

Abstract
Sexually transmitted infections with Chlamydia trachomatis (CT) and/or Neisseria gonorrhoeae (GC) and rates of pelvic inflammatory disease (PID) in women continue to rise, with reinfection being common because of poor adaptive immunity. Diagnosis remains imprecise, and pathogenesis data are derived primarily from monoinfections of mice with CT or GC. By comparing blood mRNA responses of women with CT and/or GC-induced PID and histologic endometritis to those from women with CT and/or GC infection limited to their cervix and asymptomatic uninfected women determined via microarray, we discovered important pathogenic mechanisms in PID and response differences that provide a pathway to biomarker discovery. Women with GC and/or CT-induced PID exhibit overexpression of myeloid cell genes and suppression of protein synthesis, mitochondrial oxidative phosphorylation, and T-cell specific genes. Coinfected women exhibited the greatest activation of cell death pathways and suppression of responses essential for adaptive immunity. Women solely infected with CT expressed elevated levels of type I and type II interferon genes, and enhanced type I interferon-induced chemokines in cervical secretions were associated with ascension of CT to the endometrium. Blood microarrays reveal discrete pathobiological endotypes in women with PID that are driven by pathogen invasion of the upper genital tract.
INTRODUCTION
Pelvic Inflammatory Disease (PID) is a spectrum of female upper genital tract inflammatory disorders that includes endometritis, salpingitis, tubo-ovarian abscess, and pelvic peritonitis. PID results from uterine and fallopian tube bacterial infection and can lead to serious reproductive sequelae, such as infertility, ectopic pregnancy and chronic pelvic pain. Over 750,000 women experience a PID episode each year in the United States (1) resulting in ~2 billion dollars spent on treatment of PID and its associated sequelae annually (2). Chlamydia trachomatis (CT) and Neisseria gonorrheoae (GC) are well-recognized PID pathogens, and recent data also implicate Mycoplasma genitalium (3-5). Coinfection with these pathogens is common (6), but little is known of possible synergistic or antagonistic impacts of co-infection on disease development. Vaginal microbiota (e.g., anaerobes, Gardnerella vaginalis, Haemophilus influenzae) have also been associated with PID (7, 8). Host genetics (9), physiologic effects mediated by menstrual cycling (10), or the use of hormonal contraceptives (11) can also contribute to disease severity.
The non-specific signs and symptoms that are used for clinical recognition of the syndrome challenge accurate diagnosis. Subtle or mild symptoms may not be recognized as PID but women with subclinical upper genital tract inflammation, a condition termed “silent” or “subclinical PID”, remain at increased risk for reproductive sequelae (12). The accuracy of laparoscopy or noninvasive imaging studies is variable (13) (14) and current diagnostics cannot reliably predict PID. Whether PID is a single endotype, defined by a distinct functional or pathobiological mechanism, or a syndrome comprised of several distinct endotypes is a key question. Transcriptional profiling has been used to identify diagnostic and prognostic biomarkers for cancer and other diseases (15-17). Gene-centric, fold-change-based approaches were initially favored and focused on the implications of expression differences between single genes (18-20) but advances in high-throughput genomic technologies have made it possible to define “networks” of transcriptional interactions. Network analysis has identified groups of coordinately expressed genes (modules) characteristic of specific disorders and disease mechanisms (21-23). Using this approach to investigate the pathophysiological mechanisms underlying PID is valuable, because the subtle and varied nature of this clinical syndrome suggests that disease differentiation and outcome is unlikely to be dependent on expression changes in any individual gene. Furthermore, improved understanding of pathways leading to PID will enable identification of biomarkers to improve diagnosis and effectively target treatment.
METHODS
Study population
Study participants were selected from two independent cohorts recruited at the University of Pittsburgh. The Anaerobes and Clearance of Endometritis (ACE) cohort is comprised of women who participated in a clinical trial (NCT01160640) comparing antibiotic regimens for the treatment of clinically diagnosed PID (1). The T cell Response Against Chlamydia (TRAC) cohort was comprised of women without PID who were predominantly asymptomatically infected with CT and were recruited with recognized risk factors for STI (11). Exclusion criteria for both studies included pregnancy, uterine procedure or miscarriage in the preceding 60 days, menopause, hysterectomy, antibiotic therapy in the preceding 14 days, and allergy to study medications. Participants provided informed consent at the time of enrollment and agreed to attend follow-up visits according to study protocols.
At enrollment, study personnel obtained demographic data and a standardized medical history. Participants completed a questionnaire regarding obstetric/gynecologic history, behavioral practices, sex exposure, contraceptive methods, and symptoms. General physical and pelvic exams were performed; vaginal fluid was collected for pH measurement, whiff testing and microscopy to detect clue cells was used to diagnose bacterial vaginosis using Amsel’s criteria (24). Vaginal swabs were collected for culture and molecular testing for Trichomonas vaginalis (APTIMA TV; Gen-Probe Inc., San Diego, CA). Nucleic acid amplification tests were performed on cervical swabs for detection of CT and GC (APTIMA Combo 2, Gen-Probe Inc., San Diego, CA), and M. genitalium (APTIMA MG: Gen-Probe Inc., San Diego, CA). Serum was collected for analysis of anti-chlamydial antibody titers, HIV antibody, and syphilis testing. Blood was collected in Tempus™ Blood RNA tubes (ThermoFisher Scientific, Waltham, MA) for subsequent transcriptional profiling and stored at −80°C prior to processing.
Study participants underwent endometrial sampling at enrollment. After cleaning the cervix with betadine, a sterile endometrial sampler (Unimar Pipelle de Cornier, CooperSurgical, Shelton, Connecticut) was placed into the endometrial cavity and a tissue sample collected. This tissue was discharged into a sterile petri dish after the sample device was removed. Tissue most proximal to the sampling portal of the cannula was placed in 10% formalin fixative and adjacent tissue was placed in RNAlater® solution (Thermo Scientific). Distal tissue was used for microbiologic culture and a swab absorbed 5 mm of tissue most distal to the portal for qualitative NAAT (APTIMA). Two pathologists independently scored hematoxylin and eosin stained sections of endometrial biopsy tissue as normal, acute endometritis (presence of ≥1 plasma cell per 120× field in the endometrial stroma and ≥5 neutrophils per 400× field in the endometrial surface epithelium) or chronic endometritis (presence of ≥1 plasma cell per 120× field in the endometrial stroma) (25). Discrepant evaluations were resolved after re-review and discussion.
Study Design
Comparisons to determine a disease signature were made between women with clinical symptoms meeting PID diagnostic criteria (PID+) associated with endometrial GC, GC and CT, or CT infection and histologic evidence of chronic endometritis (infected cases) and women without pelvic pain (PID-) who were co-infected with GC and CT or singly infected with CT at their cervix with normal endometrial histology (infected controls). Subsequent comparisons performed to determine if the signature was pathogen driven were made between infected cases and women with clinical PID and chronic endometritis who were negative for GC or CT, as well as women with clinical PID and normal endometrial histology who were negative for GC or CT. In addition, women with CT-induced chronic endometritis were compared to women with CT-induced acute endometritis to investigate kinetics of the response. Infected cases were also compared to women without pelvic pain who were not infected with GC or CT and had normal histology (uninfected controls). Finally, an independent group of asymptomatic women with histologically confirmed endometritis and upper genital tract infection (subclinical PID) (11) were assessed for conservation of disease-associated module networks. Subsequent evaluation of cervical inflammatory markers and bacterial load was performed for all TRAC participants for whom samples were available.
Microarray data acquisition
Total RNA was extracted from whole blood collected in Tempus™ Blood RNA tubes according to the manufacturer’s directions (Applied Biosystems/ThermoFisher Scientific, Waltham, MA). Alpha and beta globin mRNA was removed from total RNA using Globinclear (Ambion/ThermoFisher Scientific) before being analyzed on Illumina HT-12 v4 Expression BeadChips at the Genomics Research Core of the University of Pittsburgh. All microarray data have been deposited in the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) GEO accession number: GSE110106.
In situ RNA hybridization of endometrial tissues
Formalin-fixed paraffin-embedded 5-μm thick endometrial tissue sections from 14 infected and 15 uninfected patients were analyzed for IFN-β or CXCL-1 mRNA and chlamydial ribosomal RNA by in-situ hybridization using RNAScope™ 2.5 HD colorimetric duplex Assay Kit (Advanced Cell Diagnostics, Hayward, CA), as per manufacturer’s protocol. Tissue sections were deparaffinized in xylene for 5 min twice, then dehydrated in 100% ethanol twice for 1 min. The sections were then dried, treated with H2O2 for 10 min at room temperature and incubated in target retrieval buffer at 100°C-103°C for 15 min using a hot plate. After rinsing in deionized water, the sections were incubated with RNAScope Protease Plus for 30 min at 40°C in a HybEZ hybridization oven (Advanced Cell Diagnostics). Subsequent hybridization with target probes, preamplifier, amplifier, label probes, and chromogenic detection were performed as described by the manufacturer. Positive (hCyclophillin-C1, hPol2ra-C2) and negative (Bacillus subtilis DapB C1&C2) control probes in dual channels were used to assay for robustness of the sample and performance of the kit. The probes for chlamydial rs16 RNA-C1 (green), hCXCL1 mRNA-C2 (red), and hIFN-β mRNA-C2 (red) were used alone or in combination. The stained tissue sections along with their corresponding H&E slides were digitally imaged using 40× (20× with 2× mag changer) in the Aperio ScanScope XT using line-scan camera technology (Leica Biosystems) at UNC Translational Pathology Laboratory Core. Digital images were stored and analyzed within Aperio eSlide Manager software. The mRNA spots were scored using a five grade (0-4) semiquantiative scoring guideline provided by the manufacturer.
Detection of cytokines in cervical secretions
Cervical secretions were collected by placement of an ophthalmic sponge (Beaver-Visitec International Inc., Waltham, MA) within the endocervix of each patient recruited into the TRAC cohort for 30 seconds. Upon retrieval, the sponge was placed into a cryovial on ice until transfer to the lab where the vials were stored in liquid nitrogen until sponges were removed and secretions were eluted for protein assays as described with slight modifications (26). The cryovials and ophthalmic sponges were weighed to estimate the volume of secretions absorbed onto the sponge. Corning® Costar® Spin-X® centrifuge tubes containing 0.45 μm filters (Millipore Sigma, St. Louis, MO) were equilibrated with 500μl of blocking buffer (PBS, 2% BSA, and 0.05% Tween-20) for 30 minutes at room temperature. Filters were then washed 3 times with 100μl PBS. Sponges were equilibrated using 300μl of elution buffer (PBS, 0.5% BSA, 0.05% Tween-20, and protease inhibitor) before being placed in Spin-X tubes and incubated on ice for 10 minutes. Spin-X tubes containing sponges were placed in a pre-weighed cryovial and centrifuged at 10,000 rpm for 1 hour at 4°C to separate cervical secretions from the sponge matrix. Cervical secretions were stored at −80°C prior to analysis. A dilution factor was calculated based on the estimated volume of the secretion and pre- and post- weight of the collection tube: [(x-y)+ 0.36g of elution buffer]/(x-y), where x equals the weight of the sponge + cryovial tube and y is the average weight of the dry cryovial, based on independently weighing three dry cryovials. Cytokine levels in cervical secretions of 246 women recruited into TRAC were determined using Milliplex Magnetic Bead Assay Kits (Millipore Sigma) by the same operator at the Duke University Immunology Core Lab, according to manufacturers’ instructions, using a Luminex® fluorescence reader (Millipore Sigma) Kits included lyophilized standards that were reconstituted and diluted at 7 serial concentrations following manufacturers’ instructions for generation of standard curves. Standards included all recombinant cytokines tested and were considered as positive controls for the procedure.
Determination of cervical CT burden
Chlamydial cervical burden from these same patients for whom secretions were analyzed was estimated via quantitative PCR using genomic DNA extracted from reserved cervical swab eluates as template (27).
Statistics
Demographic data analysis
ANOVA was used to compare ages among groups, and equality of variances were examined by Fligner-Killeen’s test. Fisher’s exact test (two-sided) was conducted to assess the distribution of categorical variables: race, marital status, education and insurance across groups. Multiple testing p value was adjusted by Bonferroni correction.
Microarray data processing and analysis
Quantile normalization with RMA (28) was performed on raw transcript data which was subsequently log2 transformed. Genes were filtered according to expression and variance thresholds, set at μ > 6.8 and σ2 > 0.25 respectively. Batch effects were evaluated by guided principal component analysis (29), and corrected using empirical Bayes methods (30). We applied unsupervised cluster analysis and principal component analysis (PCA), to identify reliable disease subgroups. For hierarchical clustering, genes were clustered via Pearson correlation, while samples were clustered using Spearman’s rank correlation with average linkage criterion. All analyses were conducted in R (version 3.1.2). A total of 4952 transcripts that passed initial filtering criteria were used in network analysis. Samples collected and processed from women recruited with M. genitalium monoinfection, or with M. genitalium co-infection were excluded from these transcriptional analyses because they were too few to yield conclusive results.
Differential network analysis and module preservation analysis
Using WGCNA package in R, the analysis was performed as described previously (31, 32). Briefly, to construct Weighted Gene Co-expression Network, a similarity co-expression matrix was calculated with Pearson’s correlation for all genes, and transformed into matrices of connection strengths using a power function. The power of β = 9 were chose to obtain a scale-free network in this study. Hierarchical average linkage clustering based on topology overlap was used to identify gene co-expression modules, which were group of genes with similar patterns of expression. By using dynamic tree cutting, different number of clusters (modules) were obtained from the tree. The resulting modules contained genes that are densely interconnected. Two different networks, one using the reference groups and the other using the test group, were constructed. Then differential network analysis was conducted to identify genes and pathways that were differentially connected and differentially expressed between reference and test networks. Differential connectivity DiffK was defined as the difference between intramodular connectivity in one network, scaled relative to the maximally connected gene in that network, and intramodular connectivity in the other network, similarly scaled (33). Differential expression for each gene between two networks was measured by Welch’s two-sample t test. A comparison matrix was constructed via a sector plot using DiffK as the x-axis and t statistics as the y-axis. To assign a significance level (p value) to a gene’s DiffK value or to its membership in a particular sector, 1000 random permutations were performed. The algorithm is described in detail elsewhere (31, 32) but briefly, the permutation test contrasts networks built by randomly partitioning all samples into two groups while maintaining the group sample sizes as cases and controls, and reconstructs each network accordingly. This permutation process was repeated 1000 times (no.perm = 1000), each time noting the number of genes that could be found in each of the m = 8 sectors as . Similarly, sector counts for unpermuted, observed data were noted as . Then, we found a value Nm for each sector representing the number of iterations for which . The empirical p-value for each sector was found as . Subsequent functional enrichment analysis was restricted to networks detected within statistically significant sectors. Modules (clusters of highly correlated genes) in significant sectors that were differentially expressed and/or connected were analyzed as input for functional annotation as previously described (21, 31, 34). WGCNA module Preservation R function was conducted for module preservation analysis (35). A composite preservation statistic Zsummary (35, 36) was used to determine if a module present in a reference network (clinical PID) was represented in an independent test network (subclinical PID). Thresholds for Zsummary was based on simulation studies described previously (36).
Functional annotation of highly differentially expressed and/or connected modules
Biological annotation and function of genes that were highly different between two groups were explored with Ingenuity Pathway Analysis (IPA) using Fisher’s exact test. Significance of annotated pathways e.g. top 10 Pathway analysis (p-value <0.05) were adjusted by Bonferroni correction. To gather detailed information on cellular and functional components and to confirm our IPA pathway findings, we used blood modular transcriptional repertoires predefined by Chaussabel et al. (37). ANOVA was used to compare expression of cell-specific genes among multiple groups, two-sided Student’s t-Test for pairwise comparison followed significant ANOVA tests. P value for testing multiple cellular modules was adjusted by Bonferroni correction. The list of probes that make up the modules were retrieved online (37).
Analysis of cervical cytokine secretion and bacterial load
Values for cytokines in cervical secretions were log2 transformed. ANOVA was used to compare cytokine levels among multiple groups, two-sided Student’s t-Test for pairwise comparison followed significant ANOVA tests. Normality of the residuals and equality of variances were checked by Shapiro–Wilk test and Fligner-Killeen’s test respectively. Chi-square test was used to measure association between bacterial burden (high versus low) and ascension outcome. All Statistical analyses were performed using R version 3.1.1.
Study Approval
Our study complied with the guidelines of the Declaration of Helsinki. The Institutional Review Boards for Human Subject Research at the University of Pittsburgh and the University of North Carolina approved the study and each patient gave written informed consent prior to initiation of study procedures.
RESULTS
A Blood PID Transcriptional Profile is Driven by Pathogen Invasion of the Endometrium
Women included in this study were drawn from two cohorts of women at high risk for STI; women with a clinical diagnosis of PID (N=231) who were enrolled in an antibiotic treatment trial (NCT01160640), and asymptomatic women who screened positive for CT or who reported exposure to a sex partner with STI (N=247) (11). Although whole blood mRNA array data were available for all of the women in both cohorts, we restricted this analysis to 68 participants with histologically evaluable endometrial tissue (Table 1). Women were categorized according to clinical, microbiological, and histologic diagnoses (normal histology or acute or chronic endometritis) for all analyses (Table 1 and Fig. S1).
Table 1.
Characteristics of participants in this study
| All | PID+ | PID− | PID+ | PID+ | PID+ | PID− | Subclinical PID | ||
|---|---|---|---|---|---|---|---|---|---|
| GC and/or
CT endometrial infection |
CT or CT &
GC cervical infection |
CT endometrial infection |
no STI | no STI | no STI | CT endometrial infection |
P value* |
||
| chronic endometritis |
no endometritis |
acute endometritis |
chronic endometritis |
no endometritis |
no endometritis |
chronic endometritis |
|||
| Number of participants | n=68 | n=14 | n=16 | n=5 | n=7 | n=8 | n=12 | n=6 | |
| Age, y, median (range) | 21 (16-37) | 22 (18-37) | 20 (18-35) | 20 (19-24) | 23 (21-32) | 25 (16-32) | 22 (18-31) | 20 (18-25) | 0.589 |
| Race/ethnicity | |||||||||
| African American | 37 (54) | 8 (57) | 9 (56) | 3 (60) | 3 (43) | 4 (50) | 6 (50) | 4 (67) | 0.743 |
| White | 16 (24) | 4 (29) | 2 (13) | 1 (20) | 4 (57) | 2 (25) | 1 (8) | 2 (33) | |
| Hispanic or Latino | 3 (4) | 1 (7) | 1 (6) | 0 (0) | 0 (0) | 0 (0) | 1 (8) | 0 (0) | |
| Multiracial | 12 (18) | 1 (7) | 4 (25) | 1 (20) | 0 (0) | 2 (25) | 4 (33) | 0 (0) | |
| Marital status | |||||||||
| Married | 3 (4) | 0 (0) | 0 (0) | 0 (0) | 2 (29) | 1 (12.5) | 0 (0) | 0 (0) | 0.014 |
| Living with partner for ≥4 months | 13 (19) | 1 (7) | 3 (19) | 1 (20) | 4 (57) | 1 (12.5) | 3 (25) | 0 (0) | |
| Single | 52 (76) | 13 (93) | 13 (81) | 4 (80) | 1 (14) | 6 (75) | 9 (75) | 6 (100) | |
| Education level | |||||||||
| Less than high | 14 (21) | 2 (14) | 5 (31) | 1 (20) | 2 (29) | 2 (25) | 0 (0) | 2 (33) | 0.531 |
| school graduate | |||||||||
| High school graduate or GED degree | 24 (35) | 5 (36) | 7 (44) | 2 (40) | 0 (0) | 2 (25) | 5 (42) | 3 (50) | |
| Vocational training | 4 (6) | 2 (14) | 0 (0) | 0 (0) | 1 (14) | 1 (12.5) | 0 (0) | 0 (0) | |
| Some college | 21 (31) | 3 (21) | 4 (25) | 1 (20) | 4 (57) | 3 (37.5) | 5 (42) | 1 (17) | |
| College graduate | 5 (7) | 2 (14) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 2 (17) | 0 (0) | |
| Health Insurance | |||||||||
| Private | 12 (18) | 3 (21) | 1 (6) | 2 (40) | 0 (0) | 1 (12.5) | 3 (25) | 2 (33) | 0.549 |
| Medicaid | 37 (54) | 7 (50) | 8 (50) | 2 (40) | 6 (86) | 6 (75) | 6 (50) | 2 (33) | |
| Other | 3 (4) | 0 (0) | 1 (6) | 1 (20) | 0 (0) | 0 (0) | 1 (8) | 0 (0) | |
| None | 16 (24) | 4 (29) | 6 (38) | 0 (0) | 1 (14) | 1 (12.5) | 2 (17) | 2 (33) |
ANOVA was used to compare ages among groups, and Fisher’s two-sided exact test was used for categorical variables. The cutoff P value=0.05/5=0.01 after Bonferroni multiple testing correction.
The majority of participants were young (median age, 21 years), African American, single, with some high school education and insured by Medicaid (Table 1). Four women (6%) reported having an intrauterine device and 13 (21%) were receiving hormonal contraceptive therapy. Trichomonas vaginalis infection was rare (8%). Age, race, education, insurance, and contraceptive status did not differ significantly between the groups. We conducted unsupervised hierarchical clustering using 4952 transcripts that passed filtering criteria to determine if the profiles generated for participants grouped according to pathogen and/or extent of disease (Fig. 1A), and to obtain an overview of gene expression patterns. We included women with STI limited to their cervix as controls in order to enhance detection of changes associating with disease rather than infection. Transcriptional response profiles of 11 of 14 women with GC and/or CT-induced PID and chronic endometritis were tightly clustered. Two of 7 women with clinical PID and chronic endometritis, but negative for GC and CT infection, also grouped in this cluster. In contrast, women with CT-induced clinical PID and acute endometritis (N=5) did not cluster with women with GC and/or CT PID and chronic endometritis but were interspersed amongst cervically-infected (N=16) and uninfected (N=12) women without pelvic pain who had normal histology. Women with clinical symptoms of pelvic pain who were negative for GC and/or CT without endometritis (N=8) were interspersed among the subgroups with normal histology indicating that pelvic pain in the absence of infection and cervical infection alone did not alter the blood transcriptional profile compared to asymptomatic uninfected women with normal histology.
Fig. 1. Women with GC and/or CT PID and chronic endometritis exhibit a distinct whole-blood transcriptional signature.
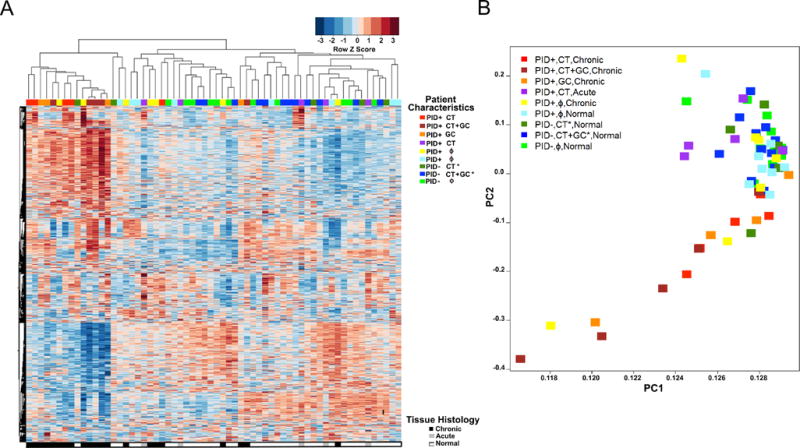
(A) Unsupervised hierarchical clustering reveals expression patterns of 4952 genes in women with GC and/or CT PID and chronic endometritis as well as six other clinical groups indicated in the graph key and represented in the colored bars at the top of the heat map. Heat map rows indicate genes; columns, individual participants. Clinical groups were determined by symptoms, infection status and endometrial histology. CT*, CT+GC* indicate infection was localized to the cervix. (B) Principal component (PC) analysis of the variance in mRNA expression of the participants depicted in 2A, using the same color scheme with each colored square depicting one subject. The x-axis represents the first principal component, PC1, which accounts for the largest variance of mRNA expression, and the y axis, PC2, explains the second largest variance.
Principal component analysis separated participants into a pattern consistent with that observed via hierarchical clustering (Fig. 1A-B). Women with GC and/or CT PID and chronic endometritis grouped together, and were mostly separated from women with acute endometritis or normal histology regardless of pain or infection status. Women with PID and chronic endometritis without GC or CT that clustered with women with GC and/or CT chronic endometritis in the heat map (Fig. 1A), also clustered with this group by PCA (Fig. 1B). Thus, a disease-related gene expression profile or endotype occurs in women with chronic endometritis and this endotype is largely pathogen driven.
Women with STI-induced PID and Chronic Endometritis Exhibit Significant Differences in Gene Connectivity and Expression Compared to Women with Normal Histology Who Have Cervical Infection or Pelvic Pain
Initial comparisons were made between responses of infected cases and women who were not infected with GC or CT, reported no pain and had normal histology (uninfected controls, N=12) (Fig. S1 and Fig. 2A-B). WGCNA identified differentially connected (measured by DiffK) and/or differentially expressed genes (measured by t-test statistic) with significance level (p value) assigned by permutation test. Plotting these values revealed how differences in connectivity related to differences in gene expression (Fig. 2). Eight sectors with high absolute values of DiffK (> 0.4) and/or t-statistic values (> 1.96) are shown in Fig. 2A (observed data) and Fig. 2B (permuted data). Based on 1000 random permutations, genes in sectors 2, 3, 5 and 6 were significantly differentially connected and/or expressed (p<0.001) when infected cases were compared with uninfected controls. We detected sub-groupings of genes with highly correlated expression, termed modules, within these sectors. Individual modules are designated by color (gray denoting differentially expressed genes outside modules). We derived 13 modules for women with GC and/or CT PID (Fig. 2) using hierarchical clustering.
Fig. 2. Women with GC and/or CT PID and chronic endometritis exhibit significant differences in gene connectivity and expression when compared to other clinical groups.
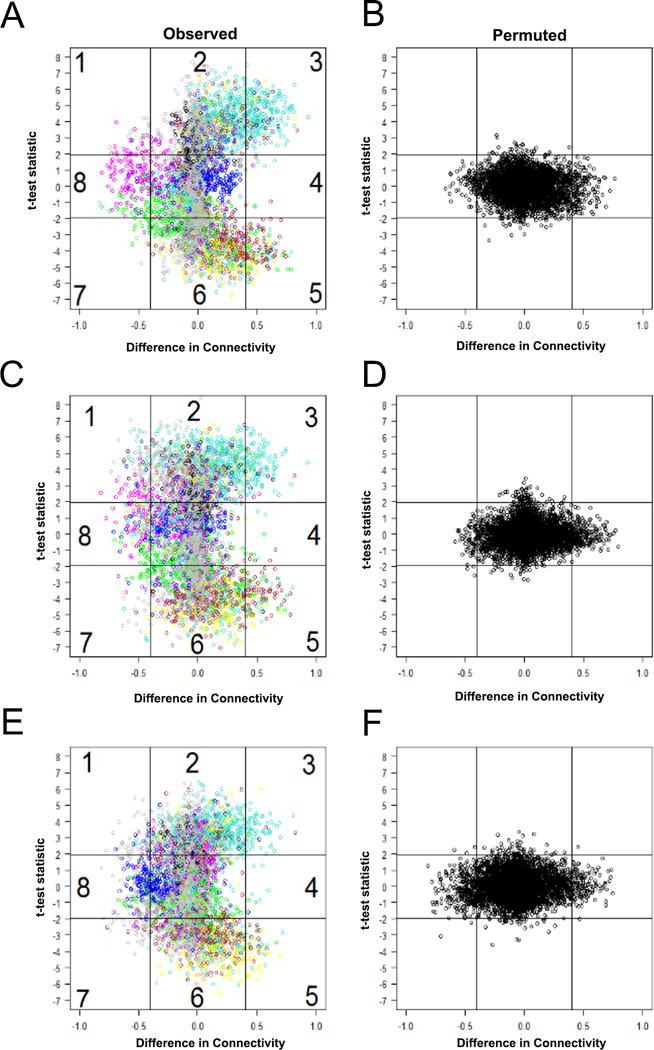
Scatterplot of differences in connectivity, DiffK vs. expression level, t statistic, for transcriptional responses of women with GC and/or CT PID and chronic endometritis versus: (A-B), responses of asymptomatic women who were uninfected with GC and/or CT with normal histology; (C-D), responses of asymptomatic women with GC and CT, or CT cervical infection with normal histology; or (E-F), women with clinical PID who were not infected with GC and/or CT with normal histology. (A, C, and E), observed data; (B, D, and F), permuted data. Horizontal lines depict sector boundaries based on t-statistic values; vertical lines depict boundaries based on DiffK. Numbers mark eight sectors. Colors reflect individual modules. Sectors 2, 3, 5, and 6 were significantly different, P < 0.005 for A versus B, C versus D, and E versus F. Sectors 1, 4, 7 and 8 were not significantly different for any group comparison. P value derived after 1000 permutations.
The mRNA profiles of women with GC and/or CT PID and chronic endometritis (infected cases, N=14) were compared to women who had cervical infection with CT or CT and GC without pelvic pain and had normal endometrial histology (infected controls, N=16) to identify disease-associated genes (Fig. S1 and Fig. 2C-D). Gene expression and/or connectivity levels in sectors 2, 3, 5 and 6 were significantly different (p<0.001) confirming that upper tract infection and inflammation induced a distinct transcriptional response that was detected in peripheral blood. Finally, we compared infected cases to women who had symptoms consistent with PID, but who were not infected with GC and/or CT and had normal histology (N=8) (Fig.S1 and Fig. 2E-F). Expression and/or connectivity of genes mapping to sectors 2, 3, 5 and 6 were also significantly altered (p<0.001). Thus, a unique blood transcriptional profile distinguished women with pelvic pain and endometrial inflammation caused by recognized sexually transmitted pathogens from women with pelvic pain of unknown cause without endometrial inflammation.
The STI-induced PID and Chronic Endometritis Blood Transcriptional Profile is Characterized by Overexpression of Myeloid Cell and Interferon Genes with Suppression of T cell, Protein Synthesis, and Mitochondrial Oxidative Phosphorylation Genes
Ingenuity Pathway Analysis (IPA) using all modular genes that mapped to sectors 2, 3, 5 and 6 when comparing infected cases to infected controls (Fig. 2C-D) was used to identify disease-associated pathways. The top IPA canonical pathways significantly enriched for genes in sectors 2 and 3 (up-regulated) within the turquoise co-expression module included multiple genes involved in neutrophil and monocyte activation and signaling (Fig. 3A and Table S1). Pathways that were significantly upregulated after multiple testing correction are listed in Table S2. Transcription of multiple TLRs and the TLR adaptor molecule, MyD88 was up-regulated, as was expression of transcription factors and kinases important for TLR, cytokine, and growth factor receptor signaling. Transcription of inflammasome-driven interleukin-1β (IL1B) and its receptor molecule, IL1R2 was also elevated in infected cases versus infected controls. Further evidence of inflammasome activation was suggested by detection of increased transcripts encoding inflammasome-associated molecules (38). Expression of molecules important for inflammatory cell migration and adhesion, cytoskeleton remodeling in response to extracellular stimulus and phagocytosis, and transcription of oncostatin M (OSM), a member of the IL-6 family that facilitates leukocyte adhesion and rolling was enhanced. Genes for interferon (IFN) signaling, including IFIT3, OAS1, PIAS1, IFNGR2, IRF9, IFNGR1, IFI35, PSMB8, IFNAR2, TAP1, IFITM3, IFIT1, IFI6, STAT1, IFNAR1, IFITM1 (p = 5.01E-15, Ratio = 4.44E-01), were also detected in sectors 2 and 3 but did not assort with the turquoise module, indicating that expression levels were not consistent across all PID cases. Nevertheless, IFN signaling was the pathway with the lowest P value and highest enrichment ratio after multiple testing correction (Table S2).
Fig. 3. Top differentially expressed canonical pathways in women with GC and/or CT PID and chronic endometritis compared to asymptomatic women with cervical infection only and no endometritis.
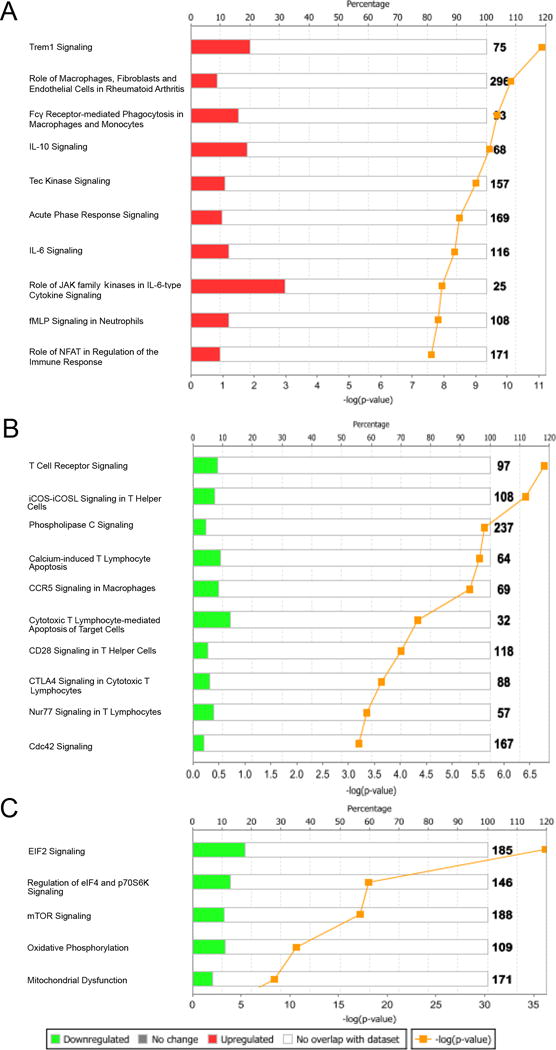
IPA showed (A) monocyte and neutrophil signaling pathways were upregulated, while pathways associated with (B) T cell signaling, (C) protein synthesis, and mitochondrial respiration were downregulated.
The top IPA canonical pathways significantly enriched for genes in sectors 5 and 6 and assigned to the yellow co-expression module (Fig. 2C) (down-regulated in GC and/or CT PID with chronic endometritis) are depicted in Fig. 3B and include genes encoding components of CD3 and CD8 T cell co-receptors, and genes for kinases involved in T cell signaling (Table S1).
The top IPA canonical pathways significantly enriched for genes in sectors 5 and 6 and assigned to the brown co-expression module (Fig. 2C) (down-regulated in GC and/or CT PID with chronic endometritis) are depicted in Fig. 3C and include genes encoding multiple eukaryotic translation factors and ribosomal proteins; all important for protein synthesis. In addition, expression of genes encoding proteins essential for mitochondrial oxidative phosphorylation was decreased (Table S1). Pathways significantly downregulated after multiple testing correction are listed in Table S3. These pathways, with reduced expression of genes encoding T cell structural components, suggested that the STI-induced PID transcriptional profile reflected altered cell number and differential gene activation in response to immune signaling.
Modular Repertoire Analysis of CT and/or GC PID Confirms Overexpression of Innate Immune Response Genes and Suppression of Adaptive Immune Response Genes
To determine the extent to which the STI-induced PID blood transcriptional profile was influenced by relative abundance of leukocyte subsets, we performed modular repertoire analysis to examine genes specific to these cells, applying an analytical framework of modular repertoires which reflect coherent functional relationships and are coordinately expressed in multiple disease datasets (37, 39) to all 2779 genes in sectors 2, 3, 5 and 6 (Fig. 4). Women with chronic endometritis and PID due to CT and/or GC displayed significant overexpression of genes of inflammatory modules (M3.2, M4.2, M4.6, M4.13, M5.1, M5.7), cell death (M6.13), and IFN (M1.2, M3.4, M5.12) with slight increases in genes within the platelet (M1.1), monocyte (M4.14), and neutrophil (M5.15) modules (Fig. S2). Modular analysis also detected decreased expression of T cell (M4.1, M4.15) module genes, consistent with detection of decreased transcripts for T cell structural molecules and T cell activation genes determined by WGCNA. Furthermore, gene expression in cell cycle (M4.7), mitochondrial respiration and stress (M5.10, M6.2), and protein synthesis (M4.3, M4.5, M5.9) modules was decreased (Fig. S2), as previously observed by WGCNA. We detected minor decreases in representation of genes within the plasma cell module (M4.11) in the circulation, although these cells define chronic endometritis in tissue. B cell (M4.10), and cytotoxic/NK cell (M3.6) gene expression was also decreased.
Fig. 4. Whole-blood transcriptional modular analysis of CT and/or GC PID reveals overexpression of innate immune response genes and suppression of adaptive immune response genes.
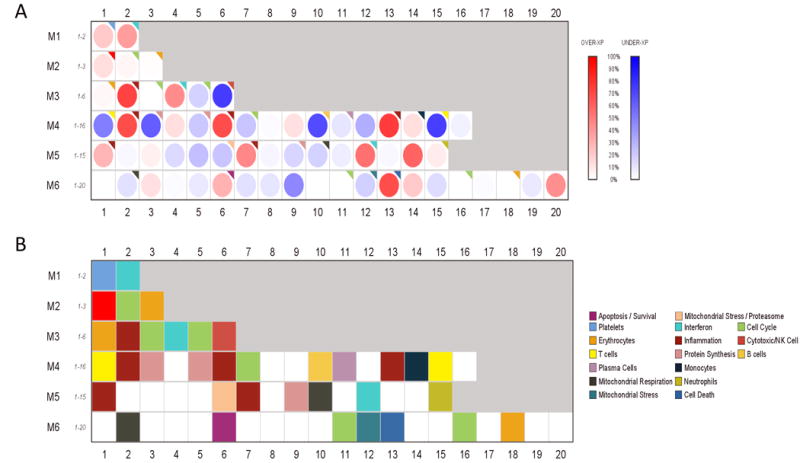
(A) Gene expression of CT and/or GC PID versus cervical infection only and no endometritis mapped within a pre-defined modular framework (37, 39). Spot intensity (red, increased; blue, decreased; white, no difference) indicates transcript abundance. (B) Functional interpretations determined by unbiased literature profiling shown by color-coded grid.
The Profile of Women with PID and Chronic Endometritis due to GC is Similar to Women with GC/CT Co-infection But Interferon Responses Distinguish Women Singly Infected with CT
To resolve the relative contributions of GC and CT to this profile, we compared the responses of co-infected women to women infected with a single pathogen (Fig. S1). Comparing sector plots of GC/CT co-infected patients to those with GC alone did not identify genes with statistically significant differences in connectivity and/or expression (Fig. 5A-B).
Fig. 5. Gene response networks are similar in women with PID and chronic endometritis due to GC/CT co-infection or GC alone, but differ from women with CT alone.
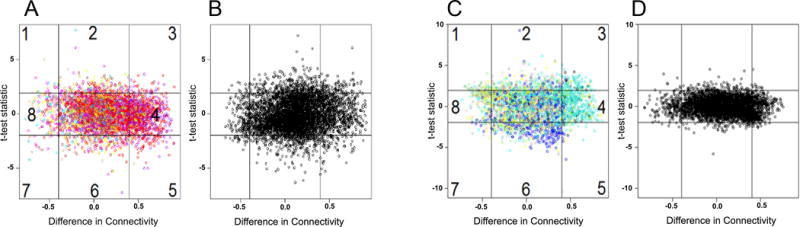
(A-B) Scatterplot of differences in connectivity, DiffK vs. expression level, t statistic, for transcriptional responses of women with GC and/or CT PID and chronic endometritis versus women with PID and chronic endometritis infected with GC alone and (C-D) CT alone. (A and C), observed data; (B and D), permuted data (×1000). Horizontal lines depict sector boundaries based on t-statistic values, and vertical lines depict boundaries based on DiffK. Numbers mark eight sectors. Colors reflect individual modules. No sectors were significantly different for A versus B. Sectors 3 and 6 were significantly different for C versus D, P = 0.01 for both, with all other sectors being not significantly different. P value derived after 1000 permutations.
In contrast, sector comparisons of women with PID and chronic endometritis due to GC/CT co-infection and women with CT alone revealed up-regulated (sector 3) and down-regulated (sector 6) gene expression (Fig. 5C). Slight but statistically significant transcriptional increases were detected for genes involved in Rho signaling, guanine nucleotide binding proteins and various actin and myosin related proteins (Fig. S2A and Table S4), suggesting co-infection drives increased activation of signaling pathways related to cellular migration.
Genes in sector 6, (Fig. 5C) that were transcriptionally down-regulated with co-infection are depicted in Fig. S2B and molecules are listed in Table S4. The EIF2 signaling pathway includes ribosomal protein genes and eukaryotic translation initiation factors. Additionally, transcripts important for Class II antigen presentation to helper T cells and T helper cell signaling were decreased in GC/CT co-infected women compared to women with CT only. Finally, multiple genes for proteins involved in type I IFN signaling were decreased in co-infected women compared to women infected with CT alone.
Modular analysis confirmed that increased expression of IFN-related genes (M1.2, M3.4 and M5.12) occurred exclusively in women with PID and chronic endometritis due to CT alone (Fig. 6). The most profound increases were detected in IFN modules 1.2 and 3.4. Genes in M1.2 are induced more by type I than type II IFNs, while genes represented in M3.4 and M5.12 are similarly induced by type I and type II IFNs (40). These data indicate that CT elicits strong type I and type II IFN responses that are not induced in women with GC monoinfection and are ablated in women with GC/CT coinfection. For every module examined, other than IFN, coinfection intensified the expression change when compared to women with monoinfection. We noted significant increases in expression of genes related to cell death and inflammatory pathways and reductions in genes associated with mitochondrial respiration. Cell cycle gene expression was most reduced in co-infected women (Fig. 7A). Cellular modules revealed that co-infection associated with the most profound reductions observed in adaptive immune cell genes and genes associated with NK cells. Genes associated with myeloid cell modules were not different among the subgroups (Fig. 7B).
Fig. 6. Interferon signaling responses in the STI-induced PID transcriptional signature are driven by CT infection.
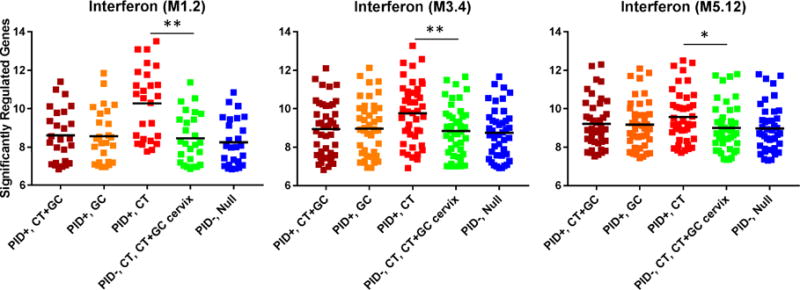
Median expression levels of interferon signaling genes among women with PID and chronic endometritis due to CT+GC, GC only, and CT only; and women without PID or endometritis with cervical infection due to CT or CT+GC, and asymptomatic uninfected women. Dots represent the median expression value for each individual transcript (log2 transformed expression) in all five subject groups in interferon modules (M1.2, M3.4, and M5.12) while the black bar indicates the group mean. Pairwise comparisons followed significant ANOVA after Bonferroni correction for all three modules. Genes related to interferon were significantly overexpressed in women with PID and chronic endometritis induced by CT alone compared to women with cervical infection due to GC or CT+GC; (p < 0.001 for M1.2, p = 0.004 for M3.4, p = 0.045 for M5.12); whereas expression levels for women with PID and chronic endometritis due to CT+GC or GC alone were not different than infected cases or uninfected controls. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Fig. 7. CT/GC co-infection leads to the most robust enhancement of genes within cell death and inflammation modules, and most significant decrease in lymphocyte and NK cell genes compared to other patient subgroups.
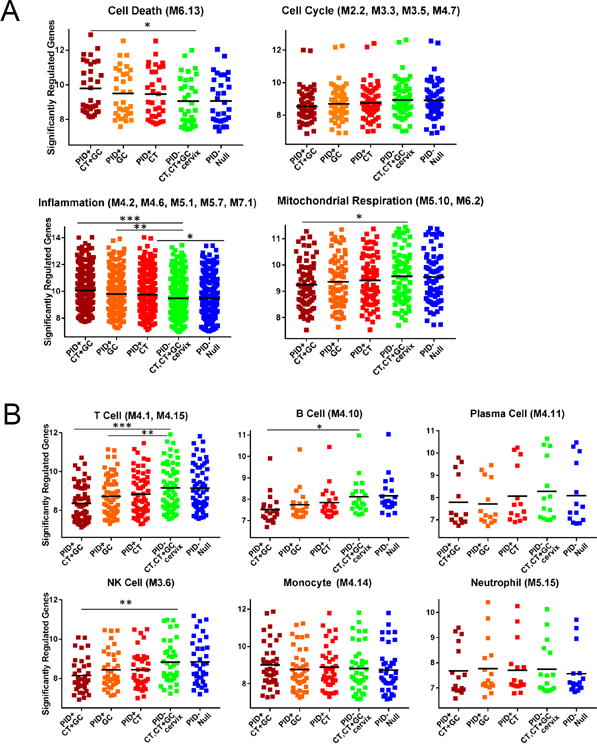
(A). Different median expression of genes within cell death, cell cycle, inflammation and mitochondrial respiration modules among indicated clinical subgroups. (B) Different median expression of genes within T cell, B cell, plasma cell, NK cell, monocyte and neutrophil modules. Dots represent the median expression value for each individual transcript (log2 transformed expression) while the black bar indicates the group mean. (A) and (B) subgroups include women with chronic endometritis and CT/GC co-infection, GC only, CT only, asymptomatic cervical CT+ GC infection, and no infection. ANOVA p< 0.05 after Bonferroni correction for cell death, inflammation, mitochondrial respiration, T cells, B cells, and NK cells modules. Pairwise comparisons after ANOVA were conducted via Student’s t-Test in these 6 modules. *, p < 0.05, ** p < 0.01, *** p < 0.005.
Transcriptional Profiling Reveals Acute Endometritis Due to CT Represents an Early Stage in the Course of PID
Marked differences in expression levels were observed for genes in sectors 2, 3, 5, and 6 after permutation when co-expression modules were compared between women with CT PID and chronic endometritis and women without pelvic pain who had cervical CT infection only and no endometritis (Fig. S3A). In comparison, when women with CT-induced PID and acute endometritis were compared to women with cervical CT infection, (Fig. S3A), sectors 2 and 6 which reflect differential expression levels were not significant although genes in sectors 3 and 5 exhibited statistically significant connectivity and altered gene expression after permutation (p < 0.001), while genes in sector 4 were significantly increased in connectivity (p < 0.001). Modular analysis did not detect overexpression of myeloid cell genes or suppression of genes associated with protein synthesis, mitochondrial oxidative phosphorylation, or T-cell specific genes. However, we detected a nascent IFN response in women with acute endometritis (Fig. S3B and Table S5) consistent with the presence of neutrophils in their endometrial tissue, a hallmark of early inflammation.
Validation of Disease Associated Modules in an Independent Cohort of Women with CT-induced Subclinical PID
We did not have sufficient study participants with clinical PID and biopsy-confirmed endometrial infection and chronic endometritis to serve as an independent validation set. As an alternative, we selected a new group of study participants comprising six asymptomatic women with endometrial chlamydial infection and chronic endometritis. We anticipated that STI PID associated modules found in women with clinical PID associated with GC and/or CT-induced chronic endometritis (N=14 original participants) would be reproduced in this independent cohort of women with subclinical STI PID if they are involved in development of STI-induced upper genital tract disease. Using a method implemented in the WGCNA package to calculate Zsummary preservation scores (36), we tested for module preservation between women with biopsy confirmed clinical PID and women with biopsy confirmed subclinical PID (Fig. 8A). Nine of 13 modules were highly conserved with Zsummary scores above 10, including all three disease associated modules [turquoise (myeloid cell activation and cell death pathways were increased), yellow (T cell activation pathways were decreased), and brown (protein synthesis and mitochondrial respiration pathways were decreased) and the permutated P values were smaller than the Bonferroni-corrected threshold p=0.05/13=0.0038); the last 4 modules were weakly to moderately preserved.
Fig. 8. Conservation of PID-associated disease modules were detected in the mRNA transcript profiles of women with CT-induced subclinical PID.
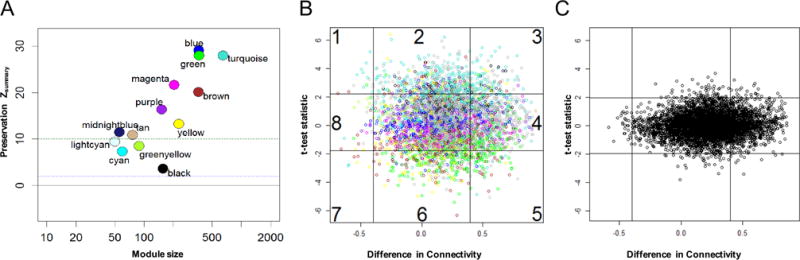
(A) Zsummary statistic was computed for each module as an overall measure of its preservation with regards to density and connectivity. Circles, (colors corresponding with the previously characterized co-expression modules identified via WGCNA) denote Zsummary preservation scores. A Zsummary score < 2 indicates no preservation, a score > 10 (green dotted line) indicates strong preservation and scores of 2-10 indicate weak to moderate evidence of preservation. Observed (B) and permuted (C) scatterplots of differences in connectivity, DiffK vs. expression level, t statistic, for transcriptional responses of women with subclinical PID associated with CT infection compared to women with cervical STI and and normal histology. p = 0.01 for sector 2; p = 0.02 for sector 3; p = 0.04 for sector 5; and p = 0.02 for sector 6. All other sectors were not significant. P value derived after 1000 permutations.
We also conducted differential network analysis between women with subclinical PID and women who had cervical infection with CT or CT and GC without pelvic pain and normal endometrial histology (N=16), infected controls that were previously used to identify disease-associated genes. Sector plots (Fig. 8B-C) were generated based on 1000 random permutations. Genes in sectors 2, 3, 5 and 6 were significantly differentially connected and/or expressed (p<0.05). To examine whether the disease related modules we had associated with clinical CT PID were reproduced in subclinical CT PID, we used the module colors previously defined in women with GC and/or CT PID and chronic endometritis (N=14 original participants). Not only were the three disease associated modules maintained (Table S6), but functional annotation detected up-regulation of IFN pathway genes in women with CT-induced subclinical PID (p = 1.8E-4 which is significant after Bonferroni multiple testing correction).
RNA in situ Hybridization Reveals IFN-β mRNA in Endometrial Tissues of GC and/or CT-infected Women with Endometritis
Since CT infection is limited to the genital tract mucosa, we hypothesized that the likely source(s) of the type I IFN signature that we detected in the peripheral blood of women with CT-induced PID were neutrophils or monocytes that gained access to the vascular system after engulfing IFN-β secreting CT-infected cells or adjacent, activated epithelium. Endometrial tissue sections from patients with GC and/or CT-induced endometritis and uninfected women with normal histology were probed by RNA-in situ hybridization for both IFN-β message and CT ribosomal RNA (rRNA) (Fig. 9). In five of five tissue sections from women with CT-induced endometritis where multiple microscope fields were positive for CT rRNA, we observed multiple epithelial and stromal cells staining positively for IFN-β mRNA. In sections from an additional five patients with CT-induced endometritis, where CT rRNA was not detected in the tissue sections available, IFN-β mRNA-specific staining was detected, albeit it sparsely, in epithelial and stromal cells of two patients (not shown). Sections from five women with GC/CT-induced endometritis stained positively for CT rRNA, but cells positive for IFN-β transcripts were detected in only two of the five patients in this group. Two of four tissue sections from patients with GC-induced endometritis revealed rare cells positive for IFN-β mRNA. Tissue sections from fifteen uninfected women without endometritis were negative for CT rRNA and/or IFN-β mRNA.
Fig. 9. Endometrial tissues from women with GC and/or CT contain epithelial and stromal cells expressing IFN-β mRNA.
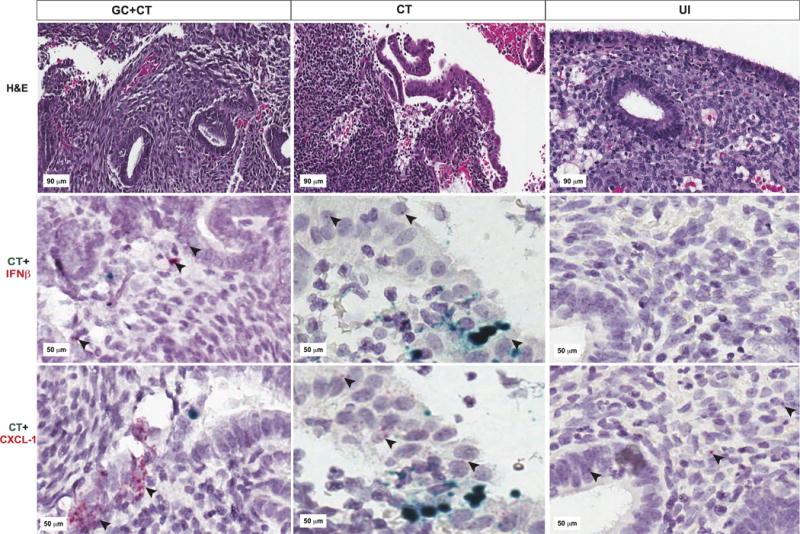
Representative images of endometrial tissue sections obtained from patients with endometritis due to GC/CT (first column); CT (2nd column); or from uninfected patients without endometritis (3rd column) are pictured. Tissue sections cut from the same endometrial biopsy were used for hematoxylin & eosin staining (top row); RNAScope for CT rs16 rRNA (green) and IFN-β mRNA (red indicated by black arrows) (middle row); or RNAScope for CT rs16 rRNA (green) and CXCL-1 mRNA (red indicated by black arrows) (bottom row).
Additional tissue sections from the same patients were costained for CT rRNA and for mRNA of the neutrophil chemokine, CXCL-1/KC. Although the cytokine, IL-1β, is a key inducer of CXCL-1 during inflammation, CXCL-1 is also produced by endometrial epithelial cells in response to vascular endothelial growth factor and is important for decidual vascularization and implantation (29). Tissue sections from uninfected patients were all weakly positive for CXCL-1 mRNA. In contrast, all of the tissues but one from women with GC and/or CT-induced endometritis were moderately or strongly positive for CXCL-1 mRNA. The majority of CXCL-1 expression was detected in epithelial cells (Fig. 9).
Cervical Secretions of Women with Endometrial CT Infection Contain Higher Levels of the Type I IFN-induced Chemokine CXCL-10 Compared to Women with Cervical CT Infection Only
To expand our investigation of the extent to which the elevated type I IFN response correlated with genital inflammatory responses and promotion of CT disease, cervical secretions collected from women recruited into the non-PID, T cell response against Chlamydia (TRAC) cohort were assayed for immune modulators. We used quantitative PCR to estimate CT burden in cervical swab eluates that remained after diagnostic testing. Although women with a cervical CT burden > 104 CT copies/swab had 3-fold greater odds of having endometrial infection (p = 0.002), a subgroup (54 of 110 women) with CT burdens > 104 were negative for endometrial infection. This suggested a suboptimal protective response in women with high cervical CT burden with ascending infection when compared to women with infection limited to the cervix. Although IFN-α, IFN-β and IFN-γ were undetectable or extremely low in cervical secretions of all patients (data not shown), CXCL-10 was significantly higher in secretions of endometrially-infected women when compared to women with cervical infection only, (p = 0.004) (Fig. 10). In contrast, although not statistically significant, cytokines IL-12p40 (p = 0.199) and IL-12p70 (p = 0.067) that induce production of IFN-γ were elevated in secretions of women with cervical CT infection only. The proinflammatory cytokines, TNF-α and IL-1β, and the Th17-related cytokine IL-17-α, were significantly increased in both high cervical burden groups when compared to participants with low cervical burden and uninfected women, but were not different between women with endometrial infection versus cervix only (Fig. 10). Our prior studies revealed that CXCL-10 production is primarily and directly dependent on IFN-β during in vitro chlamydial infection of murine cells and during in vivo murine chlamydial genital infection (41, 42). Thus, detection of higher levels of CXCL-10 in the cervical secretions of CT-infected women with endometrial infection linked the type I IFN response to increased extent of CT infection.
Fig. 10. Interferon inducible protein-10 (IP-10/CXCL-10) is increased in cervical secretions of women with endometrial infection and IL-12 is higher in women with cervical infection only.
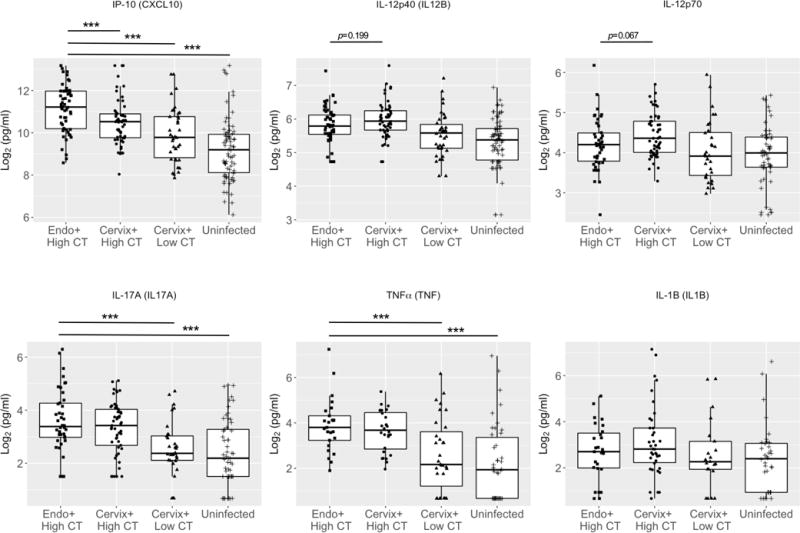
Box and whisker plots were used to display the distribution of IP-10, IL-12p40, IL-12p70, IL-17, TNFα, and IL-1β levels in cervical secretions of women enrolled in TRAC. The boundaries of the box indicate the 25th and 75th percentiles; the line within the box marks indicates the median; whiskers indicate the 10th and 90th percentiles. Data were log2 transformed prior to logistic regression analysis. Endo+ = PCR detection of CT in the endometrium. Cervix+ = PCR detection of CT only in the cervix. High CT = > 104 CT copies/swab; Low CT = < 104 CT copies/swab; Uninfected = no detection of CT copies/swab. Cervical CT burden was estimated by qPCR for CT 16s rDNA. Group means for each cytokine were analyzed using ANOVA. All cytokines with the exception of IL-1β were significant via ANOVA p < 0.05. Pairwise comparisons after ANOVA were conducted via Student’s t-Test. *** p < 0.005.
DISCUSSION
We determined that among women with a clinical diagnosis of PID, those with GC and/or CT endometrial infection and chronic endometritis had a blood borne transcriptional profile that was easily distinguished from those without upper tract infection and inflammation using microarray analysis. Principal component analysis confirmed that GC and/or CT infection-induced PID and chronic endometritis resulted in a blood endotype distinct from women with clinical PID who had normal histology and were not infected. Although 2 of 7 women with PID and chronic endometritis not associated with active GC and/or CT infection clustered with our infected cases, 5 of 7 did not. None of these women had M. genitalium or T. vaginalis and cultivation based evaluation of the endometrial samples did not yield other PID pathogens (data not shown). It is possible that these women had STI infection prior to enrollment and had residual endometritis. Six of seven had documented prior GC or CT infection and two had a history of PID. Consequently, while immune cells remained within the endometrial tissue, the systemic response was relatively quiescent. Thus, pain and/or chronic endometritis were insufficient to induce a distinct blood endotype in the absence of active GC or CT endometrial infection. Finally, transcriptional response profiles of asymptomatic women with GC or GC/CT infection limited to their cervix were indistinguishable from asymptomatic uninfected women, indicating cervical infection alone does not drive a mRNA response detectable in peripheral blood. Taken together, these data indicate that both upper tract GC and/or CT infection and histologic evidence of endometritis are required to elicit a distinct blood transcriptional profile.
Pathways activated in women with GC and/or CT induced PID and chronic endometritis included multiple innate immune response pathways that lead to migration and activation of monocytes and neutrophils, e.g., TLRs, NLRs, FcγRs, fMLPR and their downstream adaptors and transcription factors. Transcription of IL-1β and other genes involved in inflammasome activation were also increased. Cellular and immune repertoire modular analysis (39) confirmed genes involved in inflammation were increased in women with PID and chronic endometritis regardless of infection with GC, CT, or both. Modular analysis also revealed increased transcription of genes associated with cell death pathways, with intensity being highest in co-infected women.
These data agree with published results from in vitro and animal model studies of CT and GC. For example, lipooligosaccharide released from GC in the form of outer membrane blebs activates both NLRP3-induced IL-1β secretion and pyronecrosis of cells in vitro (43). Infection of human fallopian tube organoids ex vivo with CT revealed induction of IL-1 that led to direct cellular damage (44). The mouse model of CT infection reveals neutrophil infiltration correlates directly with oviduct tissue damage (45-47), and mouse GC infection induces Th17 cells that drive a significant influx of neutrophils (48). Although the IL-17 signaling pathway was not one of the top canonical pathways identified, we detected increased transcription for IL-17 receptor A (IL-17RA) (1.3 fold), which was highest when GC was present (data not shown). The association of these pathways with disease pathogenesis in animal models suggests that the models appropriately reflect responses ongoing in the endometrium of women with GC and/or CT PID that can be detected in the peripheral blood.
GC and/or CT PID also led to significant down-regulation of TCR/T cell signaling and protein synthesis pathways. This was confirmed by modular analysis, which revealed decreased transcription for T cell, B cell, NK cell, cell cycle and protein synthesis genes, all of which were most decreased in co-infected women. These data are consistent with observations in humans where active GC infection drives transient reductions in numbers of peripheral blood T cells (49) and neither GC or CT infection leads to an effective adaptive immune response, with high rates of reinfection being commonly observed after infection with either pathogen (50). However, it is possible that the decrease in abundance of genes associated with lymphocyte proliferation and activation resulted primarily from changes in T cell numbers in the peripheral blood as revealed previously in a transcriptional response study of patients with active tuberculosis (51).
Interferon response pathways are enhanced in the peripheral blood of women infected with CT alone and ablated in women co-infected with GC. The up-regulation of interferon genes within M3.4 and M5.12 in women singly infected with CT indicate enhanced transcription of genes that are induced by either type I or type II IFNs (40). Interferon-γ is a key protective mediator during CT infection, inhibiting chlamydial growth and replication, and IFN-γ activates phagocytic cells to kill GC. Thus, abrogation of this response by GC favors pathogen survival. Genes involved primarily in type I IFN signaling found in module M1.2 were significantly up-regulated in women with CT PID compared to women with cervical CT infection only. Additionally, we detected IFN-β mRNA in endometrial tissues of women with CT-induced chronic endometritis and higher levels of CXCL-10, an IFN-β-induced protein, in women with endometrial infection. Thus, as previously observed in patients with Mycobacterium tuberculosis (51) or M. leprae (52), the detection of type I IFNs in women with CT is associated with a greater extent of infection and disease. Although type I IFNs are generally beneficial during viral infection, they can play either a beneficial or detrimental role during bacterial infection (53). High and prolonged levels of type I IFN decreases the expression of IFNGR1 and IFNGR2 and inhibits IFN-γ induced activation of MHC class II expression, oxidative burst, and bactericidal activity in macrophages (54, 55). Using the mouse model of genital chlamydial infection, we determined that mice deficient in type I IFN signaling exhibited reduced bacterial burden and pathology, and an accelerated protective CD4 T cell response (42). This suggests that the type I IFN response suppresses the development of a protective response in women with CT infection leading to ongoing infection and enhanced disease.
Since cell sorting was not performed in our study, we were unable to identify the source of the IFN signature. Neutrophils were identified as the source of the IFN signature in persons with active tuberculosis (51) and these cells are candidates here given both M. tuberculosis and CT occupy an intracellular niche and because we detected neutrophil pathway activation. Although CT replicates within an intracellular protective vacuole, CT DNA can leak into the cytosol and interact with the cytosolic DNA sensor, cyclic GMP-AMP synthase (cGAS), resulting in the formation of 2′-3′-cGAMP that acts via the downstream signaling adaptor, Stimulator of IFN Genes (STING), to induce IFN-β production. Furthermore, CT-induced cyclic GMP-AMP migrates via gap junctions into adjacent uninfected cells to stimulate their expression of IFN-β (56). Macrophages exposed to gonococci in vitro are stimulated to express IFN-β via intracellular interaction of GC DNA with cGAS/STING post-invasion, and extracellularly via lipooligosaccharide interaction with the TLR4/TRIF pathway (57). Despite our inability to detect an IFN signature in the peripheral blood of GC or GC/CT-infected women with endometritis, RNA in-situ hybridization studies confirmed IFN-β transcription in endometrial tissue biopsies. However, endometrial cells positive for IFN-β were most consistently observed in microscopic fields of tissues positive for CT rRNA. The cytotoxicity of GC may prevent IFN-β-producing epithelial or stromal cells in the endometrium from surviving long enough to be detected in the peripheral blood via uptake by recirculating neutrophils or monocytes. In addition, neutrophils and monocytes that traffic to GC-infected endometrium and engulf GC directly, either fail to transcribe sufficient IFN-β or fail to egress into the bloodstream in sufficient numbers to produce a detectable type I IFN signature.
It is well recognized that GC impairs the induction of adaptive responses. GC induces apoptosis of antigen presenting cells (43) and inhibition of dendritic cell-mediated T cell proliferation (58). GC Opa proteins that bind CEACAM1 down-regulate proliferation of activated CD4 T cells and B cells (59, 60). We observed that in asymptomatic women with CT, co-infection with GC is a leading risk factor for ascending and recurrent CT infection (11). Finally, despite lack of detection of a systemic type I IFN response in GC/CT co-infected women, IFN-β mRNA was observed in infected endometrial tissues likely further promoting infection, disease, and hampering the development of adaptive immunity to CT.
To our knowledge, this is the first investigation to utilize a network-based systems biology approach to investigate the development of human PID due to STI. Balamuth, et al (61) performed blood transcriptional arrays on adolescent females with clinical PID and compared them to asymptomatic controls. They failed to identify canonical pathways related to PID, and identified few differentially expressed genes. Possible explanations for their observations include a lack of information regarding upper genital tract infection and inflammation in their PID patients and their use of uninfected healthy girls for controls. Our study included women with STI limited to their cervix as controls, which allows for detection of disease-associated changes in gene expression as opposed to infection-associated gene expression changes. Unique to our study is that clinical, microbiological, and histological data were available for multi-level in silico analysis from two unique cohorts comprised of young women at high risk for STI. We are the first to investigate the distinct molecular endotypes of PID and identify signatures unique to GC and/or CT-induced PID, indicating the possibility of using blood transcriptomic profiles as biomarkers for STI-induced PID. Heterogeneous endotypes of PID may confound the identity of biomarkers and our study provides guidance on how to classify the subject groups for a more powerful prediction.
There are several limitations to our study. Sample sizes for histologic evaluation were small because of the challenges associated with collection of adequate endometrial tissue for evaluation. Although we did not have access to samples from a second set of women with documented STI-induced clinical PID and chronic endometritis to use as a validation set, the availability of whole blood microarray data from women with subclinical PID and documented CT-induced chronic endometritis provided validation of top disease associated gene pathways. These results suggest blood transcriptional signatures can be used as biomarkers to identify STI-infected women at enhanced risk of upper genital tract infection and disease. Independent cohorts with larger sample size are warranted to confirm our findings in this study.
These investigations have enhanced our knowledge of the pathogenesis of PID by providing evidence that women with CT and acute endometritis are in an early stage of disease that will likely progress to chronic endometritis if the patient remains untreated. Our data support a role for activation of myeloid inflammatory pathways as contributors to GC and/or CT-induced chronic endometritis, and a role for type I IFN responses in progression of CT infection and disease. Finally, GC/CT co-infection may place women at particularly increased risk through enhanced activation of cell death pathways and inhibition of responses essential for development of adaptive immunity. Ongoing investigations of cervical, endometrial and specific cellular mRNA responses should enhance our understanding of the local tissue response and provide a comparator to the blood transcriptional signature.
Supplementary Material
Acknowledgments
We thank the women who agreed to participate in this study; Allison Collins, Abi Jett, Melinda Petrina, Carol Priest, Ingrid Macio and Lorna Rabe for their efforts collecting and processing samples; Antonio J. Amortegi and Giuliana Trucco for pathological assessments, the staff at the Allegheny County Health Department STD Clinic and staff of the Emergency Departments at Mercy Hospital of Pittsburgh and Magee-Womens Hospital of UPMC; and the staff of the Genomics Core at the University of Pittsburgh for their efforts; and Taylor Poston for his thoughtful review of the manuscript. We thank Bentley Midkiff in the UNC Translational Pathology Laboratory for expert technical assistance. Biomarker profiling was performed under the management of Drs. Heather E. Lynch and Andrew Macintyre and direction of Dr. Gregory Sempowski.
This work was supported by the National Institute of Allergy and Infectious Diseases, U19 AI084024 (TD), AI098660 (TD), AI119164 (TD), and U19 AI113170 (XZ). The UNC Translational Pathology Lab is supported in part, by grants from the National Cancer Institute (2-P30-CA016086-40), and the National Institute Environmental Health Sciences (2-P30ES010126-15A1). Dr. Gregory D. Sempowski in the Immunology Unit of the Duke Regional Biocontainment Laboratory received support from the National Institute of Allergy and Infectious Diseases (UC6-AI058607).
Dr. Hillier receives research funding from Becton, Dickinson and Co. and Cephid.
Footnotes
All remaining authors declare no competing interests.
AUTHOR CONTRIBUTIONS
XZ, CMO and TD designed the research. CMO catalogued and prepared samples for genomic analysis and ANR organized and analyzed the clinical data. HCW directed the clinical core and SLH directed microbiological analyses. XZ and WJ performed biostatistical analyses. MT performed all RNAscope analyses. DEL performed cervical secretion analyses. UN was involved in discussions of data analysis and interpretation. XZ, CMO, and TD wrote the paper. All the authors discussed the results and commented on the manuscript.
References
- 1.Workowski KA, Bolan GA, C. Centers for Disease, and Prevention Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1–137. [PMC free article] [PubMed] [Google Scholar]
- 2.Rein DB, Kassler WJ, Irwin KL, Rabiee L. Direct medical cost of pelvic inflammatory disease and its sequelae: decreasing, but still substantial. Obstet Gynecol. 2000;95:397–402. doi: 10.1016/s0029-7844(99)00551-7. [DOI] [PubMed] [Google Scholar]
- 3.Cohen CR, Mugo NR, Astete SG, Odondo R, Manhart LE, Kiehlbauch JA, Stamm WE, Waiyaki PG, Totten PA. Detection of Mycoplasma genitalium in women with laparoscopically diagnosed acute salpingitis. Sex Transm Infect. 2005;81:463–466. doi: 10.1136/sti.2005.015701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haggerty CL, Totten PA, Astete SG, Ness RB. Mycoplasma genitalium among women with nongonococcal, nonchlamydial pelvic inflammatory disease. Infect Dis Obstet Gynecol. 2006;2006:30184. doi: 10.1155/IDOG/2006/30184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurstrand M, Jensen JS, Magnuson A, Kamwendo F, Fredlund H. A serological study of the role of Mycoplasma genitalium in pelvic inflammatory disease and ectopic pregnancy. Sex Transm Infect. 2007;83:319–323. doi: 10.1136/sti.2007.024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forward KR. Risk of coinfection with Chlamydia trachomatis and Neisseria gonorrhoeae in Nova Scotia. Can J Infect Dis Med Microbiol. 2010;21:e84–86. doi: 10.1155/2010/760218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haggerty CL, Totten PA, Tang G, Astete SG, Ferris MJ, Norori J, Bass DC, Martin DH, Taylor BD, Ness RB. Identification of novel microbes associated with pelvic inflammatory disease and infertility. Sex Transm Infect. 2016;92:441–446. doi: 10.1136/sextrans-2015-052285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haggerty CL, Hillier SL, Bass DC, Ness RB, Evaluation PID, i. Clinical Health study Bacterial vaginosis and anaerobic bacteria are associated with endometritis. Clin Infect Dis. 2004;39:990–995. doi: 10.1086/423963. [DOI] [PubMed] [Google Scholar]
- 9.Darville T, Hiltke TJ. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis. 2010;201(Suppl 2):S114–125. doi: 10.1086/652397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Perez H, Harlow SD, Erdmann CA, Denman C. Pelvic pain and associated characteristics among women in northern Mexico. Int Perspect Sex Reprod Health. 2010;36:90–98. doi: 10.1363/ipsrh.36.090.10. [DOI] [PubMed] [Google Scholar]
- 11.Russell AN, Zheng X, O’Connell CM, Taylor BD, Wiesenfeld HC, Hillier SL, Zhong W, Darville T. Analysis of factors driving incident and ascending infection and the role of serum antibody in Chlamydia trachomatis genital tract infection. J Infect Dis. 2016;213:523–531. doi: 10.1093/infdis/jiv438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiesenfeld HC, Hillier SL, Meyn LA, Amortegui AJ, Sweet RL. Subclinical pelvic inflammatory disease and infertility. Obstet Gynecol. 2012;120:37–43. doi: 10.1097/AOG.0b013e31825a6bc9. [DOI] [PubMed] [Google Scholar]
- 13.Jaiyeoba O, Soper DE. A practical approach to the diagnosis of pelvic inflammatory disease. Infect Dis Obstet Gynecol. 2011;2011:753037. doi: 10.1155/2011/753037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darville T, C. Pelvic Inflammatory Disease Workshop Proceedings Pelvic inflammatory disease: identifying research gaps–proceedings of a workshop sponsored by Department of Health and Human Services/National Institutes of Health/National Institute of Allergy and Infectious Diseases, November 3-4, 2011. Sex Transm Dis. 2013;40:761–767. doi: 10.1097/OLQ.0000000000000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendrick DL. Transcriptional profiling to identify biomarkers of disease and drug response. Pharmacogenomics. 2011;12:235–249. doi: 10.2217/pgs.10.184. [DOI] [PubMed] [Google Scholar]
- 16.Mintz MB, Sowers R, Brown KM, Hilmer SC, Mazza B, Huvos AG, Meyers PA, Lafleur B, McDonough WS, Henry MM, Ramsey KE, Antonescu CR, Chen W, Healey JH, Daluski A, Berens ME, Macdonald TJ, Gorlick R, Stephan DA. An expression signature classifies chemotherapy-resistant pediatric osteosarcoma. Cancer Res. 2005;65:1748–1754. doi: 10.1158/0008-5472.CAN-04-2463. [DOI] [PubMed] [Google Scholar]
- 17.Rizner TL. Noninvasive biomarkers of endometriosis: myth or reality? Expert Rev Mol Diagn. 2014;14:365–385. doi: 10.1586/14737159.2014.899905. [DOI] [PubMed] [Google Scholar]
- 18.Thykjaer T, Workman C, Kruhoffer M, Demtroder K, Wolf H, Andersen LD, Frederiksen CM, Knudsen S, Orntoft TF. Identification of gene expression patterns in superficial and invasive human bladder cancer. Cancer Res. 2001;61:2492–2499. [PubMed] [Google Scholar]
- 19.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Hertoghs KM, Moerland PD, van Stijn A, Remmerswaal EB, Yong SL, van de Berg PJ, van Ham SM, Baas F, ten Berge IJ, van Lier RA. Molecular profiling of cytomegalovirus-induced human CD8+ T cell differentiation. J Clin Invest. 2010;120:4077–4090. doi: 10.1172/JCI42758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci U S A. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, Abajian C, Beckmann CF, Bernard A, Bertagnolli D, Boe AF, Cartagena PM, Chakravarty MM, Chapin M, Chong J, Dalley RA, David Daly B, Dang C, Datta S, Dee N, Dolbeare TA, Faber V, Feng D, Fowler DR, Goldy J, Gregor BW, Haradon Z, Haynor DR, Hohmann JG, Horvath S, Howard RE, Jeromin A, Jochim JM, Kinnunen M, Lau C, Lazarz ET, Lee C, Lemon TA, Li L, Li Y, Morris JA, Overly CC, Parker PD, Parry SE, Reding M, Royall JJ, Schulkin J, Sequeira PA, Slaughterbeck CR, Smith SC, Sodt AJ, Sunkin SM, Swanson BE, Vawter MP, Williams D, Wohnoutka P, Zielke HR, Geschwind DH, Hof PR, Smith SM, Koch C, Grant SGN, Jones AR. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue Z, Huang K, Cai C, Cai L, Jiang CY, Feng Y, Liu Z, Zeng Q, Cheng L, Sun YE, Liu JY, Horvath S, Fan G. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500:593–597. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amsel R, Totten PA, Spiegel CA, Chen KC, Eschenbach D, Holmes KK. Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am J Med. 1983;74:14–22. doi: 10.1016/0002-9343(83)91112-9. [DOI] [PubMed] [Google Scholar]
- 25.Kiviat NB, Wolner-Hanssen P, Eschenbach DA, Wasserheit JN, Paavonen JA, Bell TA, Critchlow CW, Stamm WE, Moore DE, Holmes KK. Endometrial histopathology in patients with culture-proved upper genital tract infection and laparoscopically diagnosed acute salpingitis. Am J Surg Pathol. 1990;14:167–175. doi: 10.1097/00000478-199002000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Rohan LC, Edwards RP, Kelly LA, Colenello KA, Bowman FP, Crowley-Nowick PA. Optimization of the weck-Cel collection method for quantitation of cytokines in mucosal secretions. Clin Diagn Lab Immunol. 2000;7:45–48. doi: 10.1128/cdli.7.1.45-48.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Russell AN, Zheng X, O’Connell CM, Wiesenfeld HC, Hillier SL, Taylor BD, Picard MD, Flechtner JB, Zhong W, Frazer LC, Darville T. Identification of Chlamydia trachomatis antigens recognized by T cells from highly exposed women who limit or resist genital tract infection. J Infect Dis. 2016;214:1884–1892. doi: 10.1093/infdis/jiw485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 29.Reese SE, Archer KJ, Therneau TM, Atkinson EJ, Vachon CM, de Andrade M, Kocher JP, Eckel-Passow JE. A new statistic for identifying batch effects in high-throughput genomic data that uses guided principal component analysis. Bioinformatics. 2013;29:2877–2883. doi: 10.1093/bioinformatics/btt480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 31.Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18:463–472. doi: 10.1007/s00335-007-9043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 33.Southworth LK, Owen AB, Kim SK. Aging mice show a decreasing correlation of gene expression within genetic modules. PLoS Genet. 2009;5:e1000776. doi: 10.1371/journal.pgen.1000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukund K, Subramaniam S. Dysregulated mechanisms underlying Duchenne muscular dystrophy from co-expression network preservation analysis. BMC Res Notes. 2015;8:182. doi: 10.1186/s13104-015-1141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, Stichweh D, Blankenship D, Li L, Munagala I, Bennett L, Allantaz F, Mejias A, Ardura M, Kaizer E, Monnet L, Allman W, Randall H, Johnson D, Lanier A, Punaro M, Wittkowski KM, White P, Fay J, Klintmalm G, Ramilo O, Palucka AK, Banchereau J, Pascual V. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–164. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun. 2015;6:8761. doi: 10.1038/ncomms9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaussabel D, Baldwin N. Democratizing systems immunology with modular transcriptional repertoire analyses. Nat Rev Immunol. 2014;14:271–280. doi: 10.1038/nri3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiche L, Jourde-Chiche N, Whalen E, Presnell S, Gersuk V, Dang K, Anguiano E, Quinn C, Burtey S, Berland Y, Kaplanski G, Harle JR, Pascual V, Chaussabel D. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014;66:1583–1595. doi: 10.1002/art.38628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagarajan UM, Ojcius DM, Stahl L, Rank RG, Darville T. Chlamydia trachomatis induces expression of IFN-gamma-inducible protein 10 and IFN-beta independent of TLR2 and TLR4, but largely dependent on MyD88. J Immunol. 2005;175:450–460. doi: 10.4049/jimmunol.175.1.450. [DOI] [PubMed] [Google Scholar]
- 42.Nagarajan UM, Prantner D, Sikes JD, Andrews CW, Jr, Goodwin AM, Nagarajan S, Darville T. Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun. 2008;76:4642–4648. doi: 10.1128/IAI.00629-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF, Ting JP. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182:6460–6469. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.May A, Huehns ER. The mechanism and prevention of sickling. Br Med Bull. 1976;32:223–233. doi: 10.1093/oxfordjournals.bmb.a071367. [DOI] [PubMed] [Google Scholar]
- 45.Prantner D, Darville T, Sikes JD, Andrews CW, Jr, Brade H, Rank RG, Nagarajan UM. Critical role for interleukin-1beta (IL-1beta) during Chlamydia muridarum genital infection and bacterial replication-independent secretion of IL-1beta in mouse macrophages. Infect Immun. 2009;77:5334–5346. doi: 10.1128/IAI.00883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramsey KH, Sigar IM, Schripsema JH, Shaba N, Cohoon KP. Expression of matrix metalloproteinases subsequent to urogenital Chlamydia muridarum infection of mice. Infect Immun. 2005;73:6962–6973. doi: 10.1128/IAI.73.10.6962-6973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riley MM, Zurenski MA, Frazer LC, O’Connell CM, Andrews CW, Jr, Mintus M, Darville T. The recall response induced by genital challenge with Chlamydia muridarum protects the oviduct from pathology but not from reinfection. Infect Immun. 2012;80:2194–2203. doi: 10.1128/IAI.00169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feinen B, Jerse AE, Gaffen SL, Russell MW. Critical role of Th17 responses in a murine model of Neisseria gonorrhoeae genital infection. Mucosal Immunol. 2010;3:312–321. doi: 10.1038/mi.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anzala AO, Simonsen JN, Kimani J, Ball TB, Nagelkerke NJ, Rutherford J, Ngugi EN, Bwayo JJ, Plummer FA. Acute sexually transmitted infections increase human immunodeficiency virus type 1 plasma viremia, increase plasma type 2 cytokines, and decrease CD4 cell counts. J Infect Dis. 2000;182:459–466. doi: 10.1086/315733. [DOI] [PubMed] [Google Scholar]
- 50.Goldstein BY, Steinberg JK, Aynalem G, Kerndt PR. High Chlamydia and gonorrhea incidence and reinfection among performers in the adult film industry. Sex Transm Dis. 2011;38:644–648. doi: 10.1097/OLQ.0b013e318214e408. [DOI] [PubMed] [Google Scholar]
- 51.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, Modlin RL. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovarik P, Castiglia V, Ivin M, Ebner F. Type I Interferons in Bacterial Infections: A Balancing Act. Front Immunol. 2016;7:652. doi: 10.3389/fimmu.2016.00652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med. 2010;207:327–337. doi: 10.1084/jem.20091746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, Nagarajan UM. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during Chlamydia trachomatis infection. J Immunol. 2014;193:2394–2404. doi: 10.4049/jimmunol.1302718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andrade WA, Agarwal S, Mo S, Shaffer SA, Dillard JP, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, Golenbock DT. Type I interferon induction by Neisseria gonorrhoeae: dual requirement of cyclic GMP-AMP synthase and Toll-like receptor 4. Cell reports. 2016;15:2438–2448. doi: 10.1016/j.celrep.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu W, Ventevogel MS, Knilans KJ, Anderson JE, Oldach LM, McKinnon KP, Hobbs MM, Sempowski GD, Duncan JA. Neisseria gonorrhoeae suppresses dendritic cell-induced, antigen-dependent CD4 T cell proliferation. PLoS One. 2012;7:e41260. doi: 10.1371/journal.pone.0041260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boulton IC, Gray-Owen SD. Neisserial binding to CEACAM1 arrests the activation and proliferation of CD4+ T lymphocytes. Nat Immunol. 2002;3:229–236. doi: 10.1038/ni769. [DOI] [PubMed] [Google Scholar]
- 60.Pantelic M, Kim YJ, Bolland S, Chen I, Shively J, Chen T. Neisseria gonorrhoeae kills carcinoembryonic antigen-related cellular adhesion molecule 1 (CD66a)-expressing human B cells and inhibits antibody production. Infect Immun. 2005;73:4171–4179. doi: 10.1128/IAI.73.7.4171-4179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balamuth F, Zhang Z, Rappaport E, Hayes K, Mollen C, Sullivan KE. RNA biosignatures in adolescent patients in a pediatric emergency department with pelvic inflammatory disease. Pediatr Emerg Care. 2015;31:465–472. doi: 10.1097/PEC.0000000000000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.