

ABSTRACT
Chemoreceptors are localized at the cell poles of Escherichia coli and other rod-shaped bacteria. Over the years, different mechanisms have been put forward to explain this polar localization, including stochastic clustering, membrane curvature-driven localization, interactions with the Tol-Pal complex, and nucleoid exclusion. To evaluate these mechanisms, we monitored the cellular localization of the aspartate chemoreceptor Tar in different deletion mutants. We did not find any indication for either stochastic cluster formation or nucleoid exclusion. However, the presence of a functional Tol-Pal complex appeared to be essential to retain Tar at the cell poles. Interestingly, Tar still accumulated at midcell in tol and in pal deletion mutants. In these mutants, the protein appears to gather at the base of division septa, a region characterized by strong membrane curvature. Chemoreceptors, like Tar, form trimers of dimers that bend the cell membrane due to a rigid tripod structure. The curvature approaches the curvature of the cell membrane generated during cell division, and localization of chemoreceptor tripods at curved membrane areas is therefore energetically favorable, as it lowers membrane tension. Indeed, when we introduced mutations in Tar that abolish the rigid tripod structure, the protein was no longer able to accumulate at midcell or the cell poles. These findings favor a model where chemoreceptor localization in E. coli is driven by strong membrane curvature and association with the Tol-Pal complex.
IMPORTANCE Bacteria have exquisite mechanisms to sense and adapt to the environment they live in. One such mechanism involves the chemotaxis signal transduction pathway, in which chemoreceptors specifically bind certain attracting or repelling molecules and transduce the signals to the cell. In different rod-shaped bacteria, these chemoreceptors localize specifically to cell poles. Here, we examined the polar localization of the aspartate chemoreceptor Tar in E. coli and found that membrane curvature at cell division sites and the Tol-Pal protein complex localize Tar at cell division sites, the future cell poles. This study shows how membrane curvature can guide localization of proteins in a cell.
KEYWORDS: chemotaxis, MCP, membrane curvature, polar localization
INTRODUCTION
Bacteria use specific chemotaxis systems to sense chemical changes in their environment and respond accordingly. One of the best-known systems is that of Escherichia coli, which comprises five different membrane-spanning chemoreceptors. The cytoplasmic domains of chemoreceptors associate with the adaptor protein CheW and with the histidine kinase CheA. The presence of repellents or absence of attractants results in increased CheA activity and, subsequently, increased levels of phosphorylated CheY, which acts on the flagellar motor to change rotation direction. Sensitivity of the chemoreceptors is tuned by methylation and demethylation, for which the methylesterase CheB and methyltransferase CheR are responsible. The chemoreceptors are therefore also referred to as methyl-accepting chemotaxis proteins (MCPs). For in-depth reviews on the chemotaxis system see, e.g., the work of Porter et al. and Parkinson et al. (1, 2).
MCPs form large protein clusters together with CheW, CheY, CheA, CheB, and CheR at the cell poles of different bacteria, including those of the Gram-negative model system E. coli and the Gram-positive model system Bacillus subtilis (3, 4). Several mechanisms have been proposed for this polar localization. In long filamentous E. coli cells, yellow fluorescent protein (YFP)-labeled CheR clusters were found to assemble with a certain periodicity along the cell axis, corresponding to the positions of future division sites. This model is referred to as the “stochastic nucleation model” (5–7). Another theory postulated that MCPs assemble preferentially at the curved membrane of cell poles (8). Chemoreceptors form membrane-spanning trimers of dimers that interact at their cytoplasmic domain at a slight angle, thereby forming a tripod-like configuration (9). Consequently, the trimers of dimers prefer bent membrane areas, due to the reduced curvature mismatch (10, 11). This model was recently supported by mechanical bending of whole E. coli cells in curved microchambers (12), and was also shown to be the main mechanism by which the chemoreceptor TlpA of B. subtilis is localized (13). However, another study suggested that polar curvature is not crucial for the localization of chemoreceptor proteins in E. coli, but that this localization requires interaction with the Tol-Pal complex (14). The transenvelope Tol-Pal complex is a widely conserved component of the cell envelope of Gram-negative bacteria and is involved in several processes among which cell division (15, 16). In agreement to this, another recent study showed that, at least for the serine chemoreceptor Tsr, the Tol-Pal complex, together with the nucleoid, is required for polar localization, and that nucleoid exclusion is a driving force for polar localization of MCPs (17). Here, using the aspartate chemoreceptor Tar, we evaluated the different polar localization models in E. coli. We found evidence for neither periodic clustering nor nucleoid exclusion, but both membrane curvature and the Tol-Pal system appeared to be required for polar localization of Tar in E. coli.
RESULTS
Stochastic nucleation.
The stochastic nucleation model was based on the formation of large YFP-CheR and CheY-YFP clusters that were regularly spaced, with a periodicity of approximately 1 μm (5). To confirm that MCPs also produce these regular clusters, the E. coli chemoreceptor Tar was C-terminally fused with a monomeric green fluorescent protein (GFP) variant (mGFP). Since GFP tends to form weak dimers, monomeric GFP was chosen to prevent possible localization artifacts (18). To reduce potential artifacts related to protein overexpression, a low-copy-number plasmid with a weakened IPTG (isopropyl-β-d-1-thiogalactopyranoside)-inducible promoter (pTRC99A [19]) was used to express the fusion protein. As shown in Fig. 1A, Tar-mGFP shows a classical septal and polar localization pattern. This localization does not depend on interaction with other chemoreceptors, since expression of the fusion protein in an MCP deletion strain shows the same localization pattern (see Fig. S2C in the supplemental material). The Tar-mGFP fusion protein was active according to a swarming plate assay (14) (see Fig. S1 in the supplemental material). The expression level was approximately twice that of Tar-mGFP expressed from its native promoter, but this increased expression did not change the localization pattern (see Fig. S2 in the supplemental material). We noticed that the use of the native promoter resulted in a substantial variation in expression levels between cells, and therefore we continued with the IPTG-inducible construct.
FIG 1.
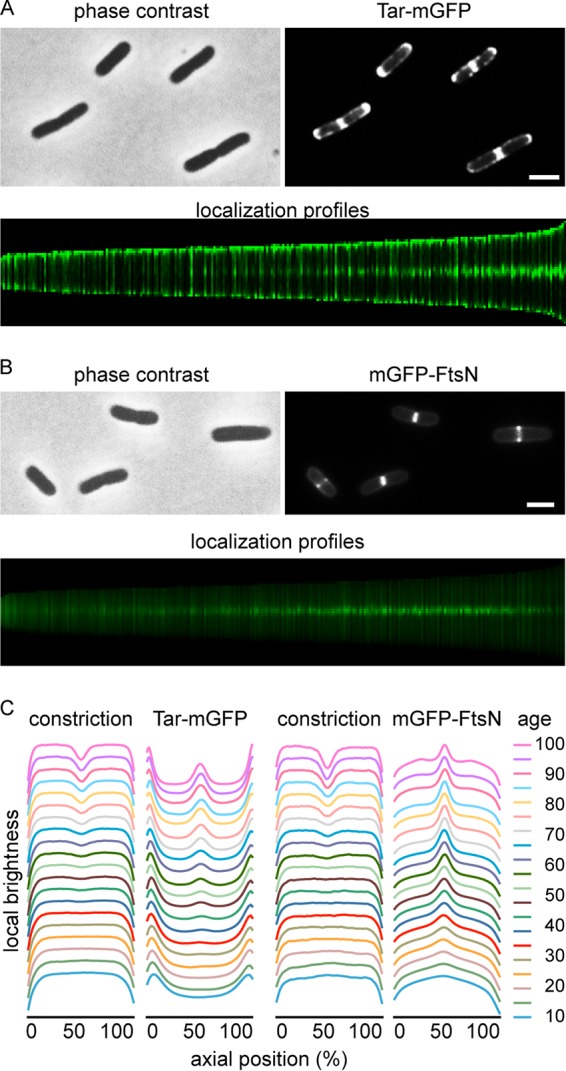
Localization of Tar-mGFP. (A) Fluorescence microscopy image of wild-type E. coli cells expressing Tar-mGFP. The lower panel shows sorted axial fluorescence profiles indicative of Tar-mGFP localization during the cell cycle. (B) Fluorescence microscopy image and cell cycle localization profile of cells expressing mGFP-FtsN. (C) Graphical presentation of cell constriction and fluorescence signals during the cell cycle, calculated from the localization profiles. Age refers to age classes divided in 5% (cell cycle) bins (see Materials and Methods for details). Totals of 5,074 and 6,437 cells were used to construct the cell cycle localization profiles for mGFP-FtsN and Tar-mGFP, respectively. Strains used in panels A and B were TSE29 and TSE48, respectively. Bars, 2 μm.
According to the stochastic nucleation model, chemotaxis proteins form large protein clusters prior to the initiation of cell division. To determine at what time in the cell cycle Tar accumulates at midcell, we performed a virtual time-lapse approach by sorting cells by size (Fig. 1A, lower panel) and plotting the related fluorescence intensities and cell constriction (Fig. 1C) (20). As a timer for cell division, we followed the localization of GFP-labeled FtsN, an essential cell division protein and part of the cell division machinery (21) (Fig. 1B). Comparison of the localization profiles indicates that Tar appears later at midcell compared to FtsN, suggesting that clustering of the chemotaxis proteins does not precede recruitment of FtsN (or the division machinery). To further confirm this, we used immunofluorescence and costained FtsZ and FtsN in cells expressing Tar-mGFP (see Fig. S3 in the supplemental material). This double-labeling experiment clearly showed that Tar-mGFP accumulated at midcell after FtsZ and FtsN.
To determine whether Tar forms regularly spaced clusters with a periodicity of around 1 μm in filamentous nondividing cells, we blocked cell division using the antibiotic cephalexin, which inactivates the cell division protein, FtsI, which is required for septum synthesis (22). As shown in Fig. 2A and B, no large regularly spaced fluorescent clusters were observed along the lateral wall of filamentous cells, but the polar clustering remained. Based on these data, it seems unlikely that Tar uses stochastic clustering to accumulate at cell poles.
FIG 2.
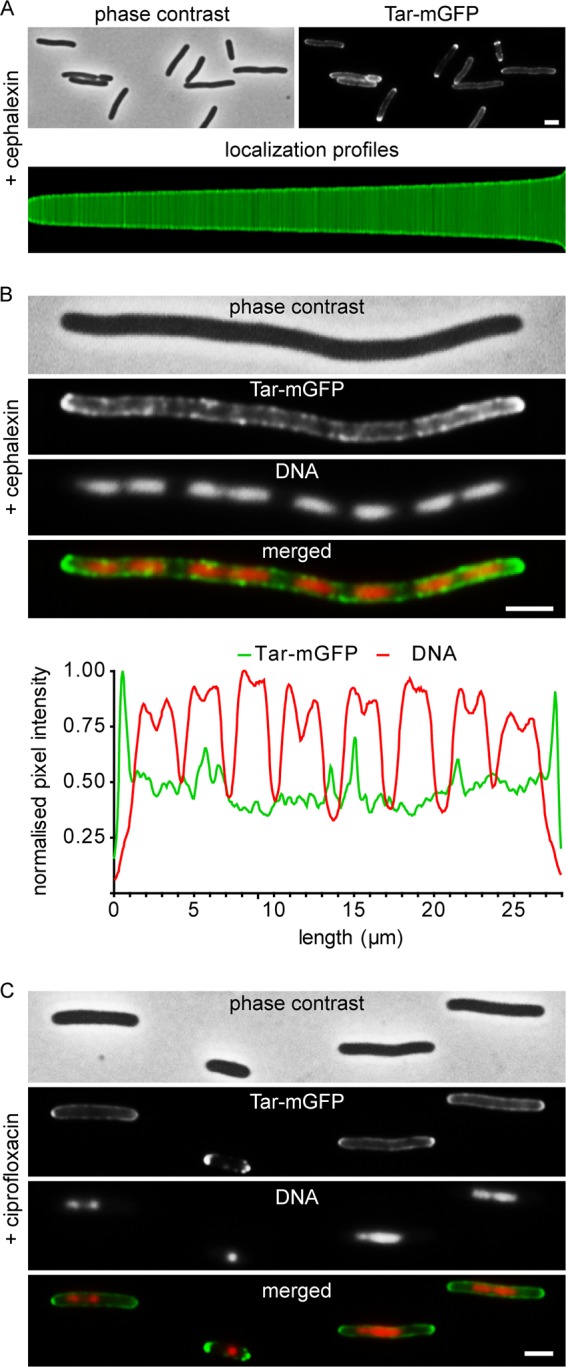
Tar-mGFP clustering and relation to nucleoid position. (A) Fluorescence microscopy image and localization profile of Tar-mGFP-expressing cells treated with 15 μg/ml cephalexin for 1 h. A total of 1,872 cells were used to construct the localization profile. (B) Fluorescence microscopy image of Tar-mGFP-expressing cells treated with 15 μg/ml cephalexin for 3 h. Nucleoids were stained with DAPI. Line scans for the GFP and DAPI signals are presented below. More examples are shown in Fig. S4 in the supplemental material. (C) Fluorescence microscopy image of Tar-mGFP-expressing cells treated with 0.035 μg/ml ciprofloxacin for 1 h. Nucleoids were stained with DAPI. Strain used was TSE29. Bars, 2 μm.
Nucleoid exclusion.
In a recent report, it was suggested that the serine MCP Tsr of E. coli is also driven to cell poles by the “volume exclusion” effect of the nucleoid (17). To examine whether nucleoids influence the distribution of Tar, we stained the cephalexin-treated cells with the fluorescence DNA dye DAPI (4′,6-diamidino-2-phenylindole) to visualize the nucleoids (Fig. 2B). We could not detect any correlation between the position of the nucleoids and the density of Tar-mGFP clusters along the lateral wall (see also line scans in Fig. 2B and S4). To corroborate this, cells were treated with ciprofloxacin, which inhibits DNA gyrase and blocks DNA replication, resulting in a dense nucleoid at the center of long cell, since the activated SOS-response also inhibit cell division (Fig. 2C) (23, 24). Under these conditions, the Tar-mGFP signal was also not reduced at the area occupied by the nucleoid. Thus, at least for Tar, nucleoid exclusion does not seem to be important for polar localization.
CheA-stimulated clustering.
The interaction of MCP trimers of dimers is stimulated by dimerization of the kinase CheA, which binds to the cytoplasmic termini of MCP trimers (2). In fact, we have found that CheA is essential to maintain polar localization of the chemoreceptor TlpA in B. subtilis (13). However, it was shown some time ago that in E. coli, CheA is not necessary for the polar clustering of chemoreceptors (25). Indeed, when we expressed Tar-mGFP in a cheA deletion mutant background, the protein accumulated at midcell and cell poles (Fig. 3A). There is some delay in Tar-mGFP accumulation at midcell compared to wild-type cells (compare with Fig. 1A, and see also Fig. S3 and S5 in the supplemental material). This is likely due to reduced clustering of MCP trimers and consequently to decreased retention of trimers at midcell.
FIG 3.
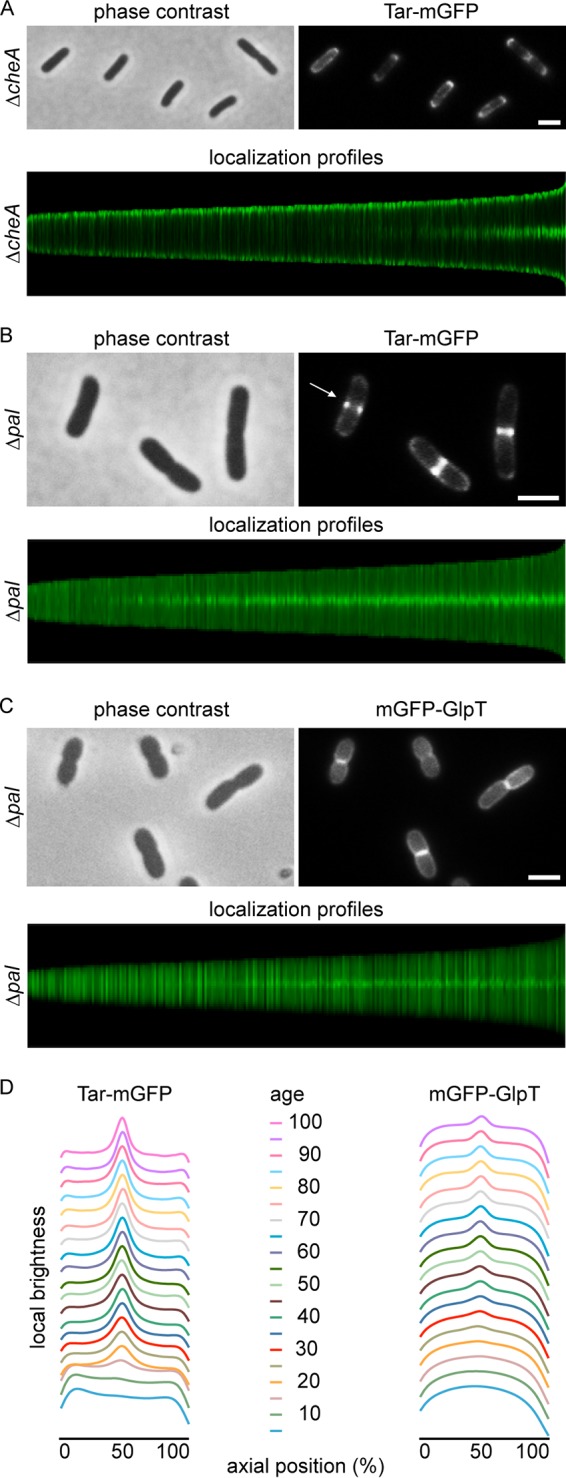
Effect of cheA and pal deletion mutants on Tar-mGFP localization. (A) Fluorescence microscopy image and localization profile of Tar-mGFP-expressing cheA deletion mutant. A total of 5,948 cells were used to construct the localization profile. (B) Fluorescence microscopy image and localization profile of Tar-mGFP-expressing pal deletion mutant. The arrow indicates Tar-mGFP foci. A total of 9,044 cells were used to construct the localization profile. (C) Fluorescence microscopy image and localization profile of mGFP-GlpT expressed in the pal deletion mutant. A total of 5,214 cells were used to construct the localization profile. (D) Graphical presentation of fluorescence signals during the pal deletion mutant cell cycle, calculated from the localization profiles. Age refers to age classes divided in 5% (cell cycle) bins. Strains used in panels A, B, and C are TSE38, TSE31, and TSE71, respectively. Bars, 2 μm.
Role of Tol-Pal.
The transenvelope Tol-Pal complex, which accumulates at midcell and assists in the division of the outer cell membrane (16), has been shown to be required for the polar localization of chemoreceptor proteins, and pulldown experiments suggested an interaction between TolA and chemoreceptors (14). To verify this, we expressed the Tar-mGFP fusion in a pal deletion mutant. Indeed, the polar accumulation of the fusion protein was completely abolished; however, there was still a strong accumulation at midcell (Fig. 3B). When the fusion protein was expressed in a tolA deletion mutant, a similar localization pattern was observed (Fig. S6).
During cell division, the cell membrane will cover the nascent division septum, thereby creating two flat membranes in close proximity between daughter cells. This will give a higher fluorescent membrane signal at midcell, so even when Tar is unable to localize and diffuses freely throughout the cell membrane, the extra cell membranes at the division sites could, in theory, account for an increase in GFP signal at midcell. To assess this, we followed the localization of a general transmembrane protein, the glycerol-3-phosphate transporter GlpT (26), throughout the cell cycle in the pal mutant. Indeed, the GFP signal showed a slight accumulation at midcell when cells started to divide (Fig. 3C); however, the signal intensity was much lower than that of Tar-mGFP (Fig. 3D), indicating that the double cell membrane at the division site is not responsible for the strong Tar-mGFP fluorescence signal at midcell in tol-pal mutant cells.
Membrane curvature.
A closer inspection of the pal mutant revealed that the Tar-mGFP signal often appears as two fluorescent dots at midcell (Fig. 3B), suggesting that Tar accumulates as a ring at midcell at the base of the nascent septum. We never observed Tar-mGFP signals of smaller sizes, reminiscent of constricting Z-rings, at midcell, indicating that the fusion protein does not follow the leading edge of the ingrowing septum. The Tol-Pal complex is recruited to the division site, and links invagination of the outer membrane with that of the cell membrane during cell division (16). Inactivation of Tol-Pal delays invagination of the outer membrane and cell separation, resulting in the formation of a division septum that resembles the septal cross walls in Gram-positive bacteria (27). The consequence of such a mode of division is that the cell membrane at the transition from the lateral wall to the nascent septal wall is strongly curved (28). This is where the Tar-mGFP fluorescence signal seems to accumulate in the pal deletion mutant (Fig. 3B). Presumably, Tar localizes at this region because of the curvature mismatch generated by the tripod configuration of the trimer of dimers in combination with the stiffness of the dimers (29). Tension in the membrane is released when these tripods locate in regions of the cell with a corresponding membrane curvature, such as those found at cell division sites. To confirm this, we introduced an N379R mutation in the trimerization site of Tar, corresponding to the N381R mutation in Tsr, which has been shown to abolish trimerization (30). Since single-membrane-spanning MCP dimers will not deform the membrane, they should therefore not accumulate at cell division sites when membrane curvature is the main driver for localization (Fig. 4A). Indeed, the N379R mutation resulted in the absence of clear septal and polar fluorescent signals (Fig. 4B). When we increased the flexibility of the dimers by introducing two glycines at positions 249 and 250, creating a stretch of 3 glycines in the HAMP domain (G248G-D249G-L250G), Tar was also no longer able to accumulate at midcell and cell poles (Fig. 4C and D), in line with the assumption that the unstructured glycine stretch eliminates the membrane curvature preference of the trimer (Fig. 4A) (12, 13). This delocalization was not a consequence of protein degradation, since Western blot analysis indicated that the point mutations did not affect the stability of the Tar-mGFP fusions (Fig. S7). Finally, when the dimerization and HAMP domain mutants were expressed in a pal mutant, a comparable delocalization was observed (Fig. S8). Thus, membrane curvature at the division septum seems to drive the localization of Tar trimers of dimers.
FIG 4.
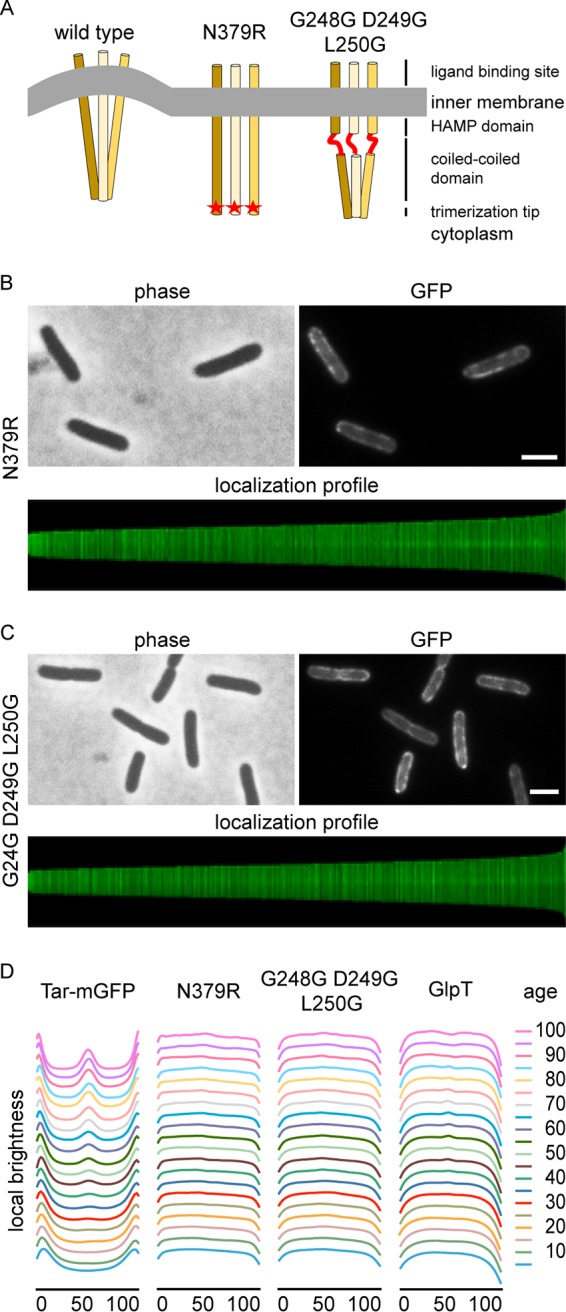
Membrane curvature is important for localization. (A) Schematic presentation of the effects of dimerization mutation N379R and the triple glycine substitution (G248G-D249G-L250G) on membrane curvature mismatch. (B) Fluorescence microscopy image and localization profile of Tar(N379R)-mGFP-expressing cells. A total of 7,331 cells were used to construct the localization profile. (C) Fluorescence microscopy image and localization profile of Tar(G248G D249G L2450G)-mGFP expressing cells. A total of 7,856 cells were used to construct the localization profile. (D) Graphical presentation of fluorescence signals during the cell cycle calculated from the localization profiles. The Tar-mGFP panel is the same as that in Fig. 1C. Age refers to age classes divided in 5% (cell cycle) bins. Strains used in panels B and C are TSE42 and TSE41, respectively, and strains used in panel D are TSE29, TSE42, TSE41 and TSE67. Bars, 2 μm.
DISCUSSION
Our data suggest that the MCPs of E. coli and B. subtilis arrive at cell poles by a comparable mechanism, which begins with cell division. First, they accumulate at midcell, where the cell membrane is bent inwards and strongly curved due to synthesis of the nascent cell division septum. Based on a composite crystal structure, the curvature of one trimer of dimers was calculated to amount to a radius of approximately 37 nm (8). Therefore, the membrane curvature mismatch is reduced considerably when a trimer of dimers is located at the base of the nascent division septum. After septation is completed, B. subtilis chemoreceptor clusters are maintained at the newly formed poles by forming large protein clusters that require CheA. However, in E. coli, the Tol-Pal complex, instead of CheA, is required to keep chemoreceptors clustered at the newly formed cell poles. Coimmunoprecipitation experiments have suggested an interaction between this complex and chemoreceptor proteins (14). Since lateral diffusion of the large transenvelope Tol-Pal complex is likely to be hampered by the peptidoglycan layer, interactions between Tol-Pal and chemoreceptor proteins might anchor MCPs and maintain their polar localization.
Several papers have argued that the curvature of the E. coli cell pole is sufficient to attract MCP trimers (8, 12, 31). However, the polar localization of Tar-mGFP is completely abolished when Tol-Pal is absent. This indicates that the curvature at the cell pole is not sufficient to markedly reduce the membrane curvature mismatch created by the Tar trimer of dimers. This is maybe not so surprising, since the cell pole has a curvature with a radius of approximately 500 nm, which is much larger than the 37 nm radius of MCP trimer of dimers (8). Moreover, the cylindrical lateral wall has a radius that is comparable to that of the cell pole, which makes the perceived curvature increase of cell poles even smaller.
Over the years, different mechanisms have been postulated, and contradictory results have been obtained in the research of polar localization of E. coli chemoreceptors. One possible explanation is that different groups use different protein reporters for chemoreceptors. In many studies, the cytoplasmic CheR was used as a proxy for chemoreceptor clusters (5, 12, 14), while others looked directly at the localization of MCPs (17). Another reason might be the use of fluorescent protein reporters with a tendency to dimerize, such as YFP and GFP. This characteristic has been shown to cause localization artifacts, especially when used with proteins that form multimers (18). Finally, we cannot exclude the possibility that different chemoreceptors use different mechanisms for localization. Nevertheless, in both E. coli and in B. subtilis it appears that the strong curvature generated during cell division is a key driving force for the localization of MCPs.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains used in this study are listed in Table S1 in the supplemental material. Strains were grown in GB1 minimal medium [6.33 g/liter K2HPO4·3H2O, 2.95 g/liter KH2PO4, 1.05 g/liter (NH4)2SO4, 0.10 g/liter MgSO4·7H2O, 28 mg/liter FeSO4·7H2O, and 7.10 mg/liter Ca(NO3)2·4H2O] supplemented with vitamin B1 (4 mg/ml), with 0.4% glucose as the carbon source, as previously described (32, 33). Auxotrophic BW25113 cells required arginine (50 μg/ml), glutamine (50 μg/ml), uracil (20 μg/ml), and thymidine (2 μg/ml). Either 100 μg/ml or 5 μg/ml (in the cases of pal or tolA mutants) of ampicillin was added to the growth medium to maintain plasmids.
Plasmid construction.
Purified DNA amplicons were used in a 1:10 molar ratio of vector to insert(s) in a Gibson assembly reaction (20 μl) at 50°C for 60 min. A total of 5 μl of each Gibson assembly reaction mix was used to transform ultracompetent E. coli TOP10 cells. Ultracompetent E. coli TOP10 cells were prepared as described by Hanahan et al. (34). Plasmids were sequenced to confirm constructs. Plasmids were transformed into chemically competent BW23115 wild-type or mutant cells prepared as described by Maniatis et al. (35). Transformants were selected on selective LB agar plates containing the appropriate concentration of antibiotic. Oligonucleotides (see Table S3) and plasmids (see Table S2) used in this study are listed in the supplemental material.
To construct the Tar-mGFP fusion, the point mutation GFP(A206K) was introduced in plasmid pBAD24-Tar-GFP (36) to prevent dimerization of GFP (18). The mutation was made by quick change, using primer pair GFP(A206K)-for/GFP(A206K)-rev and resulting in plasmid pBAD24-Tar-mGFP. To express Tar-mGFP from a weakened isopropyl-β-d-1-thiogalactoside (IPTG)-inducible promoter (19) and low-copy-number plasmid, the pBAD promoter was replaced by the pTRC99A promoter from pSAV57 (32), and the pUC19ori was replaced with the pSC101 ori from pSEN29 (37). First, pBAD24-Tar-mGFP was linearized by PCR amplification using the primer pair TerS327/TerS328, and then the pSC101 origin was amplified with the primer pair TerS425/TerS426. Subsequently, both products were ligated by Gibson assembly (38), resulting in plasmid pTNV107 (pBAD24-Tar-mGFP-pSC101 ori). To obtain the weak IPTG-inducible low-copy-number plasmid, plasmid pTNV107 was linearized with the primer pair TerS425/TerS507, and the pTRC99A promoter was amplified from pSAV057 using the primer pair TerS328/TerS506. The products were ligated using Gibson assembly, resulting in pTNV149 (pTRC99A-Tar-mGFP-pSC101 ori). To express Tar-mGFP using the native promoter from the same low-copy-number plasmid, the pBAD promoter in plasmid pTNV107 was replaced with the native promoter of tar. To this end, pTNV107 was linearized with the primer pair TerS457/TerS508, and a fragment of tar, including its promoter (39), was amplified from genomic DNA using the primer pair TerS509/TerS510. The products were ligated using Gibson assembly, resulting in pTNV148 (Ptar-Tar-mGFP-pSC101 ori).
To test if curvature caused by trimers of dimers is essential for Tar-mGFP localization, we introduced an N379R point mutation in Tar that abolished the interaction between dimers. The primer sets TerS328/TerS517 and TerS425/457 were used to introduce N379R in pTNV149 (pTRC99A-Tar-mGFP-pSC101 ori) using Gibson assembly, resulting in plasmid pTNV154 [pTRC99A-Tar(N379R)-mGFP-pSC101 ori)] We also introduced a stretch of 3-glycines in the HAMP domain of Tar to make the dimers flexible. Primer pairs TerS328/TerS516 and TerS425/515 were used to introduce the glycines G248G-D249G-L250G in Tar in pTNV149 (pTRC99A-Tar-mGFP-pSC101 ori), resulting in plasmid pTNV153 [(pTRC99A-Tar(G248G D249G L250G)-mGFP-pSC101 ori].
To compare midcell localization of Tar-mGFP to divisome assembly, we used the late-cell-division protein FtsN fused to monomeric GFP. The mGFP-FtsN fusion was constructed by PCR amplifications of pTNV149 with the primer pair TerS418/520, a monomeric variant of gfp from pTNV100 with the primer pair TerS362/521, and ftsN from E. coli genomic DNA with primer pair TerS523/541, followed by Gibson assembly, resulting in the plasmid pTNV155 (pTRC99A-mGFP-FtsN-pSC101 ori).
As a control for membrane localization, we constructed a glycerol-3-phosphate transporter GlpT-GFP fusion. mGFP-GlpT was made by PCR amplifications of pTNV149 with the primer pair TerS418/520, a monomeric variant of gfp from pTNV100 with the primer pair TerS362/521, and glpT from E. coli genomic DNA with the primer pair TerS544/545, followed by Gibson assembly, resulting in the plasmid pTNV162 (pTRC99A-mGFP-GlpT-pSC101 ori).
Functionality and stability of Tar-mGFP fusions.
To test whether the Tar-mGFP fusions were functional, we performed a motility swarming experiment according to the method of Santos et al. (14). The motility of Δmcp cells expressing the wild type and mutant Tar fusions was tested by spotting 10 μl of culture with a comparable optical density at 600 nm (OD600) of ∼0.5 on LB plates containing 0.25% agar, including 15 μM IPTG for the plasmid-containing strains.
To check that the Tar-mGFP mutant proteins were not degraded, we performed a Western blot analysis using GFP antibody. The relevant strains were grown, and expression of the fusion protein was induced with 15 μM IPTG for 2 h. After correction for optical density, cells were boiled in Laemmli sample buffer for 15 min. A total of 15 μl of each sample was used for SDS-PAGE.
Microscopy and image analysis.
The virtual time-lapse analysis is based on the fact that during steady-state growth the average mass of all cells and their age frequency distribution are constant allowing precise spatiotemporal information on protein localization during the cell cycle, as described in reference 40. Steady state was obtained by growing cells in GB1 medium at 30°C under shaking (210 rpm) while keeping optical density at 450 nm (OD450) below 0.2 by regular dilution in prewarmed medium for 3 days to reach steady-state growth. At steady state, Tar-mGFP was induced with 15 μM IPTG for at least two doubling times. Steady-state cells were centrifuged at 1,000 rpm for 2 min to bring the OD450 to ∼0.4.
Samples consisting of 0.3 μl of cells were spotted onto a microscope slide covered with a thin layer of 1.3% agarose. When applicable, cells were treated with 15 μg/ml cephalexin for 1 to 4 h or with 0.035 μg/ml ciprofloxacin for 1 h. The presence of the bla gene, which encodes beta-lactamase, on the plasmids did not interfere with the activity of the beta-lactam antibiotic cephalexin. Images were acquired with a 500-ms exposure time for the GFP channel. Fluorescence microscopy was carried out with a Nikon Eclipse Ti equipped with a CFI Plan Intensilight HG 130-W lamp, a C11440-22CU Hamamatsu ORCA camera, and NIS-Elements software version 4.20.01. Images were analyzed using ImageJ version 1.50i (https://imagej.nih.gov/ij/) and the ImageJ plug-in ObjectJ version 03p (40).
To construct the localization profiles of the fluorescent signals, images were analyzed using Coli-Inspector in the advanced mode (40). First, phase contrast and fluorescent channels were aligned using the accompanying macro, and then the backgrounds of the fluorescent channels were subtracted using the built-in commands. Second, individual cells were selected and manually confirmed. Finally, selected cells were sorted by length, and the related fluorescent signal along the cell axis was also obtained.
Immunofluorescence labeling of FtsZ and FtsN.
Immunofluorescence staining of FtsZ and FtsN was performed as described before (40). Briefly, 12.2 ml of cells were fixed with 2.8% formaldehyde and 0.04% glutaraldehyde for 15 min. Cells were harvested by centrifugation, washed, and resuspended in 150 μl of phosphate-buffered saline (PBS) buffer. To permeabilize the cells, the fixed cells were treated with 0.1% Triton X-100–PBS at room temperature for 45 min, followed by a triple washing step with PBS. Cells were then treated with 100 μg/ml lysozyme and 5 mM EDTA at room temperature for 45 min, and subsequently washed three times with PBS. Prior to labeling, nonspecific binding sites were blocked by incubating cells in 0.5% (wt/vol) blocking reagents (F. Hoffmann-La Roche) in PBS at 37°C for 30 min. FtsZ and FtsN were immune labeled using purified primary polyclonal antibodies against each protein (40) and diluted in 1:500 blocking buffer for an hour at 37°C. Cells were washed three times with PBS and subsequently incubated with secondary donkey anti-rabbit CY3 antibody diluted 1:600 in blocking buffer at 37°C for 30 min. Cells were washed three times with PBS−0.05% Tween 20 and once with PBS. After resuspension in PBS, they were stored in the fridge, ready for immunofluorescence microscopy.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Bacterial Cell Biology group for useful discussions, especially Tanneke den Blaauwen for discussions on protein localization in E. coli and for providing antibodies against FtsZ and FtsN. We thank Séverin Ronneau (Imperial College London) for constructing the initial monomeric GFP reporter, and Ikuro Kawagishi (Hosei University), Joen Luirink (VU University), Pierre Genevaux (CNRS), and Tom Shimizu (AMOLF) for kindly providing us with strains and plasmids.
This research was funded by the Biotechnology and Biological Sciences Research Council (BBSRC) (grant BB/I01327X/1), Marie Curie Career Integration Grant DIVANTI (618452), and NWO STW-Vici (grant 12128).
T.N.V.S., H.S., and L.W.H designed the research; T.N.V.S. performed the research; T.N.V.S., H.S., and L.W.H analyzed the data and wrote the paper.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00658-17.
REFERENCES
- 1.Porter SL, Wadhams GH, Armitage JP. 2011. Signal processing in complex chemotaxis pathways. Nat Rev Micro 9:153–165. doi: 10.1038/nrmicro2505. [DOI] [PubMed] [Google Scholar]
- 2.Parkinson JS, Hazelbauer GL, Falke JJ. 2015. Signaling and sensory adaptation in Escherichia coli chemoreceptors: 2015 update. Trends Microbiol 23:257–266. doi: 10.1016/j.tim.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alley MRK, Maddock JR, Shapiro L. 1992. Polar localization of a bacterial chemoreceptor. Genes Dev 6:825–836. doi: 10.1101/gad.6.5.825. [DOI] [PubMed] [Google Scholar]
- 4.Maddock Shapiro L. 1993. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science 259:1717–1723. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- 5.Thiem S, Kentner D, Sourjik V. 2007. Positioning of chemosensory clusters in E. coli and its relation to cell division. EMBO J 26:1615–1623. doi: 10.1038/sj.emboj.7601610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiem S, Sourjik V. 2008. Stochastic assembly of chemoreceptor clusters in Escherichia coli. Mol Microbiol 68:1228–1236. doi: 10.1111/j.1365-2958.2008.06227.x. [DOI] [PubMed] [Google Scholar]
- 7.Greenfield D, McEvoy AL, Shroff H, Crooks GE, Wingreen NS, Betzig E, Liphardt J. 2009. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol 7:e1000137. doi: 10.1371/journal.pbio.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endres RG. 2009. Polar chemoreceptor clustering by coupled trimers of dimers. Biophys J 96:453–463. doi: 10.1016/j.bpj.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim KK, Yokota H, Kim S-H. 1999. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature 400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 10.Derganc J. 2007. Curvature-driven lateral segregation of membrane constituents in Golgi cisternae. Phys Biol 4:317–324. doi: 10.1088/1478-3975/4/4/008. [DOI] [PubMed] [Google Scholar]
- 11.Aimon S, Callan-Jones A, Berthaud A, Pinot M, Toombes GES, Bassereau P. 2014. Membrane shape modulates transmembrane protein distribution. Dev Cell 28:212–218. doi: 10.1016/j.devcel.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Draper W, Liphardt J. 2017. Origins of chemoreceptor curvature sorting in Escherichia coli. Nat Commun 8:14838. doi: 10.1038/ncomms14838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strahl H, Ronneau S, González BS, Klutsch D, Schaffner-Barbero C, Hamoen LW. 2015. Transmembrane protein sorting driven by membrane curvature. Nat Commun 6:8728. doi: 10.1038/ncomms9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos TMA, Lin T-Y, Rajendran M, Anderson SM, Weibel DB. 2014. Polar localization of Escherichia coli chemoreceptors requires an intact Tol-Pal complex. Mol Microbiol 92:985–1004. doi: 10.1111/mmi.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sturgis JN. 2001. Organisation and evolution of the tol-pal gene cluster. J Mol Microbiol Biotechnol 3:113–122. [PubMed] [Google Scholar]
- 16.Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PAJ. 2007. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol Microbiol 63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neeli-Venkata R, Startceva S, Annila T, Ribeiro AS. 2016. Polar localization of the serine chemoreceptor of Escherichia coli is nucleoid exclusion-dependent. Biophys J 111:2512–2522. doi: 10.1016/j.bpj.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. 2012. Segregation of molecules at cell division reveals native protein localization. Nat Meth 9:480–482. doi: 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amann E, Ochs B, Abel KJ. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- 20.den Blaauwen T, Buddelmeijer N, Aarsman M, Hameete CM, Nanninga N. 1999. Timing of FtsZ assembly in Escherichia coli. J Bacteriol 181:5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Addinall SG, Cao C, Lutkenhaus J. 1997. FtsN, a late recruit to the septum in Escherichia coli. Mol Microbiol 25:303–309. doi: 10.1046/j.1365-2958.1997.4641833.x. [DOI] [PubMed] [Google Scholar]
- 22.Botta GA, Park JT. 1981. Evidence for involvement of penicillin-binding protein 3 in murein synthesis during septation but not during cell elongation. J Bacteriol 145:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeBel M. 1988. Ciprofloxacin: chemistry, mechanism of action, resistance, antimicrobial spectrum, pharmacokinetics, clinical trials, and adverse reactions. Pharmacotherapy 8:3–30. doi: 10.1002/j.1875-9114.1988.tb04058.x. [DOI] [PubMed] [Google Scholar]
- 24.Khodursky AB, Cozzarelli NR. 1998. The mechanism of inhibition of topoisomerase IV by quinolone antibacterials. J Biol Chem 273:27668–27677. doi: 10.1074/jbc.273.42.27668. [DOI] [PubMed] [Google Scholar]
- 25.Skidmore JM, Ellefson DD, McNamara BP, Couto MMP, Wolfe AJ, Maddock JR. 2000. Polar clustering of the chemoreceptor complex in Escherichia coli occurs in the absence of complete CheA function. J Bacteriol 182:967–973. doi: 10.1128/JB.182.4.967-973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oswald F, Varadarajan A, Lill H, Peterman EJG, Bollen YJM. 2016. MreB-dependent organization of the E. coli cytoplasmic membrane controls membrane protein diffusion. Biophys J 110:1139–1149. doi: 10.1016/j.bpj.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meury J, Devilliers G. 1999. Impairment of cell division in tolA mutants of Escherichia coli at low and high medium osmolarities. Biol Cell 91:67–75. doi: 10.1111/j.1768-322X.1999.tb01085.x. [DOI] [PubMed] [Google Scholar]
- 28.Lenarcic R, Halbedel S, Visser L, Shaw M, Wu LJ, Errington J, Marenduzzo D, Hamoen LW. 2009. Localisation of DivIVA by targeting to negatively curved membranes. EMBO J 28:2272–2282. doi: 10.1038/emboj.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S-H, Wang W, Kim KK. 2002. Dynamic and clustering model of bacterial chemotaxis receptors: Structural basis for signaling and high sensitivity. Proc Natl Acad Sci U S A 99:11611–11615. doi: 10.1073/pnas.132376499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gosink KK, Zhao Y, Parkinson JS. 2011. Mutational analysis of N381, a key trimer contact residue in Tsr, the Escherichia coli serine chemoreceptor. J Bacteriol 193:6452–6460. doi: 10.1128/JB.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haselwandter CA, Wingreen NS. 2014. The role of membrane-mediated interactions in the assembly and architecture of chemoreceptor lattices. PLoS Comput Biol 10:e1003932. doi: 10.1371/journal.pcbi.1003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexeeva S, Gadella TWJ, Verheul J, Verhoeven GS, den Blaauwen T. 2010. Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol Microbiol 77:384–398. doi: 10.1111/j.1365-2958.2010.07211.x. [DOI] [PubMed] [Google Scholar]
- 33.Aarsman MEG, Piette A, Fraipont C, Vinkenvleugel TMF, Nguyen-Distèche M, Blaauwen den T. 2005. Maturation of the Escherichia coli divisome occurs in two steps. Mol Microbiol 55:1631–1645. doi: 10.1111/j.1365-2958.2005.04502.x. [DOI] [PubMed] [Google Scholar]
- 34.Hanahan D, Jessee J, Bloom FR. 1991. Plasmid transformation of Escherichia coli and other bacteria. Methods Enzymol 204:63–113. doi: 10.1016/0076-6879(91)04006-A. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J, Maniatis T. 1989. Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 36.Shiomi D, Yoshimoto M, Homma M, Kawagishi I. 2006. Helical distribution of the bacterial chemoreceptor via colocalization with the Sec protein translocation machinery. Mol Microbiol 60:894–906. doi: 10.1111/j.1365-2958.2006.05145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genevaux P, Keppel F, Schwager F, Langendijk-Genevaux PS, Hartl FU, Georgopoulos C. 2004. In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep 5:195–200. doi: 10.1038/sj.embor.7400067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Meth 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 39.Kundu TK, Kusano S, Ishihama A. 1997. Promoter selectivity of Escherichia coli RNA polymerase sigmaF holoenzyme involved in transcription of flagellar and chemotaxis genes. J Bacteriol 179:4264–4269. doi: 10.1128/jb.179.13.4264-4269.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vischer NOE, Verheul J, Postma M, van den Berg van Saparoea B, Galli E, Natale P, Gerdes K, Luirink J, Vollmer W, Vicente M, den Blaauwen T. 2015. Cell age dependent concentration of Escherichia coli divisome proteins analyzed with ImageJ and ObjectJ. Front Microbiol 6:586. doi: 10.3389/fmicb.2015.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.