

Abstract
Background
South China encompasses complex and diverse landforms, giving rise to high biological diversity and endemism from the Hengduan Mountains to Taiwan Island. Many species are widely distributed across South China with similar disjunct distribution patterns. To explore the causes of these disjunct distribution patterns and their genetic consequences, we investigated the endemic species Père David’s Chinese Vole (Eothenomys melanogaster) by integrating geological and ecological factors. We analysed the genetic structure and divergence time of E. melanogaster based on fast-evolving mitochondrial and nuclear markers using Bayesian trees and coalescent species tree approaches. Historical scenarios of distribution range and demography were reconstructed based on spatial interpolations of genetic diversity and distance, extended Bayesian skyline plots, phylogeographic diffusion analysis, and ecological niche modelling (ENM) during different periods. We also assessed the relationships between geographical distance/ecological vicariance and genetic distance (isolation by distance, IBD; isolation by environment, IBE).
Results
The genetic analysis revealed three deeply divergent clades—Southeast, Southwest and Central clades, centred on the Wuyi Mountains, the Yunnan-Guizhou Plateau (YGP) and the mountains around the Sichuan Basin, respectively—that have mostly developed since the Pleistocene. IBD played an important role in early divergence, and geological events (sedimentation of plains and linking of palaeo-rivers) and IBE further reinforced genetic differentiation. ENM shows the importance of suitable habitats and elevations.
Conclusions
Our results suggest that the primary cause of the disjunct distribution in E. melanogaster is the high dependence on middle-high-altitude habitat in the current period. Mountains in the occurence range have served as “sky islands” for E. melanogaster and hindered gene flow. Pleistocene climatic cycles facilitated genetic admixture in cold periods and genetic diversification in warm periods for inland clades. During cold periods, these cycles led to multiple colonization events between the mainland and Taiwan and erased genetic differentiation.
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1168-3) contains supplementary material, which is available to authorized users.
Keywords: Disjunct distribution, Distinct intraspecific diversification, Eothenomys melanogaster, Genetic divergence, Isolation by distance, Isolation by environment, Sky islands, South China
Background
South China encompasses complex and diverse landforms. The high-altitude Hengduan Mountains (HM) and Yunnan-Guizhou Plateau (YGP) are in the west, and the terrain gradually descends to hilly areas in the east. The HM and adjacent areas are characterized by a series of parallel alpine ridges with dramatic ecological stratification and environmental heterogeneity [1, 2]. This complex topography supports one of the major biodiversity centres in the world, the southwest mountains of China [3, 4]. The mountain regions function as “sky islands” for montane species that depend exclusively on high-elevation environments [5, 6]. Dispersal of these species is strongly limited by low-elevation habitats and consequently results in genetic drifts among sky islands [7]. The southeast hilly areas, such as the Wuyi Mountains and Taiwan Island, are also described as hotspots for speciation and have high degrees of endemism [8, 9]. These regions have been influenced by tremendous climatic changes, with long-term cooler and drier tendencies during the Late Cenozoic [10, 11], and the uplift of the Qinghai-Tibetan Plateau (QTP).
The intraspecific diversification of montane species of South China is related to the uplift of the QTP since the Pliocene and the topographical complexity of the hilly areas [9, 12]. Previous studies have found that montane species migrate to low-elevation areas during glacial periods and exchange genes [13, 14]. Therefore, climatic fluctuations may also play an important role in shaping the genetic structure of montane species [8, 9]. Many species are widely distributed across South China [12], but montane species dwell at high elevations and are discontinuously distributed in this area [15, 16]. Disjunct distributions are very common in this area [9, 16]; however, in contrast to the extensively studied Southwest China, few studies have focused on the entirety of South China, particularly regarding the geographical and ecological vicariance of species within their geographical ranges and their geographical patterns of genetic diversity.
Eothenomys is a typical endemic genus of the South China mountainous regions [17]. Most species within this genus are distributed within a very narrow range and represent one of the rapid radiations of the QTP and HM (also see [18]). However, E. melanogaster is unique in this genus; it has a much wider distribution range across the whole of South China, extending to Taiwan Island. As a montane specialist, E. melanogaster has a known altitudinal range from 700 to 3,000 m [19]; environments with too low or too high elevations are not suitable. Therefore, E. melanogaster has a prominently disjunct distribution in South China (Fig. 1), which makes it an interesting species for investigating effects of geological and climatic events on the genetic patterns of species across South China.
Fig. 1.
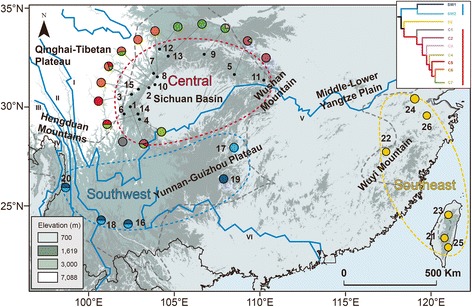
Sampling sites and extant distribution of E. melanogaster, with the major geographical features within the distributed area labelled. Great rivers in this area are indicated by Roman numerals: I, Jinsha River; II, Lancang River; III, Nu River; IV, Dulong River; V, Yangtze River; and VI, Pearl River. The locality codes and coordinates are presented in Additional file 1: Table S1. The elevation is shown with a legend in the bottom left. The colours of the sampling sites indicate mitochondrial clades and correspond to the colours in the phylogenetic tree shown in Fig. 2 (shown here in the top-right frame). The altitude information is freely available at http://www.worldclim.org/, and the base map from Esri Content Packages
In this study, we investigated the population genetic structure of E. melanogaster, historical divergence scenarios (including patterns of diffusion and demographic variation, divergence time between clades, and divergence scenarios based on ecological niche modelling, ENM) and the effects of isolation by distance (IBD) and isolation by environment (IBE) by assessing the relationships between geographical distance/ecological vicariance and genetic distance in E. melanogaster. We aimed to answer the following questions: 1) What is the main cause of the disjunct distribution of E. melanogaster: geological events or climatic oscillations? How is the genetic structure of E. melanogaster shaped among and within different areas (such as the YGP and Taiwan Island)? 2) As a representative of the South China hotspot, can E. melanogaster inform us about concerted evolution within this community in terms of the relative roles of geological events vs. climatic oscillations? Considering the cold-adaption trait, is genetic diversity within E. melanogaster representative of the diversification of extant cold-adapted species through this region? Based on these two broader questions and previous studies, we hypothesized that the tectonic events since the Late Pliocene induced intraspecific divergence followed by gene admixture during the climatic oscillations.
Methods
Ethics statement
We carried out this study strictly following to the animal research protocol IOZ-2006 approved by the Animal Care Committee of Institute of Zoology, Chinese Academy of Sciences (IOZ, CAS). Eothenomys melanogaster individuals were captured with permission of the local Protection and Research Centre and the Forestry Administration of China. All of the individuals were captured using cages and then immediately euthanized by cervical dislocation. All efforts were made to minimize potential pain and suffering. Intact skulls of all the captured individuals were collected and preserved in 100% ethanol in the field and cleaned after the field survey for further taxonomy identification. Muscle tissues to be employed for molecular analysis were collected and preserved in 100% ethanol. All specimens and skulls collected in the field were deposited in the mammal collections of the National Zoological Museum of China (IOZ, CAS), Sichuan Academy of Forestry, and Kunming Natural History Museum of Zoology (Institute of Zoology, Chinese Academy of Sciences) (KIZ, CAS).
Sampling, DNA extraction and gene sequencing
We sampled 241 individuals at 26 localities (Fig. 1 and Additional file 1: Table S1). We amplified a total of six genes: the mitochondrial cytochrome b (cytb) gene, the nuclear breast cancer (BRCA1) gene, the external transcribed spacer 2 (ETS2) gene, the growth hormone receptor (GHR) gene, intron 1 of the glucose-6-phosphate dehydrogenase (G6pd) gene, and exon 1 of the interphotoreceptor retinoid-binding protein (IRBP) gene. In addition, we used two sequences of cytb obtained from GenBank (accession numbers: AY426681 and AY426682 [17]). The primers used were as follows: cytb: H15985/L14734 [20]; BRCA1: F180_arv/R1240_arv [21]; ETS2: ETS2F/ETS2R [22]; GHR: GHR5 forward/GHR6 reverse [23]; G6pd: G6pdint1L/G6pd-int1H [24]; and IRBP: IRBP217/ IRBP1531 [25]. cytb from museum collections was amplified with primers described by Petrova et al. [26].
Polymerase chain reaction (PCR) amplifications were performed in a 25 μl volume with 0.25 μl Taq polymerase (Takara Shuzo Co. Ltd., Otsu, Japan), 3 μl of DNA template (~ 60 ng/μl), 2.5 μl 10× PCR buffer, 2 μl dNTPs (2.5 μM), and 1 μl primers (10 μM). The reactions were adjusted to a final volume of 25 μl with ddH2O. The PCRs were performed using the following procedure all the markers: 94 °C for 10 min; 35 cycles of 1 min at 94 °C, of 45 s at 54 °C, and of 90 s at 72 °C; followed by a final extension step at 72 °C for 10 min. The complete sequences were assembled using BIOEDIT 7.2.5 [27] and aligned using MUSCLE implemented in MEGA6 [28]. Considering that mitochondrial DNA may experience substitution saturation because of its rapid evolutionary rate, we assessed substitution saturation using the test described by Xia et al. [29] as implemented in DAMBE 6 (reference: Assessing substitution saturation with DAMBE) [30].
Phylogenetic and divergence time analysis
To evaluate the intraspecific phylogenetic relationships among the samples, individual gene trees were reconstructed based on cytb, each nDNA, and concatenated cytb and nDNA genes using Bayesian inference (BI) methods in MrBayes 3.2.5 [31] and maximum likelihood (ML) methods in RAxML v.8.2.10 [32]. The best fit model of nucleotide substitution for each locus was selected using MrModeltest v2.3 [33]. Two parallel runs of one cold and three heated Markov chain Monte Carlo (MCMC) analyses were performed for 15 million generations or more, with trees sampled every 1,000 generations to produce convergence (SD < 0.01). The first 25% of the Markov chain samples (N = 20,000) were discarded as burn-in, and the remaining samples were used to generate majority rule consensus trees. ML phylogenies were inferred using a GTRGAMMAI model of evolution and 1,000 bootstrapping replicates. Final trees were then viewed in FigTree 1.4.2 (available at http://tree.bio.ed.ac.uk/software/fig-tree/). To better visualize the relationships among haplotypes, median-joining (MJ) networks [34] were generated using cytb in POPART 1.7 [35]. Gaps and missing sites in the sequences were excluded from the analysis. The pairwise genetic distances of cytb sequences were calculated with MEGA6 using the Kimura two-parameter (K2P) distance between and within each clade.
We estimated divergence time using BEAST 1.8.2 [36] to determine the relationship between genetic divergence and geological events. The analysis was performed using the cytb gene because of its higher degree of variation. We calibrated with the soft bounds of three fossil calibration points as follows, and the parameters were set to log-normal distributions with a 95% interval boundary. Fossil calibration point 1: The oldest fossil of E. melanogaster is dated at 2.03 Ma [37]. We assigned a minimum age of 2.03 Ma [offset = 1.806; log (mean) = 0.37; median = 2.030; 97.5% quantile = 3.399]. Fossil calibration point 2: The occurrence time of Eothenomys is dated at the Late Pliocene (3.60–2.58 Ma) [37]. We assigned a minimum age of 3 Ma [offset = 3.0; log (mean) = 0.6; median = 3.364; 97.5% quantile = 5.583]. Fossil calibration point 3: A Russian fossil of the most recent common ancestor (MRCA) of Myodes dated to at least 2.6 Ma [38] used by Kohli et al. [39]. We assigned a minimum age of 2.60 Ma [offset = 2.60; log (mean) = 0.20; median = 2.721; 97.5% quantile = 3.461]. The outgroups are described in Additional file 1: Table S2. Prior to divergence calibration, the cytb clock model was selected based on marginal likelihood estimated (MLE) from stepping stone and path sampling [40] with 100 path steps, 1 million iterations and samples every 1,000 generations and was run twice to ensure convergence. According to the MLE analysis, a relaxed log-normal clock (log marginal likelihood = − 6056.96/− 6058.59) was favoured over a strict clock (log marginal likelihood = − 6128.77/− 6131.26) for the cytb dataset.
To estimate the species tree and test the clade divergence revealed in the BI tree, concatenated cytb and nDNA sequences were analysed using *BEAST [41], which was implemented in BEAST 1.8.2. The substitution models were unlinked, and the substitution parameters were set according to the MrModeltest results. We chose Yule Process species tree priors. Nuclear gene clock models were assessed using the ucld.stdev parameter based on preliminary runs in which the uncorrelated relaxed log-normal clock prior was applied and the resulting distribution examined, as recommended in the program documentation [42, 43]. The uncorrelated relaxed log-normal clock was set for all loci. The MCMC chains were run for 200 million generations, with sampling every 5,000 generations. The convergence of the MCMC chains was examined in Tracer 1.6, and the first 25% of runs were discarded as burn-in. The trees and posteriors were displayed and edited in FigTree 1.4.2. To test the species delimitation, we used BP&P 3.2 [44] with all samples and all loci. Reversible-jump Markov chain Monte Carlo (rjMCMC) analyses were run for 100,000 generations with a burn-in phase of 8,000. We used a gamma prior G (2, 1,000) on the population size parameters (θs) and a gamma prior G (2, 2,000) on the age of the root in the species tree (τ0). The other parameters were assigned as the prior defaults [45]. Each analysis was run twice with different starting seeds to ensure consistency. In addition, the species tree analyses were performed to avoid incomplete lineage sorting if we observed congruent structure between the concatenated gene tree (mtDNA+nDNA) and the species tree.
Demographic analyses and genetic indices visualization
The general patterns of diversity in the mtDNA and nuclear loci, we calculated the nucleotide diversity (π), the mean number of pairwise differences (k), the number of haplotypes (nh) and the haplotype diversity (h) for each clade of mtDNA and all individuals of mtDNA and nuclear loci using DnaSP 5.10.01 [46]. The significance was tested using 10,000 permutations in Arlequin 3.0 [47]. Tajima’s D [48] and Fu’s Fs [49] were conducted to test the neutrality and demographic history based on cytb and nuclear loci in DnaSP with 10,000 bootstrap replicates. Significant negative values for Tajima’s D and Fu’s Fs generally indicated a recent demographic expansion.
To better visualize the relationship between geographical pattern and genetic diversity/genetic divergence, the nucleotide diversity (π) and pairwise genetic distance were spatially interpolated using the Kriging method [50] implemented in the ‘Spatial Analyst Tools’ of ArcGIS 10.0 (ESRI, Redlands, CA, USA). We sampling the localities contained within a buffer of 1° radius around each sampling locality to include at least two individuals per locality in each genetic diversity calculation. We thus sampled a total of 25 localities. The results were masked with current suitable habitat estimated in MAXENT with the “10% logistic threshold” (see below).
We used BEAST to estimate molecular-based demographical fluctuation based on mtDNA only. The demographic variation was estimated using extended Bayesian skyline plots (EBSP), with three clades analysed separately. The analyses were run for 50,000,000 generations for each clade; each clade was sampled every 5,000 generations. All samples were included, and the ucld.mean parameter was used as previously estimated in the time calibrating analysis. The final convergence was assessed with effective sample size (ESS) in Tracer v1.6 [40]. An ESS value above 200 was considered acceptable.
Reconstruction of historical scenarios
We implemented the discrete phylogeographical analysis in BEAST v 2.3.7 [51] to examine range expansion among the study sites through time. We used haplotype data sets with one individual per haplotype per locality to achieve better convergence and reduced the dataset to 104 individuals with cytb. The time calibrating also used the ucld.mean parameter same as for the EBSP analyses. The analysis was run for 30,000,000 generations, with sampling every 3,000 generations. Convergence of runs and thus support for the inferred ages of migration events was achieved by ensuring that the ESS for the ‘geotreelikelihood’ prior was greater than 200 in the log file. The spatial-temporal diffusion pattern was then reconstructed using Time Slicer in SPREAD v1.0.6 [52]; and GOOGLE EARTH (Google, California, USA, available at http://google.com/earth/) was used for the final visualization.
We reconstructed range variation during the Last Glacial Maximum (LGM) to estimate the climatic effect of Pleistocene ice periods. We obtained climatic data from the current conditions and the LGM (~ 21,000 years before present). We used two models of the LGM climate: the community climate system model [CCSM ver. 3; [53]] and the model for interdisciplinary research on climate [MIROC ver. 3.2; [54]]. Nineteen bioclimatic variables at a resolution of 2.5 arc-min for each period were downloaded from the WorldClim database version 1.4 (available at http://www.worldclim.org/) [55]. To reduce the amount of computation, we masked all variables to include only 60° to 125°E and 15° to 35°N. All species occurrence data were collected from our sampling localities, museum records from the National Zoological Museum of China, and occurrence records from the Global Biodiversity Information Facility database (available at http://www.gbif.org/). Finally, 139 localities were used in analysis. As the influence of the effect of over-fitting, we firstly tested the correlation among climatic variables and removed one variables if two variables were highly correlated (Pearson’s correlation > 0.8). We further performed model fitness analysis in ENMTools 1.4.4 [56] by examining beta regulation values from 1 to 20. We finally kept the climatic variables of BIO1, BIO2, BIO4, BIO12, BIO14, BIO15, BIO18 and BIO19, with a beta value of two. Then, we constructed the model by randomly selecting 80% of the occurrence data, and 20% of the data were left to test the accuracy of the model in MAXENT v3.3.3 k [57]. The setting of parameters used the default convergence threshold (10− 5), 2,000 maximum iterations and 10 replications. MAXENT was performed to test the model by calculating the average area over ten replications under the receiver operating characteristic curve (AUC) and the binominal probabilities indicating the predictive ability of the model. We calculated the average elevation of suitable habitat during the LGM and the current period in ArcGIS 10.0. The area of suitable habitat was based on a 10% logistic threshold from the MAXENT result.
Examination of the effects of IBD and IBE
The effective population size (Θ = effective population size, Ne x mutation rate, μ) and effective migration rates (M = migration rate, m/mutation rate, μ) between sample sites were calculated using MIGRATE 3.6.11 [58, 59] to estimate the gene flow among the three maternal clades that were found. To eliminate bias caused by unequal population size, we subsampled the dataset to include 45 individuals, with 15 individuals per clade estimated based on all loci. MIGRATE used BI with long chains (500,000 steps sampled, 5,000 steps recorded) and 1,000 burn-in per chain. The mutation scaled M entering and leaving each population per generation and the mutation scaled Θ were estimated applying the Bayesian search strategy to determine whether there was asymmetrical gene flow between populations. Nem was calculated by multiplying M and Θ.
We implemented two analyses to assess the relationships between geographical distance/ecological vicariance and genetic distance. We first examined the effect of IBD, which was estimated by the correlation between the genetic distance and geographic distance (Euclidean distance) of each pair of localities. A Mantel test was applied to calculate correlations using the package VEGAN [60] in R [61]. Second, we tested ecological vicariance (IBE) between clades using spatial evolutionary and ecological vicariance analysis (SEEVA; [62]). We evaluated four ecological characters: mean annual temperature (BIO1), annual precipitation (BIO12), temperature seasonality (standard deviation*100, BIO4), and precipitation seasonality (coefficient of variation, BIO15). Climatic traits of each locality were extracted using ArcGIS 10.0. SEEVA was then used to divide these quantitative traits into four quartiles and calculate the correlations between ecological shifts and phylogenetic splits represented by Fisher’s exact tests and divergence indices (D).
Results
Phylogenetic and divergence time analysis
A total of 241 individuals from 26 localities were sampled, including data from GenBank (Fig. 1). We obtained six genetic markers from PCR: 1,074 base pairs (bp) of the cytb gene, 930 bp of the BRCA1 gene, 935 bp of the ETS2 gene, 777 bp of the GHR gene, 633 bp of intron 1 of the G6pd gene, and 1,210 bp of exon 1 of the IRBP gene. The sequence dataset generated herein is available in GenBank, and the accession numbers are listed in Additional file 1: Table S1. The saturation test showed that none of the genes we used indicated significant saturation (Iss < Issc, P < 0.001, data not shown).
BI and the ML trees based on cytb only, concatenated cytb and nDNA clearly indicated three clades: Southwest (SW), Southeast (SE) and Central (C) clade (Fig. 2a). The SE clade and C clade showed a closer relationship. The haplotype network showed the same result (Fig. 2b). In the C clade, the distribution of subclades was similar to a ring pattern around the Sichuan Basin, with early-diverged subclades at both ends (subclades C1, C2, C3, and C4) and a recent diversification in the northwest margin of the basin (subclades C5, C6, and C7; Fig. 1). However, the phylogenetic relationships based on different nuclear loci were reconstructed with low support except for that those based on ETS2 (see Additional file 2: Figure S1 for details). K2P analysis showed that the cytb genetic distance between different clades ranged from 6.44% to 8.04%.
Fig. 2.
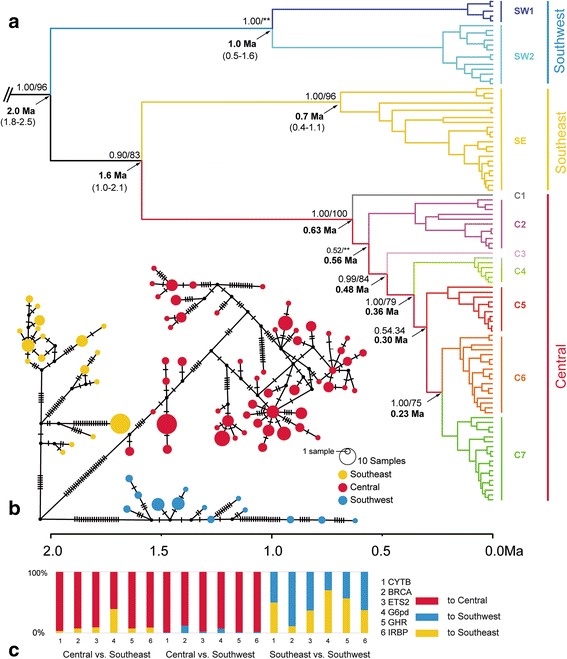
a Phylogenetic tree and divergence time estimation based on BEAST analysis of the cytb haplotypes of E. melanogaster. The numbers beside the nodes are posterior probabilities and bootstrap, and those below the nodes are divergence times estimated by BEAST (Ma, 95% HPD in brackets). b Median-joining network based on cytb with node sizes proportional to the frequencies of haplotypes. The number of hatch marks between haplotypes is proportional to the number of mutational changes. Dots represent undetected haplotypes. c Gene flow between maternal clades as estimated by MIGRATE (represented in %)
The divergence time data indicated that a hierarchical pattern had occurred since the most recent ancestor of the entire in-group, which was estimated to have existed at 2.0 Ma [1.8–2.5 Ma, 95% highest posterior density (HPD); Fig. 2a], placing the ancestry of all living representatives of the species in the Early Pleistocene. The SW clade was the first to diverge, and the subsequent divergence events of the SE and C clades occurred at 1.6 Ma (1.0–2.1 Ma, 95% HPD).
In the BP&P species delimitation analysis, three clades were strongly supported (posterior = 1.0). The *BEAST and BP&P tree showed inconsistent structure with the BI and ML tree for concatenated cytb and nDNA (Additional file 3: Figure S2). Based on the *BEAST and BP&P tree, the SW and C clades showed a closer relationship to each other than to the SE clade, and the SE clade was the first to diverge. However, the posterior probabilities were low for this structure in these two analyses.
Demographic analyses and genetic indices visualization
The SE clade had the highest mitochondrial nucleotide diversity (2.47%), the C clade had the second highest (1.79%), and the SW clade had the lowest (1.54%) (Table 1). The genetic diversity of nuclear loci ranged from 0.6% to 0.23%. The interpolated genetic diversity showed an upward trend from east (0.062%) to west (6.52%), whereas genetic distance showed deeper divergence from north to south (Fig. 3a). The genetic distance plot illustrates the degree of divergence between each clade. The values of Tajima’s D and Fu’s Fs were mostly negative but non-significant, with the only exception being the D value (significantly negative) of IRBP and the Fs values (significantly negative) of BRCA1, ETS2, and IRBP (Table 1). The demographic scenario obtained by the EBSP showed that the three clades have generally remained stable since 0.01 Ma, although the C clade showed a slight decline (Fig. 3b).
Table 1.
Genetic diversity and neutrality test estimates based on mtDNA and nDNA for all individuals and each clade
| Marker | Clade | Number | nh | s | Pi (%) | h | Pairwise difference (k) | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|---|---|
| cytb | all | 241 | 86 | 91 | 4.32 | 0.98 | 31.38 | −0.13 | −1.43 |
| SE | 49 | 20 | 128 | 2.47 | 0.88 | 25.52 | −0.11 | −0.44 | |
| C | 163 | 61 | 54 | 1.79 | 0.98 | 16.30 | −0.09 | −0.57 | |
| SW | 29 | 14 | – | 1.54 | 0.91 | 12.16 | −0.11 | − 0.04 | |
| BRCA1 | 62 | 19 | 19 | 0.23 | 0.86 | 2.02 | −0.08 | − 0.08 | |
| ETS2 | 71 | 24 | 32 | 0.60 | 0.87 | 4.86 | −0.08 | − 0.36 | |
| G6pd | 48 | 8 | 7 | 0.32 | 0.81 | 1.51 | −0.09 | 0.27 | |
| GHR | 68 | 16 | 21 | 0.59 | 0.87 | 4.37 | −0.10 | −0.16 | |
| IRBP | 65 | 32 | 39 | 0.40 | 0.96 | 3.51 | −0.07 | −0.31 |
The main clades, numbers of individuals (n), number of haplotypes (nh), number of polymorphic (segregating) sites (s), nucleotide diversity (Pi), haplotype diversity (h), and mean number of pairwise nucleotide differences (k). Values in bold indicate significance based on the P value
Fig. 3.
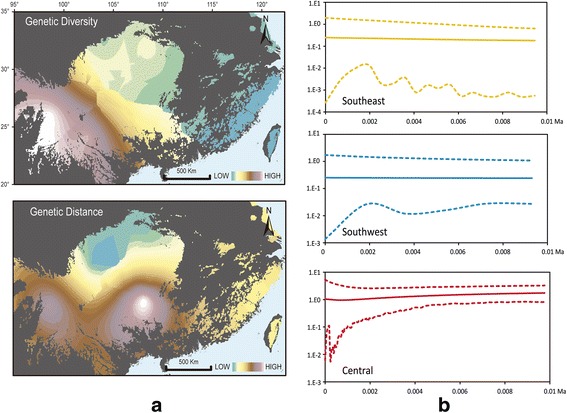
a Interpolated distribution pattern of genetic diversity and genetic divergence. The distribution range was masked with habitat estimated by MAXENT with a range of 10% logistic threshold. b Extended Bayesian Skyline plots for all the main clades of E. melanogaster. The solid lines indicate the median value of effective population size; the dashed lines denote the 95% highest posterior probability interval. The base map comes from Esri Content Packages
Reconstruction of historical scenarios
The phylogeographic diffusion analysis reconstructed a pattern suggesting that E. melanogaster originated near the YGP (Fig. 4) and that the three clades (SW, C, and SE) then likely derived from YGP, Sichuan Basin and Wuyi Mountains, respectively. The earliest fossil record was found in the Wushan Mountains, near YGP. Many locations experienced multiple colonization events and all three clades spread rapidly in the meantime. E. melanogaster was inferred to have migrated twice to Taiwan Island during the glaciation periods. The ancestors of the twice colonization have appeared at the end of penultimate glaciation period (130 Ka) (Fig. 4c) and the colonization events have finished at the end of last glaciation period (10.4 Ka) (Fig. 4e) [63].
Fig. 4.
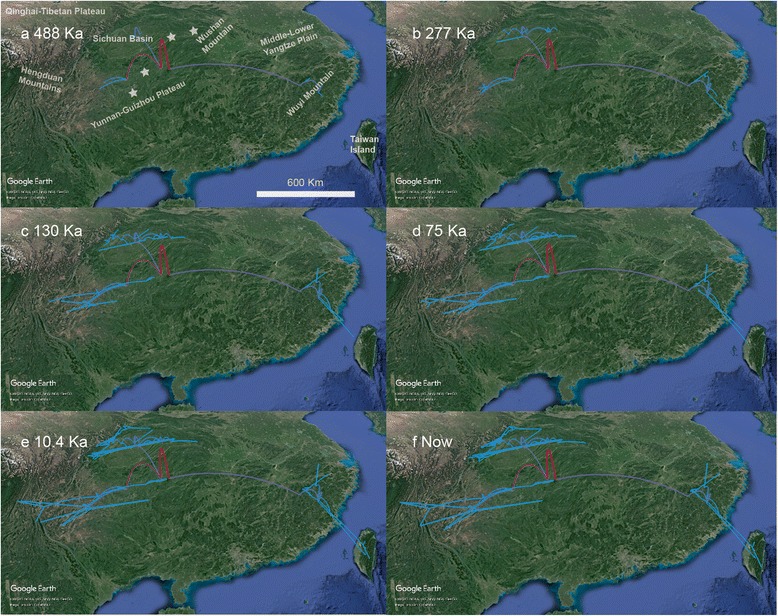
Results of the phylogeographic diffusion analysis of time points at (a) 488 Ka when E. melanogaster originally covered its extant distribution range, (b) the beginning of the penultimate glaciation period (277 Ka), (c) the end of the penultimate glaciation period (130 Ka, (d) the beginning of the last glaciation period (75 Ka), (e) the end of the last glaciation period (10.4 Ka), and (f) present. Asterisks represent the fossil records of E. melanogaster. The red line represents the ancient time and the blue line represents the current time. The map used here is from Google Earth
In the current ENM, the area under the ROC (Receiver operating characteristic) curve (AUC; Area under curve) was close to one (0.997 ± 0.002), indicating better than random prediction (0.5 = random, 1 = maximum), and the binomial probability was P < 0.001. The ENM shows the importance of suitable habitats and elevations (Fig. 5). The suitable habitats showed more fragmentation in current conditions, especially in low-latitude regions. E. melanogaster currently tends to live at higher elevations (mean elevation calculated based on 10% logistic threshold: now: 1,397.033 m), whereas its suitable habitat was lower in the north and expanded southward during the LGM (MIROC 1,134.238 m, CCSM: 1,236.908 m).
Fig. 5.
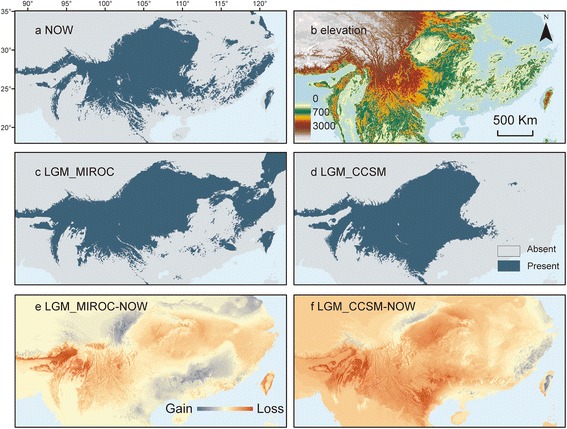
Ecological niche modelling of suitable habitats for E. melanogaster generated by MAXENT. The simulations were conducted based on the climatic conditions of (a) the current climate, (c) the Last Glacial Maximum (LGM) of the MIROC model and (d) the LGM of the CCSM model. b is the elevation distribution of this area. The continental margin in the LGM differed from the others because of the lower sea level at the time. The suitable habitat was based on the 10% logistic training threshold estimated by MAXENT. Differences in occurrence between the two periods are shown in (e) and (f). Gains (blue) mean the increase of suitable habitat in current condition, and losses (red) mean opposite. The altitude and bio-climate information are freely available at http://www.worldclim.org/, and the base map from Esri Content Packages
Examination of the effects of IBD and IBE
Gene flow among the three clades showed similar patterns for all six genes: the SW and SE clades produced migrants entering the C clade, and the reverse gene flows from the C clade to the other two clades were much lower (Fig. 2c and Additional file 1: Table S3). Gene flow between the SW and SE clades was bidirectional. A Mantel test showed strong correlations between geographical distance and genetic distance (r = 0.4363, P = 0.0001).
Based on SEEVA the three clades all showed significant divergence in all the examined ecological variables (Fig. 6). The C clade was generally found at a lower level of mean annual temperature with higher seasonal variation and a lower level of precipitation with higher seasonal variation, whereas the SW and SE clades lived in habitats with a higher mean annual temperature and precipitation with less seasonal variation. Southern populations were located at higher elevations than northern populations (Fig. 6), a pattern that is possibly related to the latitudinal gradient of the distribution of each clade.
Fig. 6.
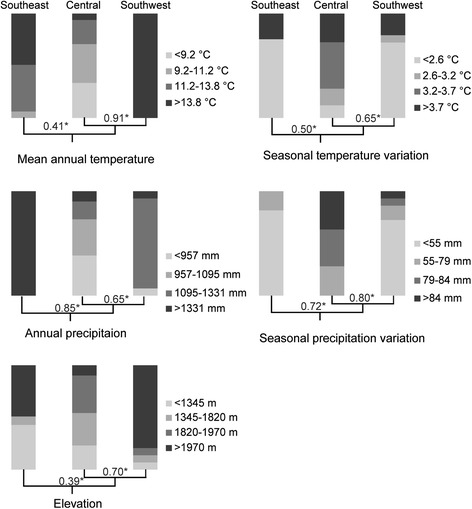
The results of the spatial evolutionary and ecological vicariance analysis (SEEVA) of E. melanogaster using mean annual temperature, annual precipitation, seasonal climatic variation and elevation coded as four quantitative section states. The total height of each histogram bar equals 100% of the observations for each clade, and the greyscale of the histograms represents the four different states. The underlined phylogeny was based on the Bayesian mitochondrial phylogeny and the species tree estimated by BP&P and *BEAST. The nodal values represent the Index of divergence (D), and asterisks indicate significant differences between sister groups using a Bonferroni criterion of P ≤ 0.05
Discussion
Intraspecific diversification with disjunct distribution
Our study performed comprehensive phylogeographic analyses to infer the evolutionary history of E. melanogaster throughout the Quaternary and to obtain insights into the processes that shaped its disjunct distribution pattern. The mtDNA analyses revealed a clear hierarchical pattern of divergence with geographical structure for E. melanogaster (Fig. 2a). Three reciprocally monophyletic maternal clades were supported: SE clade (consisting of individuals from Taiwan Island and southeast coastal China), C clade (individuals from mountainous areas surrounding the Sichuan Basin and the southeast edge of the QTP) and SW clade (individuals from the YGP and the southern part of the HM) (Fig. 1). The analyses, which included analyses of nuclear loci, all identified these three genetic clades but showed incongruence between the cytb and nDNA trees. This discordance in inference between mtDNA and nDNA genes is likely attributed to recent mitochondrial admixture or sex-biased gene flow in this species [64, 65]. However, E. melanogaster shows no significant recorded difference in dispersal capability between males and females [66]. Our nDNA tree indicated a geographical structure similar to that obtained based on mtDNA, and the support values of the three clades were fairly high, arguing against sex-biased gene flow [67]. Furthermore, the detected bidirectional gene flow between the SW and SE clades also argues against sex-biased gene flow. The only difference in structure between the mtDNA and nDNA phylogenies was in the position of the SE and SW clades and secondary mitochondrial admixture is the most likely reason (Fig. 2 and Additional file 2: Figure S1). Furthermore, incomplete lineage sorting was not observed in our study, as congruent structure was observed between the concatenated gene tree (mtDNA + nDNA) and species tree (BP&P and *BEAST; Additional file 2: Figure S1 and Additional file 3: Figure S2).
The phylogeographic diffusion analysis reconstructed a pattern showing that E. melanogaster originated from around the YGP (Fig. 4a), which is consistent with the earliest fossil record (2.03 Ma) of E. melanogaster [37]. The first divergence between the SW clade and the other two clades occurred during the Early Pleistocene (2.0 Ma). The SW clade could communicate with the C clade until the late Early Pleistocene, when the rapid uplift of the QTP and the YGP facilitated the development of the palaeo-Jinsha River that drained into a palaeolake in the Sichuan Basin [68]. During the boundary of the Early Pleistocene and Middle Pleistocene (0.7 Ma at the latest), the Wushan Mountains were intersected, which resulted in the formation of the modern Yangtze River [1]. This time period coincided with the Kunlun-Huanghe Movements, when the QTP and adjacent mountainous regions were strongly uplifted [69]. The connection of the palaeo-Yangtze River contributed to the split and further prevented gene flow between the SW and C clades. The SE and C clades subsequently diverged at approximately 1.6 Ma, when the elevation of the QTP during that time was low (< 2,000 m) [70, 71] and the plains in the southeast were at a higher elevation (the Middle-Lower Yangtze Plain had subsided approximately 20–360 m during the Quaternary) [1]. Moreover, a Mantel test showed a strong correlation between geographical distance and genetic distance. Therefore, the divergence of the SE clade was more likely a consequence of long-distance dispersal than of the vicariance induced by the landscape. The SE clade resided in the Wuyi Mountains, flourished and then spread to adjacent areas as the mountains in the southeast continuously rose.
The results of the phylogeographic diffusion analysis indicated that the distribution of E. melanogaster had already covered its extant distribution range by approximately 488 Ka (Fig. 4a). Along with the continuous uplift of the mountains and sedimentation of the plains and basins of South China, E. melanogaster gradually adapted to medium-high elevations and a cold environment, maintained and centred on the YGP, the mountainous areas surrounding the Sichuan Basin and the Wuyi Mountains (Fig. 1). Populations from lower elevations might have become extinct or subdivided because of unsuitable environments [72]. Ecological divergence further reinforced the genetic differentiation that resulted from the geographical isolation [43]. The IBE results indicated distinct vicariance of ecological traits among these three clades, especially between the C clade and each of the other two (Fig. 6). The C clade showed wider ecological niche utilization. In contrast to the patterns of species that live at lower elevations, such as the grey-cheeked fulvetta and the red-headed tree babbler [9, 73], the diversification of the high-altitude cold-adapted community represented by E. melanogaster is deeply influenced by the Pleistocene interglacial within warm-phase refugia, followed by secondary contact during cold phases [4, 74–76]. This pattern is also counter to the general dogma of glacial refugia and secondary contact during warm phases [77, 78]. The extant disjunct distribution pattern of E. melanogaster formed in response to the combination of these conditions and adds an important example to the growing literature for a full understanding of Pleistocene dynamics as a seesaw mechanism of diversification.
Genetic admixture during Pleistocene climatic oscillations
Although the main divergence occurred as a result of IBD and geological events, Pleistocene climatic changes were profoundly imprinted in the genetic structure, as was clearly supported by the ENM results (Fig. 5). Unlike some other thermophilic species, such as four species of Passeriformes birds [2], Quasipaa boulengeri [12], and Mus musculus [79], E. melanogaster exhibited traits (either psychrophilic or environment dependent) that led to expansion in the glacial period and contraction in the interglacial period. This result is similar to results found for some previously studied species in this region [3]. The middle elevational environment remained suitable in each period, whereas the suitability of low and high areas fluctuated (Fig. 5e and f, respectively).
During the ice ages, populations of E. melanogaster shifted to lower elevations because of the expansion of habitat in low-altitude areas such as the Sichuan Basin (elevation range from 200 to 750 m) and Middle-Lower Yangtze Plain and degradation of habitat in high-altitude areas such as large mountains in the northwest of Sichuan Basin, Wuyi Mountains and the Central Mountains of Taiwan (elevations over 3,000 m). The mean elevation shifts of E. melanogaster-applicable habitat between the LGM and current conditions intuitively reflected the tendency that facilitated genetic exchange. In warm periods, high-altitude localities acted as “sky islands” for E. melanogaster and resulted in genetic divergence [7]. The high levels of subclade diversity of the C clade (Fig. 2a) can be explained by this scenario. The genetic divergence that occurred during the interglacial periods and the secondary contact that occurred during the glacial periods shaped the current genetic structure. Although the analysis of nuclear data did not yield well-resolved relationships within the C clade, the weak consistency of the nuclear data with mitochondrial subdivision implied gene flow and genetic admixture.
Populations from different sites in the SE clade showed a paraphyletic structure. According to the results of the phylogeographic diffusion analysis, the whole SE clade population originated and expanded from the Wuyi Mountains area (Fig. 4). Taiwan Island is situated 230 km from the Chinese mainland. During the Pleistocene glaciations, the sea level of the Taiwan Strait fell sufficiently to create a land bridge between Taiwan and the mainland many times, which promoted contact between the organisms on both sides [1]. Populations from Taiwan Island were not isolated and repeatedly experienced colonization via the land bridge during cold periods. Similar cases of gene flow between populations in Taiwan and the mainland have been reported for two bird species [9].
Two long-distant branches with weak geographic association are found within the SW clade (Fig. 1). This pattern is associated with two distinct colonization events of two different ancestors, as indicated by the results of the phylogeographic diffusion analysis (Fig. 4). This particular genetic structure was possible due to the stable environment of the mountains of Southwest China during the Pleistocene. The phylogenetic relationships and divergence pattern revealed that the SW clade has the greatest extent of ancestral polymorphisms [77], which was also generated by the asymmetric gene flow among the three clades (Fig. 2c) [78]. Moreover, because E. melanogaster lives between 700 m and 3,000 m above sea level, we suspect that large mountain ridges (over 3,000 m) can act not only as corridors for this species but also as barriers. Therefore, a stable environment led to low diversification, and the dual effects of mountains shaped distant phylogenetic relationships within clades.
Demographic history and diversity patterns
Haplotype diversity (h) and nucleotide diversity (Pi) can reveal the general demographic history of populations [80]. The C clade showed high h and low Pi values, which suggests rapid population growth from an ancestral population (Table 1). Moreover, a similar star-shaped network and small recent fluctuations suggests that the C clade might have experienced recent expansion [14]. In contrast, the SE clade has low h and high Pi values due to a short bottleneck in a large ancestral population [81]. However, the EBSP shows that the SE clade was stable during the last 0.1 Ma (Fig. 3b). In addition, low h and high Pi implies possible admixture of samples from small, geographically subdivided populations [82]. This finding is congruent with the repeated colonization and weak phylogeographic pattern. However, both the SE and C clades show low genetic diversity within localities, which likely indicate that the isolation effect of low elevations led to greater genetic exchange within localities rather than among them [83].
Among the three clades, the SW clade shows the lowest overall genetic diversity and genetic divergence (Table 1); however, the genetic diversity and genetic divergence of each locality within this clade are the highest (Fig. 3a). The complex terrain of Southwest China and stable historical environment facilitated stable species evolution without severe fluctuations [3, 9] and preserved the high levels of genetic diversity. The EBSP analysis indicate that the SW clade has remained stable over the last 0.1 Ma (Fig. 3b). However, further sample collections in the area of the SW clade are needed to facilitate the understanding of the intra-clade phylogeographic pattern and demographic history.
Conclusions
In this study, comprehensive phylogeographic analyses were performed to infer the evolutionary history of E. melanogaster throughout the Quaternary and to obtain insights into the processes that shaped this species’ disjunct distribution pattern. The genetic analysis revealed three deeply divergent clades, Southeast, Southwest and Central, which derived from the Wuyi Mountains, the YGP and the mountains around the Sichuan Basin, respectively—mostly since the Pleistocene. IBD played important roles in early divergence, and geological events (the sedimentation of plains and the joining of palaeo-rivers) and IBE further reinforced genetic differentiation. The primary cause of the disjunct distribution in E. melanogaster was the preference for middle-high-altitude habitats in the current period. High-altitude localities acted as “sky islands” for E. melanogaster and blocked genetic exchange among populations. Pleistocene climatic cycles had various impacts on the diversification of different clades. Genetic admixture in cold periods and genetic diversification in warm periods were facilitated in inland clades but led to multiple colonization events between the mainland and Taiwan and erased genetic differentiation during cold periods.
Additional files
Table S1. Sample information and the GenBank accession numbers. Sampling information and the GenBank accession numbers of all sequences of Eothenomys melanogaster used in the study. Table S2. Outgroups of phylogenetic analysis. Outgroups used for fossil calibration and phylogenetic analyses of E. melanogaster. Table S3. Gene flow of three maternal clades. Gene flow of three maternal clades of E. melanogaster estimated by MIGRATE. C represents the Central Clade, SE represents the Southeast Clade and SW represents the Southwest Clade. (DOCX 32 kb)
Figure S1. Bayesian phylogenetic trees of nuclear loci and species tree. Bayesian phylogenetic trees of nuclear loci and species tree estimated by BP&P and *BEAST for E. melanogaster. Red indicates individuals belonging to the Central clade; blue indicates individuals belonging to the Southwest clade; and yellow indicates individuals belonging to the Southeast clade. Values beside branches indicate Bayesian posterior probability. (TIFF 24617 kb)
Figure S2. Phylogenetic trees of concatenated cytb and nDNA genes. Phylogenetic trees of concatenated cytb and nDNA genes for E. melanogaster. Values beside branches indicate posterior probability and bootstrap. (TIFF 28922 kb)
Acknowledgements
We sincerely thank Yanni Wang from Zhejiang Normal University and the staff of Anhui Qingliangfeng Nature Reserve for their help in obtaining samples during our field work. We sincerely thank Professor Hongcan Yu from Institute of Zoology, National Taiwan University; Professor Yuchun Li from Shandong University, Weihai; and Dr. Song Li from Kunming Institute of Zoology, Chinese Academy of Sciences. Their generous provision of samples from Taiwan, Fujian and Yunnan greatly enriched our study.
Funding
This research was supported by grants from the National Natural Science Foundation of China (31372177 to L.X.), Newton Advanced Fellowship of the Royal Society of the United Kingdom (Ref. NA150142 to D.G.), and the Key Laboratory of Zoological Systematics and Evolution of the Chinese Academy of Sciences (No. Y229YX5105).
Availability of data and materials
The sequences generated in the study have been deposited into GenBank under the accession numbers listed in Additional file 1: Table S1.
Abbreviations
- BI
Bayesian inference
- C
Central
- HM
Hengduan Mountains
- IBD
Isolation by distance
- IBE
Isolation by environment
- IOZCAS
Institute of Zoology, Chinese Academy of Sciences
- ML
maximum likelihood
- MLE
Marginal likelihood estimated
- QTP
Qinghai-Tibetan Plateau
- SE
Southeast
- SW
Southwest
- YGP
Yunnan-Guizhou Plateau
Authors’ contributions
QY, LX and SL conceived the study; XL, JC, YC and YM collected samples; XL and YM performed the laboratory work; XL, ZW, JC and DG analysed the data; and JC and XL wrote the first draft of the manuscript. All authors contributed to the interpretation of the results and commented on the final version of this manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Our study was carried out in strict accordance with the animal research protocol IOZ-2006 approved by the Animal Care Committee of Institute of Zoology, Chinese Academy of Sciences (CAS).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1168-3) contains supplementary material, which is available to authorized users.
Contributor Information
Xue Lv, Email: bettycatherine@126.com.
Jilong Cheng, Email: chengjilong@ioz.ac.cn.
Yang Meng, Email: mengyang@scu.edu.cn.
Yongbin Chang, Email: changyongbin@ioz.ac.cn.
Lin Xia, Email: xial@ioz.ac.cn.
Zhixin Wen, Email: wenzx@ioz.ac.cn.
Deyan Ge, Email: gedy@ioz.ac.cn.
Shaoying Liu, Email: Shaoyliu@163.com.
Qisen Yang, Email: yangqs@ioz.ac.cn.
References
- 1.Zhang LS. Palaeogeography of China: the formation of Chinese natural evironment. Beijing: Science Press; 2012. [Google Scholar]
- 2.Qu YH, Ericson PGP, Quan Q, et al. Long-term isolation and stability explain high genetic diversity in the eastern Himalaya. Mol Ecol. 2014;23:705–720. doi: 10.1111/mec.12619. [DOI] [PubMed] [Google Scholar]
- 3.Wang YH, Jiang WM, Comes HP, et al. Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): insights into Plio-Pleistocene range dynamics of evergreen forest in subtropical China. New Phytol. 2015;206:852–867. doi: 10.1111/nph.13261. [DOI] [PubMed] [Google Scholar]
- 4.He K, Hu NQ, Chen X, et al. Interglacial refugia preserved high genetic diversity of the Chinese mole shrew in the mountains of Southwest China. Heredity. 2016;116:23–32. doi: 10.1038/hdy.2015.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browne RA, Ferree PM. Genetic structure of southern Appalachian 'sky island' populations of the southern red-backed vole (Myodes gapperi) J Mammal. 2007;88:759–768. doi: 10.1644/06-MAMM-A-049R1.1. [DOI] [Google Scholar]
- 6.Wen ZX, Yang QS, Quan Q, et al. Multiscale partitioning of small mammal-diversity provides novel insights into the quaternary faunal history of Qinghai-Tibetan plateau and hengduan mountains. J Biogeogr. 2016;43:1412–1424. doi: 10.1111/jbi.12706. [DOI] [Google Scholar]
- 7.He K, Jiang XL. Sky islands of Southwest China. I: an overview of phylogeographic patterns. Chin Sci Bull. 2014;59:585–597. doi: 10.1007/s11434-013-0089-1. [DOI] [Google Scholar]
- 8.Dong L, Heckel G, Liang W, et al. Phylogeography of silver pheasant (Lophura nycthemera L.) across China: aggregate effects of refugia, introgression and riverine barriers. Mol Ecol. 2013;22:3376–3390. doi: 10.1111/mec.12315. [DOI] [PubMed] [Google Scholar]
- 9.Qu YH, Song G, Gao B, et al. The influence of geological events on the endemism of east Asian birds studied through comparative phylogeography. J Biogeogr. 2015;42:179–192. doi: 10.1111/jbi.12407. [DOI] [Google Scholar]
- 10.Howard WR. Palaeoclimatology - a warm future in the past. Nature. 1997;388:418–419. doi: 10.1038/41201. [DOI] [Google Scholar]
- 11.Zachos J, Pagani M, Sloan L, et al. Trends, rhythms, and aberrations in global climate 65 ma to present. Science. 2001;292:686–693. doi: 10.1126/science.1059412. [DOI] [PubMed] [Google Scholar]
- 12.Yan F, Zhou WW, Zhao HT, et al. Geological events play a larger role than Pleistocene climatic fluctuations in driving the genetic structure of Quasipaa boulengeri (Anura: Dicroglossidae) Mol Ecol. 2013;22:1120–1133. doi: 10.1111/mec.12153. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Yan HF, Ge XJ. Phylogeographic analysis and environmental niche modeling of widespread shrub Rhododendron simsii in China reveals multiple glacial refugia during the last glacial maximum. J Syst Evol. 2012;50:362–373. doi: 10.1111/j.1759-6831.2012.00209.x. [DOI] [Google Scholar]
- 14.Lanier HC, Olson LE. Deep barriers, shallow divergences: reduced phylogeographical structure in the collared pika (Mammalia: Lagomorpha: Ochotona collaris) J Biogeogr. 2013;40:466–478. doi: 10.1111/jbi.12035. [DOI] [Google Scholar]
- 15.Cun YZ, Wang XQ. Phylogeography and evolution of three closely related species of Tsuga (hemlock) from subtropical eastern Asia: further insights into speciation of conifers. J Biogeogr. 2015;42:315–327. doi: 10.1111/jbi.12421. [DOI] [Google Scholar]
- 16.Kou YX, Cheng SM, Tian S, et al. The antiquity of Cyclocarya paliurus (Juglandaceae) provides new insights into the evolution of relict plants in subtropical China since the late early Miocene. J Biogeogr. 2016;43:351–360. doi: 10.1111/jbi.12635. [DOI] [Google Scholar]
- 17.Luo J, Yang DM, Suzuki H, et al. Molecular phylogeny and biogeography of oriental voles: genus Eothenomys (Muridae, Mammalia) Mol Phylogenet Evol. 2004;33:349–362. doi: 10.1016/j.ympev.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Lu L, Ge DY, Chesters D, et al. Molecular phylogeny and the underestimated species diversity of the endemic white-bellied rat (Rodentia: Muridae: Niviventer) in Southeast Asia and China. Zool Scr. 2015;44:475–494. doi: 10.1111/zsc.12117. [DOI] [Google Scholar]
- 19.Kaneko Y. Morphological variation and geographical and altitudinal distribution in Eothenomys melanogaster and E. mucronatus (Rodentia, Arvicolinae) in China, Taiwan, Burma, India, Thailand, and Vietnam. Mammal Study. 2002;27:31–63. doi: 10.3106/mammalstudy.27.31. [DOI] [Google Scholar]
- 20.Ohdachi S, Dokuchaev NE, Hasegawa M, et al. Intraspecific phylogeny and geographical variation of six species of northeastern asiatic Sorex shrews based on the mitochondrial cytochrome b sequences. Mol Ecol. 2001;10:2199–2213. doi: 10.1046/j.1365-294X.2001.01359.x. [DOI] [PubMed] [Google Scholar]
- 21.Bannikova AA, Sighazeva AM, Malikov VG, et al. Genetic diversity of Chionomys genus (Mammalia, Arvicolinae) and comparative phylogeography of snow voles. Russ J Genet. 2013;49:561–575. doi: 10.1134/S1022795413050025. [DOI] [PubMed] [Google Scholar]
- 22.Lyons LA, Laughlin TF, Copeland NG, et al. Comparative anchor tagged sequences (CATS) for integrative mapping of mammalian genomes. Nat Genet. 1997;15:47–56. doi: 10.1038/ng0197-47. [DOI] [PubMed] [Google Scholar]
- 23.Galewski T, Tilak MK, Sanchez S, et al. The evolutionary radiation of Arvicolinae rodents (voles and lemmings): relative contribution of nuclear and mitochondrial DNA phylogenies. BMC Evol Biol. 2006;6:80. doi: 10.1186/1471-2148-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwasa MA, Suzuki H. Evolutionary significance of chromosome changes in northeastern asiatic red-backed voles inferred with the aid of intron 1 sequences of the g6pd gene. Chromosom Res. 2002;10:419–428. doi: 10.1023/A:1016809921433. [DOI] [PubMed] [Google Scholar]
- 25.Stanhope MJ, Czelusniak J, Si JS, et al. A molecular perspective on mammalian evolution from the gene encoding interphotoreceptor retinoid binding protein, with convincing evidence for bat monophyly. Mol Phylogenet Evol. 1992;1:148–160. doi: 10.1016/1055-7903(92)90026-D. [DOI] [PubMed] [Google Scholar]
- 26.Petrova TV, Zakharov ES, Samiya R, et al. Phylogeography of the narrow-headed vole Lasiopodomys (Stenocranius) gregalis (Cricetidae, Rodentia) inferred from mitochondrial cytochrome b sequences: an echo of Pleistocene prosperity. J Zool Syst Evol Res. 2015;53:97–108. doi: 10.1111/jzs.12082. [DOI] [Google Scholar]
- 27.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
- 28.Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia X, Xie Z, Salemi M, et al. An index of substitution saturation and its application. Mol Phylogenet Evol. 2003;26:1–7. doi: 10.1016/S1055-7903(02)00326-3. [DOI] [PubMed] [Google Scholar]
- 30.Xia X, Lemey P. Assessing substitution saturation with dambe. 2009. pp. 615–630. [Google Scholar]
- 31.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 32.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nylander J. MrModeltest v2. Program distributed by the author. Evolutionary biology Centre, Uppsala University. 2004. [Google Scholar]
- 34.Bandelt H-J, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 35.Leigh JW, Bryant D. POPART: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6:1110–1116. doi: 10.1111/2041-210X.12410. [DOI] [Google Scholar]
- 36.Drummond AJ, Suchard MA, Xie D, et al. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng S. Quaternary rodents of Sichuan and Guizhou area, China. Beijing: Science Press; 1993. [Google Scholar]
- 38.Repenning CA. Arvicolid rodent biochronology of the northern hemisphere. 1990. pp. 385–418. [Google Scholar]
- 39.Kohli BA, Fedorov VB, Waltari E, et al. Phylogeography of a holarctic rodent (Myodes rutilus): testing high-latitude biogeographical hypotheses and the dynamics of range shifts. J Biogeogr. 2015;42:377–389. doi: 10.1111/jbi.12433. [DOI] [Google Scholar]
- 40.Baele G, Lemey P, Bedford T, et al. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol. 2012;29:2157–2167. doi: 10.1093/molbev/mss084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2009;27:570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drummond AJ, Ho SY, Rawlence N, et al. A rough guide to BEAST 1.4. 2007. [Google Scholar]
- 43.Cheng J, Lv X, Xia L, et al. Impact of orogeny and environmental change on genetic divergence and demographic history of Dipus sagitta (Dipodoidea, Dipodinae) since the Pliocene in inland East Asia. J Mamm Evol. 2017; 10.1007/s10914-017-9397-6.
- 44.Yang ZH, Rannala B. Bayesian species delimitation using multilocus sequence data. Proc Natl Acad Sci U S A. 2010;107:9264–9269. doi: 10.1073/pnas.0913022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang ZH, Rannala B. Unguided species delimitation using DNA sequence data from multiple loci. Mol Biol Evol. 2014;31:3125–3135. doi: 10.1093/molbev/msu279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rozas J, Sanchez-DelBarrio JC, Messeguer X, et al. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- 47.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinforma. 2005;1:47–50. doi: 10.1177/117693430500100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu Y-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oliver MA, Webster R. Kriging: a method of interpolation for geographical information systems. Int. J. Geogr. Inf. Syst. 1990;4:313–332. doi: 10.1080/02693799008941549. [DOI] [Google Scholar]
- 51.Bouckaert R, Heled J, Kuhnert D, et al. BEAST2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bielejec F, Rambaut A, Suchard MA, et al. SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics. 2011;27:2910–2912. doi: 10.1093/bioinformatics/btr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otto-Bliesner BL, Marsha SJ, Overpeck JT, et al. Simulating arctic climate warmth and icefield retreat in the last interglaciation. Science. 2006;311:1751–1753. doi: 10.1126/science.1120808. [DOI] [PubMed] [Google Scholar]
- 54.Sugiyama M, Shiogama H, Emori S. Precipitation extreme changes exceeding moisture content increases in MIROC and IPCC climate models. Proc Natl Acad Sci U S A. 2010;107:571–575. doi: 10.1073/pnas.0903186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hijmans RJ, Cameron SE, Parra JL, et al. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol. 2005;25:1965–1978. doi: 10.1002/joc.1276. [DOI] [Google Scholar]
- 56.Warren DL, Glor RE, Turelli M. ENMTools: a toolbox for comparative studies of environmental niche models. Ecography. 2010;33:607–611. doi: 10.1111/j.1600-0587.2009.06041.x. [DOI] [Google Scholar]
- 57.Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol Model. 2006;190:231–259. doi: 10.1016/j.ecolmodel.2005.03.026. [DOI] [Google Scholar]
- 58.Beerli P. Comparison of bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics. 2006;22:341–345. doi: 10.1093/bioinformatics/bti803. [DOI] [PubMed] [Google Scholar]
- 59.Beerli P. How to use MIGRATE or why are Markov chain Monte Carlo programs difficult to use. 2009. pp. 42–79. [Google Scholar]
- 60.Oksanen J, Blanchet FG, Kindt R, et al. Package ‘vegan’. Community ecology package. 2013. [Google Scholar]
- 61.R Core Team . R: a language and environment for statistical computing [internet] Vienna: R Foundation for Statistical Computing; 2016. [Google Scholar]
- 62.Struwe L, Smouse PE, Heiberg E, et al. Spatial evolutionary and ecological vicariance analysis (SEEVA), a novel approach to biogeography and speciation research, with an example from Brazilian Gentianaceae. J Biogeogr. 2011;38:1841–1854. doi: 10.1111/j.1365-2699.2011.02532.x. [DOI] [Google Scholar]
- 63.Yi CL, Cui ZJ, Xiong HG. Numerical periods of quaternary glaciations in China. Quat Sci. 2005;25:609–619. [Google Scholar]
- 64.Ballard JWO, Chernoff B, James AC. Divergence of mitochondrial DNA is not corroborated by nuclear DNA, morphology, or behavior in Drosophila simulans. Evolution. 2002;56(3):527–545. doi: 10.1111/j.0014-3820.2002.tb01364.x. [DOI] [PubMed] [Google Scholar]
- 65.Qu YH, Zhang RY, Quan Q, et al. Incomplete lineage sorting or secondary admixture: disentangling historical divergence from recent gene flow in the vinous-throated parrotbill (Paradoxornis webbianus) Mol Ecol. 2012;21:6117–6133. doi: 10.1111/mec.12080. [DOI] [PubMed] [Google Scholar]
- 66.Bao Y, Zhuge Y. An ecological study on Eothenomys melanogaster. Acta Theriol Sinica. 1986;6:297–305. [Google Scholar]
- 67.Redenbach Z, Taylor EB. Evidence for historical introgression along a contact zone between two species of char (Pisces: Salmonidae) in northwestern North America. Evolution. 2002;56:1021–1035. doi: 10.1111/j.0014-3820.2002.tb01413.x. [DOI] [PubMed] [Google Scholar]
- 68.Yang DY, Li XS. Study on the eastward flow of the Jinsha river. J Nanjing University (Natural Sciences) 2001;37:317–322. [Google Scholar]
- 69.Cui ZJ, Wu YQ, Liu GN, et al. On Kunlun-yellow river tectonic movement. Sci China Ser D. 1998;41:592–600. doi: 10.1007/BF02878741. [DOI] [Google Scholar]
- 70.Li JJ, Fang XM. The uplift of Qinghai-Xizang (Tibetan) plateau and the studies of environmental changes. Chin Sci Bull. 1998;43:1569–1574. [Google Scholar]
- 71.Shi YF, Li JJ, Li BY, et al. Uplift of the Qinghai-Xizang (Tibetan) plateau and East Asia environmental change during late Cenozoic. Acta Geogr Sinica. 1999;54:12–22. [Google Scholar]
- 72.Lopez-Pujol J, Zhang FM, Sun HQ, et al. Centres of plant endemism in China: places for survival or for speciation? J Biogeogr. 2011;38:1267–1280. doi: 10.1111/j.1365-2699.2011.02504.x. [DOI] [Google Scholar]
- 73.Cheng R, Jiang N, Yang XS, et al. The influence of geological movements on the population differentiation of Biston panterinaria (Lepidoptera: Geometridae) J Biogeogr. 2016;43:691–702. doi: 10.1111/jbi.12676. [DOI] [Google Scholar]
- 74.Lv X, Xia L, Ge DY, et al. Continental refugium in the Mongolian plateau during quaternary glacial oscillations: phylogeography and niche modelling of the endemic desert hamster, Phodopus roborovskii. PLoS One. 2016;11:e0148182. [DOI] [PMC free article] [PubMed]
- 75.Yuan SL, Lin LK, Oshida T. Phylogeography of the mole-shrew (Anourosorex yamashinai) in Taiwan: implications of interglacial refugia in a high-elevation small mammal. Mol Ecol. 2006;15:2119–2130. doi: 10.1111/j.1365-294X.2006.02875.x. [DOI] [PubMed] [Google Scholar]
- 76.Wang WJ, McKay BD, Dai CY, et al. Glacial expansion and diversification of an east Asian montane bird, the green-backed tit (Parus monticolus) J Biogeogr. 2013;40:1156–1169. doi: 10.1111/jbi.12055. [DOI] [Google Scholar]
- 77.Jezkova T, Riddle BR, Card DC, et al. Genetic consequences of postglacial range expansion in two codistributed rodents (genus Dipodomys) depend on ecology and genetic locus. Mol Ecol. 2015;24:83–97. doi: 10.1111/mec.13012. [DOI] [PubMed] [Google Scholar]
- 78.Inoue K, Lang BK, Berg DJ. Past climate change drives current genetic structure of an endangered freshwater mussel species. Mol Ecol. 2015;24:1910–1926. doi: 10.1111/mec.13156. [DOI] [PubMed] [Google Scholar]
- 79.Jing M, Yu HT, Bi X, et al. Phylogeography of Chinese house mice (Mus musculus musculus/castaneus): distribution, routes of colonization and geographic regions of hybridization. Mol Ecol. 2014;23:4387–4405. doi: 10.1111/mec.12873. [DOI] [PubMed] [Google Scholar]
- 80.Avise JC. Phylogeography: the history and formation of species. London: Harvard University Press; 2000.
- 81.Nei M, Maruyama T, Chakraborty R. Bottleneck effect and genetic-variability in populations. Evolution. 1975;29:1–10. doi: 10.1111/j.1558-5646.1975.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 82.Grant WS, Bowen BW. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered. 1998;89:415–426. doi: 10.1093/jhered/89.5.415. [DOI] [Google Scholar]
- 83.Galbreath KE, Hafner DJ, Zamudio KR. When cold is better: climate-driven elevation shifts yield complex patterns of diversification and demography in an alpine specialist (American pika, Ochotona princeps) Evolution. 2009;63:2848–2863. doi: 10.1111/j.1558-5646.2009.00803.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sample information and the GenBank accession numbers. Sampling information and the GenBank accession numbers of all sequences of Eothenomys melanogaster used in the study. Table S2. Outgroups of phylogenetic analysis. Outgroups used for fossil calibration and phylogenetic analyses of E. melanogaster. Table S3. Gene flow of three maternal clades. Gene flow of three maternal clades of E. melanogaster estimated by MIGRATE. C represents the Central Clade, SE represents the Southeast Clade and SW represents the Southwest Clade. (DOCX 32 kb)
Figure S1. Bayesian phylogenetic trees of nuclear loci and species tree. Bayesian phylogenetic trees of nuclear loci and species tree estimated by BP&P and *BEAST for E. melanogaster. Red indicates individuals belonging to the Central clade; blue indicates individuals belonging to the Southwest clade; and yellow indicates individuals belonging to the Southeast clade. Values beside branches indicate Bayesian posterior probability. (TIFF 24617 kb)
Figure S2. Phylogenetic trees of concatenated cytb and nDNA genes. Phylogenetic trees of concatenated cytb and nDNA genes for E. melanogaster. Values beside branches indicate posterior probability and bootstrap. (TIFF 28922 kb)
Data Availability Statement
The sequences generated in the study have been deposited into GenBank under the accession numbers listed in Additional file 1: Table S1.