DOI] [PubMed] [Google Scholar]
- 725.Wade R, Eddy R, Shows TB, Kedes L. cDNA sequence, tissue-specific expression, and chromosomal mapping of the human slow-twitch skeletal muscle isoform of troponin I. Genomics. 1990;7:346–357. doi: 10.1016/0888-7543(90)90168-T. [DOI] [PubMed] [Google Scholar]
- 726.Wagner KR, Cohen JB, Huganir RL. The 87K postsynaptic membrane protein from Torpedo is a protein-tyrosine kinase substrate homologous to dystrophin. Neuron. 1993;10:511–522. doi: 10.1016/0896-6273(93)90338-r. [DOI] [PubMed] [Google Scholar]
- 727.Wahl JK, Sacco PA, McGranahan-Sadler TM, Sauppe LM, Wheelock MJ, Johnson KR. Plakoglobin domains that define its association with the desmosomal cadherins and the classical cadherins: Identification of unique and shared domains. J Cell Sci. 1996;109:1143–1154. doi: 10.1242/jcs.109.5.1143. [DOI] [PubMed] [Google Scholar]
- 728.Walklate J, Ujfalusi Z, Geeves MA. Myosin isoforms and the mechanochemical cross-bridge cycle. J Exp Biol. 2016;219:168–174. doi: 10.1242/jeb.124594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729.Wallimann T, Tokarska-Schlattner M, Schlattner U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids. 2011;40:1271–1296. doi: 10.1007/s00726-011-0877-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 730.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 731.Wear MA, Yamashita A, Kim K, Maéda Y, Cooper JA. How capping protein binds the barbed end of the actin filament. Curr Biol. 2003;13:1531–1537. doi: 10.1016/S0960-9822(03)00559-1. [DOI] [PubMed] [Google Scholar]
- 732.Weber A, Pennise CR, Babcock GG, Fowler VM. Tropomodulin caps the pointed ends of actin filaments. J Cell Biol. 1994;127:1627–1635. doi: 10.1083/jcb.127.6.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Weber FE, Vaughan KT, Reinach FC, Fischman DA. Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Differential expression, conserved domain structure and chromosome assignment. Eur J Biochem. 1993;216:661–669. doi: 10.1111/j1432-1033.1993.tb18186.x. [DOI] [PubMed] [Google Scholar]
- 734.Wegner A. Equilibrium of the actin-tropomyosin interaction. J Mol Biol. 1979;131:839–853. doi: 10.1016/0022-2836(79)90204-3. [DOI] [PubMed] [Google Scholar]
- 735.Wegner A. Kinetic analysis of actin assembly suggests that tropomyosin inhibits spontaneous fragmentation of actin filaments. J Mol Biol. 1982;161:217–227. doi: 10.1016/0022-2836(82)90149-8. [DOI] [PubMed] [Google Scholar]
- 736.Wei YJ, Cui CJ, Huang YX, Zhang XL, Zhang H, Hu SS. Upregulated expression of cardiac ankyrin repeat protein in human failing hearts due to arrhythmogenic right ventricular cardiomyopathy. Eur J Heart Fail. 2009;11:559–566. doi: 10.1093/eurjhf/hfp049. [DOI] [PubMed] [Google Scholar]
- 737.Weins A, Schwarz K, Faul C, Barisoni L, Linke WA, Mundel P. Differentiation- and stress-dependent nuclear cytoplasmic redistribution of myopodin, a novel actin-bundling protein. J Cell Biol. 2001;155:393–404. doi: 10.1083/jcb.2000.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738.Weiskirchen R, Pino JD, Macalma T, Bister K, Beckerle MC. The cysteine-rich protein family of highly related LIM domain proteins. J Biol Chem. 1995;270:28946–28954. doi: 10.1074/jbc.270.48.28946. [DOI] [PubMed] [Google Scholar]
- 739.Weisleder N, Soumaka E, Abbasi S, Taegtmeyer H, Capetanaki Y. Cardiomyocyte-specific desmin rescue of desmin null cardiomyopathy excludes vascular involvement. J Mol Cell Cardiol. 2004;36:121–128. doi: 10.1016/j.yjmcc.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 740.Weiss A, Leinwand LA. The mammalian myosin heavy chain gene family. Annu Rev Cell Dev Biol. 1996;12:417–439. doi: 10.1146/annurev.cellbio.12.1.417. [DOI] [PubMed] [Google Scholar]
- 741.Weith AE, Previs MJ, Hoeprich GJ, Previs SB, Gulick J, Robbins J, Warshaw DM. The extent of cardiac myosin binding protein-C phosphorylation modulates actomyosin function in a graded manner. J Muscle Res Cell Motil. 2012;33:449–459. doi: 10.1007/s10974-012-9312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 742.Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, Carragher BO, Milligan RA, Sweeney HL. Myosin VI is an actin-based motor that moves backwards. Nature. 1999;401:505–508. doi: 10.1038/46835. [DOI] [PubMed] [Google Scholar]
- 743.Whitmore C, Morgan J. What do mouse models of muscular dystrophy tell us about the DAPC and its components? Int J Exp Pathol. 2014;95:365–377. doi: 10.1111/iep.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Willis MS, Ike C, Li L, Wang D-Z, Glass DJ, Patterson C. Muscle Ring Finger 1, but not Muscle Ring Finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–459. doi: 10.1161/01.res.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 746.Winokur ST, Chen YW, Masny PS, Martin JH, Ehmsen JT, Tapscott SJ, van der Maarel SM, Hayashi Y, Flanigan KM. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet. 2003;12:2895–2907. doi: 10.1093/hmg/ddg327. [DOI] [PubMed] [Google Scholar]
- 747.Witt CC, Burkart C, Labeit D, McNabb M, Wu Y, Granzier H, Labeit S. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. Embo j. 2006;25:3843–3855. doi: 10.1038/sj.emboj.7601242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748.Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. Embo j. 2008;27:350–360. doi: 10.1038/sj.emboj.760.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J Mol Biol. 2005;350:713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 750.Witt SH, Labeit D, Granzier H, Labeit S, Witt CC. Dimerization of the cardiac ankyrin protein CARP: Implications for MARP titin-based signaling. J Muscle Res Cell Motil. 2005;26:401–408. doi: 10.1007/s10974-005-9022-9. [DOI] [PubMed] [Google Scholar]
- 751.Wooten EC, Hebl VB, Wolf MJ, Greytak SR, Orr NM, Draper I, Calvino JE, Kapur NK, Maron MS, Kullo IJ, Ommen SR, Bos JM, Ackerman MJ, Huggins GS. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2013;6:10–18. doi: 10.1161/circgenetics.112.965277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Xia H, Winokur ST, Kuo WL, Altherr MR, Bredt DS. Actinin-associated LIM protein: identification of a domain interaction between PDZ and spectrin-like repeat motifs. J Cell Biol. 1997;139:507–515. doi: 10.1083/jcb.139.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Xu H, Wu XR, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin α 2 (Lama2) gene. Nat Genet. 1994;8:297–302. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]
- 754.Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, Zhu H, Ma J, Han R. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. 2016;24:564–569. doi: 10.1038/mt.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 755.Xu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998;125:327–337. doi: 10.1242/dev.125.2.327. [DOI] [PubMed] [Google Scholar]
- 756.Yamamoto K. Characterization of H-protein, a component of skeletal muscle myofibrils. J Biol Chem. 1984;259:7163–7168. [PubMed] [Google Scholar]
- 757.Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181–1188. doi: 10.1161/01.RES.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 758.Yamashiro S, Gokhin DS, Kimura S, Nowak RB, Fowler VM. Tropomodulins: Pointed-end capping proteins that regulate actin filament architecture in diverse cell types. Cytoskeleton. 2012;69:337–370. doi: 10.1002/cm.21031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759.Yamashita A, Maeda K, Maéda Y. Crystal structure of CapZ: Structural basis for actin filament barbed end capping. Embo j. 2003;22:1529–1538. doi: 10.1093/emboj/cdg167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Yang Q, Hewett TE, Klevitsky R, Sanbe A, Wang X, Robbins J. PKA-dependent phosphorylation of cardiac myosin binding protein C in transgenic mice. Cardiovasc Res. 2001;51:80–88. doi: 10.1016/s0008-6363(01)00273-5. [DOI] [PubMed] [Google Scholar]
- 761.Yang YG, Makita T. Immunocytochemical localization of desmin in skeletal muscle of swine. J Vet Med Sci. 1995;57:475–479. doi: 10.1267/ahc.30.157. [DOI] [PubMed] [Google Scholar]
- 762.Yar S, Chowdhury SA, Davis RT, 3rd, Kobayashi M, Monasky MM, Rajan S, Wolska BM, Gaponenko V, Kobayashi T, Wieczorek DF, Solaro RJ. Conserved Asp-137 is important for both structure and regulatory functions of cardiac α-tropomyosin (α-TM) in a novel transgenic mouse model expressing α-TM-D137L. J Biol Chem. 2013;288:16235–16246. doi: 10.1074/jbc.M113.458695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 763.Yasuda M, Koshida S, Sato N, Obinata T. Complete primary structure of chicken cardiac C-protein (MyBP-C) and its expression in developing striated muscles. J Mol Cell Cardiol. 1995;27:2275–2286. doi: 10.1016/s0022-2828(95)91731-4. [DOI] [PubMed] [Google Scholar]
- 764.Young P, Ehler E, Gautel M. Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol. 2001;154:123–136. doi: 10.1083/jcb.2001.02110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765.Young P, Ferguson C, Banuelos S, Gautel M. Molecular structure of the sarcomeric Z-disk: Two types of titin interactions lead to an asymmetrical sorting of α-actinin. Embo j. 1998;17:1614–1624. doi: 10.1093/emboj/17.6.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Yuen M, Sandaradura SA, Dowling JJ, Kostyukova AS, Moroz N, Quinlan KG, Lehtokari VL, Ravenscroft G, Todd EJ, Ceyhan-Birsoy O, Gokhin DS, Maluenda J, Lek M, Nolent F, Pappas CT, Novak SM, D’Amico A, Malfatti E, Thomas BP, Gabriel SB, Gupta N, Daly MJ, Ilkovski B, Houweling PJ, Davidson AE, Swanson LC, Brownstein CA, Gupta VA, Medne L, Shannon P, Martin N, Bick DP, Flisberg A, Holmberg E, Van den Bergh P, Lapunzina P, Waddell LB, Sioboda DD, Bertini E, Chitayat D, Telfer WR, Laquerriere A, Gregorio CC, Otten-heijm CAC, Bonnemann CG, Pelin K, Beggs AH, Hayashi YK, Romero NB, Laing NG, Nishino I, Wallgren-Pettersson C, Melki J, Fowler VM, MacArthur DG, North KN, Clarke NF. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest. 2014;124:4693–4708. doi: 10.1172/JCI75199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 767.Zemljic-Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, Ross RS. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007;27:7522–7537. doi: 10.1128/mcb00728-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 768.Zemljic-Harpf AE, Ponrartana S, Avalos RT, Jordan MC, Roos KP, Dalton ND, Phan VQ, Adamson ED, Ross RS. Heterozygous inactivation of the vinculin gene predisposes to stress-induced cardiomyopathy. Am J Pathol. 2004;165:1033–1044. doi: 10.1016/s0002-9440(10)63364-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 769.Zhang X, Xu R, Zhu B, Yang X, Ding X, Duan S, Xu T, Zhuang Y, Han M. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development. 2007;134:901–908. doi: 10.1242/dev.02783. [DOI] [PubMed] [Google Scholar]
- 770.Zhang Z, Mu Y, Veevers J, Peter AK, Manso AM, Bradford WH, Dalton ND, Peterson KL, Knowlton KU, Ross RS, Zhou X, Chen J. Postnatal loss of kindlin-2 leads to progressive heart failure. Circ Heart Fail. 2016;9:e003129. doi: 10.1161/circheartfailure.116.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 771.Zheng M, Cheng H, Banerjee I, Chen J. ALP/Enigma PDZ-LIM domain proteins in the heart. J Mol Cell Biol. 2010;2:96–102. doi: 10.1093/jmcb/mjp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Zheng M, Cheng H, Li X, Zhang J, Cui L, Ouyang K, Han L, Zhao T, Gu Y, Dalton ND, Bang ML, Peterson KL, Chen J. Cardiac-specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Hum Mol Genet. 2009;18:701–713. doi: 10.1093/hmg/ddn400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Zhi G, Ryder JW, Huang J, Ding P, Chen Y, Zhao Y, Kamm KE, Stull JT. Myosin light chain kinase and myosin phosphorylation effect frequency-dependent potentiation of skeletal muscle contraction. Proc Natl Acad Sci U S A. 2005;102:17519–17524. doi: 10.1073/pnas.0506846102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Zhou J, Qu J, Yi XP, Graber K, Huber L, Wang X, Gerdes AM, Li F. Upregulation of γ-catenin compensates for the loss of β-catenin in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;292:H270–H276. doi: 10.1152/ajpheart.00576.2006. [DOI] [PubMed] [Google Scholar]
- 775.Zhou Q, Chu PH, Huang C, Cheng CF, Martone ME, Knoll G, Shelton GD, Evans S, Chen J. Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. J Cell Biol. 2001;155:605–612. doi: 10.1083/jcb.2001.07092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 776.Zhou X, Boren J, Akyurek LM. Filamins in cardiovascular development. Trends Cardiovasc Med. 2007;17:222–229. doi: 10.1016/j.tcm.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 777.Zieseniss A, Terasaki AG, Gregorio CC. Lasp-2 expression, localization, and ligand interactions: A new Z-disc scaffolding protein. Cell Motil Cytoskeleton. 2008;65:59–72. doi: 10.1002/cm.20244. [DOI] [PubMed] [Google Scholar]
- 778.Zolk O, Caroni P, Bohm M. Decreased expression of the cardiac LIM domain protein MLP in chronic human heart failure. Circulation. 2000;101:2674–2677. doi: 10.1161/01.CIR.101.23.2674. [DOI] [PubMed] [Google Scholar]
- 779.Zolk O, Frohme M, Maurer A, Kluxen FW, Hentsch B, Zubakov D, Hoheisel JD, Zucker IH, Pepe S, Eschenhagen T. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochem Biophys Res Commun. 2002;293:1377–1382. doi: 10.1016/s0006-291x(02)00387-x. [DOI] [PubMed] [Google Scholar]
- 780.Zou Y, Evans S, Chen J, Kuo HC, Harvey RP, Chien KR. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development. 1997;124:793–804. doi: 10.1242/dev.124.4.793. [DOI] [PubMed] [Google Scholar]
- 781.Zubrzycka-Gaarn EE, Bulman DE, Karpati G, Burghes AH, Belfall B, Klamut HJ, Talbot J, Hodges RS, Ray PN, Worton RG. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature. 1988;333:466–469. doi: 10.1038/333466a0. [DOI] [PubMed] [Google Scholar]