

Abstract
By means of a novel methodology that can statistically derive patterns of co‐alterations distribution from voxel‐based morphological data, this study analyzes the patterns of brain alterations of three important psychiatric spectra—that is, schizophrenia spectrum disorder (SCZD), autistic spectrum disorder (ASD), and obsessive‐compulsive spectrum disorder (OCSD). Our analysis provides five important results. First, in SCZD, ASD, and OCSD brain alterations do not distribute randomly but, rather, follow network‐like patterns of co‐alteration. Second, the clusters of co‐altered areas form a net of alterations that can be defined as morphometric co‐alteration network or co‐atrophy network (in the case of gray matter decreases). Third, within this network certain cerebral areas can be identified as pathoconnectivity hubs, the alteration of which is supposed to enhance the development of neuronal abnormalities. Fourth, within the morphometric co‐atrophy network of SCZD, ASD, and OCSD, a subnetwork composed of eleven highly connected nodes can be distinguished. This subnetwork encompasses the anterior insulae, inferior frontal areas, left superior temporal areas, left parahippocampal regions, left thalamus and right precentral gyri. Fifth, the co‐altered areas also exhibit a normal structural covariance pattern which overlaps, for some of these areas (like the insulae), the co‐alteration pattern. These findings reveal that, similarly to neurodegenerative diseases, psychiatric disorders are characterized by anatomical alterations that distribute according to connectivity constraints so as to form identifiable morphometric co‐atrophy patterns.
Keywords: alteration propagation, anatomical covariance, autism spectrum disorder, co‐alteration, co‐athropy, damage spread, obsessive‐compulsive spectrum disorder, pathoconnectivity, schizophrenia spectrum disorder, transdiagnostic approach
1. INTRODUCTION
The anatomical co‐alteration networking analysis is an intriguing and promising new field of connectomics, which can be defined as the study of the network patterns according to which structural alterations distribute across the brain (Yates, 2012). In other words, the networking analysis of brain disorders aims to give a description of networks formed by co‐altered (or co‐atrophic, in the case of grey matter (GM) decreases) cerebral regions. Recent studies in this field show that brain alterations are rarely confined to a single cerebral area, but rather tend to distribute to many different sites. Often brain areas in which alterations co‐occur also exhibit patterns of anatomical covariance (Evans, 2013). Furthermore, converging evidence suggests that pathological alterations caused by brain disorders are likely to follow patterns of distribution that strongly have a network‐like architecture, which depends on the organization of both the structural and functional connectivity (Cauda, et al., 2017; Cauda, et al., 2012b; Crossley, et al., 2016; Crossley, et al., 2014; Fornito, Zalesky, & Breakspear, 2015; Menon, 2013; Raj, Kuceyeski, & Weiner, 2012; Saxena & Caroni, 2011; Seeley, Crawford, Zhou, Miller, & Greicius, 2009; Yates, 2012; Zhou, Gennatas, Kramer, Miller, & Seeley, 2012).
This new field of research is destined to provide invaluable insight in the understanding and diagnosis of brain disorders. Thus far the diagnosis of neuropsychiatric conditions relies entirely on the observation of constellations of behavioral signs and symptoms. However, these diagnostic procedures do not seem to have clear relations to the underlying biological processes that should be the targets of medical treatments (Poldrack & Farah, 2015). What is more, this approach can result in heterogeneity within diagnostic categories and in poor inter‐rater reliability for many clinical evaluations, which in turn produces a negative impact on clinical outcomes (Freedman, et al., 2013).
Even the fifth recent version of the DSM still lacks a rigorous neurobiological basis. This is why the US National Institute of Mental Health Research Domain Criteria (RDoC, http://www.nimh.nih.gov/research-priorities/rdoc/index.shtml; Insel, 2010, 2014) has suggested to construct a map of relationships between symptoms’ manifestations and wide biological realms, so as to improve both the homogeneity and reliability of brain disorders’ classification. The aim of RDoC is to go beyond the old imprecise pathological categories and give the clinicians more reliable diagnostic tools. Recent studies that are consistent with this line of thought have already tried to identify important biomarkers capable of defining classes of patients independently of their symptomatic manifestations (Clementz, et al., 2016; Marquand, Rezek, Buitelaar, & Beckmann, 2016).
The case of schizophrenia (SCZD), autistic (ASD) and obsessive‐compulsive (OCSD) spectrum disorders is paradigmatic of the current neuroscientific climate as well as of the difficulties in clinical diagnosis (Luciano, Keller, Politi, Aguglia, & Magnano, 2014). For instance, some authors tend to recognize a clear clinical connection between SCZD and ASD (Bolte, Rudolf, & Poustka, 2002; Hommer & Swedo, 2015; Nylander, Lugnegård, & Unenge Hallerbäck, 2008; Ornitz, 1969; Rapoport, Chavez, Greenstein, Addington, & Gogtay, 2009; Sporn, et al., 2004; Starling & Dossetor, 2009; Stone & Iguchi, 2011). This idea may be supported by recent epidemiologic, genetic, molecular, and brain imaging evidence suggesting an underlying shared neurobiological substrate for ASD and SCZD (Arnone, et al., 2009; Biamino, et al., 2016; Cheung, et al., 2010; de Lacy & King, 2013; King & Lord, 2011; Stone & Iguchi, 2011). In turn, it has been hypothesized that a common neurobiological mechanism might be at the basis of the repetitive behavior in both ASD and OCSD (Langen, Durston, Kas, van Engeland, & Staal, 2011a; Langen, Kas, Staal, van Engeland, & Durston, 2011b). Also it has been proposed that OCSD and SCZD might share similar pathogenetic underpinnings (Owashi, Ota, Otsubo, Susa, & Kamijima, 2010). Of note, other brain structural (Goodkind, et al., 2015) and genetic studies (The Cross‐Disorder Group of the Psychiatric Genomics Consortium, 2013; The Network & Pathway Analysis Subgroup of the Psychiatric Genomics Consortium, 2015) provide further evidence for transdiagnostic overlaps between psychiatric conditions. These findings suggest significant comorbidities between syndromes as well as relevant overlap of their symptomatology (Buckley, Miller, Lehrer, & Castle, 2009; DeVylder, Burnette, & Yang, 2014; Gorun, et al., 2015; Kessler, et al., 1994; Markon, 2010; Vaidyanathan, Patrick, & Iacono, 2012).
Therefore, the need to understand brain disorders in terms of neurobiological features is ever more compelling (Wang & Krystal, 2014). In particular, with regard to mental illness it has been proposed that metabolic and microstructural modifications in certain sets of brain regions might be associated with many different conditions (Buckholtz & Meyer‐Lindenberg, 2012; Crossley, et al., 2016; Crossley, Scott, Ellison‐Wright, & Mechelli, 2015; Goodkind, et al., 2015; McTeague, Goodkind, & Etkin, 2016). These abnormalities develop as morphological alterations of gray matter (GM) or white matter (WM) density, which appear as increased or decreased values in voxel‐based morphometry (VBM) investigations or as dysfunctional patterns in brain activity (Baker, et al., 2014; Ellison‐Wright & Bullmore, 2010; Etkin & Wager, 2007; Goodkind, et al., 2015; Hamilton, et al., 2012). These studies challenge the intuitive view that each and every brain disorder should exhibit a specific pattern of brain alterations as well as a specific constellation of clinical symptoms (Buckholtz & Meyer‐Lindenberg, 2012; Caspi, et al., 2014; Goodkind, et al., 2015; McTeague, et al., 2016).
In line with these data a recent study by our research group demonstrated that SCZD, OCSD and ASD do not show distinctive patterns of GM alterations; rather, these three spectra showed a common pattern, which can be divided into two clusters of alterations extending across the insulae, medial thalamic and the cingulate cortices (Cauda, et al., 2017). This finding was also recently supported by a study on functional alterations (Sprooten, et al., 2017), which has showed that very few functional differences can be statistically observed in a variety of psychiatric conditions (i.e., schizophrenia, bipolar disorder, major depressive disorder, anxiety disorders, and obsessive compulsive disorder). Of note, this specific pattern of alterations common to SCZD, OCSD, and ASD is probably not only shared by these three spectra, but also by other psychiatric disorders (Buckholtz & Meyer‐Lindenberg, 2012; Caspi, et al., 2014; Crossley, et al., 2014; McTeague, et al., 2016). In fact, it encompasses a set of “core areas” that exhibit higher functional diversity (Andersson, Kinnison, & Pessoa, 2013; or entropy) and have been found to be active in a number of important cognitive functions during fMRI studies (Cauda, et al., 2012b). These core areas are thought to be part of the salience network (Palaniyappan & Liddle, 2012), which is in turn part of the cognitive control network (Cole & Schneider, 2007; Niendam, et al., 2012), and are characterized by abundant populations of Von Economo neurons (VEN; Cauda, Geminiani, & Vercelli, 2014b; Cauda, et al., 2013). VEN are large, spindle‐shaped projection neurons present in layer V of the frontoinsular and cingulate cortex, supposed to be involved in the pathogenesis of specific neurological and psychiatric diseases (Cauda, et al., 2014b).
In light of these findings, we devised an innovative meta‐analytic method for performing an anatomical co‐alteration networking analysis of brain disorders. In fact, although this meta‐analysis focuses on SCZD, ASD, and OCSD, our methodology can be generally applied for studying every disease capable of producing appreciable neuropathological alterations.
In particular, the present study aimed to investigate and address the following issues:
Do neuronal alterations distribute coherently across the brain areas structurally affected by SCZD, ASD, and OCSD in a network like manner?
In case of a positive answer, can the anatomical co‐alteration network of SCZD, ASD, and OCSD be identified?
Within this co‐alteration network are there brain areas the alteration of which could lead to a faster and more diffuse distribution of neuronal abnormalities?
Can distinct clusters and/or subclusters of co‐altered brain areas be identified within the co‐alteration patterns of neuroanatomical alterations produced by SCZD, ASD, and OCSD?
Finally, can co‐altered brain areas exhibit a normal pattern of anatomical covariance? If so, how similar is this pattern to the co‐alteration pattern of the same areas?
2. MATERIALS AND METHODS
2.1. Selection of studies
We identified the pool of all eligible experiments in the BrainMap database (http://www.brainmap.org; Fox & Lancaster, 2002; Laird, et al., 2009; Laird, et al., 2005) which reported GM/WM changes within the brain parenchyma. At the time of the selection phase, the BrainMap database was made up by coordinates and associated meta‐data of 3,076 publications and 15,243 neuroimaging experiments. For the present meta‐analysis, only the brain studies reporting GM and WM changes in standard stereotaxic space were retrieved. As the first step, we identified (separately for each spectrum) all experiments that featured at least one focus of GM or WM change. WM data were not analyzed in this study but, for the sake of completeness, we reported them in the tables.
Criteria for including studies in an anatomical likelihood estimation (ALE) meta‐analysis may be influenced by knowledge of the results of the set of potential studies, leading to inclusion bias. To avoid bias in location and selection of studies, the following additional measures were taken: (a) assessment of the bibliographies of each study in BrainMap so as to identify additional studies that might have not been included in the database and (b) search on PubMed of the literature whose temporal boundaries are not included in the BrainMap database (https://www.ncbi.nlm.nih.gov/pubmed). With regard to this last step, a systematic search strategy was used to identify relevant studies, published until 15 July 2016, involving SCZD, ASD, and OCSD. The search algorithm have been constructed so as to match for: “autism spectrum disorder” (ASD); “obsessive‐compulsive disorder” (OCD); “schizophrenia”; “schizoaffective disorder”; diffusion tensor imaging (DTI); and VBM, respectively (for more details on literature search and algorithms, see the online Supporting Information).
Up until 15 July 2016, 1419 papers had been indexed on PubMed with the selected search terms. In particular, all the articles were reviewed in order to ensure: (1) both the presence of the healthy control group and the pathological sample; (2) that the results were reported by using the Talairach/Tournoux or Montreal Neurological Institute (MNI) coordinates; (3) that the foci of interest had a significance of at least <.05; (4) that the studies described cerebral structural changes visible with VBM or DTI (only FA technique); (5) that the studies were original works; and (6) that original diagnosis was made on the basis of DSM criteria and clinical test batteries.
We adopted the definition of meta‐analysis accepted by the Cochrane Collaboration (Green, et al., 2008) and the “PRISMA Statement” international guidelines in order to ensure a transparent and complete report of data selection (Liberati, et al., 2009; Moher, Liberati, Tetzlaff, & Altman, 2009).
Studies from BrainMap database and those from PubMed were compared looking for cases of multiple references to the same datasets across articles so as to make sure that only one reference to the same data contributed to the coordinates for the present meta‐analysis (see Table 1 and Supporting Information, Graph S1). Then, the studies were examined to detect dissimilarities or discrepancies. The researchers who carried out this research stage have reached substantial agreement as regards inclusion and exclusion of studies (Cohen's K = .7409).
Table 1.
Synopsis of the selection procedure with number of articles identified at each stage
| BrainMap | PubMed | Screening | Eligibility | ASD | OCSD | SCZD |
|---|---|---|---|---|---|---|
| Articles | Articles | Abstract exclusions | Full‐text exclusions | Selected studies | Selected studies | Selected studies |
| 242 | 1419 | 44 | 49 | 110 | ||
| ⇓ | ⇓ | Sample (N) | Sample (N) | Sample (N) | ||
| Additional records | Additional records | 1719 | 1738 | 5236 | ||
| 0 | 15 | |||||
| Eligibility for full‐text lecture | Selected studies 203 | |||||
| Phase 1 | Phase 2 | Phase 3 | Phase 4 | Phase 5 | ||
| ⇓ | ||||||
| 242 records | 1434 records | Data extraction | ||||
Meta‐data were extracted from each selected article. For all articles that possessed the information, we evaluated the diagnostic clusters of each psychiatric spectrum, classifying them on the basis of what was indicated in the section “Subjects and Methods” by the authors of the selected articles. In case the authors did not specified the clinical type described in their study, the “mixed” label was used (Cauda, et al., 2017). The description of the sample composition and the distribution of the three psychiatric spectra are viewable on the online Supplementary Materials. In order to facilitate analysis, coordinates from MNI space were converted into Talairach coordinates by using Lancaster transformation (Lancaster, et al., 2007).
Given that many experiments do not report GM increased values, we decided to focus our meta‐analysis prevalently on GM decreased values, performing analysis on 1171 foci of interest (see Table 2). To have also information about GM increases, their number of foci (which was not sufficiently large to be statistically analyzed) and the number of foci associated with GM decreases were summed and analyzed. We then compared the results of this analysis to those of the analysis performed only on GM decreases data. Since most of our networking analyses principally concern GM decreases data, henceforth we will refer to the co‐alteration network as co‐atrophy network. In fact GM decreased VBM values indicate a volume reduction of neurons, which can be interpreted as brain atrophy (Table 1).
Table 2.
White matter and gray matter variations with relative numbers of foci for each of the selected psychiatric spectra. The items shown in the table are the result of the entire selection process as shown in PRISMA (2009) flow chart on the Supporting Information
| SPECTRUM | Diagnostic Label |
Foci (N) |
White matter changes | Gray matter Changes | ||
|---|---|---|---|---|---|---|
| ⇑ | ⇓ | ⇑ | ⇓ | |||
| ASD (N = 1,719) | Mixed form | 312 | 27 | 66 | 125 | 94 |
| Primary autism | 186 | 7 | 57 | 93 | 29 | |
| Asperger | 54 | 11 | 14 | 2 | 27 | |
| High‐functionality autism | 53 | 5 | 20 | 16 | 12 | |
| Pervasive developmental disorder | 16 | 0 | 11 | 0 | 5 | |
| OCSD (N = 1,738) | Obsessive‐compulsive disorder | 364 | 25 | 77 | 67 | 195 |
| Tourette syndrome | 70 | 13 | 33 | 5 | 19 | |
| Trichotillomania | 4 | 0 | 4 | 0 | 0 | |
| SCZD (N = 5,236) | Schizophrenia simplex | 190 | 0 | 4 | 4 | 182 |
| Paranoia | 20 | 0 | 17 | 1 | 2 | |
| Auditory hallucination | 34 | 7 | 0 | 0 | 27 | |
| First episode psychosis | 92 | 3 | 45 | 4 | 40 | |
| Mixed form | 633 | 18 | 160 | 22 | 433 | |
| First episode schizophrenia | 65 | 5 | 19 | 0 | 41 | |
| Acute psychosis with no hallucination | 21 | 0 | 21 | 0 | 0 | |
| Early onset symptoms of psychosis | 52 | 0 | 8 | 0 | 44 | |
| Hallucination | 21 | 0 | 16 | 0 | 5 | |
| Paranoia with schizophrenia symptoms | 26 | 0 | 9 | 1 | 16 | |
| Foci (Total) | 121 | 581 | 340 | 1,171 | ||
| 702 | 1,511 | |||||
2.2. Anatomical likelihood estimation and modeled activation creation map
We performed an ALE (Eickhoff, Bzdok, Laird, Kurth, & Fox, 2012; Eickhoff, et al., 2009; Turkeltaub, et al., 2012) to statistically show the commonalities between the selected experiments on patients with SCZD, ASD, and OCSD. The ALE is a quantitative voxel‐based meta‐analysis technique, which provides information about the anatomical reliability of results by comparing them with a sample of reference studies obtained from the existing literature. Each focus of every study is considered as the central point of a three‐dimensional Gaussian probability distribution:
where d is the Euclidean distance between the voxels and the considered focus, and e is the spatial uncertainty. The standard deviation is easily obtained through the Full‐Width Half‐Maximum (FWHM) as:
For each experiment, we determined a modeled activation (MA) map, resulting from the union of the Gaussian probability distribution of every focus of each experiment. Then we determined the ALE map, derived from the union of the MA maps.
Statistical significance of the activation within the ALE map was calculated by cluster‐level inference, as suggested by Eickhoff et al. (2012, 2017, 2016). Given a particular cluster forming threshold, a null distribution of cluster sizes was obtained by simulating a long series of experiments using the same characteristic of the real data and then by calculating an ALE map. The obtained score histogram was eventually used to assign a threshold p values.
2.3. Construction of the morphometric co‐atrophy network
In order to identify the distribution of brain alterations we developed an innovative methodology capable of constructing the anatomical co‐alteration networks of brain disorders. Our analysis can find out whether or not the structural alteration of brain area A is statistically concatenated with the alteration of one or more other brain areas (B, C, etc.). The analysis therefore results in the construction of a morphometric co‐atrophy network (MCN) formed by the brain areas that occur to be altered together, in which it is possible to examine (i) how an altered brain area is statistically connected to other altered areas and (ii) which areas appear to be parts of a more extended web of alterations.
2.4. Node creation
We created a set of nodes, localized in the points of the ALE map, derived from the union of the MA maps as described in the previous section, where the probability of alteration was higher (peaks of ALE values). In particular, we used a peak detection algorithm to determine the set of local maxima of the ALE values. We selected the voxels showing an ALE peak value greater than a given threshold. To avoid an excessive number of regions of interest (ROIs) a threshold was set at the 75 percentile of the peak values distribution. We then calculated the distance between peaks, thus obtaining a distance matrix for each peak. This was done to avoid ROIs superimposition. Indeed we excluded all peaks within a distance of 10 mm from other peaks. We obtained then a definitive set of peaks. Finally, around every peak we designed a ROI of 10 mm2.
The rationales behind these methodological choices are the following. (i) The dimension of the nodes is based on the work of Eickhoff, et al. (2009) that empirically analyzed meta‐analytical imaging data providing quantitative estimates to explicitly model the spatial uncertainty associated with the reported coordinate. Since this analysis evidenced an uncertainty in spatial location with a mean of 10.2 mm with an SD of .4 mm, we chose a radius of 10 mm for our nodes. (ii) We chose the 75 percentile because if the signal is noisy there is the possibility to detect false peaks considering the Chebyshev's inequality (Kotz, Balakrishnan, & Johnson, 2000), which shows that, independently of the type or form of the probability distribution, the proportion of the observation falling within k standard deviations of the population mean is at least , which, with , correspond to the 75 percentile. (iii) Although several Authors pointed out that the lack of a gold standard makes the definition of nodes arbitrary, as shown by Zalesky and colleagues (2010) it is possible to make any comparison between networks, if the node parcellation was made at the same spatial scale. This is why we paid particular attention to perform all the analyses at a comparable spatial scale. For a schema depicting the node detection pipeline see Figure 1.
Figure 1.
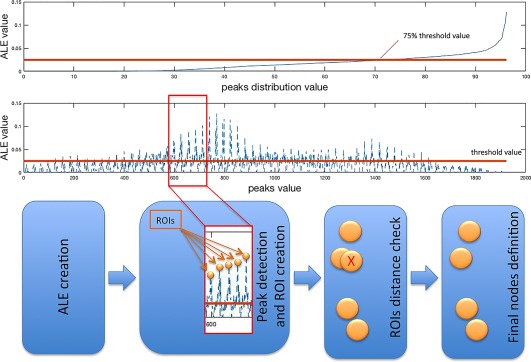
Schema depicting the pipeline utilized for the ROI (nodes) detection [Color figure can be viewed at http://wileyonlinelibrary.com]
2.5. Co‐atrophy distribution and connectivity
To study the co‐atrophy pattern, we created a co‐alteration matrix using the previously defined set of nodes. In a N × M matrix each row represents an experiment, while each column represents a network node; in our particular case the matrix is 127 (experiments) × 33 (nodes). For each experiment we reported a node (ROI) as being altered if the experiment MA map reported 20% or more of the voxels within the ROI.
From this matrix we obtained the strength of the co‐alteration between the nodes by using the Patel's k index (Patel, Bowman, & Rilling, 2006). In fact, starting from a Bernoulli generation model of data it is possible to construct a probability distribution of joint alteration values for each pair of nodes. Given two nodes (a and b), we can describe their conjoint state of alteration through two binary variables representing four cases: (i) a and b both altered; (ii) a and b both unaltered; (iii) a altered and b unaltered; and (iv) a unaltered and b altered (Table 3). Therefore, frequencies of the different combinations through all experiments give the following four probabilities:
which represent the conjoint state frequencies of two nodes (a and b) in all their four possible combinations. Marginal probabilities are illustrated by the following table:
Table 3.
Marginal probabilities between altered and unaltered nodes
| Voxel a | |||||||
|---|---|---|---|---|---|---|---|
| Altered | Unaltered | ||||||
| Voxel b | Altered |
|
|
|
|||
| Unaltered |
|
|
|
||||
|
|
|
1 | |||||
Considering these four probabilities, we can apply the two indices proposed by Patel et al. (2006) for the calculus of connectivity and directionality, called k and τ, respectively. These indices have been validated by Smith et al. (2011) with simulated data. With regard to Patel's τ, however, a recent study has criticized its efficacy (Wang, David, Hu, & Deshpande, 2017). For the sake of caution, we decided therefore to limit our analysis to the connectivity aspect by solely using Patel's k index, even though it is worth noting that the criticism by Wang et al. is typically directed to problems (i.e., deconvolution of the hemodynamic response, temporal resolution) related to the application of empirical Bayesian techniques to fMRI data rather than to anatomical morphometric data, which are to be considered in the present study.
With regard to Patel's k, this index measures the probability that two nodes (say, a and b) appear to be co‐altered compared to the probability that a and b are independently altered. This index is defined as follows:
where
In the fraction the numerator represents the difference between the probability that a and b are co‐altered and the expected probability that a and b are independently altered, whereas the denominator represents a weighted normalizing constant. indicates the maximum value of conjoint probability , given and , while indicates the minimum value of , given and . Patel's k index ranges from −1 and 1. A value of |k| close to 1 is evidence of high connectivity. The statistical significance of k is evaluated by simulating, through a Monte Carlo algorithm, a multinomial, generative model of data, which can take into account alterations of all nodes. In particular, the Monte Carlo statistical procedure consists in obtaining an estimate of by sampling a Dirichlet distribution and determining the samples’ proportion in which , where is the threshold of statistical significance.
2.6. Topological analysis
Some network‐based analysis techniques were employed to analyze the neural web of co‐atrophy area. The network submitted to these analysis was a connectivity matrix between the previously selected set of nodes. In this matrix the edges between the nodes are constituted by the values of the thresholded Patel's k described in the previous section. The analysis of complex networks is a powerful technique for quantifying both brain structure and functional architecture. A network is defined as a system of nodes connected by a series of links. In our case the link is the strength of the co‐alteration between nodes.
2.7. Node degree
The node degree is the number of connections linking a node with other nodes. We used the degree distribution to compare the node degree of different networks. By using the degree distribution we compared a randomly generated network with those exhibited by the three psychiatric spectra. The degree distribution is the fraction of nodes with degree k, which is defined as:
2.8. Edge betweenness
The edge betweenness consists in the number of the shortest paths going through an edge within a graph or a network (Girvan & Newman, 2002). Edges with high values of betweenness are involved in a large number of shortest paths, so that their removal may affect communication between many pairs of nodes.
2.9. Network clustering
We employed the k‐core decomposition algorithm (Alvarez‐Hamelin, Dall'asta, Barrat, & Vespignani, 2005; Bader & Hogue, 2003) to disentangle the hierarchical structures of our concordance network by progressively focusing on their central cores. A k‐core of a graph G is the highest connected subgraph of G, in which all the vertices (at least) present a degree of k. In fact, the k‐core decomposition of a network recursively removes all the vertices having a degree less than k, until in the graph all vertices with degree k or more remain. This procedure allowed us to consider our concordance network graph as the central most densely cluster or connected subgraph.
2.10. Anatomical dataset (normative population)
2.10.1. Subjects and image acquisition
For the anatomical covariance we employed the Beijing dataset which has been publicly released within the “1000 Functional Connectomes” Project. This dataset consists of 198 subjects (76 males and 122 female) with age ranging from 18 to 26 years, mean 21.16, SD 1.83, that underwent structural and resting‐state scans. All subjects were right‐handed and had no history of neurological or psychiatric disorders. Written informed consent was obtained from each participant, and the study was approved by the Institutional Review Board of Beijing Normal University Imaging Center for Brain Research.
MRI data were acquired using a SIEMENS TRIO 3‐Tesla scanner in the Beijing Normal University Imaging Center for Brain Research. Participants lay supine with the head fixed by straps and foam pads so as to minimize movements. During the resting‐state session, participants were instructed to be as still as possible and let their mind roam. Functional images were obtained using an EPI sequence with the following parameters: 33 axial slices, thickness/gap = 3/.6 mm, in‐plane resolution = 64 × 64, TR = 2,000 ms, TE = 30 ms, flip angle = 90°, FOV = 200 × 200 mm. Furthermore, a T1‐weighted sagittal three‐dimensional magnetization‐prepared rapid gradient echo (MPRAGE) sequence was acquired, which covered the entire brain: 128 slices, TR = 2,530 ms, TE = 3.39 ms, slice thickness = 1.33 mm, flip angle = 7°, inversion time = 1,100 ms, FOV = 256 × 256 mm, and in‐plane resolution = 256 × 192.
2.10.2. Structural covariance analysis on normative population
The structural covariance analysis was conducted on the anatomical dataset of normative population previously described with an optimized protocol using the FSL Tools (Douaud, et al., 2007; Good, et al., 2001; Smith, et al., 2004). First, structural images were brain‐extracted and GM‐segmented before being registered to the MNI 152 standard space using non‐linear registration (Andersson, Jenkinson, & Smith, 2007). The obtained images were averaged and flipped along the x‐axis to create a left‐right symmetric, study‐specific GM template. Second, all native GM images were non‐linearly registered to this study‐specific template and “modulated” to correct for local expansion (or contraction) due to the non‐linear component of the spatial transformation (Good, et al., 2001). The modulated GM images were then smoothed with an isotropic Gaussian kernel with a sigma of 3 mm (Andersson, et al., 2007; Douaud, et al., 2007; Good, et al., 2001; Smith, et al., 2004). The GM images were also merged to obtain a 4D image: the usual x, y, and z coordinates of the brain standard space (MNI coordinates) and along the 4th dimension of the various subjects. For the node coordinates obtained from the ALE analysis we extracted a set of time series to form a matrix of dimension subjects × nodes. To do so, for each ROI we averaged the time courses of all voxel belonging to this ROI. From this matrix we calculated the correlation between the columns, thus obtaining a correlation matrix of dimension nodes × nodes. The correlation matrix was compared with the co‐alteration matrix using a BRAMILA tool to perform a Mantel test (Glerean, et al., 2016; Mantel, 1967). This procedure is a type of randomization in which the columns of the matrices are permutated and the correlation between the distances is consequently calculated (5,000 times).
2.11. Reliability
To better understand the contribution of each spectrum to the MCN we tried to determine the co‐alteration patterns associated with each spectrum. However, the number of experiments of two spectra (ASD and OCSD) was not sufficient for a valid statistical analysis; indeed the statistics on ASD or OCSD data only did not produce any significant results. We therefore decided to conduct four different analyses. (1) One spectrum was left out in alternation to study how its removal could change the MCN. (2) We statistically compared (correlation) the Patel's k values of each edge between the three spectra. (3) We created a Euclidean distance matrix between the MA maps obtained from all the three spectra and performed a k‐mean clustering of the same maps to visualize the different contributions of experiments to the MCN (see Supporting Information for methodological details). (4) To further investigate the possibility that the greater amount of SCZD data could somehow drive our results, we constructed the MCN with an equal number of experiments for each spectrum. We did not adopt this procedure in the main analysis because we wanted to use as much data as possible for statistical needs. Inevitably, a single sampling, with the inclusion of some SCZD data and the exclusion of others, would have produced biased and less representative results. To avoid this issue we used a bootstrap technique by randomly selecting a number of SCZD experiments (from 25 to 30 studies; this interval corresponds to the sample range of ASD and OCSD) that was equal to the average of the number of experiments about ASD and OCSD. We then constructed the co‐alteration matrix for this whole new dataset (for more details see Section 2). This type of analysis was repeated with 1,000 permutations (each time by selecting a different sample from SCZD data), so as to assess the stability of the results (SE). We subsequently evaluated with the Dice coefficient (DC) the overlapping value between the averaged MCN obtained with this new analysis and the MCN obtained with the whole original dataset. By comparing two samples (in our case the co‐alteration matrices), the DC can measure their similarity, as follows:
where and are the number of elements of the two samples, respectively.
3. RESULTS
3.1. Common patterns of anatomical alterations
We performed an ALE that pooled the outcomes of all the experiments taken into consideration in this meta‐analysis. This statistic analysis revealed significant GM density decreased values mainly in the dorsal lateral prefrontal cortex, insulae, medial thalami, ventromedial prefrontal, orbitofrontal, precentral, and cingulate areas. In contrast, GM density increased values were found mainly in frontal poles, posterior lateral thalami, caudate nuclei, putamen, posterior parietal, cerebellar and inferior temporal cortices (Figure 2).
Figure 2.
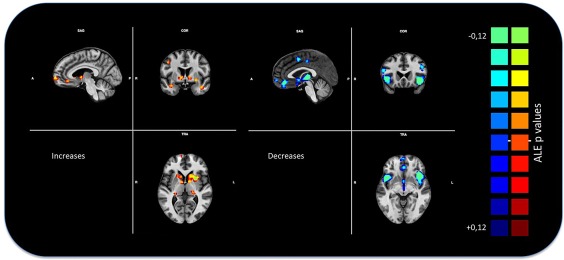
Gray matter anatomical likelihood estimation (ALE) results. The image summarizes the results of all the experiments considered in this meta‐analysis. Colors from red to yellow show gray matter increases, colors from blue to green show gray matter decreases (ALE maps were computed at a threshold of p < .001, cluster‐level corrected for multiple comparison (Eickhoff et al., 2016) and visualized using BrainVoyager QX. Only decreases have been utilized for the subsequent analysis [Color figure can be viewed at http://wileyonlinelibrary.com]
3.2. Co‐atrophy network
Our meta‐analysis prevalently focuses on GM decreased values, as many experiments do not report GM increased values.
The left panel of Figure 3 and Table 4 show the regions of interest forming the MCN, while the right panel of Figure 3 illustrates the distance matrix showing the k values between nodes. In turn, Figure 4 illustrates the MCN: edges colored from blue to red indicate increasing k values.
Figure 3.
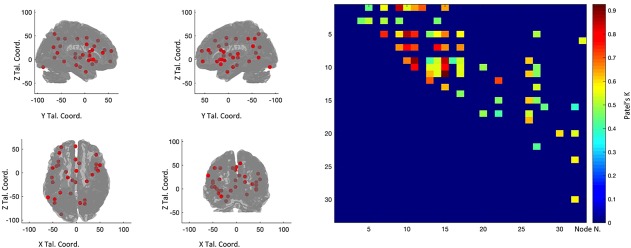
The left panels shows the nodes that entered the co‐atrophy calculation. The right panel shows the co‐atrophy matrix. Colors from blue to red indicates increasing Patel's k values (i.e., increasing co‐alteration probabilities [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 4.
Nodes employed for the co‐atrophy analysis
| Tal Coord | |||
|---|---|---|---|
| Node Name | X | Y | Z |
| Cingulum_Ant_R | 6 | 20 | 28 |
| Cingulum_Mid_L | −8 | −16 | 44 |
| Frontal_Inf_Oper_L | −50 | 10 | 16 |
| Frontal_Inf_Oper_R | 42 | 10 | 8 |
| Frontal_Inf_Orb_L | −38 | 24 | −4 |
| Frontal_Inf_Tri_R | 50 | 16 | 20 |
| Frontal_Mid_L | −36 | 42 | 20 |
| Frontal_Mid_Orb_L | −32 | 54 | −4 |
| Frontal_Mid_R | 42 | 38 | 14 |
| Frontal_Sup_Medial_L | −2 | 56 | 18 |
| Frontal_Sup_Medial_L_1 | −2 | 28 | 40 |
| Hippocampus_L | −18 | −10 | −16 |
| Insula_L | −38 | 16 | −10 |
| Insula_L_1 | −42 | 14 | 0 |
| Insula_R | 42 | 8 | −2 |
| Insula_R_1 | 42 | 18 | 0 |
| Insula_R_2 | 34 | −4 | 10 |
| Lingual_L | −32 | −88 | −16 |
| Lingual_L_1 | −12 | −44 | −4 |
| ParaHippocampal_L | −22 | 2 | −26 |
| ParaHippocampal_R | 22 | −18 | −12 |
| Parietal_Inf_L | −46 | −56 | 44 |
| Parietal_Inf_L_1 | −34 | −42 | 44 |
| Precentral_R | 46 | 4 | 32 |
| Precuneus_R | 18 | −58 | 14 |
| Precuneus_R_1 | 18 | −66 | 26 |
| Precuneus_R_2 | 8 | −64 | 54 |
| Supp_Motor_Area_L | 0 | 4 | 46 |
| SupraMarginal_L | −60 | −52 | 28 |
| Temporal_Mid_L | −46 | −64 | 4 |
| Temporal_Sup_L | −52 | −10 | 4 |
| Temporal_Sup_L_1 | −50 | −20 | 12 |
| Thalamus_L | −2 | −20 | 4 |
Figure 4.
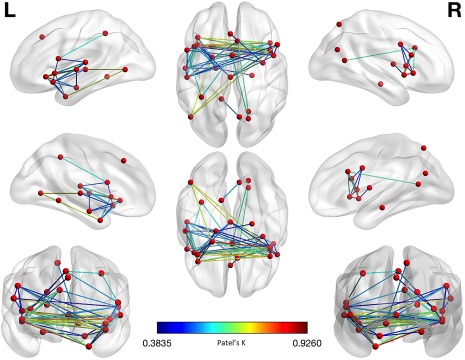
Morphometric co‐atrophy network results. Colors from blue to red indicates increasing Patel's k values (i.e., increasing co‐alteration probabilities) [Color figure can be viewed at http://wileyonlinelibrary.com]
The node creation reported 33 nodes placed in prefrontal, insular, cingulate, hippocampal, lateral and medial parietal, temporal and thalamic brain areas. Eight nodes out of 33 were found to be unconnected (i.e., they do not exhibit any significant edge of co‐atrophy).
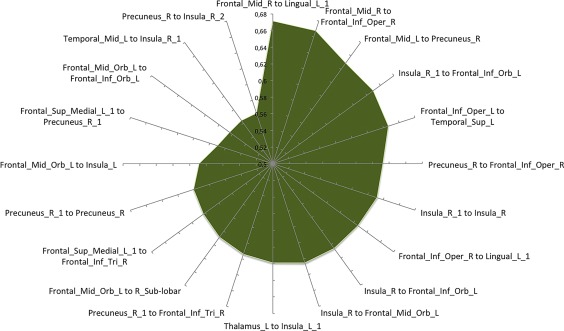
The co‐atrophy network, as evidenced in Table 5 and Figure 4, shows 70 edges, among which the 20 ones with the highest Patel's k values (i.e., the couples of most strongly co‐altered nodes) are shown in Figure 6. Most of them involves insulo‐frontal, insulo‐insular, insulo‐hippocampal and frontoparietal co‐altered nodes. Patel's k values range from .92 of the insular and fronto‐orbital edges to .37 of the insulo‐supplementary motor areas’ edges (see the k value graph of S4 in Supporting Information).
Table 5.
Edge anatomical co‐atrophy strength between co‐altered nodes (Patel's k)
| X | Y | Z | Node 1 | X | Y | Z | Node 2 | Patel's k |
|---|---|---|---|---|---|---|---|---|
| −18 | −10 | −16 | Hippocampus_L | 22 | −18 | −12 | ParaHippocampal_R | .4574 |
| −22 | 2 | −26 | ParaHippocampal_L | −38 | 16 | −10 | Insula_L | .4439 |
| −18 | −10 | −16 | Hippocampus_L | −38 | 16 | −10 | Insula_L | .5129 |
| −22 | 2 | −26 | ParaHippocampal_L | −38 | 24 | −4 | Frontal_Inf_Orb_L | .5516 |
| −18 | −10 | −16 | Hippocampus_L | −38 | 24 | −4 | Frontal_Inf_Orb_L | .4961 |
| −38 | 16 | −10 | Insula_L | −38 | 24 | −4 | Frontal_Inf_Orb_L | .7499 |
| −22 | 2 | −26 | ParaHippocampal_L | 42 | 8 | −2 | Insula_R | .8412 |
| −18 | −10 | −16 | Hippocampus_L | 42 | 8 | −2 | Insula_R | .4695 |
| −38 | 16 | −10 | Insula_L | 42 | 8 | −2 | Insula_R | .6024 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | 42 | 8 | −2 | Insula_R | .6568 |
| −22 | 2 | −26 | ParaHippocampal_L | −42 | 14 | 0 | Insula_L_1 | .5438 |
| −38 | 16 | −10 | Insula_L | −42 | 14 | 0 | Insula_L_1 | .8574 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | −42 | 14 | 0 | Insula_L_1 | .7781 |
| 42 | 8 | −2 | Insula_R | −42 | 14 | 0 | Insula_L_1 | .6787 |
| −22 | 2 | −26 | ParaHippocampal_L | 42 | 18 | 0 | Insula_R_1 | .6509 |
| −38 | 16 | −10 | Insula_L | 42 | 18 | 0 | Insula_R_1 | .7408 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | 42 | 18 | 0 | Insula_R_1 | .7202 |
| 42 | 8 | −2 | Insula_R | 42 | 18 | 0 | Insula_R_1 | .884 |
| −42 | 14 | 0 | Insula_L_1 | 42 | 18 | 0 | Insula_R_1 | .7877 |
| −22 | 2 | −26 | ParaHippocampal_L | −46 | −64 | 4 | Temporal_Mid_L | .706 |
| −38 | 16 | −10 | Insula_L | −2 | −20 | 4 | Thalamus_L | .4655 |
| 42 | 8 | −2 | Insula_R | −2 | −20 | 4 | Thalamus_L | .4183 |
| −42 | 14 | 0 | Insula_L_1 | −2 | −20 | 4 | Thalamus_L | .5438 |
| 42 | 18 | 0 | Insula_R_1 | −2 | −20 | 4 | Thalamus_L | .6326 |
| −46 | −64 | 4 | Temporal_Mid_L | −2 | −20 | 4 | Thalamus_L | .6959 |
| −18 | −10 | −16 | Hippocampus_L | −52 | −10 | 4 | Temporal_Sup_L | .4574 |
| −38 | 16 | −10 | Insula_L | −52 | −10 | 4 | Temporal_Sup_L | .5666 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | −52 | −10 | 4 | Temporal_Sup_L | .8068 |
| 42 | 8 | −2 | Insula_R | −52 | −10 | 4 | Temporal_Sup_L | .5277 |
| −42 | 14 | 0 | Insula_L_1 | −52 | −10 | 4 | Temporal_Sup_L | .5438 |
| 42 | 18 | 0 | Insula_R_1 | −52 | −10 | 4 | Temporal_Sup_L | .5358 |
| −22 | 2 | −26 | ParaHippocampal_L | 42 | 10 | 8 | Frontal_Inf_Oper_R | .6509 |
| −38 | 16 | −10 | Insula_L | 42 | 10 | 8 | Frontal_Inf_Oper_R | .6775 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | 42 | 10 | 8 | Frontal_Inf_Oper_R | .6628 |
| 42 | 8 | −2 | Insula_R | 42 | 10 | 8 | Frontal_Inf_Oper_R | .8412 |
| −42 | 14 | 0 | Insula_L_1 | 42 | 10 | 8 | Frontal_Inf_Oper_R | .7877 |
| 42 | 18 | 0 | Insula_R_1 | 42 | 10 | 8 | Frontal_Inf_Oper_R | .926 |
| −2 | −20 | 4 | Thalamus_L | 42 | 10 | 8 | Frontal_Inf_Oper_R | .6326 |
| 42 | 8 | −2 | Insula_R | 34 | −4 | 10 | Insula_R_2 | .4183 |
| −38 | 16 | −10 | Insula_L | −50 | −20 | 12 | Temporal_Sup_L_1 | .5666 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | −50 | −20 | 12 | Temporal_Sup_L_1 | .6629 |
| 42 | 8 | −2 | Insula_R | −50 | −20 | 12 | Temporal_Sup_L_1 | .5277 |
| −42 | 14 | 0 | Insula_L_1 | −50 | −20 | 12 | Temporal_Sup_L_1 | .5438 |
| 42 | 18 | 0 | Insula_R_1 | −50 | −20 | 12 | Temporal_Sup_L_1 | .5358 |
| −52 | −10 | 4 | Temporal_Sup_L | −50 | −20 | 12 | Temporal_Sup_L_1 | .4662 |
| −42 | 14 | 0 | Insula_L_1 | −50 | 10 | 16 | Frontal_Inf_Oper_L | .4874 |
| −50 | −20 | 12 | Temporal_Sup_L_1 | −50 | 10 | 16 | Frontal_Inf_Oper_L | .4883 |
| −22 | 2 | −26 | ParaHippocampal_L | 50 | 16 | 20 | Frontal_Inf_Tri_R | .413 |
| −46 | −64 | 4 | Temporal_Mid_L | 50 | 16 | 20 | Frontal_Inf_Tri_R | .7108 |
| 42 | 10 | 8 | Frontal_Inf_Oper_R | 50 | 16 | 20 | Frontal_Inf_Tri_R | .4654 |
| −50 | −20 | 12 | Temporal_Sup_L_1 | 50 | 16 | 20 | Frontal_Inf_Tri_R | .413 |
| −38 | 16 | −10 | Insula_L | 6 | 20 | 28 | Cingulum_Ant_R | .4807 |
| 42 | 8 | −2 | Insula_R | 6 | 20 | 28 | Cingulum_Ant_R | .6127 |
| 42 | 18 | 0 | Insula_R_1 | 6 | 20 | 28 | Cingulum_Ant_R | .6194 |
| 42 | 10 | 8 | Frontal_Inf_Oper_R | 6 | 20 | 28 | Cingulum_Ant_R | .6194 |
| −50 | −20 | 12 | Temporal_Sup_L_1 | 6 | 20 | 28 | Cingulum_Ant_R | .5301 |
| 18 | −58 | 14 | Precuneus_R | 6 | 20 | 28 | Cingulum_Ant_R | .6271 |
| −38 | 16 | −10 | Insula_L | 46 | 4 | 32 | Precentral_R | .5005 |
| −42 | 14 | 0 | Insula_L_1 | 46 | 4 | 32 | Precentral_R | .4744 |
| 42 | 18 | 0 | Insula_R_1 | 46 | 4 | 32 | Precentral_R | .4654 |
| −2 | −20 | 4 | Thalamus_L | 46 | 4 | 32 | Precentral_R | .5329 |
| 42 | 10 | 8 | Frontal_Inf_Oper_R | 46 | 4 | 32 | Precentral_R | .4654 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | 46 | 4 | 32 | Precentral_R | .4225 |
| −50 | 10 | 16 | Frontal_Inf_Oper_L | −34 | −42 | 44 | Parietal_Inf_L_1 | .6016 |
| 42 | 8 | −2 | Insula_R | 0 | 4 | 46 | Supp_Motor_Area_L | .3729 |
| 34 | −4 | 10 | Insula_R_2 | 0 | 4 | 46 | Supp_Motor_Area_L | .3835 |
| −50 | 10 | 16 | Frontal_Inf_Oper_L | 0 | 4 | 46 | Supp_Motor_Area_L | .5369 |
| 18 | −66 | 26 | Precuneus_R_1 | 0 | 4 | 46 | Supp_Motor_Area_L | .5881 |
| −34 | −42 | 44 | Parietal_Inf_L_1 | 0 | 4 | 46 | Supp_Motor_Area_L | .5881 |
| −12 | −44 | −4 | Lingual_L_1 | 8 | −64 | 54 | Precuneus_R_2 | .5359 |
Figure 6.
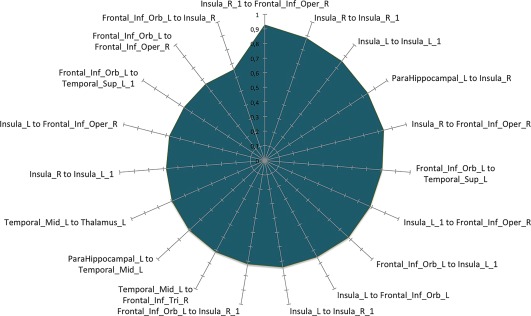
Morphometric co‐atrophy network results. This graph shows the 20 edges showing the highest Patel's k values (i.e., the couples of most strongly co‐altered nodes) [Color figure can be viewed at http://wileyonlinelibrary.com]
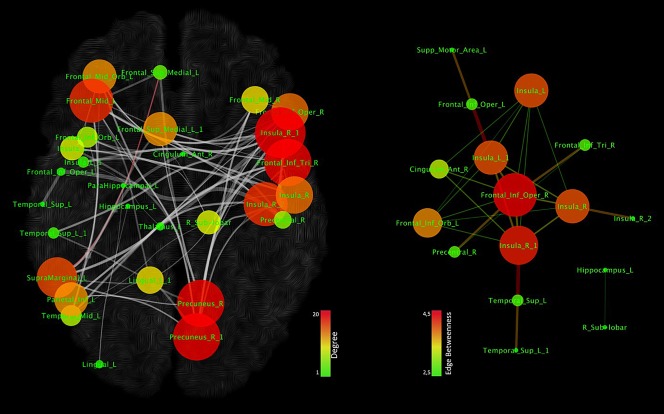
Figure 5 shows a representation of the MCN in which the nodes are characterized by sizes and colors proportionally to their degree values. In other words, according to their low or high degree values, nodes are proportionally represented in small or big sizes and in green or red colors. Among the nodes the insular cortices and inferior frontal areas show the highest degree values, followed by the superior temporal, thalamic, parahippocampal, precentral hippocampal and cingulate regions. Node degree values range from 1 to 8, while node betweenness values range from 0 to .15. Co‐alterations between nodes exhibit a network‐like pattern because the connectivity matrix is obtained using the Patel's k, which is calculated by means of a Monte Carlo simulation based on the conjoint probability distribution of altered and unaltered nodes. Indeed in this procedure, the null hypothesis is that no network‐like architecture is present, which is to say that all connections are casual or random. Figure 6 shows the 20 edges with the highest Patel's k values (i.e., the couples of most strongly co‐altered nodes).
Figure 5.
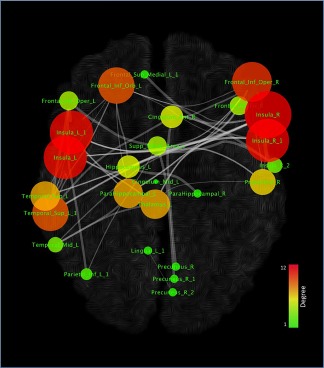
Topological analysis of the morphometric co‐atrophy network. Colors and dimensions of nodes indicates their topological degree (smaller node = lower degree; from green to red = from lower to higher values) [Color figure can be viewed at http://wileyonlinelibrary.com]
As the MCN is rather large, we investigated the possibility of identifying within it the central most densely connected subnetworks. The analysis conducted with the k‐core decomposition algorithm reported a subnetwork (shown in Figure 7) composed of eleven nodes located in the insulae, inferior frontal gyrus (IFG), superior temporal gyrus (STG), thalamus and right precentral gyrus. Similarly to Figure 5, Figure 7 right panel shows the node degree and the edge betweenness. Node degree values range from 1 to 8, while node betweenness values range from 2.5 to 4.5. Low or high degree values of nodes are proportionally represented in small or big sizes and in green or red colors. In addition, edges with low or high betweenness are represented in green or red colors and proportionally vary in thickness. Although the nodes exhibiting the highest degree are the insular and inferior frontal areas, the connections showing the highest values of edge betweenness link the insular and precentral/thalamic regions (Figure 7, right panel).
Figure 7.
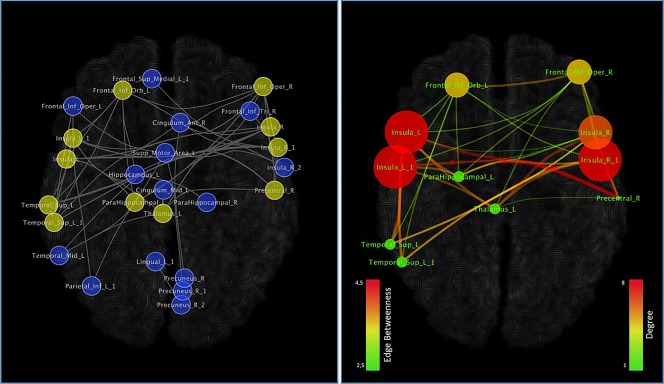
Network clustering. On the basis of our data, the k‐core algorithm reported a subnetworks evidenced in the left panel (yellow nodes) and in the right panel. Colors and dimensions of nodes indicates their topological degree (smaller node = lower degree; from green to red = from lower to higher values). Colors and dimensions of edges indicates the degree of edge betweenness (smaller edge = lower degree; from green to red = from lower to higher values) [Color figure can be viewed at http://wileyonlinelibrary.com]
Since several ROIs fall within the insular cortices (Figure 8, upper panel), in order to inspect the pattern of connectivity of these nodes we calculated for each of them the resting state functional connectivity (see Supporting Information for a brief description of the connectivity methods). We found that the five different insular nodes express three different patterns of connectivity (Figure 8, middle panel): one sensorimotor (Insula_R2), three ventral attentional (Insula_R, Insula_R1, Insula_L1), and one showing part of the default mode network (DMN) (Insula_L). The attentional nodes are all placed in the anterior insulae and the sensorimotor one in the right posterior insula. The DMN one is also the most ventral among the five. The two nodes characterized by a ventral attentional connectivity pattern are located in the most anterior dorsal regions. The lower panel of Figure 8 shows the network composed by the first connected nodes of these three different patterns (Insula_R2; Insula_R, Insula_R1, Insula_L1; Insula_L). All the three patterns show interconnections between insular ROIs; aside from these insular connections, Insula_R2 is connected with superior frontal gyrus and the supplementary motor area.
Figure 8.
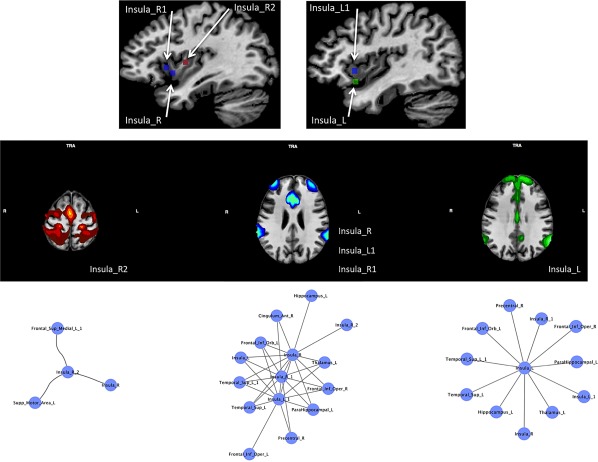
The upper panel shows the location of the insular nodes. The middle panel shows the meta‐analytic connectivity modeling (MACM) associated to these nodes (p < .001 cluster‐level corrected for multiple comparison). Nodes are associated on the basis of their patterns of connectivity. The lower panel shows the first‐step nodes of the morphometric co‐atrophy network connected to the insular nodes pertaining to each connectivity pattern [Color figure can be viewed at http://wileyonlinelibrary.com]
3.3. Structural covariance analysis
The structural co‐atrophy/co‐alteration data can be considered a form of pathological anatomical structural covariance. To better compare the normal structural covariance and the pathological alteration of the nodes that, within the MCN, appear to be the most frequently characterized by GM reduction, we calculated the former in a normative population constituted by a set of healthy subjects. Figure 9 (upper panels) shows the results of this analysis. The investigated nodes exhibit a rich pattern of anatomical covariance that is fairly correlated with the co‐atrophy pattern (r = .2059). This value is statistically significant (p < .0076). The anatomical covariance edges have r values ranging from .67 to .31 (Table 6). The 20 edges with the highest covariance values are shown in Figure 10 and mostly exhibit fronto‐parietal, insulo‐frontal, insulo‐parietal and temporo‐frontal correlations. The nodes with the highest degree are localized in medial and inferior frontal cortices, right insula, right precuneus and left supramarginal cortices (Figure 11, left panel).
Figure 9.
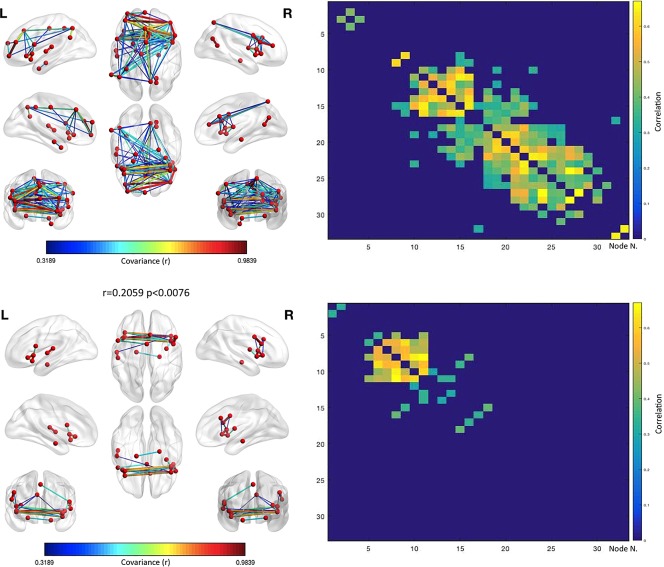
Anatomical covariance results. The upper left panels shows the structural covariance network results for the same set of nodes employed in the co‐atrophy analysis. The upper right panel shows the anatomical covariance matrix. The lower left panels shows the logic AND between the co‐atrophy results and the structural covariance network results for the same set of nodes employed in the co‐atrophy analysis (i.e., the edges represented are those that are statistically significant in the co‐atrophy AND in the anatomical covariance analysis). The lower right panel shows the logic AND between the anatomical covariance matrix and the co‐atrophy co‐alteration matrix. The correlation between the two matrices is .2059 (p < .0076). Colors from blue to red indicates increasing correlation values (r) [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 6.
Edge anatomical covariance strength between nodes (r)
| X | Y | Z | Node 1 | X | Y | Z | Node 2 | r |
|---|---|---|---|---|---|---|---|---|
| 42 | 38 | 14 | Frontal_Mid_R | −12 | −44 | −4 | Lingual_L_1 | .6716 |
| 42 | 38 | 14 | Frontal_Mid_R | 42 | 10 | 8 | Frontal_Inf_Oper_R | .668 |
| −36 | 42 | 20 | Frontal_Mid_L | 18 | −58 | 14 | Precuneus_R | .6495 |
| 42 | 18 | 0 | Insula_R_1 | −38 | 24 | −4 | Frontal_Inf_Orb_L | .6493 |
| −50 | 10 | 16 | Frontal_Inf_Oper_L | −52 | −10 | 4 | Temporal_Sup_L | .6463 |
| 18 | −58 | 14 | Precuneus_R | 42 | 10 | 8 | Frontal_Inf_Oper_R | .6324 |
| 42 | 18 | 0 | Insula_R_1 | 42 | 8 | −2 | Insula_R | .6321 |
| 42 | 10 | 8 | Frontal_Inf_Oper_R | −12 | −44 | −4 | Lingual_L_1 | .6264 |
| 42 | 8 | −2 | Insula_R | −38 | 24 | −4 | Frontal_Inf_Orb_L | .6258 |
| 42 | 8 | −2 | Insula_R | −32 | 54 | −4 | Frontal_Mid_Orb_L | .6253 |
| −2 | −20 | 4 | Thalamus_L | −42 | 14 | 0 | Insula_L_1 | .6189 |
| 18 | −66 | 26 | Precuneus_R_1 | 50 | 16 | 20 | Frontal_Inf_Tri_R | .6141 |
| −32 | 54 | −4 | Frontal_Mid_Orb_L | 22 | −18 | −12 | ParaHippocampal_R | .6089 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 50 | 16 | 20 | Frontal_Inf_Tri_R | .6028 |
| 18 | −66 | 26 | Precuneus_R_1 | 18 | −58 | 14 | Precuneus_R | .5998 |
| −32 | 54 | −4 | Frontal_Mid_Orb_L | −38 | 16 | −10 | Insula_L | .5886 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 18 | −66 | 26 | Precuneus_R_1 | .5701 |
| −32 | 54 | −4 | Frontal_Mid_Orb_L | −38 | 24 | −4 | Frontal_Inf_Orb_L | .5643 |
| −46 | −64 | 4 | Temporal_Mid_L | 42 | 18 | 0 | Insula_R_1 | .5641 |
| 18 | −58 | 14 | Precuneus_R | 34 | −4 | 10 | Insula_R_2 | .5639 |
| 42 | 8 | −2 | Insula_R | −38 | 16 | −10 | Insula_L | .5582 |
| 18 | −66 | 26 | Precuneus_R_1 | 42 | 10 | 8 | Frontal_Inf_Oper_R | .5536 |
| −38 | 16 | −10 | Insula_L | 22 | −18 | −12 | ParaHippocampal_R | .5422 |
| 42 | 38 | 14 | Frontal_Mid_R | 18 | −58 | 14 | Precuneus_R | .5406 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | 22 | −18 | −12 | ParaHippocampal_R | .5381 |
| 18 | −66 | 26 | Precuneus_R_1 | 42 | 38 | 14 | Frontal_Mid_R | .5261 |
| −46 | −56 | 44 | Parietal_Inf_L | 18 | −66 | 26 | Precuneus_R_1 | .5032 |
| 18 | −58 | 14 | Precuneus_R | −12 | −44 | −4 | Lingual_L_1 | .5018 |
| −60 | −52 | 28 | SupraMarginal_L | −2 | 56 | 18 | Frontal_Sup_Medial_L | .4981 |
| 18 | −66 | 26 | Precuneus_R_1 | −36 | 42 | 20 | Frontal_Mid_L | .4848 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | 18 | −58 | 14 | Precuneus_R | .4763 |
| 42 | 18 | 0 | Insula_R_1 | −32 | 54 | −4 | Frontal_Mid_Orb_L | .4731 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | 34 | −4 | 10 | Insula_R_2 | .4642 |
| −60 | −52 | 28 | SupraMarginal_L | 18 | −66 | 26 | Precuneus_R_1 | .463 |
| −46 | −56 | 44 | Parietal_Inf_L | 18 | −58 | 14 | Precuneus_R | .4606 |
| 18 | −66 | 26 | Precuneus_R_1 | −12 | −44 | −4 | Lingual_L_1 | .4585 |
| 18 | −58 | 14 | Precuneus_R | 42 | 18 | 0 | Insula_R_1 | .4568 |
| 34 | −4 | 10 | Insula_R_2 | −32 | 54 | −4 | Frontal_Mid_Orb_L | .4566 |
| −46 | −64 | 4 | Temporal_Mid_L | −32 | 54 | −4 | Frontal_Mid_Orb_L | .4565 |
| 18 | −66 | 26 | Precuneus_R_1 | 34 | −4 | 10 | Insula_R_2 | .4556 |
| −60 | −52 | 28 | SupraMarginal_L | −36 | 42 | 20 | Frontal_Mid_L | .4532 |
| −60 | −52 | 28 | SupraMarginal_L | 42 | 10 | 8 | Frontal_Inf_Oper_R | .4514 |
| −46 | −64 | 4 | Temporal_Mid_L | −38 | 24 | −4 | Frontal_Inf_Orb_L | .4496 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 34 | −4 | 10 | Insula_R_2 | .4469 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 18 | −58 | 14 | Precuneus_R | .4428 |
| 42 | 18 | 0 | Insula_R_1 | −38 | 16 | −10 | Insula_L | .4418 |
| −60 | −52 | 28 | SupraMarginal_L | 18 | −58 | 14 | Precuneus_R | .4415 |
| −50 | −20 | 12 | Temporal_Sup_L_1 | −12 | −44 | −4 | Lingual_L_1 | .4414 |
| −36 | 42 | 20 | Frontal_Mid_L | 50 | 16 | 20 | Frontal_Inf_Tri_R | .439 |
| −18 | −10 | −16 | Hippocampus_L | −32 | −88 | −16 | Lingual_L | .4294 |
| 18 | −66 | 26 | Precuneus_R_1 | −32 | 54 | −4 | Frontal_Mid_Orb_L | .4286 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 42 | 10 | 8 | Frontal_Inf_Oper_R | .4285 |
| 18 | −58 | 14 | Precuneus_R | 42 | 8 | −2 | Insula_R | .4272 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 46 | 4 | 32 | Precentral_R | .4258 |
| −38 | 24 | −4 | Frontal_Inf_Orb_L | −38 | 16 | −10 | Insula_L | .4241 |
| 42 | 8 | −2 | Insula_R | 22 | −18 | −12 | ParaHippocampal_R | .4195 |
| −36 | 42 | 20 | Frontal_Mid_L | −2 | 56 | 18 | Frontal_Sup_Medial_L | .4165 |
| −36 | 42 | 20 | Frontal_Mid_L | 42 | 10 | 8 | Frontal_Inf_Oper_R | .412 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | −36 | 42 | 20 | Frontal_Mid_L | .4078 |
| 34 | −4 | 10 | Insula_R_2 | 42 | 8 | −2 | Insula_R | .4075 |
| 18 | −66 | 26 | Precuneus_R_1 | 42 | 8 | −2 | Insula_R | .4063 |
| −46 | −56 | 44 | Parietal_Inf_L | 42 | 10 | 8 | Frontal_Inf_Oper_R | .4032 |
| −32 | −88 | −16 | Lingual_L | −22 | 2 | −26 | ParaHippocampal_L | .4014 |
| −60 | −52 | 28 | SupraMarginal_L | 42 | 38 | 14 | Frontal_Mid_R | .4013 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 42 | 18 | 0 | Insula_R_1 | .398 |
| 6 | 20 | 28 | Cingulum_Ant_R | −60 | −52 | 28 | SupraMarginal_L | .3915 |
| −46 | −56 | 44 | Parietal_Inf_L | 42 | 38 | 14 | Frontal_Mid_R | .3883 |
| 34 | −4 | 10 | Insula_R_2 | −38 | 16 | −10 | Insula_L | .385 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | 42 | 10 | 8 | Frontal_Inf_Oper_R | .3831 |
| −60 | −52 | 28 | SupraMarginal_L | −12 | −44 | −4 | Lingual_L_1 | .3811 |
| 42 | 18 | 0 | Insula_R_1 | 22 | −18 | −12 | ParaHippocampal_R | .3802 |
| −2 | 56 | 18 | Frontal_Sup_Medial_L | 18 | −58 | 14 | Precuneus_R | .3774 |
| 46 | 4 | 32 | Precentral_R | 50 | 16 | 20 | Frontal_Inf_Tri_R | .3773 |
| −36 | 42 | 20 | Frontal_Mid_L | 42 | 38 | 14 | Frontal_Mid_R | .3755 |
| −46 | −56 | 44 | Parietal_Inf_L | −12 | −44 | −4 | Lingual_L_1 | .3751 |
| 18 | −66 | 26 | Precuneus_R_1 | −38 | 16 | −10 | Insula_L | .3724 |
| 42 | 38 | 14 | Frontal_Mid_R | −50 | −20 | 12 | Temporal_Sup_L_1 | .369 |
| −46 | −56 | 44 | Parietal_Inf_L | −60 | −52 | 28 | SupraMarginal_L | .3674 |
| −46 | −56 | 44 | Parietal_Inf_L | −2 | 56 | 18 | Frontal_Sup_Medial_L | .3668 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | 42 | 8 | −2 | Insula_R | .3645 |
| −50 | −20 | 12 | Temporal_Sup_L_1 | 42 | 10 | 8 | Frontal_Inf_Oper_R | .3595 |
| −36 | 42 | 20 | Frontal_Mid_L | 34 | −4 | 10 | Insula_R_2 | .3593 |
| −46 | −56 | 44 | Parietal_Inf_L | −36 | 42 | 20 | Frontal_Mid_L | .3556 |
| 34 | −4 | 10 | Insula_R_2 | 42 | 10 | 8 | Frontal_Inf_Oper_R | .3517 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | −32 | 54 | −4 | Frontal_Mid_Orb_L | .3508 |
| 46 | 4 | 32 | Precentral_R | 34 | −4 | 10 | Insula_R_2 | .3501 |
| 18 | −66 | 26 | Precuneus_R_1 | 42 | 18 | 0 | Insula_R_1 | .3465 |
| −36 | 42 | 20 | Frontal_Mid_L | −46 | −64 | 4 | Temporal_Mid_L | .3445 |
| −46 | −64 | 4 | Temporal_Mid_L | 22 | −18 | −12 | ParaHippocampal_R | .3404 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | 42 | 8 | −2 | Insula_R | .3388 |
| −60 | −52 | 28 | SupraMarginal_L | 50 | 16 | 20 | Frontal_Inf_Tri_R | .3388 |
| −42 | 14 | 0 | Insula_L_1 | 42 | 8 | −2 | Insula_R | .3371 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | 42 | 18 | 0 | Insula_R_1 | .3345 |
| −2 | −20 | 4 | Thalamus_L | 42 | 18 | 0 | Insula_R_1 | .3334 |
| 34 | −4 | 10 | Insula_R_2 | 42 | 18 | 0 | Insula_R_1 | .3312 |
| −46 | −56 | 44 | Parietal_Inf_L | 50 | 16 | 20 | Frontal_Inf_Tri_R | .3297 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | −46 | −64 | 4 | Temporal_Mid_L | .3285 |
| −2 | 28 | 40 | Frontal_Sup_Medial_L_1 | −60 | −52 | 28 | SupraMarginal_L | .3272 |
| −36 | 42 | 20 | Frontal_Mid_L | −12 | −44 | −4 | Lingual_L_1 | .3266 |
| −36 | 42 | 20 | Frontal_Mid_L | 42 | 18 | 0 | Insula_R_1 | .3239 |
| 46 | 4 | 32 | Precentral_R | −50 | 10 | 16 | Frontal_Inf_Oper_L | .3238 |
| 42 | 18 | 0 | Insula_R_1 | −42 | 14 | 0 | Insula_L_1 | .3231 |
| −46 | −56 | 44 | Parietal_Inf_L | 34 | −4 | 10 | Insula_R_2 | .3228 |
| 18 | −58 | 14 | Precuneus_R | −32 | 54 | −4 | Frontal_Mid_Orb_L | .3191 |
| 50 | 16 | 20 | Frontal_Inf_Tri_R | 22 | −18 | −12 | ParaHippocampal_R | .3132 |
| −60 | −52 | 28 | SupraMarginal_L | 34 | −4 | 10 | Insula_R_2 | .3126 |
| 42 | 18 | 0 | Insula_R_1 | 46 | 4 | 32 | Precentral_R | .3109 |
Figure 10.
Anatomical covariance results. This graph shows the 20 edges showing the highest correlation values (i.e., the couples of most strongly structurally covariant nodes) [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 11.
Anatomical covariance results. The left panel shows the anatomical covariance network results for the same set of nodes employed in the co‐atrophy analysis (GM decreases). Colors and dimensions of nodes indicates their topological degree (smaller node = lower degree; from green to red = from lower to higher values). The right panel shows a spring embedded visualization of the logic AND between the co‐atrophy results and the anatomical covariance network results for the same set of nodes employed in the co‐atrophy analysis (i.e., the edges represented are those that are statistically significant in the co‐atrophy AND in the anatomical covariance analysis). Colors and dimensions of nodes indicates their topological degree (smaller node = lower degree; from green to red = from lower to higher values). Colors and dimensions of edges indicates the degree of edge betweenness (smaller edge = lower degree; from green to red = from lower to higher values) [Color figure can be viewed at http://wileyonlinelibrary.com]
Areas showing both anatomical covariance and co‐atrophy (AND logic between anatomical covariance and co‐atrophy values; see Figure 9 lower panels, Figure 11 right panel for comparison between edges) mainly exhibit insulo‐insular, interemispheric, fronto‐insular, cingulo‐insular and insulo‐temporal connections, all of which are present both in the anatomical covariance and in the pathological co‐atrophy analyses. Among the nodes showing both anatomical covariance and pathological co‐atrophy, the ones with the highest degree are those placed in orbitofrontal, opercular and insular regions (Figure 11, right panel). Edges showing the highest edge betweenness values are those linking fronto‐insular and temporo‐insular nodes (Figure 11, right panel).
3.4. Co‐alteration networks of GM decreases and GM increases
As already said, most of the studies examined in this meta‐analysis do not report GM increase data. However, since we wanted to obtain information from these data as well, we performed a co‐alteration analysis on a dataset that was the sum of GM decreased and increased values. The rationale for doing this was that GM increase data were not on their own sufficient to be statistically examined.
Specifically, we conducted two supplementary analyses using as inputs both GM decrease and GM increase data. In the first analysis we employed the same set of nodes previously used for the MCN (see Figure 12, left panel, Table 4). In the second analysis we created a new set of nodes keeping the same node detection parameters already employed.
Figure 12.
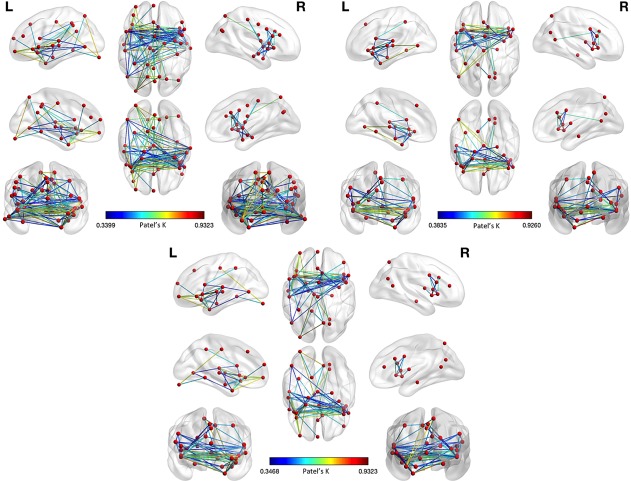
Results comparison of the morphometric co‐alteration network for GM increases plus GM decreases, and for GM decreases only. The left panel shows the morphometric co‐alteration network results for GM increases plus GM decreases using an ad hoc developed set of nodes. The central panel shows the morphometric co‐alteration network results for GM increases plus GM decreases using the set of nodes previously employed for the co‐atrophy analysis (see these nodes in Figure 2). The right panel shows the morphometric co‐alteration network results for GM decreases. Colors from blue to red indicates increasing Patel's k values (i.e., increasing co‐alteration probabilities) [Color figure can be viewed at http://wileyonlinelibrary.com]
The first analysis of data (GM decrease plus GM increase) with the same set of nodes of the MCN shows a co‐alteration pattern that is somewhat similar to the pattern obtained from the analysis of GM decrease data only (Figure 12, right and middle panels). It can be observed that in both analyses the group of main edges linking the insular, cingulate, temporal and parietal cortices is quite constant, even though it is slightly thicker when the sum of GM decrease and GM increase is considered. However, some prefrontal and parietal connections, which are only present when GM decrease data are considered, are lost. In the results obtained from both GM decrease and GM increase data, Patel's k values range from .93 to .34.
The second analysis of data (GM decrease plus GM increase), conducted with a new set of nodes, shows a much thicker network (Figure 12, left panel). New nodes, which were not present in the results obtained from GM decrease data only, now emerge, while the nodes that were lost in the first analysis return to be part of the network. Also in this case Patel's k values range from .93 to .34.
Figure 13 upper panels show the pattern of anatomical covariance in healthy subjects associated with the new set of nodes used in the second analysis of data (GM decrease plus GM increase); Figure 13 lower panels show the comparison between the co‐alteration pattern obtained from both GM decrease and GM increase data and the anatomical covariance of the same nodes in healthy subjects. In the case of the new set of nodes r values range from .25 to .78; while in the case of the previous set of nodes r values range from .34 to .93.
Figure 13.
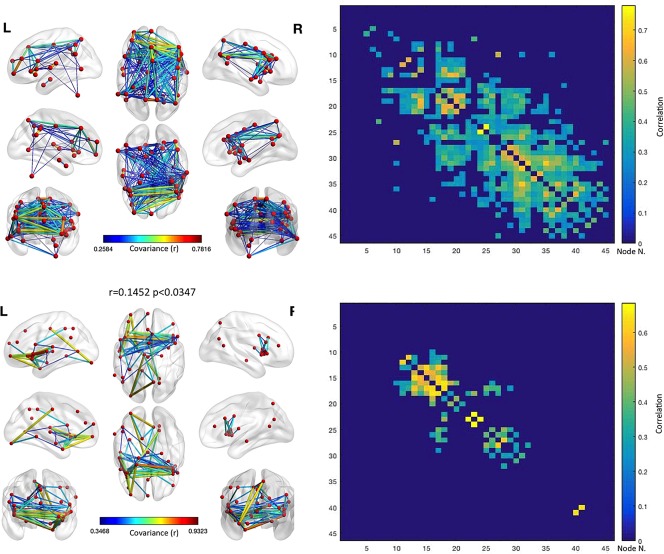
Anatomical covariance results for the GM increases plus GM decreases set of nodes. The upper left panel shows the anatomical covariance network results for the same set of nodes employed in the co‐alteration analysis of GM increases plus GM decreases. The upper right panel shows the anatomical covariance matrix. The lower left panel shows the logic AND between the co‐alteration results of GM increases plus GM decreases and the anatomical covariance network results for the same set of nodes employed in the analysis of GM decreases (i.e., the edges represented are those that are statistically significant in the co‐atrophy AND in the anatomical covariance analysis). The lower right panel shows the logic AND between the anatomical covariance matrix and the co‐alteration matrix. The correlation between the two matrices is .1452 (p < .0347). Colors from blue to red indicates increasing correlation values (r) [Color figure can be viewed at http://wileyonlinelibrary.com]
Just at first sight it is evident that the pattern of anatomical covariance and the co‐alteration pattern are less similar when GM decrease and GM increase data are analyzed together than when only GM decrease data are analyzed. In fact in this case the correlation value is r = .1452 (p < .0347). This is probably so because the new nodes obtained by adding the GM increase data are more anatomically covariant than anatomically co‐altered. The nodes and edges shared by the anatomical covariance matrix and the matrix obtained from both GM decrease and GM increase data are those associated with the connections between the insular cortices and between the insular and prefrontal/cingulate cortices already found in the MCN. In addition to these connections, other ones occur between the cingulate and posterior parietal cortices. Therefore, similarities between the anatomical covariance matrix and the matrix obtained from both GM decrease and GM increase are less statistically relevant, but they extend beyond the insular cortices, involving also prefrontal, cingulate and parietal regions, which are not present when the anatomical covariance matrix and the MCN matrix are compared.
3.5. Reliability
Analysis 1 reveals that certain edges linking the insulae, temporal lobes and cingulate cortices are always present in the MCN, independently of which spectrum is removed. These edges connect the “core” areas already described in Cauda, et al. (2017). Most of the other edges are present when ASD and OCSD are removed, but not when SCZD is left out of the analysis. In this case, the MCN is significantly less distributed and less relevant edges are present (see also Supporting Information, Figure S7).
This phenomenon can be better understood in light of analysis 2, which reveals that the correlation results between the Patel's k values of the edges associated to each spectrum suggest a good similarity between ASD and SCZD, and between OCSD and SCZD: for ASD versus SCZD r = .37, and for OCSD versus SCZD r = .56, respectively. In contrast, correlation values between the ASD matrix and OCSD matrix appear to be less similar: for ASD versus OCSD r = .13.
As already pointed out in Cauda, et al. (2017), analysis 3 reveals that the structural alterations (GM decreases) caused by the three spectra can be clearly subsumed under two clusters. Neither of the two clusters can be specifically associated with a spectrum, as all three psychiatric disorders distribute almost equally within them (see Supporting Information, Figure S3). Figure 14 summarizes the results of these three analyses.
Figure 14.
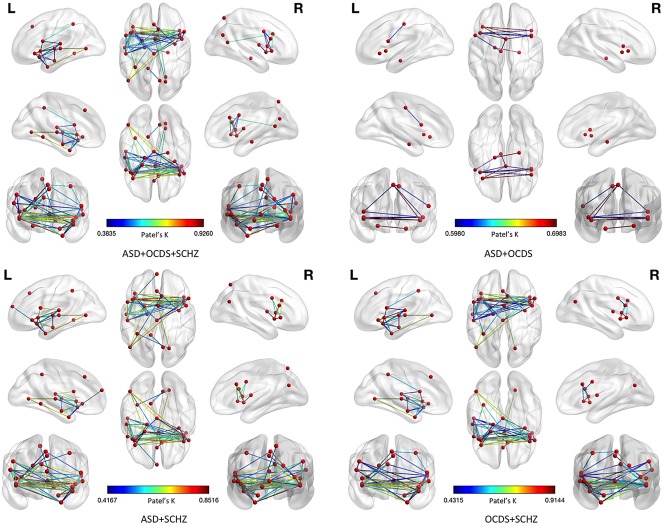
Results of the morphometric co‐atrophy network by leaving one spectrum out in alternation. This figure shows the comparison between the results of the morphometric co‐atrophy network of the three spectra together (upper left panel) and each of the morphometric co‐atrophy network obtained from leaving one spectrum out in alternation. Colors from blue to red indicates increasing Patel's k values (i.e., increasing co‐alteration probabilities) [Color figure can be viewed at http://wileyonlinelibrary.com]
As Figure 14 illustrates, when we leave SCZD out, the number of edges substantially diminishes; this, along with the fact that ASD and OCSD have more inhomogeneous sample data, as shown in Cauda et al. (2017), explains why the correlation value between the matrix constructed with the SCZD data only and the matrix constructed with the whole dataset is low. Moreover, it is worth noting that, even though results are significantly reduced when SCZD is left out, the remaining edges are those that form the most connected part of the MCN, which is also the core of the pattern constructed with the SCZD data only (please see both the upper right panel of Figure 14 and the upper panel of Supporting Information, Figure S5).
Finally, analysis 4 shows a high degree of similarity between the MCN constructed with the same amount of data for each spectrum and the MCN constructed with the original dataset. In fact, the DC is significantly high: .7969 (Figure 15, left and middle panels). Moreover, the evaluation of the stability of the result has been assessed by repeating the analysis with 1,000 permutations (each time with a different sample selection from SCZD data). Of note, the standard error has very low values, ranging from 0 to .04 (Figure 15, right panel). The result of analysis 4 shows that, even though the reduced sample leads to the loss of certain edges and the decrease of Patel's k values, the new MCN largely overlaps the original MCN obtained with the whole dataset, thus providing evidence that the predominance of SCZD data does not bias the validity of our original outcome.
Figure 15.

Results of the morphometric co‐atrophy network (MCN) obtained with an equal number of experiments fro each psychiatric spectrum. The left panel shows the results obtained with the original dataset. Colors from blue to red indicates increasing Patel's k values (i.e., increasing co‐alteration probabilities). The central panel shows the overlap between the MCN calculated with the equalized dataset and the MCN calculated with the original dataset. The right panel shows the stability of the equalized results (analyses were repeated with 1,000 permutations, each time by randomly selecting a different sample of schizophrenia data) expressed in standard error values. Colors from blue to red indicates increasing standard error values [Color figure can be viewed at http://wileyonlinelibrary.com]
4. DISCUSSION
The analysis of the co‐atrophy network of SCZD, ASD, and OCSD reveals that alterations in certain GM sites appear to be statistically related to alterations of other GM regions. Although this finding has already been proved to be the case in neurodegenerative diseases, it has never been found before in psychiatric conditions. As our study dealt mainly with GM decreases, we propose to define the ensemble of co‐altered areas as morphometric co‐atrophy network or MCN and the structural and functional pathways linking these areas as pathoconnectivity.
Our analysis of VBM data has revealed that alterations in the GM density of patients with SCZD, ASD, and OCSD do not develop randomly but rather follow identifiable patterns of co‐alteration. In particular, our results indicate that a small number of brain areas show a high degree of pathoconnectivity (Yates, 2012); in other words, only a few cerebral areas appear to be particularly co‐altered with several other regions. Many of these areas also exhibit a normal pattern of anatomical covariance that can be partly altered by the progressive impact of SCZD, ASD, and OCSD. Clearly, these brain sites play an important role in the formation and development of the MCN and, as a consequence, can be thought of as pathoconnectivity hubs. For instance, the left lingual gyrus appears to be co‐altered only with one particular area (i.e., the right precuneus), whereas the left insula (ROI Insula_L1) appears to be co‐altered with eleven other regions. Thus, the co‐alteration patterns of these two areas contribute differently in shaping and developing the MCN across the brain. In fact, neuronal alterations are supposed to distribute more quickly and diffusely from cerebral regions showing a high degree of pathoconnectivity. These results were obtained by calculating the network degree or level of connectivity for each altered area.
Overall, brain sites with the highest network degree were found to be the insulae and the prefrontal cortices, which are also densely connected with each other. These regions are therefore pathoconnectivity hubs and can be considered as primary altered areas, whereas the other brain regions, which have a lower network degree and appear to be connected only with pathoconnectivity hubs, can be considered as secondary altered areas.
The network clustering analysis developed and employed in this study was able to identify within the MCN a “core” subnetwork composed of eleven nodes located in the insulae, IFG, STG, thalamus, and right precentral gyrus. Some of these regions are involved in supporting the salience network, which is an essential part of the frontoparietal control system. The insular cortices are pivotal components of this important circuitry, which has been found to be altered in a great variety of brain disorders (Cole, Repovs, & Anticevic, 2014; McTeague, et al., 2016; Sprooten, et al., 2017). In particular, the disruption of the functional integrity of this network would account for the executive deficits that are frequently observed across several psychiatric conditions (Power, et al., 2011). That the insula might be thought of as a pathoconnectivity hub is not surprising, as this brain area has vast and extensive connections to both several cortical areas and the limbic system. The insula has also been found to be involved in important brain functions—that is, integration of external sensory stimuli with emotions, the conscious perception of error, the generation and maintenance of a state of awareness associated with the body's condition (Cauda, et al., 2012a; Cauda, et al., 2011a; Klein, Ullsperger, & Danielmeier, 2013; Vercelli, et al., 2016; Wylie & Tregellas, 2010).
Our analysis reveals that particularly the anterior part of the insular cortex seems to be mostly involved in the formation of the MCN associated with SCZD, ASD, and OCSD. In fact only one node was located within the posterior insula (i.e., Insula_R2). This node has been found to be part of a network with sensorimotor functions. Overall, these findings provide evidence that the insula is not only an important brain hub supporting functional connectivity during rest as well as task activities, but also a pathoconnectivity hub lying at the center of co‐alteration networks produced by a variety of brain disorders. Indeed the idea that anatomically defined subsets of brain regions might be hotspots for abnormality of GM volume is supported by the fact that these core areas are more functionally valuable for higher‐order cognitive tasks and adaptive behavior, and thereby also more likely to be associated with a wide range of pathological processes (Crossley, et al., 2016; Crossley, et al., 2014).
The STG multimodal areas are involved in cortical integration of both sensory and limbic information at the highest level; this makes them key regions implicated in the social perceptual skills. Moreover, STG is thought to process biological motion (Jou, et al., 2011a; Jou, et al., 2011b) and has been associated with some verbal and non‐verbal communication impairments observed in patients with ASD (Radua, Via, Catani, & Mataix‐Cols, 2011).
Precentral and inferior frontal gyri are involved in the mirror neuron system; GM thinning in regions associated with the mirror‐neuron system have been correlated with social and communication deficits in patients with ASD (Cattaneo & Rizzolatti, 2009; Keller, Bugiani, Fantin, & Pirfo, 2011; Kilner, Friston, & Frith, 2007; Rizzolatti & Craighero, 2004).
The disruption of the thalamus has been variously associated with SCZD and ASD. For instance, a reduced GM density in the thalamus, right cerebellum hemisphere and left temporoparietal cortex is related to intellectual disabilities in ASD (Spencer, et al., 2006). Moreover, other findings suggest a relationship between hypoconnectivity disturbances in the thalamofrontal system and ASD (Cheon, et al., 2011). As we will see, the thalamus is also supposed to play an important role in inflammatory processes.
It is worth noting that the brain areas altered by the three spectra (the nodes of our analyses) have a normal pattern of anatomical covariance (Evans, 2013). This pattern overlaps partially with the MCN (r = .2059, p < .0076). Similarities are mainly found in the insulo‐insular as well as cingulo‐cingulate intraparietal connections. This result suggests that the normal anatomical covariance of insular and cingulate areas tends to be progressively altered toward the development of a pathological anatomical covariance (co‐atrophy). Our finding is in line with the frequent observation that the patterns of brain co‐alterations match in part the patterns of brain connectivity (Cauda, et al., 2017; Cauda, et al., 2012b; Crossley, et al., 2016; Crossley, et al., 2014; Evans, 2013; Fornito, et al., 2015; Menon, 2013; Raj, et al., 2012; Saxena & Caroni, 2011; Seeley, et al., 2009; Yates, 2012; Zhou, et al., 2012). In contrast, when different edges are involved, as it is the case of the other altered or co‐atrophic areas of the MCN, especially those with a lower network degree, the normal pattern of anatomical covariance does not overlap with the alteration pattern.
The fact that neuronal abnormalities caused by SCZD, ASD, and OCSD converge on a set of core areas that are associated with cognitive control functions (Buckholtz & Meyer‐Lindenberg, 2012; McTeague, et al., 2016) is also consistent with previous evidence showing that in brain disorders GM alterations and WM alterations tend to exhibit concordant patterns of distribution, which are influenced by brain connectivity (Iturria‐Medina & Evans, 2015; Raj, et al., 2012; Voytek & Knight, 2015; Zhou, et al., 2012). As already pointed out, this finding is further supported by the good correlation between the patterns of anatomical covariance and co‐atrophy, especially with regard to the set of “core areas”.
The inclusion in the MCN analysis of the GM increase data (GM decrease plus GM increase) leads to a pattern that is fairly similar to the one found on the basis of GM decrease data only; however, the former is significantly less similar than the latter to the normal pattern of anatomical covariance associated with the same nodes. This suggests that GM increase data may contribute to the MCN by adding a series of edges that are not present in the anatomical covariance pattern. However, it must be observed that, given the little knowledge with regard to the processes at the root of the increase and decrease of GM density, the conjoint use of GM decrease and GM increase data is still a debated issue, which is reflected by different approaches and positions in the scientific literature (Eickhoff, et al., 2012; Radua & Mataix‐Cols, 2009).
To date, at least four mechanisms have been suggested to explain how brain abnormalities propagate (Fornito, et al., 2015; Zhou, et al., 2012), two of which call attention to the role that may be played by brain architecture. The first mechanism hypothesizes a shared vulnerability (caused in particular by the co‐expression of genes) between certain brain areas (Cioli, Abdi, Beaton, Burnod, & Mesmoudi, 2014; French, Tan, & Pavlidis, 2011; Lichtman & Sanes, 2008; Prieto, Risueno, Fontanillo, & De las Rivas, 2008; Wolf, Goldberg, Manor, Sharan, & Ruppin, 2011; Zhou, et al., 2012). The second mechanism posits a transneuronal spread of misfolded proteins along axonal pathways connecting different brain regions (Chevalier‐Larsen & Holzbaur, 2006; Fornito, et al., 2015; Goedert, Clavaguera, & Tolnay, 2010; Guest, et al., 2011; Iturria‐Medina, Sotero, Toussaint, & Evans, 2014; Zhou, et al., 2012). The third mechanism proposes a trophic failure in the neuronal metabolism, that is, a dysfunction of trophic factors that would disrupt the development and maintenance of neural wiring (Appel, 1981; Fornito, et al., 2015; Salehi, et al., 2006; Zhou, et al., 2012). Finally, the fourth mechanism hypothesizes that the network hubs might be more exposed to nodal stress and, thereby, be more susceptible to pathological modifications (Crossley, et al., 2014; Saxena & Caroni, 2011; Zhou, et al., 2012). Probably certain pathological processes are preferentially involved in some disorders than in others, at least at their inception. However, it is important to highlight that these four mechanisms are not mutually exclusive, as different pathological factors may operate simultaneously.
Of note, all the three psychiatric spectra examined in this study have been linked to genetic dysfunction. Recently, the risk of developing SCZD has been associated with variation in the major histocompatibility complex locus, in particular in the complement component four genes (Sekar, et al., 2016). In turn, a number of altered genetic mechanisms have been associated with ASD. In patients with ASD, alterations of gene expressions might disrupt the autoregulatory feedback of neuronal loops, the brain electrical activity, the concentration of signaling molecules, as well as the mechanisms of synaptic excitation and inhibition (Mullins, Fishell, & Tsien, 2016). With regard to OCSD, it has been suggested that complex patterns of molecular dysfunctions due to genetic factors (regarding in particular the serotonergic and dopaminergic systems) may account for the familial occurrences of this disorder (Nestadt, Grados, & Samuels, 2010; Pauls, 2010; Tukel, et al., 2016). Therefore, studies that aim to understand the genetic patterns of brain disorders are extremely important and future investigations are needed to find out whether altered co‐expressions of genes can affect the areas that this meta‐analysis has put forward as key nodes in the anatomical co‐alteration networking of SCZD, ASD, and OCSD.
Furthermore, proteins such as astrotactins have been suggested as a common genetic link among these three psychiatric spectra, because they are fundamental in guiding neurons migration during brain development (Lionel, Tammimies, Vaags, Rosenfeld, & Ahn, 2014). Oxytocin too—which is a hormone not only associated with childbirth, milk let‐down, and maternal care, but also with the regulation of social behavior and the formation of pair bonds—has been found to be involved in multiple psychiatric disorders, including ASD, SCZD, and OCSD. Indeed, in patients with ASD oxytocin appears to be related to social recognition, attachment, and stereotyped behaviors, whereas in patients with SCZD it has been associated with a potential antipsychotic effect. Interestingly, in patients with OCSD this hormone has been found to have higher values in patients that respond to serotonin reuptake inhibitor treatment, but lower values in patients who have autistic traits (Cochran, Fallon, Hill, & Frazier, 2013; Humble, Uvnas‐Moberg, Engstrom, & Bejerot, 2013; Romano, Tempesta, Micioni Di Bonaventura, & Gaetani, 2015; Shin, et al., 2015; Strauss, et al., 2015).
There are significant symptomatic overlaps between ASD, OCSD, and SCZD. Between ASD and OCSD common symptoms are insistence on sameness, tics, ritual and repetitive behaviors. Within SCZD a subtype of schizophrenia (schizo‐obsessive) is particularly characterized by obsessive symptoms (Bleich‐Cohen, et al., 2014). Of note, the first diagnostic criterion of DSM‐5 for ASD is strictly related to the negative symptoms of SCZD, while the second diagnostic criterion is similar to the OCSD symptomatology. The relative symptomatic similarity between ASD and SCZD is consistent with a neurobiological model that suggests a common basis for SCZD and ASD, with a number of genetic alterations (SHANK 3 variations, DISC 1, dysregulation of CYFIP1, SCN2A, NRXN1 neurexin gene or RELN), cytoarchitectural abnormalities (about proliferation, migration and lamination defects), neuropsychological deficit, neuroimaging investigations (about GM/WM abnormalities and structural/functional connectivity alterations), and clinical observations (Baribeau & Anagnostou, 2013; Cauda, et al., 2017; Cauda, et al., 2014a; Cauda, et al., 2011b; Cheung, et al., 2010; Chisholm, Lin, Abu‐Akel, & Wood, 2015; de Lacy & King, 2013; King & Lord, 2011; Pathania, et al., 2014).
The psychotic manifestations of SCZD, especially the negative and catatonic ones, are so much associated with the social withdraw and communicative difficulty in ASD that even specific tests, such as the Autism Diagnostic Observation Schedule (ADOS), sometimes fail and produce false positives if performed on adolescent patients with SCZD (Bertelli, et al., 2015). Furthermore, psychotic manifestations and ASD are frequently observed in the clinical practice, and it is particularly worth noting that some patients, diagnosed with ASD in their childhood, show a schizophrenic development in adolescence or adulthood, accompanied by psychotic and neurodegenerative aspects (Keller, Piedimonte, Bianco, Bari, & Cauda, 2016). These clinical data might be accounted for by finding out common genetic roots at the basis of neurodevelopment disorders, which bring about phenotypic expressions with different timings and modalities, due to epigenetic factors affecting the production of proteins with regulatory function over the brain organization and development (Di Gregorio, et al., 2017). This hypothesis is supported by the clinical examination of families of patients with ASD, in which phenotypic expressions bear psychiatric disorders different from ASD, OCSD, and SCZD (Biamino, et al., 2016). The relationship between genes, epigenetic and environmental factors could typically emerge from the specific patterns of structural alterations that our analysis has discovered. In particular, brain hubs are likely to be the areas in which this relationship appears to be stronger.
Compared to OCSD, structural abnormalities in SCZD and ASD are especially located in brain areas with a high degree of pathoconnectivity—that is, areas where altered voxel exhibit a higher degree of connectivity than unaltered voxel—albeit not necessarily in network hubs with the highest degree of connections. Presumably, factors being specific to the disorder are likely to determine the regions to be first affected and how anatomical alterations distribute across the brain (Crossley, et al., 2016; Crossley, et al., 2014).
As pointed out, frontal and insular cortices are essential parts of the MCN shared by OCSD, ASD, and SCZD. In particular, fronto‐striatal regions are involved in OCSD, thus supporting the hypothesis that an orbitofrontal‐striatal circuit may be abnormal in OCSD (Cavedini, Riboldi, Keller, D'annucci, & Bellodi, 2002; Nakao, Okada, & Kanba, 2014).
In both ASD and SCZD, disruption of the loop system of the basal ganglia is thought to explain impaired sensorimotor access, which reflects the ability of an organism to filter out irrelevant stimuli. In turn, the neural circuit composed of the caudate nucleus, the orbitofrontal cortex, and the anterior cingulate cortex has been reported to play an important role in OCSD (Chamberlain, Blackwell, Fineberg, Robbins, & Sahakian, 2005). What is more, the impairment of the anterior cingulate cortex is supposed to be relevant for causing awareness deficits in bipolar disorder (Palermo, et al., 2015). Finally, hippocampal disruption has been associated with both ASD and SCZD (Wible, 2012).
From the clinical point of view, alteration of the areas forming the MCN may lead to a disruption of social cognition, which is frequently associated with ASD and SCZD (Bicks, Koike, Akbarian, & Morishita, 2015). Approaches of functional connectivity reveal that specific parameters of connectivity networks present heritability and are associated with familial risk for psychopathology, suggesting a genetic role not only with regard to psychiatric categories tout court but also with regard to the brain inter‐regional synchronization, thus confirming liability to broad dimensions of symptomatically related disorders (Buckholtz & Meyer‐Lindenberg, 2012; Caspi, et al., 2014). What is more, mental illness is generally characterized by polygenic inheritance, which constantly causes genetic liability. This defies the validity of categorical models of psychiatric illness and risk, as it implies that brain disorders can be viewed as the extreme manifestations of normally distributed quantitative traits (Cauda et al 2017).
The neurobiological substrate of these common alterations may involve a neurochemical unbalance, especially an alteration in the ratios of GABA‐glutamate on the one hand, and oxytocin‐vasopressin on the other, which could be the targets of specific pharmacological therapies (Ford, Nibbs, & Crewther, 2017; LeBlanc & Fagiolini, 2011).
If we draw our attention to the altered areas that are not shared by the three spectra, we see that they can account for differences in symptomatology. For instance, the more involvement of posterior areas (sensorimotor and occipito‐temporal) in ASD than in SCZD can lead to a peculiar hypersensitivity for auditory, tactile, and visual stimuli and/or hyposensitivity for pain stimuli. In turn, a more intense fronto‐striatal alteration characterizes OCSD, thus supporting the hypothesis that an orbitofrontal‐striatal circuit may be abnormal in OCSD (Benzina, Mallet, Burguiere, N'diaye, & Pelissolo, 2016; Cauda, et al., 2017; Cavallaro, et al., 2003; Cavedini, et al., 2002).
Several studies have supported a role of neuroinflammation in the etiology of ASD, SCZD and other brain disorders. In fact, an increased inflammatory response in the central nervous system is supposed to activate microglial cells, the activity of which leads to the release of pro‐inflammatory cytokines, including interleukin (IL)‐1β, IL‐6, and tumor necrosis factor‐α. In turn, pro‐inflammatory cytokines aggravate and propagate neuroinflammation, thus degenerating healthy neurons and impairing brain functions. Thus, the activated microglia may contribute to the generation of GM abnormalities and, consequently, to the pathogenesis of psychiatric disorders (Hong, Kim, & Im, 2016).
With regard to SCZD, cortical immune activation and immune‐related markers have been reported in the prefrontal cortex, along with deficits in the basilar dendritic spines of layer 3 pyramidal neurons and disturbances in inhibitory inputs to pyramidal neurons. Importantly, microglia is supposed to regulate excitatory and inhibitory input to pyramidal neurons (Volk, 2017). Furthermore, differently from SCZD, the development of ASD appears to be more related to cerebellar dysfunction and subsequent thalamic hyperactivation in early childhood. In contrast, SCZD seems to be triggered by thalamic hyperactivation in late adolescence, whereas hippocampal aberration can possibly originate in childhood. A possible culprits could be found in the metal homeostasis disturbances, which can induce dysfunction of blood–cerebrospinal fluid barrier. Thalamic hyperactivation is thought to be produced by microglia‐mediated neuroinflammation as well as by abnormalities of the intracerebral environment. Consequently, it is likely that thalamic hyperactivation leads to the dysregulation of the circuit formed by dorsolateral prefrontal cortex and lower brain regions related to social cognition (Nakagawa & Chiba, 2016).
All these considerations provide further support for the hypothesis of a common neurobiological substrate at the basis of diagnostically different psychiatric conditions (Buckholtz & Meyer‐Lindenberg, 2012; Caspi, et al., 2014; Crossley, et al., 2014; McTeague, et al., 2016). Intriguingly, our findings are consistent with similar results obtained by Goodkind et al. (2015), which have highlighted the importance of the anterior insula/dorsal anterior cingulate network as a key circuit that is impaired by different brain disorders. Both our results and those of Goodkind et al. are in line with the theoretical proposals suggesting the need for transdiagnostic accounts of neuropathological diseases (Buckholtz & Meyer‐Lindenberg, 2012; Caspi, et al., 2014; Crossley, et al., 2014; Douaud, et al., 2014; Etkin & Cuthbert, 2014; McTeague, et al., 2016; Voytek & Knight, 2015). The co‐alteration networking analysis developed in this study could play a major role in order to investigate the pathological brain as well as to lead us to a unifying perspective on neuropathology.
4.1. Relationship between the co‐alteration matrix and diffusion matrix
At first sight the relationship between the structural co‐alteration pattern and the propagation pattern of alterations may seem unclear, as the two concepts are likely to be considered unrelated. In fact, the former is straightforwardly associated with a picture of a static event, which is based on anatomical evidence, while the latter is typically conceived as implying a causal event characterized by a precise temporal directionality, which can be investigated by longitudinal studies. The propagation of neuronal abnormalities is a dynamic phenomenon, which has its onset in certain cerebral areas and then proceeds to affect other parts of the brain. Cerebral areas appear therefore to be altered in different temporal sequences, thus originating different propagation patterns. These patterns, however, can be mathematically described by a Laplacian matrix. What we propose to show is that this type of matrix can be also obtained from co‐alteration meta‐analytical data. Strictly speaking, therefore, although this does not allow to speak about propagation, it does allow us to interpret the co‐alteration pattern as having a network‐like architecture.
If we consider two brain areas (populations of neurons): A1 (altered) and A2 (unaltered) that are structurally connected, then we can say that the alteration factor, which propagates to A2, is the product of the concentration of the alteration factor and the strength of the effective connectivity . Consequently, at a certain time the concentration of the alteration in A2 is going to increase by a factor of , where is the diffusion constant that controls the speed of alterations’ spread. This model can be mathematically described in differential terms, such as:
Abdelnour, Voss, and Raj, (2014) have shown that it is possible to construct a network of cerebral nodes by developing this model with the following equation:
where L is the Laplacian matrix, which is defined as:
In this equation D is the degree matrix, which is a diagonal matrix containing information about the number of edges attached to every node, and A is the adjacency matrix, which is a square matrix containing information about whether or not pairs of nodes are adjacent or connected.
It can be shown that this formula is the heat equation, which in turn is a particular case of the diffusion equation, generalized to complex networks (Kondor & Lafferty, 2002). The diffusion equation has the following explicit solution:
| (1) |
This formula defines the evolution of an initial configuration of a diffusion process.
The Laplacian matrix, needed for the solution of the diffusion equation, can be derived from the co‐alteration matrix of meta‐analytical data. In fact the co‐alteration matrix, which can be calculated in several ways (Crossley et al., 2013), represents the relationship between the structural alterations of different nodes of a complex network. In our case we determined the co‐alteration matrix by using the Patel's k, which is an index capable of describing statistically the degree of association between nodes (Patel et al., 2006) see Section 2 for more details). The final result is a square and symmetric matrix, which is tantamount to the adjacency matrix A containing in its rows and columns the nodes of the complex network. This is nothing but a connection matrix, from which it is possible to obtain the degree matrix D and, thereby, derive the Laplacian matrix L, which eventually allows to construct the diffusion dynamics of the complex network.
We can see clearly now that there is a strict mathematical relationship between propagation pattern (causation) and co‐alteration pattern, as it is possible to obtain from co‐alteration data the diffusion matrix of the alterations. In fact, the Laplacian matrix of Equation (1) may allow us to create a temporal description of the alterations’ spread. What we need is just an initial node, in which alterations begin to accumulate (start condition ); from that point of onset we therefore could see how alterations propagate in time and space through the sequence of the progressively altered nodes. Still, the analysis of meta‐analytical data of co‐alterations cannot permit to identify the onset of the alteration process (initial nodes). Therefore, we can say that alterations diffuse from nodes to nodes, but we cannot describe the progression of this diffusion. This is why in the article we refer to the diffusion of alterations in the terms of “distribution” rather than “spread” or “propagation”. However, on the grounds of the mathematical considerations stated above, we can confidently hold that the co‐alteration pattern resulted from our analyses is to be interpreted as a network‐like phenomenon.
4.2. Limitations
The meta‐analysis presented here has been carefully designed. However, some critical aspects should be recognized. The first aspect regards the literature search, in which a good number of potential studies have been identified but others could have been missed. The second aspect regards the heterogeneity of group subjects (different for age and gender) that participated in the studies retrieved for this meta‐analysis, which could be a possible confounding factor.
The third aspect concerns the fact that the sample used to calculate the anatomical covariance obviously differ from the sample used to calculate the MCN; in other words, it must always bear in mind that these analyses are derived from two different populations and, importantly, from different types of data, native ones on the one hand, and meta‐analytical ones on the other. As meta‐analytical data are aggregated for groups of subjects, we are not able to distinguish possible differences due to either the healthy population or the pathological one. However, it is very unlikely that these differences could increase the number of false positives; in contrast, it is much more likely that they could increase the number of false negatives.
The fourth aspect concerns the greater representativeness of SCZD with respect to the other two spectra, which could lead to think that our results are considerably driven by SCZD data. However, the further analyses discussed in the paragraph “Reliability” address this limitation and make it unlikely for the following considerations. (1) Edges co‐alteration values of ASD and OCSD are somewhat similar to those of SCZD. (2) The MCN calculated without SCZD data is very similar to the MCN calculated by using all the data of the three spectra. (3) GM alterations are not specific to a particular spectrum but tend to be subsumed into two clusters within which they distribute rather homogeneously (see Supporting Information, Figure S3). (4) Apart from the inevitable differences due to the reduced sample, the MCN obtained by equalizing the number of experiments for each psychiatric spectrum largely overlaps the MCN obtained by using the whole original dataset (Dice coefficient = .7969; see Figure 15). This result is very encouraging, as the new MCN overlaps for the most part the original MCN, thus making much less likely that the analysis on the whole dataset is largely driven by SCZD data.
Of course we cannot completely exclude that the numerousness of the SCZD sample somehow influenced the MCN pattern but, taken together, all the reliability results lead us to think that most of the MCN actually represents an alteration pattern that is common to the three neuropsychiatric spectra. Finally, it is worth noting that one of the main goals of our work is to demonstrate that it is possible to investigate anatomical co‐alteration patterns related to neuropsychiatric/neurological conditions by using techniques that are based on the conditional probability. Given that the application of this methodology is new, we preferred to test it on as many data as possible; the union of these data was allowed by transdiagnostic considerations as well as by a previous publication of our research group (Cauda et al., 2017), in which we showed that SCZD, ASD and OCSD share a significant set of neuronal alterations. Still, other studies are needed to confirm the hypothesis that different psychiatric conditions share brain alteration patterns with the help of different methodologies and other data whose source is not meta‐analytical.
5. CONCLUSIONS
This meta‐analysis was able to address the following important issues.
In SCZD, ASD, and OCSD GM alterations do not distribute randomly across the brain but rather follow identifiable patterns of co‐alteration.
These patterns exhibit a network‐like architecture, forming an ensemble of co‐altered areas that can be defined as morphometric co‐atrophy network or MCN, along with the structural and functional pathways linking these areas, which in turn can be defined as pathoconnectivity.
Within the MCN it is possible to identify certain cerebral areas, which can be thought of as pathoconnectivity hubs according to their network degree or level of connectivity with every other co‐altered area. The alteration of these hubs is supposed to lead to a faster and more diffuse distribution of neuronal abnormalities across the brain.
Within the MCN it is also possible to identify a “core” subnetwork of co‐altered areas, encompassing insular, prefrontal, thalamic, parahippocampal, superior temporal and precentral regions.
A normal pattern of anatomical covariance is also associated with the MCN areas. In part the MCN and the anatomical covariance pattern overlap, which suggests that brain disorders may influence and alter the development of the anatomical covariance by modifying preexisting structural covariances as well as producing other pathological ones (co‐alterations).
The new methodology of morphometric co‐alteration network analysis developed in this study, as well as the possibility to implement this analysis on huge databases, promises to open exciting prospects for the understanding of the pathological brain. A growing body of research is developing around the hypothesis that the brain is altered on the basis of its neural organization. Therefore, the development of a valuable connectomic approach to the co‐alteration of structural and functional neuronal abnormalities is essential for understanding better the dynamic of brain disorders (Fornito et al., 2015). Also, from the etiological point of view, neuroanatomical alterations may be associated with a pathologically increased neuroinflammatory response. Future investigations into this line of research are promising to shed new light on how the brain responds to illness as well as on how brain diseases can be influenced by the cerebral structural and functional organization. This important knowledge will help the clinical practice to achieve a better predictive diagnostic power and improve medical care and treatment.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information
AKNOWLEDGMENTS
This study was supported by the Fondazione Carlo Molo, Turin, NIH/NIMH grant MH074457 (P Fox, PI) and CDMRP grant W81XWH‐14‐1‐0316 (P Fox, PI).
Cauda F, Nani A, Costa T, et al. The morphometric co‐atrophy networking of schizophrenia, autistic and obsessive spectrum disorders. Hum Brain Mapp. 2018;39: 1898–1928. 10.1002/hbm.23952
Funding information Fondazione Carlo Molo, Turin, NIH/NIMH, Grant Number: MH074457; Congressionally Directed Medical Research Programs, Grant Number: W81XWH‐14‐1‐0316
REFERENCES
- Abdelnour, F. , Voss, H. U. , & Raj, A. (2014). Network diffusion accurately models the relationship between structural and functional brain connectivity networks. NeuroImage, 90, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Hamelin, I. , Dall'asta, L. , Barrat, A. , & Vespignani, A. (2005). K‐Core decomposition: A tool for the visualization of large scale networks. Advances in Neural Information Processing Systems, 18, 41. arXiv:cs/0504107 [cs.NI]. [Google Scholar]
- Andersson, J. L. R. , Jenkinson, M. , & Smith, S. (2007). Non‐linear registration aka Spatial normalisation. Oxford, UK: FMRIB Centre.
- Andersson, M. L. , Kinnison, J. , & Pessoa, L. (2013). Describing functional diversity of brain regions and brain networks. NeuroImage, 73, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel, S. H. (1981). A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease. Annals of Neurology, 10, 499–505. [DOI] [PubMed] [Google Scholar]
- Arnone, D. , Cavanagh, J. , Gerber, D. , Lawrie, S. M. , Ebmeier, K. P. , & McIntosh, A. M. (2009). Magnetic resonance imaging studies in bipolar disorder and schizophrenia: Meta‐analysis. British Journal of Psychiatry, 195, 194–201. mental science, [DOI] [PubMed] [Google Scholar]
- Bader, G. D. , & Hogue, C. W. (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, J. T. , Holmes, A. J. , Masters, G. A. , Yeo, B. T. , Krienen, F. , Buckner, R. L. , & Ongur, D. (2014). Disruption of cortical association networks in schizophrenia and psychotic bipolar disorder. JAMA Psychiatry, 71, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribeau, D. A. , & Anagnostou, E. (2013). A comparison of neuroimaging findings in childhood onset schizophrenia and autism spectrum disorder: A review of the literature. Frontiers in Psychiatry, 4, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzina, N. , Mallet, L. , Burguiere, E. , N'diaye, K. , & Pelissolo, A. (2016). Cognitive dysfunction in obsessive‐compulsive disorder. Current Psychiatry Reports, 18, 80. [DOI] [PubMed] [Google Scholar]
- Bertelli, M. O. , Piva Merli, M. , Bradley, E. , Keller, R. , Varrucciu, N. , Del Furia, C. , & Panocchia, N. (2015). The diagnostic boundary between autism spectrum disorder, intellectual developmental disorder and schizophrenia spectrum disorders. Advances in Mental Health and Intellectual Disabilities, 9, 243–264. [Google Scholar]
- Biamino, E. , Di Gregorio, E. , Belligni, E. F. , Keller, R. , Riberi, E. , Gandione, M. , … Brusco, A. (2016). A novel 3q29 deletion associated with autism, intellectual disability, psychiatric disorders, and obesity. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 171, 290–299. [DOI] [PubMed] [Google Scholar]
- Bicks, L. K. , Koike, H. , Akbarian, S. , & Morishita, H. (2015). Prefrontal cortex and social cognition in mouse and man. Frontiers in Psychology, 6, 1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleich‐Cohen, M. , Hendler, T. , Weizman, R. , Faragian, S. , Weizman, A. , & Poyurovsky, M. (2014). Working memory dysfunction in schizophrenia patients with obsessive‐compulsive symptoms: An fMRI study. European Psychiatry: The Journal of the Association of European Psychiatrists, 29, 160–166. [DOI] [PubMed] [Google Scholar]
- Bolte, S. , Rudolf, L. , & Poustka, F. (2002). The cognitive structure of higher functioning autism and schizophrenia: A comparative study. Comprehensive Psychiatry, 43, 325–330. [DOI] [PubMed] [Google Scholar]
- Buckholtz, J. W. , & Meyer‐Lindenberg, A. (2012). Psychopathology and the human connectome: Toward a transdiagnostic model of risk for mental illness. Neuron, 74, 990–1004. [DOI] [PubMed] [Google Scholar]
- Buckley, P. F. , Miller, B. J. , Lehrer, D. S. , & Castle, D. J. (2009). Psychiatric comorbidities and schizophrenia. Schizophrenia Bulletin, 35, 383–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi, A. , Houts, R. M. , Belsky, D. W. , Goldman‐Mellor, S. J. , Harrington, H. , Israel, S. , … Moffitt, T. E. (2014). The p factor: One general psychopathology factor in the structure of psychiatric disorders? Clinical Psychological Science: A Journal of the Association for Psychological Science, 2, 119–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo, L. , & Rizzolatti, G. (2009). The mirror neuron system. Archives of Neurology, 66, 557–560. [DOI] [PubMed] [Google Scholar]
- Cauda, F. , Costa, T. , Nani, A. , Fava, L. , Palermo, S. , Bianco, F. , … Keller, R. (2017). Are schizophrenia, autistic, and obsessive spectrum disorders dissociable on the basis of neuroimaging morphological findings?: A voxel‐based meta‐analysis. Autism Research. [DOI] [PubMed] [Google Scholar]
- Cauda, F. , Costa, T. , Palermo, S. , D'agata, F. , Diano, M. , Bianco, F. , … Keller, R. (2014a). Concordance of white matter and gray matter abnormalities in autism spectrum disorders: A voxel‐based meta‐analysis study. Human Brain Mapping, 35, 2073–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauda, F. , Costa, T. , Torta, D. M. , Sacco, K. , D'agata, F. , Duca, S. , … Vercelli, A. (2012a). Meta‐analytic clustering of the insular cortex: Characterizing the meta‐analytic connectivity of the insula when involved in active tasks. NeuroImage, 62, 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauda, F. , D'agata, F. , Sacco, K. , Duca, S. , Geminiani, G. , & Vercelli, A. (2011a). Functional connectivity of the insula in the resting brain. NeuroImage, 55, 8–23. [DOI] [PubMed] [Google Scholar]
- Cauda, F. , Geda, E. , Sacco, K. , D'agata, F. , Duca, S. , Geminiani, G. , & Keller, R. (2011b). Grey matter abnormality in autism spectrum disorder: An activation likelihood estimation meta‐analysis study. Journal of Neurology, Neurosurgery, and Psychiatry, 82, 1304–1313. [DOI] [PubMed] [Google Scholar]
- Cauda, F. , Geminiani, G. C. , & Vercelli, A. (2014b). Evolutionary appearance of von Economo's neurons in the mammalian cerebral cortex. Frontiers in Human Neuroscience, 8, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauda, F. , Torta, D. M. , Sacco, K. , D'agata, F. , Geda, E. , Duca, S. , … Vercelli, A. (2013). Functional anatomy of cortical areas characterized by Von Economo neurons. Brain Structure and Function, 218, 1–20. [DOI] [PubMed] [Google Scholar]
- Cauda, F. , Torta, D. M. , Sacco, K. , Geda, E. , D'agata, F. , Costa, T. , … Amanzio, M. (2012b). Shared “core” areas between the pain and other task‐related networks. PloS One, 7, e41929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro, R. , Cavedini, P. , Mistretta, P. , Bassi, T. , Angelone, S. M. , Ubbiali, A. , & Bellodi, L. (2003). Basal‐corticofrontal circuits in schizophrenia and obsessive‐compulsive disorder: A controlled, double dissociation study. Biological Psychiatry, 54, 437–443. [DOI] [PubMed] [Google Scholar]
- Cavedini, P. , Riboldi, G. , Keller, R. , D'annucci, A. , & Bellodi, L. (2002). Frontal lobe dysfunction in pathological gambling patients. Biological Psychiatry, 51, 334–341. [DOI] [PubMed] [Google Scholar]
- Chamberlain, S. R. , Blackwell, A. D. , Fineberg, N. A. , Robbins, T. W. , & Sahakian, B. J. (2005). The neuropsychology of obsessive compulsive disorder: The importance of failures in cognitive and behavioural inhibition as candidate endophenotypic markers. Neuroscience and Biobehavioral Reviews, 29, 399–419. [DOI] [PubMed] [Google Scholar]
- Cheon, K. A. , Kim, Y. S. , Oh, S. H. , Park, S. Y. , Yoon, H. W. , Herrington, J. , … Schultz, R. T. (2011). Involvement of the anterior thalamic radiation in boys with high functioning autism spectrum disorders: A diffusion tensor imaging study. Brain Research, 1417, 77–86. [DOI] [PubMed] [Google Scholar]
- Cheung, C. , Yu, K. , Fung, G. , Leung, M. , Wong, C. , Li, Q. , … McAlonan, G. (2010). Autistic disorders and schizophrenia: Related or remote? An anatomical likelihood estimation. PloS One, 5, e12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier‐Larsen, E. , & Holzbaur, E. L. (2006). Axonal transport and neurodegenerative disease. Biochimica Et Biophysica Acta, 1762, 1094–1108. [DOI] [PubMed] [Google Scholar]
- Chisholm, K. , Lin, A. , Abu‐Akel, A. , & Wood, S. J. (2015). The association between autism and schizophrenia spectrum disorders: A review of eight alternate models of co‐occurrence. Neuroscience and Biobehavioral Reviews, 55, 173–183. [DOI] [PubMed] [Google Scholar]
- Cioli, C. , Abdi, H. , Beaton, D. , Burnod, Y. , & Mesmoudi, S. (2014). Differences in human cortical gene expression match the temporal properties of large‐scale functional networks. PloS One, 9, e115913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz, B. A. , Sweeney, J. A. , Hamm, J. P. , Ivleva, E. I. , Ethridge, L. E. , Pearlson, G. D. , … Tamminga, C. A. (2016). Identification of distinct psychosis biotypes using brain‐based biomarkers. American Journal of Psychiatry, 173, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran, D. M. , Fallon, D. , Hill, M. , & Frazier, J. A. (2013). The role of oxytocin in psychiatric disorders: A review of biological and therapeutic research findings. Harvard Review of Psychiatry, 21, 219–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, M. W. , Repovs, G. , & Anticevic, A. (2014). The frontoparietal control system: A central role in mental health. The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 20, 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, M. W. , & Schneider, W. (2007). The cognitive control network: Integrated cortical regions with dissociable functions. NeuroImage, 37, 343–360. [DOI] [PubMed] [Google Scholar]
- Crossley, N. A. , Mechelli, A. , Ginestet, C. , Rubinov, M. , Bullmore, E. T. , & McGuire, P. (2016). Altered hub functioning and compensatory activations in the connectome: A meta‐analysis of functional neuroimaging studies in schizophrenia. Schizophrenia Bulletin, 42, 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley, N. A. , Mechelli, A. , Scott, J. , Carletti, F. , Fox, P. T. , McGuire, P. , & Bullmore, E. T. (2014). The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain, 137, 2382–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley, N. A. , Mechelli, A. , Vertes, P. E. , Winton‐Brown, T. T. , Patel, A. X. , Ginestet, C. E. , … Bullmore, E. T. (2013). Cognitive relevance of the community structure of the human brain functional coactivation network. Proceedings of the National Academy of Sciences of the United States of America, 110, 11583–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley, N. A. , Scott, J. , Ellison‐Wright, I. , & Mechelli, A. (2015). Neuroimaging distinction between neurological and psychiatric disorders. British Journal of Psychiatry, 207, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lacy, N. , & King, B. H. (2013). Revisiting the relationship between autism and schizophrenia: Toward an integrated neurobiology. Annual Review of Clinical Psychology, 9, 555–587. [DOI] [PubMed] [Google Scholar]
- DeVylder, J. E. , Burnette, D. , & Yang, L. H. (2014). Co‐occurrence of psychotic experiences and common mental health conditions across four racially and ethnically diverse population samples. Psychological Medicine, 44, 3503–3513. [DOI] [PubMed] [Google Scholar]
- Di Gregorio, E. , Riberi, E. , Belligni, E. F. , Biamino, E. , Spielmann, M. , Ala, U. , … Brusco, A. (2017). Copy number variants analysis in a cohort of isolated and syndromic developmental delay/intellectual disability reveals novel genomic disorders, position effects and candidate disease genes. Clinical Genetics, 92, 415–422. [DOI] [PubMed] [Google Scholar]
- Douaud, G. , Groves, A. R. , Tamnes, C. K. , Westlye, L. T. , Duff, E. P. , Engvig, A. , … Johansen‐Berg, H. (2014). A common brain network links development, aging, and vulnerability to disease. Proceedings of the National Academy of Sciences of Sciences, 111, 17648–17653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douaud, G. , Smith, S. , Jenkinson, M. , Behrens, T. , Johansen‐Berg, H. , Vickers, J. , … James, A. (2007). Anatomically related grey and white matter abnormalities in adolescent‐onset schizophrenia. Brain, 130, 2375–2386. a journal of neurology, [DOI] [PubMed] [Google Scholar]
- Eickhoff, S. B. , Bzdok, D. , Laird, A. R. , Kurth, F. , & Fox, P. T. (2012). Activation likelihood estimation meta‐analysis revisited. NeuroImage, 59, 2349–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff, S. B. , Laird, A. R. , Fox, P. M. , Lancaster, J. L. , & Fox, P. T. (2017). Implementation errors in the GingerALE Software: Description and recommendations. Human Brain Mapping, 38, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff, S. B. , Laird, A. R. , Grefkes, C. , Wang, L. E. , Zilles, K. , & Fox, P. T. (2009). Coordinate‐based activation likelihood estimation meta‐analysis of neuroimaging data: A random‐effects approach based on empirical estimates of spatial uncertainty. Human Brain Mapping, 30, 2907–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff, S. B. , Nichols, T. E. , Laird, A. R. , Hoffstaedter, F. , Amunts, K. , Fox, P. T. , … Eickhoff, C. R. (2016). Behavior, sensitivity, and power of activation likelihood estimation characterized by massive empirical simulation. NeuroImage, 137, 70–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison‐Wright, I. , & Bullmore, E. (2010). Anatomy of bipolar disorder and schizophrenia: A meta‐analysis. Schizophrenia Research, 117, 1–12. [DOI] [PubMed] [Google Scholar]
- Etkin, A. , & Cuthbert, B. (2014). Beyond the DSM: Development of a transdiagnostic psychiatric neuroscience course. Academic Psychiatry, 38, 145–150. [DOI] [PubMed] [Google Scholar]
- Etkin, A. , & Wager, T. D. (2007). Functional neuroimaging of anxiety: A meta‐analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. American Journal of Psychiatry, 164, 1476–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, A. C. (2013). Networks of anatomical covariance. NeuroImage, 80, 489–504. [DOI] [PubMed] [Google Scholar]
- Ford, T. C. , Nibbs, R. , & Crewther, D. P. (2017). Glutamate/GABA+ ratio is associated with the psychosocial domain of autistic and schizotypal traits. PloS One, 12, e0181961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito, A. , Zalesky, A. , & Breakspear, M. (2015). The connectomics of brain disorders. Nature Reviews. Neuroscience, 16, 159–172. [DOI] [PubMed] [Google Scholar]
- Fox, P. T. , & Lancaster, J. L. (2002). Opinion: Mapping context and content: The BrainMap model. Nature Reviews. Neuroscience, 3, 319–321. [DOI] [PubMed] [Google Scholar]
- Freedman, R. , Lewis, D. A. , Michels, R. , Pine, D. S. , Schultz, S. K. , Tamminga, C. A. , … Yager, J. (2013). The initial field trials of DSM‐5: New blooms and old thorns. The American Journal of Psychiatry, 170, 1–5. [DOI] [PubMed] [Google Scholar]
- French, L. , Tan, P. P. , & Pavlidis, P. (2011). Large‐scale analysis of gene expression and connectivity in the rodent brain: Insights through data integration. Frontiers in Neuroinformatics, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan, M. , & Newman, M. E. (2002). Community structure in social and biological networks. Proceedings of the National Academy of Sciences of the United States of, 99, 7821–7826. America, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glerean, E. , Pan, R. K. , Salmi, J. , Kujala, R. , Lahnakoski, J. M. , Roine, U. , … Jaaskelainen, I. P. (2016). Reorganization of functionally connected brain subnetworks in high‐functioning autism. Human Brain Mapping, 37, 1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert, M. , Clavaguera, F. , & Tolnay, M. (2010). The propagation of prion‐like protein inclusions in neurodegenerative diseases. Trends in Neurosciences, 33, 317–325. [DOI] [PubMed] [Google Scholar]
- Good, C. D. , Johnsrude, I. S. , Ashburner, J. , Henson, R. N. , Friston, K. J. , & Frackowiak, R. S. (2001). A voxel‐based morphometric study of ageing in 465 normal adult human brains. NeuroImage, 14, 21–36. [DOI] [PubMed] [Google Scholar]
- Goodkind, M. , Eickhoff, S. B. , Oathes, D. J. , Jiang, Y. , Chang, A. , Jones‐Hagata, L. B. , … Etkin, A. (2015). Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry, 72, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorun, A. , Cieslak, K. , Harkavy‐Friedman, J. , Deptula, A. , Goetz, D. , Goetz, R. , & Malaspina, D. (2015). Frequent Comorbidity and Predictors of Social Anxiety in Persons With Schizophrenia: A Retrospective Cohort Study. The primary care companion for CNS disorders, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, S. , Higgins, J. P. T. , Alderson, P. , Clarke, M. , Mulrow, C. D. , & Oxman, A. D. (2008). Introduction In: Higgins J.T.P., Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions: The Cochrane Collaboration. Chichester, West Sussex; Hoboken NJ: John Wiley & Sons, Ltd. [Google Scholar]
- Guest, W. C. , Silverman, J. M. , Pokrishevsky, E. , O'neill, M. A. , Grad, L. I. , & Cashman, N. R. (2011). Generalization of the prion hypothesis to other neurodegenerative diseases: An imperfect fit. Journal of Toxicology and Environmental Health. Part A, 74, 1433–1459. [DOI] [PubMed] [Google Scholar]
- Hamilton, J. P. , Etkin, A. , Furman, D. J. , Lemus, M. G. , Johnson, R. F. , & Gotlib, I. H. (2012). Functional neuroimaging of major depressive disorder: A meta‐analysis and new integration of base line activation and neural response data. American Journal of Psychiatry, 169, 693–703. [DOI] [PubMed] [Google Scholar]
- Hommer, R. E. , & Swedo, S. E. (2015). Schizophrenia and autism‐related disorders. Schizophrenia Bulletin, 41, 313–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, H. , Kim, B. S. , & Im, H. I. (2016). Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders. International Neurourology Journal, 20, S2–S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humble, M. B. , Uvnas‐Moberg, K. , Engstrom, I. , & Bejerot, S. (2013). Plasma oxytocin changes and anti‐obsessive response during serotonin reuptake inhibitor treatment: A placebo controlled study. BMC Psychiatry, 13, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel, T. R. (2010). Rethinking schizophrenia. Nature, 468, 187–193. [DOI] [PubMed] [Google Scholar]
- Insel, T. R. (2014). The NIMH research domain criteria (RDoC) project: Precision medicine for psychiatry. The American Journal of Psychiatry, 171, 395–397. [DOI] [PubMed] [Google Scholar]
- Iturria‐Medina, Y. , & Evans, A. C. (2015). On the central role of brain connectivity in neurodegenerative disease progression. Frontiers in Aging Neuroscience, 7, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturria‐Medina, Y. , Sotero, R. C. , Toussaint, P. J. , & Evans, A. C. (2014). Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Computational Biology, 10, e1003956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou, R. J. , Jackowski, A. P. , Papademetris, X. , Rajeevan, N. , Staib, L. H. , & Volkmar, F. R. (2011a). Diffusion tensor imaging in autism spectrum disorders: Preliminary evidence of abnormal neural connectivity. The Australian and New Zealand Journal of Psychiatry, 45, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou, R. J. , Mateljevic, N. , Kaiser, M. D. , Sugrue, D. R. , Volkmar, F. R. , & Pelphrey, K. A. (2011b). Structural neural phenotype of autism: Preliminary evidence from a diffusion tensor imaging study using tract‐based spatial statistics. AJNR. American Journal of Neuroradiology, 32, 1607–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, R. , Bugiani, S. , Fantin, P. , & Pirfo, E. (2011). Mirror neurons and autism. Giorn Ital Psicopatol, 17, 404–412. [Google Scholar]
- Keller, R. , Piedimonte, A. , Bianco, F. , Bari, S. , & Cauda, F. (2016). Diagnostic characteristics of psychosis and autism spectrum disorder in adolescence and adulthood. A Case Series. Autism Open Access, 6, [Google Scholar]
- Kessler, R. C. , McGonagle, K. A. , Zhao, S. , Nelson, C. B. , Hughes, M. , Eshleman, S. , … Kendler, K. S. (1994). Lifetime and 12‐month prevalence of DSM‐III‐R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Archives of General Psychiatry, 51, 8–19. [DOI] [PubMed] [Google Scholar]
- Kilner, J. M. , Friston, K. J. , & Frith, C. D. (2007). The mirror‐neuron system: A Bayesian perspective. Neuroreport, 18, 619–623. [DOI] [PubMed] [Google Scholar]
- King, B. H. , & Lord, C. (2011). Is schizophrenia on the autism spectrum? Brain Research, 1380, 34–41. [DOI] [PubMed] [Google Scholar]
- Klein, T. A. , Ullsperger, M. , & Danielmeier, C. (2013). Error awareness and the insula: Links to neurological and psychiatric diseases. Frontiers in Human Neuroscience, 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondor, R. I. , & Lafferty, J. (2002). Diffusion kernels on graphs and other discrete input spaces. 19th International Conf. on Machine Learning.
- Kotz, S. , Balakrishnan, N. , & Johnson, N. L. (2000). Continuous Multivariate Distributions. Vol. 1, Models and Applications, 2nd Edition, New York: Wiley. [Google Scholar]
- Laird, A. R. , Eickhoff, S. B. , Kurth, F. , Fox, P. M. , Uecker, A. M. , Turner, J. A. , … Fox, P. T. (2009). ALE meta‐analysis workflows via the brainmap database: Progress towards a probabilistic functional brain atlas. Frontiers in Neuroinformatics, 3, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird, A. R. , Fox, P. M. , Price, C. J. , Glahn, D. C. , Uecker, A. M. , Lancaster, J. L. , … Fox, P. T. (2005). ALE meta‐analysis: Controlling the false discovery rate and performing statistical contrasts. Human Brain Mapping, 25, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster, J. L. , Tordesillas‐Gutierrez, D. , Martinez, M. , Salinas, F. , Evans, A. , Zilles, K. , … Fox, P. T. (2007). Bias between MNI and Talairach coordinates analyzed using the ICBM‐152 brain template. Human Brain Mapping, 28, 1194–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen, M. , Durston, S. , Kas, M. J. , van Engeland, H. , Staal, W. G. (2011a). The neurobiology of repetitive behavior: …and men. Neuroscience and Biobehavioral Reviews, 35, 356–365. [DOI] [PubMed] [Google Scholar]
- Langen, M. , Kas, M. J. , Staal, W. G. , van Engeland, H. , & Durston, S. (2011b). The neurobiology of repetitive behavior of mice. Neuroscience and Biobehavioral Reviews, 35, 345–355. [DOI] [PubMed] [Google Scholar]
- LeBlanc, J. J. , & Fagiolini, M. (2011). Autism: A “critical period” disorder? Neural Plasticity, 2011, 921680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati, A. , Altman, D. G. , Tetzlaff, J. , Mulrow, C. , Gotzsche, P. C. , Ioannidis, J. P. , … Moher, D. (2009). The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: Explanation and elaboration. Plos Medicine, 62, e1–34. [DOI] [PubMed] [Google Scholar]
- Lichtman, J. W. , & Sanes, J. R. (2008). Ome sweet ome: What can the genome tell us about the connectome? Current Opinion in Neurobiology, 18, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionel, A. C. , Tammimies, K. , Vaags, A. K. , Rosenfeld, J. A. , Ahn, J. W. , … Scherer, S. W. (2014). Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Human Molecular Genetics, 23, 2752–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano, C. , Keller, R. , Politi, P. , Aguglia, E. , Magnano, F. , … Berardi, D. (2014). Misdiagnosis of high function autism spectrum disorders in adults: An Italian case series. Autism: The International Journal of Research and Practice, 4, 131. [Google Scholar]
- Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Research, 27, 209–220. [PubMed] [Google Scholar]
- Markon, K. E. (2010). Modeling psychopathology structure: A symptom‐level analysis of Axis I and II disorders. Psychological Medicine, 40, 273–288. [DOI] [PubMed] [Google Scholar]
- Marquand, A. F. , Rezek, I. , Buitelaar, J. , & Beckmann, C. F. (2016). Understanding heterogeneity in clinical cohorts using normative models: Beyond case‐control studies. Biological Psychiatry, 80, 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTeague, L. M. , Goodkind, M. S. , & Etkin, A. (2016). Transdiagnostic impairment of cognitive control in mental illness. Journal of Psychiatric Research, 83, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon, V. (2013). Developmental pathways to functional brain networks: Emerging principles. Trends in Cognitive Sciences, 17, 627–640. [DOI] [PubMed] [Google Scholar]
- Moher, D. , Liberati, A. , Tetzlaff, J. , & Altman, D. G. (2009). Preferred reporting items for systematic reviews and meta‐analyses: The PRISMA statement. Journal of Clinical Epidemiology, 62, 1006–1012. [DOI] [PubMed] [Google Scholar]
- Mullins, C. , Fishell, G. , & Tsien, R. W. (2016). Unifying views of autism spectrum disorders: A consideration of autoregulatory feedback loops. Neuron, 89, 1131–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, Y. , & Chiba, K. (2016). Involvement of neuroinflammation during brain development in social cognitive deficits in autism spectrum disorder and schizophrenia. The Journal of Pharmacology and Experimental Therapeutics, 358, 504–515. [DOI] [PubMed] [Google Scholar]
- Nakao, T. , Okada, K. , & Kanba, S. (2014). Neurobiological model of obsessive‐compulsive disorder: Evidence from recent neuropsychological and neuroimaging findings. Psychiatry and Clinical Neurosciences, 68, 587–605. [DOI] [PubMed] [Google Scholar]
- Nestadt, G. , Grados, M. , & Samuels, J. F. (2010). Genetics of obsessive‐compulsive disorder. The Psychiatric Clinics of North America, 33, 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niendam, T. A. , Laird, A. R. , Ray, K. L. , Dean, Y. M. , Glahn, D. C. , & Carter, C. S. (2012). Meta‐analytic evidence for a superordinate cognitive control network subserving diverse executive functions. Cognitive, Affective, & Behavioral Neuroscience, 12, 241–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander, L. , Lugnegård, T. , & Unenge Hallerbäck, M. (2008). Autism spectrum disorders and schizophrenia spectrum disorders in adults – Is there a connection? A literature review and some suggestions for future clinical research. Clinical Neuropsychiatry, 5, 43–54. [Google Scholar]
- Ornitz, E. M. (1969). Disorders of perception common to early infantile autism and schizophrenia. Comprehensive Psychiatry, 10, 259–274. [DOI] [PubMed] [Google Scholar]
- Owashi, T. , Ota, A. , Otsubo, T. , Susa, Y. , & Kamijima, K. (2010). Obsessive‐compulsive disorder and obsessive‐compulsive symptoms in Japanese inpatients with chronic schizophrenia – A possible schizophrenic subtype. Psychiatry Research, 179, 241–246. [DOI] [PubMed] [Google Scholar]
- Palaniyappan, L. , & Liddle, P. F. (2012). Does the salience network play a cardinal role in psychosis? An emerging hypothesis of insular dysfunction. Journal of Psychiatry & Neuroscience, 37, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo, S. , Cauda, F. , Costa, T. , Duca, S. , Gallino, G. , Geminiani, G. , … Amanzio, M. (2015). Unawareness of bipolar disorder: The role of the cingulate cortex. Neurocase, 21, 438–447. [DOI] [PubMed] [Google Scholar]
- Patel, R. S. , Bowman, F. D. , & Rilling, J. K. (2006). A Bayesian approach to determining connectivity of the human brain. Human Brain Mapping, 27, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania, M. , Davenport, E. C. , Muir, J. , Sheehan, D. F. , Lopez‐Domenech, G. , & Kittler, J. T. (2014). The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Translational Psychiatry, 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls, D. L. (2010). The genetics of obsessive‐compulsive disorder: A review. Dialogues in Clinical Neuroscience, 12, 149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poldrack, R. A. , & Farah, M. J. (2015). Progress and challenges in probing the human brain. Nature, 526, 371–379. [DOI] [PubMed] [Google Scholar]
- Power, J. D. , Cohen, A. L. , Nelson, S. M. , Wig, G. S. , Barnes, K. A. , Church, J. A. , … Petersen, S. E. (2011). Functional network organization of the human brain. Neuron, 72, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto, C. , Risueno, A. , Fontanillo, C. , & De las Rivas, J. (2008). Human gene coexpression landscape: Confident network derived from tissue transcriptomic profiles. PloS One, 3, e3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radua, J. , & Mataix‐Cols, D. (2009). Voxel‐wise meta‐analysis of grey matter changes in obsessive‐compulsive disorder. British Journal of Psychiatry, 195, 393–402. [DOI] [PubMed] [Google Scholar]
- Radua, J. , Via, E. , Catani, M. , & Mataix‐Cols, D. (2011). Voxel‐based meta‐analysis of regional white‐matter volume differences in autism spectrum disorder versus healthy controls. Psychological Medicine, 41, 1539–1550. [DOI] [PubMed] [Google Scholar]
- Raj, A. , Kuceyeski, A. , & Weiner, M. (2012). A network diffusion model of disease progression in dementia. Neuron, 73, 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport, J. , Chavez, A. , Greenstein, D. , Addington, A. , & Gogtay, N. (2009). Autism spectrum disorders and childhood‐onset schizophrenia: Clinical and biological contributions to a relation revisited. Journal of the American Academy of Child and Adolescent Psychiatry, 48, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolatti, G. , & Craighero, L. (2004). The mirror‐neuron system. Annual Review of Neuroscience, 27, 169–192. [DOI] [PubMed] [Google Scholar]
- Romano, A. , Tempesta, B. , Micioni Di Bonaventura, M. V. , & Gaetani, S. (2015). From autism to eating disorders and more: The role of oxytocin in neuropsychiatric disorders. Frontiers in Neuroscience, 9, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi, A. , Delcroix, J. D. , Belichenko, P. V. , Zhan, K. , Wu, C. , Valletta, J. S. , … Mobley, W. C. (2006). Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron, 51, 29–42. [DOI] [PubMed] [Google Scholar]
- Saxena, S. , & Caroni, P. (2011). Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron, 71, 35–48. [DOI] [PubMed] [Google Scholar]
- Seeley, W. W. , Crawford, R. K. , Zhou, J. , Miller, B. L. , & Greicius, M. D. (2009). Neurodegenerative diseases target large‐scale human brain networks. Neuron, 62, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar, A. , Bialas, A. R. , de Rivera, H. , Davis, A. , Hammond, T. R. , Kamitaki, N. , … McCarroll, S. A. (2016). Schizophrenia risk from complex variation of complement component 4. Nature, 530, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, N. Y. , Park, H. Y. , Jung, W. H. , Park, J. W. , Yun, J. Y. , Jang, J. H. , … Kwon, J. S. (2015). Effects of oxytocin on neural response to facial expressions in patients with schizophrenia. Neuropsychopharmacology: Official publication of the American College of Neuropsychopharmacology. [DOI] [PMC free article] [PubMed]
- Smith, S. M. , Jenkinson, M. , Woolrich, M. W. , Beckmann, C. F. , Behrens, T. E. , Johansen‐Berg, H. , … Matthews, P. M. (2004). Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage, 23(Suppl 1), S208–S219. [DOI] [PubMed] [Google Scholar]
- Smith, S. M. , Miller, K. L. , Salimi‐Khorshidi, G. , Webster, M. , Beckmann, C. F. , Nichols, T. E. , … Woolrich, M. W. (2011). Network modelling methods for FMRI. NeuroImage, 54, 875–891. [DOI] [PubMed] [Google Scholar]
- Spencer, M. D. , Moorhead, T. W. , Lymer, G. K. , Job, D. E. , Muir, W. J. , Hoare, P. , … Johnstone, E. C. (2006). Structural correlates of intellectual impairment and autistic features in adolescents. NeuroImage, 33, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Sporn, A. L. , Addington, A. M. , Gogtay, N. , Ordonez, A. E. , Gornick, M. , Clasen, L. , … Rapoport, J. L. (2004). Pervasive developmental disorder and childhood‐onset schizophrenia: Comorbid disorder or a phenotypic variant of a very early onset illness? Biological Psychiatry, 55, 989–994. [DOI] [PubMed] [Google Scholar]
- Sprooten, E. , Rasgon, A. , Goodman, M. , Carlin, A. , Leibu, E. , Lee, W. H. , & Frangou, S. (2017). Addressing reverse inference in psychiatric neuroimaging: Meta‐analyses of task‐related brain activation in common mental disorders. Human Brain Mapping, 38, 1846–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling, J. , & Dossetor, D. (2009). Pervasive developmental disorders and psychosis. Current Psychiatry Reports, 11, 190–196. [DOI] [PubMed] [Google Scholar]
- Stone, W. S. , & Iguchi, L. (2011). Do apparent overlaps between schizophrenia and autistic spectrum disorders reflect superficial similarities or etiological commonalities? North American Journal of Medicine & Science, 4, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss, G. P. , Keller, W. R. , Koenig, J. I. , Gold, J. M. , Frost, K. H. , & Buchanan, R. W. (2015). Plasma oxytocin levels predict social cue recognition in individuals with schizophrenia. Schizophrenia Research, 162, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cross‐Disorder Group of the Psychiatric Genomics Consortium . (2013). Identification of risk loci with shared effects on five major psychiatric disorders: A genome‐wide analysis. Lancet, 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Network and Pathway Analysis Subgroup of the Psychiatric Genomics Consortium . (2015). Psychiatric genome‐wide association study analyses implicate neuronal, immune and histone pathways. Nature Neuroscience, 18, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukel, R. , Alkas, E. , Gurvit, H. , Aslantas Ertekin, B. , Ertekin, E. , Baran, B. , … Saruhan Direskeneli, G. (2016). Serotonin transporter promoter polymorphism is associated with executive function impairments in patients with obsessive compulsive disorder. The Clinical Neuropsychologist, 30, 536–546. [DOI] [PubMed] [Google Scholar]
- Turkeltaub, P. E. , Eickhoff, S. B. , Laird, A. R. , Fox, M. , Wiener, M. , & Fox, P. (2012). Minimizing within‐experiment and within‐group effects in activation likelihood estimation meta‐analyses. Human Brain Mapping, 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan, U. , Patrick, C. J. , & Iacono, W. G. (2012). Examining the overlap between bipolar disorder, nonaffective psychosis, and common mental disorders using latent class analysis. Psychopathology, 45, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercelli, U. , Diano, M. , Costa, T. , Nani, A. , Duca, S. , Geminiani, G. , … Cauda, F. (2016). Node detection using high‐dimensional fuzzy parcellation applied to the insular cortex. Neural Plasticity, 2016, 1938292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk, D. W. (2017). Role of microglia disturbances and immune‐related marker abnormalities in cortical circuitry dysfunction in schizophrenia. Neurobiology of Disease, 99, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voytek, B. , & Knight, R. T. (2015). Dynamic network communication as a unifying neural basis for cognition, development, aging, and disease. Biological Psychiatry, 77, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. J. , & Krystal, J. H. (2014). Computational psychiatry. Neuron, 84, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , David, O. , Hu, X. , & Deshpande, G. (2017). Can Patel's tau accurately estimate directionality of connections in brain networks from fMRI? Magnetic Resonance in Medicine. [DOI] [PubMed] [Google Scholar]
- Wible, C. G. (2012). Hippocampal temporal‐parietal junction interaction in the production of psychotic symptoms: A framework for understanding the schizophrenic syndrome. Frontiers in Human Neuroscience, 6, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, L. , Goldberg, C. , Manor, N. , Sharan, R. , & Ruppin, E. (2011). Gene expression in the rodent brain is associated with its regional connectivity. PLoS Computational Biology, 7, e1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie, K. P. , & Tregellas, J. R. (2010). The role of the insula in schizophrenia. Schizophrenia Research, 123, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates, D. (2012). Neurodegenerative networking. Nature Reviews. Neuroscience, 13, 288. [DOI] [PubMed] [Google Scholar]
- Zalesky, A. , Fornito, A. , Harding, I. H. , Cocchi, L. , Yucel, M. , Pantelis, C. , & Bullmore, E. T. (2010). Whole‐brain anatomical networks: Does the choice of nodes matter?. NeuroImage, 50, 970–983. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Gennatas, E. D. , Kramer, J. H. , Miller, B. L. , & Seeley, W. W. (2012). Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron, 73, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information