

Abstract
A microfluidic system that combines membraneless microfluidic dialysis and dielectrophoresis to achieve label-free isolation and concentration of bacteria from whole blood is presented. Target bacteria and undesired blood cells are discriminated on the basis of their differential susceptibility to permeabilizing agents that alter the dielectrophoretic behavior of blood cells but not bacteria. The combined membraneless microdialysis and dielectrophoresis system isolated 79 ± 3% of Escherichia coli and 78 ± 2% of Staphylococcus aureus spiked into whole blood at a processing rate of 0.6 mL h−1. Collection efficiency was independent of the number of target bacteria up to 105 cells. Quantitative PCR analysis revealed that bacterial 16S rDNA levels were enriched more than 307-fold over human DNA in the fraction recovered from the isolation system compared with the original specimen. These data demonstrate feasibility for an instrument to accelerate the detection and analysis of bacteria in blood by first isolating and concentrating them in a microchamber.
Introduction
Serious trauma and certain medical procedures, like radio-and chemotherapy, intravenous transfusion or implantation of devices can cause bacterial bloodstream infections (BSI) and lead to severe sepsis.1–4 Despite advances in antimicrobial therapies, BSI are a major cause of death worldwide with an approximately 50% mortality rate.5–7 Early intervention with effective antimicrobial chemotherapy greatly reduces the risk of mortality and morbidity.8 However, in early stages of BSI there may only be 1–10 bacterial cells per milliliter of whole blood.9,10 Culture techniques are the most definitive and cost-effective strategy for detecting low abundance bacteria in blood but are significantly limited by long incubation periods, ranging from one to seven days. Molecular detection assays offer detection limits down to as few as a single colony forming unit CFU mL−1 in just a few hours but are too expensive and technically difficult to implement for routine clinical diagnostics and hemovigilance, and have failed to supplant culture methods in most clinical microbiology laboratories.11–13
There is an urgent need for a cost-effective, automated system that shortens the time to confirm infection and determine bacterial drug-resistance from days to just a few hours. Advances in microfluidics and microelectronics offer opportunities for achieving rapid detection and characterization of low abundance bacteria. Engineering at the micro- and nano-scale opens new opportunities to significantly improve separation and detection assays by enhancing transport phenomena, substantially reducing reagent consumption, and providing a means to harness forces for manipulating target analytes and cells that cannot be exploited over the macro-scale.14,15 A multitude of lab-on-chip bacterial detection technologies have been reported that capitalize on the advantages gained through miniaturization,16–24 but many of these advanced technologies cannot be readily applied to clinical specimens because they can only process tiny volumes. Herein, a microfluidic system is reported that isolates and concentrates bacteria from whole blood into microliter volumes by harnessing microscale diffusion and exploiting their intrinsic cytoelectric properties via dielectrophoresis (DEP) (Fig. 1).
Fig. 1.
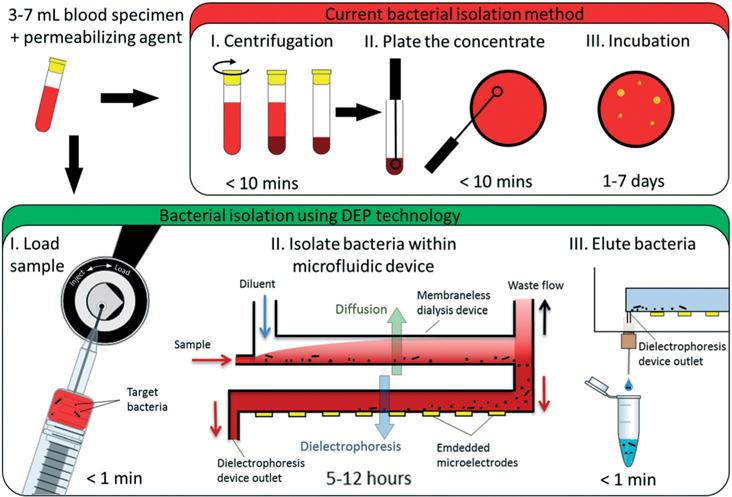
Overview of the current and proposed methods for isolating bacteria from blood. It is standard practice to collect blood via venipuncture directly into a tube containing saponin, which permeabilizes blood cells. Current methods (the red box) 1) concentrate target bacteria before 2) plating the sample on solid media and 3) incubating the plates for hours or some times days until bacterial colonies are visible. The proposed method (green box) is based on novel combination of microfluidic technologies. The blood specimen mixed with a permeabilizaing agent is 1) injected into the machine and 2) pumped into the microfluidic apparatus which desalts the sample by inline membraneless dialysis and isolates and concentrates target bacteria within a microfluidic chamber by dielectrophoresis (DEP). 3) Then, after the whole sample is processed, the dielectrophoresis signal is simply shut off and target bacteria are eluted for off-line detection and characterization.
The electrodynamic phenomena underlying DEP as a motive force for isolating target cells provide several compelling advantages over other reported methods.25 Cytoelectric properties emerge from the structural form and composition of cellular membranes and from dielectric contrast between the cell interior and the suspending medium. Using DEP to capture target cells makes it unnecessary to use expensive bio-reagents to discriminate between target bacteria and background blood cells (BCs). In this work, additives were used that alter the cytoelectric properties of BCs, and thereby allowed for bacteria to be discriminated from BCs using DEP. This strategy of selectively altering the cytoelectric properties of undesired BCs was incorporated into a scalable and continuous-mode microfluidic system that allows operators to enrich for target bacteria directly from undiluted whole human blood. Experiments with blood samples spiked with model organisms representing Gram-negative and Gram-positive bacteria demonstrated feasibility for using this microfluidic system to harness this electrokinetic phenomenon to isolate and concentrate bacteria from blood specimens in order to enable downstream bioanalysis of pathogens.
Results
Chemically-mediated dielectrophoretic separation of bacteria from blood
The cell membrane separates the cell interior from the suspending medium and provides a barrier to the flow of ions. Applying an electric field to the cell suspension causes charge carriers to accumulate at the membrane interfaces, and the cells become electrically polarized.26 If the applied electric field is spatially non-uniform, then the cell experiences a DEP force that either repels it from (negative DEP: nDEP) or attracts it toward (positive DEP: pDEP) electric field maxima located near the edges of microelectrodes (Fig. 2A and B). The magnitude and direction of the induced polarization depend upon the intrinsic cytoelectric properties of the cell, its suspending medium, and the frequency of the applied field (Note S1†). Using a co-planar quadrupole microelectrode geometry embedded at the bottom of a small well, it was possible to manipulate BCs and bacteria suspended in aqueous media via DEP. It was observed via fluorescent microscopy that blood cells and E. coli (labelled with the fluorescent dye SYTO9) collected at the electrode edges by pDEP at frequencies between 0.5 and 30 MHz when the ionic conductivity was 40 mS m−1 and below (Fig. 2C).
Fig. 2.
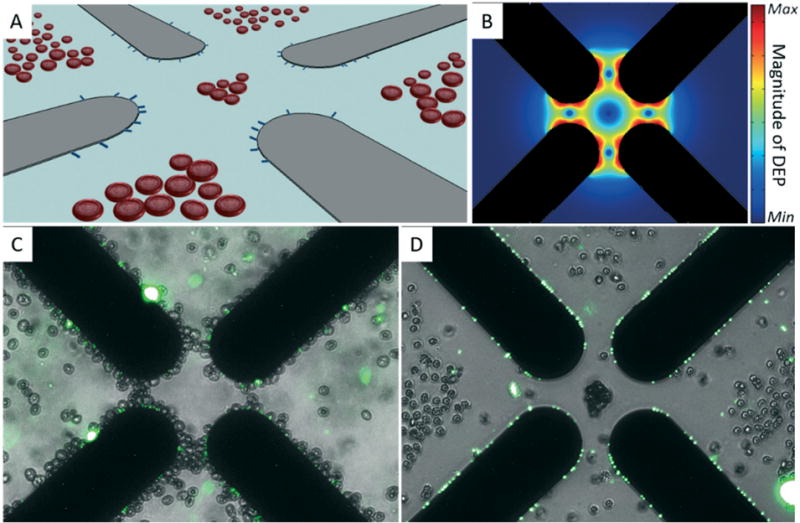
Discrimination of bacteria and human blood cells via chemically mediated dielectrophoresis. (A) Illustration of co-planar quadrupole microelectrodes (gray objects) used to separate bacteria (blue rods) from blood cells by DEP. Blood cells are repelled away from electric field maxima located at the electrode edges while bacteria are pulled toward these regions. (B) Simulated magnitude of the DEP force near the quadrupole micro-electrode tips (black shadows). (C) Merged microphotographs of E. coli labeled with the fluorescent dye SYTO9 (green channel) and erythrocytes (bright-field channel) suspended in 40 mS m−1 280 μM D-mannitol. Both cell types were collected by pDEP at microelectrode edges using a sinusoidal signal of 20 MHz and 4 Vpp. (D) Merged microphotographs of labeled E. coli (green channel) and erythrocytes (bright-field channel) suspended in 40 mS m−1 280 μM D-mannitol and 260 nM monensin. Bacteria and blood cells were separated using the same DEP signal as in (C).
Ionophores and pore-forming agents in low doses can facilitate the transport of ions across cell membranes.27 Low doses of such agents can control the loss of ions from the cell interior while avoiding cell lysis and minimize the release of molecular contaminants that could interfere with downstream bioanalysis. This concept was demonstrated by adding the ionophore monensin to a mixture of BCs and fluorescently-labelled Escherichia coli (E. coli) suspended above a quadrupole microelectrode array. When the concentration of monensin was 2.6 μM, BCs could not be collected by pDEP at frequencies from 0.5 to 50 MHz suggesting that the addition of monensin to the cell suspension changes the frequency-dependent DEP behavior of BCs almost immediately. At 260 nM monensin, the majority of BCs could be collected at frequencies between 0.5 and 10 MHz but were repelled at frequencies up to 50 MHz (the upper limit of the signal generator used). Adding monensin to a suspension of BCs and fluorescently-labelled E. coli to a final concentration of 260 nM allowed for their complete separation using a DEP signal of 20 MHz and 4 Vpp when the conductivity was 40 mS m−1. Bacteria collected at the microelectrode edges by pDEP, while BCs were repelled away from the edges by nDEP and concentrated in the gaps (Fig. 2D). In all experiments, the DEP behavior of E. coli was not affected by monensin and could be collected by pDEP at conductivities below 50 mS m−1 and frequencies between 0.5–50 MHz. The frequency window that produced a differential DEP response between BCs and bacteria was found to widen with increasing concentrations of monensin, suggesting that the compound’s effect on BCs cytoelectric properties is dose dependent. Collectively, these observations demonstrated that it is possible to use small quantities of additives to create contrast in the cytoelectric properties of target bacteria and blood cells enabling discrimination using DEP at low conductivities.
Continuous-mode dielectrophoretic enrichment of bacteria from blood
Accelerating the detection and analysis of bacteria in clinical blood specimens using microfluidic biosensing technologies demands that several milliliters be processed and target bacteria concentrated into microliter volumes. As a proof-of-concept, we exploited ion loss induced by targeted membrane permeabilization to achieve continuous-mode isolation of bacteria from blood within a microfluidic DEP trapping device. The flow-through DEP microchamber was 1.5 cm wide × 5 cm long × 0.0058 cm high (43.5 μL), and had co-planar parallel microelectrodes 50 microns wide and spaced embedded on the bottom wall (Fig. 3A). This narrow channel height allowed for BCs and bacteria suspended in the flow to be physically confined close to the microelectrode array, where the DEP force is strongest (Fig. 3B). Exposure to a high concentration of saponin is a common method used in clinical labs to lyse BCs.28,29 This treatment results is loss of most cellular contents including ions and has been exploited for sample preparation in bioimpedance analysis and electro-physiology.30,31 Like monensin, at low concentrations saponin permeabilizes the plasma membrane and ions leak into the medium when a concentration gradient exists. The encapsulating membranes of most bacterial pathogens are resistant to saponin, allowing them to retain ions and be collected by pDEP. Since this compound is routinely used in clinical microbiology laboratories, we selected it over monensin as the permeabilizing agent in subsequent experiments. We found that low doses of saponin (<0.5 mg mL−1) were sufficient to facilitate ion loss across the membrane of BCs. As with monensin, exposure to saponin rendered BCs less electrically polarizable than their suspending medium (40 mS m−1 0.28 mM D-mannitol) and they were repelled by nDEP at frequencies greater than 0.5 MHz.
Fig. 3.
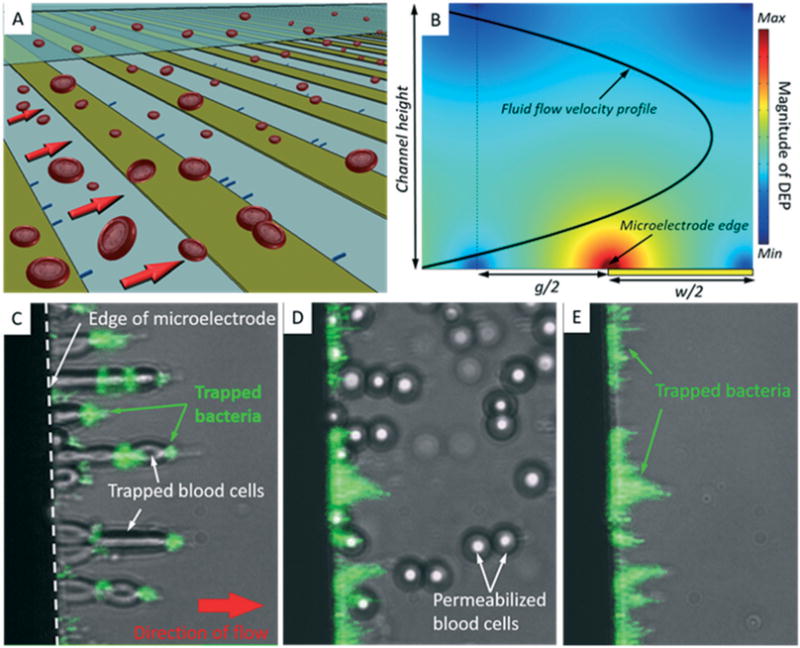
Continuous-mode dielectrophoretic separation of bacteria from blood cells. (A) Illustration of the flow-through DEP separation microchamber. Co-planar and parallel microelectrodes embedded within a microfluidic channel create the DEP force field to collect target bacteria and repel permeabilized BCs, which are swept to waste by fluid flow. (B) The DEP force field strength was simulated within a cross-sectional view of the microfluidic DEP device. The calculations reveal the location of the DEP trapping region near the electrode edge. The parabolic fluid velocity profile is also shown (bold black curve). The DEP force is symmetric about the middle plane of the electrode gaps (broken line). (C)–(E) Microphotographs (20× magnification) of BCs and fluorescently labeled E. coli within the microfluidic DEP device. The direction of flow is left to right, and all images are composites between bright-field and fluorescence images taken of the same field of view. (C) BCs and bacteria co-collect in the electric high field region at the edge of the microelectrode using a DEP signal of 1 MHz and 15 Vpp. (D) Saponin caused blood cells to be released from electrodes. Some bacteria were temporarily released but redistribute along microelectrodes downstream. (E) E. coli remained trapped by DEP.
To assess the use of saponin to facilitate separation of bacteria from BCs by DEP, 1 mL aliquots of blood spiked with E. coli or Staphylococcus aureus (S. aureus) were resuspended in a low conductivity (5 mS m−1) solution containing 280 mM D-mannitol. Fifty microliters of sample were processed through a DEP microfluidic device at 10 μL min−1. Initially, both BCs and bacteria collected in the elec tric high field region by pDEP using a sinusoidal waveform having a frequency of 1 MHz and amplitude of 15 Vpp (Fig. 3C). BCs appeared elongated because they were distorted by the combined action of fluid drag and pDEP. Using fluorescence microscopy, bacteria labeled with a SYTO9 were observed among BCs trapped by pDEP. After the entire sample was processed, an aliquot of the DEP buffer containing 5 mg mL−1 saponin was loaded into the sample loop and injected into the microchamber. Almost immediately after the saponin buffer entered the microchamber, the DEP behavior of blood cells changed significantly. BCs rapidly became spherical in shape, presumably due to rapid influx of water through the perforated cell membrane. Following this morphological change blood cells quickly released from the microelectrode edge by the imposed fluid flow and carried toward the device outlet (Fig. 3D). Bacteria remained confined within the chamber by pDEP in the presence of saponin (Fig. 3E). After all BCs had cleared, the isolated bacteria were eluted from the chamber by simply shutting off the DEP signal. Recovered E. coli and S. aureus could be cultured on lysogeny broth (LB) agar, indicating that they remained viable during the separation process.
Integrated microfluidic dialysis-dielectrophoresis isolation system
Preliminary tests with E. coli and S. aureus strains indicated that the conductivity of the suspending medium needed to be reduced 100× below physiological levels (1.5 S m−1 for whole blood) to realize efficient pDEP on bacteria. This reduction is necessary to establish physicochemical conditions that allow for pDEP on bacteria, and to minimize Joule heating within the DEP microdevice and to mitigate power loss between the signal amplifier and the device. In the context of isolating rare cells, achieving this reduction by dilution leads to exceedingly long sample processing times. Here, a membraneless microdialysis device (MMD) developed earlier32 was adapted to reduce the electrical conductivity of blood specimens. MMDs exploit rapid diffusion over micro-scale distances, much like H-filter microfluidic devices (Note S2†). In order to deplete ions from blood, two MMDs were connected in series such that the sample stream outlet of one MMD was connected to the input of a second MMD via a short length of PEEK tubing (Fig. 4A). The hydrodynamic resistances of the waste ports of each microdialysis device were adjusted using various lengths of PEEK tubing to control fluid splitting. Both devices were operated at a flow rate of 100 μL min−1 (10 μL min−1 sample processing rate) so that the specimen ion concentration was reduced 10× in each device. A syringe pump was used to drive two syringes with diluent and a separate pump injected the sample. The cascaded MMDs were connected by PEEK tubing to the DEP trapping stage so that the subsystems could be operated continuously as a single instrument. An in-line conductivity probe placed at the output of the DEP device confirmed that the conductivity of the specimen stream fell from 1.5 S m−1 to 15–17 mS m−1.
Fig. 4.
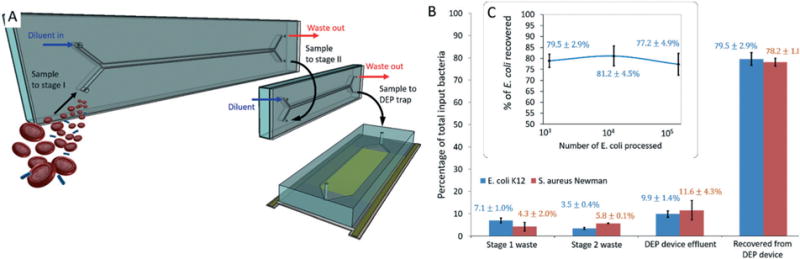
Combined dialysis-DEP isolation system. (A) Illustration of the microfluidic apparatus used to isolate bacteria from whole blood using DEP. Two MMDs were operated inline to achieve sample deionization so that target bacteria could be collected by pDEP. (B) The total trapping efficiency for two model organisms was evaluated by culturing fluid collected at all outlets of the apparatus and comparing the total colony forming unit counts to that of the input specimen. (C) The capture efficiency was estimated using culture methods as a function of the total number of E. coli spiked into input blood samples.
Two hundred microliters of saponin-treated blood spiked with 1000 CFUs of either E. coli or S. aureus were loaded into a 6-port Rheodyne valve with a 450 μL sample loop. To estimate the separation efficiency, fluid was collected at the waste outlets of each microdialysis and DEP device during operation. Once the sample was fully processed, the DEP field was turned off and captured bacteria were eluted. CFUs in collected fractions were compared to those measured in the original sample to estimate the recovery efficiency. These fluid samples were plated on LB agar and incubated under aerobic conditions at 37 °C. The number of colonies were counted after 16 hours to estimate CFUs. Eighty percent of target bacteria spiked into whole blood were isolated and recovered using the saponin treatment and processing through the combined membraneless dialysis-DEP microfluidic system (Fig. 4B). The trapping efficiency of the DEP device was also measured for suspensions having varying concentrations of E. coli. Data from these tests indicate that the recovery efficiency of the instrument is independent of number of target bacteria below 105 total cells (maximum number of bacteria loaded) (Fig. 4C).
Quantitative PCR targeting human 60S ribosomal protein L30 gene and the bacterial 16S ribosomal RNA gene (16S rDNA) was used to estimate the relative levels of host and bacterial DNA contained in the original input sample and one mL fraction collected after the sample had been processed. To recover the trapped bacteria into this final 1 mL fraction, the DEP signal was shut off so that the bacteria would elute from the microfluidic device. Human DNA could not be detected in 77% of final one mL fractions, and so a conservative estimate of the mean concentration in these samples was found by fitting the data to a Poisson distribution. Compared with the original sample, the relative DNA levels in the final fraction were reduced 84-fold and the levels of bacterial 16S rDNA were enriched 3.66-fold. These data demonstrate that trapping bacteria within the DEP device provides an approximately 307-fold enrichment of bacterial DNA over human DNA compared with the original sample.
Discussion
Current methods for isolating and detecting low-level bacteria in blood samples are time-consuming, laborious and costly. Despite significant public and private investment in diagnostics and treatment, sepsis-related mortality and morbidity rates are staggeringly high, even in the most developed regions of the world. Rapid, cost-effective and reliable diagnostics are a necessary component of a broader solution to this medical emergency. Earlier embodiments of DEP and other electrokinetic microfluidic technologies for isolating and concentrating bacteria relied on either membrane filtration, centrifugation, or dilution to interface DEP microfluidic components to biofluid specimens. Diluting specimens is a straightforward approach to achieve conditions that allow for pDEP concentration of target bacteria, and simultaneously reduces confounding effects such as particle–particle interactions and Joule heating. However, these sample preconditioning strategy, particularly sample dilution, are impractical in a clinical setting where the goal is to accelerate sample processing. The limitations of those strategies were circumvented by combining the selective permeabilization strategy with in-line deionization via MMDs. Permeabilization of undesired cells minimized the confounding effects of particle–particle interactions and prevented undesired BCs from collecting in the DEP stage. MMDs were successfully integrated in-line to pre-condition blood and enable efficient DEP trapping of target bacteria without diluting the sample. Importantly, the combination of using small quantities of additives and inline deonization using MMD technology provides far greater operational flexibility compared with previous embodiments of DEP-based bacterial isolation systems.
The most important challenge in applying microfluidic technology to recover rare cells from large volume specimens is achieving sufficient volumetric throughput. In its current state, with the volume of the final DEP microchamber being 43.5 μL, then the concentration factor within the DEP device would be about 160× (7000 μL/43.5 μL) for an adult specimen and 69× (3000 μL/43.5 μL) for a neonate whole blood specimen. The system requires minimal operator intervention and it is feasible to run multiple instruments in parallel. The approach is label-free, antibody-independent, and the MMD and DEP stages could be integrated and made of disposable materials, opening the possibility for deploying the system in both high and low infrastructure settings. As a result of bacterial cell enrichment using combined microdialysis-DEP, contaminating cells or nucleic acids are depleted from the sample thus improving the signal to noise ratio for downstream molecular analysis. The availability of a label-free isolation and concentration technology to enrich bacteria will aid in enhancing bacterial genetic analyses, especially when the identity and antimicrobial resistance profile of etiological agents is not known.
In its current the prototype can process blood specimens faster than culture methods, which typically require 18 hours or more to confirm the presence of low-level bacterial pathogens (if coupled to a sufficiently sensitive detection method). However, typical blood specimens are 3 mL for neonates and about 7 mL for adults, and the entire specimen must be processed within just a few hours so that clinicians can make informed decisions regarding antimicrobial chemotherapy. The literature in microfluidics is rife with reports of creative technologies that harness microscale physics but do not offer a feasible route to scaling up, other than parallelization. The MMD and DEP microfluidic devices were designed such that the forces that drive separations are applied perpendicular to the Poiseuille flow gradient. In this way, the microfluidic devices can be scaled in the other two dimensions to support higher processing rates and volumes demanded for rapid sample analysis. Work is now under way to realize scaled-up microfluidic devices capable of supporting greater volumetric processing rates so a single clinical blood specimen can be processed in just an hour.
Overall, the progress reported herein represents important advances toward realizing a microfluidic instrument for improving routine clinical microbiological analysis. The prototypes used in this research are currently being applied to other biofluid specimens, including urine, saliva, and stool preparations, to deplete contaminants from the host matrix and enrich for targeted microbes and microbial metabolites/ biomolecules. Charting the relationships and functions within microbiomes would be greatly facilitated if complex microbial communities could be isolated prior to biomolecular compositional analysis.
In this work, proof of concept for technology to isolate and concentrate target bacteria from whole blood to enhance downstream pathogen detection and analysis was achieved. A strategy to selectively alter the cytoelectric properties of undesired cells in order to facilitate the isolation of target bacteria by DEP was demonstrated and successfully incorporated into a microfluidic instrument.
Experimental
Preparation of test suspensions and specimens
For continuous-mode separations, a DEP buffer was made of 51 mg mL−1 D-mannitol, 5 mg mL−1 saponin, 0.1 mg mL−1 pluronic and 0.1 mg mL−1 bovine serum albumin (all obtained from Sigma-Aldrich, Saint Louis, MO, USA). This buffer was used both as a carrier fluid and diluent in multistage microfluidic system, and was also used as the suspending fluid in experiments to demonstrate the effect of saponin on blood cells. For the purposes of evaluating the effect of the ionophore monensin on the DEP behavior of blood cells and bacteria, small volumes of monensin solution were added suspensions of 100 μL of blood and a suspension of fluorescently labeled E. coli diluted in 500 μL of DEP buffer that did not contain saponin. All other experiments were carried out on whole blood. E. coli K12 and S. aureus strain Newman were used as model organisms of Gram-negative and Gram-positive bacteria, respectively. Both were grown overnight at 37 °C using a mechanical shaker in lysogeny broth, which contained 10 mg mL−1 tryptone, 5 mg mL−1 yeast extract, and 10 mg mL−1 sodium chloride. Whole blood was obtained via venipuncture from healthy and consenting volunteers. These experiments were conducted under the approval of Institutional Review Board for Human Research at the Baylor College of Medicine following Protocol H-29645, and were done in compliance with regulations codified in the United States Code of Federal Regulations at 45 CFR 46 and 21 CFR 56. One mL aliquots of whole blood were treated with saponin (final concentration of 5 mg mL−1) and spiked with either E. coli or S. aureus cells labeled with a fluorescent nucleic acid stain (STYO 9, Life Technologies, Carlsbad, CA, USA). In separate experiments to demonstrate the effect of saponin, blood suspensions spiked with bacteria were centrifuged at 8000 RPM for 8 minutes and resuspended in DEP buffer four times. Bacterial concentrations were estimated using optical density readings at 600 nm and by standard plating techniques. No dilution steps were required for experiments using whole blood. All experiments were done following strict sterile guidelines.
Microfluidic dialysis device fabrication
The mold for a dialysis device 10 cm in length, 3 mm in height and 250 μm in width was designed using Mastercam software package (CNC Software, Inc, Tolland, CT, USA), which produced machine code to control a Benchman 2000 CNC Mill (intelitek, Inc, Manchester, NH, USA). An angled fluid path was first carved into a 5 mm thick poly-methylmethacrylate (PMMA) work piece using a 1/16″ carbide round bottom end mill (Harvey Tool Company, LLC, Rowley, MA, USA). A second tool path used a 1/32″ carbide square bottom end mill with a flute length of 6 mm to define the microchannels. A feed plane of 100 μm (50 μm for the angled segment), a feed rate of 150%, 60% step over and a spindle rate of 4000 RPMs were determined to be the adequate settings for the machining process. Polydimethylsiloxane (PDMS) (Sylgard® 184, Dow Corning Corporation, Midland, MI, USA) in a 5 : 1 silicone to cross-linker ratio was poured in the mold and cured in at 80 °C. This ratio produced a more rigid cast than the typical 10 : 1 ratio used in standard soft lithography techniques. The firmer cast was easier to manipulate during the bonding step. Fluid inlets and outlets are drilled in a 5 mm PMMA sheet laminated with a double-coated polyester sheet with differential silicone/acrylic adhesive (#D757, Specialty Tapes Manufacturing, Franksville, WI, USA). The silicone cast defined the top and side walls of the microfluidic channel and was bonded to the PMMA base to form a complete MMD. The double sided adhesive system provides a strong bond between the PDMS top and PMMA base. In preliminary studies the remaining silicone adhesive exposed within the chamber interior was not found to significantly affect fluid flow. PEEK tubing (IDEX Health & Science LLC, Oak Harbor, WA, USA) is inserted into the inlets and outlets and sealed using a quick set epoxy. The electrical conductance of test suspensions was measured using an inline probe and conductivity meter (EC Meter 19101-00, Cole-Parmer Instrument Co., Chicago, IL, USA).
DEP device fabrication
All microelectrode arrays were patterned in gold-chromium on 2–5 mm thick glass substrates using standard photolithography to create planar DEP microelectrode arrays. The metallization layer thickness was between 100–300 nm. In experiments to determine the maximum conductivity needed to achieve pDEP on E. coli and S. aureus, a castellated microelectrode geometry 20 μm wide and spaced was used. The continuous-mode DEP devices had interdigitated parallel bar electrodes 50 μm wide and spaced. Annealed poly-methyl methacrylate (PMMA) (Acrylite® FF, US Plastics Inc, Lima, OH, USA) ∼5 mm thick was used as the microfluidic chamber top. Fluid inlets and outlets drilled to create the fluid ports using 1/16″ drill bit such that 1/16″ PEEK tubing (Upchurch Scientific, Oak Harbor, WA, USA) could be dry fit without fluid leaking. The final microfluidic DEP device was assembled by bonding the PMMA chamber top and the microelectrode array substrate with a 2.3 mil thick acrylic adhesive (Adhesive Transfer Tape Double Linered 7952MP, 3M, USA). The channel design was cut out of the adhesive sheet with a CO2 laser engraving and cutting system (Versalaser, Universal Laser Systems, Scottsdale, AZ, USA). A compact red pitaya arbitrary waveform generator (Line Technologies, Slovenia) connected to an amplifier (ZHL-3A, MiniCircuits, NY, USA) energized the microelectrode array with a DEP signal. Prior to separation experiments, 5 mg mL−1 bovine serum albumin solution was loaded into the DEP separation apparatus for one hour to minimize cell adhesion to the accumulation wall. The addition of pluronic to the DEP buffer helped to minimize undesirable cell adhesion to the DEP microelectrode surface. Experiments to measure the DEP behavior of blood cells and bacteria exposed to monensin used a quadrupole microelectrode array embedded in the bottom of a small well made by bonding a rubber O-ring to the electrode glass substrate. These microelectrodes were fabricated in the same way as the co-planar parallel electrode arrays.
DNA extraction and quantitative PCR
Targeted qPCR was used to measure the relative levels of human and bacterial DNA. One mL of carrier flow was collected at the device outlet while a 1 MHz and 20 Vpp DEP signal was applied to trap bacteria. This fraction contained permeabilized blood cells. An additional one mL of carrier flow was allowed to flow through the chamber, and then isolated bacteria were recovered from the microchamber by shutting off the DEP signal and collecting one final mL. DNA was extracted from 200 μL of these fractions using UltraClean BloodSpin DNA Isolation Kit (MoBio, Carlsbad, CA, USA) according to the manufacturer’s instructions. Bacteria-directed primers (1369F 5′-CGGTGAATACGTTCYCGG and 1492R 5′-GGWTACCTTGTTACGACTT) targeting the 16S ribosomal RNA gene V9 region, and human-directed primers targeting the 60S ribosomal protein L30 (RPL30; 5′-GCCCGTTCAGTCTCTTCGATT forward and 5′-CAAGGCAAAGCGAAATTGGT reverse), were used for qPCR analysis. Each 20 μL reaction mixture consisted of 10 μL of Power SYBR Green 2× Master Mix (Applied Biosystems, Foster City, CA, USA), 5 μL of Ultrapure HyClone molecular biology grade water (Thermo Fisher Scientific, Waltham, MA, USA), 1 μL of forward primer 1369F (5 μM), 1 μL of reverse primer 1492R (5 μM) and 3 μL of DNA template. Purified standards and no-template controls were included in all qPCR runs. The same volumes and concentrations were used for the RPL30 forward and reverse primers, which come in the NEBNext Microbiome Enrichment Kit (New England Biolabs, Ipswich, MA, USA). Thermal cycling parameters were as follows: hold at 95 °C for 10 min to achieve initial denaturation, followed by 40 cycles of: 10 s hold at 95 °C to denature, ramp-down to 60 °C for primer annealing and extension occurring through a 35 s ramp-up to 95 °C. In this study, all samples were analyzed in triplicate.
Numerical simulations
Finite element methods were used to simulate forces and calculate trajectories of particles within the microfluidic devices used in this research (COMSOL Multiphysics, Stockholm, Sweden). All other calculations were performed using Matlab (Mathworks, Natick, MA, USA).
Conclusions
Bacterial bloodstream infections are a major threat to public safety. Contributing to this issue is the fact that current methods to detect and profile bacteria causing life-threatening infections in blood can take days, greatly exceeding the time within which clinicians must administer effective therapy. The microfluidic platform technology presented herein can isolate and concentrate target bacteria from whole blood specimens and requires no prior knowledge of the etiological agent. By addressing these urgent technological gaps, the microfluidic system allows for next generation assays to be used far more effectively in the clinic for confirming infections and profiling antibiotic-resistance biomarkers.
Supplementary Material
Acknowledgments
The authors would like to thank Jeffery Tarrand, MD, for sharing his knowledge of clinical microbiology, and Thomas Anderson for helping design the microfluidic devices. The quadrupole microelectrodes were gifted by Hywel Morgan, PhD and Nicolas Green, PhD, from the Centre for Hybrid Bio-devices at the University of Southampton. Funding was provided by the Cancer Prevention Research Institute of Texas (RP130476) and the Baylor College of Medicine Alkek Center for Metagenomics and Microbiome Research. LD was supported in part by the Whitaker Foundation Summer Research grant and the National Institute of Biomedical Imaging and Bioengineering Imaging Sciences training grant (grant number 1 T32 EB 7507-1 A2).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c6lc01277a
References
- 1.Murray CK. J Trauma. 2008;64:S232–238. doi: 10.1097/TA.0b013e318163c3f5. [DOI] [PubMed] [Google Scholar]
- 2.Tsuji E, Hiki N, Nomura S, Fukushima R, Kojima J, Ogawa T, Mafune K, Mimura Y, Kaminishi M. Int J Cancer. 2003;107:303–308. doi: 10.1002/ijc.11196. [DOI] [PubMed] [Google Scholar]
- 3.Widmer AF. Clin Infect Dis. 2001;33(Suppl 2):S94–106. doi: 10.1086/321863. [DOI] [PubMed] [Google Scholar]
- 4.Rao PL, Strausbaugh LJ, Liedtke LA, Srinivasan A, Kuehnert MJ. Transfusion. 2007;47:1206–1211. doi: 10.1111/j.1537-2995.2007.01269.x. [DOI] [PubMed] [Google Scholar]
- 5.Jaimes F. Rev Panam Salud Publica. 2005;18:163–171. doi: 10.1590/s1020-49892005000800003. [DOI] [PubMed] [Google Scholar]
- 6.Engel C, Brunkhorst FM, Bone HG, Brunkhorst R, Gerlach H, Grond S, Gruendling M, Huhle G, Jaschinski U, John S, Mayer K, Oppert M, Olthoff D, Quintel M, Ragaller M, Rossaint R, Stuber F, Weiler N, Welte T, Bogatsch H, Hartog C, Loeffler M, Reinhart K. Intensive Care Med. 2007;33:606–618. doi: 10.1007/s00134-006-0517-7. [DOI] [PubMed] [Google Scholar]
- 7.Wiens MO, Kumbakumba E, Kissoon N, Ansermino JM, Ndamira A, Larson CP. Clin Epigenetics. 2012;4:319–325. doi: 10.2147/CLEP.S35693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Kumar A, Cheang M. Crit Care Med. 2006;34:1589–1596. doi: 10.1097/01.CCM.0000217961.75225.E9. [DOI] [PubMed] [Google Scholar]
- 9.Yagupsky P, Nolte FS. Clin Microbiol Rev. 1990;3:269–279. doi: 10.1128/cmr.3.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kellogg JA, Manzella JP, Bankert DA. J Clin Microbiol. 2000;38:2181–2185. doi: 10.1128/jcm.38.6.2181-2185.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wellinghausen N, Kochem AJ, Disqué C, Mühl H, Gebert S, Winter J, Matten J, Sakka SG. J Clin Microbiol. 2009;47:2759–2765. doi: 10.1128/JCM.00567-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt M, Sireis W, Seifried E. Transfus Med Hemother. 2011;38:259–265. doi: 10.1159/000330305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rood IGH, Pettersson A, de Korte D, Savelkoul PHM. Lab Med. 2008;39:553–558. [Google Scholar]
- 14.Squires T, Quake S. Rev Mod Phys. 2005;77:977–1026. [Google Scholar]
- 15.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 16.Boedicker JQ, Li L, Kline TR, Ismagilov RF. Lab Chip. 2008;8:1265–1272. doi: 10.1039/b804911d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boehm DA, Gottlieb PA, Hua SZ. Sensors Actuators B Chem. 2007;126:508–514. [Google Scholar]
- 18.Lee H, Sun E, Ham D, Weissleder R. Nat Med. 2008;14:869–874. doi: 10.1038/nm.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mairhofer J, Roppert K, Ertl P. Sensors. 2009;9:4804–4823. doi: 10.3390/s90604804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Microchannel S, Yoon S, Lim M, Park S, Shin J, Kim Y, Been THAS. J Microelectromech Syst. 2008;17:590–598. [Google Scholar]
- 21.Puttaswamy S, Lee BD, Sengupta S. J Clin Microbiol. 2011;49:2286–2289. doi: 10.1128/JCM.00369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suehiro J, Shutou M, Hatano T, Hara M. Sensors Actuators B Chem. 2003;96:144–151. [Google Scholar]
- 23.Zhou R, Wang P, Chang HC. Electrophoresis. 2006;27:1376–1385. doi: 10.1002/elps.200500329. [DOI] [PubMed] [Google Scholar]
- 24.Bisceglia E, Cubizolles M, Trainito CI, Berthier J, Pudda C, Français O, Mallard F, Le Pioufle B. Sensors Actuators B Chem. 2015;212:335–343. [Google Scholar]
- 25.Hou HW, Bhagat AAS, Lee WC, Huang S, Han J, Lim CT. Micromachines. 2011;2:319–343. [Google Scholar]
- 26.Pohl HA, Crane JS. Biophys J. 1971;11:711–727. doi: 10.1016/S0006-3495(71)86249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonso MA, Carrasco L. Eur J Biochem. 1982;127:567–569. doi: 10.1111/j.1432-1033.1982.tb06909.x. [DOI] [PubMed] [Google Scholar]
- 28.Wright AJ, Finglas PM, Southon S. Clin Chem. 2000;46:1978–1986. [PubMed] [Google Scholar]
- 29.Cockerill F, 3rd, Torgerson C, Reed G, Vetter E, Weaver A, Dale J, Roberts G, Henry N, Ilstrup D, Rosenblatt J. J Clin Microbiol. 1996;34:20. doi: 10.1128/jcm.34.1.20-24.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han X, van Berkel C, Gwyer J, Capretto L, Morgan H. Anal Chem. 2012;84:1070–1075. doi: 10.1021/ac202700x. [DOI] [PubMed] [Google Scholar]
- 31.Hoettges KF, Ogin SL, Hughes MP. IEE Proc: Nanobiotechnol. 2005;152:150–154. doi: 10.1049/ip-nbt:20050011. [DOI] [PubMed] [Google Scholar]
- 32.Shim S, Stemke-Hale K, Tsimberidou AM, Noshari J, Anderson TE, Gascoyne PRC. Biomicrofluidics. 2013;7:11807. doi: 10.1063/1.4774304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.