

Abstract
The design, synthesis and biological evaluation of stilbene-based novobiocin analogs is reported. Replacement of the biaryl amide side chain with a triazole side chain produced compounds that exhibited good anti-proliferative activities. Hsp90 inhibition was observed when N-methyl piperidine was replaced with acyclic tertiary amines on the stilbene analogs that also contain a triazole-derived side chain. These studies revealed that ~24 Å is the optimal length for compounds that exhibit good anti-proliferative activity as a result of Hsp90 inhibition.
Keywords: Heat Shock Protein 90, Stilbene, C-terminal inhibitors, Anti-cancer agents
Table of Contents
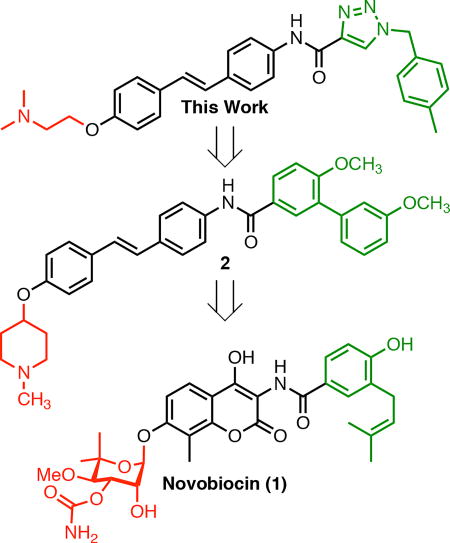
This work focused on the design and biological evaluation of stilbene-based novobiocin analogs. Intial efforts revealed the stilbene moiety as the optimal core. These studies revealed that replacement of the biaryl amide side chain with triazole containing analogs and tertiary amines on the stilbene core produced more efficacious compounds as confirmed by EC50 values and western blot analyses.
Introduction
Heat shock protein 90 kDa (Hsp90) is a molecular chaperone that folds polypeptides and denatured proteins into their bioactive conformation.[1] In humans, Hsp90 exists as four isoforms: Hsp90α, Hsp90β, glucose-regulated protein 94 kDa (Grp94), and tumor necrosis receptor-associated protein 1 (Trap1). Hsp90α and Hsp90β are localized to the cytoplasm, whereas Grp94 resides in the endoplasmic reticulum, and Trap1 is localized within the mitochondria.[2,3] Structurally, these isoforms are homodimers, whose monomeric unit consists of an N-terminal ATP-binding motif, a middle domain, and a C-terminal dimerization motif.[4,5] Hsp90 is responsible for the activation/maturation of more than 300 client proteins, many of which are associated with the 10 hallmarks of cancer.[6–8] Since Hsp90 inhibition results in the simultaneous degradation of multiple oncogenic targets, it has emerged as a novel target for to development of anti-cancer agents. Additionally, studies have shown that Hsp90 is overexpressed in cancer, which provides an additional opportunity to selectively target Hsp90 in cancer cells versus normal tissue.[9–11]
Initial efforts to develop Hsp90 inhibitors for the treatment of cancer focused on compounds that bind the Hsp90 N-terminus. These efforts led to the identification of twenty Hsp90 N-terminal inhibitors that have now been investigated in a clinical setting.[12–15] To date, no Hsp90 inhibitor has been approved by the FDA for the treatment of cancer. Unfortunately, these studies were plagued by dosing difficulties and some toxicity issues associated with induction of the pro-survival heat shock response at the same concentration that inhibits client protein folding.[16] In order to address these issues, Hsp90 isoformselective inhibitors have been sought to limit and identify potential on-target toxicities.[17] Recently, it was demonstrated that selective inhibition of Hsp90β can inhibit a number of cancers without concomitant induction of Hsp90.[18]
Hsp90 C-terminal inhibitors have been shown to induce Hsp90 client protein degradation without concomitant induction of the heat shock response.[19] Therefore, this approach represents an attractive alternative to Hsp90 N-terminal inhibition. Although there is no co-crystal structure of the Hsp90 C-terminus bound to inhibitors,[20] drug discovery efforts have led to the identification of Hsp90 C-terminal inhibitors that exhibit excellent anti-proliferative activity against a variety of cancers.[21] Those Hsp90 C-terminal inhibitors have been developed by elucidation and evaluation of structure-activity relationship (SAR) studies on novobiocin (1), which was the first Hsp90 C-terminal inhibitor identified.[22–24] Previous studies also determined the optimal distance and angle between the N-methyl piperidine and biaryl amide side chain[25] which revealed that the most efficacious anti-proliferative and Hsp90 inhibitory activities were obtained when the distance was between 7.7 to 11.8 Å and the angle was ~180° (Figure 1). In fact, stilbene 2 was identified as the most efficacious inhibitor that exhibited both potent anti-proliferative and Hsp90 inhibitory activity. Therefore, the focus of this work was to optimize 2, identify structure-activity relationships, and obtain more efficacious compounds.
Figure 1.

Previous Hsp90 inhibitors
Results and Discussion
Synthesis and biological evaluation of stilbene core analogs
Initial efforts to modify 2 consisted of three approaches (Figure 2): The first approach was to restrict rotation of the phenyl rings within the stilbene core, thus prohibiting the entropic penalty observed upon binding. Such compounds could provide insight into preferred orientation of the phenyl ring upon binding Hsp90. The first ring-constrained analogs were comprised of a 6-phenyl-7,8-dihydronaphthalene core, which allowed rotation of one phenyl ring. The second approach sought to investigate the electronic effects by enlisting a Topliss tree approach[26] to dictate subsequent modification. The final approach was to modify orientation of the N-methyl piperidine and biaryl amide side chain through the incorporation of a cyclopropane ring or inclusion of a saturated phenyl ring.
Figure 2.

Initial analogs of 2
Synthesis of ring-constrained analogs
Synthesis of 3 commenced via a Suzuki coupling between vinyl triflate 8[27] and 4-hydroxyphenylboronic acid to produce 9 in 45% yield (Scheme 1). N-methyl-4-piperidinol (10) was then coupled with 9 via a Mitsunobu etherification.[28] Following removal of the Boc-protecting group, the resulting aniline was coupled with acid chloride 12 to yield 3 in 39% over both steps. Compound 3 was subsequently subjected to hydrogenation conditions to yield the saturated derivative, 13.
Scheme 1.

Synthesis of 3
Preparation of 4 was initiated by installation of a methoxymethyl (MOM) protecting group onto 6-hydroxy-2-tetralone (Scheme 2). Tetralone 14 was first converted to the vinyl triflate and then immediately coupled with 4-nitrophenylboronic acid, to form 16. Upon removal of the protecting group present in 16, the resulting phenol was coupled with N-methyl-4-piperidinol through a Mitsunobu etherification reaction. Following reduction of the nitro group in 17 to the corresponding aniline, the aniline was treated with acid chloride 12 to form 4 in 50% yield. Hydrogenation of 4 led to saturated derivative 18, in 58% yield.
Scheme 2.

Synthesis of 4
Synthesis of analogs to study electronic effects
The first stilbene derivatives synthesized contained a chloride group in the ortho- or meta-position of the amide substituted phenyl ring (See supporting information for synthesis). Since the chloro-containing derivatives were less active than the parent compound, methoxy-substituted stilbenes were synthesized according to the Topliss approach (Scheme 3). Stilbenes 20a/b were synthesized via a Heck coupling between styrene 19[29] and the corresponding nitrobenzenes. Upon reduction of the nitro groups in 20a/b, the resulting anilines were coupled with carboxylic acid 21 to form amides 22a/b. Following removal of the MOM group, the corresponding phenols were coupled with N-methyl-4-piperidinol.
Scheme 3.

Synthesis of 23a/b
Synthesis of saturated analogs of 2
Synthesis of the cyclopropane analog[30] began via cyclopropanation of stilbene 24.[31,32] (Scheme 4) Following demethylation of 25, a methoxymethyl group was placed onto the resulting phenol. Aryl bromide 26 was coupled[33] with amide 27 via Hartwig-Buchwald amidation to form 28. After removal of the methoxymethyl ether, the corresponding phenol was coupled with N-methyl-4-piperidinol to form the final product, 6.
Scheme 4.

Synthesis of cyclopropane analog
Preparation of the saturated analog of 2 commenced via a Horner-Wadsworth-Emmons reaction between phosphonate 29 and aldehyde 30,[34] to form 31 (Scheme 5). After removal of the ketal on 31, the resulting ketone 32 was subjected to a deprotection-protection protocol to produce 33. Syn alcohol 34 was obtained via the treatment of 33 with L-Selectride. Following activation of the alcohol with methanesulfonyl chloride, the resulting mesylate was displaced to form azide 35. Amide 36 was then formed by reduction of azide 35 to form the corresponding amine, which was subsequently coupled with carboxylic acid 21 to give 36. Removal of the toluenesulfonate ester and coupling of the resulting phenol with N-methyl-4-piperidinol completed the preparation of 7.
Scheme 5.

Synthesis of saturated analog of 2
These initial analogs were evaluated against three cancer cell lines: MCF-7 (estrogen receptor positive breast cancer cells), SKBr3 (estrogen receptor negative, HER2 overexpressing breast cancer cells) and HCT-116 (human colon cancer cells). (Table 1) Restriction of rotation of either phenyl ring resulted in a significant or complete loss of anti-proliferative activity across all three cancer cell lines. These results suggest that the addition of two carbons at the center of the stilbene core may cause a steric clash with Hsp90. The addition of a chloro or methoxy group also caused a significant loss in anti-proliferative activity, as compared to the parent compound. The saturated analogs manifested the most efficacious anti-proliferative activities, and the activity of 7 was comparable to parent compound 2. In order to determine whether the anti-proliferative activities manifested by the saturated analogs were the result of Hsp90 inhibition, western blot analyses were performed on MCF-7 cell lysates treated with 6 and 7 for 24 hours (Figure 1S). Neither compound was able to induce Hsp90 dependent client protein degradation at concentrations that mirror the IC50 values. Overall, the results from these compounds show that stilbene 2 remains the optimal core for the design of future analogs.
Table 1.
Evaluation of anti-proliferative activity
| ||||
|---|---|---|---|---|
| Compound | Structure | MCF-7[a] | SKBr3[a] | HCT-116[a] |
| 2 |
|
1.3±0.59 | 0.68±0.10 | 3.68±0.41 |
| 3 |
|
>50 | >50 | >50 |
| 13 |
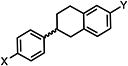
|
>50 | >50 | >50 |
| 4 |
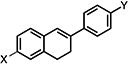
|
2.61±0.98 | 3.54±0.43 | 3.57±0.19 |
| 18 |
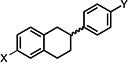
|
7.36±0.82 | 10.59±1.61 | 6.43±0.18 |
| 23a |
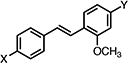
|
3.59±0.068 | 3.64±0.20 | 4.50±0.99 |
| 23b |
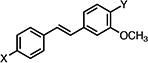
|
>50 | >50 | >50 |
| 37a |
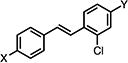
|
>50 | >50 | >50 |
| 37b |
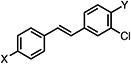
|
>50 | 27.46±0.057 | >50 |
| 6 |
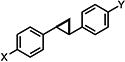
|
2.64±1.04 | 1.31±0.251 | 2.90±0.69 |
| 7 |
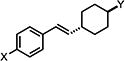
|
0.814±0.126 | 0.894±0.004 | 0.801±0.091 |
= Values are in µM, which represent mean standard deviation for at least two separate experiments performed in triplicate.
Second-generation stilbene analogs
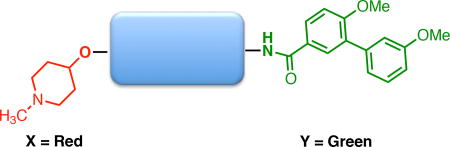
It was important to change either the N-methyl piperidine or biaryl amide side chain based on the information gained from the first generation of compounds. Since the N-methyl piperidine and biaryl amide side chain were optimized for the coumarin and biphenyl scaffold, appendages on both sides of the stilbene core were investigated.
Investigation of replacements for the N-methyl piperidine and biaryl amide side chain
Previous optimization studies on the noviose sugar and prenylated amide side chain of novobiocin (1) provided insight towards the inclusion of N-methyl piperidine and the biaryl amide side chain. For example, such studies revealed that the best anti-proliferative activities were obtained when the noviose sugar was replaced with a tertiary amine.[22b] Subsequent studies have shown that the tertiary amine (cyclic versus acyclic) can play a significant role in binding the Hsp90 C-terminal domain.[35] Therefore, various acyclic tertiary amines were investigated with this stilbene core. In terms of the biaryl amide side chain, work has been done to probe the utility of each aromatic ring on the amide side chain and alternative aromatic systems have been identified that can replace the biaryl amide side chain. Of the systems investigated from the literature, a triazole-containing amide side chain was shown to manifest the most efficacious activity against multiple cancer cell lines.[22c] Therefore, triazole-based amide side chains were investigated as replacements for the biaryl amide side chain in this study.
Scheme 6 illustrates the general synthetic route used to construct the triazole-based stilbene analogs. 4-Nitro-4’-hydroxystilbene was protected with a methoxymethyl group to yield 39. Next, the nitro group was reduced to the corresponding aniline, which was then coupled with the requisite carboxylic acid to form the resulting amide. The protecting group on the amide was removed before the appropriate tertiary amine was then coupled with the phenol via a Mitsunobu etherification or SN2 reaction.
Scheme 6.

General Scheme for the preparation of second-generation stilbene analogs
Replacement of the biaryl amide side chain with the triazole moiety caused a significant increase in anti-proliferative activity against all of the investigated cell lines (Table 2). Regardless of R, compounds containing the triazole and 4-methylbenzyl group (C) were more potent than the benzyl (B) substituted derivatives. When N-methyl-piperidine (X) was replaced with 3-(dimethylamino)propane (Y), the activity decreased as in 47, while the activity of 46 was similar to 44. Interestingly, when the shorter 2-(dimethylamino)ethane (Z) was added onto the stilbene core, an improvement in anti-proliferative activity was observed for both triazole-containing compounds (48 and 49). This data suggests that a limited amount of space exists within the binding pocket. Once a compound exceeds that defined space, then the anti-proliferative activities decrease. For example, the activities manifested by 49 are better than the activities exhibited by 47, suggesting that 47 is too large to fit within the Hsp90 C-terminal binding pocket. In order to independently probe the effect of the acyclic amine on anti-proliferative activity, 50 was tested against all of the cancer cell lines. Since 50 is significantly less active than 2, it is clear that the increase in anti-proliferative activity is due to replacement of the biaryl amide side chain with the triazole moiety.
Table 2.
Evaluation of biological activities of second-generation stilbene analogs
| Compound | R4 | R2 | MCF-7[a] | SKBr3[a] | HCT-116[a] |
|---|---|---|---|---|---|
| 2 | X | A | 1.3±0.59 | 0.68±0.10 | 3.68±0.41 |
| 44 | X | B | 0.326±0.081 | 0.231±0.035 | 0.301±0.043 |
| 45 | X | C | 0.092±0.005 | 0.099±0.020 | 0.186±0.035 |
| 46 | Y | B | 0.325±0.034 | 0.382±0.101 | 0.350±0.064 |
| 47 | Y | C | 0.494±0.053 | 0.184±0.071 | 0.740±0.164 |
| 48 | Z | B | 0.234±0.052 | 0.183±0.003 | 0.350±0.064 |
| 49 | Z | C | 0.150±0.008 | 0.141±0.028 | 0.179±0.050 |
| 50 | Y | A | 5.86±1.23 | 9.68±0.209 | 5.51±0.225 |
= Values are in µM, which represent mean standard deviation for at least two separate experiments performed in triplicate.
Western blot analyses of MCF-7 cell lysates incubated with 44 and 45 for 24 hours were conducted to determine whether the observed anti-proliferative activities resulted from Hsp90 inhibition (Figure 3). For 44, a dose-dependent degradation of Hsp90-dependent client proteins was observed, whereas no degradation of Hsp90 client proteins was observed for 45. The western blot analysis of 46 and 47 showed that 46, but not 47, induced client protein degradation (Figure 4). On the other hand, western blot analysis of 48 and 49 revealed that 49 was able to induce Hsp90 client protein degradation. This data supports that there is an overall optimal length for occupancy of the Hsp90 C-terminal binding pocket. As shown in Figure 5, the best compounds (46 and 49) are ~ 24 Å in length. If the compound is too long (47) or too short (48), then the compound is unable to inhibit Hsp90 and induce client protein degradation. The improvement in Hsp90 inhibition by 46 and 49 reveals that the incorporation of a flexible 3-(dimethylamino)alkyl group can produce better interactions with Hsp90 for this particular scaffold.
Figure 3.


Western blot analysis of Hsp90 client protein degradation in MCF-7 cells. Geldanamycin (GDA 500 nm) represents a positive control, while DMSO, represents the negative control. A. Analysis of cells treated with 44. B. Analysis of cells treated with 45.
Figure 4.


Western blot analysis of Hsp90 client protein degradation in MCF-7 cells treated with (A) 37 and 38 or (B) 39 and 40. Geldanamycin (GDA 500 nm) represents a positive control, while DMSO, represents the negative control. L represents a concentration 1/2XIC50 value while H represents a concentration of 5XIC50 value.
Figure 5.

3D structures of 46–49 with the length (Å) of each compound. A simple MM2 minimization was done in Chem3D[36] for all of the compounds. The distance of the minimized structures were measured in Maestro.[37]
Conclusions
In an effort to optimize 2, structure-activity relationships were elucidated for 17 analogs, which revealed the stilbene core to be optimal for these compounds. Replacement of the biaryl amide side chain with a triazole-containing group improved anti-proliferative activity. In addition, replacement of the N-methyl piperidine with a (dimethylamino)alkyl group led to Hsp90 inhibition. The overall length of the best Hsp90 inhibitors was 24 Å.
Experimental Section
1H NMR were recorded at 400 or 500 MHz (Bruker DRX- 400 Bruker with a H/C/P/F QNP gradient probe) spectrometer and 13C NMR spectra were recorded at 125 MHz (Bruker DRX 500 with broadband, inverse triple resonance, and high resolution magic angle spinning HR-MA probe spectrometer); chemical shifts are reported in δ (ppm) relative to the internal reference CDCl3 (CDCl3, 7.26 ppm). FAB (HRMS) spectra were recorded with a LCT Premier (Waters Corp., Milford, MA) spectrometer and IR spectra were recorded on a Magna FT-IR spectrometer (Nicolet Instrument Corporation, Madison, WI). The purity of all compounds was determined to be >95% as determined by 1H NMR and 13C NMR spectra, unless otherwise noted. TLC was performed on glass- backed silica gel plates (Uniplate) with spots visualized by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure. Detailed experimental procedures can be found in the supporting information.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge support of this project by the National Cancer Institute of the National Institutes of Health, CA120458 and CA167079 (KMB) and NIH Dynamic Aspects of Chemical Biology, T32 GM08545 (CNK).
Footnotes
Anuj Khandewal and the life he lived
References
- 1.Trepel J, Mollapour M, Giaccone G, Neckers L. Nature Rev. Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sreedhar AS, Kalmar E, Csermely P, Shen Y. FEBS Lett. 2004;562:11–15. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 3.Seo YH. Arch. Pharm. Res. 2015;38:1582–1590. doi: 10.1007/s12272-015-0636-1. [DOI] [PubMed] [Google Scholar]
- 4.Schopf FH, Biebl MM, Buchner J. Nature Rev. Mol. Cell Biol. 2017;18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 5.Pearl LH. Biopolymers. 2016;105:594–607. doi: 10.1002/bip.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 8.Miyata Y, Nakamoto H, Neckers L. Current Pharmaceutical Design. 2013;19:347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Picard D. Cell. Mol. Life Sci. 2002;59:1640–1648. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Issacs JS, Xu W, Neckers L. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]; c) Pratt WB, Toft DO. Exp. Biol. Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]; d) Whitesell L, Lindquist SL. Nature Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 10.Moser C, Lang SA, Stoeltzing O. Anticancer Res. 2009;29:2031–42. [PubMed] [Google Scholar]
- 11.Koga F, Kihara K, Nechers L. Anticancer Res. 2009;29:797–807. [PubMed] [Google Scholar]
- 12.Franke J, Eichner S, Zeilinger C, Kirschning A. Nat. Prod. Rep. 2013;30:1299–1323. doi: 10.1039/c3np70012g. [DOI] [PubMed] [Google Scholar]
- 13.Khandelwal A, Crowley V, Blagg BSJ. Med. Res. Rev. 2015;35:92–118. doi: 10.1002/med.21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M, Shen A, Zhang C, Song Z, Ai J, Liu H, Sun L, Ding J, Geng M, Zhang A. J. Med. Chem. 2016;59:5563–5586. doi: 10.1021/acs.jmedchem.5b01106. [DOI] [PubMed] [Google Scholar]
- 15.Jhaveri K, Modi S. Onco Targets Ther. 2015;8:1849–1858. doi: 10.2147/OTT.S65804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong Banerji U, Tavana B, George GC, Aaron J, Kurzock R. Cancer Treatment Reviews. 2013;39:375–387. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 17.a) Ernst JT, Neubert T, Liu M, Sperry S, Zuccola H, Turnbull A, Fleck B, Kargo W, Woody L, Chiang P, Tran D, Chen W, Snyder P, Alcacio T, Nezami A, Reynolds J, Alvi K, Goulet L, Stamos D. J. Med. Chem. 2014;57:3382–3400. doi: 10.1021/jm500042s. [DOI] [PubMed] [Google Scholar]; b) Mishra SJ, Ghosh S, Stothert AR, Dickey CA, Blagg BSJ. ACS Chem. Biol. 2017;12:244–253. doi: 10.1021/acschembio.6b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O. J. Am. Chem. Soc. 2012;134:9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, Gewirth DT, Chiosis G. Nat. Chem. Biol. 2013;9:677–684. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Crowley VM, Khandelwal A, Mishra S, Stothert AR, Huard DJE, Zhao J, Muth A, Duerfeldt AS, Kizziah JL, Lieberman RL, Dickey CA, Blagg BSJ. J. Med. Chem. 2016;59:3471–3488. doi: 10.1021/acs.jmedchem.6b00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khandelwal A, Kent CN, Balch M, Peng S, Mishra SJ, Deng J, Day VW, Liu W, Holzbeierlein JM, Matts R, Blagg BSJ. Nat. Commun. 2017 doi: 10.1038/s41467-017-02013-1. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Shelton SN, Shawgo ME, Comer SB, Lu Y, Donnelly AC, Szabla K, Tanol M, Vielhauer GA, Rajewski RA, Matts RL, Blagg BS, Robertson JD. Mol. Pharmacol. 2009;76:1314–1322. doi: 10.1124/mol.109.058545. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Conde R, Belak ZR, Nair M, O’Carroll RF, Ovsenek N. Biochem. Cell Biol. 2009;87:845–851. doi: 10.1139/o09-049. [DOI] [PubMed] [Google Scholar]
- 20.a) Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BSJ. ACS Chem. Biol. 2011;6:800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moroni E, Zhao H, Blagg BSJ, Colombo G. J. Chem. Inf. Model. 2014;54:195–208. doi: 10.1021/ci4005767. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sattin S, Tao J, Vettoretti G, Moroni E, Pennati M, Lopergolo A, Morelli L, Bugatti A, Zuehlke A, Moses M, Prince T, Kijima T, Beebe K, Rusnati M, Neckers L, Zaffaroni N, Agard DA, Bernardi A, Colombo G. Chem. Eur. J. 2015;21:13598–15608. doi: 10.1002/chem.201502211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.a) Strocchia M, Terracciano S, Chini MG, Vassallo A, Vaccaro MC, Dal Piaz F, Leone A, Riccio R, Bruno I, Bifulco G. Chem Commun. 2015;51:3850–3853. doi: 10.1039/c4cc10074c. [DOI] [PubMed] [Google Scholar]; b) Terracciano S, Foglia A, Chini MG, Vaccaro MC, Russo A, Dal Piaz F, Saturnino C, Riccio R, Bifulco G, Bruno I. RSC Adv. 2016;6:82330–82340. [Google Scholar]; c) Wang Y, McAlpine SR. Org. Biomol. Chem. 2015;13:4627–4631. doi: 10.1039/c5ob00044k. [DOI] [PubMed] [Google Scholar]; d) Wang Y, McAlpine SR. Chem. Commun. 2015;51:1410–1403. doi: 10.1039/c4cc07284g. [DOI] [PubMed] [Google Scholar]; e) Gavenonis J, Jonas NE, Kritzer JA. Bioorg. Med. Chem. 2014;22:3989–3993. doi: 10.1016/j.bmc.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Garg G, Zhao H, Blagg BS. J. ACS Med. Chem. Lett. 2015;6:204–209. doi: 10.1021/ml5004475. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hall JA, Forsberg LK, Blagg BSJ. Future Med. Chem. 2014;6:1587–1605. doi: 10.4155/fmc.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhao J, Zhao H, Hall JA, Brown D, Brandes E, Bazzill J, Grogan PT, Subramanian C, Vielhauer G, Cohen MS, Blagg BSJ. MedChemComm. 2014;5:1317–1323. doi: 10.1039/C4MD00102H. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhao H, Moroni E, Colombo G, Blagg BS. J. ACS Med. Chem. Lett. 2014;5:84–88. doi: 10.1021/ml400404s. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhao H, Yan B, Peterson LB, Blagg BS. J. ACS Med. Chem. Lett. 2012;3:327–331. doi: 10.1021/ml300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kusuma BR, Peterson LB, Zhao H, Vielhauer G, Holzbeierlein, Blagg BSJ. J. Med. Chem. 2011;54:6234–6253. doi: 10.1021/jm200553w. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J. Am. Chem. Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]; h) Burlison JA, Blagg BS. J. Org. Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]; i) Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J. Am. Chem. Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]; j) Davis RE, Zhang Z, Blagg BSJ. MedChemComm. 2017;8:593–598. doi: 10.1039/c6md00377j. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Garg G, Zhao H, Blagg BS. J. Bioorg. Med. Chem. 2017;25:451–457. doi: 10.1016/j.bmc.2016.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Zhao H, Kusuma BR, Blagg BSJ. ACS Med. Chem. Lett. 2010;1:311–315. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao H, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J. Med. Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Zhao H, Garg G, Zhao J, Moroni E, Girgis A, Franco LS, Singh S, Colombo G, Blagg BSJ. Eur. J. Med. Chem. 2015;89:442–466. doi: 10.1016/j.ejmech.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao H, Anyika M, Girgis A, Blagg BSJ. Bioorg. Med. Chem. Lett. 2014;24:3633–3637. doi: 10.1016/j.bmcl.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byrd KM, Subramanian C, Sanchez J, Motiwala HF, Liu W, Cohen MS, Holzbeierlein J, Blagg BSJ. Chem. Eur. J. 2016;22:6921–6931. doi: 10.1002/chem.201504955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Topliss JG. J. Med. Chem. 1972;15:1006–1011. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 27.Boinapally S, Huang B, Abe M, Katan C, Noguchi J, Watanabe S, Kasai H, Xue B, Kobayashi T. J. Org. Chem. 2014;79:7822–7830. doi: 10.1021/jo501425p. [DOI] [PubMed] [Google Scholar]
- 28.Tsunoda T, Yamamiya Y, Kawamura Y, Itô S. Tetrahedron Lett. 1995;36:2529–2530. [Google Scholar]
- 29.Singh M, Argade N. Synthesis. 2012;44:2895–2902. [Google Scholar]
- 30.The authors tried using 4-nitro-4’-hydroxystilbene as the starting material for the cyclopropanation but none of the standard reactions work. Due to safety concerns, diazomethane was not used for the cyclopropanation of this compound.
- 31.Skhiri A, Salem RB, Soulé J, Doucet H. Synthesis. 2016;48:3097–3106. [Google Scholar]
- 32.Pitts CR, Ling B, Synder JA, Bragg AE, Lectka T. J. Am. Chem. Soc. 2016;138:6598–6609. doi: 10.1021/jacs.6b02838. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J. Am. Chem. Soc. 2003;125:6653–6655. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]
- 34.Kitbunnadaj R, Hoffmann M, Fratantoni SA, Bongers G, Bakker RA, Wieland K, Jilali A, De Esch IJP, Menge WMPB, Timmerman H, Leurs R. Bioorg. Med. Chem. 2005;13:6309–6323. doi: 10.1016/j.bmc.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Garg G, Forsberg LF, Zhao H, Blagg BSJ. Chem. Eur. J. 2017:10. doi: 10.1002/chem.201703206. [DOI] [Google Scholar]
- 36.ChemBioUltra Version 13.0 (Cambridgesoft)
- 37.Maestro Version 11.2014 (Schrodinger)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.