

Supplemental Digital Content is available in the text
Keywords: CDKN2A, clinical features, expression, methylation, PCa, prognosis
Abstract
Background:
Reduction of cyclin-dependent kinase inhibitor 2A (CDKN2A) (p16 and p14) expression through DNA methylation has been reported in prostate cancer (PCa). This meta-analysis was conducted to assess the difference of p16 and p14 methylation between PCa and different histological types of nonmalignant controls and the correlation of p16 or p14 methylation with clinicopathological features of PCa.
Methods:
According to the preferred reporting items for systematic reviews and meta-analyses (PRISMA) statement criteria, articles were searched in PubMed, Embase, EBSCO, Wanfang, and CNKI databases. The strength of correlation was calculated by the pooled odds ratios (ORs) and their corresponding 95% confidence intervals (95% CIs). Trial sequential analysis (TSA) was used to estimate the required population information for significant results.
Results:
A total of 20 studies published from 1997 to 2017 were identified in this meta-analysis, including 1140 PCa patients and 530 cases without cancer. Only p16 methylation in PCa was significantly higher than in benign prostatic lesions (OR = 4.72, P = .011), but had a similar level in PCa and adjacent tissues or high-grade prostatic intraepithelial neoplasias (HGPIN). TSA revealed that this analysis on p16 methylation is a false positive result in cancer versus benign prostatic lesions (the estimated required information size of 5116 participants). p16 methylation was not correlated with PCa in the urine and blood. Besides, p16 methylation was not linked to clinical stage, prostate-specific antigen (PSA) level, and Gleason score (GS) of patients with PCa. p14 methylation was not correlated with PCa in tissue and urine samples. No correlation was observed between p14 methylation and clinical stage or GS. CDKN2A mutation and copy number alteration were not associated with prognosis of PCa in overall survival and disease-free survival. CDKN2A expression was not correlated with the prognosis of PCa in overall survival (492 cases) (P > .1), while CDKN2A expression was significantly associated with a poor disease-free survival (P < .01).
Conclusion:
CDKN2A methylation may not be significantly associated with the development, progression of PCa. Although CDKN2A expression had an unfavorable prognosis in disease-free survival. More studies are needed to confirm our results.
1. Introduction
According to GLOBOCAN estimates, prostate cancer (PCa) is the most frequent human malignancy and the 3rd-leading cause of cancer-related deaths among men in developed countries. And this disease is the 4th most common cancer and the 6th-leading cause of cancer-related deaths among men in developing countries.[1] Although therapeutic strategies have marked advances in recent years for PCa patients, patients with advanced stage easily develop metastasis, with a poor survival rate.[2–4]
Increasing evidence suggests that epigenetic modifications are an early biotic regulation mechanism in human cancers.[5–7] As a frequent mechanism of epigenetic alterations, DNA methylation is closely linked to cancer initiation and progression.[8,9] Aberrantly methylated tumor suppressor genes (TSGs) have been reported in PCa, such as RAS association domain family protein 1 A,[10]miR-23b,[11] and glutathione S-transferase P1.[12] Mapped to human chromosome 9p21, the p16 and p14 genes, are encoded by the cyclin-dependent kinase inhibitor 2A (CDKN2A) and play a key role in cell cycle regulation.[13,14] As a common TSG, downregulation or loss of CDKN2A (p16 and p14) expression through DNA methylation frequently occurs in malignant tumors.[15–17] The p16 or p14 gene has been reported to be methylated in PCa.[18–21]
A previous meta-analysis only reported the correlation of p16 gene methylation between PCa and controls.[22] However, there are some inconsistent and conflicting results regarding p16 or p14 methylation in PCa. For example, Yaqinuddin 2013 et al[23] reported that the p16 gene was not methylated in tissue samples of PCa patients. Although Ameri 2011 et al[24] reported that the p16 gene was frequently methylated in tissue samples of patients with PCa. Hence, we systematically gathered all available publications to evaluate the correlation of p16 or p14 methylation between PCa and different pathological types of nonmalignant tissue samples (adjacent tissues, benign prostatic lesions, or high-grade prostatic intraepithelial neoplasias [HGPIN]). In addition, we also evaluated whether p16 or p14 methylation was associated with PCa in blood or urine samples. Finally, we assessed the relationship of p16 or p14 methylation with the clinicopathological features of patients with PCa.
2. Materials and methods
2.1. Literature search strategy
A comprehensive search of PubMed, Embase, EBSCO, Wanfang, and CNKI databases was performed to get the eligible studies before March 7, 2017. The following search terms and key words were used: “P16 OR CDKN2A OR INK4A OR cyclin-dependent kinase inhibitor 2A,” “p14 OR p14ARF,” “methylation OR methylated OR epigene∗,” “prostate cancer OR PCa OR prostate adenocarcinoma OR prostate tumor OR prostate carcinoma OR prostate neoplasm.” The eligible studies were retrieved to identify other potential publications.
2.2. Inclusion criteria
All eligible studies had to meet the following selection criteria in this meta-analysis: all patients were confirmed with primary PCa; studies reported the information of p16 or p14 methylation in PCa, with no restriction of sample types (tissue, urine, or blood); studies provided sufficient data to evaluate the association between p16 or p14 methylation and PCa in cancer versus different control types (adjacent tissues, benign prostatic lesions, or HGPIN, etc); and studies provided sufficient data to assess the relationship of p16 or p14 methylation with clinicopathological features of patients with PCa, including clinical stage, prostate-specific antigen (PSA) level, and Gleason score (GS). The most recent or complete paper was included when duplicate publications were reported using the same study population.
2.3. Ethical statement
This meta-analysis was not primary research involving human samples, but rather a secondary analysis of human subject data published in the public domain.
2.4. Data extraction
Data extraction was performed by 2 independent authors (ZC and LW). Disagreements were discussed to resolve the dispute by all authors. The relevant information was carefully scanned and extracted from all eligible publications: first author's name, year of publication, country, ethnicity, detection method, sample types, age, the number of the study population, control groups, methylation frequency, and the clinicopathological features. Control groups included adjacent tissues, benign prostatic lesions, HGPIN, and blood or urine samples without cancer. The clinicopathological features consisted of tumor stage (stage 3–4 vs stage 1–2), PSA level (PSA > 8 vs PSA ≤ 8), and GS (GS ≥ 7 vs GS ≤ 6).
2.5. Statistical analysis
The statistical analyses were conducted by Stata 12.0 software (Stata Corporation, College Station, TX). The strength of association between p16 or p14 methylation and PCa risk was estimated by calculating combined odds ratios (ORs) and their corresponding 95% confidence intervals (CIs). The heterogeneity among the included publications was investigated with the Q-test statistic.[25,26] This meta-analysis was performed using a random-effects model. A P value of less than .10 was considered as substantial heterogeneity. We performed a sensitivity analysis to analyze the influence of an individual study on the pooled result and heterogeneity by omitting one study.[27,28] For the analyses with more than 8 studies, the potential publication bias was measured with Egger line regression test.[29,30] For significant results, to decrease the type I and II error rates, the trial sequential meta-analysis (trial sequential analysis [TSA]) was conducted to estimate the needed sample information.[31,32] The type I error rate was defined as 5%, and the type II error rate was set as 20% (a statistical power of 80%). The relative risk reduction (RRR) rate was considered as 20%. When the cumulative Z-curve crossed the trial sequential monitoring boundary or the required information size, which suggested that the evidence is conclusive. Otherwise, a false positive result is achieved.[33,34]
3. Results
3.1. Study characteristics
After searching a range of online databases, 285 articles were initially searched, as shown in Fig. 1, according to the above-mentioned selection criteria, final 20 articles published from 1997 to 2017[18–21,23,24,35–48] were included in the present meta-analysis, including 1140 patients with PCa and 530 cases without cancer. Of the eligible studies, 17 studies evaluated the relationship between p16 methylation and PCa in cancer versus different control groups.[18–21,23,24,35,39–48] 12 studies including 748 patients with PCa assessed the correlation of p16 methylation with the clinicopathological characteristics of PCa.[18,19,21,24,36–38,41–43,45,46] Seven publications analyzed the correlation between p14 methylation and PCa.[20,21,37,41,42,44,45] The basic characteristics of the eligible publications are listed in Table S1.
Figure 1.
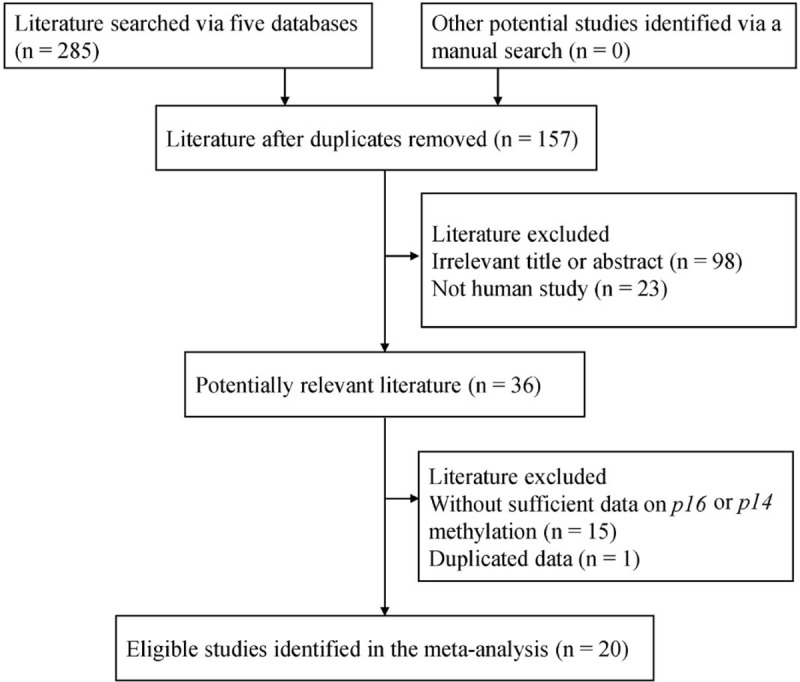
Flow diagram of the procedure for selecting literature.
3.2. Correlation between p16 methylation and PCa in cancer versus different control types
When cancerous tissues were compared to noncancerous tissue samples, the data included 9 studies with 637 PCa patients and 228 benign prostatic lesions, 5 studies with 219 PCa patients and 78 adjacent tissue samples, and 3 studies with 167 PCa patients and 55 patients with HGPIN (Fig. 2). The results of p16 methylation demonstrated that a significant relationship was found in PCa versus benign prostatic lesions (OR = 4.72, 95% CI = 1.42–15.70, P = .011), but no association was observed in PCa versus adjacent tissues or HGPIN (OR = 1.68, 95% CI = 0.24–11.85, P = .602; OR = 1.30, 95% CI = 0.41–4.17, P = .657, respectively).
Figure 2.
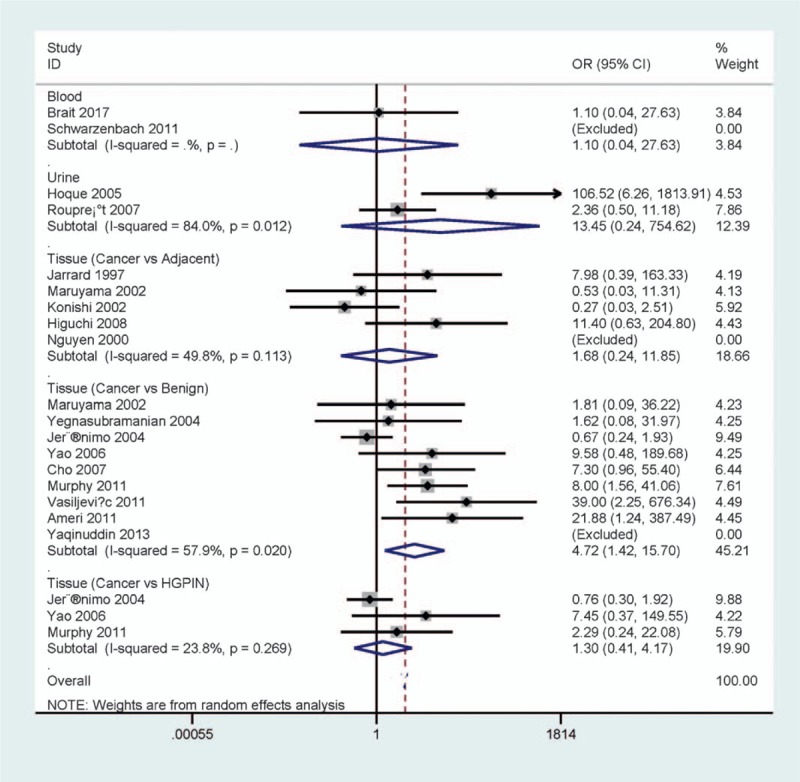
Forest plot of the pooled OR between p16 methylation and PCa in cancer versus different types of noncancerous tissues, urine, and blood samples. OR = odds ratio, PCa = prostate cancer.
We analyzed whether p16 methylation was associated with PCa in the urine and blood (Fig. 2). The result from 2 studies with 97 PCa and 40 cases without cancer showed that p16 methylation was not correlated with PCa in the blood (OR = 1.10, 95% CI = 0.04–27.63, P = .956). The result from 2 studies involving 147 PCa and 129 cases without cancer demonstrated that p16 methylation was not associated with PCa in the urine (OR = 13.45, 95% CI = 0.24–754.62, P = .206).
3.3. Subgroup analysis of p16 methylation in PCa versus benign prostatic lesions
According to ethnicity (Caucasians and Asians) and testing method (methylation-specific polymerase chain reaction [MSP] and non-MSP), subgroup analysis of ethnicity showed a trend toward the correlation between p16 methylation and Caucasians with PCa (OR = 4.16, 95% CI = 0.91–19.16, P = .067) (Fig. 3). A significant correlation was found between p16 methylation and Asians with PCa (OR = 7.95, 95% CI = 1.49–42.54, P = .015) (Fig. 3).
Figure 3.
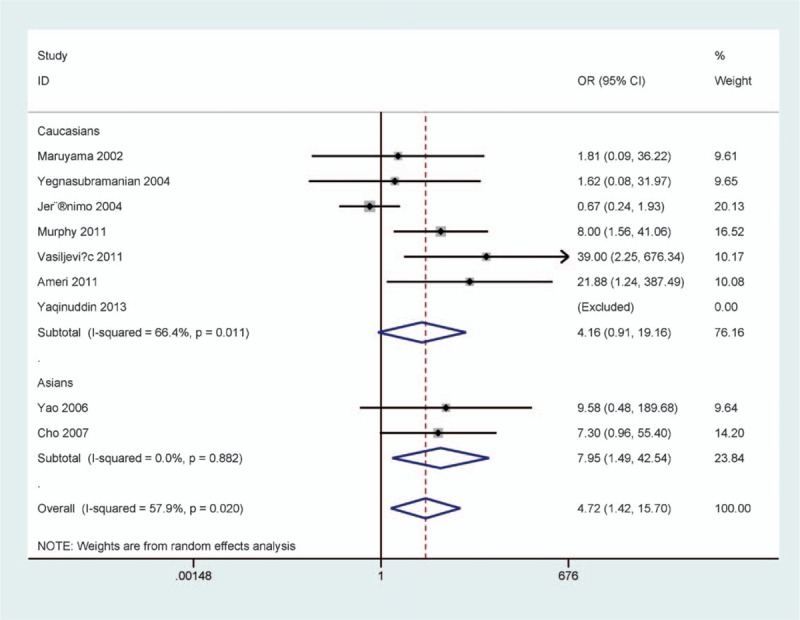
Forest plot of subgroup analysis by ethnicity in prostate cancer (PCa) versus benign prostatic lesions.
Subgroup analysis of detection method demonstrated that p16 methylation was correlated with PCa in the MSP method (OR = 7.40, 95% CI = 2.01–27.26, P = .003), but not in the non-MSP subgroup (OR = 3.63, 95% CI = 0.53–24.83, P = .189) (Fig. 4).
Figure 4.
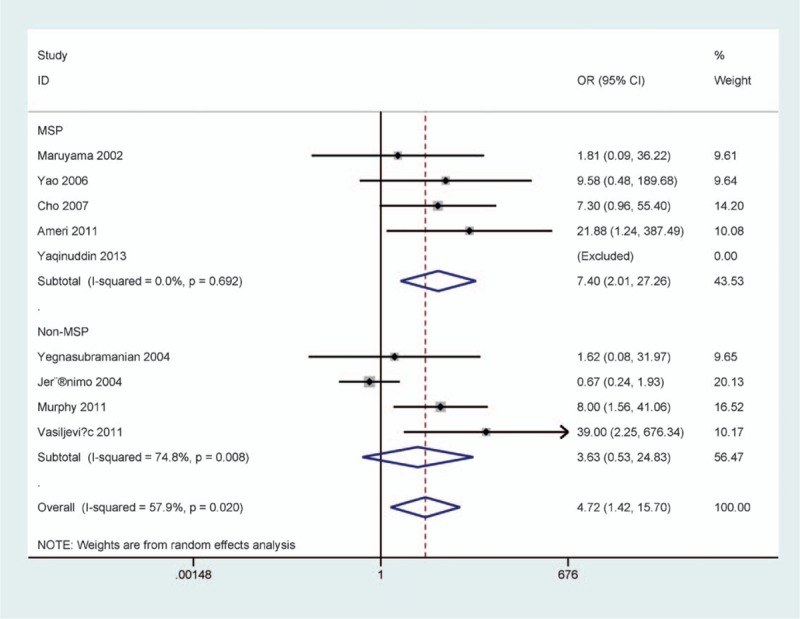
Forest plot of subgroup analysis by the testing method in prostate cancer (PCa) versus benign prostatic lesions.
3.4. Sensitivity analysis of p16 methylation in PCa versus benign prostatic lesions
A substantial heterogeneity was detected in the comparison of PCa and benign prostatic lesions (P = .020 < .1). One study (Jerónimo 2004 et al)[20] was deleted, the pooled OR was recalculated (OR = 7.80, 95% CI = 3.13–19.45, P < .001), with no evidence of heterogeneity (P = .693).
3.5. Correlation of p16 methylation with clinicopathological features of patients with PCa
The result demonstrated that p16 methylation was not associated with clinical stage, PSA level or GS of patients with PCa (all Ps > .1) (Fig. 5). These data included 5 studies with 547 PCa patients on clinical stage, 4 studies with 211 PCa patients on PSA level and 12 studies with 747 PCa patients on GS.
Figure 5.
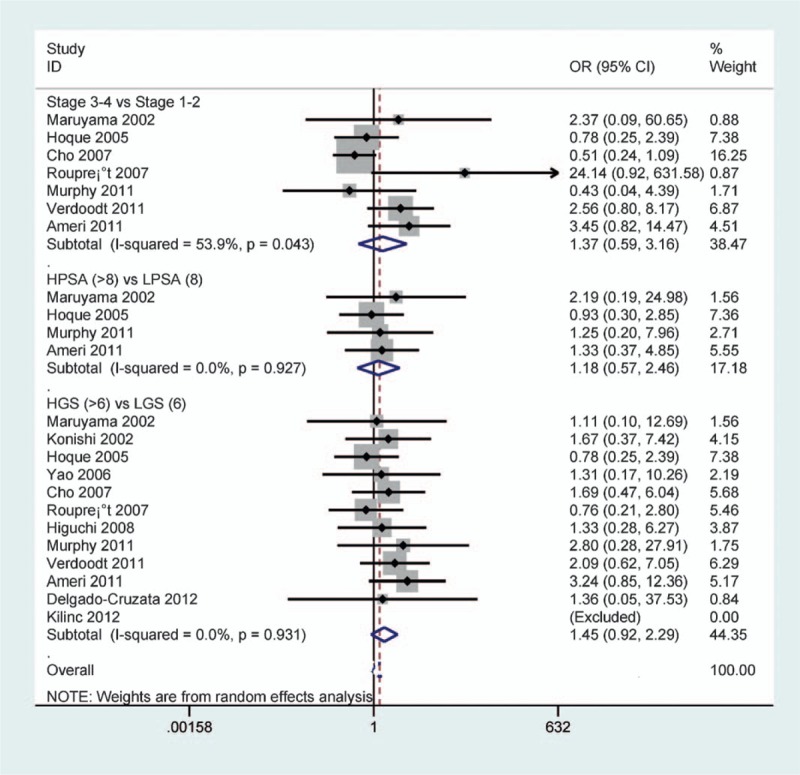
Forest plot of the association between p16 methylation and clinical stage, PSA level, or GS of patients with PCa (all P > .1). GS = Gleason score, PCa = prostate cancer, PSA = prostate-specific antigen.
3.6. Publication bias
Egger test was used to estimate the potential publication bias in PCa versus benign prostatic lesions and GS (Fig. 6). No obvious evidence of publication bias was found in PCa versus benign prostatic lesions and GS (P > .5).
Figure 6.
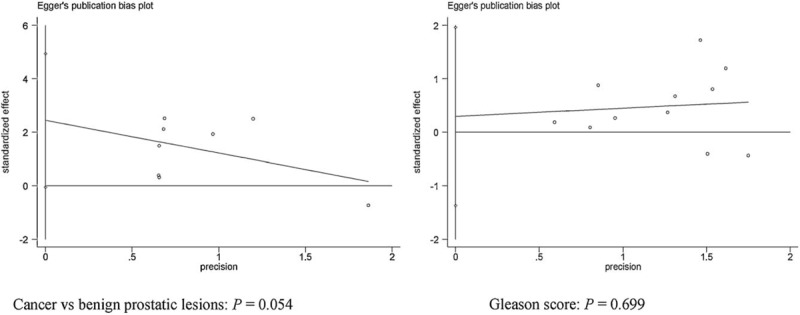
Forest plot of publication bias using Egger test in PCa versus benign prostatic lesions and GS (P > .05). GS = Gleason score, PCa = prostate cancer.
3.7. Correlation between p14 methylation and PCa in cancer versus different control types
No correlation was found between p14 methylation and different control types in cancer versus adjacent, benign tissue samples, or urine controls without cancer among 2 studies, respectively (all Ps > .5) (Fig. 7). Because the sample sizes were small in the current meta-analysis, additional studies with a large population are needed.
Figure 7.
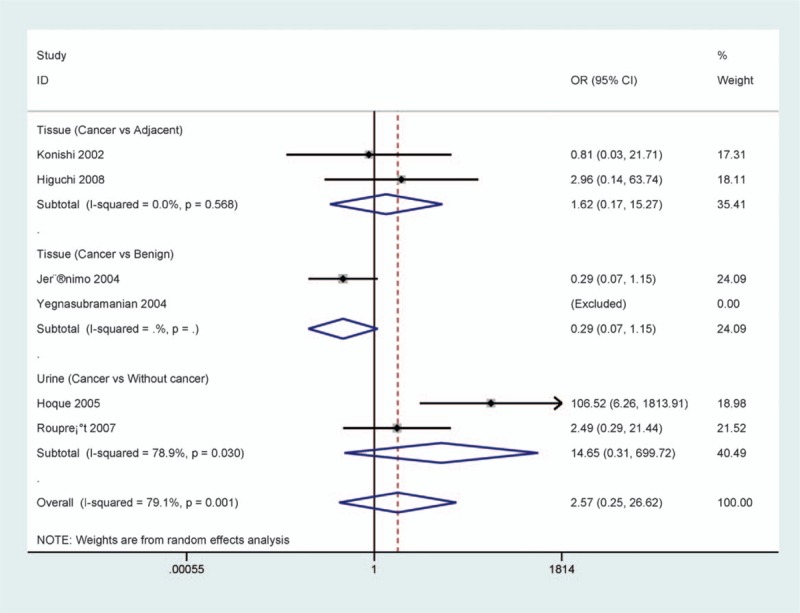
Forest plot of the association between p14 methylation and prostate cancer (PCa) in cancer versus different control types (all P > .05).
3.8. Correlation of p14 methylation with clinicopathological features of patients with PCa
The result from 3 studies involving 238 PCa patients shown no correlation between p14 methylation and tumor stage (P = .666) (Fig. 8). No relationship was found between p14 methylation and GS among 5 studies (P = .269), including 323 PCa patients (Fig. 8).
Figure 8.
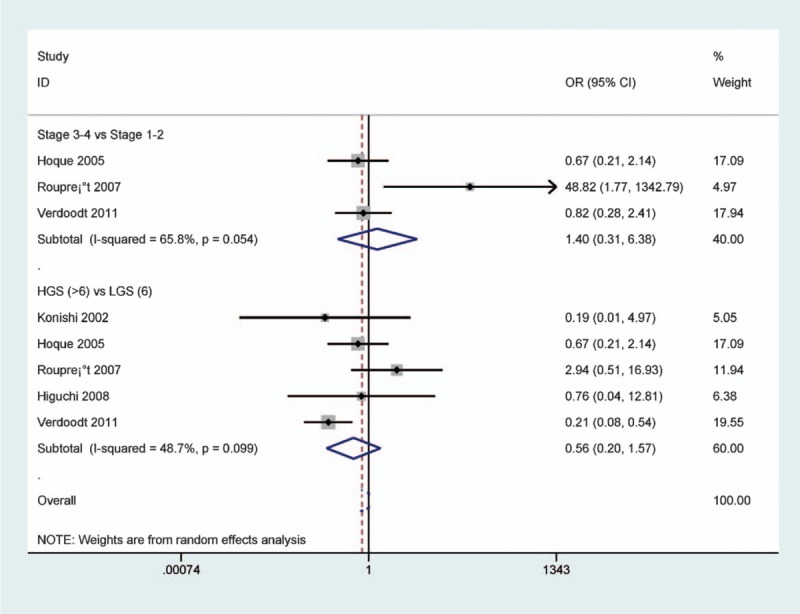
Forest plot of the association of p14 methylation with clinicopathological features of patients with prostate cancer (PCa) (all P > .1).
3.9. TSA
TSA was performed using the priori anticipated information size (APIS) method in PCa versus benign lesions, as shown in Fig. 9, the required information size was 5116 participants. The result showed that the cumulative Z-curve was not more than the trial sequential monitoring boundary, suggesting a false positive result.
Figure 9.
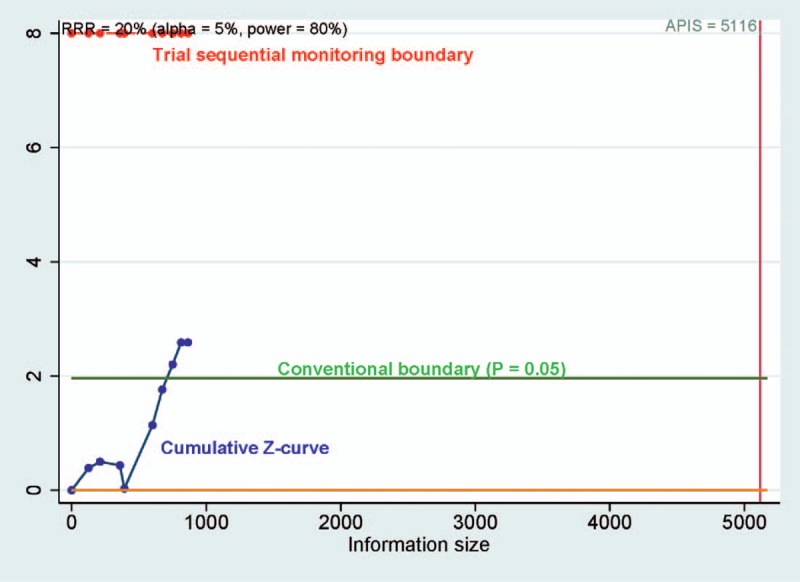
Trial sequential analysis assessing the required sample information in prostate cancer (PCa) versus benign lesions, the cumulative Z-curve crossed the conventional boundary (Z = 1.96, P = .05), did not cross the trial sequential monitoring boundary, suggesting that the cumulative evidence is inconclusive.
3.10. Prognostic role of CDKN2A
No survival information or data on CDKN2A (p16 and p14) methylation was reported in PCa. The data from GEPIA database[49] demonstrated that no association was found between CDKN2A expression and the prognosis of PCa in overall survival (492 cases) (P > .1) (Fig. 10), while CDKN2A expression was significantly associated with a poor disease-free survival (P < .01) (Fig. 11).
Figure 10.
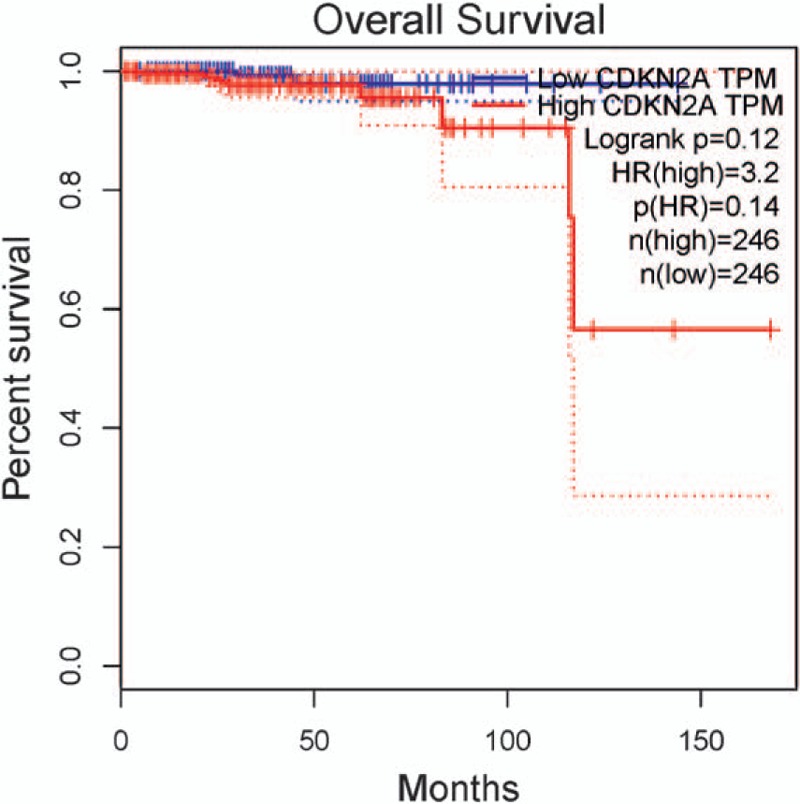
The correlation between CDKN2A expression and the prognosis of PCa in overall survival (P > .1). CDKN2A = cyclin-dependent kinase inhibitor 2A, PCa = prostate cancer.
Figure 11.
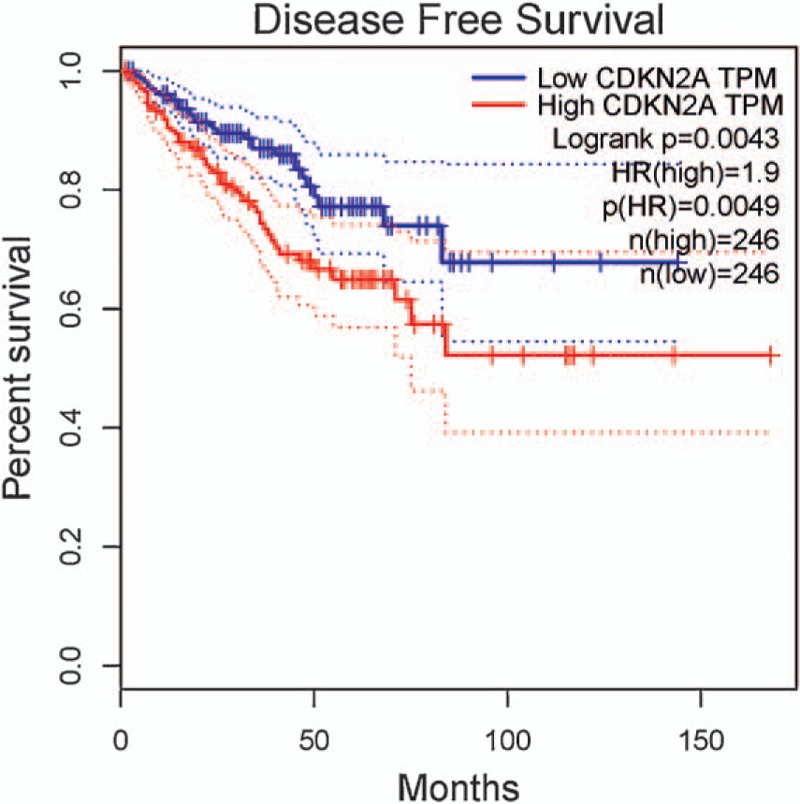
The correlation between CDKN2A expression and the prognosis of PCa in disease-free survival (P < .01). CDKN2A = cyclin-dependent kinase inhibitor 2A, PCa = prostate cancer.
4. Discussion
DNA methylation of TSGs leads to loss of gene expression and plays an essential role in the development and progression of human cancers.[50–52] Some studies suggest that promoter methylation of TSGs is closely associated with PCa.[53,54] As an important TSG, p16 methylation has been reported in PCa, the relationship between p16 methylation and its expression was observed in PCa, with loss of p16 expression.[18,48]p16 methylation may play a crucial role in the tumorigenesis of PCa.[24,43]p14 methylation was significantly higher in PCa than in urine samples without cancer by Hoque et al.[21] However, the exact association between p16 or p14 methylation and PCa is still controversial. Therefore, we conducted this meta-analysis to resolve these inconsistent results on the role of p16 or p14 methylation in PCa.
PCa develops from normal epithelium into benign lesions (benign prostatic hyperplasias), then progresses from premalignant (HGPIN) to malignant (prostate carcinoma) lesions via multiple sequences.[55–57] When cancerous tissue samples were compared to different types of noncancerous tissue samples, no association between p16 methylation and PCa was found in cancer versus adjacent tissue samples among all studies.[41,45–48] Moreover, no relationship between p16 methylation and PCa was observed in cancer versus and HGPIN among all studies.[18–20] Four studies reported that significant correlation between p16 methylation and PCa was found in cancer versus benign prostatic lesions,[18,24,39,43] but the other studies showed no correlation.[19,20,23,44,46] Our meta-analysis involving more eligible articles showed that p16 methylation was notably higher in PCa than in benign prostatic lesions (OR = 4.72, P = .011), but p16 methylation had a similar OR value in PCa and adjacent normal tissue samples or HGPIN. In addition, further TSA revealed that the result in the comparison of PCa and benign lesions was false positive, additional studies are necessary to confirm it. Our results suggested that p16 methylation may not be significantly correlated with PCa development. Because the sample sizes were small, more studies are needed in PCa versus adjacent normal tissue samples and HGPIN.
We also determined whether p16 methylation was correlated with PCa in urine and blood samples, p16 methylation was not associated with PCa in the blood.[35,40] One study reported the correlation between p16 methylation and PCa in urine samples,[21] but another study showed no association between p16 methylation and PCa in urine samples.[42] Our results suggested that p16 methylation was not correlated with PCa in the blood and urine. Additional studies with large sample sizes are needed to confirm whether p16 methylation may be a noninvasive biomarker using blood or urine samples in the future.
No correlation of p14 methylation was found in the tissue (cancer vs adjacent tissues)[41,45] and (cancer vs benign tissues).[20,44] The current results indicated no relationship between p14 methylation and PCa in tissue samples, which were consistent with an individual study. p14 methylation showed a higher frequency in PCa than in urine samples without cancer,[21] but another study demonstrated a similar frequency in PCa and control urine samples without cancer.[42] Our results showed that p14 methylation was not associated with PCa in the urine. The results of p14 methylation should be carefully considered because small sample sizes were included in this meta-analysis.
We analyzed whether p16 or p14 methylation was correlated with the clinicopathological features, including tumor stage, prostate-specific antigen level, and GS. For eligible studies, no significant relationship was observed between p16 methylation and tumor stage,[18,21,24,37,42,43,46] prostate-specific antigen level,[18,21,24,46] or GS.[18,19,21,24,36–38,41–43,45,46] Our results comprising more studies with a large population showed that p16 methylation was not correlated with tumor stage, prostate-specific antigen level, and GS (P > .1). Our results were consistent with an individual study regarding p16 methylation. One study reported the relationship between p14 methylation and clinical stage,[42] but the other studies showed no association.[21,37]p14 methylation was negatively associated with GS by Verdoodt 2011 et al,[37] while no correlation was reported among the remaining 4 studies.[21,41,42,45] The results demonstrated that p14 methylation was not linked to clinical stage and GS of patients with PCa. The above analyses suggested that p16 and p14 methylation may not be linked to the clinicopathological features of PCa patients.
Interestingly, we found that CDKN2A expression was significantly associated with a reduced prognosis of PCa in disease-free survival. The cBioportal database[58,59] (TCGA, Provisional, 499 samples) was also used to explore the possible prognostic impact of CDKN2A mutation and copy number alteration in PCa. CDKN2A mutation was shown in 1 (0.2%) of 498 samples and was not associated with the prognosis in overall survival and disease-free survival (data not shown). CDKN2A copy number alteration (deep deletion) was shown in 8 (1.6%) of 498 samples and was not correlated with the prognosis in overall survival and disease-free survival (data not shown). The other mechanisms such as allelic loss or amplification may lead to CDKN2A expression. And therefore CDKN2A expression may influence the prognosis of PCa in disease-free survival. Additional studies are necessary in the future.
The current meta-analysis compared favorably with this previous meta-analysis by Feng et al,[22] the previous study only reported that p16 methylation was correlated with PCa in cancer versus controls. The number of study population included in our meta-analysis (n = 1670 samples) was larger than in the previous study (n = 1296 samples). A further TSA was conducted in our study, and we found this false positive result in PCa versus benign lesions. Additionally, this previous meta-analysis did not evaluate the exact correlation between p16 methylation and the sequence of histological changes of the prostate ([adjacent normal tissues-benign lesions (benign prostatic hyperplasias)-premalignant lesions (HGPIN)-malignant prostate]). Finally, this previous meta-analysis did not analyze the association between p16 methylation and clinicopathological features of PCa, the association between p14 methylation and PCa, and the potential prognostic effect of CDKN2A methylation, expression, mutation, or copy number alteration in PCa.
A slight heterogeneity was measured in PCa versus benign prostatic lesions (p16 gene: P = .020 < .1). When we removed this study (Jerónimo 2004 et al)[20] and recalculated the overall OR from the remaining studies. The pooled OR did not significantly change (OR = 7.80, P < .001), with no heterogeneity (P = .693). The possible reasons for substantial heterogeneity were not very clear, possibly because of the use of the inappropriate conditions in the detection of p16 methylation. The present meta-analysis had several limitations. First, only articles published in English or Chinese were selected in this study. Publications in other languages were excluded based on insufficient information, which may cause a selection bias. Second, only 2 studies of blood or urine samples were analyzed. Third, sample sizes were small among different subgroups and in the comparison of PCa and different control groups. More studies with large populations are necessary to validate these results.
5. Conclusions
Our findings suggest that only p16 methylation may have a higher frequency in PCa than in benign prostatic lesions. p16 methylation was not associated with PCa risk in PCa versus adjacent tissues or HGPIN. No correlation between p16 methylation and PCa was found in blood or urine samples. In addition, p16 methylation was not correlated with clinical stage, PSA level, and GS of patients with PCa. p14 methylation was not correlated with PCa in the tissue and urine, and no relationship was observed between p14 methylation and clinical stage and GS. No correlation was found between CDKN2A mutation and copy number alteration and the prognosis of PCa in overall survival and disease-free survival. No association was observed between CDKN2A expression and the prognosis of PCa in overall survival, while CDKN2A expression was significantly associated with a poor prognosis in disease-free survival. Further large-scale and well-designed trials are essential to confirm our findings in the future.
6. Author contributions
Conceptualization: Z. Cao, X. Yao.
Formal analysis: Z. Cao, L. Wei, W. Zhu, X. Yao.
Methodology: Z. Cao, X. Yao.
Software: Z. Cao, L. Wei, W. Zhu, X. Yao.
Supervision: X. Yao.
Validation: Z. Cao, X. Yao.
Writing – original draft: Z. Cao, L. Wei, W. Zhu, X. Yao.
Writing – review & editing: Z. Cao, L. Wei, X. Yao.
Supplementary Material
Footnotes
Abbreviations: CI = confidence interval, CDKN2A = cyclin-dependent kinase inhibitor 2A, GS = Gleason score, HGPIN = high-grade prostatic intraepithelial neoplasias, MSP = methylation-specific polymerase chain reaction, OR = odds ratio, PCa = prostate cancer, TSA = trial sequential analysis, TSG = tumor suppressor gene.
Authorship: XY and ZC contributed to the conception and design of this research. ZC, LW, and WZ contributed to the drafting of the article and final approval of the submitted version. ZC, LW, WZ, and XY contributed to data analyses and interpretation and completion of the figures and table. All authors read and approved the final manuscript.
The authors have no funding and conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- [2].Fossa SD, Nilssen Y, Kvale R, et al. Treatment and 5-year survival in patients with nonmetastatic prostate cancer: the Norwegian experience. Urology 2014;83:146–52. [DOI] [PubMed] [Google Scholar]
- [3].Heidenreich A, Bastian PJ, Bellmunt J, et al. EAU guidelines on prostate cancer. part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur Urol 2014;65:124–37. [DOI] [PubMed] [Google Scholar]
- [4].Sharifi N, Dahut WL, Figg WD. The genetics of castration-resistant prostate cancer: what can the germline tell us? Clin Cancer Res 2008;14:4691–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Khan SA, Reddy D, Gupta S. Global histone post-translational modifications and cancer: biomarkers for diagnosis, prognosis and treatment? World J Biol Chem 2015;6:333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Esteller M. Epigenetics in cancer. N Engl J Med 2008;358:1148–59. [DOI] [PubMed] [Google Scholar]
- [7].Ye M, Huang T, Ni C, et al. Diagnostic capacity of RASSF1A promoter methylation as a biomarker in tissue, brushing, and blood samples of nasopharyngeal carcinoma. EBioMedicine 2017;18:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ye M, Huang T, Ying Y, et al. Detection of 14-3-3 sigma (sigma) promoter methylation as a noninvasive biomarker using blood samples for breast cancer diagnosis. Oncotarget 2017;8:9230–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hattori N, Ushijima T. Epigenetic impact of infection on carcinogenesis: mechanisms and applications. Genome Med 2016;8:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Agarwal S, Amin KS, Jagadeesh S, et al. Mahanine restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in prostate cancer cells. Mol Cancer 2013;12:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Majid S, Dar AA, Saini S, et al. miR-23b represses proto-oncogene Src kinase and functions as methylation-silenced tumor suppressor with diagnostic and prognostic significance in prostate cancer. Cancer Res 2012;72:6435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zelic R, Fiano V, Zugna D, et al. Global hypomethylation (LINE-1) and gene-specific hypermethylation (GSTP1) on initial negative prostate biopsy as markers of prostate cancer on a rebiopsy. Clin Cancer Res 2016;22:984–92. [DOI] [PubMed] [Google Scholar]
- [13].Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer 2012;130:1715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lukas J, Parry D, Aagaard L, et al. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature 1995;375:503–6. [DOI] [PubMed] [Google Scholar]
- [15].Gamell C, Gulati T, Levav-Cohen Y, et al. Reduced abundance of the E3 ubiquitin ligase E6AP contributes to decreased expression of the INK4/ARF locus in non-small cell lung cancer. Sci Signal 2017;10: [DOI] [PubMed] [Google Scholar]
- [16].Shima K, Nosho K, Baba Y, et al. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expression in 902 colorectal cancers: cohort study and literature review. Int J Cancer 2011;128:1080–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ribas C, Colleoni GW, Felix RS, et al. p16 gene methylation lacks correlation with angiogenesis and prognosis in multiple myeloma. Cancer Lett 2005;222:247–54. [DOI] [PubMed] [Google Scholar]
- [18].Murphy TM, Sullivan L, Lane C, et al. In silico analysis and DHPLC screening strategy identifies novel apoptotic gene targets of aberrant promoter hypermethylation in prostate cancer. Prostate 2011;71:1–7. [DOI] [PubMed] [Google Scholar]
- [19].Yao Q, He XS, Zhang JM, et al. [Promotor hypermethylation of E-cadherin, p16 and estrogen receptor in prostate carcinoma]. Zhonghua Nan Ke Xue 2006;12:28–31. [PubMed] [Google Scholar]
- [20].Jerónimo C, Henrique R, Hoque MO, et al. A quantitative promoter methylation profile of prostate cancer. Clin Cancer Res 2004;10:8472–8. [DOI] [PubMed] [Google Scholar]
- [21].Hoque MO, Topaloglu O, Begum S, et al. Quantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J Clin Oncol 2005;23:6569–75. [DOI] [PubMed] [Google Scholar]
- [22].Feng W, Han Z, Zhu R, et al. Association of p16 gene methylation with prostate cancer risk: a meta-analysis. J BUON 2015;20:1074–80. [PubMed] [Google Scholar]
- [23].Yaqinuddin A, Qureshi SA, Pervez S, et al. Frequent DNA hypermethylation at the RASSF1A and APC gene loci in prostate cancer patients of Pakistani origin. ISRN Urol 2013;2013:627249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ameri A, Alidoosti A, Hosseini SY, et al. Prognostic value of promoter hypermethylation of retinoic acid receptor beta (RARB) and CDKN2 (p16/MTS1) in prostate cancer. Chin J Cancer Res 2011;23:306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Coory MD. Comment on: heterogeneity in meta-analysis should be expected and appropriately quantified. Int J Epidemiol 2010;39:932.author reply 933. [DOI] [PubMed] [Google Scholar]
- [26].Patsopoulos NA, Evangelou E, Ioannidis JP. Sensitivity of between-study heterogeneity in meta-analysis: proposed metrics and empirical evaluation. Int J Epidemiol 2008;37:1148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med 1997;127:820–6. [DOI] [PubMed] [Google Scholar]
- [28].Han S, Zong S, Shi Q, et al. Is Ep-CAM expression a diagnostic and prognostic biomarker for colorectal cancer? A systematic meta-analysis. EBioMedicine 2017;20:61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Egger M, Davey Smith G, Schneider M, et al. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315:629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Han S, Yang W, Zong S, et al. Clinicopathological, prognostic and predictive value of CD166 expression in colorectal cancer: a meta-analysis. Oncotarget 2017;8:64373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kulinskaya E, Wood J. Trial sequential methods for meta-analysis. Res Synth Methods 2014;5:212–20. [DOI] [PubMed] [Google Scholar]
- [32].Brok J, Thorlund K, Gluud C, et al. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta-analyses. J Clin Epidemiol 2008;61:763–9. [DOI] [PubMed] [Google Scholar]
- [33].Holst LB, Petersen MW, Haase N, et al. Restrictive versus liberal transfusion strategy for red blood cell transfusion: systematic review of randomised trials with meta-analysis and trial sequential analysis. BMJ 2015;350:h1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wetterslev J, Thorlund K, Brok J, et al. Estimating required information size by quantifying diversity in random-effects model meta-analyses. BMC Med Res Methodol 2009;9:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Brait M, Banerjee M, Maldonado L, et al. Promoter methylation of MCAM, ERalpha and ERbeta in serum of early stage prostate cancer patients. Oncotarget 2017;8:15431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kilinc D, Ozdemir O, Ozdemir S, et al. Alterations in promoter methylation status of tumor suppressor HIC1, SFRP2, and DAPK1 genes in prostate carcinomas. DNA Cell Biol 2012;31:826–32. [DOI] [PubMed] [Google Scholar]
- [37].Verdoodt B, Sommerer F, Palisaar RJ, et al. Inverse association of p16 INK4a and p14 ARF methylation of the CDKN2a locus in different Gleason scores of prostate cancer. Prostate Cancer Prostatic Dis 2011;14:295–301. [DOI] [PubMed] [Google Scholar]
- [38].Delgado-Cruzata L, Hruby GW, Gonzalez K, et al. DNA methylation changes correlate with Gleason score and tumor stage in prostate cancer. DNA Cell Biol 2012;31:187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vasiljevic N, Wu K, Brentnall AR, et al. Absolute quantitation of DNA methylation of 28 candidate genes in prostate cancer using pyrosequencing. Dis Markers 2011;30:151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schwarzenbach H, Chun FK, Isbarn H, et al. Genomic profiling of cell-free DNA in blood and bone marrow of prostate cancer patients. J Cancer Res Clin Oncol 2011;137:811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Higuchi T, Nakamura M, Shimada K, et al. HRK inactivation associated with promoter methylation and LOH in prostate cancer. Prostate 2008;68:105–13. [DOI] [PubMed] [Google Scholar]
- [42].Roupret M, Hupertan V, Yates DR, et al. Molecular detection of localized prostate cancer using quantitative methylation-specific PCR on urinary cells obtained following prostate massage. Clin Cancer Res 2007;13:1720–5. [DOI] [PubMed] [Google Scholar]
- [43].Cho NY, Kim BH, Choi M, et al. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol 2007;211:269–77. [DOI] [PubMed] [Google Scholar]
- [44].Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 2004;64:1975–86. [DOI] [PubMed] [Google Scholar]
- [45].Konishi N, Nakamura M, Kishi M, et al. DNA hypermethylation status of multiple genes in prostate adenocarcinomas. Jpn J Cancer Res 2002;93:767–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Maruyama R, Toyooka S, Toyooka KO, et al. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res 2002;8:514–9. [PubMed] [Google Scholar]
- [47].Nguyen TT, Nguyen CT, Gonzales FA, et al. Analysis of cyclin-dependent kinase inhibitor expression and methylation patterns in human prostate cancers. Prostate 2000;43:233–42. [DOI] [PubMed] [Google Scholar]
- [48].Jarrard DF, Bova GS, Ewing CM, et al. Deletional, mutational, and methylation analyses of CDKN2 (p16/MTS1) in primary and metastatic prostate cancer. Genes Chromosomes Cancer 1997;19:90–6. [PubMed] [Google Scholar]
- [49].Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45:W98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ye M, Huang T, Li J, et al. Role of CDH13 promoter methylation in the carcinogenesis, progression, and prognosis of colorectal cancer: a systematic meta-analysis under PRISMA guidelines. Medicine (Baltimore) 2017;96:e5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Maziveyi M, Alahari SK. Breast cancer tumor suppressors: a special emphasis on novel protein nischarin. Cancer Res 2015;75:4252–9. [DOI] [PubMed] [Google Scholar]
- [52].Jin M, Piao Z, Kim NG, et al. p16 is a major inactivation target in hepatocellular carcinoma. Cancer 2000;89:60–8. [DOI] [PubMed] [Google Scholar]
- [53].Pan J, Chen J, Zhang B, et al. Association between RASSF1A promoter methylation and prostate cancer: a systematic review and meta-analysis. PLoS One 2013;8:e75283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Van Neste L, Herman JG, Otto G, et al. The epigenetic promise for prostate cancer diagnosis. Prostate 2012;72:1248–61. [DOI] [PubMed] [Google Scholar]
- [55].Zeng Y, Kakehi Y, Nouh MA, et al. Gene expression profiles of lysophosphatidic acid-related molecules in the prostate: relevance to prostate cancer and benign hyperplasia. Prostate 2009;69:283–92. [DOI] [PubMed] [Google Scholar]
- [56].Thomas LN, Douglas RC, Lazier CB, et al. Type 1 and type 2 5alpha-reductase expression in the development and progression of prostate cancer. Eur Urol 2008;53:244–52. [DOI] [PubMed] [Google Scholar]
- [57].Henrique R, Costa VL, Cerveira N, et al. Hypermethylation of Cyclin D2 is associated with loss of mRNA expression and tumor development in prostate cancer. J Mol Med (Berl) 2006;84:911–8. [DOI] [PubMed] [Google Scholar]
- [58].Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:l1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.