

Abstract
Abelson helper integration site 1 (AHI1) is associated with several neuropsychiatric and brain developmental disorders, such as schizophrenia, depression, autism, and Joubert syndrome. Ahi1 deficiency in mice leads to behaviors typical of depression. However, the mechanisms by which AHI1 regulates behavior remain to be elucidated. Here, we found that down-regulation of expression of the rate-limiting enzyme in dopamine biosynthesis, tyrosine hydroxylase (TH), in the midbrains of Ahi1-knockout (KO) mice is responsible for Ahi1-deficiency–mediated depressive symptoms. We also found that Rev-Erbα, a TH transcriptional repressor and circadian regulator, is up-regulated in the Ahi1-KO mouse midbrains and Ahi1-knockdown Neuro-2a cells. Moreover, brain and muscle Arnt-like protein 1 (BMAL1), the Rev-Erbα transcriptional regulator, is also increased in the Ahi1-KO mouse midbrains and Ahi1-knockdown cells. Our results further revealed that AHI1 decreases BMAL1/Rev-Erbα expression by interacting with and repressing retinoic acid receptor–related orphan receptor α, a nuclear receptor and transcriptional regulator of circadian genes. Of note, Bmal1 deficiency reversed the reduction in TH expression induced by Ahi1 deficiency. Moreover, microinfusion of the Rev-Erbα inhibitor SR8278 into the ventral midbrain of Ahi1-KO mice significantly increased TH expression in the ventral tegmental area and improved their depressive symptoms. These findings provide a mechanistic explanation for a link between AHI1-related behaviors and the circadian clock pathway, indicating an involvement of circadian regulatory proteins in AHI1-regulated mood and behavior.
Keywords: depression, dopamine, circadian clock, Rev-ErbA alpha (NR1D1), biosynthesis, Abelson helper integration site 1, behavioral disorder, RAR-related orphan receptor alpha, transcriptional regulation, tyrosine hydroxylase
Introduction
Abelson helper integration site 1 (AHI1)3 is associated with mental disorders and neural development. Mutations in AHI1 are identified as a frequent factor of an autosomal recessive Joubert syndrome that is characterized by abnormal neurodevelopment and mental disturbance (1–3). Moreover, many lines of evidence indicate the linkage between AHI1 and neuropsychiatric disorders, including schizophrenia (SCZ) (4–7), depression (8), and autism (9). In an animal model, Ahi1 knockout (KO) mice exhibit typical depressive behaviors, sharing common neurological characters in human psychiatric disorders, accompanied by decreases of neurotransmitters such as dopamine (DA) and serotonin (5-HT) in various brain regions (10–12). These findings suggest that AHI1 plays an essential role in mood and mood-related behavior regulation. However, the molecular pathways involved in the regulation of AHI1-related depressive behavior are still largely unknown.
Monoamines, including neurotransmitters DA, 5-HT, and noradrenaline, are disturbed in subjects with mood disorders (13, 14). Especially, mesolimbic DA pathways are important for brain activities in emotion, including reward, motivation, and hedonic tone. DA dysfunction has been implicated in many mental disorders, including depression, SCZ, autism spectrum disorder, obsessive compulsive disorder, and attention deficit-hyperactivity disorder (15–17). A decreased DA function in midbrain is closely related to pathophysiology of depression, whereas increased DA activity is involved in mania (15, 17, 18).
Circadian rhythms have important roles in mood regulation. Polymorphisms in many clock genes are associated with various psychiatric disorders (19–21). Circadian gene products regulate daily oscillation of monoamine neurotransmitters, by controlling either their biosynthesis or metabolism (19, 20, 22). Tyrosine hydroxylase (TH), a rate-limiting enzyme for DA biosynthesis, is directly repressed by circadian nuclear receptor Rev-Erbα (23, 24). Rev-Erbα binds to RRE (REV-ERBs/retinoic acid receptor–related orphan nuclear receptor response element (RORE))/NBRE (NGFI-B response element) elements in the TH promoter to repress TH expression via competing with nuclear receptor–related protein 1 (NURR1), a major transcriptional factor of TH (23, 25). TH expression is also negatively regulated by circadian locomotor output cycle kaput (CLOCK) that is heterodimerized with brain and muscle Arnt-like protein 1 (BMAL1) (26–28). Clock components BMAL1, neuronal PAS domain protein 2 (NPAS2; a paralog of CLOCK), and PERIOD 2 (PER2) also participate in mood regulation by transcriptionally regulating the expression of monoamine oxidase A (MAOA), an enzyme that degrades DA and 5-HT (29).
Here, we demonstrate that AHI1 affects mood and behavior through circadian protein-mediated TH expression. AHI1 binds to RORα and represses BMAL1 expression, subsequently inhibits Rev-Erbα expression, and in turn up-regulates TH expression. Loss of AHI1 increases BMAL1 and Rev-Erbα levels and leads to TH expression inhibition.
Results
Decreased TH expression in Ahi1 KO mouse midbrains and Ahi1 knockdown cells
The previous studies demonstrated that Ahi1 KO mice present depression-like behaviors with a significant decrease of DA and 5-HT levels in many brain regions (10, 11). Considering that there are no differences in behaviors and AHI1 expression between Ahi1 heterozygous (Ahi1+/−) and wildtype mice (10, 11), we used Ahi1+/− mice as controls and investigated the depressive behaviors of Ahi1 KO mice. Ahi1 KO mice showed a significant increase of immobility as compared with controls in tail suspension test (TST) and forced swim test (FST) (Fig. 1, A and B), indicating that Ahi1 deficiency indeed leads to depression in mice. To identify how AHI1 affects production or release of DA and 5-HT, we measured mRNA levels of the key enzymes and transporters that are involved in production or release of DA and 5-HT in Ahi1 KO mouse midbrains and the littermate controls, including 5-HT biosynthetic enzymes tryptophan hydroxylase 1 and 2 (TPH1 and TPH2), DA biosynthetic enzyme TH and DOPA decarboxylase (DDC), 5-HT and DA metabolic enzymes MAOA and MAOB, DA metabolic enzyme catechol-O-methyltransferase (COMT), 5-HT transporter (5-HTT), DA transporter (DAT), and vesicular monoamine transporter 2 (VMAT2). Interestingly, TH mRNA levels but not others were significantly decreased in Ahi1 KO mouse midbrains (Fig. 1C). Consistently, TH protein levels but not other detected proteins were decreased in Ahi1 KO mouse midbrains (Fig. 1D). Interestingly, stereological analysis revealed that there is no difference of TH-positive neuron numbers in the ventral tegmental area (VTA) between Ahi1 KO mice and the littermate controls using diaminobenzidine (DAB) staining (Fig. 1, E and F). However, immunofluorescence staining showed that the TH fluorescence intensity was significantly decreased in VTA of Ahi1 KO mice compared with the littermate controls (Fig. 1, G and H). These data suggest that Ahi1 KO mice have lower TH protein expression levels but no changes in the cell numbers of TH neurons.
Figure 1.

Decreased TH expression in Ahi1 KO mouse midbrains and Ahi1 knockdown cells. A, immobility time in TST was measured in control mice (n = 10) and Ahi1 KO mice (n = 7). **, p < 0.01. B, immobility time in FST was measured in control mice (n = 10) and Ahi1 KO mice (n = 6). **, p < 0.01. C, relative mRNA levels of the indicated genes of Ahi1 KO mouse midbrains and littermate controls were performed by real-time qPCR. **, p < 0.01 (n = 5). D, the indicated protein abundance of Ahi1 KO mouse midbrains and the littermate controls was performed by Western blot analysis. The levels of the indicated proteins relative to GAPDH are shown on the right. **, p < 0.01 (n = 3). E, representative images of TH-DAB staining in VTA of Ahi1 KO mice and the littermate controls are shown at AP −3.5 mm. F, quantification of the TH-positive cells from (E) was shown. ns, no statistical significance (n = 3). G, representative images of TH-fluorescence staining in VTA of Ahi1 KO mice and the littermate controls are shown at AP −3.5 mm. H, intensity of TH immunofluorescence signals in G was analyzed. ***, p < 0.001 (n = 3). I, N2a cells were transfected with the indicated siRNAs. Seventy-two h after transfection, real-time qPCR was performed. *, p < 0.001 (n = 3). J, N2a cells were transfected with indicated siRNAs. Seventy-two h after transfection, the total cell lysates were subjected to immunoblot analysis. The intensities of AHI1 or TH relative to GAPDH (right) were analyzed. ***, p < 0.001 (n = 3). K, TH protein abundance of Ahi1 KO mouse midbrain and littermate controls at CT12 and CT00 by Western blot analysis. L, the relative ratios of TH to GAPDH in I were analyzed from density analysis by one-way ANOVA. Data are presented as means ± S.E. (error bars). **, p < 0.01 (n = 3).
Next, we verified the regulation of TH by AHI1 in cultured cells. In Neuro-2a (N2a) cells, knockdown of Ahi1 by two targeted siRNAs significantly decreased both TH mRNA and TH protein levels (Fig. 1, I and J). Meanwhile, overexpression of exogenous EGFP-AHI1 markedly increased TH protein levels (Fig. S1A). To further identify whether AHI1 regulates TH promoter activity, we cloned a 2-kb promoter fragment of murine TH gene and inserted it into a luciferase reporter vector. Overexpression of EGFP-AHI1 in cells dramatically increased TH promoter-driven luciferase reporter activity (Fig. S1B).
Many studies indicated that expression of TH is a time-of-day circadian oscillating pattern, and its expression is lowered at daytime and raised at night in mouse brain (23, 28, 30). Therefore, we tested whether AHI1 affects TH expression at different times of the day. As shown in Fig. 1K, TH protein levels are higher at subjective dawn (circadian time 00, CT00) than at subjective dusk (CT12) in both Ahi1+/− and Ahi1−/− mouse midbrains. Moreover, TH protein levels are significantly lower in Ahi1 KO mice midbrain than littermate controls at both CT00 and CT12 (Fig. 1, K and L). Taken together, our results suggested that Ahi1 KO mice exhibit depression-like behaviors, at least in part, by decreasing TH transcriptional expression.
Increased Rev-Erbα expression in Ahi1 KO mouse midbrains and Ahi1 knockdown cells
NURR1 is a major transcriptional factor of TH, and the circadian nuclear receptor Rev-Erbα represses TH transcription via competing with NURR1 to bind to RRE/NBRE elements of the TH promoter (23, 25). Therefore, we measured whether AHI1 has an effect on expression of NURR1 or Rev-Erbα. In Ahi1 KO mouse midbrains, Rev-Erbα protein levels were dramatically increased compared with the littermate controls; however, NURR1 protein levels kept unchanged (Fig. 2, A and B). Real-time qPCR analysis showed that Rev-Erbα but not Nurr1 mRNA levels were elevated in Ahi1 KO mouse midbrains (Fig. 2C). In cultured N2a cells, knockdown of Ahi1 using siRNAs markedly increased Rev-Erbα protein as well as Rev-Erbα mRNA levels but did not affect NURR1 protein and mRNA levels (Fig. 2, D and E). We next identified the correlation between the decrease of TH and increase of Rev-Erbα expression caused by AHI1 deficiency. Knockdown of Rev-erα increases TH protein levels in cells (Fig. S2), consistent with a study by other investigators (23). We next cloned a mutant TH promoter that lacks a Rev-Erbα–binding site and investigated the effect of AHI1 on its activity (Fig. 2F). Interestingly, EGFP-AHI1 lost the ability to influence the mutant TH promoter activity (Fig. 2G). These results suggest that AHI1-mediated TH expression is dependent on negatively regulating Rev-Erbα but not NURR1 expression. In contrast to the TH circadian pattern (Fig. 1, K and L), Rev-Erbα protein levels are lower at CT00 than at CT12, and it is increased in Ahi1 KO mouse midbrain compared with littermate controls at both CT00 and CT12 (Fig. 2, H and I).
Figure 2.

Increased Rev-Erbα expression in Ahi1 KO mouse midbrains and Ahi1 knockdown cells. A, the indicated protein abundance of Ahi1 KO mouse midbrains and littermate controls was examined using immunoblot analysis (n = 3). B, the relative levels of the indicated proteins to GAPDH in A were analyzed. ***, p < 0.001; ns, no statistical significance (n = 3). C, relative mRNA levels of Rev-Erbα and TH of Ahi1 KO mouse midbrains and littermate controls were examined using real-time qPCR. ***, p < 0.001; ns, no statistical significance (n = 5). D, N2a cells were transfected with the indicated siRNAs. Seventy-two h after transfection, the cell lysates were subjected to immunoblot analysis. The relative levels of Rev-Erbα and NURR1 to GAPDH (bottom) were analyzed. ***, p < 0.001; ns, no statistical significance (n = 3). E, N2a cells were transfected with the indicated siRNAs. Seventy-two h after transfection, real-time qPCR was performed. ***, p < 0.001; ns, no statistical significance (n = 3). F, schematic representation of mutant TH (mTH) promoter that lacks the Rev-Erbα binding motif in PGL3-Basic vector. G, HEK293 cells were transfected with PGL3-TH-Luc or PGL3-mTH-Luc along with EGFP or EGFP-AHI1, respectively. Forty-eight h after transfection, luciferase reporter assays were performed. ***, p < 0.001; ns, no statistical significance (n = 3). H, Rev-Erbα protein abundance of Ahi1 KO mouse midbrain and littermate controls at CT12 and CT00 by Western blot analysis. I, the relative ratios of Rev-Erbα to GAPDH in H were analyzed from density analysis by one-way ANOVA. Data are presented as means ± S.E. (error bars). ***, p < 0.001; **, p < 0.01 (n = 3).
Induction of Rev-Erbα expression by Ahi1 deficiency is BMAL1-dependent
BMAL1/CLOCK binds to the E-box element of the Rev-Erbα promoter to activate its transcription (31, 32). We therefore constructed two luciferase reporter vectors, a fragment of promoter in the Rev-Erbα gene (−1482/+502) (WT-Luc) and its deletion mutant (ΔCACATG (+24/+29)) (mutant-Luc), which lacks the BMAL1/CLOCK-binding site (Fig. 3A), to identify whether BMAL1 is involved in the regulation of Rev-Erbα by AHI1. Overexpression of EGFP-AHI1 repressed both basal and BMAL1/CLOCK-induced activities of the Rev-Erbα promoter (Fig. 3A). However, AHI1 did not affect activity of the mutant Rev-Erbα promoter (Fig. 3A). As the BMAL1/CLOCK-binding site in the Rev-Erbα promoter is involved in AHI1-regulated Rev-Erbα transcription, we examined whether BMAL1 and CLOCK are regulated by AHI1. In Ahi1 knockdown cells, BMAL1 but not CLOCK protein levels were increased (Fig. 3B). Similar to results from in vitro assays, BMAL1 but not CLOCK protein levels were increased in Ahi1 KO mouse midbrains (Fig. 3C). Moreover, Bmal1 but not Clock mRNA levels were also increased in Ahi1 knockdown cells (Fig. 3D), suggesting that AHI1 may transcriptionally regulate BMAL1 expression. Other clock genes, such as Periods (Per1, Per2, and Per3) and Crytochromes (Cry1 and Cry2), are also transcriptionally controlled by BMAL1 (33). We therefore examined Pers and Crys mRNA levels and found that Pers and Crys genes were also up-regulated in Ahi1 KO mouse midbrains (Fig. S4). BMAL1 transcription is activated by transcriptional factor RORα (34). We next performed immunoprecipitation assays to examine the interactions between AHI1 and RORα. FLAG-RORα was co-precipitated when EGFP-AHI1 but not EGFP was precipitated using anti-GFP antibodies (Fig. 3E). We also performed immunocytochemistry staining to detect the distribution of AHI1 and RORα. Although EGFP-AHI1 was mainly distributed in cytoplasm, it partly localized in the nucleus and co-localized with FALG-RORα (Fig. 3F). Next, we examined whether AHI1 influences Bmal1 promoter activity through RORα. The schematic diagrams of Bmal1 promoter luciferase reporter and its deletion mutant lacking two ROREs that are responsible for RORα binding are shown (Fig. 3G). EGFP-AHI1 significantly repressed basal activity and RORα-induced activity of the Bmal1 promoter (Fig. 3G). However, EGFP-AHI1 did not repress activity of the mutant Bmal1 promoter (Fig. 3G). These data suggest that AHI1 interacts with RORα to repress Bmal1 transcription.
Figure 3.

BMAL1-dependent induction of Rev-Erbα expression by Ahi1 deficiency. A, schematic representation of wildtype (WT-Luc) and mutant Rev-Erbα promoters (mutant-Luc) that were constructed into PGL3-Basic vector. HEK293 cells were transfected with WT or mutant Rev-Erbα-Luc along with EGFP, EGFP-AHI1 or HA, HA-BMAL1/HA-CLOCK (B/C), respectively. Forty-eight h after transfection, a luciferase reporter assay was performed. ***, p < 0.001; ns, no statistical significance (n = 3). B, N2a cells were transfected with the indicated siRNAs. Seventy-two h after transfection, the cell lysates were subjected to immunoblot analysis. The intensities of BMAL1 and CLOCK relative to GAPDH (bottom) were analyzed. **, p < 0.01 (n = 3). C, the BMAL1 and CLOCK protein levels of Ahi1 KO mouse midbrains and the littermate controls were examined using immunoblot analysis (n = 3). The intensity of BMAL1 relative to GAPDH (bottom) was analyzed. **, p < 0.01 (n = 3). D, N2a cells were transfected with the indicated siRNAs. Seventy-two h after transfection, real-time qPCR was performed. ***, p < 0.001; ns, no statistical significance (n = 3). E, HEK293 cells were transfected with FLAG-RORα along with EGFP or EGFP-AHI1. Forty-eight h after transfection, an immunoprecipitation assay was performed with anti-GFP antibodies. F, HEK293 cells were transfected with FLAG-RORα and EGFP-AHI1 (green). Forty-eight h after transfection, immunofluorescence was performed with anti-FLAG (red). The nuclei were stained with DAPI (blue). G, left, a schematic representation shows the WT and mutant Bmal1 promoter luciferase reporter. Right, HEK293 cells were transfected with WT-Luc or mutant-Luc of Bmal1 promoter along with EGFP, EGFP-AHI1 or FLAG, FLAG-RORα, respectively. Forty-eight h after transfection, a luciferase reporter assay was performed. ***, p < 0.001; ns, no statistical significance (n = 3). Error bars, S.E. IP, immunoprecipitation; IB, immunoblotting.
Regulation of TH by AHI1 is BMAL1-dependent
We have shown that AHI1 deficiency reduces TH expression and up-regulates Rev-Erbα levels and that overexpression of AHI1 decreases BMAL1 expression. We wondered whether the regulation of TH expression by AHI1 is mediated by BMAL1-regulated Rev-Erbα expression. In Bmal1 knockdown cells, increase of TH and decrease of Rev-Erbα protein levels were observed (Fig. 4A). Moreover, TH and Rev-Erbα mRNA levels were altered simultaneously in Bmal1 knockdown cells (Fig. 4B). We next examined the effects of BMAL1/CLOCK on TH promoter activity. BMAL1/CLOCK dramatically repressed TH promoter activity, and BMAL1/CLOCK had no significant effect on the mutant TH promoter lacking the Rev-Erbα–binding site, indicated by reporter gene assays (Fig. S3). Moreover, increase of Rev-Erbα and decrease of TH protein levels induced by Ahi1 knockdown were completely eliminated by BMAL1 deficiency (Fig. 4C), further suggesting that AHI1-induced TH expression is mediated by the BMAL1/CLOCK/Rev-Erbα circadian pathway.
Figure 4.

BMAL1-dependent regulation of TH by AHI1. A, N2a cells were transfected with si-Ctrl or si-Bmal1. Seventy-two h after transfection, the cell lysates were subjected to immunoblot analysis. The intensities of BMAL1, Rev-Erbα, or TH relative to GAPDH (right) were analyzed. ***, p < 0.001; **, p < 0.01 (n = 3). B, N2a cells were transfected with the indicated siRNAs. Seventy-two h after transfection, real-time qPCR was performed. ***, p < 0.001 (n = 3). C, N2a cells were transfected with the indicated siRNAs. Seventy-two h after transfection, the cell lysates were subjected to immunoblot analysis. The intensities of BMAL1, Rev-Erbα, or TH relative to GAPDH (right) were analyzed. ***, p < 0.001; **, p < 0.01; *, p < 0.05; ns, no statistical significance (n = 3). Error bars, S.E.
Rev-Erbα inhibition prevents TH reduction and improves depressive behaviors of Ahi1 KO mice
As Rev-Erbα up-regulation contributes to the decreased TH expression induced by AHI1 deficiency, we wondered whether Rev-Erbα inhibition has impacts on TH expression and behavior of Ahi1 KO mice. We microinfused SR8278, a Rev-Erbα inhibitor, into the VMB of Ahi1 KO mice or control mice to inhibit midbrain Rev-Erbα activity. We examined TH expressions in VTA after SR8278 microinfusion. Although SR8278 did not significantly affect TH levels in control mice, TH levels were dramatically increased in VTA in Ahi1 KO mice after SR8278 administration (Fig. 5, A and B). In addition, SR8278 administration significantly improved the performance of Ahil KO mice in TST but did not significantly affect the control mice (Fig. 5C).
Figure 5.

Restoration of TH levels and improvement of depressive behaviors of Ahi1 KO mice by Rev-Erbα inhibition. A–C, SR8278 (16 μg/mouse) or an equal volume of DMSO was microinfused into the VMB of control mice or Ahi1 KO mice. After 2 days, immunohistochemistry and TST were performed. A, fluorescence-TH staining of representative images in VTA were shown at AP −3.5 mm. B, intensity of TH immunofluorescence signals in A was analyzed. *, p < 0.05; ns, no statistical significance (n = 3). C, immobility time in TST was measured. Data are presented as means ± S.E. (error bars). **, p < 0.01; ns, no statistical significance (n = 6–8).
Discussion
In the present study, we revealed that AHI1 regulates TH transcriptional expression through the circadian RORα/BMAL1/Rev-Erbα pathway, to participate in mood and behavior regulation. TH is a rate-limiting enzyme of biosynthesis of catecholamines, such as DA, noradrenaline, and epinephrine, by converting tyrosine to l-3,4-dihydroxyphenylalanine (l-DOPA) (35). It has been reported that the TH gene is associated with depressive disorder or bipolar disorder (36, 37). Our study demonstrates that AHI1-regulated TH expression may contribute to mood and behavior regulation. Decreased TH levels in the Ahi1 KO mouse midbrain explain the decreased DA levels responsible for animal depressive behaviors.
AHI1 has comprehensive roles in mental regulation. Ahi1 KO mice also show depressive phenotypes (10, 11). AHI1 is abundant in the hypothalamus and amygdale, the regions that are important for emotional regulation, and loss of AHI1 influences TrkB signaling involved in depression behaviors (11, 38). In our study, we further found that AHI1 deficiency induces decreases of TH levels in VTA. As DA in VTA is important for emotional regulation, our study suggests an involvement of DA in AHI1-deficiency–induced depressive behavior.
TH expression is activated by NURR1 or repressed by circadian clock protein Rev-Erbα through competing for binding to the NBRE motif in the TH promoter (23, 25). In our observation, AHI1 does not affect NURR1 expression, but AHI1 deficiency induces Rev-Erbα expression. In addition, TH promoter activity that lacks the NBRE motif cannot be regulated by AHI1, further suggesting that Rev-Erbα is involved in AHI1-regulated TH expression. It was reported that loss of AHI1 impairs TrkB signaling (11). Interestingly, TrkB signaling activates TH expression through activating its promoter (39). In addition, TrkB activity is also regulated by the circadian clock pathway, showing that TrkB activity is higher at night and lower during the day (40). Whether AHI1-mediated circadian rhythm regulation influences TrkB signaling and whether TrkB participates in AHI1-mediated TH regulation remain to be evaluated.
The core circadian proteins CLOCK and BMAL1 form heterodimers to activate the transcription of several clock genes, such as PERs, CRYs, RORα, and Rev-Erbα. In turn, PERs and CRYs form complexes to repress the transcriptional activity of CLOCK/BMAL1. On the other hand, RORα and Rev-Erbα competitively bind to ROREs in the BMAL1 promoter to activate or repress its transcription, respectively (33).
The association between circadian rhythms and mood regulation as well as behavior processes has been well-documented (41). Disruption of biological rhythms is considered as a hallmark for and one of the key contributors to several mental disorders, such as major depression (42, 43), bipolar disorder (44), and SCZ (45). Polymorphisms in several circadian genes have been identified to associate with mental disorders (20, 45, 46). Recently, many studies showed that circadian gene deficiency is associated with mood and behavior abnormalities in animals (47). For example, mice harboring the Clock mutant gene exhibit mania-like behavior with increased dopaminergic activity (26, 28), and Per2 and Bmal1 KO mice also display mania-like response (29, 48, 49). Interestingly, inhibition of Rev-Erbα by gene KO or inhibitor leads to emotional instability with mania-like behavior and hyperactivity by increasing TH protein levels and dopamine production (23). We also found that microinfusion of Rev-Erbα inhibitor SR8278 into the VMB of Ahi1 KO mice significantly increases TH expression and reduces animal immobility time in TST. However, SR8278 only slightly increases TH expression and improves the control mouse performance in TST. The reason may be that Rev-Erbα is abundant in Ahi1 KO mice as compared with control mice; thus, Ahi1 KO mice are more sensitive to SR8278. This phenomenon is in accordance with the fact that SR8278 significantly reduces immobility time in Rev-Erbα high expression periods in a day rather than that in Rev-Erbα low expression periods (23). Thus, deficient Rev-Erbα activity in Rev-Erbα KO mice induces mania-like behavior, but increased Rev-Erbα levels in Ahi1 KO mice result in depressive behavior; both show associations with alterations of TH and DA.
Although several E-box motifs are present in human, mouse, and rat TH promoters (23, 35), TH promoter activity cannot be activated by BMAL1/CLOCK in our observations. Conversely, BMAL1/CLOCK dramatically represses TH promoter activity, consistent with a reported study indicating that TH mRNA levels are augmented in Clock Δ19 mutant mice (26). BMAL1/CLOCK can bind to the E-box of PER1 and activating transcription factor 5 (ATF5) promoters but not TH promoter by an electrophoretic mobility shift assay (30). These data suggest that BMAL1/CLOCK negatively regulate TH transcription by an indirect pathway. Considering that BMAL1/CLOCK activates Rev-Erbα transcription and Rev-Erbα directly represses TH expression (23, 31) and that Rev-Erbα and TH mRNA levels are attenuated and increased in Clock Δ19 mice (20, 26, 28), we propose that BMAL1/CLOCK inhibits TH expression via activating Rev-Erbα expression.
In conclusion, we identified that AHI1 binds to circadian protein RORα to negatively regulate BMAL1 and Rev-Erbα expressions, which eliminates Rev-Erbα–repressive effects and subsequently up-regulates TH expression, suggesting that AHI1 facilitates TH expression and DA biosynthesis through its regulating circadian clock pathway. Loss of AHI1 leads to an increase of Rev-Erbα that negatively regulates TH expression (Fig. 6). In conclusion, the present study demonstrates that the mood-related protein AHI1 links the molecular circadian clock pathway to regulate TH expression.
Figure 6.
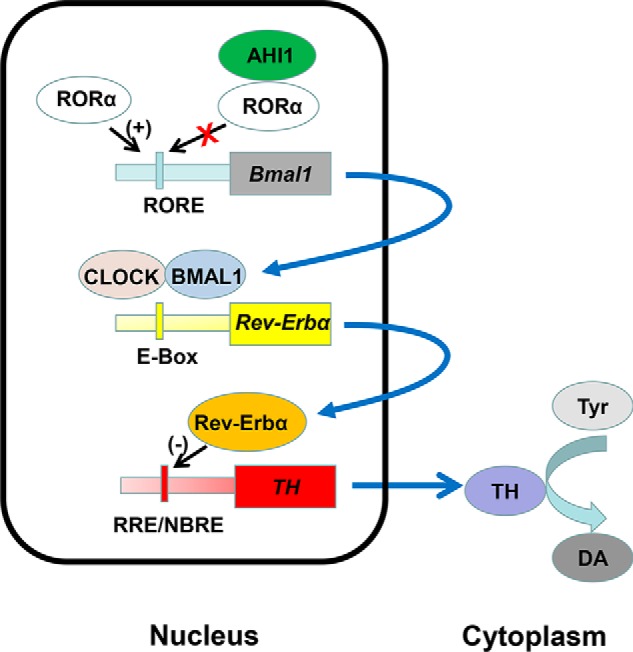
Schematic diagram of the pathway of TH expression and DA biosynthesis regulated by AHI1. AHI1 binds to the core circadian clock protein RORα and inhibits BMAL1 transcriptional expression. Decrease of BMAL1 results in down-regulation of Rev-Erbα expression and in turn up-regulates TH transcription and facilitates DA biosynthesis. Loss of AHI1 leads to increases of BMAL1/Rev-Erbα expressions, thereby repressing TH expression and DA biosynthesis, which contributes to animal depressive behaviors.
Experimental procedures
Plasmid constructs
pEGFP-C3-AHI1 was a gift from Joseph Gleeson (Addgene plasmid 30494) (50). HA-CLOCK, HA-BMAL1, FLAG-RORα, wildtype mouse Bmal1-luciferase (nucleotides −383 to +83), and mutant Bmal1-luciferase (mutation in RORE1 and RORE2) plasmids were kindly provided by Dr. Ying Xu (CAM-SU Genomic Resource Center at Soochow University). A 2.0-kb fragment of the Rev-Erbα promoter (nucleotides −1482 to +502) was amplified from a human genomic DNA library with the primers 5′-CGACGCGTGCTGCCTGTGGAGAAGGGCTT-3′ and 5′-CCCTCGAGGAAGCACCCTGCAGCAAGGTC-3′ and inserted into PGL3-Basic vector (Promega) at MluI/XhoI sites. Mutated Rev-Erbα promoter was constructed by deleting the BMAL1-binding E-box motif (CACATG (+24/+29)) (26, 27, 31) via site-directed mutagenesis with PGL3-Rev-Erbα-luciferase plasmid as template with primers 5′-GTACCTGCTCCAGTGCCG-3′ and 5′-ATCCCAGGGAGCGCCTCG-3′. A 2.0-kb fragment of the TH promoter (nucleotides −2001 to +0) was amplified from a mouse genomic DNA library with the primers 5′-GGGGTACCCAGCACCCTCTAAAGGAG-3′ and 5′-CCAAGCTTAGTGCAAGCTGGTGGTCC-3′ and inserted into PGL3-Basic vector at KpnI/HindIII sites. Mutated TH promoter that lacks the Rev-Erbα and NURR1 binding site (ΔGTCAGGTCA (−1535/−1526)) was generated by site-directed mutagenesis using PGL3-TH-luciferase plasmid as template with primers 5′-GCAGGGGAGGTTAGGGAGT-3′ and 5′-CCCCACAGTTCCTGTTCCAG-3′. The fidelities of all constructs were confirmed by sequencing.
Animals
Ahi1+/− and Ahi1 homozygous (Ahi1−/−) littermate mice were derived from Ahi1+/− and Ahi1−/− mice that were described previously (10). All animals were used according to the institutional guidelines for animal use and care, and all procedures were approved by the ethical committee of Soochow University. 8–10-week-old mice were housed in quarters under a 12-h light/12-h dark photoperiod (light on at 9:00 a.m.) and fed abundant food and water. Mice were sacrificed for collecting brain tissues at 2:00–4:00 pm except where specially indicated. CT00 was defined as lights on in the previous light/dark schedule when the mice were kept in constant darkness for 2 days.
Depression-like behaviors
The TST and FST were performed at 2:00–4:00 p.m. as described previously (10, 11). For TST, mice were hung in the 40-cm-high shelves by taping the tail (1–2 cm from tip). The immobility time was measured for 6 min. Mice were considered immobile when they gave up escaping or hung passively. For FST, mice were placed into a transparent beaker (16-cm diameter, 23-cm height) containing water (21–25 °C) at a depth of 15 cm. After a habituation period of 2 min, immobile time was measured within 4 min. Mice were considered as immobile when they floated or had slight limb movements.
Immunofluorescence and immunohistochemistry
Immunostaining was carried out as described previously (51). Mice were perfused with saline, followed by PBS (pH 7.4) containing 4% paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight at 4 °C and then dehydrated with 30% sucrose for 2 days at 4 °C. After dehydration, the brain was cut into 30-μm-thick coronal sections including the VTA with freezing microtome (CM1900, Leica). Slices were incubated with anti-TH (AB152, Millipore) antibodies for 4 h. For immunofluorescence, slices were stained with rhodamine (red)-conjugated secondary antibody (Invitrogen) for 2 h. Finally, the slices were observed with an inverted system microscope Ti2-E (Nikon, Japan). TH immunofluorescence signals were also quantified by ImageJ software (National Institutes of Health). For immunohistochemistry, slices were incubated with anti-TH antibodies for 4 h, followed by the DAB staining using a non-biotin detection system (GTVision III Anti-Mouse/Rabbit-HRP; Gene Tech), and then slices were observed with an inverted system microscope Ti2-E.
Stereology and image analysis
To determine cell numbers and intensity of TH-immunoreactive neurons in the VTA, an unbiased stereological method was performed according to the optical fractionator principle (52). Briefly, every fifth section (120-μm interval) was selected from each mouse and processed for immunostainings for TH using DAB staining. All images were acquired under the same conditions. The numbers of TH-immunoreactive neurons in the VTA were counted after outlining cell bodies and processes using ImageJ software.
Drug application
Mice were anesthetized by intraperitoneal injection of 4% chloral hydrate. Mice were placed prone, and heads were mounted in a stereotaxic apparatus (RWD Life Science Co, Shenzhen, China) by hooking incisors and inserting the ear stick into external auditory canal. Mouse skulls were exposed by cutting the scalp and corroding the meninges with H2O2. Bregma's location was set to the following: AP 0.0 mm, ML 0.0 mm, DV 0.0 mm. SR8278 was dissolved in DMSO to a concentration of 32 μg/μl and loaded into a 2.5-μl Hamilton syringe, which was fixed on a stereotaxic apparatus. SR8278 was infused into the VMB (AP −3.5 mm, ML ±1.2 mm, DV −4.5 mm) (16 μg/mouse; a dose was performed according to the previous study (23)) at a rate of 0.1 μl/min. After injection, the injector needle was slowly pulled out at a uniform speed in 2 min to avoid bleeding. The mouse scalp was sutured, and mice were placed in a warm environment until they woke up naturally. TST and immunohistochemistry were executed 2 days after microinfusion.
Cell culture and plasmid transfection
Mouse neuroblastoma N2a cells and human embryonic kidney 293 (HEK293) cells were grown in Dulbecco's modified Eagle's medium (Gibco) containing 10% fetal bovine serum (Gibco) with streptomycin (100 μg/ml) and penicillin (100 units/ml) (Gibco). For plasmid transfection, cells were transfected with plasmids using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions.
Immunocytochemistry
HEK293 cells transfected with EGFP-AHI1 and FLAG-RORα were washed with PBS twice and fixed with 4% paraformaldehyde for 10 min at room temperature. After treatment with 0.25% Triton X-100 for 15 min, the cells were incubated with 4% fetal bovine serum and anti-FLAG antibody (F3165, Sigma) in PBS overnight at 4 °C. Next, the cells were incubated with Alexa Fluor 594 donkey anti-mouse secondary antibodies (Life Technologies, Inc.) for 2 h, and then the nuclei were stained with DAPI (Sigma) for 10 min. Finally, the cells were observed with an inverted system microscope Ti2-E (Nikon, Japan).
siRNA knockdown
siRNAs against the following mouse genes were synthesized with the following sequences, respectively: Ahi1 1, 5′-GCCACCUCAAUAUCAUUUATT-3′ and 5′-UAAAUGAUAUUGAGGUGGCTT-3′; Ahi1 2, 5′-GAUUUCUCACCCAAUGGUAAATT and 5′-UUUACCAUUGGGUGAGAAAUCTT-3′; Rev-Erbα, 5′-GCAUCGUUGUUCAACGUGATT-3′ and 5′-UCACGUUGAACAACGAUGCAA-3′; Bmal1, 5′-CAGUAAAGGUGGAAGAUAATT-3′ and 5′-UUAUCUUCCACCUUUACUGTT-3′. Cells were transfected with siRNAs using the Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the manufacturer's instructions.
Luciferase reporter assay
HEK293 cells were cotransfected with luciferase reporter and expression plasmids along with Renilla luciferase vector pRL-CMV as an internal control for normalization. The total amount of plasmid DNA was held constantly by the addition of empty plasmid. Cell extracts were prepared with Passive Lysis Buffer (Promega) 48 h after transfection, and the luciferase activities were measured with a Dual-Luciferase assay kit (Promega) using a microplate reader, Infinite M1000 Pro (Tecan), according to the manufacturer's instructions.
Immunoprecipitation assay
Cells were lysed in cell lysis buffer (50 mm Tris-HCl, pH 7.5, buffer containing 150 mm NaCl, 1% Nonidet P-40, and 0.5% deoxycholate) supplemented with the protease inhibitor mixture (Roche Applied Science) at 4 °C. After centrifugation at 12,000 × g for 15 min, the supernatants were used for immunoprecipitation with appropriate antibodies coupled to protein G-Sepharose beads (Roche Applied Science). The immunoprecipitants were then washed with cell lysis buffer and subjected to immunoblot analysis. The input represents 10% of the supernatant used in the co-immunoprecipitation experiments.
Immunoblot analysis
Cell extracts and midbrain tissue homogenates were lysed in cell lysis buffer. About 20 μg of proteins were electrophoresed and electrotransferred to a polyvinylidene difluoride membrane (Millipore). Blots were incubated with the following primary antibodies: polyclonal anti-AHI1 antibodies that were described previously (38), rabbit polyclonal anti-TH (AB152, Millipore), anti-NURR1 (sc-991, Santa Cruz Biotechnology, Inc.), anti-HA (sc-805, Santa Cruz Biotechnology), and anti-histone H2B (ab45695, Abcam) antibodies; rabbit monoclonal anti-TPH1 (ab52924, Abcam), anti-DDC (ab131282, Abcam), anti-MAOA (ab126751, Abcam), and anti-MAOB (ab125010, Abcam) antibodies; and mouse monoclonal anti-GFP (sc-9996, Santa Cruz Biotechnology), anti-FLAG (F3165, Sigma), anti-GAPDH (MAB374, Millipore), anti-CLOCK (sc-271603, Santa Cruz Biotechnology), anti-Rev-Erbα (sc-100910, Santa Cruz Biotechnology), and anti-BMAL1 (sc-365645, Santa Cruz Biotechnology) antibodies. The following secondary antibodies were used: horseradish peroxidase–conjugated sheep anti-mouse and anti-rabbit antibodies (Amersham Biosciences). The proteins were visualized with an ECL detection kit (Thermo) using a chemiluminescence imaging system (Bioshine ChemiQ 4800).
Real-time quantitative PCR (qPCR)
Total RNA was extracted from cells or midbrain tissues using TRIzol reagent (Invitrogen). Five hundred ng of each RNA sample was reverse-transcribed into cDNA for PCR assays with a PrimeScript RT Master Mix (Takara). Real-time PCR analysis was performed for quantitative measurement of the target RNA abundance with power SYBR Green PCR Master Mix (Applied Biosystems) using a 7500 real-time PCR system (Applied Biosystems). Mouse primer sequences used for real-time PCR were as follows: 5′-GACAGGAGAACAAGTGGCAATG-3′ and 5′-ATCAGTGGTCAGCACGAACGA-3′ for Ahi1; 5′-GACCATCTTCCGAGAGCTAA-3′ and 5′-GGATGTTGTCTTCCCGATAG-3′ for Tph1; 5′-GTGGCTACAGGGAAGACAAC-3′ and 5′-AAGTCTCTTGGGCTCAGGTA-3′ for Tph2; 5′-GGTATACGCCACGCTGAAGG-3′ and 5′-TAGCCACAGTACCGTTCCAGA-3′ for TH; 5′-AAGAGCTGGGTTAATTGGTG-3′ and 5′-CAGTGTAGCGACCACAAAGA-3′ for Ddc; 5′-GCTGCTGTCTCATTGGGTCTC-3′ and 5′-CGAACTCAAACCAACCAATAGCC-3′ for Comt; 5′-GAGGCTCCAATTTCAATCACTCTG-3′ and 5′-ATGTAGTTTAGCAAGTCGTTCAGC-3′ for Maoa; 5′-AAGCGATGTGATCGTGGTGG-3′ and 5′-ACACTGAGGCCACAATCATGC-3′ for Maob; 5′-CTTCTCCTCTGGCTTCGTTGT-3′ and 5′-CAGGGTAGATGATGAAGATCAACC-3′ for Dat; 5′-GCGAGCATCTCTTATCTCATTGG-3′ and 5′-AAATGCTGATCCCAACAACTATC-3′ for Vmat2; 5′-ATCACGCTGGGTTTGGATAG-3′ and 5′-ATCACGCTGGGTTTGGATAG-3′ for 5-htt; 5′-AGGGCACAAGCAACATTACC-3′ and 5′-CACAGGCGTGCACTCCATAG-3′ for Rev-Erbα; 5′-TGAAGAGAGCGGACAAGGAGATC-3′ and 5′-TCTGGAGTTAAGAAATCGGAGCTG-3′ for Nurr1; 5′-CAAGCACCTTCCTTCCAATG-3′ and 5′-GATTGCAGTCCACACCACTG-3′ for Bmal1; and 5′-GCTACAGCTTCACCACCACA-3′ and 5′-TCTCCAGGGAGGAAGAGGAT-3′ for β-actin as an internal control. Relative gene expression was calculated by the 2−ΔΔCT method.
Statistical analysis
Densitometric analysis of immunoblots from three independent experiments was calculated using Photoshop version 7.0 software (Adobe). The data were analyzed using Origin version 6.0 software (Originlab). Statistical analysis was performed by one-way analysis of variance (ANOVA). Student's t tests were used for comparing two groups. A p value of < 0.05 was considered statistically significant. All results are presented as means ± S.E.
Author contributions
D. G. data curation; D. G., H. S., and Z. H. software; D. G., Xingyun Xu, and Z. H. formal analysis; D. G., S. Z., and Xingyun Xu methodology; D. G. and H. R. writing-original draft; S. Z. validation; S. Z., H. S., and C. M. investigation; C. M., G. W., and H. R. supervision; C. M. project administration; Xingshun Xu resources; G. W. and H. R. conceptualization; G. W. and H. R. funding acquisition; G. W. and H. R. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. Ying Xu (CAM-SU Genomic Resource Center at Soochow University) for providing HA-CLOCK, HA-BMAL1, FLAG-RORα, wildtype mouse Bmal1-luciferase, and mutant Bmal1-luciferase plasmids. We thank Dr. Joseph Gleeson for providing the pEGFP-C3-AHI1 plasmid.
This work was supported by National Natural Sciences Foundation of China Grants 81761148024, 31330030, and 31471012; National Key Scientific R&D Program of China Grants 2016YFC1306000 and 2012CB947602; Suzhou Clinical Research Center of Neurological Disease Grant Szzx201503; and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- AHI1
- Abelson helper integration site 1
- 5-HT
- serotonin
- BMAL1
- brain and muscle Arnt-like protein 1
- CLOCK
- locomotor output cycle kaput
- DA
- dopamine
- KO
- knockout
- FST
- forced swim test
- MAOA and MAOB
- monoamine oxidase A and B, respectively
- RORα
- retinoic acid receptor–related orphan receptor α
- NURR1
- nuclear receptor-related protein 1
- TH
- tyrosine hydroxylase
- TST
- suspension test
- VMB
- ventral midbrain
- VTA
- ventral tegmental area
- AP
- anteroposterior
- ML
- medial lateral
- DV
- dorsal ventral
- SCZ
- schizophrenia
- RRE
- REV-ERBs/RORE
- RORE
- retinoic acid receptor–related orphan nuclear receptor response element
- NBRE
- NGFI-B response element
- DAB
- diaminobenzidine
- CT
- circadian time
- HEK293
- human embryonic kidney 293
- ANOVA
- analysis of variance
- qPCR
- quantitative PCR.
References
- 1. Ferland R. J., Eyaid W., Collura R. V., Tully L. D., Hill R. S., Al-Nouri D., Al-Rumayyan A., Topcu M., Gascon G., Bodell A., Shugart Y. Y., Ruvolo M., and Walsh C. A. (2004) Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 36, 1008–1013 10.1038/ng1419 [DOI] [PubMed] [Google Scholar]
- 2. Utsch B., Sayer J. A., Attanasio M., Pereira R. R., Eccles M., Hennies H. C., Otto E. A., and Hildebrandt F. (2006) Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr. Nephrol. 21, 32–35 10.1007/s00467-005-2054-y [DOI] [PubMed] [Google Scholar]
- 3. Valente E. M., Brancati F., Silhavy J. L., Castori M., Marsh S. E., Barrano G., Bertini E., Boltshauser E., Zaki M. S., Abdel-Aleem A., Abdel-Salam G. M., Bellacchio E., Battini R., Cruse R. P., Dobyns W. B., et al. (2006) AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann. Neurol. 59, 527–534 10.1002/ana.20749 [DOI] [PubMed] [Google Scholar]
- 4. Amann-Zalcenstein D., Avidan N., Kanyas K., Ebstein R. P., Kohn Y., Hamdan A., Ben-Asher E., Karni O., Mujaheed M., Segman R. H., Maier W., Macciardi F., Beckmann J. S., Lancet D., and Lerer B. (2006) AHI1, a pivotal neurodevelopmental gene, and C6orf217 are associated with susceptibility to schizophrenia. Eur. J. Hum. Genet. 14, 1111–1119 10.1038/sj.ejhg.5201675 [DOI] [PubMed] [Google Scholar]
- 5. Ingason A., Giegling I., Cichon S., Hansen T., Rasmussen H. B., Nielsen J., Jürgens G., Muglia P., Hartmann A. M., Strengman E., Vasilescu C., Mühleisen T. W., Djurovic S., Melle I., Lerer B., et al. (2010) A large replication study and meta-analysis in European samples provides further support for association of AHI1 markers with schizophrenia. Hum. Mol. Genet. 19, 1379–1386 10.1093/hmg/ddq009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ingason A., Sigmundsson T., Steinberg S., Sigurdsson E., Haraldsson M., Magnusdottir B. B., Frigge M. L., Kong A., Gulcher J., Thorsteinsdottir U., Stefansson K., Petursson H., and Stefansson H. (2007) Support for involvement of the AHI1 locus in schizophrenia. Eur. J. Hum. Genet. 15, 988–991 10.1038/sj.ejhg.5201848 [DOI] [PubMed] [Google Scholar]
- 7. Rivero O., Reif A., Sanjuán J., Moltó M. D., Kittel-Schneider S., Nájera C., Töpner T., and Lesch K. P. (2010) Impact of the AHI1 gene on the vulnerability to schizophrenia: a case-control association study. PLoS One 5, e12254 10.1371/journal.pone.0012254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porcelli S., Pae C. U., Han C., Lee S. J., Patkar A. A., Masand P. S., Balzarro B., Alberti S., De Ronchi D., and Serretti A. (2014) Abelson helper integration site-1 gene variants on major depressive disorder and bipolar disorder. Psychiatry Investig. 11, 481–486 10.4306/pi.2014.11.4.481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alvarez Retuerto A. I., Cantor R. M., Gleeson J. G., Ustaszewska A., Schackwitz W. S., Pennacchio L. A., and Geschwind D. H. (2008) Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum. Mol. Genet. 17, 3887–3896 10.1093/hmg/ddn291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ren L., Qian X., Zhai L., Sun M., Miao Z., Li J., and Xu X. (2014) Loss of Ahi1 impairs neurotransmitter release and causes depressive behaviors in mice. PLoS One 9, e93640 10.1371/journal.pone.0093640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu X., Yang H., Lin Y. F., Li X., Cape A., Ressler K. J., Li S., and Li X. J. (2010) Neuronal Abelson helper integration site-1 (Ahi1) deficiency in mice alters TrkB signaling with a depressive phenotype. Proc. Natl. Acad. Sci. U.S.A. 107, 19126–19131 10.1073/pnas.1013032107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weng L., Lin Y. F., Li A. L., Wang C. E., Yan S., Sun M., Gaertig M. A., Mitha N., Kosaka J., Wakabayashi T., Xu X., Tang B., Li S., and Li X. J. (2013) Loss of Ahi1 affects early development by impairing BM88/Cend1-mediated neuronal differentiation. J. Neurosci. 33, 8172–8184 10.1523/JNEUROSCI.0119-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blier P. (2013) Neurotransmitter targeting in the treatment of depression. J. Clin. Psychiatry 74, 19–24 10.4088/JCP.12084su1c.04 [DOI] [PubMed] [Google Scholar]
- 14. Meyer J. H. (2008) Applying neuroimaging ligands to study major depressive disorder. Semin. Nucl. Med. 38, 287–304 10.1053/j.semnuclmed.2008.02.007 [DOI] [PubMed] [Google Scholar]
- 15. Southwick S. M., Vythilingam M., and Charney D. S. (2005) The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu. Rev. Clin. Psychol. 1, 255–291 10.1146/annurev.clinpsy.1.102803.143948 [DOI] [PubMed] [Google Scholar]
- 16. Charney D. S. (2004) Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am. J. Psychiatry 161, 195–216 10.1176/appi.ajp.161.2.195 [DOI] [PubMed] [Google Scholar]
- 17. Dunlop B. W., and Nemeroff C. B. (2007) The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry 64, 327–337 10.1001/archpsyc.64.3.327 [DOI] [PubMed] [Google Scholar]
- 18. Ashok A. H., Marques T. R., Jauhar S., Nour M. M., Goodwin G. M., Young A. H., and Howes O. D. (2017) The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol. Psychiatry 22, 666–679 10.1038/mp.2017.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bellivier F., Geoffroy P. A., Etain B., and Scott J. (2015) Sleep- and circadian rhythm-associated pathways as therapeutic targets in bipolar disorder. Expert Opin. Ther. Targets 19, 747–763 10.1517/14728222.2015.1018822 [DOI] [PubMed] [Google Scholar]
- 20. Albrecht U. (2017) Molecular Mechanisms in mood regulation involving the circadian clock. Front. Neurol. 8, 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bunney B. G., Li J. Z., Walsh D. M., Stein R., Vawter M. P., Cartagena P., Barchas J. D., Schatzberg A. F., Myers R. M., Watson S. J., Akil H., and Bunney W. E. (2015) Circadian dysregulation of clock genes: clues to rapid treatments in major depressive disorder. Mol. Psychiatry 20, 48–55 10.1038/mp.2014.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korshunov K. S., Blakemore L. J., and Trombley P. Q. (2017) Dopamine: a modulator of circadian rhythms in the central nervous system. Front. Cell Neurosci. 11, 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chung S., Lee E. J., Yun S., Choe H. K., Park S. B., Son H. J., Kim K. S., Dluzen D. E., Lee I., Hwang O., Son G. H., and Kim K. (2014) Impact of circadian nuclear receptor REV-ERBα on midbrain dopamine production and mood regulation. Cell 157, 858–868 10.1016/j.cell.2014.03.039 [DOI] [PubMed] [Google Scholar]
- 24. Jager J., O'Brien W. T., Manlove J., Krizman E. N., Fang B., Gerhart-Hines Z., Robinson M. B., Klein P. S., and Lazar M. A. (2014) Behavioral changes and dopaminergic dysregulation in mice lacking the nuclear receptor Rev-erbα. Mol. Endocrinol. 28, 490–498 10.1210/me.2013-1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zetterström R. H., Solomin L., Jansson L., Hoffer B. J., Olson L., and Perlmann T. (1997) Dopamine neuron agenesis in Nurr1-deficient mice. Science 276, 248–250 10.1126/science.276.5310.248 [DOI] [PubMed] [Google Scholar]
- 26. McClung C. A., Sidiropoulou K., Vitaterna M., Takahashi J. S., White F. J., Cooper D. C., and Nestler E. J. (2005) Regulation of dopaminergic transmission and cocaine reward by the Clock gene. Proc. Natl. Acad. Sci. U.S.A. 102, 9377–9381 10.1073/pnas.0503584102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sidor M. M., Spencer S. M., Dzirasa K., Parekh P. K., Tye K. M., Warden M. R., Arey R. N., Enwright J. F. 3rd, Jacobsen J. P., Kumar S., Remillard E. M., Caron M. G., Deisseroth K., and McClung C. A. (2015) Daytime spikes in dopaminergic activity drive rapid mood-cycling in mice. Mol. Psychiatry 20, 1406–1419 10.1038/mp.2014.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roybal K., Theobold D., Graham A., DiNieri J. A., Russo S. J., Krishnan V., Chakravarty S., Peevey J., Oehrlein N., Birnbaum S., Vitaterna M. H., Orsulak P., Takahashi J. S., Nestler E. J., Carlezon W. A. Jr., and McClung C. A. (2007) Mania-like behavior induced by disruption of CLOCK. Proc. Natl. Acad. Sci. U.S.A. 104, 6406–6411 10.1073/pnas.0609625104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hampp G., Ripperger J. A., Houben T., Schmutz I., Blex C., Perreau-Lenz S., Brunk I., Spanagel R., Ahnert-Hilger G., Meijer J. H., and Albrecht U. (2008) Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr. Biol. 18, 678–683 10.1016/j.cub.2008.04.012 [DOI] [PubMed] [Google Scholar]
- 30. Lemos D. R., Goodspeed L., Tonelli L., Antoch M. P., Ojeda S. R., and Urbanski H. F. (2007) Evidence for circadian regulation of activating transcription factor 5 but not tyrosine hydroxylase by the chromaffin cell clock. Endocrinology 148, 5811–5821 10.1210/en.2007-0610 [DOI] [PubMed] [Google Scholar]
- 31. Triqueneaux G., Thenot S., Kakizawa T., Antoch M. P., Safi R., Takahashi J. S., Delaunay F., and Laudet V. (2004) The orphan receptor Rev-erbalpha gene is a target of the circadian clock pacemaker. J. Mol. Endocrinol. 33, 585–608 10.1677/jme.1.01554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Preitner N., Damiola F., Lopez-Molina L., Zakany J., Duboule D., Albrecht U., and Schibler U. (2002) The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251–260 10.1016/S0092-8674(02)00825-5 [DOI] [PubMed] [Google Scholar]
- 33. Partch C. L., Green C. B., and Takahashi J. S. (2014) Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 24, 90–99 10.1016/j.tcb.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akashi M., and Takumi T. (2005) The orphan nuclear receptor RORα regulates circadian transcription of the mammalian core-clock Bmal1. Nat. Struct. Mol. Biol. 12, 441–448 10.1038/nsmb925 [DOI] [PubMed] [Google Scholar]
- 35. Tekin I., Roskoski R. Jr, Carkaci-Salli N., and Vrana K. E. (2014) Complex molecular regulation of tyrosine hydroxylase. J. Neural Transm. 121, 1451–1481 10.1007/s00702-014-1238-7 [DOI] [PubMed] [Google Scholar]
- 36. Furlong R. A., Rubinsztein J. S., Ho L., Walsh C., Coleman T. A., Muir W. J., Paykel E. S., Blackwood D. H., and Rubinsztein D. C. (1999) Analysis and metaanalysis of two polymorphisms within the tyrosine hydroxylase gene in bipolar and unipolar affective disorders. Am. J. Med. Genet. 88, 88–94 10.1002/(SICI)1096-8628(19990205)88:1%3C88::AID-AJMG16%3E3.0.CO%3B2-J [DOI] [PubMed] [Google Scholar]
- 37. Serretti A., Macciardi F., Verga M., Cusin C., Pedrini S., and Smeraldi E. (1998) Tyrosine hydroxylase gene associated with depressive symptomatology in mood disorder. Am. J. Med. Genet. 81, 127–130 10.1002/(SICI)1096-8628(19980328)81:2%3C127::AID-AJMG1%3E3.0.CO%3B2-T [DOI] [PubMed] [Google Scholar]
- 38. Sheng G., Xu X., Lin Y. F., Wang C. E., Rong J., Cheng D., Peng J., Jiang X., Li S. H., and Li X. J. (2008) Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J. Clin. Invest. 118, 2785–2795 10.1172/JCI35339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fukuchi M., Fujii H., Takachi H., Ichinose H., Kuwana Y., Tabuchi A., and Tsuda M. (2010) Activation of tyrosine hydroxylase (TH) gene transcription induced by brain-derived neurotrophic factor (BDNF) and its selective inhibition through Ca2+ signals evoked via the N-methyl-d-aspartate (NMDA) receptor. Brain Res. 1366, 18–26 10.1016/j.brainres.2010.10.034 [DOI] [PubMed] [Google Scholar]
- 40. Jang S. W., Liu X., Pradoldej S., Tosini G., Chang Q., Iuvone P. M., and Ye K. (2010) N-Acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. U.S.A. 107, 3876–3881 10.1073/pnas.0912531107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karatsoreos I. N. (2014) Links between circadian rhythms and psychiatric disease. Front. Behav. Neurosci. 8, 162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCarthy M. J., and Welsh D. K. (2012) Cellular circadian clocks in mood disorders. J. Biol. Rhythms 27, 339–352 10.1177/0748730412456367 [DOI] [PubMed] [Google Scholar]
- 43. Lamont E. W., Legault-Coutu D., Cermakian N., and Boivin D. B. (2007) The role of circadian clock genes in mental disorders. Dialogues Clin. Neurosci. 9, 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dallaspezia S., and Benedetti F. (2015) Chronobiology of bipolar disorder: therapeutic implication. Curr. Psychiatry Rep. 17, 606 [DOI] [PubMed] [Google Scholar]
- 45. Lamont E. W., Coutu D. L., Cermakian N., and Boivin D. B. (2010) Circadian rhythms and clock genes in psychotic disorders. Isr. J. Psychiatry Relat. Sci. 47, 27–35 [PubMed] [Google Scholar]
- 46. Wulff K., Gatti S., Wettstein J. G., and Foster R. G. (2010) Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat. Rev. Neurosci. 11, 589–599 10.1038/nrn2868 [DOI] [PubMed] [Google Scholar]
- 47. Barandas R., Landgraf D., McCarthy M. J., and Welsh D. K. (2015) Circadian clocks as modulators of metabolic comorbidity in psychiatric disorders. Curr. Psychiatry Rep. 17, 98 10.1007/s11920-015-0637-2 [DOI] [PubMed] [Google Scholar]
- 48. De Bundel D., Gangarossa G., Biever A., Bonnefont X., and Valjent E. (2013) Cognitive dysfunction, elevated anxiety, and reduced cocaine response in circadian clock-deficient cryptochrome knockout mice. Front. Behav. Neurosci. 7, 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kondratova A. A., Dubrovsky Y. V., Antoch M. P., and Kondratov R. V. (2010) Circadian clock proteins control adaptation to novel environment and memory formation. Aging 2, 285–297 10.18632/aging.100142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lancaster M. A., Louie C. M., Silhavy J. L., Sintasath L., Decambre M., Nigam S. K., Willert K., and Gleeson J. G. (2009) Impaired Wnt-β-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat. Med. 15, 1046–1054 10.1038/nm.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gu C., Zhang Y., Hu Q., Wu J., Ren H., Liu C. F., and Wang G. (2017) P7C3 inhibits GSK3β activation to protect dopaminergic neurons against neurotoxin-induced cell death in vitro and in vivo. Cell Death Dis. 8, e2858 10.1038/cddis.2017.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gundersen H. J., Bagger P., Bendtsen T. F., Evans S. M., Korbo L., Marcussen N., Møller A., Nielsen K., Nyengaard J. R., and Pakkenberg B. (1988) The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS 96, 857–881 10.1111/j.1699-0463.1988.tb00954.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.