

ABSTRACT
Microbes in biofilms face the challenge of substrate limitation. In particular, oxygen often becomes limited for cells in Pseudomonas aeruginosa biofilms growing in the laboratory or during host colonization. Previously we found that phenazines, antibiotics produced by P. aeruginosa, balance the intracellular redox state of cells in biofilms. Here, we show that genes involved in denitrification are induced in phenazine-null (Δphz) mutant biofilms grown under an aerobic atmosphere, even in the absence of nitrate. This finding suggests that resident cells employ a bet-hedging strategy to anticipate the potential availability of nitrate and counterbalance their highly reduced redox state. Consistent with our previous characterization of aerobically grown colonies supplemented with nitrate, we found that the pathway that is induced in Δphz mutant colonies combines the nitrate reductase activity of the periplasmic enzyme Nap with the downstream reduction of nitrite to nitrogen gas catalyzed by the enzymes Nir, Nor, and Nos. This regulatory relationship differs from the denitrification pathway that functions under anaerobic growth, with nitrate as the terminal electron acceptor, which depends on the membrane-associated nitrate reductase Nar. We identified the sequences in the promoter regions of the nap and nir operons that are required for the effects of phenazines on expression. We also show that specific phenazines have differential effects on nap gene expression. Finally, we provide evidence that individual steps of the denitrification pathway are catalyzed at different depths within aerobically grown biofilms, suggesting metabolic cross-feeding between community subpopulations.
IMPORTANCE An understanding of the unique physiology of cells in biofilms is critical to our ability to treat fungal and bacterial infections. Colony biofilms of the opportunistic pathogen Pseudomonas aeruginosa grown under an aerobic atmosphere but without nitrate express a denitrification pathway that differs from that used for anaerobic growth. We report that the components of this pathway are induced by electron acceptor limitation and that they are differentially expressed over the biofilm depth. These observations suggest that (i) P. aeruginosa exhibits “bet hedging,” in that it expends energy and resources to prepare for nitrate availability when other electron acceptors are absent, and (ii) cells in distinct biofilm microniches may be able to exchange substrates to catalyze full denitrification.
KEYWORDS: nitrate reductase, nitrite reductase, Nap, Nir, RpoS, Anr, denitrification, biofilm physiology
INTRODUCTION
The opportunistic pathogen Pseudomonas aeruginosa readily forms biofilms, communities of cells encased in self-produced matrices (1). These crowded environments are characterized by the formation of chemical gradients and microniches that uniquely affect cellular physiology. We have used a colony morphology assay to study the pathways of metabolic electron flow in biofilms of P. aeruginosa PA14. In this system, oxygen becomes limited for cells at depth in the biofilm due to its consumption by those closer to the periphery. Oxidant limitation promotes colony wrinkling, which increases the access to oxygen in the atmosphere for resident cells (2). The production of phenazines, endogenous antibiotics that can shuttle electrons to other oxidants, balances the intracellular redox state and inhibits colony wrinkling. Amendment of the growth substrate with nitrate, which is an alternate electron acceptor for P. aeruginosa metabolism, also induces both of these effects (3, 4).
Pseudomonad nitrate metabolism has been studied extensively using well-mixed liquid cultures. These bacteria have three nitrate reductases: Nas, which enables Pseudomonas spp. to use nitrate as a nitrogen source (5); Nap, a periplasmic enzyme that is expressed during the stationary phase of growth in aerobic cultures (6–9); and Nar, a membrane-associated enzyme that is expressed during anaerobic growth with nitrate as the sole electron acceptor (8–10). During anaerobic growth by nitrate respiration, Nar works with the enzymes Nir, Nor, and Nos to catalyze denitrification, the full reduction of nitrate to nitrogen gas. The activities of transcription factors regulating the expression of these enzymes are controlled so that the pathways function under anoxic conditions and in the presence of nitrogenous oxides. These regulators include Anr, which is thought to respond to the shift from oxic to anoxic conditions (11–13), Dnr, which responds to nitric oxide (12, 14), and the two-component system NarXL, which responds to nitrate (15) (see Fig. 2A).
FIG 2.
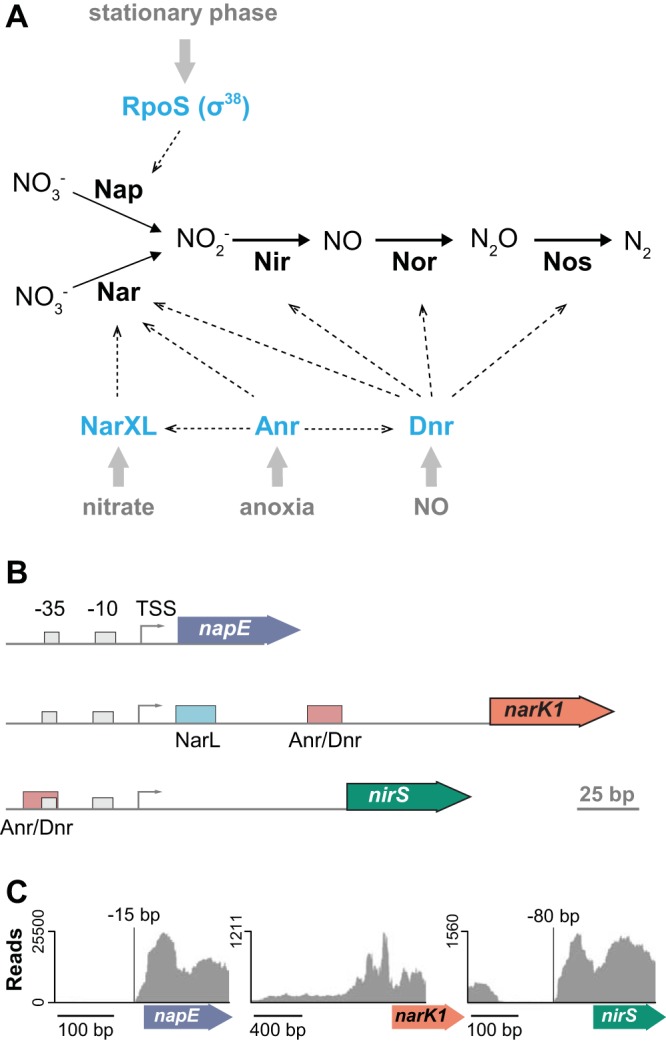
Transcriptional regulation of genes involved in denitrification. (A) Model showing the enzymes that catalyze each step of the denitrification pathway, the regulators (in blue) that control their expression, and the conditions (in gray) that influence their activities. (B) Cartoons depicting the promoters and loci encoding, from top to bottom, the periplasmic nitrate reductase Nap, the membrane-associated nitrate reductase Nar, and the nitrite reductase Nir. TSS, transcriptional start site. NarL and Anr/Dnr transcription factor binding sites are indicated by colored squares. (C) Graphical representation of RNA-seq results obtained for mRNA samples collected from Δphz mutant colonies grown without nitrate. The y axes represent read coverage for each location in the transcript. The protein-coding regions of napE, narK1, and nirS are represented by colored arrows under the x axes. napE and nirS show well-defined transcriptional start sites relative to the start codon.
In our prior studies of nitrate utilization by colony biofilms of strain PA14, we found that Nap, Nir, and Nor, but not Nar, were required for nitrate-dependent repression of colony wrinkling under aerobic conditions (4). This result suggested that the unique conditions of biofilms induced a Nap-dependent denitrification pathway. While such a pathway had been shown to support anaerobic growth in P. aeruginosa suppressor mutants (6, 16), we were intrigued by the possibility that it could operate naturally in wild-type biofilms. In the present study, we conducted a proteomic analysis comparing wild-type and phenazine-null mutant colony biofilm samples. Though these colonies were grown aerobically and in the absence of nitrate, we observed that Nir and Nos were among the proteins showing the greatest increases in abundance in the phenazine-null mutant relative to that in phenazine producers. Here, we describe our characterization of redox-driven regulation of strain PA14 loci involved in denitrification. As P. aeruginosa biofilms formed during chronic pulmonary colonization often have access to nitrate (17), these results can inform models for the utilization of redox metabolisms in this environment.
RESULTS AND DISCUSSION
Enzymes involved in denitrification are induced in phenazine-null biofilms.
Several years ago, we discovered that phenazine production profoundly affects the morphogenesis of PA14 colony biofilms (18, 19) and oxidizes the cellular redox state for oxygen-limited cells (3, 4). Early in colony biofilm development, a defect in phenazine production leads to a more reduced cellular redox state, which is followed by an induction of biofilm matrix production and colony wrinkling (20). Colony wrinkling increases the colony surface area and access to oxygen for the cells within (2), ultimately leading to a more oxidized (i.e., wild-type-like) cellular redox state at later stages of development (4). Colony wrinkling and phenazine-mediated electron shuttling are alternate strategies that can serve to balance the intracellular redox state for oxidant-limited cells in colonies.
We sought to investigate the physiological changes associated with phenazine production during biofilm development and analyzed the proteomes of colonies formed by the wild type, the Δphz mutant, which is unable to produce any phenazines, and a mutant called “BigBlue,” which overproduces 5-methylphenazine-1-carboxylate (5-Me-PCA) and pyocyanin (Fig. 1A) (18). These methylated phenazines strongly impact cellular redox state, because they have higher oxidizing potentials than other P. aeruginosa phenazines that have been characterized (Fig. 1A; see also Fig. S1 in the supplemental material) (21–23). We harvested three colonies of each strain after 36 h of growth, which typically marks the onset of wrinkling by the phenazine-null mutant (4). High-resolution mass spectrometry detected an average of 2,465 (standard deviation [SD], 96) individual proteins for each sample, representing ∼41% of the P. aeruginosa proteome. We used hierarchical clustering to group the proteins according to their abundance differences between strains and identified two clusters in which protein abundance was either correlated (“cluster 1”) or anticorrelated (“cluster 2”) with the production of methylated phenazines (Fig. 1B; see also Table S1). We were particularly intrigued to find an inverse correlation between methylated phenazine production and the abundance of denitrification enzymes, even though the growth medium for our colony biofilm samples did not contain nitrate and colonies were grown under an oxic atmosphere (Fig. 1C and E).
FIG 1.

Inverse correlation between the production of phenazines and denitrification enzymes in strain PA14 colonies. (A) The phenazine biosynthetic pathway. PCA, phenazine-1-carboxylic acid; 5-Me-PCA, 5-methyl-PCA; PCN, phenazine-1-carboxamide; PYO, pyocyanin. Redox potentials, E1/2 at pH 7 (mV/NHE), of the phenazines are in parentheses (21, 22). (B) Left: representative images of BigBlue, WT, and Δphz strains grown for 3 days in the colony morphology assay (medium: 1% tryptone, 1% agar containing the dyes Congo red and Coomassie blue). Center: heat map generated from proteomic data obtained for BigBlue, WT, and Δphz mutant colonies. Rows and columns represent independent samples (3 biological replicates for each strain) and proteins, respectively. Red indicates higher protein abundance, while blue indicates lower protein abundance. The dendrogram represents results from hierarchical clustering and depicts similarities (Pearson correlation) between protein levels under the tested conditions. Cluster 1 contains 209 proteins that were upregulated with increasing production of methylated phenazines, while cluster 2 contains 284 proteins that were downregulated with increasing production of methylated phenazines (see Table S1 in the supplemental material). Right: percentages of proteins in cluster 1 and cluster 2 that perform functions falling into the listed categories. (C) Average changes in protein levels for each of the indicated complexes when WT and Δphz colony samples are compared to those collected for BigBlue. Error bars represent standard deviations from biological replicates. (D) Top left: NADH/NAD+ over time for colony biofilms harvested at the indicated time points. Error bars represent the standard deviations from biological triplicates. Bottom left: representative images of colonies grown in parallel with those subjected to NADH/NAD+ measurement and harvested for the proteomics analysis time course. Right: mass spectrometry signal intensities for NirS and NosZ detected in WT and Δphz mutant samples taken over the course of colony development. (E) Graphical representation of chromosomal loci encoding each of the complexes involved in denitrification.
We previously reported that the cellular redox potential in Δphz mutant colonies becomes more reducing (i.e., they have a higher NADH/NAD+ ratio) after 2 days of growth compared to that of wild-type colonies. This condition leads to an increased production of extracellular matrix, which in turn causes the characteristic hyperwrinkling phenotype (4). Given these dynamics, we hypothesized that the induction of denitrification enzymes in the Δphz mutant would follow the increase in NADH/NAD+, and we compared the proteomes of wild-type and Δphz strains over the course of colony development. We grew three colonies of each strain in parallel and harvested them at 36-, 39-, 42-, and 54-h time points. Each colony sample was split so that the NADH/NAD+ ratio could be assessed for the same colonies that would be subjected to proteomic analysis. As described previously, we found that NADH/NAD+ for cells in wild-type colonies remained fairly constant over time (4). However, for Δphz mutant colonies, NADH/NAD+ showed an increase between 36 and 39 h that coincided with the onset of colony wrinkling and gradually decreased to a wild-type level as colony wrinkling continued over the next day (Fig. 1D). In the proteome analysis, the membrane-associated nitrate reductase Nar, the periplasmic nitrate reductase Nap, and the nitric oxide reductase Nor were below the detection limit or detected only at low levels in our samples. However, subunits of the Nir and Nos complexes showed higher levels or increasing levels over time in the Δphz mutant, while remaining relatively constant in the wild type (Fig. 1D; see also Table S2), suggesting a regulatory link to the redox state dynamics in the Δphz mutant. As the expression of the nir and nos operons is known to be regulated by Anr and Dnr (9) (Fig. 2A), previous studies have focused on their induction in response to shifts from oxic to anoxic conditions and in response to nitrogenous oxides (15, 17, 24). Our results suggest that an altered phenazine availability can also affect the expression of these enzymes.
We chose to follow up on our proteomics results by analyzing the effects of phenazines on the expression of denitrification genes at the transcriptional level. To design strains that report denitrification gene expression, we first carried out transcriptome sequencing (RNA-seq) to identify the transcriptional start sites (TSSs) upstream of the nap, nir, and nar loci. The schematics of these promoter regions are shown in Fig. 2B. RNA-seq was conducted on biofilm samples grown in the absence of nitrate. The RNA-seq results showed that the nap and nir operons have defined TSSs at bp −15 and −80, respectively, relative to their start codons. In contrast, the nar operon did not show a defined TSS, likely due to the absence of nitrate, which is required for the induction of nar expression by the transcription factor NarXL (15) (Fig. 2C). We note that a previous study (16) reported a TSS for the nap operon in anaerobically grown P. aeruginosa PAO1 at bp −118 relative to the start codon. The difference between our TSS analysis and that by Van Alst et al. (16) may indicate alternative TSSs that operate under growth- or strain-specific conditions. Taking this information regarding the nap and nir TSSs into account, we engineered strains that express green fluorescent protein (GFP) under the control of the promoter sequences for each of these loci. For the corresponding reporter strain for the nar locus, we used a conservative 800-bp region upstream of the TSS.
When we grew the GFP reporter strains in aerobic, shaken liquid cultures, we observed a strong induction of nap in stationary phase, with greater expression in the Δphz mutant than in the wild type. Under this condition, we saw little or no expression of nar, nir, nor, and nos (Fig. 3A). To examine the effect of oxygen limitation on expression, we repeated this experiment with cultures that were covered with mineral oil. Under these microaerobic conditions (Fig. 3B), we once again observed stationary-phase expression of nap, which was further enhanced in the absence of phenazines, and little or no expression of nar. However, we also observed an induction of nir, nor, and nos in stationary phase. Both nir and nos showed further increases in expression in the Δphz background; the expression levels of nor were too low to determine whether phenazine deficiency had a significant effect at the transcriptional level (Fig. 3B). The profiles provided by our transcriptional reporters show that nap expression is influenced by phenazines or the cellular redox state and confirm that this dependence is also the case for the nir and nos operons. It has been reported that the expression of the periplasmic nitrate reductase Nap is controlled by the sigma factor RpoS (25) (Fig. 2B). Interestingly, both the Nap and Nir/Nos portions of the denitrification pathway are similarly influenced by phenazines, as they are known to be controlled by different regulators (i.e., RpoS versus Anr/Dnr) (Fig. 2A) and show different expression dynamics over time in liquid culture (i.e., a relatively gradual versus a relatively abrupt induction) (Fig. 3).
FIG 3.
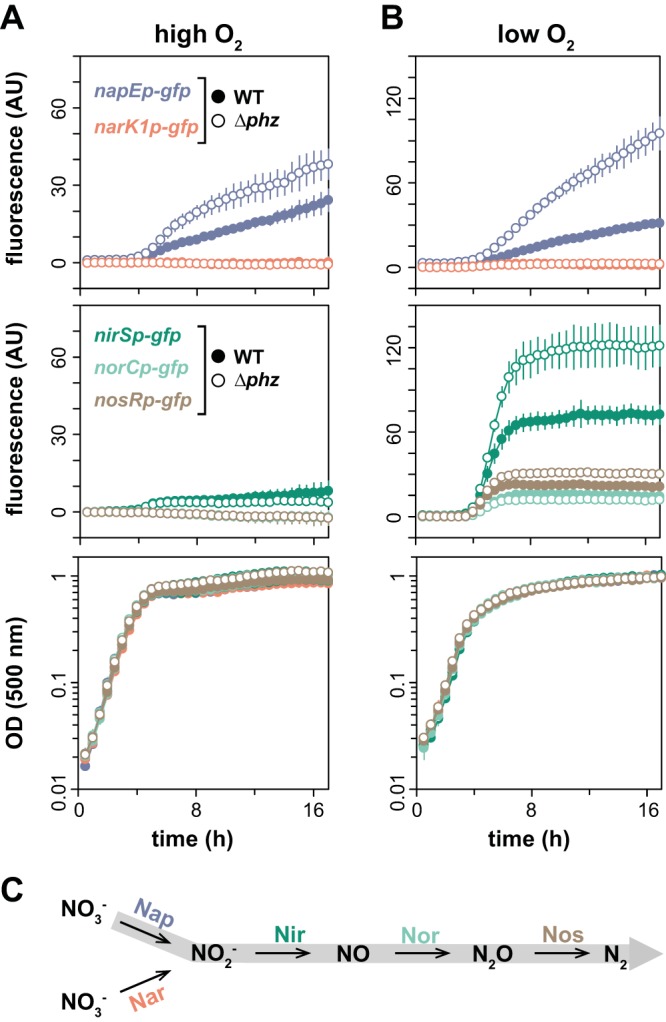
Phenazine deficiency induces a Nap-dependent denitrification pathway, particularly under low-oxygen conditions. (A) Expression of denitrification genes, as reported by promoter-gfp fusions, in shaken liquid cultures (1% tryptone medium). (B) Expression of denitrification genes, as reported by promoter-gfp fusions, in shaken liquid cultures (1% tryptone medium) covered with mineral oil to limit oxygen availability. Corresponding growth curves are shown in the bottom panels. Error bars represent the standard deviations from biological triplicates. (C) Graphical representation of the Nar- and Nap-dependent denitrification pathways. The Nap-dependent pathway operating in colonies grown under an oxic atmosphere is shaded gray.
As nitrate has been shown to induce the expression of genes involved in denitrification (15, 26), we next tested whether it affected expression in the context of phenazine deficiency. We did not observe effects on nap expression when nitrate was added to oxygen-limited liquid cultures (Fig. 4A). However, we found that nitrate induced nar expression in both the wild-type and Δphz backgrounds and revealed a phenazine-dependent enhancement of nar expression under this condition (Fig. 4B). We also found that nitrate strongly induced nir expression, though this effect was specific to the Δphz background (Fig. 4C).
FIG 4.

Transcription of the nar and nir operons is induced by nitrate, whereas transcription of the nap operon is not strongly affected by nitrate. Expression of the nap (A), nar (B), and nir (C) loci, as reported by promoter-gfp fusions, in shaken liquid cultures (1% tryptone with or without 40 mM KNO3) covered with mineral oil. Expression is reported as fluorescence and shown in the top panels. Corresponding growth curves are shown in the bottom panels. Error bars represent standard deviations from biological triplicates.
Specific phenazine deficiencies induce Nap expression.
In a previous study, we observed that specific phenazines differentially affect colony morphology and the expression of the mexGHI-opmD operon (“mex”), the latter of which is regulated by the redox-sensing transcription factor SoxR and encodes a phenazine transporter (23) (Fig. 5A and B). We had evaluated this using mutants with disruptions in the phenazine biosynthetic pathway and found that the production of phenazine-1-carboxylate (PCA), a phenazine with a relatively low redox potential, moderately induces mex, while the production of 5-Me-PCA, which has a much higher redox potential, yields a strong induction of mex. To test whether the effect on nap expression was specific to individual phenazines, we examined reporter fluorescence in these mutants. We found that the ability to produce PCA leads to a moderate decrease in nap expression relative to that in the Δphz mutant, but that the production of 5-Me-PCA is required for the repression of nap to the expression level seen in the wild type (Fig. 5C). This pattern of nap operon repression in response to specific phenazine deficiencies is the inverse of the pattern of mex induction in the same strains. This result suggests that a regulator acts either to enhance nap expression in response to a reduced cellular redox state or to repress nap expression in response to phenazines or an oxidized cellular redox state.
FIG 5.
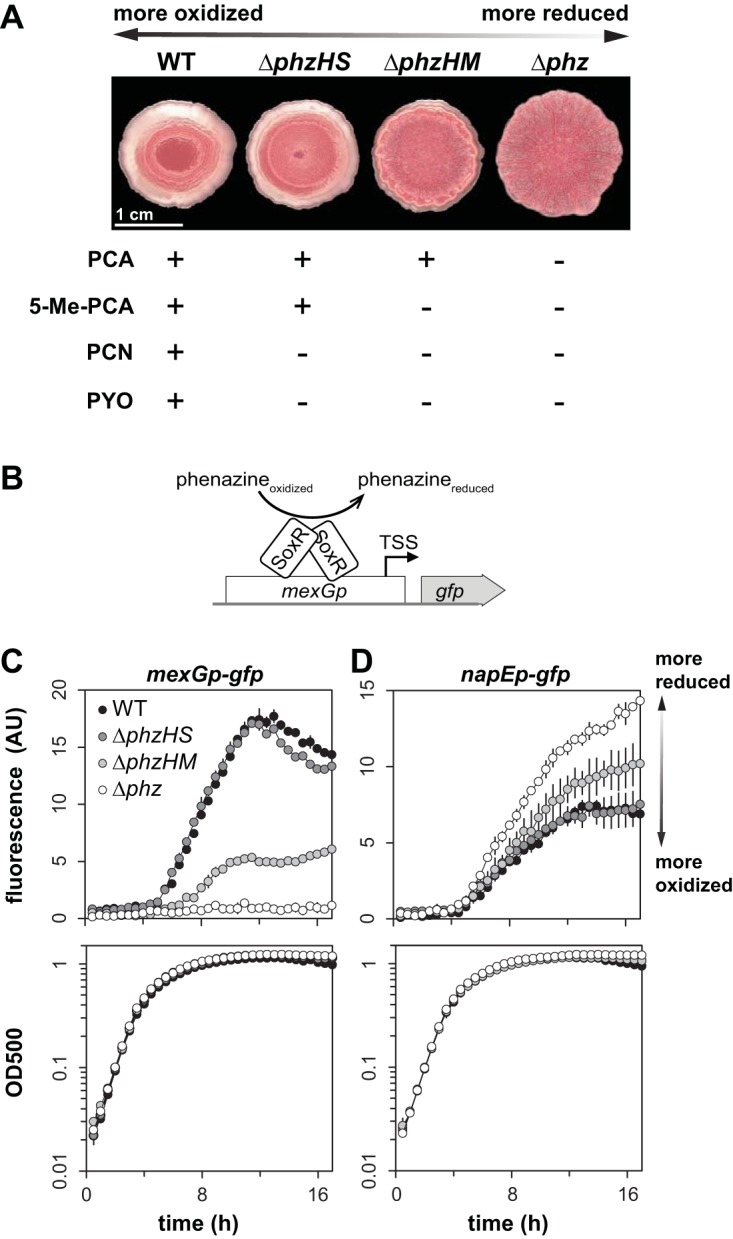
Specific phenazines differentially affect nap expression. (A) Colony morphologies and phenazines produced by various biosynthetic mutants. The production of more methylated phenazines corresponds to a more oxidized cellular redox state, and vice versa, as indicated by the double-headed arrow. Colonies were grown for 3 days on 1% tryptone, 1% agar containing Congo red and Coomassie blue. PCA, phenazine-1-carboxylic acid; 5-Me-PCA, 5-methyl-PCA; PCN, phenazine-1-carboxamide; PYO, pyocyanin. (B) Graphical representation of mexGp-gfp as a biosensor for phenazines. SoxR acts as the transcription factor that senses oxidizing phenazines and activates transcription of the mexGHI-opmD operon. (C and D) Expression of mexGHI-opmD (C) and the nap operon (D), as reported by promoter-gfp fusions, in various phenazine biosynthetic mutant backgrounds during growth in liquid cultures (medium: MOPS plus 20 mM glucose plus 20% LB). Error bars represent the standard deviations from biological triplicates.
Native sequence between the −10 and −35 promoter sites, but not the up- or downstream sequence, is required for the phenazine-dependent effects on nap expression.
We hypothesized that the sequences upstream of the transcription start sites in the nap and nir promoter regions contain regulatory regions that confer sensitivity to phenazines or the cellular redox state. To identify these regions, we began by creating reporter strains containing portions of the nap promoter fused to GFP and compared the fluorescence in the wild-type and Δphz backgrounds. None of the regions tested showed an effect on the phenazine-dependent difference in induction (Fig. 6A and B).
FIG 6.

Sequence between the −10 and −35 regions of the nap promoter confers phenazine sensitivity. (A) Schematics of promoter-gfp fusion reporter constructs. napEp, promoter of the nap operon; TSS, transcription start site; RBS, ribosome-binding site. Orange, yellow, and green boxes indicate regions of the promoter sequences being shuffled. Expression reported by promoter-gfp fusions of truncated (B) and shuffled (C) nap promoter sequences in WT and Δphz backgrounds. Strains were grown in shaken liquid culture (MOPS plus 20 mM glucose plus 20% LB medium). Error bars represent standard deviations from biological triplicates. (D) Graphical representation of the sigma factor RpoS and sequence elements conferring redox sensitivity.
Next, we generated constructs in which the sequence between the RNA polymerase binding sites at bp −35 and −10 upstream of the transcriptional start site was shuffled and fused to gfp (Fig. 6A). We found that, though these constructs still showed stationary-phase induction of nap, the phenazine-specific effect on expression was abolished (Fig. 6C). This suggests that the region between the RNA polymerase binding sites confers sensitivity to phenazines and/or the cellular redox state, perhaps via the activity of a redox-sensing transcription factor. Though some transcription factors, such as SoxR (27), are known to act by binding between the −35 and −10 sites of target promoters, we were not able to identify any known transcription factor consensus sequences or palindromic motifs in the corresponding region for the nap promoter.
The Anr/Dnr site is sufficient for phenazine-dependent effects on Nir expression.
To identify the portion of the nir promoter region required for phenazine sensitivity, we created two constructs (Fig. 7A). In “shuffle-A,” a portion of the predicted Anr/Dnr binding site (12) (−137 to −131 of the start codon, i.e., the sequence that did not overlap the predicted −35 site) was shuffled. In “shuffle-B,” three portions of the sequence were shuffled: an 86-bp region upstream of the Anr/Dnr site, the sequence between the −35 and −10 sites, and a 93-bp region between the −10 site and the ribosome-binding site. We found that expression from the nir promoter was abolished when the shuffle-A construct was used, demonstrating that the Anr/Dnr site is absolutely required for this. However, we were surprised to find that the shuffle-B construct showed expression levels that were comparable to those observed for the intact sequence both in the presence and absence of nitrate (Fig. 4C and 7A), indicating that the Anr/Dnr site alone is sufficient for normal nir expression. This suggests that Anr and/or Dnr mediates the phenazine dependence of nir operon expression.
FIG 7.

Though Anr and Dnr are required for full expression of nir, Anr alone is sufficient for low levels of phenazine-regulated expression. (A) Top: schematics of nir promoter-gfp fusion constructs showing the shuffled regions (yellow boxes). The pink rectangle indicates the position of the Anr/Dnr binding site in the native nir promoter. RBS, ribosome-binding site. Bottom: expression of shuffled nir promoter-gfp reporters. (B) Expression of native nir promoter-gfp reporters in combinatorial mutants lacking Anr, Dnr, and/or phenazines. Shaken liquid cultures were grown in 1% tryptone medium, amended with 1 mM KNO3 where indicated, and covered with mineral oil. Error bars represent the standard deviations from biological triplicates.
Both Anr and Dnr are required for full expression of the nir operon.
To examine Anr- and Dnr-dependent expression in the context of phenazine deficiency, we moved a nir reporter construct into mutants with combinatorial anr, dnr, and phz deletions (Fig. 7B). We found that the anr deletion abolished nir expression, consistent with Anr's status as a master regulator of denitrification. In the Δdnr mutant, nir expression levels were decreased by ∼90% compared to levels observed in the wild-type background. Interestingly, however, a phenazine-dependent effect on expression was still visible (Fig. 7B, right panels), suggesting that this effect may be mediated by Anr. Overall, these results confirm the previous findings that both Anr and Dnr are required for the full expression of the nir operon (9). They also show that Anr-dependent activation of denitrification genes can be modulated by phenazines. This effect is more pronounced under microaerobic conditions and suggests that not only oxygen but also the cellular redox state may regulate Anr activity.
Differential expression of Nap and Nir throughout biofilm depth suggests cross-feeding between subpopulations.
The results of our colony proteome and reporter expression analyses suggest that growth in biofilms, particularly in the absence of phenazines, promotes the synthesis of a Nap-dependent denitrification pathway. However, loci involved in this pathway are regulated by different cues and thus may be differentially affected by the variations in conditions that develop throughout the biofilm depth. To examine this, we grew colony biofilms of our reporter strains, prepared thin sections of biofilm samples, and imaged them by fluorescence microscopy. We also performed microsensor measurements throughout the depth of colony biofilms to evaluate whether the presence of nitrate affects oxygen levels. Consistent with our liquid culture expression profiling, we found that the nap and nir loci were induced in the Δphz biofilms relative to the wild-type biofilms and that provision of nitrate in the medium induced nir expression in both wild-type and Δphz biofilms (Fig. 8A). Intriguingly, however, we found that nap and nir are maximally expressed at different depths: nap expression is more pronounced in the upper, more oxygenated portion of the biofilm, while nir expression is higher in the lower, microoxic/anoxic portion (Fig. 8A and B). The high nap expression at the top of the biofilm is consistent with previous reports of high rpoS expression in this region (28, 29), while high nir expression in the lower portion is consistent with regulation by Anr under microoxic/anoxic conditions. The sharp transition of nir expression at a depth of 40 to 50 μm (Fig. 8A), corresponding to 80 to 100 μmol/liter oxygen (Fig. 8B), highlights the tight regulation of the Anr regulon under microoxic conditions. However, as oxygen levels were not significantly different in wild-type and Δphz mutant colonies (Fig. 8B), the difference in nap and nir expression between these strains cannot be attributed to oxygen-dependent effects. Thus, these results further suggest that different redox-related cues control the expression of the denitrification machinery. In the case of nap, the fact that nitrate does not influence expression in liquid cultures (Fig. 4A) or biofilms suggests that the effects on expression that we have observed in the Δphz mutant compared to that in the wild type are oxidant specific. Finally, given that Nap and Nir are functionally linked in denitrification, it is noteworthy that their maximal expression is spatially segregated throughout the colony depth. This finding indicates that substrate cross-feeding occurs between subpopulations present in the upper and lower portions of the biofilm (Fig. 8C).
FIG 8.
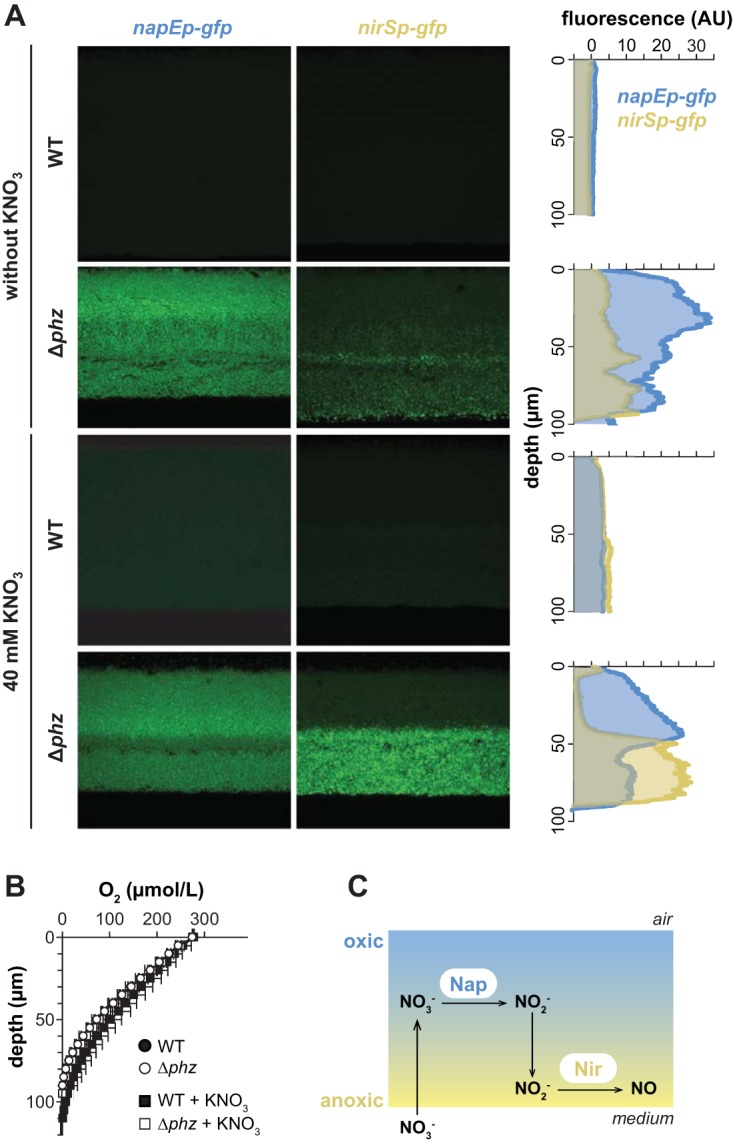
nap and nir are induced in Δphz mutant biofilms and are differentially expressed throughout biofilm depth. (A) Fluorescence images of colony biofilm thin sections showing expression of GFP from nap and nir promoter fusions. Plots at right show fluorescence intensity as a function of depth, with fluorescence from MCS-gfp, a promoterless GFP reporter, subtracted as background. Colonies were grown for 3 and 2 days for WT and Δphz strains, respectively, on 1% tryptone, 1% agar with or without 40 mM KNO3. (B) Oxygen profiles in colony biofilms as a function of depth. (C) Cartoon representing a biofilm, shown along the z axis, and the cross-feeding model for the denitrification pathway.
Concluding remarks.
P. aeruginosa cells in communities can employ several different mechanisms, including the reduction of endogenous antibiotics (phenazines) and denitrification using exogenously available nitrate (4, 30), to balance the intracellular redox state when electron donors and nutrients are available in excess. We have uncovered unexpected regulatory links between these metabolic strategies. While previous studies showed that denitrification genes can be induced in stationary phase and by an oxic-to-anoxic shift (8, 9), our results show that they can also be induced by phenazine deficiency. In the context of our earlier observations that phenazines and nitrate reduction both oxidize the cellular redox state and inhibit colony wrinkling in P. aeruginosa (4), we interpret the induction of denitrification genes in the Δphz background as a bet-hedging strategy, in which phenazine-deficient cells expend energy to produce the denitrification machinery in case the substrate becomes available. While the induction of the biofilm matrix and colony wrinkling in response to oxygen, nitrate, and/or phenazine limitation has been well known (19, 20), the induction of denitrification genes in the phenazine-null background represents an entirely new component to the integrated network of redox-balancing mechanisms in P. aeruginosa communities.
How do phenazines affect the expression of the nap and nir loci? The sensitivity of these operons to phenazine production adds a layer of complexity to their conditional responses. Studies in Escherichia coli, Paracoccus pantotrophus, and purple phototrophs have shown that nap expression can be controlled by diverse environmental cues, including oxygen, nitrate, molybdenum, iron, carbon source, and cellular redox potential (31–38). Considerable variability exists in the regulation of nap operons in different organisms, and detailed mechanisms are not conserved. In P. aeruginosa, nap is traditionally recognized as an RpoS target and therefore a stationary phase-specific gene (25), though experiments conducted in microaerobic cultures have suggested that electron acceptor limitation can promote its induction during active growth (24). The enhanced expression of nap observed in this study could be effected via the same mechanism if it operates specifically in response to reduced cellular redox states arising from oxygen and/or phenazine limitation.
The nir operon is regulated by Anr and Dnr (9). According to current models, Anr induces the expression of Dnr in response to oxygen limitation, and Dnr induces nir expression in response to nitric oxide (Fig. 2A). Our observations suggest that Anr is less active in the presence of phenazines or an oxidized cellular redox state (Fig. 8A) and expand our model for the specific cues determining the activities of this regulator. Anr is a homologue of E. coli Fnr, a well-characterized Fe-S-cluster protein. Fnr can be present in three states depending on the oxidation state of the Fe-S cluster: reduced ([4Fe-4S]2+) clusters support the assembly of an active DNA-binding dimer; oxidation of the clusters to the [2Fe-2S]2+ form leads to the separation of the subunits to form inactive independent monomers; and further oxidation promotes cluster disassembly and renders monomeric Fnr an apoprotein (39, 40) (Fig. 9A). Our results indicate that phenazines can affect the balance between active and inactive Anr by oxidizing the Fe-S cluster directly or indirectly. This proposed redox modulation of Anr is in line with studies demonstrating the reversibility of Fnr activity in vivo and in vitro (39–41), as well as with the fact that redox-cycling compounds, such as phenazines, can activate the redox-active transcription factor SoxR (42–45) by oxidizing its Fe-S cluster.
FIG 9.
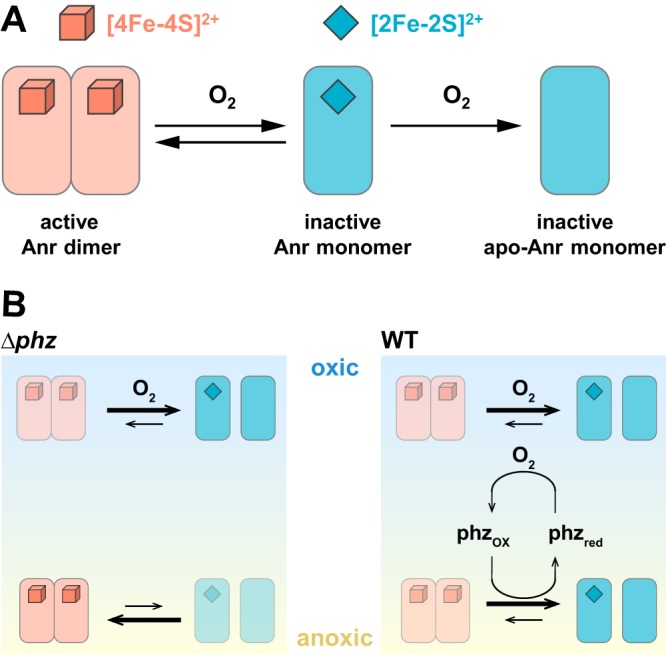
Effects of oxygen and phenazines on Anr activity. (A) Literature-based model for the effect of oxygen on Anr activity (40, 65). The [4Fe-4S]2+ cluster supports the formation of the dimer, which is capable of binding DNA. Oxidation of the cluster to the [2Fe-2S]2+ form causes the protein to separate into monomers. Further oxidation of the cluster leads to disassembly, leaving the Anr apoprotein. (B) Results from this study suggest that in the upper portion of a colony biofilm, Anr is inactive (in either the monomeric or apo form) due to the presence of oxygen in both Δphz and WT strain biofilms. In the lower (microoxic/anoxic) portion of Δphz mutant biofilms, Anr is active due to electron acceptor limitation. In the lower portion of WT biofilms, Anr activity is dampened specifically by phenazines.
In biofilm thin sections, we observed an increased expression of the nap and nir operons in the Δphz strain compared to that in the wild type. The expression of nir was further enhanced by growth on nitrate. We also found that the zones of maximum nap and nir expression were spatially segregated, occurring at different depths (Fig. 8A). This pattern of strain- and condition-dependent differences results from a combination of effects, including an apparent phenazine-dependent inactivation of Anr. These results raise the intriguing possibility that Anr is controlled by redox cues other than oxygen, such as phenazines or the intracellular redox state (Fig. 9). Because Anr oxidation, either through direct interaction or indirect oxidation of the cellular potential, would require oxidized phenazines, our results also provide evidence that phenazines are redox cycled in colony biofilms, an activity that we have inferred from many independent observations but that has been challenging to demonstrate experimentally (30). In this context, we also note that the addition of nitrate to stationary-phase aerobically grown Δphz liquid cultures has the effect of oxidizing the cellular redox state (3). We would therefore expect the cells in Δphz mutant colonies grown on nitrate to be relatively oxidized, and their high expression of nir suggests that an oxidized cellular redox state is not sufficient to deactivate Anr.
Because Nap and Nir catalyze two sequential steps in denitrification, our results suggest that there is cross-feeding of nitrite—the product of Nap and the substrate for Nir—between cells in adjacent microenvironments. The segregation of metabolic activities and substrate cross-feeding have been identified in many multispecies communities (46, 47). Indeed, cross-feeding of nitrite has been shown to reduce interenzyme competition and accelerate nitrite consumption in Pseudomonas stutzeri in planktonic cultures (48). From an evolutionary perspective, substrate cross-feeding maximizes energy generation, which provides a selective advantage (49–51). Our studies indicate that substrate cross-feeding could take place in a single-species biofilm, presumably to maximize metabolic efficiency and survival on a community level.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Unless otherwise indicated, P. aeruginosa strain UCBPP-PA14 (52) was routinely grown in lysogeny broth ([LB] 1% tryptone, 1% NaCl, 0.5% yeast extract) (53) at 37°C with shaking at 250 rpm. Overnight cultures were grown for 16 ± 1 h. For genetic manipulation, the strains were typically grown on LB solidified with 1.5% agar. The strains used in this study are listed in Table S3 in the supplemental material. In general, liquid precultures served as the inocula for experiments. The overnight precultures for biological replicates were started from separate clonal source colonies on streaked agar plates.
Proteomic analysis.
Each colony was scraped from the agar-solidified growth medium with a sterile razor blade, transferred to a microcentrifuge tube, washed with 1 ml of phosphate-buffered saline (PBS), and resuspended in 500 μl of ice-cold lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, and 0.3% SDS supplemented with cOmplete Mini protease inhibitor cocktail [Roche]). Homogenized colonies were sonicated for 120 s (4 × 30 s) on ice with the microtip of a Branson Sonifier 250 (50% duty cycle, 50% output). To extract protein from each sonicated sample, 650 μl of methanol and 187.5 μl of chloroform were added to the sample, which was then vigorously vortexed for ∼1 min. After centrifuging at 16,873 × g for 5 min for phase separation, a white disc of protein formed at the interface and the top methanol layer of each sample was carefully removed. Then, 562.5 μl of methanol was added and gently mixed to precipitate the protein, which was pelleted at 16,873 × g for 5 min. The protein pellets were stored at −80°C. Prior to mass spectrometry analysis, each sample was solubilized with 0.1% RapiGest at 95°C for 5 min, digested with trypsin (1:50 [wt/wt]) at 37°C for 4 h, and lyophilized with 2% formic acid. The peptides were separated by using a reverse-phase nanoflow high-performance liquid chromatography (LC) system (Eksigent NanoLC 2DPlus) with a 15-cm Agilent ZORBAX 300 StableBond C18 reverse-phase column (5065-9911; 150-mm length, 75-μm inner diameter, 3.5-μm particle, 300-Å pore size) with the gradient of 2 to 90% acetonitrile and 0.1% formic acid over 240 min. LC-eluted peptides were injected in-line into an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific) for tandem mass spectrometry analysis. Data-dependent analysis was performed in the Fourier transform (FT) mass analyzer at a resolution of 60,000 in profile mode, and automatic gain control was set to 1E6. Dynamic exclusion was set to 90 s if the mass to charge ratio (m/z) acquisition was repeated within a 45-s interval. MS2 data were collected in the ion trap (IT) mass analyzer in centroid mode, and the automatic gain control was set to 3E4. The injection time was 100 ms, the isolation window was 2 m/z, and the normalized collision energy was at 35 V. In each scan cycle, the top 20 precursor ions (MS) were subjected to fragmentation (MS/MS) via collision-induced dissociation. Each sample was injected two to four times to obtain technical replicates.
Proteomics data analysis.
Raw data were processed using the MaxQuant software (1.3.0.5) (54), combining technical replicates and searching against the P. aeruginosa PA14 sequence database and a list of commonly observed contaminants supplied by MaxQuant. Protein identification was performed using 20 ppm tolerance at the MS level (FT mass analyzer) and 0.5 Da at the MS/MS level (ion trap analyzer). Up to two missed trypsin cleavages were allowed, and the oxidation of methionine and N-terminal acetylation were searched as variable posttranslational modifications, with cysteine carbamidomethylation as a fixed modification. The minimal required peptide length was seven amino acids. The false discovery rate was set to 0.25 at the peptide level and 0.1 at the protein level. We used label-free quantification (LFQ) intensities obtained from the proteingroups.txt output file for quantitation of the proteins. Protein identifications and quantitative information are provided in Data Set S1, showing the MaxQuant output files for the two proteomics experiments. After filtering contaminants and reversed sequences, we quantified a total of 2,772 and 2,089 proteins across the mutant and time series experiments, respectively. Protein abundances were normalized so that the total LFQ intensity within each sample and set to 1,000,000. The protein heat map in Fig. 1B was generated by first filtering the normalized data to require at least six valid values for each protein and log-transforming and row-median centering the data, followed by hierarchical clustering with complete linkage, using the Pearson correlation as a similarity measure. The resulting tree was split into 20 clusters. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (55) partner repository with the data set identifier PXD008924.
NAD(H) extraction.
The extraction and quantification of NADH and NAD+ were carried out according to the methods described by San et al. (56) and Bernofsky and Swan (57). For cultures grown in LB, two 1-ml samples of culture were placed in two separate microcentrifuge tubes and centrifuged at 16,000 × g for 1 min. Colonies grown on agar-solidified growth medium (1% tryptone and 1% agar plates amended with 40 μg/ml Congo red and 20 μg/ml Coomassie blue) were scraped off the agar at the indicated time points using sterile razor blades and each resuspended in 1 ml of 1% tryptone. The colonies were disrupted using a pellet disrupter. For each resuspended colony, two 450-μl samples were placed into two separate microcentrifuge tubes. NADH and NAD+ were then extracted from the liquid culture or colony-derived samples, and relative or absolute quantification was carried out using an enzyme-cycling assay, as described by Price-Whelan et al. (3).
Construction of mutant P. aeruginosa strains.
For making markerless deletion mutants in P. aeruginosa PA14 (Table S3), ∼1-kb flanking sequences from each side of the target gene were amplified using the primers listed in Table S4 and inserted into pMQ30 through gap repair cloning in Saccharomyces cerevisiae InvSc1 (58). Each plasmid listed in Table S3 was transformed into Escherichia coli strain UQ950, verified by sequencing, and moved into strain PA14 using biparental conjugation. PA14 single recombinants were selected on LB agar plates containing 100 μg/ml gentamicin. Double recombinants (markerless deletions) were selected on sucrose plates (1% tryptone, 0.5% yeast extract, 10% sucrose, and 1.5% agar). The genotypes of deletion mutants were verified by PCR. Combinatorial mutants were constructed by using single mutants as parent strains.
Construction of GFP reporter strains.
Transcriptional reporter constructs for the operons napEFDABC, narK1K2GHJI, nirSMCFLGHJEN, norCBD, and nosRZDFYL were made by fusing the upstream promoter sequences with gfp using the primers listed in Table S4. The respective primers were used to amplify promoter regions (as indicated in Table S4) and to add SpeI digest sites to the 5′ ends of the promoters and XhoI digest sites to the 3′ ends of the promoters. Purified PCR products were digested and ligated into the multiple cloning site of the allelic replacement vector pLD2477 (pMQ30 derivative) (58) or pLD2722 (pYL122 derivative) (59), upstream of the gfp sequence. Both pLD2477 and pLD2722 contain identical ribosome-binding sites between the multiple-cloning sites MCS and gfp sequences. The plasmids were transformed into E. coli strain UQ950, verified by sequencing, and moved into PA14 using biparental conjugation. The conjugative transfer of pLD2477 or pLD2722 was conducted with E. coli strain BW29427 or S17-1, respectively. PA14 single recombinants of pLD2477 were selected on LB agar plates containing 100 μg/ml gentamicin. PA14 single recombinants of pLD2722 were selected on M9 minimal medium agar plates (47.8 mM Na2HPO4·7H2O, 22 mM KH2PO4, 8.6 mM NaCl, 18.6 mM NH4Cl, 1 mM MgSO4, 0.1 mM CaCl2, 20 mM sodium citrate dihydrate, 1.5% agar) containing 100 μg/ml gentamicin. pLD2477 double recombinants with GFP reporter insertions were selected on sucrose plates (1% tryptone, 0.5% yeast extract, 10% sucrose, 1.5% agar). The genotypes of gfp insertion mutants were confirmed by PCR. For pLD2722, the plasmid backbone was resolved from that of PA14 using Flp-FRT recombination by the introduction of the pFLP2 plasmid (60) and selected on M9 minimal medium agar plates containing 300 μg/ml carbenicillin and further on sucrose plates. The presence of gfp in the final clones was confirmed by PCR.
Shuffled promoter sequences.
The shuffled nirS and napE promoter sequences were designed using “Sequence Manipulation Suite (SMS): Shuffle DNA” (61) (Table S5). Promoter-shuffle sequences were amplified from strain PA14 genomic DNA by PCR using primers that contained the shuffle sequences (Table S4). These PCR fragments of napEp were joined and integrated into pLD2477 by gap repair cloning in S. cerevisiae InvSc1 (58). Shuffled promoters for nirS were synthesized by GenScript and subcloned into pLD2929 by replacing the native nirS promoter.
Thin-sectioning analyses.
To produce bilayer plates, a bottom layer of medium (1% agar,1% tryptone) was poured to a depth of 4.5 mm and allowed to solidify before pouring a 1.5-mm layer on top. Precultures were incubated overnight, diluted 1:100 in LB, and cultured until early-mid-exponential phase (optical density at 500 nm [OD500] of ∼0.5). Subcultures were spotted onto the top layer of agar-solidified medium in 5-μl aliquots and incubated in the dark at 25°C with >90% humidity (Percival CU-22L) for up to 3 days. Colonies were sacrificed for thin sectioning at specified time points by first covering them with a 1.5-mm-thick layer of 1% agar, which sandwiches each colony between two 1.5-mm layers of solidified agar. Colonies were lifted from the bottom layer of agar and soaked in 50 mM l-lysine in PBS (pH 7.4) at 4°C for 4 h, fixed in 4% paraformaldehyde, 50 mM l-lysine, PBS (pH 7.4) at 4°C for 4 h, and then incubated overnight at 37°C. The fixed colonies were washed twice in PBS and dehydrated through a series of ethanol washes (25%, 50%, 70%, 95% ethanol in PBS, 3 times in 100% ethanol) for 60 min each and then cleared via three 60-min washes in Histo-Clear II (HS-202; National Diagnostics). The cleared colonies were infiltrated with paraffin wax (50-276-89, Electron Microscopy Sciences; Fisher Scientific) at 55°C twice for 2 h each. The infiltrated colonies were solidified by an overnight incubation at 4°C. Sections were cut perpendicularly to the base of the colony in 10-μm slices using an automatic microtome (905200ER; Thermo Fisher Scientific), floated over a water bath at 45°C, collected onto slides, and air dried overnight. The dried slides were heat fixed on a hotplate at 45°C for 1 h, and then rehydrated in PBS by reversing the dehydration steps listed above. The sections were then immediately mounted beneath a coverslip in Tris-buffered Fluoro-Gel with DAPI 4′,6-diamidino-2-phenylindole (50-246-93 Electron Microscopy Sciences; Fisher Scientific). Differential interference contrast (DIC) and fluorescent confocal images were captured from at least three biological replicates of each strain using an LSM700 confocal microscope (Zeiss).
Liquid culture growth assays.
Overnight (16 h) precultures were diluted 1:100 in a clear-bottom, polystyrene, black 96-well plate (82050-756; VWR), with each well containing 200 μl of medium. Cultures were then incubated at 37°C with continuous shaking at medium speed in a Biotek Synergy 4 plate reader. The media used for reporter strains made with pLD2477 (pMQ37) and pLD2722 (pSEK103) vectors were MOPS (morpholinepropanesulfonic acid) plus 20 mM glucose plus 20% LB or 1% tryptone, respectively. When specified, 50 μl of light mineral oil (470301-505; VWR) was added to each well of the 96-well plate to prevent evaporation and to create microaerobic conditions. The expression of GFP was assessed by taking fluorescence readings at excitation and emission wavelengths of 480 nm and 510 nm, respectively, every 30 min for up to 24 h. Growth was assessed by taking OD readings at 500 nm simultaneously with the fluorescence readings.
RNA-seq analysis.
Δphz mutant colonies were grown on filter membranes (0.2-μm pore size, 25-mm diameter; Whatman) placed on 1% tryptone and 1.5% agar at 25°C for 76 h. Colony samples were harvested by microscopic laser dissection, and total RNA was extracted using the RNeasy Plant Minikit (Qiagen). RNA samples were sent to Genewiz for further processing, including rRNA depletion and dUTP incorporation for strand-specific sequencing, and sequenced with an Illumina HiSeq 2500 platform. Sixteen fastq files were mapped to the reference PA14 genome using Bowtie2 (62) with an ∼97% success rate to generate SAM (sequence alignment map) files. SAM files containing ∼2 × 108 reads in total were merged, sorted, and indexed with SAMtools (63). Read coverage was visualized (Fig. 2C) with Integrative Genomics Viewer (IGV) software (64).
Oxygen profiling of biofilms.
A 25-μm-tip oxygen microsensor (Unisense OX-25) was used to measure oxygen concentrations in day 3 and day 2 colony biofilms for wild-type (WT) and Δphz strains, respectively, grown as described above. The microsensor was subjected to a two-point calibration using a Unisense calibration chamber (CAL300). The first calibration was to atmospheric oxygen as water in the chamber was continuously bubbled with air. For the second calibration, the “zero” point was set using water that had been bubbled with N2 for 30 min. Oxygen measurements were then taken throughout the depth of the biofilm using a measurement time of 3 s and a wait time between measurements of 5 s, with a step size of 5 μm. A micromanipulator (Unisense MM33) was used to move the microsensor within the biofilm, and profiles were recorded using a multimeter (Unisense) and the SensorTrace Profiling software (Unisense).
Supplementary Material
ACKNOWLEDGMENTS
We thank Jeanyoung Jo for performing the oxygen microsensor measurements and Tara Rock for support with the proteomics analysis.
This work was supported by an NSF Career award and NIH/NIAID grant R01AI103369 to L.E.P.D. and an NSF Eager grant and NIH/NIGMS grant 1R01GM113237-01 to C.V.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00031-18.
REFERENCES
- 1.López D, Vlamakis H, Kolter R. 2010. Biofilms. Cold Spring Harb Perspect Biol 2:a000398. doi: 10.1101/cshperspect.a000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kempes CP, Okegbe C, Mears-Clarke Z, Follows MJ, Dietrich LEP. 2014. Morphological optimization for access to dual oxidants in biofilms. Proc Natl Acad Sci U S A 111:208–213. doi: 10.1073/pnas.1315521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price-Whelan A, Dietrich LEP, Newman DK. 2007. Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J Bacteriol 189:6372–6381. doi: 10.1128/JB.00505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietrich LEP, Okegbe C, Price-Whelan A, Sakhtah H, Hunter RC, Newman DK. 2013. Bacterial community morphogenesis is intimately linked to the intracellular redox state. J Bacteriol 195:1371–1380. doi: 10.1128/JB.02273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berks BC, Ferguson SJ, Moir JW, Richardson DJ. 1995. Enzymes and associated electron transport systems that catalyse the respiratory reduction of nitrogen oxides and oxyanions. Biochim Biophys Acta 1232:97–173. doi: 10.1016/0005-2728(95)00092-5. [DOI] [PubMed] [Google Scholar]
- 6.Carlson CA, Ferguson LP, Ingraham JL. 1982. Properties of dissimilatory nitrate reductase purified from the denitrifier Pseudomonas aeruginosa. J Bacteriol 151:162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potter L, Angove H, Richardson D, Cole J. 2001. Nitrate reduction in the periplasm of Gram-negative bacteria. Adv Microb Physiol 45:51–112. doi: 10.1016/S0065-2911(01)45002-8. [DOI] [PubMed] [Google Scholar]
- 8.Williams HD, Zlosnik JEA, Ryall B. 2007. Oxygen, cyanide and energy generation in the cystic fibrosis pathogen Pseudomonas aeruginosa. Adv Microb Physiol 52:1–71. doi: 10.1016/S0065-2911(06)52001-6. [DOI] [PubMed] [Google Scholar]
- 9.Arai H. 2011. Regulation and function of versatile aerobic and anaerobic respiratory metabolism in Pseudomonas aeruginosa. Front Microbiol 2:103. doi: 10.3389/fmicb.2011.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon SS, Hennigan RF, Hilliard GM, Ochsner UA, Parvatiyar K, Kamani MC, Allen HL, DeKievit TR, Gardner PR, Schwab U, Rowe JJ, Iglewski BH, McDermott TR, Mason RP, Wozniak DJ, Hancock REW, Parsek MR, Noah TL, Boucher RC, Hassett DJ. 2002. Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev Cell 3:593–603. doi: 10.1016/S1534-5807(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 11.Ye RW, Haas D, Ka JO, Krishnapillai V, Zimmermann A, Baird C, Tiedje JM. 1995. Anaerobic activation of the entire denitrification pathway in Pseudomonas aeruginosa requires Anr, an analog of Fnr. J Bacteriol 177:3606–3609. doi: 10.1128/jb.177.12.3606-3609.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trunk K, Benkert B, Quäck N, Münch R, Scheer M, Garbe J, Jänsch L, Trost M, Wehland J, Buer J, Jahn M, Schobert M, Jahn D. 2010. Anaerobic adaptation in Pseudomonas aeruginosa: definition of the Anr and Dnr regulons. Environ Microbiol 12:1719–1733. doi: 10.1111/j.1462-2920.2010.02252.x. [DOI] [PubMed] [Google Scholar]
- 13.Hammond JH, Dolben EF, Smith TJ, Bhuju S, Hogan DA. 2015. Links between Anr and quorum sensing in Pseudomonas aeruginosa biofilms. J Bacteriol 197:2810–2820. doi: 10.1128/JB.00182-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arai H, Igarashi Y, Kodama T. 1995. Expression of the nir and nor genes for denitrification of Pseudomonas aeruginosa requires a novel CRP/FNR-related transcriptional regulator, DNR, in addition to ANR. FEBS Lett 371:73–76. doi: 10.1016/0014-5793(95)00885-D. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber K, Krieger R, Benkert B, Eschbach M, Arai H, Schobert M, Jahn D. 2007. The anaerobic regulatory network required for Pseudomonas aeruginosa nitrate respiration. J Bacteriol 189:4310–4314. doi: 10.1128/JB.00240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Alst NE, Sherrill LA, Iglewski BH, Haidaris CG. 2009. Compensatory periplasmic nitrate reductase activity supports anaerobic growth of Pseudomonas aeruginosa PAO1 in the absence of membrane nitrate reductase. Can J Microbiol 55:1133–1144. doi: 10.1139/W09-065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer KL, Brown SA, Whiteley M. 2007. Membrane-bound nitrate reductase is required for anaerobic growth in cystic fibrosis sputum. J Bacteriol 189:4449–4455. doi: 10.1128/JB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dietrich LEP, Teal TK, Price-Whelan A, Newman DK. 2008. Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321:1203–1206. doi: 10.1126/science.1160619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madsen JS, Lin Y-C, Squyres GR, Price-Whelan A, de Santiago Torio A, Song A, Cornell WC, Sørensen SJ, Xavier JB, Dietrich LEP. 2015. Facultative control of matrix production optimizes competitive fitness in Pseudomonas aeruginosa PA14 biofilm models. Appl Environ Microbiol 81:8414–8426. doi: 10.1128/AEM.02628-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okegbe C, Fields BL, Cole SJ, Beierschmitt C, Morgan CJ, Price-Whelan A, Stewart RC, Lee VT, Dietrich LEP. 2017. Electron-shuttling antibiotics structure bacterial communities by modulating cellular levels of c-di-GMP. Proc Natl Acad Sci U S A 114:E5236–E5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Newman DK. 2008. Redox reactions of phenazine antibiotics with ferric (hydr)oxides and molecular oxygen. Environ Sci Technol 42:2380–2386. doi: 10.1021/es702290a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng H, Kim J, Liew M, Yan JK, Herrera O, Bok JW, Kelleher NL, Keller NP, Wang Y. 2015. Redox metabolites signal polymicrobial biofilm development via the NapA oxidative stress cascade in Aspergillus. Curr Biol 25:29–37. doi: 10.1016/j.cub.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakhtah H, Koyama L, Zhang Y, Morales DK, Fields BL, Price-Whelan A, Hogan DA, Shepard K, Dietrich LEP. 2016. The Pseudomonas aeruginosa efflux pump MexGHI-OpmD transports a natural phenazine that controls gene expression and biofilm development. Proc Natl Acad Sci U S A 113:E3538–E3547. doi: 10.1073/pnas.1600424113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez-Ortega C, Harwood CS. 2007. Responses of Pseudomonas aeruginosa to low oxygen indicate that growth in the cystic fibrosis lung is by aerobic respiration. Mol Microbiol 65:153–165. doi: 10.1111/j.1365-2958.2007.05772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuster M, Hawkins AC, Harwood CS, Greenberg EP. 2004. The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol Microbiol 51:973–985. doi: 10.1046/j.1365-2958.2003.03886.x. [DOI] [PubMed] [Google Scholar]
- 26.Härtig E, Schiek U, Vollack KU, Zumft WG. 1999. Nitrate and nitrite control of respiratory nitrate reduction in denitrifying Pseudomonas stutzeri by a two-component regulatory system homologous to NarXL of Escherichia coli. J Bacteriol 181:3658–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe S, Kita A, Kobayashi K, Miki K. 2008. Crystal structure of the [2Fe-2S] oxidative-stress sensor SoxR bound to DNA. Proc Natl Acad Sci U S A 105:4121–4126. doi: 10.1073/pnas.0709188105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pérez-Osorio AC, Williamson KS, Franklin MJ. 2010. Heterogeneous rpoS and rhlR mRNA levels and 16S rRNA/rDNA (rRNA gene) ratios within Pseudomonas aeruginosa biofilms, sampled by laser capture microdissection. J Bacteriol 192:2991–3000. doi: 10.1128/JB.01598-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williamson KS, Richards LA, Perez-Osorio AC, Pitts B, McInnerney K, Stewart PS, Franklin MJ. 2012. Heterogeneity in Pseudomonas aeruginosa biofilms includes expression of ribosome hibernation factors in the antibiotic-tolerant subpopulation and hypoxia-induced stress response in the metabolically active population. J Bacteriol 194:2062–2073. doi: 10.1128/JB.00022-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okegbe C, Price-Whelan A, Dietrich LEP. 2014. Redox-driven regulation of microbial community morphogenesis. Curr Opin Microbiol 18:39–45. doi: 10.1016/j.mib.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobao MM, Martínez-Luque M, Moreno-Vivián C, Castillo F. 1994. Effect of carbon and nitrogen metabolism on nitrate reductase activity of Rhodobacter capsulatus E1F1. Can J Microbiol 40:645–650. doi: 10.1139/m94-102. [DOI] [Google Scholar]
- 32.Darwin AJ, Ziegelhoffer EC, Kiley PJ, Stewart V. 1998. Fnr, NarP, and NarL regulation of Escherichia coli K-12 napF (periplasmic nitrate reductase) operon transcription in vitro. J Bacteriol 180:4192–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reyes F, Gavira M, Castillo F, Moreno-Vivián C.. 1998. Periplasmic nitrate-reducing system of the phototrophic bacterium Rhodobacter sphaeroides DSM 158: transcriptional and mutational analysis of the napKEFDABC gene cluster. Biochem J 331:897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Tseng CP, Gunsalus RP. 1999. The napF and narG nitrate reductase operons in Escherichia coli are differentially expressed in response to submicromolar concentrations of nitrate but not nitrite. J Bacteriol 181:5303–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gavira M, Roldán MD, Castillo F, Moreno-Vivián C. 2002. Regulation of nap gene expression and periplasmic nitrate reductase activity in the phototrophic bacterium Rhodobacter sphaeroides DSM158. J Bacteriol 184:1693–1702. doi: 10.1128/JB.184.6.1693-1702.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellington MJK, Sawers G, Sears HJ, Spiro S, Richardson DJ, Ferguson SJ. 2003. Characterization of the expression and activity of the periplasmic nitrate reductase of Paracoccus pantotrophus in chemostat cultures. Microbiology 149:1533–1540. doi: 10.1099/mic.0.26277-0. [DOI] [PubMed] [Google Scholar]
- 37.Ellington MJK, Fosdike WLJ, Sawers RG, Richardson DJ, Ferguson SJ. 2006. Regulation of the nap operon encoding the periplasmic nitrate reductase of Paracoccus pantotrophus: delineation of DNA sequences required for redox control. Arch Microbiol 184:298–304. doi: 10.1007/s00203-005-0044-x. [DOI] [PubMed] [Google Scholar]
- 38.Sparacino-Watkins C, Stolz JF, Basu P. 2014. Nitrate and periplasmic nitrate reductases. Chem Soc Rev 43:676–706. doi: 10.1039/C3CS60249D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel P, Trageser M, Unden G. 1991. Reversible interconversion of the functional state of the gene regulator FNR from Escherichia coli in vivo by O2 and iron availability. Arch Microbiol 156:463–470. [DOI] [PubMed] [Google Scholar]
- 40.Kiley PJ, Beinert H. 1998. Oxygen sensing by the global regulator, FNR: the role of the iron-sulfur cluster. FEMS Microbiol Rev 22:341–352. doi: 10.1111/j.1574-6976.1998.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 41.Lazazzera BA, Beinert H, Khoroshilova N, Kennedy MC, Kiley PJ. 1996. DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J Biol Chem 271:2762–2768. doi: 10.1074/jbc.271.5.2762. [DOI] [PubMed] [Google Scholar]
- 42.Dietrich LEP, Price-Whelan A, Petersen A, Whiteley M, Newman DK. 2006. The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol Microbiol 61:1308–1321. doi: 10.1111/j.1365-2958.2006.05306.x. [DOI] [PubMed] [Google Scholar]
- 43.Gu M, Imlay JA. 2011. The SoxRS response of Escherichia coli is directly activated by redox-cycling drugs rather than by superoxide. Mol Microbiol 79:1136–1150. doi: 10.1111/j.1365-2958.2010.07520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheplock R, Recinos DA, Mackow N, Dietrich LEP, Chander M. 2013. Species-specific residues calibrate SoxR sensitivity to redox-active molecules. Mol Microbiol 87:368–381. doi: 10.1111/mmi.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh AK, Shin J-H, Lee K-L, Imlay JA, Roe J-H. 2013. Comparative study of SoxR activation by redox-active compounds. Mol Microbiol 90:983–996. doi: 10.1111/mmi.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE, Flint HJ. 2006. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Environ Microbiol 72:3593–3599. doi: 10.1128/AEM.72.5.3593-3599.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramsey MM, Rumbaugh KP, Whiteley M. 2011. Metabolite cross-feeding enhances virulence in a model polymicrobial infection. PLoS Pathog 7:e1002012. doi: 10.1371/journal.ppat.1002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lilja EE, Johnson DR. 2016. Segregating metabolic processes into different microbial cells accelerates the consumption of inhibitory substrates. ISME J 10:1568–1578. doi: 10.1038/ismej.2015.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treves DS, Manning S, Adams J. 1998. Repeated evolution of an acetate-crossfeeding polymorphism in long-term populations of Escherichia coli. Mol Biol Evol 15:789–797. doi: 10.1093/oxfordjournals.molbev.a025984. [DOI] [PubMed] [Google Scholar]
- 50.Pfeiffer T, Bonhoeffer S. 2004. Evolution of cross-feeding in microbial populations. Am Nat 163:E126–E135. doi: 10.1086/383593. [DOI] [PubMed] [Google Scholar]
- 51.Costa E, Pérez J, Kreft J-U. 2006. Why is metabolic labour divided in nitrification? Trends Microbiol 14:213–219. doi: 10.1016/j.tim.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 52.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 53.Bertani G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J Bacteriol 186:595–600. doi: 10.1128/JB.186.3.595-600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 55.Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu Q-W, Wang R, Hermjakob H. 2016. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.San K-Y, Bennett GN, Berríos-Rivera SJ, Vadali RV, Yang Y-T, Horton E, Rudolph FB, Sariyar B, Blackwood K. 2002. Metabolic engineering through cofactor manipulation and its effects on metabolic flux redistribution in Escherichia coli. Metab Eng 4:182–192. doi: 10.1006/mben.2001.0220. [DOI] [PubMed] [Google Scholar]
- 57.Bernofsky C, Swan M. 1973. An improved cycling assay for nicotinamide adenine dinucleotide. Anal Biochem 53:452–458. doi: 10.1016/0003-2697(73)90094-8. [DOI] [PubMed] [Google Scholar]
- 58.Shanks RMQ, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from Gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lequette Y, Greenberg EP. 2005. Timing and localization of rhamnolipid synthesis gene expression in Pseudomonas aeruginosa biofilms. J Bacteriol 187:37–44. doi: 10.1128/JB.187.1.37-44.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 61.Stothard P. 2000. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1102–1104. [DOI] [PubMed] [Google Scholar]
- 62.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mettert EL, Kiley PJ. 14 November 2017. Reassessing the structure and function relationship of the O2 sensing transcription factor FNR. Antioxid Redox Signal doi: 10.1089/ars.2017.7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.