

Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent liver disease worldwide. It may result in several types of liver problems including impaired liver regeneration, but the mechanism for this is unknown. Because liver regeneration depends on calcium signaling, we examined the effects of NAFLD on expression of the type II inositol 1,4,5-trisphosphate receptor (ITPR2), the principle calcium release channel in hepatocytes. ITPR2 promoter activity was measured in Huh7 and HepG2 cells. ITPR2 and c-Jun protein levels were evaluated in Huh7 cells, in liver tissue from a rat model of NAFLD and in liver biopsy specimens of patients with simple steatosis and non-alcoholic steatohepatitis (NASH). Liver regeneration was assessed in wild-type and Itpr2 knockout (Itpr2-/-) mice following 67% hepatectomy. Cell proliferation was examined in ITPR2-knockout HepG2 cells generated by the CRISPR/Cas9 system. c-Jun dose-dependently decreased activity of the human ITPR2 promoter. c-Jun expression was increased and ITPR2 was decreased in fat-loaded Huh7 cells and in the livers of rats fed a high-fat, high-fructose diet. Overexpression of c-Jun reduced protein and messenger RNA expression of ITPR2 in Huh7 cells, whereas knockdown of c-Jun prevented the decrease of ITPR2 in fat-loaded Huh7 cells. ITPR2 expression was decreased and c-Jun was increased in liver biopsies of patients with steatosis and NASH compared to controls. ITPR2-knockout cells exhibited less nuclear calcium signaling and cell proliferation than control cells. Liver regeneration assessed by Ki-67 and proliferating cell nuclear antigen was markedly decreased in Itpr2-/- mice.
Conclusion
These findings suggest that fatty liver induces a c-Jun-mediated decrease in ITPR2 in hepatocytes. This may account for the impaired liver regeneration that occurs in NAFLD.
Keywords: Hepatic steatosis, Steatohepatitis, Liver regeneration, c-Jun, Calcium release channel
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease worldwide, affecting over half a billion people (1). Although the majority of NAFLD patients have simple steatosis, a subset of patients has non-alcoholic steatohepatitis (NASH), and 20% of NASH patients develop cirrhosis. In addition, NAFLD is associated with increased risk of hepatocellular carcinoma (HCC), the second most common cause of cancer death. NAFLD also results in other specific liver problems including impaired liver regeneration (2, 3). Although weight loss (4) and exercise (5) can be beneficial in NASH, the value of pharmacological treatments is less established (6). Therefore, an improved understanding of mechanisms responsible for the pathological consequences of NAFLD/NASH is a high priority.
Calcium (Ca2+) is a ubiquitous second messenger that regulates a wide range of functions in hepatocytes, including glucose (7) and lipid metabolism (8), apoptosis (9), gene transcription (10), bile secretion (11, 12) and cell proliferation (13). Ca2+ signaling results from activation of the inositol 1,4,5-trisphosphate receptor (ITPR), and the type II inositol 1,4,5-trisphosphate receptor (ITPR2) is the principal intracellular calcium release channel in hepatocytes (14). ITPR may be particularly important in lipid homeostasis, because Drosophila with Itpr mutations become obese and store excess triglycerides in their fat bodies even on a normal diet (15). The type I inositol 1,4,5-trisphosphate receptor (ITPR1) may be more important than ITPR2 in regulating mammalian hepatic lipid metabolism, because ITPR1 selectively couples to mitochondria and regulates lipid droplet formation, and liver-specific Itpr1-/- mice are resistant to NAFLD (8), whereas Itpr2-/- mice have no change in lipid metabolism (16). However, interestingly, hepatic ITPR2 expression is decreased in obese mice (16), but the mechanism of this and its pathophysiological relevance are unknown.
Experimental Procedures
Animal models
Eight-week-old male Sprague-Dawley rats (180-200 grams) were obtained from the National Laboratory Animal Center, Mahidol University. After acclimatization period, rats were assigned to receive either control diet (10% kcal as fat, Envigo) with reverse osmosis water or high-fat diet (60% kcal as fat, Envigo) with 30% w/v fructose in water for 12 weeks to develop NAFLD model.
The whole-body homozygous knockout Itpr2-/- mice in a C57BL/6 genetic background were used in this study. Nine- to ten-week-old male Itpr2-/- and their wild-type littermates underwent sham laparotomy or two-thirds hepatectomy as described previously (17).
All animals received humane care in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health and all study procedures were approved by the Institutional Animal Care and Use Committee. The study protocol for rats challenged with high-fat, high-fructose diet was approved by the Institutional Animal Care and Use Committee of Faculty of Science, Mahidol University. The study protocol for Itpr2-/- mice was approved by Yale's IACUC.
Generation of an ITPR2-knockout HepG2 cell line
An ITPR2-knockout HepG2 cell line was established using the CRISPR/Cas9 system. Control CRISPR/Cas9 plasmid, a non-targeting 20-nt scramble guide RNA designed as a negative control, IP3R-II CRISPR/Cas9 KO plasmid and IP3R-II HDR plasmid were obtained from Santa Cruz Technology. Successful deletion of ITPR2 was confirmed by immunoblot.
Nuclear calcium imaging
ITPR2 CRISPR-knockout or CRISPR-control HepG2 cells were plated onto coverslips and transfected with a nuclear targeted calcium-sensing protein, GCaMP3 plasmid (a gift from Dr. Anant Parekh, University of Oxford). Transfected nuclear-GCaMP3 cells were perfused with a HEPES buffer while stimulated with 20 μM adenosine triphosphate (ATP). This ATP concentration was used because it is sufficient to elicit nuclear Ca2+ signals in this cell type (18). Ca2+ signaling in the cell nucleus was monitored using a Zeiss 710 confocal microscope as described (18).
Human ethics statement
Human liver tissue biopsies were obtained under the auspices of protocols approved by the Institutional Review Board on the Protection of the Rights of Human Subjects (Yale University), which was conducted in accordance with the Declaration of Helsinki. The Human Investigation Committee protocol number is HIC-1304011763.
Statistical analysis
All data are expressed as means ± standard error of mean. GraphPad Prism 7 Software was used for data analysis. Statistical analyses were performed using the Student's t-test or the One-way analysis of variance (ANOVA). Differences with p < 0.05 was considered statistically significant.
Further Methodological Details
Detailed additional Materials and Methods are available in the Supporting Information.
Results
ITPR2 in hepatocytes is transcriptionally regulated by c-Jun
The transcription factor c-Jun is often elevated in inflammation, including in NAFLD (19). We recently cloned the human ITPR2 promoter and found that it contained at least one AP-1/c-Jun binding site (20). Analysis of the human ITPR2 promoter by MatInspector software revealed there are actually four putative AP-1 binding sites for c-Jun, located between i) -1279 to -1292 bp, ii) -1128 to -1148 bp, iii) -871 to -895 bp, and iv) -180 to -200 bp upstream of the transcription starting site (Fig. 1A). To understand the functional significance of this, we investigated whether c-Jun regulates ITPR2 expression. Huh7 and HepG2 cells were co-transfected with a human ITPR2 promoter luciferase construct and a c-Jun expression plasmid. Co-transfection of the ITPR2 promoter with c-Jun led to a significant down-regulation of promoter activity both in Huh7 (Fig. 1B) and HepG2 cells (Fig. 1C). To examine the direct effect of c-Jun on ITPR2 expression, Huh7 cells were transfected with varying amounts of a c-Jun expression plasmid. Overexpression of c-Jun decreased ITPR2 expression, both at the mRNA (Fig. 1D) and protein levels (Fig.1E-F) without any change in ITPR1 (Supporting Fig. S1). These findings provide evidence that c-Jun negatively regulates ITPR2 expression in hepatocytes with no compensation in ITPR1 expression.
Figure 1. ITPR2 in hepatocytes is transcriptionally regulated by c-Jun.
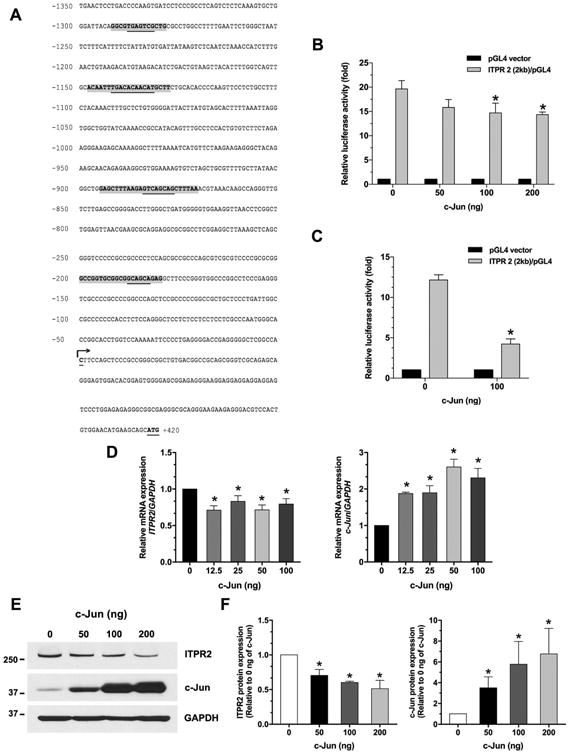
(A) DNA sequence of the proximal region (2 kb) of the human ITPR2 promoter. Potential transcriptional binding sites for c-Jun/AP-1 are shaded in gray, and underlined nucleotides are the core sequence of AP-1. Numbered positions refer to the transcription starting sites (nucleotide, +1, arrow). (B) c-Jun inhibits ITPR2 promoter activity in a dose-dependent manner in Huh7 cells and (C) in HepG2 cells. Results are presented as fold changes relative to empty luciferase vector (pGL4) from the same dual luciferase assay. *p < 0.05, compared with 0 ng of c-Jun plasmid; Huh7 (n = 6), HepG2 (n = 3). (D) Relative mRNA expressions of ITPR2 (left panel) and c-Jun (right panel) in Huh7 cells transfected with various concentrations of c-Jun plasmid for 24 hours. c-Jun overexpression directly decreases ITPR2 mRNA expression in Huh7 cells transfected with c-Jun plasmids. *p < 0.05, compared with 0 ng of c-Jun plasmid (n = 4). (E) Representative immunoblot and (F) Relative protein expressions of ITPR2 (left panel) and c-Jun (right panel) in Huh7 cells transfected with c-Jun plasmids, measured after 48 hours. *p < 0.05, compared with 0 ng of c-Jun plasmid (n = 3).
Steatosis inhibits ITPR2 expression via c-Jun in vitro
To investigate whether effects of c-Jun on ITPR2 expression are relevant in steatosis, we examined Huh7 cells loaded with palmitic acid to mimic fat accumulation in the liver. Palmitic acid-treated Huh7 cells exhibited a marked increase in fat deposition compared to BSA-treated Huh7 cells as a control group (Fig. 2A). Expression of c-Jun was significantly increased and ITPR2 expression was significantly decreased in fat-loaded Huh7 cells (Fig. 2B-C). To investigate whether this was a causal relationship, siRNA was used to knockdown c-Jun in Huh7 cells. Loss of c-Jun did not affect ITPR2 expression in control cells, but the palmitic acid-induced decrease in ITPR2 was abolished by knockdown of c-Jun (Fig. 2D-E). These findings provide evidence that steatosis decreases ITPR2 expression in liver cells, and that is mediated by c-Jun.
Figure 2. Steatosis inhibits ITPR2 expression via c-Jun in vitro.
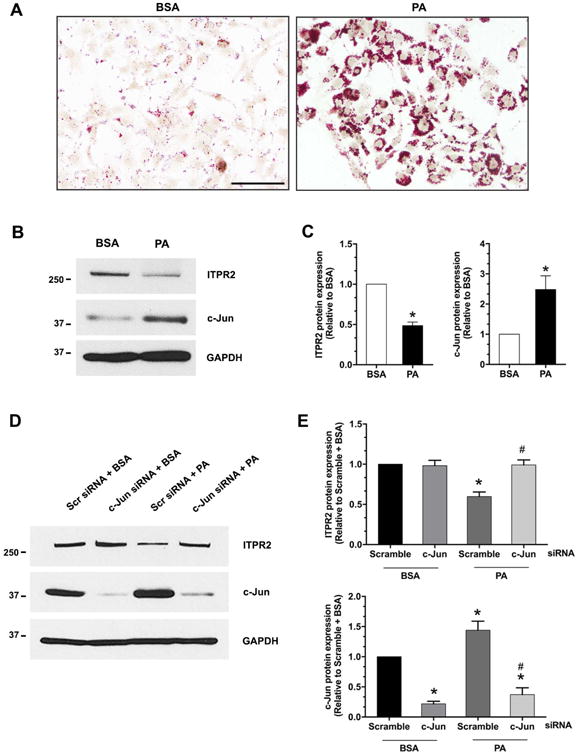
(A) Oil red O staining in Huh7 cells loaded with BSA (vehicle control) or 500 μM palmitic acid (PA) for 24 hours. Scale bar: 100 μm. (B) Representative immunoblot and (C) Densitometry analysis of protein expression of ITPR2 (left panel) and c-Jun (right panel) in Huh7 cells treated with PA demonstrates that lipid loading decreases ITPR2 and increases c-Jun expression. *p < 0.05, compared with BSA control (n = 4). (D) Representative immunoblot and (E) Relative protein expression of ITPR2 (top panel) and c-Jun (bottom panel) in Huh7 cells transfected with scrambled-siRNA or c-Jun-siRNA either in the presence or absence of PA. PA-induced decrease in ITPR2 expression requires c-Jun. *p < 0.01, compared with scramble-siRNA + BSA; #p < 0.01, compared with scramble-siRNA + PA (n = 4).
c-Jun is increased and ITPR2 expression is decreased in livers of NAFLD rats
To investigate the relevance of these findings in vivo, rats were fed a high-fat, high-fructose diet (HFFD) to mimic human NAFLD. HFFD-fed rats developed features of hepatic steatosis that included an accumulation of lipid droplets in the hepatocytes (Supporting Fig. S2A-B) and elevated liver triglyceride levels (Supporting Fig. S2C). These rats also developed liver damage, as reflected by increased serum levels of ALT (Supporting Fig. S2D) and ALP (Supporting Fig. S2E), relative to control diet-fed rats. Oxidative stress and inflammation that occurs in NAFLD condition can increase c-Jun (21), so we also measured hepatic MDA, Il-6 and Tnf-α mRNA levels. Both the oxidative stress marker MDA (Supporting Fig. S2F) and the inflammatory cytokines Il-6 (Supporting Fig. S2G) and Tnf-α (Supporting Fig. S2H) were increased significantly in HFFD-fed rats, relative to control diet-fed rats. This observation demonstrates that this particular animal model of NAFLD is associated with steatosis, mild hepatitis and associated increases in hepatic inflammatory markers. We also investigated the effects of NAFLD on c-Jun and ITPR2 expression in this animal model (Fig. 3). The expression of c-Jun was significantly increased (Fig. 3A), while ITPR2 expression was significantly decreased (Fig. 3B) in livers of HFFD-fed rats. In contrast, ITPR1 expression was unchanged (Fig. 3C). This is consistent with the recent observation that ITPR1 expression is unchanged in liver biopsies from patients with simple steatosis (8). Although this isoform is increased in patients with NASH (8), levels of ITPR1 remained unchanged in HFFD-fed rats (Fig. 3C). Finally, we examined the c-Jun NH2-terminal kinase (JNK) pathway, which is activated by oxidative stress and regulates c-Jun expression. Phospho-JNK was increased in HFFD-fed rats (Fig. 3D), consistent with its role as an intermediary in elevating c-Jun. Together, these findings support the idea that NAFLD leads to hepatic inflammation, which in turn results in a JNK- and c-Jun-mediated decrease in hepatic ITPR2 expression in an animal model.
Figure 3. c-Jun is increased and ITPR2 is decreased in the NAFLD model in rats.
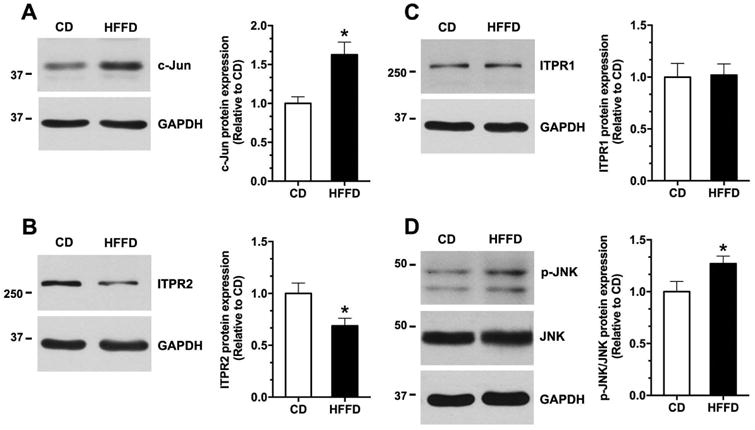
Representative immunoblot and relative protein expression of (A) c-Jun, (B) ITPR2, (C) ITPR1, and (D) phospho-JNK/JNK in the livers extracted from rats fed control diet (CD) or HFFD diet. *p < 0.05, compared with CD (n = 5).
c-Jun is increased and ITPR2 expression is decreased in livers of patients with NAFLD
To investigate the clinical relevance of these observations, we examined liver biopsies from patients with simple steatosis and NASH, plus histologically normal biopsies. Histological diagnoses were established based on examination of H&E staining of the specimens (Fig. 4A). Immunohistochemistry of these liver sections demonstrated that c-Jun expression in hepatocytes was low in normal biopsies, but was markedly and significantly increased in patients with steatosis or NASH (Fig. 4B and 4D). Moreover, c-Jun was localized to the nuclei of hepatocytes and to infiltrating inflammatory cells in the NAFLD specimens. In contrast, ITPR2 was expressed in normal human liver and was most concentrated in the peri-canalicular region (Fig. 4C), similar to what is observed in rodent livers (14, 22). Furthermore, ITPR2 expression was significantly decreased or absent in steatosis and NASH (Fig. 4C and 4E). These findings are consistent with our observations in vitro and in an animal model that NAFLD leads to a c-Jun-mediated loss of ITPR2 in hepatocytes.
Figure 4. c-Jun is increased and ITPR2 is decreased in livers of patients with NAFLD and NASH.
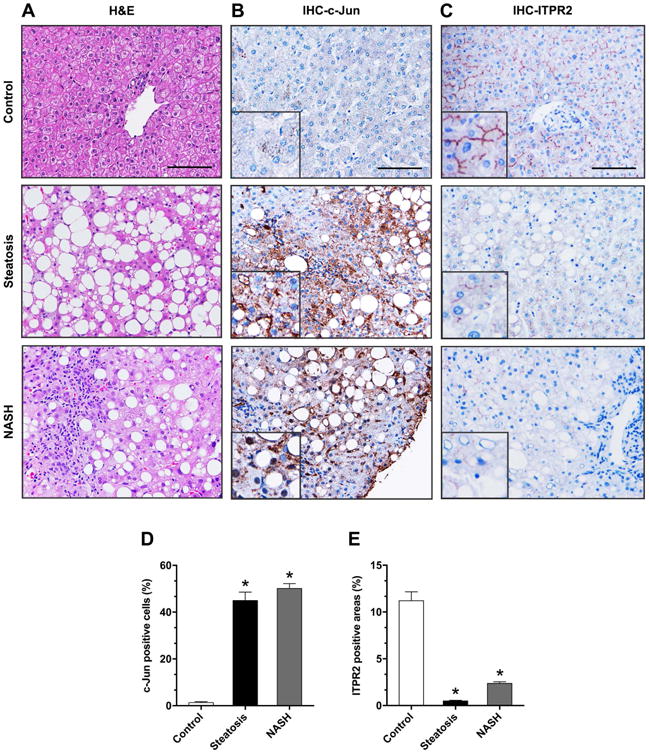
(A) Representative microscopic photographs of liver biopsy specimens from controls and patients with simple steatosis and NASH stained with H&E. Representative images of immunohistochemistry staining (IHC) of (B) c-Jun and (C) ITPR2 in liver samples. Images are representative of what was observed in 3-6 patients in each category. Scale bar: 100 μm. Quantitative analysis of IHC images stained for (D) c-Jun and (E) ITPR2. *p < 0.0001 (n = 3-6 patients).
Loss of ITPR2 impairs liver regeneration
Finally, we investigated the potential pathophysiological relevance of loss of ITPR2. Calcium signaling is important for hepatocyte proliferation (13) and liver regeneration (7, 9, 23), and liver regeneration is impaired in NAFLD (2, 24, 25), so we examined liver regeneration in Itpr2-/- mice. Male wild-type and Itpr2-/- mice that were 9 - 10 weeks of age were subjected to partial hepatectomy (PH). Histological examination 48 hours post-operatively revealed liver damage and necrosis in the Itpr2-/- but not in the wild-type mice (Fig. 5A-D). The post-hepatectomy Itpr2-/- livers were notable for diffuse steatosis, plus patchy but well-demarcated necrotic areas. The patchy necrosis was coagulative, randomly rather than zonally distributed across the hepatic lobule, and showed no signs of cholestasis. Moreover, in the necrotic areas, cell borders remained visible, and the cytoplasm and nuclei were in varying stages of disintegration, but were not swollen. There was scant neutrophilic inflammation in these foci, without macrophages, fibrosis, feathery degeneration or bile pigment deposition. These changes are not characteristic of bile infarcts, unlike what is seen after PH in TGR5- (26) or P2×4-knockout mice (27). In addition, liver regeneration assessed by either Ki-67 (Fig. 5E) or PCNA (Fig. 5F) labeling revealed a marked decrease in Itpr2-/- mice relative to wild-type mice.
Figure 5. Loss of ITPR2 impairs liver regeneration.
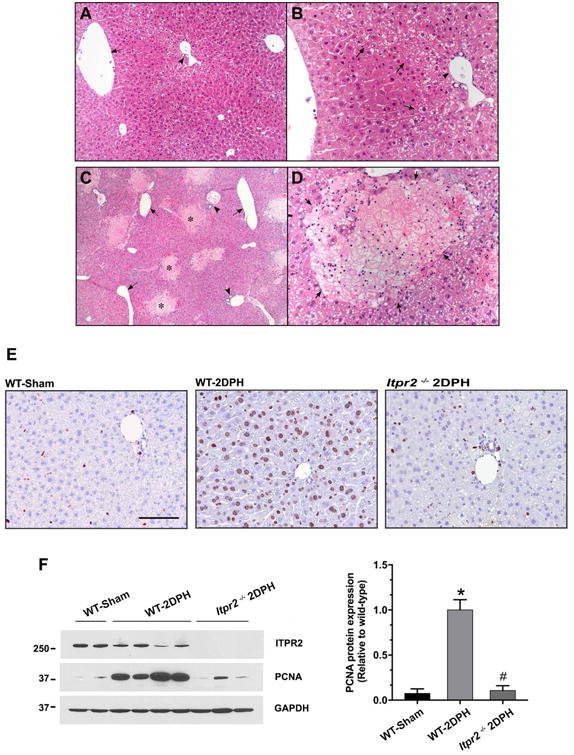
(A-D) Representative photomicrographs of H&E-stained liver sections 2 days after partial hepatectomy (2DPH). (A) Low power view (25×) of wild-type (WT)-2DPH mouse liver. Livers show mild, patchy cytoplasmic clearing that is predominantly periportal in distribution. Minimal small droplet macrovesicular steatosis is seen. There is less steatosis and no necrosis. Symbol: arrow = central vein, arrow-head = portal tract. (B) High power view (200×) of WT-2DPH mouse liver. A region of hepatocytes shows cytoplasmic clearing without swelling, more prominently associated with periportal and zone two regions. Some of the cytoplasmic clearing is due to early steatosis. Symbol: arrow = region of mild cytoplasmic clearing, arrow-head = portal tract. (C) Low power view (25×) of Itpr2-/--2DPH mouse liver. Livers show diffuse, predominantly small droplet, macrovesicular steatosis. Multiple well-circumscribed foci of necrotic hepatocytes (pale staining areas) are scattered randomly throughout the parenchyma, in a non-zonal distribution. Symbol: arrow = central vein, arrow-head = portal tract, * = areas of necrosis. (D) High power view (200×) of Itpr2-/--2DPH mouse liver. Well-circumscribed areas of bland (coagulative type) necrosis are seen (arrows). The necrotic cells show rarefaction of the cytoplasm and loss of nuclei without significant cellular swelling. Rare inflammatory cells, mostly neutrophils, are seen at the periphery and occasionally in the center of the necrotic foci. The viable surrounding liver show small droplet macrovesicular steatosis. No cholestasis is present. (n = 3-6 mice). (E) Representative images of Ki-67 immunohistochemistry staining. Itpr2-/--2DPH mice show less Ki-67-positive cells. Scale bar: 100 μm. (F) Representative immunoblot and relative protein expression of PCNA. Itpr2-/--2DPH mice show less PCNA expression. *p < 0.01, compared with WT-Sham; #p < 0.01, compared with WT-2DPH mice.
As typical immediate early genes, c-Jun and c-Fos are rapidly induced following PH (23). To investigate whether ITPR2 expression is inversely correlated with c-Jun and c-Fos soon after PH, we measured hepatic mRNA expression of Itpr-2, c-Jun and c-Fos at 1 and 2 hours after PH in C57BL/6 mice (Fig. 6). At both of these early time points following PH, hepatic mRNA levels of c-Jun and c-Fos were markedly increased, while Itpr2 was significantly decreased 2 hours after PH (Fig. 6D). This observation is consistent with the idea that increased c-Jun is an early event that is responsible for decreased ITPR2 expression after PH (28).
Figure 6. Hepatic mRNA levels of c-Jun and c-Fos are increased and Itpr2 is decreased soon after partial hepatectomy.
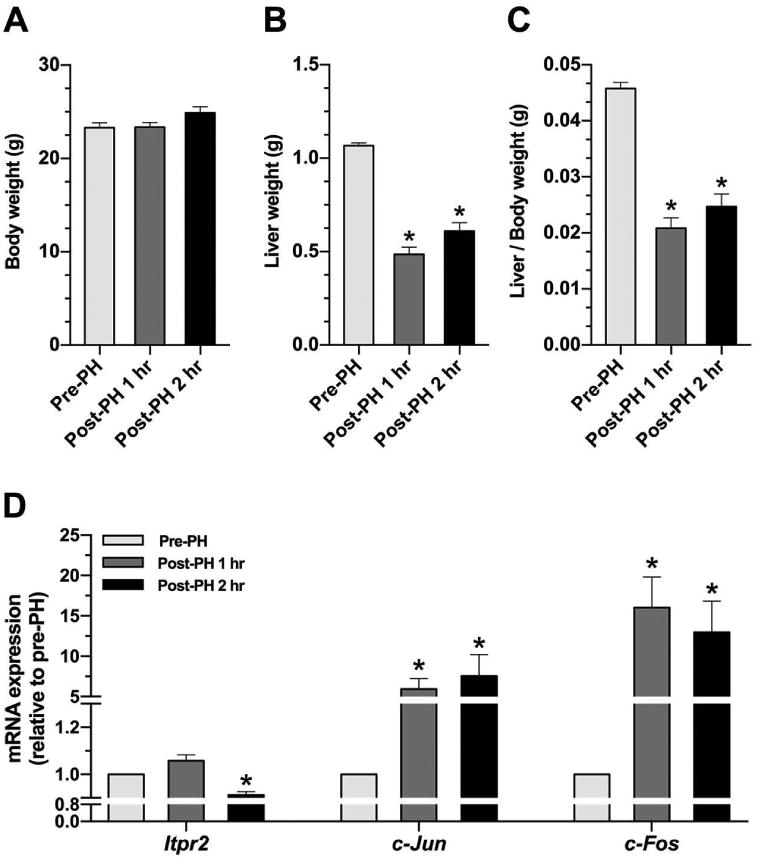
(A) Body weight, (B) Liver weight, and (C) Liver/body weight ratio in livers of wild-type mice subjected to partial hepatectomy (PH). (D) Relative hepatic mRNA expression of Itpr2, c-Jun, and c-Fos at pre-hepatectomy, 1-hour, and 2-hours after PH. *p < 0.01, compared with pre-hepatectomy (n = 4-5 mice).
Nuclear Ca2+ signaling and cell proliferation depend on ITPR2 in HepG2 cells
Because nucleoplasmic calcium is required for cell proliferation (29) and ITPR2 is the most abundant ITPR isoform in hepatocytes (14), we examined whether deletion of ITPR2 affects nuclear Ca2+ signaling and cell proliferation. ITPR2-knockout (KO) HepG2 cells were generated using the CRISPR/Cas9 system, then transfected with nuclear targeted calcium-sensing protein (GCaMP3) (30). Nuclear Ca2+ signals were monitored in cells stimulated with ATP, because ATP induces Ca2+ signals in the nucleus of HepG2 cells (18), and because ATP serves as a paracrine messenger that regulates liver regeneration (31, 32). Absence of ITPR2 in ITPR2-CRISPR-KO cells was verified (Fig. 7A). ITPR2-CRISPR-KO cells demonstrated markedly diminished amplitudes of ATP-stimulated nuclear Ca2+ signals compared to CRISPR-control cells, despite similar levels of Ca2+ stores as demonstrated by similar levels of ionomycin-induced Ca2+ signals (Fig. 7B). Similarly, the fraction of ITPR2-CRISPR-KO cells in which ATP induced a nuclear Ca2+ signal was markedly and significantly lower than that of control cells (Fig. 7C). This result supports the idea that ITPR2 regulates nuclear Ca2+ signaling. Two different assays were used to determine the relevance of ITPR2 for hepatocyte proliferation. The BrdU-ELISA assay was performed in ITPR2-CRISPR-KO and control cells 24 and 48 hours after addition of serum, and showed that proliferation was decreased by ∼50% at both time points (Fig. 7D-E). Similarly, EdU-imaging showed that proliferation of ITPR2-CRISPR-KO cells was decreased by nearly 70% relative to controls 24 hours after addition of serum (Fig. 8). These observations provide evidence that ITPR2 is important for liver regeneration because it mediates nuclear Ca2+ signaling, which in turn is necessary for cell proliferation.
Figure 7. Loss of ITPR2 impairs nuclear calcium signaling and cell proliferation in HepG2 cells.
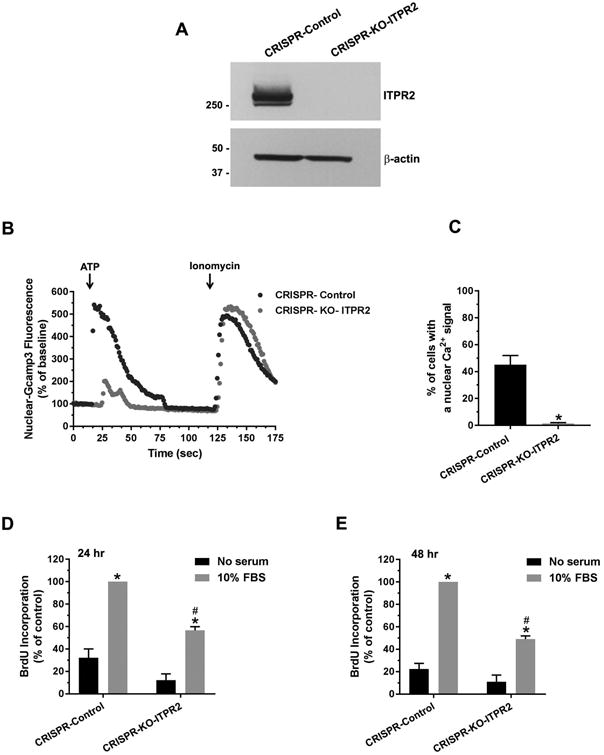
(A) Immunoblot analysis of ITPR2 expression in CRISPR-control and ITPR2-CRISPR-knockout (KO) HepG2 cells. (B) The amplitude of ATP (20 μM)-induced nuclear Ca2+ signals in ITPR2-CRISPR-KO cells is markedly lower than that of CRISPR-control cells. Tracing is representative of what was observed in 16-18 coverslips in three separate experiments. (C) The percentage of ITPR2-CRISPR-KO cells with a nuclear Ca2+ signal in response to ATP was much lower than controls. *p < 0.0001, compared with CRISPR-control (16-18 coverslips). BrdU incorporation is decreased in ITPR2-CRISPR-KO cells at (D) 24 and (E) 48 hours after addition of serum. *p < 0.0001, compared with CRISPR-control without serum; #p < 0.0001, compared with CRISPR-control with 10% FBS (n = 4 experiments).
Figure 8. Loss of ITPR2 decreases EdU-incorporation into HepG2 cells.
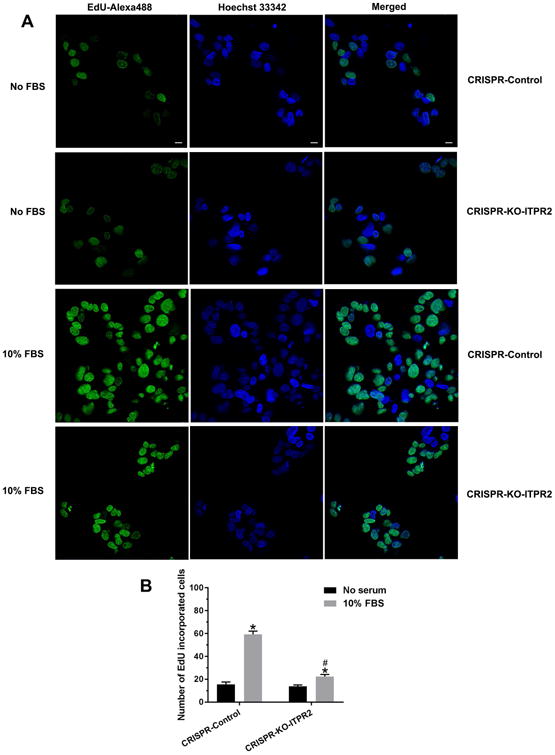
(A) Confocal imaging of cell 24 hours after addition of serum shows EdU-Alexa Fluor 488 labeling (green), indicative of proliferation, in control and ITPR2-CRISPR-KO cells. Hoechst 33342 labeling identifies all nuclei and merged image identifies proliferating cells. Scale bar: 10 μm. (B) Quantitative analysis of EdU-incorporated cells. *p < 0.05, compared with CRISPR-control without serum; # p < 0.05, compared with CRISPR-control with 10% FBS (6 coverslips in three experiments).
Discussion
Altered calcium signaling has been implicated in the pathogenesis of a range of liver diseases, including cholestasis (11, 12), NAFLD (8), and HCC (13). These disparate effects of calcium are likely because there is differential control of calcium signals in distinct subcellular regions. For example, peri-canalicular calcium signals regulate secretion (11, 12), mitochondrial calcium signals regulate lipid droplet formation (8), and nuclear calcium signals regulate cell proliferation (13). Subcellular regulation of calcium signals in turn is due at least in part to the subcellular distribution of different ITPR isoforms. ITPR2, which accounts for approximately 80% of ITPRs in hepatocytes, is most heavily concentrated in the region of the endoplasmic reticulum beneath the canalicular membrane (14, 22), and loss of this pool results in cholestasis (11, 12). Although ITPR1 accounts for only approximately 20% of ITPRs in hepatocytes, this isoform selectively couples to mitochondria and so it plays a role in lipid droplet formation and its increased expression is associated with development of NAFLD (8). There is also specificity in these effects, because loss of ITPR2 does not affect basal lipid metabolism (16), while loss of ITPR1 does not affect secretion (8). Our findings provide evidence, however, that NAFLD affects ITPR2 expression, and that cell proliferation is a previously unappreciated target of ITPR2-mediated calcium signals. Given the implications of these findings for the impaired liver regeneration that is known to occur in NAFLD (2, 24, 25), the factors that regulate ITPR2 expression are of potential clinical importance. Both nuclear factor of activated T cells (33) and cAMP/cAMP responsive element binding protein increase ITPR2 transcription (20). In fact, ITPR2 expression is increased during fasting, which may be mediated by glucagon-induced increases in cAMP (20). Although inhibition of ITPR2 expression by miRNA-133a has been described in cardiomyocytes (34), to our knowledge the current study is the first report of transcription factor-mediated inhibition of ITPR2 expression.
It is appreciated that Ca2+ signaling is important for liver regeneration, but the current work extends our understanding of this relationship in an important way. Previous work had established that cytosolic (23), nuclear (29) and mitochondrial (9) Ca2+ signals each contribute in separate ways to hepatocyte proliferation and liver regeneration. Furthermore, it had been shown that cytosolic Ca2+ signals and ITPR2 expression, in particular, are decreased after partial hepatectomy (28), and the current work provides evidence that these are mediated by an increase in c-Jun that occurs very early after the hepatectomy (Fig. 6). However, ITPR2 expression typically is reduced by only 30-50% after partial hepatectomy, and Ca2+ signals, while reduced, are not absent (28). Combined with the previous observation that hepatocyte Ca2+ signals are largely abolished in the absence of ITPR2 (8), this suggests that the remaining ITPR2's after partial hepatectomy continue to function properly. The current work further suggests there is an absolute requirement for ITPR2 in order to generate nuclear Ca2+ signals, and that cell proliferation is reduced by at least 50% without this Ca2+ release channel (Fig. 7-8). Loss of other receptors such as TGR5 (26) and P2×4 (27) also result in impaired liver regeneration, and in the relevant knockout animals there is evidence for bile infarcts in the periportal region of regenerating livers. In contrast, in regenerating livers of Itpr2-/- mice there is patchy necrosis that does not resemble bile infarcts and is not distributed in a lobular pattern (Fig. 5). This suggests that the impaired liver regeneration seen in Itpr2-/- mice is not due to biliary defects, which is not surprising because ITPR2 represents a minor fraction of the total ITPR pool in cholangiocytes (35), and loss of ITPR2 from cholangiocytes has only a mild inhibitory effect on secretion (36).
The relationship between fatty liver, ITPR2, and liver regeneration that is described here suggests an unexpected link. It has long been appreciated that fatty liver is associated with impaired liver regeneration, which is clinically relevant because patients with NAFLD have increased mortality following liver resection (2). Although a number of responsible mechanisms have been proposed (24, 25), the current observation may suggest a more fundamental explanation. AP-1/c-Jun is a well-known mediator of inflammation, and liver regeneration is thought to be more significantly impaired in NASH than in simple steatosis (2). The current work identifies AP-1/c-Jun as a negative transcriptional regulator of ITPR2 expression and provides evidence that one specific effect of fatty liver is to decrease ITPR2 expression through this mechanism. Although ITPR2 is most concentrated in the peri-canalicular region of the cytosol, where it regulates multidrug resistance-associated protein-2 (12) and bile salt export pump (11), it also is found in the nucleus (37, 38), where it is positioned to regulate calcium-mediated intra-nuclear processes. Furthermore, nuclear ITPR's are selectively activated by growth factors such as hepatocyte growth factor (HGF) (39), epidermal growth factor (EGF) (40), and insulin (7), which are specifically responsible for the calcium signals in the nucleus that regulate cell proliferation (7, 13, 39). Moreover, liver regeneration is impaired in the absence of either HGF (41) or EGF (42), and outright liver failure occurs if both of these growth factors are absent (43). Therefore, the subpopulation of ITPR2 localized to the nucleus may participate in a final common pathway that permits growth factors to elicit the calcium signals in the nucleus that are necessary for liver regeneration. Indeed, the current work demonstrates that nuclear Ca2+-signals depend on ITPR2, and that hepatocyte proliferation, which depends on nuclear Ca2+ signals (29), also depends on ITPR2 (Fig. 7-8). This in turn may explain why NAFLD, by increasing c-Jun and thus decreasing ITPR2, results in impaired liver regeneration. Further work will be needed to confirm the relative importance of this mechanism in patients with NAFLD and to determine whether this pathway can be exploited for therapeutic benefit.
Supplementary Material
Acknowledgments
The authors thank Mateus T. Guerra, Emma Kruglov, Kathy Harry, and Lei Zhang for technical assistance.
Financial support: This study was funded by the National Institutes of Diabetes and Digestive and Kidney Diseases (P01-DK57751, P30-DK34989 and R56-DK99470 to MHN), the Thailand Research Fund and Mahidol University (RSA6080072 to JW), the Science Achievement Scholarship of Thailand (SAST to TK), and the Thailand Research Fund through the Royal Golden Jubilee Ph.D. Program (PHD/4.PE.MU/54/C.1 to NC and VS).
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- HFFD
high-fat, high-fructose diet
- ITPR
inositol 1,4,5-trisphosphate receptor
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- PH
partial hepatectomy
- TNF-α
tumor necrosis factor-alpha
Footnotes
Author names in bold designate shared co-first authorship.
References
- 1.Wang FS, Fan JG, Zhang Z, Gao B, Wang HY. The global burden of liver disease: the major impact of China. Hepatology. 2014;60:2099–2108. doi: 10.1002/hep.27406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy SK, Marsh JW, Varley PR, Mock BK, Chopra KB, Geller DA, Tsung A. Underlying steatohepatitis, but not simple hepatic steatosis, increases morbidity after liver resection: a case-control study. Hepatology. 2012;56:2221–2230. doi: 10.1002/hep.25935. [DOI] [PubMed] [Google Scholar]
- 3.Kele PG, van der Jagt EJ, Gouw AS, Lisman T, Porte RJ, de Boer MT. The impact of hepatic steatosis on liver regeneration after partial hepatectomy. Liver Int. 2013;33:469–475. doi: 10.1111/liv.12089. [DOI] [PubMed] [Google Scholar]
- 4.Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, Fava JL, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan S, Kirk EP, Mittendorfer B, Patterson BW, Klein S. Randomized trial of exercise effect on intrahepatic triglyceride content and lipid kinetics in nonalcoholic fatty liver disease. Hepatology. 2012;55:1738–1745. doi: 10.1002/hep.25548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 7.Amaya MJ, Oliveira AG, Guimaraes ES, Casteluber MC, Carvalho SM, Andrade LM, Pinto MC, et al. The insulin receptor translocates to the nucleus to regulate cell proliferation in liver. Hepatology. 2014;59:274–283. doi: 10.1002/hep.26609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feriod CN, Oliveira AG, Guerra M, Nguyen L, Mitchell-Richards K, Jurczak MJ, Ruan HB, et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type I Mediates Fatty Liver. Hepatology Communications. 2017;1:23–35. doi: 10.1002/hep4.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guerra MT, Fonseca EA, Melo FM, Andrade VA, Aguiar CJ, Andrade LM, Pinheiro AC, et al. Mitochondrial calcium regulates rat liver regeneration through the modulation of apoptosis. Hepatology. 2011;54:296–306. doi: 10.1002/hep.24367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pusl T, Wu JJ, Zimmerman TL, Zhang L, Ehrlich BE, Berchtold MW, Hoek JB, et al. Epidermal growth factor-mediated activation of the ETS domain transcription factor Elk-1 requires nuclear calcium. Journal of Biological Chemistry. 2002;277:27517–27527. doi: 10.1074/jbc.M203002200. [DOI] [PubMed] [Google Scholar]
- 11.Kruglov EA, Gautam S, Guerra MT, Nathanson MH. Type 2 inositol 1,4,5-trisphosphate receptor modulates bile salt export pump activity in rat hepatocytes. Hepatology. 2011;54:1790–1799. doi: 10.1002/hep.24548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz LN, Guerra MT, Kruglov E, Mennone A, Garcia CR, Chen J, Nathanson MH. Regulation of multidrug resistance-associated protein 2 by calcium signaling in mouse liver. Hepatology. 2010;52:327–337. doi: 10.1002/hep.23625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodrigues MA, Gomes DA, Leite MF, Grant W, Zhang L, Lam W, Cheng YC, et al. Nucleoplasmic calcium is required for cell proliferation. Journal of Biological Chemistry. 2007;282:17061–17068. doi: 10.1074/jbc.M700490200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirata K, Pusl T, O'Neill AF, Dranoff JA, Nathanson MH. The type II inositol 1,4,5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology. 2002;122:1088–1100. doi: 10.1053/gast.2002.32363. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian M, Metya SK, Sadaf S, Kumar S, Schwudke D, Hasan G. Altered lipid homeostasis in Drosophila InsP3 receptor mutants leads to obesity and hyperphagia. Dis Model Mech. 2013;6:734–744. doi: 10.1242/dmm.010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feriod CN, Nguyen L, Jurczak MJ, Kruglov EA, Nathanson MH, Shulman GI, Bennett AM, et al. Inositol 1,4,5-trisphosphate receptor type II (InsP3R-II) is reduced in obese mice, but metabolic homeostasis is preserved in mice lacking InsP3R-II. Am J Physiol Endocrinol Metab. 2014;307:E1057–1064. doi: 10.1152/ajpendo.00236.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 18.Leite MF, Thrower EC, Echevarria W, Koulen P, Hirata K, Bennett AM, Ehrlich BE, et al. Nuclear and cytosolic calcium are regulated independently. Proc Natl Acad Sci U S A. 2003;100:2975–2980. doi: 10.1073/pnas.0536590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2016;64:955–965. doi: 10.1002/hep.28456. [DOI] [PubMed] [Google Scholar]
- 20.Kruglov E, Ananthanarayanan M, Sousa P, Weerachayaphorn J, Guerra MT, Nathanson MH. Type 2 inositol trisphosphate receptor gene expression in hepatocytes is regulated by cyclic AMP. Biochem Biophys Res Commun. 2017;486:659–664. doi: 10.1016/j.bbrc.2017.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorn C, Engelmann JC, Saugspier M, Koch A, Hartmann A, Muller M, Spang R, et al. Increased expression of c-Jun in nonalcoholic fatty liver disease. Lab Invest. 2014;94:394–408. doi: 10.1038/labinvest.2014.3. [DOI] [PubMed] [Google Scholar]
- 22.Nagata J, Guerra MT, Shugrue CA, Gomes DA, Nagata N, Nathanson MH. Lipid rafts establish calcium waves in hepatocytes. Gastroenterology. 2007;133:256–267. doi: 10.1053/j.gastro.2007.03.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagoudakis L, Garcin I, Julien B, Nahum K, Gomes DA, Combettes L, Nathanson MH, et al. Cytosolic calcium regulates liver regeneration in the rat. Hepatology. 2010;52:602–611. doi: 10.1002/hep.23673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inaba Y, Furutani T, Kimura K, Watanabe H, Haga S, Kido Y, Matsumoto M, et al. Growth arrest and DNA damage-inducible 34 regulates liver regeneration in hepatic steatosis in mice. Hepatology. 2015;61:1343–1356. doi: 10.1002/hep.27619. [DOI] [PubMed] [Google Scholar]
- 25.Ling J, Chaba T, Zhu LF, Jacobs RL, Vance DE. Hepatic ratio of phosphatidylcholine to phosphatidylethanolamine predicts survival after partial hepatectomy in mice. Hepatology. 2012;55:1094–1102. doi: 10.1002/hep.24782. [DOI] [PubMed] [Google Scholar]
- 26.Pean N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, Branchereau S, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology. 2013;58:1451–1460. doi: 10.1002/hep.26463. [DOI] [PubMed] [Google Scholar]
- 27.Besnard A, Gautherot J, Julien B, Tebbi A, Garcin I, Doignon I, Pean N, et al. The P2×4 purinergic receptor impacts liver regeneration after partial hepatectomy in mice through the regulation of biliary homeostasis. Hepatology. 2016;64:941–953. doi: 10.1002/hep.28675. [DOI] [PubMed] [Google Scholar]
- 28.Nicou A, Serriere V, Hilly M, Prigent S, Combettes L, Guillon G, Tordjmann T. Remodelling of calcium signalling during liver regeneration in the rat. J Hepatol. 2007;46:247–256. doi: 10.1016/j.jhep.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Rodrigues MA, Gomes DA, Leite MF, Grant W, Zhang L, Lam W, Cheng YC, et al. Nucleoplasmic calcium is required for cell proliferation. J Biol Chem. 2007;282:17061–17068. doi: 10.1074/jbc.M700490200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kar P, Mirams GR, Christian HC, Parekh AB. Control of NFAT Isoform Activation and NFAT-Dependent Gene Expression through Two Coincident and Spatially Segregated Intracellular Ca2+ Signals. Mol Cell. 2016;64:746–759. doi: 10.1016/j.molcel.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzales E, Julien B, Serriere-Lanneau V, Nicou A, Doignon I, Lagoudakis L, Garcin I, et al. ATP release after partial hepatectomy regulates liver regeneration in the rat. J Hepatol. 2010;52:54–62. doi: 10.1016/j.jhep.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thevananther S, Sun H, Li D, Arjunan V, Awad SS, Wyllie S, Zimmerman TL, et al. Extracellular ATP activates c-jun N-terminal kinase signaling and cell cycle progression in hepatocytes. Hepatology. 2004;39:393–402. doi: 10.1002/hep.20075. [DOI] [PubMed] [Google Scholar]
- 33.Sankar N, deTombe PP, Mignery GA. Calcineurin-NFATc regulates type 2 inositol 1,4,5-trisphosphate receptor (InsP3R2) expression during cardiac remodeling. J Biol Chem. 2014;289:6188–6198. doi: 10.1074/jbc.M113.495242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drawnel FM, Wachten D, Molkentin JD, Maillet M, Aronsen JM, Swift F, Sjaastad I, et al. Mutual antagonism between IP(3)RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J Cell Biol. 2012;199:783–798. doi: 10.1083/jcb.201111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirata K, Dufour JF, Shibao K, Knickelbein R, O'Neill AF, Bode HP, Cassio D, et al. Regulation of Ca(2+) signaling in rat bile duct epithelia by inositol 1,4,5-trisphosphate receptor isoforms. Hepatology. 2002;36:284–296. doi: 10.1053/jhep.2002.34432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minagawa N, Nagata J, Shibao K, Masyuk AI, Gomes DA, Rodrigues MA, Lesage G, et al. Cyclic AMP regulates bicarbonate secretion in cholangiocytes through release of ATP into bile. Gastroenterology. 2007;133:1592–1602. doi: 10.1053/j.gastro.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Echevarria W, Leite MF, Guerra MT, Zipfel WR, Nathanson MH. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nature Cell Biology. 2003;5:440–446. doi: 10.1038/ncb980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leite MF, Thrower EC, Echevarria W, Koulen P, Hirata K, Bennett AM, Ehrlich BE, et al. Nuclear and cytosolic calcium are regulated independently. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2975–2980. doi: 10.1073/pnas.0536590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomes DA, Rodrigues MA, Leite MF, Gomez MV, Varnai P, Balla T, Bennett AM, et al. c-Met must translocate to the nucleus to initiate calcium signals. Journal of Biological Chemistry. 2008;283:4344–4351. doi: 10.1074/jbc.M706550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Angelis Campos AC, Rodrigues MA, de Andrade C, de Goes AM, Nathanson MH, Gomes DA. Epidermal growth factor receptors destined for the nucleus are internalized via a clathrin-dependent pathway. Biochemical & Biophysical Research Communications. 2011;412:341–346. doi: 10.1016/j.bbrc.2011.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michalopoulos GK, Khan Z. Liver regeneration, growth factors, and amphiregulin. Gastroenterology. 2005;128:503–506. doi: 10.1053/j.gastro.2004.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paranjpe S, Bowen WC, Mars WM, Orr A, Haynes MM, DeFrances MC, Liu S, et al. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology. 2016;64:1711–1724. doi: 10.1002/hep.28721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.