

Abstract
Endogenous nitric oxide (NO) generated by inducible NO synthase (iNOS) promotes glioblastoma cell proliferation and invasion and also plays a key role in glioblastoma resistance to chemotherapy and radiotherapy. Non-ionizing photodynamic therapy (PDT) has anti-tumor advantages over conventional glioblastoma therapies. Our previous studies revealed that glioblastoma U87 cells up-regulate iNOS after a photodynamic challenge and that the resulting NO not only increases resistance to apoptosis but renders surviving cells more proliferative and invasive. These findings were largely based on the effects of inhibiting iNOS activity and scavenging NO. Demonstrating now that iNOS expression in photostressed U87 cells is mediated by NF-κB, we hypothesized that (i) recognition of acetylated lysine (acK) on NF-κB p65/RelA by bromodomain and extra-terminal (BET) protein Brd4 is crucial; and (ii) by suppressing iNOS expression, a BET inhibitor (JQ1) would attenuate the negative effects of photostress. The following evidence was obtained. (i) Like iNOS, Brd4 protein and p65-acK levels increased severalfold in photostressed cells. (ii) JQ1 at minimally toxic concentrations had no effect on Brd4 or p65-acK up-regulation after PDT but strongly suppressed iNOS, survivin, and Bcl-xL up-regulation, along with the growth and invasion spurt of PDT-surviving cells. (iii) JQ1 inhibition of NO production in photostressed cells closely paralleled that of growth/invasion inhibition. (iv) Finally, at 1% the concentration of iNOS inhibitor 1400W, JQ1 reduced post-PDT cell aggressiveness to a far greater extent. This is the first evidence for BET inhibitor targeting of iNOS expression in cancer cells and how such targeting can markedly improve therapeutic efficacy.
Keywords: glioblastoma, photodynamic therapy, nitric oxide, nitric-oxide synthase, NF-kB transcription factor, bromodomain-containing protein 4 (BRD4), acetylation
Introduction
Most established malignant tumors exist under moderate inflammatory conditions, which foster tumor cell survival, proliferation, and metastatic expansion (1–3). Low level reactive oxygen species (ROS)2 as well as nitric oxide (NO) generated by inducible nitric-oxide synthase (iNOS/NOS2) play important roles in many of these processes (3–6). There is now compelling evidence that endogenous iNOS/NO not only supports growth and progression of many tumors but also plays a key role in pro-tumor immunosuppression (7, 8) as well as resistance to chemotherapeutic and radiotherapeutic interventions (9–11). A less common intervention for solid tumors is photodynamic therapy (PDT), which employs non-ionizing radiation. PDT was introduced about 45 years ago as a minimally invasive modality involving a photosensitizing agent (PS), PS-exciting visible-to-near infrared light, and molecular oxygen (12–14). All three components (PS, light, and O2) must be engaged concurrently for PDT to lethally damage tumor cells, which often occurs via the formation of the cytotoxic ROS, singlet oxygen (1O2). Light-independent PS effects are usually negligible, and little if any damage to normal tissue occurs during PDT, which is not the case for many chemotherapeutic agents. Another advantage of PDT is site-specificity, i.e. limitation of photodynamic action to the tumor site at which light is directed, typically via fiber optic transmitters (13, 14). An oligomeric hematoporphyrin preparation, now known as Photofrin®, was the first PS to receive FDA approval for PDT, about 20 years ago, and it is now used for a variety of solid tumors (13, 14). 5-Aminolevulinic acid (ALA)-based PDT is a more recently developed alternative in which ALA (or an ALA ester) is administered as a pro-PS. ALA is metabolized to the actual PS, protoporphyrin IX (PpIX), via the heme biosynthetic pathway, with PpIX accumulating initially in the mitochondria (15, 16). As heme synthesis is enhanced in tumor cells, these cells can attain much higher levels of ALA-induced PpIX than surrounding normal cells (17), which for this type of PDT, provides a further element of tumor site specificity. The potential interference of NO with PDT was discovered by showing that Photofrin®–PDT (18, 19) or ALA–PDT (20) cure rates for various mouse-borne tumors could be significantly increased by administering NOS inhibitors, particularly for tumors with relatively high basal NO outputs. The proffered explanation was that NO-mediated dilation of tumor microvasculatures acts in opposition to the vasoconstrictive effects of PDT (19, 20). However, until relatively recently, many questions remained unanswered, e.g. as to the NOS isoform(s) involved and its/their cellular source(s).
In previous work, we showed that NO from endogenous iNOS in various human cancer lines (breast, prostate, and glioblastoma) subjected to an ALA–PDT–like challenge elicited the following negative responses: (i) increased resistance to apoptotic photokilling; and (ii) increased proliferative, migratory, and invasive aggressiveness for cells surviving the challenge (21–26). Most of this evidence was based on the strong counteractive effects of iNOS enzyme inhibitors such as 1400W and GW274150 (27, 28) or the NO scavenger cPTIO (29). Using human glioblastoma cells in the present study, we determined that basal and photostress-induced iNOS is regulated by NF-κB. Knowing this and projecting from recently published evidence (30, 31), we hypothesized that bromodomain and extra-terminal domain (BET) protein recognition of ϵ-N-acetylated lysine residue(s) (acK) on the NF-κB p65/RelA subunit played a key role in iNOS expression. BET family proteins (Brd2, Brd3, Brd4, and Brdt) “read” acK residues on histones and transcription factors (32, 33). Brd4 is important in cancer progression (30, 31, 33), but the role of iNOS in Brd4-mediated cancer progression has not been described previously. In testing our hypothesis, we found that Brd4 is a key co-activator of photostress-augmented iNOS expression. Blocking Brd4 with the BET bromodomain inhibitor JQ1 (34, 35) substantially reduced acquired cell aggressiveness and to a much greater extent than an iNOS enzymatic inhibitor at many times greater concentration. These and related findings demonstrated for the first time that suppressing iNOS expression via Brd4 inhibition can markedly increase the efficacy of an anti-tumor therapy, in this case PDT. We demonstrate this using a model system for glioblastoma PDT.
Results
JQ1 enhancement of PDT cytotoxicity
In initial experiments, we tested the sensitivity of glioblastoma U87 cells to the BET bromodomain inhibitor JQ1 and compared this with cell sensitivity to PDT and PDT combined with JQ1. As shown in Fig. 1A, CCK-8 (cell counting kit-8)-assessed cell viability decreased progressively from 0.1 to 0.5 μm with increasing concentrations of JQ1 alone, with the highest concentration reducing viability to ∼65% of the vehicle control value after 24 h of exposure. As also observed previously under similar conditions (26), ALA/light-induced PDT reduced cell viability to ∼80% (Fig. 1A). However, combining JQ1 with PDT caused a further increase in cytotoxicity; for example, JQ1 at 0.5 μm immediately after irradiation reduced viability to ∼45%, which was significantly lower than the level attained with JQ1 or PDT alone (Fig. 1A). Cytotoxicity was also examined as extent of apoptotic cell death, which for our PDT approach occurs via the intrinsic pathway, because PpIX is localized mainly in mitochondria (15, 26). As shown in Fig. 1B, JQ1 alone increased annexin V—FITC–detected apoptosis of U87 cells in a dose-dependent fashion from 0.1 to 0.5 μm, with the highest concentration inducing ∼35% apoptosis after 5 h of incubation. On the other hand, propidium iodide-assessed necrosis remained low (∼1%), barely above the control level. PDT by itself produced about the same level of apoptosis as 0.5 μm JQ1, but when combined with PDT, JQ1 enhanced apoptosis significantly such that a synergistic effect was apparent. For example, PDT combined with 0.3 μm JQ1 caused 50% apoptosis, whereas PDT alone and JQ1 alone caused 30 and 10% apoptosis, respectively (Fig. 1B). We chose 0.3 μm JQ1 for all subsequent experiments, because this was minimally cytotoxic by itself, thus emphasizing the ability of JQ1 to enhance PDT cytotoxicity. In a previous study (26), we showed that 25 μm 1400W, a competitive inhibitor of iNOS enzymatic activity, increases U87 apoptosis by ∼33% over PDT alone. As shown in Fig. 1B, JQ1 at a much lower concentration (0.3 μm) promoted apoptosis much more substantially, i.e. by ∼66%. This recognition was a strong impetus for studying the mechanism of action of JQ1 in the context of PDT.
Figure 1.
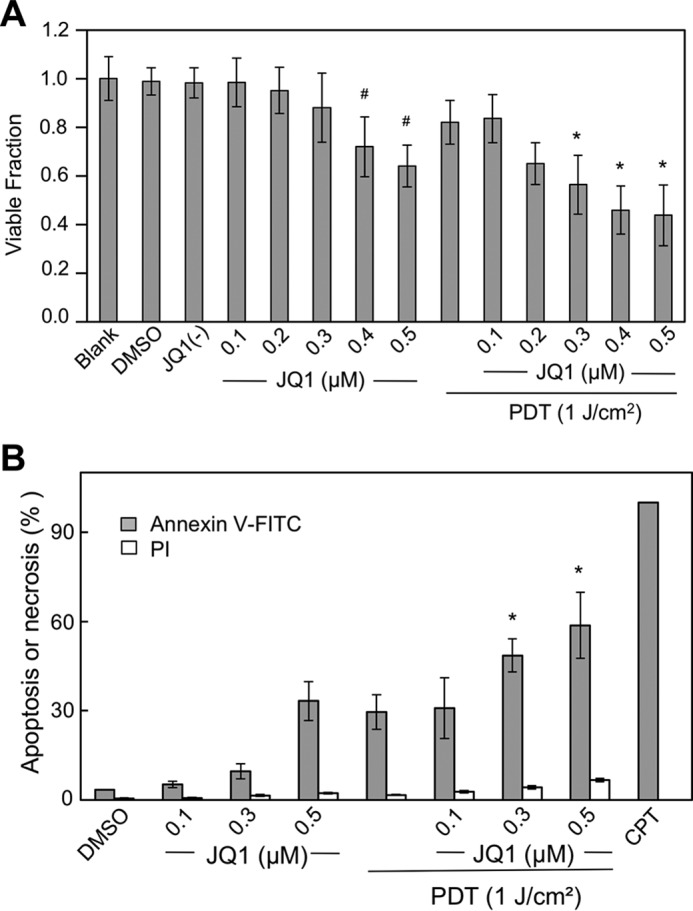
Cytotoxic effects of PDT on glioblastoma U87 cells: Enhancement by BET bromodomain inhibitor JQ1. A, cells at ∼60% confluency were either treated directly with JQ1 in increasing concentrations up to 0.5 μm or treated with JQ1 after preincubation with 1 mm ALA followed by irradiation (light fluence ∼ 1 J/cm2). Nontreated (blank), and vehicle DMSO- and 0.5 μm JQ1(−)-treated cells were studied alongside as controls. After 24 h of dark incubation, cell viability was determined by CCK-8 assay. Plotted values are means ± S.E. (n = 4); *, p < 0.05 versus PDT alone or 0.3 μm JQ1 alone; #, p < 0.05 versus blank or DMSO vehicle control. B, cells prepared as described in A were analyzed for extent of apoptosis or necrosis, 5 h after treatment with JQ1 or PDT plus JQ1, using annexin V–FITC for apoptosis and propidium iodide for necrosis. Camptothecin (CPT, 25 μm) served as an indicator for maximum apoptosis. Plotted data are means ± S.E. (n = 4); *, p < 0.01 versus PDT alone or 0.3 μm JQ1 alone.
JQ1 inhibition of iNOS expression
We showed previously that a PDT oxidative challenge results in prolonged up-regulation of pro-survival iNOS in several cancer cell lines, including glioblastoma lines (21–26). Given that NF-κB is often implicated in iNOS expression (6, 23, 36) and that Brd4 can serve as a NF-κB co-activator (30, 31), we asked whether the observed JQ1 enhancement of PDT cytotoxicity could be explained on this basis. We looked first at the effects of low-level JQ1 on U87 iNOS expression after a PDT challenge compared with that in a dark control. As shown by the immunoblot in Fig. 2A, control (ALA-only) cells expressed iNOS protein at a relatively low level, which was no different from that in untreated cells (not shown). After irradiation, ALA-primed cells exhibited a rapid and prolonged up-regulation of iNOS over at least a 24-h post-irradiation period (Fig. 2A). JQ1 (0.3 μm) strongly inhibited basal iNOS expression, as well as the robust induction of enzyme by photodynamic stress (Fig. 2B). For example, at 6 h after irradiation, the iNOS level for PDT plus JQ1 was only ∼40% of the iNOS level for PDT alone. Thus, JQ1 markedly reduced basal as well as stress-induced iNOS, the latter being associated with a strong pro-survival response in stressed cells and a switch to a more aggressive phenotype in surviving cells (21–26).
Figure 2.
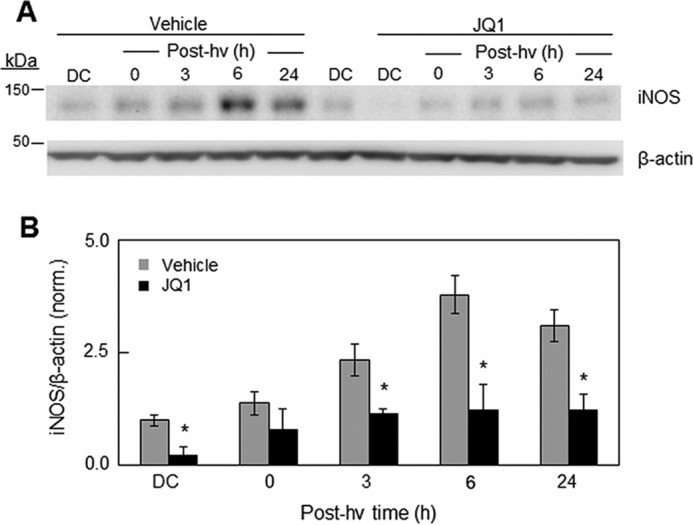
Effect of JQ1 on iNOS up-regulation in U87 cells after a PDT challenge. A, cells were sensitized by ALA treatment and then either irradiated (1 J/cm2) or returned to the incubator for 24 h as a dark control (DC). Immediately after irradiation, cells either received DMSO vehicle alone or 0.3 μm JQ1 in DMSO and were recovered for iNOS and β-actin Western blot analysis after increasing periods of dark incubation, beginning at 0 h and extending to 24 h. hν, light. B, quantification of iNOS Western blotting bands expressed as mean fold changes ± S.E. relative to β-actin and normalized to the dark control without JQ1. The Western blotting shown in A is representative of three from replicate experiments with similar results. *, p < 0.01 versus vehicle control in each pair.
Generation of NO and suppression thereof by JQ1
We used the fluorophore DAF-FM-DA to probe for NO-derived oxidant levels in photodynamically stressed U87 cells and how NO levels might be altered by JQ1 and 1400W. Fluorescence of the NO-derived triazole product of the probe (DAF-FM-triazole) (38) was monitored. Cells treated with DAF-FM-DA directly or following the addition of the JQ1 vehicle (DMSO) exhibited the same relatively low probe fluorescence after 5 and 18 h of preincubation (Fig. 3). The intensity of this background fluorescence was significantly reduced by 1400W, implying detection of iNOS-derived NO. JQ1 at a small fraction of the 1400W concentration reduced the DAF-FM-triazole signal more substantially than 1400W, i.e. by ∼80% at 18 h relative to control (Fig. 3). A striking 10–12-fold increase in DAF-FM-triazole fluorescence was observed at 5 and 18 h after cells underwent a PDT (ALA/light) challenge (Fig. 3). Whereas JQ1(−), the enantiomer of JQ1, which does not bind BET bromodomains (34), had no effect on the level of photostress-generated NO, active JQ1 reduced NO levels to ∼14% of the ALA/light value. A large decrease in NO output was also observed with 1400W, i.e. to ∼30% of the ALA/light value. Thus, the effect of 1400W was not as impressive as that of JQ1 at a far lower starting concentration. Therefore, limiting NO by presumed JQ1 inhibition of iNOS expression appeared more effective in increasing PDT cytotoxicity than limiting NO by inhibiting iNOS enzyme activity.
Figure 3.

NO accumulation in U87 cells after a PDT challenge: attenuation by JQ1 versus 1400W. U87 cells were preincubated with ALA, washed, and irradiated (light fluence ∼ 1 J/cm2). Immediately thereafter, the cells were treated with 1400W (25 μm), JQ1 (0.3 μm), or JQ1(−) (0.3 μm). Light-only controls without or with 1400W or JQ1 were prepared alongside. At 5 and 18 h after irradiation, the cells were incubated with the NO probe DAF-FM-DA (25 μm) for 20 min in the dark, after which they were viewed by fluorescence microscopy using 488 nm excitation and 650 nm emission. ImageJ software was used to quantify image intensities; integrated values are indicated above the panels for each condition. *, p < 0.05 versus DMSO vehicle control at each time point; #, p < 0.01 versus ALA/hν at each time point. Scale bar: 100 μm.
The results shown in Fig. 3 were confirmed by measuring NO-derived NO2−/NO3− (NOx) via the Griess assay. As shown in Fig. S1, the NOx output of photostressed U87 cells was significantly greater (∼2-fold) than that of dark controls, and this difference persisted from 5 to 24 h after irradiation. JQ1 reduced control U87 [NOx] by ∼50% and the elevated [NOx] from photostressed cells by at least this amount (Fig. S1), thereby supporting the imaging data obtained with DAF-FM-DA (Fig. 3).
NF-κB–regulated iNOS expression in glioblastoma cells
To establish whether NF-κB activation is necessary for iNOS expression and up-regulation in photostressed U87 cells, we first tested the effects of Bay11-7082 (Bay11), an inhibitor of the IKK complex, which phosphorylates regulatory IκB on NF-κB, leading to the release and translocation of NF-κB/p50-p65 to the nucleus, where NF-κB–mediated gene transcription ensues (39–41). As shown in Fig. 4A, Bay11 strongly attenuated not only basal iNOS expression in U87 cells but also PDT–up-regulated iNOS, suggesting control by NF-κB. As further evidence, we found that PDT (ALA/light) resulted in complete translocation of the p65/RelA subunit of NF-κB from the cytosol to the nucleus, and Bay11 prevented p65 translocation (Fig. 4B). On the other hand, JQ1 had little (if any) inhibitory effect on p65 translocation, which is consistent with JQ1 acting in the nucleus (33, 34).
Figure 4.

NF-κB involvement in post-PDT up-regulation of iNOS in glioblastoma cells. A, U87 cells were sensitized with PpIX by preincubation with 1 mm ALA for 30 min. After washing, the cells were treated with Bay11 and either dark-incubated for 24 h (DC) or irradiated (1 J/cm2) and then analyzed for iNOS and β-actin by immunoblotting after increasing post-irradiation times up to 24 h. A dark control without Bay11 was also analyzed. Numbers below the bands are iNOS/β-actin ratios relative to DC (minus Bay11). hν, light. B, NF-κB activation and subcellular distribution following PDT. U87 cells treated with PDT alone (ALA/hν), PDT plus 0.3 μm JQ1 (ALA/hν/JQ1), or PDT plus 5 μm Bay11 (ALA/hν/Bay) were homogenized at 5 h after irradiation and separated into nuclear (Nuc) and cytosolic (Cyt) fractions, each of which was immunoblotted for the p65 subunit of NF-κB along with histone H3 as a nuclear marker and α-tubulin (Tub) as a cytosolic marker. A dark control (ALA-only) was analyzed similarly.
Upstream events in photostress induction of iNOS
We learned previously that human breast cancer COH-BR1 cells underwent a rapid and robust phosphorylation–activation of the pro-survival/progression kinase Akt after an ALA/light challenge and that this subsided during prolonged post-irradiation incubation (23). Akt activation was nullified by wortmannin, an inhibitor of PI3K, which is required for Akt activation, and this also prevented iNOS induction. These and related findings, e.g. the inability of 1400W to inhibit Akt activation, suggested that Akt was an upstream mediator of iNOS induction through the phosphorylation–activation of IKK and thence the activation and nuclear translocation of NF-κB (23). To assess whether Akt might function similarly in photostressed glioblastoma cells, we monitored its phosphorylation status after ALA/light treatment and how this might be affected by JQ1. As shown in Fig. 5A, Akt was strongly activated in U87 cells (appearance of p-Akt band) 3–6 h after irradiation, with total Akt remaining the same throughout. The PI3K inhibitor LY294002 prevented this activation (Fig. 5C), but JQ1 had no effect on it (Fig. 5B) nor did 1400W (not shown). LY294002 also prevented photostress up-regulation of iNOS (Fig. 5D), implying that upstream activation of PI3K and Akt was necessary for iNOS induction, as observed previously for COH-BR1 cells (23). These results ruled out any possible JQ1 impairment of upstream signaling events leading to iNOS up-regulation, including Akt activation.
Figure 5.

Post-PDT Akt activation in the absence versus presence of LY294002 or JQ1 and iNOS up-regulation in the absence versus presence of LY294002. U87 cells were stressed photodynamically as described in the legend for Fig. 4. Immediately thereafter, the cells were treated with LY294002 (20 μm) or JQ1 (0.3 μm) and then dark-incubated for increasing periods up to 24 h. Controls containing DMSO vehicle were incubated alongside. At the indicated times, samples were recovered for Western blot analysis. A, appearance of phosphorylated Akt (p-Akt, Ser-473 epitope). hν, light. B, appearance of p-Akt in the presence of JQ1. C, LY294002 inhibition of p-Akt formation. D, LY294002 inhibition of iNOS induction. Results shown are indistinguishable from those obtained in two other replicate experiments.
Elevated Brd4 and p65-acK310 levels after photodynamic stress
We discovered that like iNOS, Brd4 was strongly up-regulated after U87 cells were subjected to photodynamic action. As shown by the Western blot analysis in Fig. 6B, the Brd4 level increased progressively after ALA/light treatment, reaching nearly 3-fold above the dark control basal level 24 h after irradiation. In contrast, there was little, if any, up-regulation of another BET family member, Brd2, after the same PDT challenge (Fig. 6A). On the other hand, the level of acK310 on the p65 subunit of NF-κB exhibited a substantial increase after PDT, rising to ∼3-fold over background after 24 h (Fig. 6C), similar to the elevation in Brd4. Total p65 expression was not altered by PDT (results not shown), so the observed acK310 response must have been due to more extensive acetylation at this particular lysine residue. As shown in Fig. 6B, JQ1 had no effect on basal or photostress-induced Brd4 protein level (Fig. 6B) or on p65-acK310 level (Fig. 6C). This finding rules out any inhibition of Brd4 expression or extent of p65 lysine 310 acetylation as a possible factor in iNOS/NO suppression by JQ1 (Figs. 2 and 3).
Figure 6.

Effect of PDT on Brd2, Brd4, and p65-acK310 expression in the absence versus presence of JQ1. U87 cells were challenged photodynamically (cf. Fig. 4) and then dark-incubated in the presence of DMSO vehicle or 0.3 μm JQ1 for increasing time periods up to 24 h. At the indicated times, samples were recovered for Western blot analysis of Brd2 (A), Brd4 (B), and p65-acK310 (C). Upper panels in A–C represent Brd2, Brd4, and p65-acK310, respectively, without JQ1 (vehicle controls). Lower panels in A–C show plots of integrated band intensities for Brd2, Brd4, and p650-acK310, respectively, in the absence versus presence of JQ1; plotted values are relative to a dark control (DC) for each analyte and are means ± S.E. (n = 3). Brd2 (A) showed no significant difference ± JQ1 or with PDT at all time points. For Brd4 (B) and p65-acK310 (C), PDT exerted a significant effect over dark control from 3 to 24 h. *, p < 0.01 (however, JQ1 had no significant effect). hν, light.
JQ1-inhibitable interaction of Brd4 and NF-κB/p65 in PDT-stressed U87 cells
Knowing that photostress induction of iNOS depended on nuclear translocation of NF-κB/p65 and that this was accompanied by up-regulation of Brd4 and acK310 on p65, we postulated that Brd4 interaction with acK310 is necessary for NF-κB/p65 activation (30, 31). To investigate this possibility, we used a pulldown approach in which p65 was immunoprecipitated, collected on protein A–linked Sepharose beads, and after release, checked for the presence of Brd4 by immunoblotting. As shown in Fig. 7A, the p65 immunoprecipitation revealed a strong Brd4 immunoblot band, the intensity of which was substantially reduced by JQ1. We deduced from this evidence that Brd4 served as a co-activator of NF-κB/p65 in photostressed cells and that JQ1 suppressed iNOS induction by targeting Brd4 and preventing its binding to p65-acK310. We went on to determine whether another BET protein, Brd2, might contribute to co-activation and possibly also be present in the p65 immunoprecipitate. As shown in Fig. 7B, a pulldown immunoblot band for Brd2 was detected, but it was very weak compared with Brd4, yet strong Brd2 and p65 bands were seen in the overall lysate. Although other BET proteins have not been interrogated similarly, we believe, in agreement with others using different cancer cells (30, 31), that Brd4 was the predominant (if not sole) co-activator for iNOS expression/overexpression in our system.
Figure 7.

BET protein interaction with NF-κB/p65. After preincubation with ALA in the dark, U87 cells were irradiated (∼1 J/cm2), incubated in the absence or presence of 0.3 μm JQ1 for 8 h, and then lysed. After determination of total protein concentration, cell lysates were subjected to immunoprecipitation (IP) using a monoclonal antibody against p65. After 16 h of incubation with antibody at 4 °C followed by 6 h with protein A–conjugated Sepharose beads, the bound proteins were eluted and subjected to immunoblotting (IB) using a monoclonal antibody against Brd2 or Brd4, with nonspecific IgG serving as a control. Overall p65, Brd2, and Brd4 levels in the lysates were also detected by immunoblotting. A, Brd4 Western blots; B, Brd2 Western blots.
Accelerated growth and invasiveness of PDT-surviving glioblastoma cells: Suppression by JQ1
Having attributed acquired photostress resistance to up-regulated iNOS/NO (24–26), we asked whether this iNOS/NO might stimulate growth and invasion of cells that could withstand a photochallenge and, if so, how JQ1 would affect these responses. Twenty-four hours after ALA/light treatment, surviving U87 cells, along with ALA-only controls, were recovered, replated at equal live-cell densities, and monitored for proliferation over a 48-h period in the absence versus presence of 1400W (25 μm) or JQ1 (0.3 μm). As shown in Fig. 8, proliferation of non-irradiated control cells was slowed somewhat by 1400W and slightly more so by JQ1. On the other hand, surviving ALA/light-treated cells exhibited a sizeable growth spurt (∼61% in 24 h) relative to ALA-only controls, and 1400W slowed this spurt much more than it did control cell growth (∼50 versus ∼10%). However, JQ1 slowed surviving cell growth to an even greater extent than 1400W, i.e. by ∼80%, and JQ1 did this at only ∼1% the 1400W concentration in bulk cell system, making JQ1 more impressive than 1400W for pharmacological arrest of iNOS-stimulated proliferation.
Figure 8.
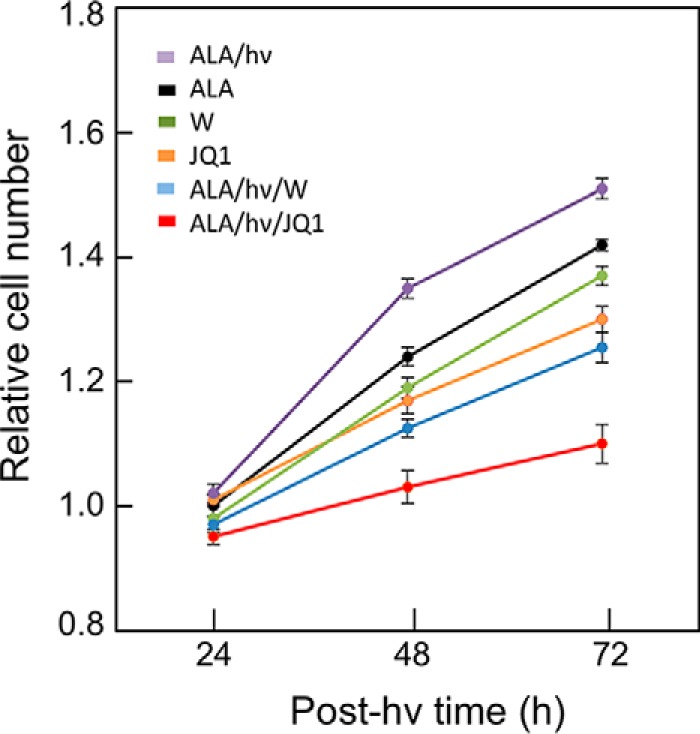
Accelerated proliferation of PDT-surviving cells: Suppression by JQ1 versus 1400W. Twenty-four hours after U87 cells were ALA/light-treated (see legend for Fig. 4), any detached cells were carefully removed by aspiration. The remaining live cells were recovered by gentle scraping and seeded into a 96-well plate along with non-irradiated control cells. A predetermined greater seeding density was used for photostressed cells to account for the portion that succumbed to this treatment. Accordingly, the initial cell count was approximately the same for all post-irradiation conditions studied. PDT-surviving cells, along with non-stressed (ALA-only) controls, in 10% serum-containing medium were dark-incubated in the presence of 0.3 μm JQ1 or 25 μm 1400W (W) and maintained at these concentrations throughout. At the indicated time points, live cell levels were determined by CCK-8 assay. The plotted numbers are means ± S.E. (n = 4). hν, light.
In addition to exploiting iNOS/NO for proliferative signaling, glioblastoma cells are known to rely on NO for migratory and invasive potency (26, 42–44). We compared the effects of 1400W and JQ1 on surviving U87 cell invasiveness after a typical ALA/light challenge. Invasion measurements were started immediately after ALA-primed cells were irradiated, with non-stressed controls analyzed similarly. Fig. 9A shows that control cell invasion rate was only moderately inhibited by JQ1 or Bay11 (∼15%), with 1400W having a smaller effect and JQ1(−) no significant effect. Cells surviving PDT exhibited a striking 35–40% increase in invasion rate, which was unaffected by JQ1(−). However, JQ1 not only abrogated the more rapid invasion, but it brought the remaining invasion rate to ∼40% that of the vehicle control (Fig. 9A). Although 1400W also eliminated the more rapid invasion, the residual rate with 1400W was ∼80% of the control rate, i.e. much greater than the rate left by JQ1. Thus, JQ1 inhibited invasion to a far greater extent than 1400W at >80-fold the JQ1 concentration. As shown in Fig. 9A, Bay11, which strongly reduced iNOS expression via inhibition of NF-κB activation (Fig. 4), suppressed post-PDT invasiveness to nearly the same extent as JQ1. Although it further supports the role of iNOS in hyperinvasiveness, this finding raises the issue of specificity, because Bay11 can inhibit other pro-growth/invasion effectors besides IκB kinase, e.g. protein tyrosine phosphatases (45).
Figure 9.
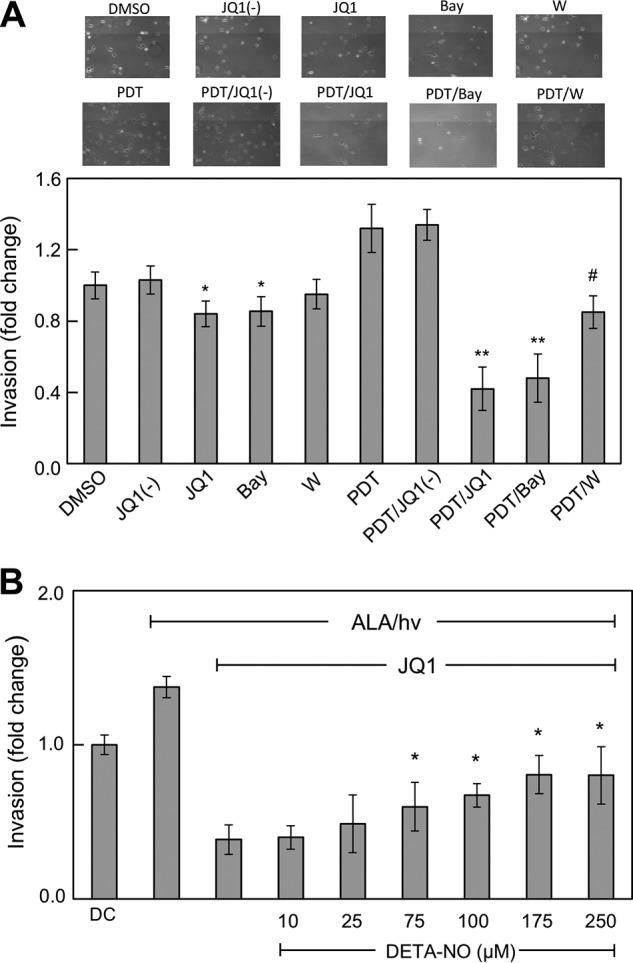
Accelerated invasion of PDT-surviving cells: Suppression by JQ1 and partial recovery induced by exogenous NO. A, U87 cells at ∼60% confluency in MEM medium on 35-mm dishes were ALA/light-challenged as described in the legend for Fig. 4. Immediately after irradiation, these cells along with non-stressed controls were harvested into MEM, transferred to the upper wells of a prewarmed 96-place Boyden-type device, and allowed to invade through Matrigel-infused polycarbonate filters using 10% serum-containing medium in the lower wells as an attractant. Invasion incubations were carried out in the absence versus presence of 0.3 μm JQ1, 0.3 μm JQ1(−), 5 μm Bay11 (Bay), or 25 μm 1400W (W) and maintained at these concentrations throughout; DMSO was included as a vehicle control for JQ1 and JQ1(−). After 24 h of dark incubation, cells adhering to the underside of the filter were centrifuged off into the 96-well plate, then stained and photographed (upper images), and finally quantified by CCK-8 assay (lower images). Plotted data for PDT-challenged cells were corrected for predetermined viability losses occurring during the 24 h of invasion incubation, and thus normalized live cell numbers are represented. Values described in A are means ± S.E. (n = 3). *, p < 0.05 versus DMSO; **, p < 0.001 versus PDT; #p < 0.01 versus PDT. B, U87 cells were subjected to ALA/light stress as described above and then examined for invasiveness in the presence of 0.3 μm JQ1 alone or JQ1 plus DETA-NO at each of the indicated starting concentrations, ranging from 10 to 250 μm. Plotted values are means ± S.E. (n = 3). *, p < 0.05 versus ALA/hν with JQ1 alone. hν, light.
We asked whether NO from an exogenous source might restore the invasiveness that JQ1 had strongly suppressed. When photostressed and JQ1-treated U87 cells were exposed to the NO donor DETA-NONOate, a concentration-dependent increase in invasion rate from a very low point was observed. This rate maximized at about twice that observed for cells exposed only to ALA, light, and JQ1 (Fig. 9B). Thus, although iNOS, the primary source of endogenous NO, was depleted (along with several other tumor-promoting effectors, see below), these cells were still able to respond to NO by becoming more invasive. The signaling mechanism behind this remarkable response remains to be elucidated.
As shown in Fig. S2, JQ1 also abolished PDT-promoted invasiveness in another human glioblastoma line, U251 cells. Similar to their U87 counterparts (Fig. 9A), U251 cells exhibited a residual invasion rate following PDT/JQ1 (∼35%) that was far below that of control cells or cells treated with 1400W after PDT (∼80%). Thus, PDT survivors in at least two different glioblastoma cell lines exhibited greater invasiveness, which could be eliminated by JQ1.
JQ1-inhibitable induction of other pro-survival/pro-invasion effector proteins in PDT-stressed cells
Realizing that the expression of other NF-κB–controlled tumor-supporting proteins besides iNOS might also be modulated by an ALA/light challenge to U87 cells, we selected five examples for monitoring: survivin, Bcl-xL (B-cell lymphoma, extra large), p21 (cyclin-dependent kinase inhibitor-1), MMP-9 (matrix metalloproteinase-9), and c-Myc protein (40, 41).
As shown by the immunoblot in Fig. 10A, survivin, a potent inhibitor of apoptosis (46), underwent a time-dependent up-regulation during post-PDT incubation, reaching ∼2-fold the ALA-only control level after 24 h. This control level was no different from that of untreated cells (not shown). A similar response was observed in our previous study (26). The presence of JQ1 not only suppressed basal survivin expression, consistent with the results of others (47, 48), but also the strong induction of survivin by PDT (Fig. 10A).
Figure 10.

Altered post-PDT expression of NF-κB–regulated proteins other than iNOS: Effects of JQ1. U87 cells were sensitized with ALA-induced PpIX as described in the legends for Figs. 1 and 2. Immediately thereafter, DMSO vehicle control or 0.3 μm JQ1 was introduced, and cells were dark-incubated for 0, 3, 6, or 24 h after which they were recovered, solubilized, examined for total protein concentration, and then subjected to Western blot analysis for survivin (A), Bcl-xL (B), p21 (C), MMP-9 (D), and c-Myc (E). Western blotting is annotated as follows: non-irradiated control at 24 h, ALA; irradiated ALA-treated cells, ALA/hν; irradiated ALA-treated cells with JQ1, ALA/hν/JQ1. Effects of JQ1 without ALA/hν are also represented. The number below each band represents band intensity relative to β-actin and normalized to the ALA-only control. Each of the Western blot analyses shown is representative of two from duplicate experiments with nearly identical results. hν, light.
Bcl-xL is another NF-κB–regulated anti-apoptotic protein that is highly expressed in glioblastoma cells (51). Bcl-xL underwent a gradual up-regulation following PDT, reaching approximately twice the control level at 24 h (Fig. 10B). A similar response was observed previously for a breast cancer cell line (52). JQ1 strongly reduced Bcl-xL expression in U87 control cells, consistent with results of others (51), and also attenuated its post-PDT up-regulation, e.g. 40% less at 24 h (Fig. 10B).
The responses of p21 were diametrically opposite to those of iNOS, surviving, and Bcl-xL. Thus, the p21 level declined progressively over 24 h of dark incubation after PDT (Fig. 10C). A remarkable reversal of this response was observed when JQ1 was present, such that p21 reached twice its control level 3 h after PDT and remained there for at least another 21 h, with JQ1 alone (without PDT) producing a similar effect (Fig. 10C). Strong induction of p21 by JQ1 has been reported for several other cell lines (47, 53, 54). Such induction could promote cell death through the arrest of cell cycle progression. The striking down-regulation of p21 observed after PDT is consistent with our evidence that surviving cells proliferated more rapidly (Fig. 8). That JQ1 not only inhibited p21 down-regulation but also elicited a long-lasting up-regulation of this protein could explain the strong suppression of this post-PDT growth spurt by JQ1 (Fig. 8). MMP-9, which catalyzes the degradation of extracellular matrices, is associated with the migratory/invasive characteristics of many tumor cells, including glioblastoma cells (55, 56).
As shown in Fig. 10D, MMP-9 underwent a slow up-regulation after PDT, reaching about 50% greater expression than its control level by 24 h. JQ1 strongly inhibited this response to photostress, reducing the MMP-9 level by nearly the extent that JQ1 did in control cells (Fig. 10D). In a previous study (26), we found that although U87 MMP-9 expression was only slightly elevated after PDT, MMP-9 activity assessed by in-gel zymography was increased by ∼80%. This increase was nearly abolished by l-NAME or 1400W, thus implicating iNOS/NO in the MMP-9 activation (26).
The oncogenic protein c-Myc, which is constitutively expressed in a variety of aggressive tumors including glioblastomas, acts not only as a transcription factor but also as a global regulator of pro-tumor epigenetic modifications (57). We found that c-Myc expression in U87 cells underwent a rapid decline after PDT, beginning immediately after irradiation, reaching a nadir at ∼3 h, and then gradually rising so that the c-Myc level at 24 h approximated that of the non-irradiated control (Fig. 10E). JQ1 not only prevented this delayed return to background c-Myc expression after PDT but, by itself (without PDT), nearly abolished all c-Myc expression, in agreement with previous studies involving glioblastoma and other cell lines (51, 58, 59). The effects of different PDT approaches on c-Myc status have been described previously for other cancer cell types, often with contrasting results. In some cases, expressed c-Myc mRNA or protein steadily increased after PDT (60, 61), whereas in other cases, it decreased (62–64), but no rational explanations were offered. In most of these studies, c-Myc was not monitored for more than 4–6 h after irradiation, whereas we tracked it over 24 h. The striking decline in c-Myc and return to constitutive level after 24 h (Fig. 10E) might reflect a unique stress accommodation response of this effector in preparation for accelerated cell division.
We concluded from these findings that although PDT stress-induced iNOS played a major role in promoting cell resistance and aggressiveness, the altered expression of survivin, Bcl-xL, p21, MMP-9, and c-Myc made a significant contribution, which could be significantly counteracted by inhibition of BET bromodomains by JQ1.
Discussion
Malignant gliomas such as glioblastoma multiforme are among the most aggressive and lethal of the primary brain tumors. Without treatment, a patient's average survival time after initial diagnosis is typically 4–6 months (65, 66). Even with the most advanced surgical techniques or surgery combined with radiotherapy or chemotherapy, survival time remains dismal at 18–24 months (66). Pre-existing or acquired resistance to conventional chemo- and radiotherapy remains a serious impediment to the benefits of these treatments, which has stimulated the development of better alternatives. One such alternative is PDT using Photofrin® (67–69) or ALA-induced PpIX (69, 70) as a photosensitizing agent. In addition to improving the average survival time relative to cisplatin-based chemotherapy, for example (69), PDT has the advantage of high tumor-site specificity, i.e. fewer negative off-target effects on normal tissue (12–14). However, like other therapeutic interventions, PDT can be antagonized by pre-existing or stress-induced factors, which could increase cell resistance to photokilling and/or provide the surviving cells with a growth and migratory advantage.
One major antagonist of PDT's antitumor effects is iNOS-derived NO, as amply demonstrated in our previous studies on several human cancer cell lines including glioblastoma lines (22–26). Three key findings emerged from these studies: (i) cancer cell iNOS undergoes a rapid and prolonged up-regulation after a photodynamic (ALA/light) insult originating in mitochondria; (ii) stress signaling by up-regulated iNOS/NO increases cell resistance to apoptotic photokilling, pre-existing iNOS typically being much less important in this regard; and (iii) induced iNOS/NO promotes growth and migration/invasion aggressiveness in cells withstanding the photodynamic stress (22–26). Highly specific inhibitors of iNOS enzymatic activity, viz. 1400W and GW274150, played a key role in our discovery of these anti-PDT responses. For example, 1400W increased the extent of apoptotic cell photokilling but decreased the hyperaggressiveness of surviving cells (22–26). Similar results were obtained with the NO scavenger cPTIO (24–26). The translational potential of iNOS inhibitors was readily apparent from these findings, viz. their ability to improve PDT outcomes by increasing tumor regression and/or suppressing greater migratory activity. Some of these inhibitors (l-NIL and GW274150) have already been safely tested in clinical trials, although these had no relationship to cancer or PDT (28, 71). As an intermediary proof of concept, we recently showed that ALA–PDT suppression of mouse-borne human breast tumor xenografts was substantially augmented by the administration of 1400W or GW274150, whereas no significant effect was observed on control tumor growth (52). Consistently, iNOS protein in tumor samples was strongly up-regulated after ALA–PDT, and NO-derived NO2− levels were also elevated relative to control levels and in 1400W-inhibitable fashion (53). However, it was apparent from our previous in vitro and in vivo studies that the modulating effects of iNOS enzyme inhibitors (e.g. increased PDT cytotoxicity or decreased survivor aggressiveness) were far from maximal at relatively high inhibitor concentrations or dosages (24–26, 53). This prompted us to ask whether possible suppression of iNOS expression with a BET bromodomain inhibitor such as JQ1 might be more effective than inhibition of expressed iNOS activity.
JQ1 and other inhibitors of epigenetic (BET-containing) reader proteins such as Brd2, Brd3, and Brd4 have emerged as highly potent and relatively selective pharmacologic suppressors of cancer cell proliferation, migration, and metastatic dissemination (32–35). BET bromodomain inhibitors function by binding to acK recognition motifs on BET proteins, thereby preventing BET bromodomains from binding to acK sites on histones and transcription factors (32, 33). Many BET bromodomain inhibitors are under clinical trial scrutiny for a variety of malignancies, including multiple myeloma, lymphoma, triple negative breast cancer, and other solid tumors (72, 73). BET-containing proteins recognize specific acK residues on histones and transcription factors such as NF-κB (32–35, 72). Recent seminal studies by Chen and colleagues (30, 31), using A549 lung cancer cells, revealed that Brd4 binding to acK310 on the p65 subunit of NF-κB maintains the latter in an active form. Brd4 knockdown by shRNA or inhibition by JQ1 suppressed expression of NF-κB target genes while inducing ubiquitination and degradation of nuclear p65, both in constitutively active and TNF-α–stimulated forms (31). Target genes such as E-selectin, A20, and IL-8 were identified in those studies, but there was no indication as to whether iNOS expression was also activated by Brd4, and if so, how JQ1 would affect it. In fact, to our knowledge there is no published prior work on how a BET bromodomain inhibitor such as JQ1 might limit cancer progression by interfering with iNOS expression. However, in macrophage-mediated immune responses to bacterial pathogens, analogous interference has been described (74).
In the present study, we found that activation and nuclear translocation of NF-κB in glioblastoma U87 cells played a key role in basal as well as PDT-stimulated iNOS expression. Using JQ1 at a concentration that was minimally toxic to these cells, we showed that this BET bromodomain inhibitor strongly suppressed iNOS expression and NO generation in both control and PDT-challenged U87 cells. Concomitantly, JQ1 caused a striking increase in apoptotic cell death when used in combination with PDT such that an overall synergistic effect was seen (∼50% apoptosis (Fig. 1B)). In contrast, relatively little necrotic cell death occurred (<5%), which is significant because clinical PDT strives to maximize apoptosis and minimize necrosis to limit nonspecific inflammatory stress from necrosis (12–14). Based on our previous evidence obtained with breast cancer cells (21–23), we predicted that PDT would activate PI3K-dependent Akt in U87 cells upstream of NF-κB activation and iNOS/NO expression. A robust phosphorylation–activation of Akt did occur following PDT (Fig. 5), and Akt presumably activated NF-κB via phosphorylation of IκB kinase (23). Importantly, JQ1 had no effect on Akt activation, suggesting that JQ1 suppression of iNOS/NO occurred entirely at the site of iNOS expression. Although the strong enhancement of U87 photokilling by JQ1 (Fig. 1) has important implications for improving clinical PDT efficacy with this BET bromodomain inhibitor, the observed JQ1 suppression of hyperaggressiveness in PDT-surviving cells (Figs. 8 and 9) has even greater significance in terms of limiting cancer progression. We discovered previously (26), and confirmed here using glioblastoma U87 and U251 cells, that cells not lethally photodamaged can grow and migrate/invade more rapidly, potentially leading to greater metastatic dissemination if occurring in vivo. By suppressing iNOS/NO up-regulation as a major contributing factor, JQ1 curbed this enhanced aggressiveness, much more effectively than an iNOS enzyme inhibitor (1400W) and at exceedingly higher concentration. Taken together, our findings suggest that by restraining iNOS expression and NO production, JQ1 could greatly improve clinical PDT outcomes, not only enhancing tumor regression but also limiting the adverse effects of surviving cells, i.e. more rapid invasion leading to metastasis.
A novel and particularly interesting observation in this study is that Brd4, like iNOS itself, was up-regulated severalfold in PDT-stressed U87 cells, whereas a Brd4 paralog, Brd2, was unaffected. Equally interesting is our observation that the level of acK310 on the p65 subunit of NF-κB also increased severalfold in these cells. Like the Brd4 response, p65-acK310 up-regulation has not, to our knowledge, been described previously for any type of cancer cell subjected to a therapy-related oxidative challenge. The explanation for the elevated p65-acK310 level is not yet clear. However, one can speculate that stress induction and/or activation of acetyltransferase p300/CBP was involved or possibly down-regulation of a p65 deacetylase such as Sirt1 (75). These different possibilities will be assessed in our ongoing studies. We postulate that post-PDT Brd4 and acK310 up-regulation are co-operative stress responses that promote p65-mediated iNOS expression and other pro-survival/expansion genes, leading to a more resistant and aggressive cell phenotype. Although JQ1 did not affect Brd4 or acK310 up-regulation, it nearly abolished basal as well as stress-activated iNOS expression, most likely by binding to Brd4 and preventing its access to p65-acK310.
It is clear from the large protective effects of an iNOS enzyme inhibitor (Figs. 3 and 8 and Ref. 26) that iNOS played a major role in glioblastoma cell resistance to photokilling as well as greater aggressiveness of surviving cells. However, based on evidence shown in Fig. 10, other NF-κB–regulated proteins such as survivin, Bcl-xL, and p21 probably contributed to these responses, survivin and Bcl-xL being up-regulated by PDT and p21 down-regulated. Each of these responses was strongly affected by JQ1, i.e. inhibited for survivin and Bcl-xL and reversed for p21. JQ1 may have down-regulated survivin directly by blocking its transcription (47, 48). However, an indirect effect due to iNOS down-regulation was also possible, because NO is known to signal for survivin induction (49, 50). Similar direct and indirect effects of JQ1 may have occurred in the case of Bcl-xL, given our recent evidence that Bcl-xL up-regulation in PDT-stressed breast cancer cells is suppressed by iNOS enzyme inhibitors (52). Whether low-level iNOS-derived NO has any influence on p21 expression is not known. Therefore, for at least two of the effectors described in Fig. 10, JQ1 could have acted directly by preventing Brd4 binding at promoter sites as well as indirectly via suppression of iNOS expression.
It is likely that other oxidative stress–based therapeutic modalities will induce pro-survival/expansion adaptations similar to PDT. For example, ionizing radiation has been reported to elicit such responses, and overexpressed iNOS/NO has been implicated (44), but the underlying regulation of iNOS expression were not investigated. Of related interest is a recent study demonstrating that JQ1 can function as a radiosensitizer, i.e. act additively or even synergistically with ionizing radiation in dispatching malignant cells (54). Numerous other examples of combining BET bromodomain inhibitors with conventional chemotherapeutic agents have been described recently, e.g. JQ1 with paclitaxel for triple negative breast cancer (77) and OTX015 with temozolomide for glioblastoma (37). A potential clinical advantage of such combined treatments is that lower than normal individual drug dosages can be used, thus causing less off-target toxicity. Another advantage is that different subcellular sites with different negative effects can be targeted. For ALA—PDT, which we described here, mitochondria are the primary targets and nuclear sites are secondary targets, albeit indirect ones through JQ1 binding/inactivation of Brd4. It remains to be seen whether other anti-cancer therapies will realize similar advantages through the use of conventional modalities combined with BET bromodomain inhibitors in moderate doses.
In conclusion, we have described a PDT-aggravated growth and invasive aggressiveness of glioblastoma cells in which NO from photostress-induced iNOS plays a major driving role. The NF-κB–dependent iNOS response was fostered by the up-regulation of epigenetic readers Brd4 and acK310 on the p65 subunit of NF-κB (Fig. 11). The Brd4 inhibitor JQ1 suppressed iNOS expression, NO production, and cell hyperaggressiveness much more powerfully than an inhibitor of iNOS enzymatic activity, suggesting that JQ1 or a related BET bromodomain inhibitor could greatly improve clinical PDT outcomes for glioblastoma and possibly other malignancies. A few examples of combining JQ1 with conventional radio- or chemotherapeutic approaches have been reported (37, 54, 77), but the present study represents the first time that JQ1 has been combined with PDT, which is recognized as one of the best treatment options for many solid tumors, including glioblastomas (67–69). Our findings from this in vitro study provide a strong incentive for more advanced work involving JQ1 in a mouse tumor PDT model, which will soon be under way.
Figure 11.
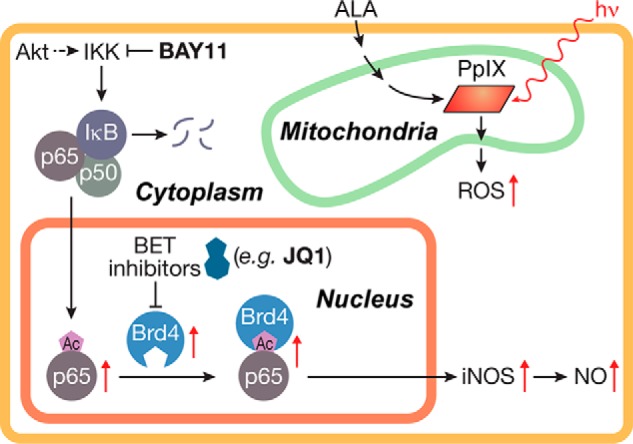
Scheme depicting key features of this study. Included are (i) ROS generation from photoactivated ALA-induced PpIX, (ii) Akt-mediated NF-κB activation and translocation to nucleus, and (iii) JQ1-inhibitable Brd4 binding to acetylated p65 at the requisite iNOS promoter site followed by iNOS expression.
Experimental procedures
General materials
ALA, BAY-11, fetal bovine serum (FBS), growth media, and other cell culture materials were obtained from Sigma-Aldrich. Cayman Chemicals (Ann Arbor, MI) supplied 1400W, DAF-FM-DA, LY294002, DETA-NONOate, reagents for the Griess NOx assay, and a rabbit polyclonal antibody against human iNOS (catalog no. 160862, lot 0502774-1). The thienodiazepine BET bromodomain inhibitor (+)-JQ1 (denoted as JQ1 here) and its inactive enantiomer (−)-JQ1 (denoted as JQ1(−) here) were also obtained from Cayman Chemicals. Stock solutions of JQ1 or JQ1(−) in DMSO were prepared immediately before experimental use. Cell-counting kit-8 (CCK-8) was obtained from Dojindo Molecular Technologies (Rockville, MD). Cell Signaling Technologies (Danvers, MA) supplied the rabbit-derived monoclonal antibodies against human p65 (catalog no. 4764S, lot 6), Brd2 (catalog no. 5848S, lot 6), Brd4 (catalog no. 13440S, lot 4), Akt (catalog no. 4691, lot 11), and p-Akt (catalog no. 2965, lot 3) and the mouse-derived monoclonal antibodies against survivin (catalog no. 2802, lot 7), β-actin (catalog no. 3700S, lot 10), and α-tubulin (catalog no. 3873, lot 6), as well as the control IgG (catalog no. 3990, lot 7) and peroxidase-conjugated IgG secondary antibodies. EMD Millipore supplied the mouse-derived monoclonal MMP-9 (catalog no. IM37, lot D00144532) antibody. Abcam (Cambridge, MA) supplied the rabbit polyclonal antibody against p65-acK310 (catalog no. ab19870, lot GR63202-1).
Cell culture
Human glioblastoma U87-MG and U251-MG cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). From this point on, these designations are shortened to U87 and U251, respectively. Cells were grown in a humidified incubator at 37 °C/5% CO2 using minimal essential medium with Earle's salts (MEM) supplemented with 10% FBS, 1% pyruvate, penicillin (100 units/ml), and streptomycin (100 μg/ml). The cells were switched to fresh supplemented medium every third day, passaged fewer than six times for all experiments, and used at no greater than ∼65% confluency.
Cell sensitization and irradiation
U87 or U251 cells at 60–65% confluency in 35-mm culture dishes were metabolically sensitized with PpIX by incubating them with 1 mm ALA in serum- and phenol red-free MEM for 30 min in the dark at 37 °C. As shown previously (26), most of the PpIX at this point was localized in mitochondria, where it originated via the heme anabolic pathway. Immediately after this step, the medium was removed, and cells were overlaid with fresh MEM lacking serum, phenol red, and ALA. Cell dishes were then placed on a translucent plastic platform over a bank of four 40W cool-white fluorescent lamps and irradiated at room temperature. The light power density (irradiance or fluence rate) at the bottom of each dish was ∼1.1 × 10−3 watts/cm2. Cells were typically irradiated for 15 min, which corresponds to a delivered light dose or fluence of ∼1 J/cm2. Immediately thereafter, the cells were overlaid with fresh 10% (v/v) FBS-supplemented medium, which either lacked or contained the BET bromodomain inhibitor JQ1 or iNOS inhibitor 1400W at a predetermined starting concentration. Immediately before use, stock solutions of JQ1 and 1400W were prepared in DMSO and PBS, respectively. A vehicle control for JQ1 was prepared and examined alongside; for 0.3 μm JQ1 (frequently used), this amounted to 0.001% (v/v) DMSO. Each inhibitor was maintained at the same starting concentration throughout all subsequent dark incubations. After various post-irradiation incubation times, cell samples were recovered for the determination of parameters such as viable fraction, extent of apoptosis, and surviving cell proliferation and invasion rate. Cells treated with ALA alone or light alone were prepared and analyzed alongside as controls. For the in vitro experiments in this study, the term PDT is defined as “photodynamic treatment” and is distinguished from PDT as photodynamic therapy, which should apply only to in vivo situations.
Measurement of viability loss and extent of apoptosis in PDT-treated cells
The effects of photodynamic stress on overall cell viability were determined by a Dojindo CCK-8 assay (76), which was typically carried out 24 h after irradiation. Light-only or ALA-only controls were analyzed as well. Early-stage apoptosis, as indicated by externalization of plasma membrane phosphatidylserine, was assessed by annexin V–FITC staining with fluorescence microscopy. A 96-well plate reader system (Biotek Synergy MX) was used with 485 nm excitation and 535 nm emission. Any necrosis due to plasma membrane disruption was assessed by propidium iodide staining. Other details were as described previously (26).
Detection of NO in photodynamically stressed cells
NO levels in photostressed glioblastoma cells were assessed using the fluorophore DAF-FM-DA (38). Upon entering the cells, DAF-FM-DA is hydrolyzed and trapped inside as DAF-FM, which fluoresces weakly. In aerobic systems, NO-derived dinitrogen trioxide (N2O3) can nitrosate DAF-FM to give highly fluorescent DAF-FM-triazole. A stock solution of 1 mm DAF-FM-DA in DMSO was prepared immediately prior to experimental use and shielded from room light. At various post-irradiation times, cells in serum-free medium were incubated in the dark for 50 min with 10 μm DAF-FM-DA and then washed and examined for DAF-FM-triazole level by fluorescence microscopy using a Nikon Eclipse TS100 microscope set at 495 nm excitation and 515 nm emission.
NO generated by photostressed cells was also determined by Griess assay. U87 cells (3 × 104/well) were seeded into a 96-well plate and allowed to attach overnight. After ALA treatment, irradiation, and washing, the cells were overlaid with serum-free medium containing either 0.3 μm JQ1 or DMSO vehicle. After dark incubation for various intervals, the medium was recovered for measurement of NO-derived nitrite (NO2−) and nitrate (NO3−) by Griess assay using a protocol recommended by the reagent supplier (Cayman Chemical Co.). The procedure included the reduction of any NO3− in the samples by nitrate reductase. Absorbance of the azo dye product at 540 nm was recorded in a plate reader, and quantification of total NO2−/NO3− (NOx) was based on a NO2− standard curve. Standardization was based on a determination of total protein in each well.
Western blot procedures
The expression of iNOS, Brd2, Brd4, MMP-9, survivin, c-Myc, Bcl-xL, and p21 in U87 cells before and after a photodynamic challenge was monitored by Western blot analysis using commercially available and authenticated primary antibodies (see “General materials” above). At various dark incubation times after irradiation, beginning immediately (0 h) and extending to 24 h, the treated cells along with appropriate controls were recovered by gentle scraping, centrifuged, and washed with ice-cold PBS. Cells were suspended in cold pH 7.4 lysis buffer (10 mm Tris, pH 8.0, 1% v/v Triton X-100, 0.1% w/v sodium deoxycholate, 0.1% w/v SDS, 140 mm NaCl, and 1 mm phenylmethylsulfonyl fluoride) containing protease inhibitors (22) and homogenized as described elsewhere (22). After centrifugation, the supernatant fraction was analyzed for total protein by BCA assay, after which samples of equal protein content (typically ∼100 μg) were separated by SDS-PAGE using appropriate acrylamide/bis-acrylamide mixtures. The separated proteins were transferred to a polyvinylidene difluoride membrane, and after blocking using 5% w/v nonfat dry milk in TBST, the membrane was treated overnight at 4 °C with a primary antibody diluted as follows: 1:250 for iNOS and 1:1000 for all other proteins. Following washing, the membrane was treated with a peroxidase-conjugated IgG secondary antibody (1:10,000) after which protein bands were analyzed using SuperSignal West Pico chemiluminescence detection (Thermo Scientific). Other details were as described previously (22, 26).
Detection of NF-κB/p65 in cytoplasmic and nuclear fractions
U87 cells were treated with ALA alone; ALA and light; ALA, Bay11 (5 μm), and light; or ALA, JQ1 (0.3 μm), and light. Irradiated cells along with controls (ALA alone) were incubated in the dark for 5 h. The cells were removed by gentle scraping into PBS, pelleted by centrifugation, and recovered. Nuclear and cytoplasmic fractions were then isolated using a NE-PERTM kit supplied by Thermo Scientific. All centrifugation and lysing steps for preparing cytoplasmic and nuclear fractions were done according to the suppliers' recommendations. After determination of total protein concentration, samples from each fraction were analyzed by Western blotting using antibodies against p65, histone H3 as a nuclear marker, and α-tubulin as a cytosolic marker.
Detection of BET protein interaction with NF-κB/p65
The possibility that post-PDT activation of NF-κB/p65 required interaction with a BET protein (Brd2 or Brd4) was investigated using an immunoprecipitation approach. After an ALA/light challenge, U87 cells were switched to 10% FBS-containing medium lacking or containing 0.3 μm JQ1 and returned to the incubator. After 8 h of incubation, cells were lysed, and total lysate protein was determined. A primary monoclonal antibody against p65 (10 μl from a 1:100 diluted stock solution from Cell Signaling Technologies (catalog no. 4764S) was added to 250 μg of total cell lysate and incubated for 16 h at 4 °C with mild agitation. After incubation, 100 μl of protein A-conjugated Sepharose bead slurry was added to each lysate followed by 6 h of additional incubation at 4 °C with agitation. After centrifugation, the beads with attached proteins were washed three times with lysis buffer to remove any contaminating proteins and then treated with 50 μl of 0.2 m glycine buffer, pH 2.6, for 10 min to release bound proteins. After centrifugation, proteins were recovered in supernatant fractions, which were brought to pH 8.0 with 20 mm Tris buffer. Samples were then mixed with SDS sample buffer in preparation for SDS-PAGE followed by p65, Brd2, and Brd4 immunoblotting using the antibody dilutions described above.
Evaluation of surviving cell proliferation
Twenty-four hours after an ALA/light challenge followed by a wash to remove detached (dead or dying) cells, the remaining (surviving) glioblastoma cells were recovered by gentle scraping along with non-irradiated controls and seeded into a 96-well plate using 10% (v/v) FBS-containing MEM. Normalization of the seeding density was based on prior knowledge of U87 viability losses, e.g. a 25% cell kill was compensated for by plating 25% more cells so that the initial cell count for each experimental condition was approximately the same. These cells, along with non-stressed controls, were dark-incubated in the presence of JQ1 or 1400W at the indicated starting concentrations. At various time points out to 72 h, the numbers of viable cells were determined by CCK-8 assay and expressed relative to the 24 h post-irradiation starting point.
Evaluation of surviving cell invasiveness
The invasiveness of U87 or U251 cells that could withstand an ALA/light challenge was examined using a 96-place Transwell device (model MBA96) from NeuroProbe (Gaithersburg, MD). Immediately after ALA/light exposure, these cells, along with ALA-only or light-only controls, were treated with JQ1, JQ1(−), Bay11, or 1400W at the indicated concentrations in serum-free medium and transferred to the upper wells of the invasion chamber (225 μl/well). Prior to this, 225 μl of 10% FBS-containing medium was added to each lower well of the invasion chamber, with the serum serving as a cell attractant. A Matrigel-infused polycarbonate filter with 8-μm pores was fitted over each lower well, after which the unit was prewarmed at 37 °C. The upper and lower wells were then clamped together, and the closed unit was placed in a 37 °C incubator. After a given incubation period (typically 24 h), the medium in the upper wells was carefully removed and cells remaining on top of the filters were gently wiped off with a cotton swab. Cells that had invaded to the filter bottoms were detached by centrifugation into 10% FBS-containing medium (400 × g for 15 min), allowed to adhere on a 96-well plate, and then either stained and photographed or quantified by CCK-8 assay.
Statistical analyses
Data are presented as means ± S.E. of values from at least three replicate experiments. Statistical significance was determined using the Student's t test in conjunction with GraphPad Prism software, and p values of <0.05 were considered statistically significant.
Author contributions
J. M. F., B. C. S., and A. W. G. conceptualization; J. M. F. data curation; J. S. S., B. C. S., and A. W. G. methodology; B. C. S. resources; B. C. S. formal analysis; B. C. S. and A. W. G. funding acquisition; B. C. S. and A. W. G. writing-review and editing; A. W. G. supervision; A. W. G. writing-original draft.
Supplementary Material
This work was supported by National Institutes of Health Grant CA70823 from NCI (to A. W. G.). This work also was supported by Grant 5520347 from the Advancing a Healthier Wisconsin Research and Education Program (to A. W. G.), BSC Grant 3308238/9 from the MCW Cancer Center (to A. W. G.), American Heart Association Scientist Development Grant 15SDG25830057 (to B. C. S.), Institutional Research Grants 14-247-29-IRG and 86-004-26-IRG from the American Cancer Society (to B. C. S.), and Grant 5520310 from the Advancing a Healthier Wisconsin Endowment (to B. C. S.). The authors declare that they have no conflicts of interest with the contents of this publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- ROS
- reactive oxygen species
- NO
- nitric oxide
- iNOS
- inducible nitric-oxide synthase
- MEM
- minimal essential medium
- ALA
- 5-aminolevulinic acid
- PDT
- photodynamic therapy/treatment
- PS
- photosensitizing agent
- PpIX
- protoporphyrin IX
- BET
- bromodomain and extra-terminal domain
- acK
- acetylated lysine
- JQ1
- (S)-tert-butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate
- JQ1(−)
- inactive JQ1 enantiomer
- 1400W
- N-[3 (aminomethyl)benzyl]acetamidine
- DAF-FM-DA
- 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate
- cPTIO
- 2-(4-carboxy-phenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide
- CCK-8
- cell counting kit-8
- PI3K
- phosphatidylinositol 3-kinase
- BAY11
- BAY11-7082 ((E)-3-(4-methylphenylsulfonyl)-2-propenenitrile)
- FBS
- fetal bovine serum
- LY294002
- 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one
- DETA-NONOate
- 2,2′-(hydroxynitrosohydrazino)bis-ethanamine.
References
- 1. Shacter E., and Weitzman S. A. (2002) Chronic inflammation and cancer. Oncology (Williston Park) 16, 217–226 [PubMed] [Google Scholar]
- 2. Marelli G., Sica A., Vannucci L., and Allavena P. (2017) Inflammation as target in cancer therapy. Curr. Opin. Pharmacol. 35, 57–65 10.1016/j.coph.2017.05.007 [DOI] [PubMed] [Google Scholar]
- 3. Burke A. J., Garrido P., Johnson C., Sullivan F. J., and Glynn S. A. (2017) Inflammation and nitrosative stress effects in ovarian and prostate pathology and carcinogenesis. Antioxid. Redox Signal. 26, 1078–1090 10.1089/ars.2017.7004 [DOI] [PubMed] [Google Scholar]
- 4. Wink D. A., Vodovotz Y., Laval J., Laval F., Dewhirst M. W., and Mitchell J. B. (1998) The multifaceted roles of nitric oxide in cancer. Carcinogenesis 19, 711–721 10.1093/carcin/19.5.711 [DOI] [PubMed] [Google Scholar]
- 5. Wink D. A., Ridnour L. A., Hussain S. P., and Harris C. C. (2008) The reemergence of nitric oxide and cancer. Nitric Oxide 19, 65–67 10.1016/j.niox.2008.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vannini F., Kashfi K., and Nath N. (2015) The dual role of iNOS in cancer. Redox. Biol. 6, 334–343 10.1016/j.redox.2015.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raber P. L., Thevenot P., Sierra R., Wyczechowska D., Halle D., Ramirez M. E., Ochoa A. C., Fletcher M., Velasco C., Wilk A., Reiss K., and Rodriguez P. C. (2014) Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer 134, 2853–2864 10.1002/ijc.28622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fionda C., Abruzzese M. P., Santoni A., and Cippitelli M. (2016) Immunoregulatory and effector activities of nitric oxide and reactive nitrogen species in cancer. Curr. Med. Chem. 23, 2618–2636 10.2174/0929867323666160727105101 [DOI] [PubMed] [Google Scholar]
- 9. Hirst D., and Robson T. (2010) Nitric oxide in cancer therapeutics: Interaction with cytotoxic chemotherapy. Curr. Pharm. Des. 16, 411–420 10.2174/138161210790232185 [DOI] [PubMed] [Google Scholar]
- 10. Saleem W., Suzuki Y., Mobaraki A., Yoshida Y., Noda S., Saitoh J. I., and Nakano T. (2011) Reduction of nitric oxide level enhances the radiosensitivity of hypoxic non-small cell lung cancer. Cancer Sci. 102, 2150–2156 10.1111/j.1349-7006.2011.02095.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matsunaga T., Yamaji Y., Tomokuni T., Morita H., Morikawa Y., Suzuki A., Yonezawa A., Endo S., Ikari A., Iguchi K., El-Kabbani O., Tajima K., and Hara A. (2014) Nitric oxide confers cisplatin resistance in human lung cancer cells through upregulation of aldo-keto reductase 1B10 and proteasome. Free Radic. Res. 48, 1371–1385 10.3109/10715762.2014.957694 [DOI] [PubMed] [Google Scholar]
- 12. Dougherty T. J., Gomer C. J., Henderson B. W., Jori G., Kessel D., Korbelik M., Moan J., and Peng Q. (1998) Photodynamic Therapy. J. Natl. Cancer Inst. 90, 889–905 10.1093/jnci/90.12.889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agostinis P., Berg K., Cengel K. A., Foster T. H., Girotti A. W., Gollnick S. O., Hahn S. M., Hamblin M. R., Juzeniene A., Kessel D., Korbelik M., Moan J., Mroz P., Nowis D., Piette J., et al. (2011) Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 61, 250–281 10.3322/caac.20114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benov L. (2015) Photodynamic therapy: Current status and future directions. Med. Princ. Pract. 24, 14–28 10.1159/000362416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peng Q., Berg K., Moan J., Kongshaug M., and Nesland J. M. (1997) 5-Aminolevulinic acid-based photodynamic therapy: Principles and experimental research. Photochem. Photobiol. 65, 235–251 10.1111/j.1751-1097.1997.tb08549.x [DOI] [PubMed] [Google Scholar]
- 16. Tetard M. C., Vermandel M., Mordon S., Lejeune J. P., and Reyns N. (2014) Experimental use of photodynamic therapy in high grade gliomas: A review focused on 5-aminolevulinic acid. Photodiagnosis Photodyn. Ther. 11, 319–330 10.1016/j.pdpdt.2014.04.004 [DOI] [PubMed] [Google Scholar]
- 17. Rollakanti K. R., Anand S., and Maytin E. V. (2015) Vitamin D enhances the efficacy of photodynamic therapy in a murine model of breast cancer. Cancer Med. 4, 633–642 10.1002/cam4.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henderson B. W., Sitnik-Busch T. M., and Vaughan L. A. (1999) Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate dependent. Photochem. Photobiol. 70, 64–71 10.1111/j.1751-1097.1999.tb01950.x [DOI] [PubMed] [Google Scholar]
- 19. Korbelik M., Parkins C. S., Shibuya H., Cecic I., Stratford M. R., and Chaplin D. J. (2000) Nitric oxide production by tumour tissue: Impact on the response to photodynamic therapy. Br. J. Cancer 82, 1835–1843 10.1054/bjoc.2000.1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reeves K. J., Reed M. W., and Brown N. J. (2010) The role of nitric oxide in the treatment of tumours with aminolaevulinic acid-induced photodynamic therapy. J. Photochem. Photobiol. B 101, 224–232 10.1016/j.jphotobiol.2010.07.007 [DOI] [PubMed] [Google Scholar]
- 21. Bhowmick R., and Girotti A. W. (2010) Cytoprotective induction of nitric oxide synthase in a cellular model of 5-aminolevulinic acid-based photodynamic therapy. Free Radic. Biol. Med. 48, 1296–1301 10.1016/j.freeradbiomed.2010.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhowmick R., and Girotti A. W. (2011) Rapid upregulation of cytoprotective nitric oxide in breast tumor cells subjected to a photodynamic therapy-like oxidative challenge. Photochem. Photobiol. 87, 378–386 10.1111/j.1751-1097.2010.00877.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhowmick R., and Girotti A. W. (2013) Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress. Free Radic. Biol. Med. 57, 39–48 10.1016/j.freeradbiomed.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhowmick R., and Girotti A. W. (2014) Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model. Cancer Lett. 343, 115–122 10.1016/j.canlet.2013.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fahey J. M., and Girotti A. W. (2015) Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: Role of nitric oxide. Nitric Oxide 49, 47–55 10.1016/j.niox.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fahey J. M., Emmer J. V., Korytowski W., Hogg N., and Girotti A. W. (2016) Antagonistic effects of endogenous nitric oxide in a glioblastoma photodynamic therapy model. Photochem. Photobiol. 92, 842–853 10.1111/php.12636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garvey E. P., Oplinger J. A., Furfine E. S., Kiff R. J., Laszlo F., Whittle B. J., and Knowles R. G. (1997) 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 272, 4959–4963 10.1074/jbc.272.8.4959 [DOI] [PubMed] [Google Scholar]
- 28. Singh D., Richards D., Knowles R. G., Schwartz S., Woodcock A., Langley S., and O'Connor B. J. (2007) Selective inducible nitric oxide synthase inhibition has no effect on allergen challenge in asthma. Am. J. Respir. Crit. Care Med. 176, 988–993 10.1164/rccm.200704-588OC [DOI] [PubMed] [Google Scholar]
- 29. Akaike T., and Maeda H. (1996) Quantitation of nitric oxide using 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO). Methods Enzymol. 268, 211–221 10.1016/S0076-6879(96)68023-9 [DOI] [PubMed] [Google Scholar]
- 30. Huang B., Yang X. D., Zhou M. M., Ozato K., and Chen L. F. (2009) Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell. Biol. 29, 1375–1387 10.1128/MCB.01365-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zou Z., Huang B., Wu X., Zhang H., Qi J., Bradner J., Nair S., and Chen L. F. (2014) Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene 33, 2395–2404 10.1038/onc.2013.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Filippakopoulos P., and Knapp S. (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356 10.1038/nrd4286 [DOI] [PubMed] [Google Scholar]
- 33. Shu S., and Polyak K. (2016) BET bromodomain proteins as cancer therapeutic targets. Cold Spring Harb. Symp. Quant. Biol. 81, 123–129 10.1101/sqb.2016.81.030908 [DOI] [PubMed] [Google Scholar]
- 34. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T., Felletar I., Philpott M., Munro S., McKeown M. R., Wang Y., Christie A. L., et al. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fu L. L., Tian M., Li X., Li J. J., Huang J., Ouyang L., Zhang Y., and Liu B. (2015) Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 6, 5501–5516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie Q. W., Kashiwabara Y., and Nathan C. (1994) Role of transcription factor NF-κ B/Rel in induction of nitric oxide synthase. J. Biol. Chem. 269, 4705–4708 [PubMed] [Google Scholar]
- 37. Berenguer-Daizé C., Astorgues-Xerri L., Odore E., Cayol M., Cvitkovic E., Noel K., Bekradda M., MacKenzie S., Rezai K., Lokiec F., Riveiro M. E., and Ouafik L. (2016) OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 139, 2047–2055 10.1002/ijc.30256 [DOI] [PubMed] [Google Scholar]
- 38. Kojima H., Urano Y., Kikuchi K., Higuchi T., Hirata Y., and Nagano T. (1999) Fluorescent indicators for imaging nitric oxide production. Angew. Chem. Int. Ed. Engl. 38, 3209–3212 10.1002/(SICI)1521-3773(19991102)38:21%3C3209::AID-ANIE3209%3E3.0.CO%3B2-6 [DOI] [PubMed] [Google Scholar]
- 39. Chaturvedi M. M., Sung B., Yadav V. R., Kannappan R., and Aggarwal B. B. (2011) NF-κB addiction and its role in cancer: “One size does not fit all”. Oncogene. 30, 1615–1630 10.1038/onc.2010.566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xia Y., Shen S., and Verma I. M. (2014) NF-κB, an active player in human cancers. Cancer Immunol. Res. 2, 823–830 10.1158/2326-6066.CIR-14-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li F., Zhang J., Arfuso F., Chinnathambi A., Zayed M. E., Alharbi S. A., Kumar A. P., Ahn K. S., and Sethi G. (2015) NF-κB in cancer therapy. Arch. Toxicol. 89, 711–731 10.1007/s00204-015-1470-4 [DOI] [PubMed] [Google Scholar]
- 42. Eyler C. E., Wu Q., Yan K., MacSwords J. M., Chandler-Militello D., Misuraca K. L., Lathia J. D., Forrester M. T., Lee J., Stamler J. S., Goldman S. A., Bredel M., McLendon K. E., Sloan A. E., Hjelmeland A. B., and Rich J. N. (2011) Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 146, 53–66 10.1016/j.cell.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kostourou V., Cartwright J. E., Johnstone A. P., Boult J. K., Cullis E. R., Whitley G., and Robinson S. P. (2011) The role of tumour-derived iNOS in tumor progression and angiogenesis. Br. J. Cancer 104, 83–90 10.1038/sj.bjc.6606034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim R. K., Suh Y., Cui Y. H., Hwang E., Lim E. J., Yoo K. C., Lee G. H., Yi J. M., Kang S. G., and Lee S. J. (2014) Fractionated radiation-induced nitric oxide promotes expansion of glioma stem-like cells. Cancer Sci. 104, 1172–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krishnan N., Bencze G., Cohen P., and Tonks N. K. (2013) The anti-inflammatory compound BAY-11–7082 is a potent inhibitor of protein tyrosine phosphatases. FEBS J. 280, 2830–2841 10.1111/febs.12283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garg H., Suri P., Gupta J. C., Talwar G. P., and Dubey S. (2016) Survivin: A unique target for tumor therapy. Cancer Cell Int. 16, 49 10.1186/s12935-016-0326-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meloche J., Potus F., Vaillancourt M., Bourgeois A., Johnson I., Deschamps L., Chabot S., Ruffenach G., Henry S., Breuils-Bonnet S., Tremblay È, Nadeau V., Lambert C., Paradis R., Provencher S., and Bonnet S. (2015) Bromodomain-containing protein 4: The epigenetic origin of pulmonary arterial hypertension. Circ. Res. 117, 525–535 10.1161/CIRCRESAHA.115.307004 [DOI] [PubMed] [Google Scholar]
- 48. Zanca C., Villa G. R., Benitez J. A., Thorne A. H., Koga T., D'Antonio M., Ikegami S., Ma J., Boyer A. D., Banisadr A., Jameson N. M., Parisian A. D., Eliseeva O. V., Barnabe G. F., Liu F., et al. (2017) Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes Dev. 31, 1212–1227 10.1101/gad.300079.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Engels K., Knauer S. K., Loibl S., Fetz V., Harter P., Schweitzer A., Fisseler-Eckhoff A., Kommoss F., Hanker L., Nekljudova V., Hermanns I., Kleinert H., Mann W., du Bois A., and Stauber R. H. (2008) NO signaling confers cytoprotectivity through the survivin network in ovarian carcinomas. Cancer Res. 68, 5159–5166 10.1158/0008-5472.CAN-08-0406 [DOI] [PubMed] [Google Scholar]
- 50. Fetz V., Bier C., Habtemichael N., Schuon R., Schweitzer A., Kunkel M., Engels K., Kovács A. F., Schneider S., Mann W., Stauber R. H., and Knauer S. K. (2009) Inducible NO synthase confers chemoresistance in head and neck cancer by modulating survivin. Int. J. Cancer 124, 2033–2041 10.1002/ijc.24182 [DOI] [PubMed] [Google Scholar]
- 51. Ishida C. T., Bianchetti E., Shu C., Halatsch M. E., Westhoff M. A., Karpel-Massler G., and Siegelin M. D. (2017) BH3-mimetics and BET-inhibitors elicit enhanced lethality in malignant glioma. Oncotarget 8, 29558–29573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fahey J. M., and Girotti A. W. (2017) Nitric oxide-mediated resistance to photodynamic therapy in a human breast tumor xenograft model: improved outcome with NOS2 inhibitiors. Nitric Oxide 62, 52–61 10.1016/j.niox.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sahni J. M., Gayle S. S., Bonk K. L., Vite L. C., Yori J. L., Webb B., Ramos E. K., Seachrist D. D., Landis M. D., Chang J. C., Bradner J. E., and Keri R. A. (2016) Bromodomain and extraterminal protein inhibition blocks growth of triple-negative breast cancers through the suppression of aurora kinases. J. Biol. Chem. 291, 23756–23768 10.1074/jbc.M116.738666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang J., Wang Y., Mei H., Yin Z., Geng Y., Zhang T., Wu G., Lin Z. (2017) The BET bromodomain inhibitor JQ1 radiosensitizes non-small cell lung cancer cells by upregulating p21. Cancer Lett. 391, 141–151 10.1016/j.canlet.2017.01.031 [DOI] [PubMed] [Google Scholar]
- 55. Stamenkovic I. (2000) Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 10, 415–433 10.1006/scbi.2000.0379 [DOI] [PubMed] [Google Scholar]
- 56. Björklund M., and Koivunen E. (2005) Gelatinase-mediated migration and invasion of cancer cells. Biochim. Biophys. Acta 1755, 37–69 [DOI] [PubMed] [Google Scholar]
- 57. Poole C. J., and van Riggelen J. (2017) MYC: Master regulator of the cancer epigenome and transcriptome. Genes (Basel) 8, E142 10.3390/genes8050142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Delmore J. E., Issa G. C., Lemieux M. E., Rahl P. B., Shi J., Jacobs H. M., Kastritis E., Gilpatrick T., Paranal R. M., Qi J., Chesi M., Schinzel A. C., McKeown M. R., Heffernan T. P., Vakoc C. R., et al. (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 10.1016/j.cell.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pérez-Salvia M., Simó-Riudalbas L., Llinàs-Arias P., Roa L., Setien F., Soler M., de Moura M. C., Bradner J. E., Gonzalez-Suarez E., Moutinho C., and Esteller M. (2017) Bromodomain inhibition shows antitumoral activity in mice and human luminal breast cancer. Oncotarget 8, 51621–51629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Luna M. C., Wong S., and Gomer C. J. (1994) Photodynamic therapy mediated induction of early response genes. Cancer Res. 54, 1374–1380 [PubMed] [Google Scholar]
- 61. Sanovic R., Krammer B., Grumboeck S., and Verwanger T. (2009) Time-resolved gene expression profiling of human squamous cell carcinoma cells during the apoptosis process induced by photodynamic treatment with hypericin. Int. J. Oncol. 35, 921–939 [DOI] [PubMed] [Google Scholar]
- 62. Cekaite L., Peng Q., Reiner A., Shahzidi S., Tveito S., Furre I. E., and Hovig E. (2007) Mapping of oxidative stress responses of human tumor cells following photodynamic therapy using hexaminolevulinate. BMC Genomics 8, 273 10.1186/1471-2164-8-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Uzdensky A., Kristiansen B., Moan J., and Juzeniene A. (2012) Dynamics of signaling, cytoskeleton and cell cycle regulation proteins in glioblastoma cells after sub-lethal photodynamic treatment: Antibody microarray study. Biochim. Biophys. Acta 1820, 795–803 10.1016/j.bbagen.2012.03.008 [DOI] [PubMed] [Google Scholar]
- 64. Li W., Xie Q., Lai L., Mo Z., Peng X., Leng E., Zhang D., Sun H., Li Y., Mei W., and Gao S. (2017) In vitro evaluation of ruthenium complexes for photodynamic therapy. Photodiagnosis Photodyn. Ther. 18, 83–94 10.1016/j.pdpdt.2017.02.001 [DOI] [PubMed] [Google Scholar]
- 65. Behin A., Hoang-Xuan K., Carpentier A. F., and Delattre J. Y. (2003) Primary brain tumours in adults. Lancet 361, 323–331 10.1016/S0140-6736(03)12328-8 [DOI] [PubMed] [Google Scholar]
- 66. Wen P. Y., and Kesari S. (2008) Malignant gliomas in adults. New Engl. J. Med. 359, 492–507 10.1056/NEJMra0708126 [DOI] [PubMed] [Google Scholar]
- 67. Whelan H. T. (2012) High-grade glioma/glioblastoma multiforme: Is there a role for photodynamic therapy? J. Natl. Compr. Canc. Netw. 10, S31–S34 10.6004/jnccn.2012.0171 [DOI] [PubMed] [Google Scholar]
- 68. Quirk B. J., Brandal G., Donlon S., Vera J. C., Mang T. S., Foy A. B., Lew S. M., Girotti A. W., Jogal S., LaViolette P. S., Connelly J. M., and Whelan H. T. (2015) Photodynamic therapy (PDT) for malignant brain tumors: Where do we stand? Photodiagnosis Photodyn Ther. 12, 530–544 10.1016/j.pdpdt.2015.04.009 [DOI] [PubMed] [Google Scholar]
- 69. Bechet D., Mordon S. R., Guillemin F., and Barberi-Heyob M. A. (2014) Photodynamic therapy of malignant brain tumours: A complementary approach to conventional therapies. Cancer Treat. Rev. 40, 229–241 10.1016/j.ctrv.2012.07.004 [DOI] [PubMed] [Google Scholar]
- 70. Olzowy B., Hundt C. S., Stocker S., Bise K., Reulen H. J., and Stummer W. (2002) Photoirradiation therapy of experimental malignant glioma with 5-aminolevulinic acid. J. Neurosurg. 97, 970–976 10.3171/jns.2002.97.4.0970 [DOI] [PubMed] [Google Scholar]
- 71. Hansel T. T., Kharitonov S. A., Donnelly L. E., Erin E. M., Currie M. G., Moore W. M., Manning P. T., Recker D. P., and Barnes P. J. (2003) A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics, FASEB J. 17, 1298–1300 10.1096/fj.02-0633fje [DOI] [PubMed] [Google Scholar]
- 72. Fujisawa T., and Filippakopoulos P. (2017) Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell. Biol. 18, 246–262 10.1038/nrm.2016.143 [DOI] [PubMed] [Google Scholar]
- 73. Andrieu G., Belkina A. C., and Denis G. V. (2016) Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 19, 45–50 10.1016/j.ddtec.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wienerroither S., Rauch I., Rosebrock F., Jamieson A. M., Bradner J., Muhar M., Zuber J., Müller M., and Decker T. (2014) Regulation of NO synthesis, local inflammation, and innate immunity to pathogens by BET family proteins. Mol. Cell. Biol. 34, 415–427 10.1128/MCB.01353-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kleszcz R., Paluszczak J., and Baer-Dubowska W. (2015) Targeting aberrant cancer metabolism: The role of sirtuins. Pharmacol. Rep. 67, 1068–1080 10.1016/j.pharep.2015.03.021 [DOI] [PubMed] [Google Scholar]
- 76. Pan L., Lin H., Tian S., Bai D., Kong Y., and Yu L. (2017) The sensitivity of glioma cells to pyropheophorbide-αmethyl ester-mediated photodynamic therapy is enhanced by inhibiting ABCG2. Lasers Surg. Med. 49, 719–726 10.1002/lsm.22661 [DOI] [PubMed] [Google Scholar]
- 77. Wali V. B., Langdon C. G., Held M. A., Platt J. T., Patwardhan G. A., Safonov A., Aktas B., Pusztai L., Stern D. F., and Hatzis C. (2017) Systematic drug screening identifies tractable targeted combination therapies in triple-negative breast cancer. Cancer Res. 77, 566–578 10.1158/1538-7445.AM2017-566,10.1158/0008-5472.CAN-16-1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.