

Abstract
Intellectual disability (ID) comprises a large group of heterogeneous disorders, often without a known molecular cause. X-linked ID accounts for 5–10% of male ID cases. We investigated a large, three-generation family with mild ID and behavior problems in five males and one female, with a segregation suggestive for X-linked inheritance. Linkage analysis mapped a disease locus to a 7.6 Mb candidate region on the X-chromosome (LOD score 3.3). Whole-genome sequencing identified a 2 bp insertion in exon 2 of the chromosome X open reading frame 56 gene (CXorf56), resulting in a premature stop codon. This insertion was present in all intellectually impaired individuals and carrier females. Additionally, X-inactivation status showed skewed methylation patterns favoring the inactivation of the mutated allele in the unaffected carrier females. We demonstrate that the insertion leads to nonsense-mediated decay and that CXorf56 mRNA expression is reduced in the impaired males and female. In murine brain slices and primary hippocampal neuronal cultures, CXorf56 protein was present and localized in the nucleus, cell soma, dendrites, and dendritic spines. Although no other families have been identified with pathogenic variants in CXorf56, these results suggest that CXorf56 is the causative gene in this family, and thus a novel candidate gene for X-linked ID with behavior problems.
Introduction
Intellectual disability (ID) is a neurodevelopmental condition affecting approximately 1–3% of the population [1, 2]. It is characterized by impaired cognitive and adaptive functioning. Many different genes for syndromic and non-syndromic ID have been identified [1–5]. Despite the characteristic inheritance pattern of X-linked ID (XLID) most non-syndromic forms are difficult to clinically classify and must be viewed as a homogeneous group that can only be distinguished when a mapped linkage area or gene defect is detected [3]. Without further research options, a large amount of patients remains genetically undiagnosed. In the past decades, many causative genes were identified, using (combinations of) different molecular techniques [4, 6–11], including linkage analysis and Sanger sequencing of candidate genes [4, 12, 13]. With the introduction of next-generation sequencing techniques, detection of causative genes within large linkage areas has become more feasible [1, 2, 14–18]. In this paper we describe the identification of a new candidate gene, CXorf56, in a large family with non-syndromic XLID and unspecific behavior problems, identified through linkage analysis followed by whole-genome sequencing (WGS). We further describe functional studies on X-inactivation, nonsense-mediated decay, and the cellular distribution of CXorf56.
Materials and methods
Patients
Written informed consent was obtained from patients and relatives. Medical history was obtained and physical examination was performed by a clinical geneticist. Blood samples were taken from the 6 index patients (five males and one female) and 18 unaffected family members. Karyotyping and CGG repeat in the FMR1 gene was normal (data not shown).
Genotyping and linkage analysis
DNA was isolated from blood samples using the PUREGENE Genomic DNA Isolation Kit (Qiagen, Germany). 18 X-linked short tandem repeat (STR) markers from the ABI Prism Linkage Mapping Set Version 2; Panel 28 (Applied Biosystems, Foster City, CA, USA) were tested, defining a ten-centimorgan (cM) resolution X-chromosome map. Markers were amplified by PCR according to standard protocol and sequenced on an ABI 3130XL automated sequencer. Data were analyzed using Gene Mapper Version 2.1 software (Applied Biosystems). Additional markers for fine mapping were selected from the UCSC Human Genome Browser (https://genome.ucsc.edu): DXS424, DXS8064, DXS8067, and DXS8059. Primers to amplify these STR’s were designed newly and are available on request. Multi-point linkage analysis was performed using Allegro v1.2c [19] and easyLINKAGE Plus v5.02 [20] as graphical interface. Parametric linkage analysis was carried out using equal allele frequencies (estimated from the observed number of alleles), a disease allele frequency of 0.001, and no phenocopies. Penetrance of the disease-associated genotype was set to 0.99 for males and 0.1 for females. Haplotypes were constructed manually, based on the minimal number of recombinations.
Whole-genome sequencing
WGS was performed by Complete Genomics (now a BGI Company, Mountain View, CA), using a sequencing-by-ligation method described previously [21]. Briefly, the human genome sequencing procedures included DNA library construction, DNA Nano-Ball (DNB) generation, DNB array self-assembling, cPAL-based sequencing, imaging, image data analyses including base calling, DNB mapping, and sequence assembly. Reads were mapped to NCBI reference genome, build 36.3. Variants were called by local de novo assembly (format version 1.3) according to the methods previously described [21], and annotated using NCBI build 36.3 and dbSNP build 130. Data were provided as lists of sequence variants (SNPs and short indels) relative to the reference genome. Analysis of the WGS data was performed using Complete Genomics analysis tools version 1.6.0.43. Mapped sequence of the four samples varied in size between 236 and 290 GB, resulting in an average coverage between 82 and 101-fold per genome. Confident variant calling could be performed for 94–95% of the reference genome in all samples. See Table S1 for details regarding quality metrics and numbers of variants per sample. Variants were filtered according to the following criteria: located in candidate linkage region on X-chromosome, present in affected males, non-synonymous exonic and splice variants and rare or absent in public databases.
PCR and Sanger sequencing for variant verification
Sanger sequencing of the CXorf56 gene was performed in all family members from whom DNA was available. Additionally the CXorf56 gene was screened in a cohort of unrelated individuals with ID (222 males, 191 females) and in a cohort of 188 healthy Dutch individuals (94 males, 94 females). All exons and exon–intron boundaries of CXorf56 (NM_022101.3) were PCR-amplified from genomic DNA using the primers indicated in Table S2 and the supplementary protocols. Nomenclature insertion: c.159_160ins TA; the A from translation start ATG from the transcript CXorf56-201 (Ensembl Transcript ID ENST00000371594; NM_022101.3 (build 37)) was taken as 1. Protein: p.(Asp54*) (Ensembl Protein ID: ENSP00000360652) (https://www.ensembl.org).
The identified CXorf56 variant has been submitted to LOVD (https://databases.lovd.nl/shared/variants/0000195713).
Determining X-chromosome inactivation status
X-inactivation and methylation status of X-chromosomes were studied using the highly polymorphic CAG repeat in the human androgen-receptor gene (MIM 313700) and determining the methylation status of two HpaII restriction sites within 100 base pairs (bp) 5′ to this repeat [22]. In this assay, methylation status of the HpaII site is a measure for activation or inactivation of either female X-chromosomes. For the procedure, see the supplementary protocols.
CXorf56 expression studies
Epstein barr virus (EBV)-transformed lymphoblasts were cultured at 37 °C and 5% CO2 in 10–20 ml RPMI plus Ultra glutamine (2 mM L-glutamine) and 15% fetal bovine serum. Cells were grown to approximately 1 × 106 cells/ml. After transformation cells were grown to the desired amount and total RNA was isolated (RNAzol B, Cinna/Biotecx). cDNA was prepared using IScript cDNA synthesis kit (Bio-Rad, The Netherlands). To validate the correct sequence, CXorf56 cDNA products were Sanger sequenced. Expression of CXorf56 was normalized to the reference gene GUSB [23]. Primers are indicated in Table S3. Samples were run as triplicates on Bio Rad CFX96 Real Time System. Data was analyzed using CFX Manager Software V1.5 (Bio-Rad). Statistical analysis using GUSB/CXorf56 ∆Ct values from each sample was done using ANOVA and TUKEY HSD.
Nonsense-mediated mRNA decay (NMD)
To study the possible presence of NMD [24, 25], EBV-transformed lymphoblasts were cultured with and without treatment of cycloheximide (100 μg/ml for 8 h) before mRNA isolation and cDNA synthesis.
Immunohistochemistry
Wild-type male c57/bl6 mice (8 weeks old) were housed under standard conditions. Brains were dissected immediately after cervical dislocation and fixed overnight in 4% paraformaldehyde. Subsequently, brains were embedded in paraffin according to standard protocols. Sections (7 µm) were cut and mounted on coated slides. Immunostainings were performed with rabbit antibodies against Cxorf56 protein (Abgent, San Diego, CA, AP5213a) followed by a three-step immunoperoxidase protocol [26]. Animal experiments were carried out with permission of the local ethics committee.
Expression vectors and primary hippocampal neuron culture
Complete human CXorf56 cDNA (NCBI NM_022101.3), encompassing start and stop sequences (total 838 bp), was obtained by RT-PCR of total RNA isolated from EBV-transformed cell line, using the Pfu DNA polymerase (Stratagene, La Jolla, USA). Primers used: forward (5′-TTCGGGGCGACCCTCTTC-3′) and reverse (5′-ACAACGTAAAGCCTTCTGCTAGGG-3′).
The product was cloned into the pCR®2.1-TOPO® (Invitrogen, The Netherlands). Sequences of the construct were verified by Sanger sequencing. The CXorf56 amplified product was excised by digestion with the restriction enzyme EcoRI and cloned in frame into the EcoRI site of the β-actin-GFP or β-actin-mCherry vector. Sanger sequencing was used to validate the sequence and to determine the right orientation.
Hippocampi were dissected from E17 wildtype c57/bl6 mouse brain and placed in DMEM (Gibco, BRL). Primary hippocampal neurons were further cultured as described [27]. After 14 days in vitro, the hippocampal neurons were transfected with the CXorf56 construct using Lipofectamine 2000 (Invitrogen). One day after transfection, cells were fixed with 4% paraformaldehyde in PBS, washed in PBS, and mounted in Mowiol 4–88 solution.
Results
Clinical features
The study includes 25 individuals from one large family (Fig. 1). Five males and one female were affected with mild to moderate ID, consistent with an X-linked inheritance pattern.
Fig. 1.

The four-generation XLID pedigree and haplotype analysis results on Xq24. The disease-linked haplotype is depicted in black. Haplotypes are indicated with X-chromosome STR markers. The proximal boundary is determined by a recombination event in individual III-3 between DXS8055 and DXS424. The distal border is determined by both a recombination event in individual IV-5 and a historical recombination in IV-7 and IV-8 that was already present in III-12 between DXS8067 and DXS8059. An arrow indicates the location of the CXorf56 gene. Black symbols represent XLID impaired individuals, open symbols represent unaffected individuals, dotted symbols represent unaffected carriers, diagonal slash means deceased
II-4 had ID (IQ below 70 on a Raven’s IQ test [28], no other IQ test is available). In his childhood he had convulsions. He worked in a sheltered workplace. Physical examination at the age of 56 years showed a long and narrow face, prominent chin, upward slant of the palpebral fissures, flat philtrum with narrow upper lip and large, everted ears with prominent cruxes.
II-5 had delayed development from early childhood. No IQ test is available. In adulthood, he worked at a sheltered workplace. Physical examination at the age of 70 years showed mild short stature (−3SD), a long narrow face, prominent chin, large ears with prominent cruxes, slightly upward slant of the palpebral fissures, and a flat philtrum with narrow upper lip.
III-3 was mildly intellectually disabled with aberrant adaptive behavior, a pervasive developmental disorder, bad articulation, and no abilities for reading or writing. He attended special education. IQ test (WAIS [29]) showed a total IQ of 53. On physical examination at age of 32 years he had height and head circumference at 5th centile, a long face with a prominent mandible, an abnormal crux superior, and upward slant of the palpebral fissures.
IV-5 was born by cesarean section. His development was delayed, with a pervasive developmental disorder, attention disorder, hyperactivity, and transgressive behavior. A personality disorder with narcistic features was established. He was institutionalized since 6 years of age. KAIT intelligence test [30] revealed a total IQ of 55. He attended special education. On physical examination at 13 years of age, an adipose boy was seen with a broad non-prominent chin and large ears for his age.
IV-7 had a mild developmental delay and attended special education. No IQ test is available. She worked on a sheltered work center. Physical examination at age 32 showed a long face.
IV-8 had delayed motor and intellectual development. His speech is slow and unintelligible and he is very anxious. At 5 years of age his IQ was assessed at <55 at the Pediatric Psychiatry Department of the Public Health Service of Rotterdam. There is no official IQ test available from a later age. He attended special education and worked at a sheltered work center. On physical examination at 20 years of age, he had a long face and a high broad forehead.
Linkage analysis
Multipoint linkage analysis using X-linked micro satellite markers resulted in one linkage peak at Xq23-24 with a significant LOD score of 3.3 for the marker DXS1001 (Fig. S1). Initially the borders of the linked region were determined by the markers DXS8055 and DXS1047, a region of 22.7 cM (https://www.bli.uzh.ch/BLI/Projects/genetics/maps/marsh.html). Haplotype analysis and fine mapping using four extra STR markers (DXS424, DXS8064, DXS8067, and DXS8059) confirmed an X-linked segregating area and defined a 7.6 cM candidate region between marker DXS8055 and DXS8059 (Fig. 1). The region corresponds with 7.6 Mb and contained 92 genes, including 6 hypothetical LOC genes and 33 pseudogenes (NCBI build 37.2).
WGS analysis
WGS was performed on DNA of three affected males (II-4, III-3, IV-8) and one unaffected male (IV-2) with average coverage of 82–101× (Table S1 for WGS quality metrics). In total 6,128,979 variants were detected, including single-nucleotide variants and small insertions, deletions and substitutions of up to about 50 bp. After filtering for the X-chromosome 145,736 variants remained. Of these, 8019 variants (both SNVs and indels) were located in the linkage region (build 36: 114.561.198–122.091.092 bp, equivalent to build 37: 114.654.942–122.263.411 bp) and 2950 variants were present in the male patients and absent in the unaffected male. After selecting protein affecting variants only (non-synonymous, exonic, and splice site variants), only four variants remained. Three of the variants (all missense variants) were present in dbSNP130 and in public databases with a high frequency. Only one exonic variant in the CXorf56 gene was left, consisting of a 2 bp TA insertion in exon 2, resulting in a frameshift and premature stop codon in the first quarter of the protein (c.159_160insTA; p.(Asp54*); Fig. S2).
Variant validation of CXorf56 in family members
Sanger sequencing of the CXorf56 gene in all 25 participating family members revealed the insertion in the five affected males, the affected female (IV-7) and the eight unaffected carrier females (II-2, II-7, III-1, III-2, III-9, III-12, III-14, and IV-9) (Fig. S3).
The c.159_160insTA variant is not reported in the 1000 genome database (https://www.1000genomes.org), the Exome Variant Server of the NHLBI Exome Sequencing Project (ESP), Seattle, WA (June 2016, https://snp.gs.washington.edu/EVS/) or the ExAC browser (https://exac.broadinstitute.org/). Sanger sequencing of all CXorf56 exons in a cohort of 188 healthy Dutch individuals (94 males, 94 females) did not identify the TA insertion, nor did it identify any other novel pathogenic variants [31]. Additionally, sequencing of all CXorf56 exons in a large cohort of sporadic individuals with ID (222 males, 191 females) from eight different care and special education centers in the Netherlands revealed no nonsense, missense, or splice variants. Finally, four other XLID families with a linkage area in chromosome Xq24 did not have pathogenic variants in CXorf56 (data not shown).
Conservation of CXorf56 (previously known as LOC63932 and FLJ22965)
CXorf56 consists of seven exons and is highly conserved among several species (Fig. S4). Although this gene is well supported by transcript data, no functional information on its protein products is currently available. Four valid protein coding transcripts of different length have been described, varying from 904 to 2372 bp (Ensembl build 37, https://www.ensembl.org) with predicted protein sizes of 173 and 222 aminoacids (Fig. S5). No functional information is currently available in databases and conserved domains are not found. The insertion is affecting all four transcripts and both proteins. The ExAC database score (pLI) for intolerance of loss-of-function for CXorf56 is extremely high (0.93) meaning that the number of observed variants is very low compared to the expected LoF variants (60,706 samples). Also in the Exome Variant Server (NHLBI, GO ESP) database no splice site, nonsense or frameshift variants are reported in CXorf56 (6500 samples). This means that the probability of a LoF variant in this gene in general populations is very unlikely. An intronless pseudogene of CXorf56, LOC728024 (669 bp), without a translated protein, is located on chromosome 8p11.23. Primers used for sequencing the X-chromosome located CXorf56 do not match the sequences of the pseudogene.
X-chromosomal inactivation status
X-chromosomal alleles were distinguished by the highly polymorphic CAG repeat in the human androgen-receptor gene [22] in all family members. The polymorphic CAG repeat was heterozygous and informative in six unaffected female carriers (III-1, III-2, III-9, III-12, III-14, and IV-9), but not informative in the affected female (IV-7). Methylation status of all six unaffected carrier females showed the maternal allele containing the variant to be inactivated for 76–93% (Table S4). The X-chromosomes in the affected female did not show a skewed inactivation pattern (54%), likely resulting in increased levels of mutant CXorf56 mRNA levels compared to the unaffected females (Table S4, Figs. S6, S7).
Expression levels of CXorf56 in EBV-transformed lymphoblasts
Messenger RNAs containing premature stop codons are generally targeted for degradation through NMD. Cycloheximide interferes with NMD through inhibition of protein synthesis and accumulation of mutated mRNA [24, 25]. In order to determine relative gene expression levels of CXorf56 and the possible contribution of mutated mRNA degradation through NMD, accounting for low mRNA levels in patients, quantification RT-qPCR data of relative gene expression were compared for EBV cell lines grown with and without cycloheximide (Fig. 2) [24, 25, 32]. mRNA levels of CXorf56 in EBV cell lines in the affected male (III-3) was reduced by 75% (p < 0.001) and in the affected female (IV-7) by 37% (p < 0.01), compared to the non-carrier healthy female (III-5) and the unaffected carrier female (III-2). No significant difference was detected in the levels of mRNA between the healthy carrier female (III-2) and the non-carrier female (III-5).
Fig. 2.
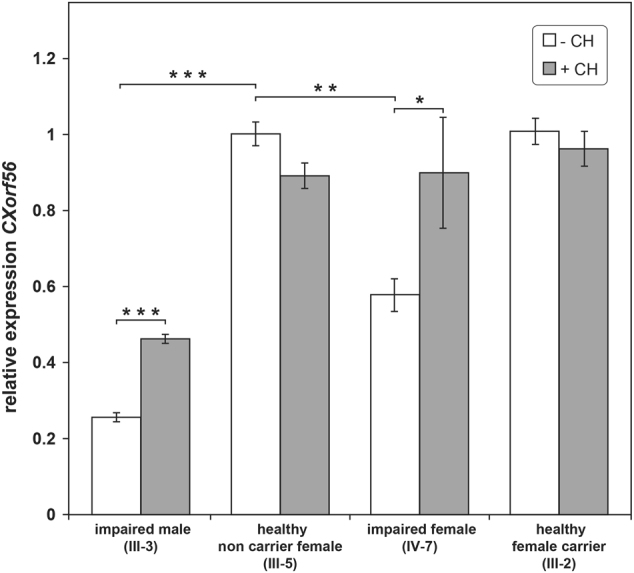
Histogram displaying results of the relative quantitative CXorf56 mRNA analysis. Mean CXorf56 mRNA level relative to the reference gene GUSB and a calibrator sample (healthy non-carrier female (III-5)) are shown (±SEM). White and gray bars represent CXorf56 expression in EBV cell lines without (−CH) and with (+CH) treatment of cycloheximide, respectively. In the cell lines from an impaired male (III-3) and impaired female (IV-7) endogenous CXorf56 expression is significantly lower. After treatment with cycloheximide expression is significantly increased. Expression levels of an unaffected female carrier (III-2) are comparable to the control sample (III-5). *p < 0.05; **p < 0.01; ***p < 0.001
Treatment with cycloheximide significantly increased the CXorf56 expression levels in the affected male (p < 0.001) and female (p < 0.05), suggesting that NMD is indeed the mechanism leading to the reduced expression level in impaired individuals. For summary of X-inactivation and expression results, see Fig. S7.
Localization studies
Immunohistochemistry staining showed endogenous CXorf56 protein to be predominantly present in the nucleus and cell soma of most neurons throughout the brain cortex and cerebellum in 8-week-old murine brain (Fig. 3a, b). Expression of CXorf56 protein was studied using transfection of full-length human CXorf56 cDNA in HEK293T cells and cultured primary hippocampal neurons. In HEK293T cells, overexpression of CXorf56 protein showed a predominant nuclear localization with exception of the nucleolus (Fig. S8). Overexpression in cultured primary hippocampal neurons showed CXorf56 protein expression in the nucleus, dendrites, and postsynaptic dendritic spines (Fig. 4a, b). Colocalization studies with Map2, a specific dendritic marker, clearly demonstrates a distribution of CXorf56 in the dendrites (Fig. 4a, b). Map2 was not present in the dendritic spines, whereas CXorf56 protein is clearly present there.
Fig. 3.
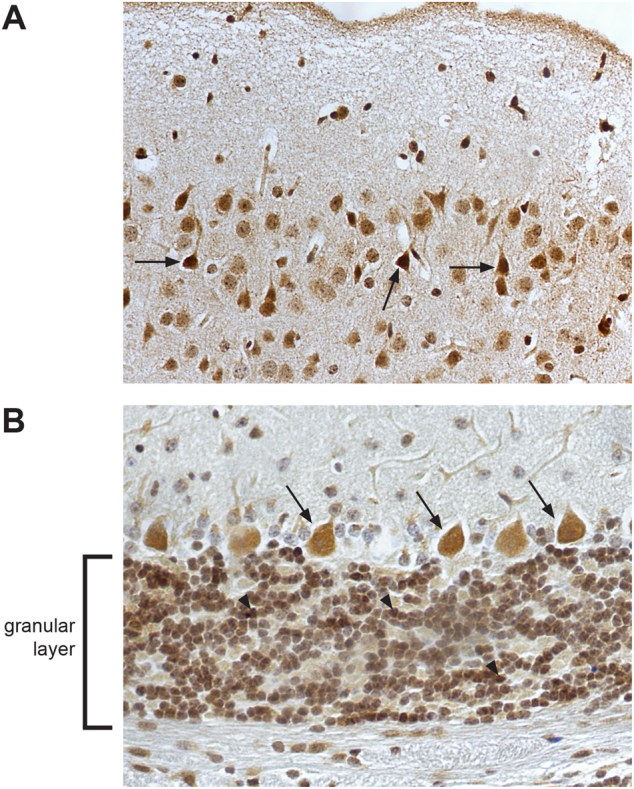
Subcellular localization of endogenous CXorf56 protein in murine brain. Immunohistochemical staining of endogenous CXorf56 protein in brain slices of 8-week-old male mice. Signal is present in the nucleus and cell soma of a pyramidal neurons in the cortex (examples indicated by black arrows) and b Purkinje cells (examples indicated by black arrows) and neurons in the granular layer (examples indicated by arrowheads) in the cerebellum. Note the nuclear as well as the cytoplasmic signal in the neurons
Fig. 4.

Localization of CXorf56 in cultured primary hippocampal neurons. a–b Primary hippocampal neuron cultures of mice E17 pups. a Colocalization of CXorf56 protein in nucleus and dendrites (red, left panel) with endogenous Map2, a dendritic marker (green, middle panel), after 14 days in vitro. Merge (right panel). b High magnification of a dendrite with spines. Note the presence of CXorf56 protein in dendritic spines (indicated with arrows)
Discussion
In this study we identified the CXorf56 gene as a new putative cause for XLID. Using linkage analysis followed by next-generation sequencing, we were able to identify a two basepair insertion in a family with mild ID and behavior problems, which is probably disease causing. Analysis of coding sequence data of the complete linkage region in four family members from three different generations, showed only one variant, a 2 bp insertion in exon 2, in the CXorf56 gene shared in three patients and absent in one related unaffected male. This insertion segregates with affected individuals and obligatory carriers and leads to a premature termination codon in exon 2.
The pathogenicity of the CXorf56 variant is supported by expression and X-inactivation studies. CXorf56 mRNA expression was decreased in affected individuals and increased after adding cycloheximide, a known NMD inhibitor [32]. This suggests NMD might be involved in the lowered expression of the mutated transcript. Although NMD also has a more general role in post-transcriptional regulation of gene expression, it is also a known translation-coupled quality-control system that recognizes and degrades aberrant mRNAs with truncated open reading frames due to the presence of a premature termination codon [24, 25]. In addition, we show skewed X-inactivation for unaffected carrier females, favoring inactivation of the affected allele in lymphocytes. In contrast, skewing was not seen in the affected female.
Although skewing of X-inactivation above 80% is suggestive of an X-linked disorder, it is sometimes also present in normal females in the general population [33]. However in the family presented here skewing is present in all six tested normal carrier females, and is a known mechanism related to the degree of severity of disease phenotype in females [33]. Presumably it results from a post-inactivation selection against cells with the mutated gene on the active X-chromosome [33–35].
Although skewing in lymphocytes is not a direct indication of the X-inactivation pattern in brain, a relation between the proportion of the wildtype active X in lymphocytes and IQ has been shown in other XLID syndromes, for example in female carriers of the fragile X syndrome [36].
The absence of LoF mutations in CXorf56 in 188 tested healthy controls and from public databases, the high pLI score of 0.93 (ExAC database), and the high conservation between species suggests that CXorf56 might have an important cellular function. To date, the family investigated in this paper is the only one described with a likely pathogenic variant in this gene. We did not find LoF of function variants in CXorf56 in other patients that we screened, including a cohort of 413 ID patients and four other ID families with overlapping linkage regions on the X-chromosome, nor in a database search or in literature. In large sequencing studies in families with ID and an X-linked pattern of transmission, no pathogenic variants have been reported in CXorf56, i.e., by Tarpey et al., who tested 208 families, and Hu et al., who tested 405 famlies with XLID [15, 18]. However, it is known that ID is extremely heterogeneous [15, 16, 37, 38] and patients and families often have their own private mutations in one of the many involved genes, each accounting for only a small number of patients with ID [16]. In addition, many ID genes are due to de novo mutations. Possibly, mutations in CXorf56 are not a frequent cause of ID either.
Very limited data is available on the function of CXorf56 protein. Unfortunately, no knock-out animal model is available, or data from knock-down experiments in cell cultures, to recapitulate the loss of CXorf56. We show endogenous CXorf56 protein expression in mouse brain cortex and cerebellar neurons in the nucleus and cell soma. In contrast, overexpression in the non-neuronal HEK293T cells shows predominantly nuclear expression. Overexpression in cultured hippocampal neurons shows expression in the nucleus, dendrites, and dendritic spines. This suggests that the CXorf56 protein might have different localization and function in different cell types. However, the overexpression might influence the subcellular localization pattern as well [39]. According to the GTEx database (https://www.gtexportal.org/home/), CXorf56 is expressed in a range of tissues including brain (Fig. S9) and our results show protein specifically in the nucleus, dendrites, and dendritic spines of neurons throughout the brain. The presence of CXorf56 protein specifically in dendritic spines might suggest a role in synaptic function [39]; however, this has to be investigated further.
Although we show CXorf56 is present in the nucleus, the protein itself has no predicted nuclear localization signal (NLS) (PredictProtein, https://www.predictprotein.org). However, CXorf56 is a binding partner of nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor-like1 (NFKBIL1). A protein–protein interaction with NFKBIL1 has been described using affinity capture mass spectrometry, where NFKBIL1 was used as a bait [40] (genBank_GI:5031194). NFKBIL1 has been implicated as a risk factor for a number of diseases [41]. CXorf56 (or FLJ22965) binds to NFKBIL1 with a high confidence score of 0.334 [40]. Interestingly, NFKBIL1 does have a NLS (PredictProtein) [42] (Fig. S10), indicating that CXorf56 might be transported to the nucleus bound to NFKBIL1. Other protein interactors are mentioned in BioGRID (https://thebiogrid.org/122000); however, what role they might play in relation to CXorf56 is at the moment unclear.
The only report that includes any information concerning function of CXorf56 is by Cox et al., who studied male mouse embryos derived from ES cells with a gene trapped allele of the mouse homolog of CXorf56 (C330007P06Rik; https://www.informatics.jax.org/image/phenoSummary/marker/MGI:1924894, https://www.informatics.jax.org/image/phenoSummary/allele/MGI:4128982) [43]. These embryos exhibited abnormal midbrain-hindbrain boundary morphology, decreased forebrain size and developmental delay. The mouse gene was expressed in brain, eyes, and neural tube [43]. This data underlines a role for CXorf56 protein in brain development. In addition, CXorf56, as well as Cactin, one of the interactors of CXorf56 (BioGRID) have been found to act as non-RNP spliceosome-associated protein in the spliceosomal complex C [44], although nothing is known about their role there. Other components of the spliceosomal complex C have been found to give rise to ID, for example a homozygous pathogenic mutation in the ZCCHC8 gene has been identified in one ID patient [45].
CXorf56 is a neighboring gene of UBE2A, a known gene involved in ID [46]. Point mutations in UBE2A lead to syndromal XLID with a broad range of other clinical features [46]. Additionally, microdeletions of Xq24, including UBE2A, CXorf56, and several other genes have been reported, leading to a similar phenotype [47, 48]. As the phenotype in the patients of the family investigated in our paper is mild and non-syndromal ID, the contribution of the presence of CXorf56 in these microdeletion patients could be small or overshadowed by the severe phenotype caused by the absence of UBE2A. However, for most microdeletion syndromes it is difficult to pinpoint one clinical feature to the absence of a specific gene in a (micro)deletion encompassing many genes [10, 49] and to determine the role of each gene independently on the phenotype.
In conclusion, we have identified CXorf56 as a new candidate XLID gene in a large family with mild ID and behavior problems. The prevalence of pathogenic variants in this gene is unknown, since no other families have been identified yet. Adding this gene to the list of (candidate) genes for ID might lead to new diagnoses in other families. CXorf56 is expressed in brain, and is specifically present in the nucleus and dendritic spines throughout the brain. Possibly, future studies might elucidate its function and the consequence of its loss for development.
Electronic supplementary material
Acknowledgements
We thank the family for their continuous cooperation, their endless patience, perseverance, and trust. We thank Diana van Rongen-Nispen and Lorette Dubbel-Hulsman for cell culture, Dicky Halley and Hans van Bokhoven for availability of DNA samples, and Edith Friesema and Theo Visser for supporting the project. Tom de Vries Lentsch is acknowledged for excellent graphical support. We thank Willem Verhoeven and collaborators of the Vincent van Gogh institute for performing IQ tests of family members.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (10.1038/s41431-017-0051-9) contains supplementary material, which is available to authorized users.
References
- 1.Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18. doi: 10.1038/nrg3999. [DOI] [PubMed] [Google Scholar]
- 2.Willemsen MH, Kleefstra T. Making headway with genetic diagnostics of intellectual disabilities. Clin Genet. 2014;85:101–10. doi: 10.1111/cge.12244. [DOI] [PubMed] [Google Scholar]
- 3.Gecz J, Shoubridge C, Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 2009;25:308–16. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Lubs HA, Stevenson RE, Schwartz CE. Fragile X and X-linked intellectual disability: four decades of discovery. Am J Hum Genet. 2012;90:579–90. doi: 10.1016/j.ajhg.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genom Hum Genet. 2010;11:161–87. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 6.Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- 7.Kaufman L, Ayub M, Vincent JB. The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord. 2010;2:182–209. doi: 10.1007/s11689-010-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Vries BB, Winter R, Schinzel A, van Ravenswaaij-Arts C. Telomeres: a diagnosis at the end of the chromosomes. J Med Genet. 2003;40:385–98. doi: 10.1136/jmg.40.6.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rooms L, Vandeweyer G, Reyniers E, et al. Array-based MLPA to detect recurrent copy number variations in patients with idiopathic mental retardation. Am J Med Genet A. 2011;155a:343–8. doi: 10.1002/ajmg.a.33810. [DOI] [PubMed] [Google Scholar]
- 10.Vissers LE, de Vries BB, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet. 2010;47:289–97. doi: 10.1136/jmg.2009.072942. [DOI] [PubMed] [Google Scholar]
- 11.Whibley AC, Plagnol V, Tarpey PS, et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet. 2010;87:173–88. doi: 10.1016/j.ajhg.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verkerk AJ, Schot R, Dumee B, et al. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. 2009;85:40–52. doi: 10.1016/j.ajhg.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Musante L, Ropers HH. Genetics of recessive cognitive disorders. Trends Genet. 2014;30:32–9. doi: 10.1016/j.tig.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet. 2012;57:621–32. doi: 10.1038/jhg.2012.91. [DOI] [PubMed] [Google Scholar]
- 15.Tarpey PS, Smith R, Pleasance E, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–43. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–12. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 17.Kuhlenbaumer G, Hullmann J, Appenzeller S. Novel genomic techniques open new avenues in the analysis of monogenic disorders. Hum Mutat. 2011;32:144–51. doi: 10.1002/humu.21400. [DOI] [PubMed] [Google Scholar]
- 18.Hu H, Haas SA, Chelly J, et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry. 2016;21:133–48. doi: 10.1038/mp.2014.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet. 2000;25:12–3. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 20.Lindner TH, Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–7. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 21.Drmanac R, Sparks AB, Callow MJ, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 22.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–39. [PMC free article] [PubMed] [Google Scholar]
- 23.de Brouwer AP, van Bokhoven H, Kremer H. Comparison of 12 reference genes for normalization of gene expression levels in Epstein-Barr virus-transformed lymphoblastoid cell lines and fibroblasts. Mol Diagn Ther. 2006;10:197–204. doi: 10.1007/BF03256458. [DOI] [PubMed] [Google Scholar]
- 24.Popp MW, Maquat LE. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell. 2016;165:1319–22. doi: 10.1016/j.cell.2016.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schweingruber C, Rufener SC, Zund D, Yamashita A, Muhlemann O. Nonsense-mediated mRNA decay—mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta. 2013;1829:612–23. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Seelaar H, Schelhaas HJ, Azmani A, et al. TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain. 2007;130:1375–85. doi: 10.1093/brain/awm024. [DOI] [PubMed] [Google Scholar]
- 27.de Vrij FM, Levenga J, van der Linde HC, et al. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–32. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raven J. The Raven’s progressive matrices: change and stability over culture and time. Cogn Psychol. 2000;41:1–48. doi: 10.1006/cogp.1999.0735. [DOI] [PubMed] [Google Scholar]
- 29.Nelson JM, Canivez GL, Watkins MW. Structural and incremental validity of the Wechsler Adult Intelligence Scale-Fourth Edition with a clinical sample. Psychol Assess. 2013;25:618–30. doi: 10.1037/a0032086. [DOI] [PubMed] [Google Scholar]
- 30.Kaufman AS, Horn JL. Age changes on tests of fluid and crystallized ability for women and men on the Kaufman Adolescent and Adult Intelligence Test (KAIT) at ages 17-94 years. Arch Clin Neuropsychol. 1996;11:97–121. [PubMed] [Google Scholar]
- 31.Collins JS, Schwartz CE. Detecting polymorphisms and mutations in candidate genes. Am J Hum Genet. 2002;71:1251–2. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noensie EN, Dietz HC. A strategy for disease gene identification through nonsense-mediated mRNA decay inhibition. Nat Biotechnol. 2001;19:434–9. doi: 10.1038/88099. [DOI] [PubMed] [Google Scholar]
- 33.Fieremans N, Van Esch H, Holvoet M, et al. Identification of intellectual disability genes in female patients with a skewed X-inactivation pattern. Hum Mutat. 2016;37:804–11. doi: 10.1002/humu.23012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orstavik KH, Orstavik RE, Naumova AK, et al. X chromosome inactivation in carriers of Barth syndrome. Am J Hum Genet. 1998;63:1457–63. doi: 10.1086/302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willard HF. X chromosome inactivation and X-linked mental retardation. Am J Med Genet. 1996;64:21–6. doi: 10.1002/(SICI)1096-8628(19960712)64:1<21::AID-AJMG2>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 36.de Vries BB, Wiegers AM, Smits AP, et al. Mental status of females with an FMR1 gene full mutation. Am J Hum Genet. 1996;58:1025–32. [PMC free article] [PubMed] [Google Scholar]
- 37.de Brouwer AP, Yntema HG, Kleefstra T, et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum Mutat. 2007;28:207–8. doi: 10.1002/humu.9482. [DOI] [PubMed] [Google Scholar]
- 38.Tzschach A, Grasshoff U, Beck-Woedl S, et al. Next-generation sequencing in X-linked intellectual disability. Eur J Hum Genet. 2015;23:1513–8. doi: 10.1038/ejhg.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tahirovic S, Bradke F. Neuronal polarity. Cold Spring Harb Perspect Biol. 2009;1:a001644. doi: 10.1101/cshperspect.a001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ewing RM, Chu P, Elisma F, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An J, Nakajima T, Shibata H, Arimura T, Yasunami M, Kimura A. A novel link of HLA locus to the regulation of immunity and infection: NFKBIL1 regulates alternative splicing of human immune-related genes and influenza virus M gene. J Autoimmun. 2013;47:25–33. doi: 10.1016/j.jaut.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 42.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–6. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox BJ, Vollmer M, Tamplin O, et al. Phenotypic annotation of the mouse X chromosome. Genome Res. 2010;20:1154–64. doi: 10.1101/gr.105106.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agafonov DE, Deckert J, Wolf E, et al. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol. 2011;31:2667–82. doi: 10.1128/MCB.05266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 46.Nascimento RM, Otto PA, de Brouwer AP, Vianna-Morgante AM. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am J Hum Genet. 2006;79:549–55. doi: 10.1086/507047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–9. doi: 10.1016/S0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- 48.Thunstrom S, Sodermark L, Ivarsson L, Samuelsson L, Stefanova M. UBE2A deficiency syndrome: a report of two unrelated cases with large Xq24 deletions encompassing UBE2A gene. Am J Med Genet A. 2015;167a:204–10. doi: 10.1002/ajmg.a.36800. [DOI] [PubMed] [Google Scholar]
- 49.Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–46. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.