

Abstract
Identifying molecular residual disease (MRD) after treatment of localized lung cancer could facilitate early intervention and personalization of adjuvant therapies. Here, we apply cancer personalized profi ling by deep sequencing (CAPP-seq) circulating tumor DNA (ctDNA) analysis to 255 samples from 40 patients treated with curative intent for stage I–III lung cancer and 54 healthy adults. In 94% of evaluable patients experiencing recurrence, ctDNA was detectable in the fi rst posttreatment blood sample, indicating reliable identifi cation of MRD. Posttreatment ctDNA detection preceded radiographic progression in 72% of patients by a median of 5.2 months, and 53% of patients harbored ctDNA mutation profi les associated with favorable responses to tyrosine kinase inhibitors or immune checkpoint blockade. Collectively, these results indicate that ctDNA MRD in patients with lung cancer can be accurately detected using CAPP-seq and may allow personalized adjuvant treatment while disease burden is lowest.
Introduction
Lung cancer is the leading cause of cancer and cancer-related mortality worldwide (1). In patients with nonmetastatic lung cancers, a subset can be cured after primary surgical resection, radiotherapy, and/or combined treatment approaches, including chemotherapy (1,2). Following curative-intent first-line therapies, current routine clinical surveillance involves serial radiographic imaging (1,2). However, such surveillance can detect only macroscopic disease recurrence and is frequently inconclusive due to posttreatment normal tissue changes (3,4). Unfortunately, outcomes are especially poor after clinical disease progression (5). Therefore, a sensitive and specific biomarker that detects molecular residual disease (MRD) before macroscopic recurrence and potentially enables initiation of adjuvant treatment while disease burden is minimal is a major unmet need.
Liquid biopsy approaches represent a promising strategy for disease surveillance in solid tumors (6). Circulating tumor DNA (ctDNA) has been shown to identify MRD shortly after completion of local therapy in patients with nonmetastatic breast and colon cancers using assays that require personalization (7,8). These studies demonstrated the ability of ctDNA to predict disease recurrence with high specificity using assays that primarily tracked a single mutation in each patient. However, ctDNA was not detected in ≥50% of patients who ultimately recurred (7,8), suggesting that increased sensitivity for ctDNA detection may be beneficial. We previously reported development of cancer personalized profiling by deep sequencing (CAPP-seq), a next-generation sequencing-based method that tracks multiple mutations per patient, can achieve lower limits of detection ∼0.002%, and does not require the creation of personalized assays (9,10). In this study, we set out to determine whether CAPP-seq ctDNA analysis can reliably identify MRD in patients with localized lung cancer. We also addressed the hypothesis that integrating multiple mutations and mutation types improves sensitivity for disease detection and explored if ctDNA analysis might guide personalized interventions such as targeted therapy or immunotherapy.
Results
We retrospectively profiled 255 blood and tissue samples from 40 patients with localized lung cancers being treated with curative-intent first-line therapies and 54 healthy adults (Supplementary Fig. S1; Supplementary Tables S1–S3). All patients had biopsy-proven non–small cell lung cancer (NSCLC; n = 37, 93%) or small cell lung cancer (SCLC; n = 3, 7%), with 7 patients (18%) having stage IB and 33 patients (82%) having stage II or III disease (Supplementary Tables S4 and S5).
Plasma samples were collected before treatment and at follow-up visits, which occurred every 2 to 6 months and were usually coincident with surveillance CT or PET/CT scans (Fig. 1A). For ctDNA analysis, we applied a 188-kb CAPP-seq selector targeting 128 genes recurrently mutated in lung cancer (Supplementary Table S6; refs. 9,10).
Figure 1.
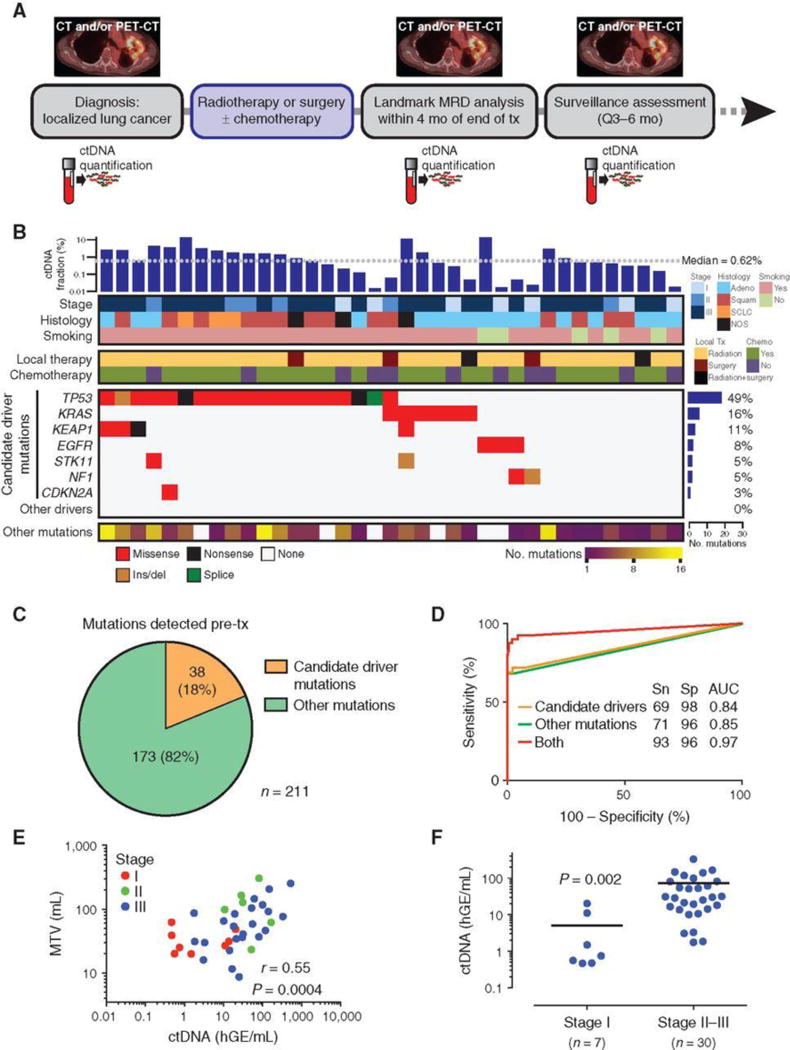
Pretreatment assessment of ctDNA in patients with localized lung cancer. A, Study schematic. Patients with biopsy- and imaging-proven nonmetastatic lung cancer were enrolled pretreatment. Plasma samples were collected before treatment and at follow-up visits, which occurred every 3–6 months and were usually coincident with surveillance scans (CT or PET/CT). B, Co-mutation plot based on pretreatment ctDNA analysis of patients with localized lung cancer. Each column represents pretreatment data from a single patient. Mutant allele fraction is shown in the top bar graph. Top heat maps indicate key patient characteristics. Mutation recurrence rate is depicted by bar graph to the right. Nonsynonymous mutations in candidate driver genes are shown in descending order of prevalence in the middle heat map. The number of other (i.e., likely passenger) mutations detected is indicated in the bottom heat map. C, Pie chart showing the number of candidate driver and other mutations detected in pretreatment plasma. D, ROC analysis of pretreatment (n = 40) and healthy control (n = 54) plasma samples using candidate driver, other, or both types of mutations. E, Scatter plot correlating ctDNA concentration (haploid genome equivalents per mL, hGE/mL) with pretreatment metabolic tumor volume (MTV) measured by PET-CT in patients with detectable ctDNA (n = 37). P value and r were calculated by Pearson correlation. F, Pretreatment ctDNA concentration in stage I (n = 7) and stage II–III (n = 30) patients with lung cancer. Data represent mean + SEM. P value was calculated by the Student t test with Welch correction. mo, months; tx, treatment; adeno, adenocarcinoma; squam, squamous cell carcinoma; NOS, not otherwise specified; Sn, sensitivity; Sp, specificity; AUC, area under the curve; PET, positron emission tomography; CT, computed tomography.
Using an optimized ctDNA detection approach we recently described (10), we detected pretreatment ctDNA in 37 patients (93%) with an average of 5 mutations per patient and median mutant allele fraction (AF) of 0.62%, nearly 10-fold lower than we previously observed in metastatic lung adenocarcinoma (11). Among the mutations we detected pretreatment were nonsynonymous mutations in the candidate driver genes TP53, KRAS, KEAP1, EGFR, STK11, NF1, and CDKN2A (Fig. 1B). Candidate driver genes were defined as genes that were found to be statistically significantly mutated in NSCLC or SCLC in prior studies (Supplementary Methods; Supplementary Table S6; refs. 12–15). The majority of mutations (82%; “other mutations”) we identified were not previously classified as driver mutations and consisted of “private” or “passenger” mutations that have no known functional impact (Fig. 1C). This matched the fraction of nonsilent passenger mutations we observed in 1,178 NSCLC tumors from The Cancer Genome Atlas (TCGA) when considering the same genomic coordinates covered by our CAPP-seq panel (9,671 out of 11,738; 82%; Supplementary Table S7).
In order to assess the clinical specificity of our approach for disease monitoring, we also applied CAPP-seq to cell-free DNA extracted from the plasma of 54 healthy adults (Supplementary Tables S2 and S8). The median age of healthy donors was 57 years (range, 27–82), which was somewhat lower than for patients (median 66.5 years; range, 47–91; P < 0.05). ROC analysis revealed an area under curve of 0.97, with maximal sensitivity and specificity of 93% and 96%, respectively, and was superior to detection by candidate driver or other mutations alone (Fig. 1D). Pretreatment ctDNA detection rates were 89% for adenocarcinoma, 93% for squamous cell carcinoma, and 100% for other NSCLC subtypes and SCLC. Among patients with ctDNA detectable before therapy, pretreatment ctDNA concentration was highly correlated with metabolic tumor volume (Pearson r = 0.55, P = 0.0004; Fig. 1E). Concentration of ctDNA in pretreatment plasma was significantly lower in patients with stage I compared with those with stage II–III tumors (P = 0.002; Fig. 1F). Baseline characteristics did not correlate with overall survival (OS; Supplementary Table S9).
To explore serial ctDNA analysis for disease surveillance during follow-up, we performed posttreatment monitoring of the 37 patients with detectable pretreatment ctDNA by both cross-sectional imaging and ctDNA analysis (Supplementary Table S1). The presence of ctDNA was evaluated by searching for the presence of previously identified tumor mutations in posttreatment plasma using CAPP-seq and a previously described Monte Carlo–based approach (see Methods). We detected ctDNA in at least one posttreatment time point in 20 patients (54%), and all 20 of these patients ultimately recurred. Both candidate driver and passenger mutations were important for ctDNA detection during surveillance, with detection of only driver mutations in 35%, only passenger mutations in 35%, and both types of mutations in 30% of patients (Fig. 2A; Supplementary Table S10). The most frequently detected mutations in surveillance samples included mutations in TP53, KRAS, EGFR, and KEAP1 (Fig. 2B). Patients with detectable ctDNA at any posttreatment time point had significantly lower freedom from progression (FFP) and survival than those in whom we did not detect ctDNA after completion of therapy (P < 0.001; Fig. 2C; Supplementary Fig. S2). Results remained highly significant when accounting for guarantee-time bias (P < 0.001; Supplementary Table S11; ref. 16).
Figure 2.
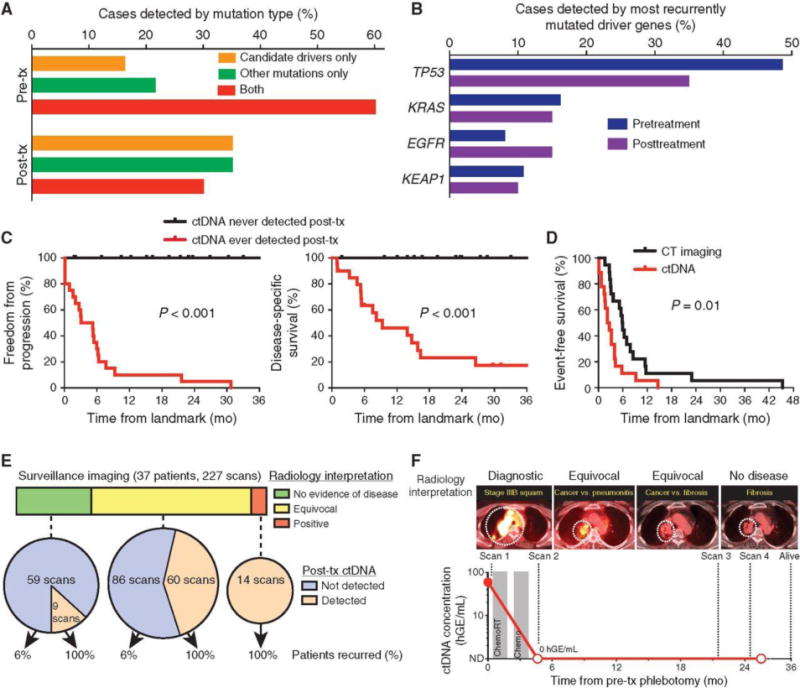
Application of ctDNA analysis for posttreatment surveillance in patients with localized lung cancer. A, Both driver and other (i.e., likely passenger) mutations are useful for detection of posttreatment ctDNA. Detection of mutation types pretreatment and at first detectable posttreatment time point is shown. B, Most recurrently mutated driver genes detected pretreatment and at first posttreatment time point. C, Kaplan–Meier analysis for freedom from progression (left) and disease-specific survival (right) stratified by ctDNA detection status during posttreatment surveillance; ever positive (n = 20) versus never positive (n = 17). Landmark analysis was performed from the first posttreatment blood draw. D, Kaplan–Meier analysis of time to ctDNA detection and time to imaging progression from the end of treatment for all patients who experienced posttreatment disease progression by RECIST 1.1 criteria (n = 18); HR = 2.4. P value was calculated by the log-rank test and HR by the Cox exp(beta) method. E, Analysis of ctDNA could aid interpretation of equivocal CT and PET-CT scans during posttreatment surveillance (n = 227 scans from 37 patients). Scans were interpreted as negative, equivocal, or positive by board-certified radiologists and compared with posttreatment ctDNA results and patient recurrence. F, Example of patient with stage IIIB NSCLC with equivocal surveillance imaging and undetectable posttreatment ctDNA who achieves long-term survival. mo, months; tx, treatment; CT, computed tomography; PET, positron emission tomography; squam, squamous cell carcinoma; hGE, haploid genome equivalents; chemoRT, chemoradiotherapy.
Detection of ctDNA preceded radiographic progression as determined by RECIST 1.1 criteria (17) in 72% of patients and by a median of 5.2 months (Fig. 2D). Although RECIST criteria are frequently used to assess efficacy of treatments in clinical trials, they are not routinely used in clinical practice, where diagnostic radiology reports usually more generally classify scans as showing (i) no evidence of disease, (ii) recurrent/persistent disease, or (iii) equivocal findings due to an inability to distinguish tumor from posttreatment tissue changes or other processes (18). We therefore systematically analyzed all posttreatment radiology reports (n = 227) for patients in our cohort and classified them into these three groups (Fig. 2E; Supplementary Table S1). Analysis of ctDNA served as a reliable predictor of ultimate outcomes in patients with negative or equivocal scans (Fig. 2E and F). These findings suggest that ctDNA analysis may be a useful adjunct to routine imaging studies.
We next asked whether ctDNA could be detected at a prespecified “MRD landmark,” which was defined as the first posttreatment blood draw within 4 months of treatment completion and generally corresponded to the time of the first follow-up scan (1). Landmark methodology was used in order to minimize guarantee-time bias (16). Thirty-two patients had their first posttreatment ctDNA assessment within 4 months of treatment completion and were thus included in this analysis (Supplementary Fig. S1). Analyzing the mutations detected in pretreatment plasma or tumor specimens, we detected ctDNA in 17 patients (53%) at the MRD landmark, with a median mutant allele fraction of 0.20% (Supplementary Fig. S3). We detected an average of 2 mutations per patient at the MRD landmark, >50% less than pretreatment, indicating that tracking of multiple mutations including drivers and passengers is beneficial for MRD detection.
We next sought to explore whether detection of ctDNA MRD was associated with outcome. FFP at 36 months after the MRD landmark was 0% in patients with detectable and 93% in patients with undetectable ctDNA MRD (P < 0.001, HR 43.4; 95% CI, 5.7–341; Fig. 3A). Only 1 patient who ultimately recurred had undetectable ctDNA at the MRD landmark, and in this patient ctDNA became detectable 8 months later, coincident with local disease recurrence (Supplementary Fig. S4). Analysis of disease-specific survival (DSS) and OS revealed similar results (Fig. 3A and B; Supplementary Fig. S5), with patients with undetectable ctDNA at the MRD landmark experiencing significantly better long-term survival than those with detectable ctDNA (P < 0.001). In contrast, radiographic response assessment by computed tomography (CT) at the MRD landmark was not prognostic in this cohort (Supplementary Fig. S6). Detection of ctDNA was strongly prognostic in both node-negative patients who predominantly received stereotactic ablative radiotherapy or surgery, and in node-positive patients who predominantly received chemoradiotherapy (Supplementary Fig. S7), and remained significant by Cox regression with multiple covariates (P < 0.001; Supplementary Table S12). Results remained highly significant when considering only patients with NSCLC (Supplementary Fig. S8). Because some patients had already progressed clinically or radiographically by the prespecified MRD landmark, we also performed a post hoc subset analysis in which we assessed patients at an earlier landmark of 6 weeks after treatment. Thirteen patients had blood drawn by this time point and were thus eligible for this analysis. Kaplan–Meier analysis revealed similar results with significantly higher FFP and OS in patients with undetectable posttreatment ctDNA compared with those with detectable ctDNA at this early posttreatment time point (Supplementary Fig. S9).
Figure 3.
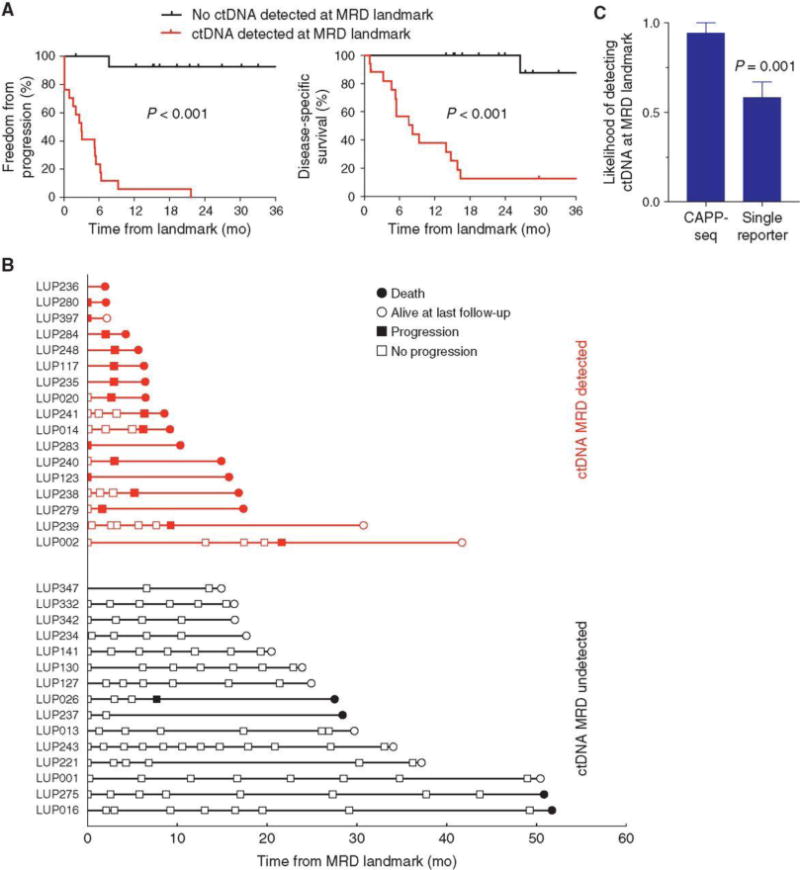
Detection of MRD in patients with localized lung cancer. Kaplan–Meier analysis of (A) freedom from progression (left) and disease-specific survival (right) stratified by detection of ctDNA at the MRD landmark (first posttreatment blood draw within 4 months of treatment completion); ctDNA MRD detected (n = 17), not detected (n = 15). P value was calculated by the log-rank test and HR by the Cox exp(beta) method. B, Event chart showing progression by RECIST 1.1 criteria and survival of patients with ctDNA detected at the MRD landmark (red) and patients with no ctDNA detected at the MRD landmark (black). C, Likelihood of detecting ctDNA at the MRD landmark (mean + SEM) by simultaneously tracking all known mutations (n = 65; CAPP-seq), or tracking each mutation separately (n = 65; single reporter). Data represent mean + SEM. P values were calculated by the Student t test. mo, months; tx, treatment.
To quantify the impact of tracking multiple variants on the sensitivity of MRD detection, we compared our approach to tracking a single mutation. With single-mutation tracking, the MRD detection rate was 58% on average, significantly lower than the 94% detection rate when using all variants (P = 0.001; Fig. 3C). Therefore, tracking of multiple mutations maximizes sensitivity of lung cancer MRD detection.
The ability to detect MRD could facilitate testing if early intervention, prior to clinical recurrence, could improve outcomes. We therefore explored types of treatments that could potentially have been offered to patients in our cohort at the time of MRD detection. In three patients, we identified EGFRL858R mutations in ctDNA at the MRD landmark, preceding clinical progression by an average of 3 months. For example, patient LUP20 was an 81-year-old who received stereotactic ablative radiotherapy (SABR) for stage IB disease and had an excellent radiographic response (Fig. 4A). However, this patient presented with symptomatic brain metastases 3 months later for which she refused treatment, and she died shortly thereafter. Of note, she did not have pretreatment brain MRI. A PET-CT 2 weeks after diagnosis of brain metastases demonstrated increased size and FDG avidity in the right adrenal gland, suspicious for metastasis. We detected EGFRL858R at the MRD landmark prior to the development of symptoms, suggesting that this patient could potentially have been offered early initiation of an EGFR TKI and/or brain MRI surveillance.
Figure 4.
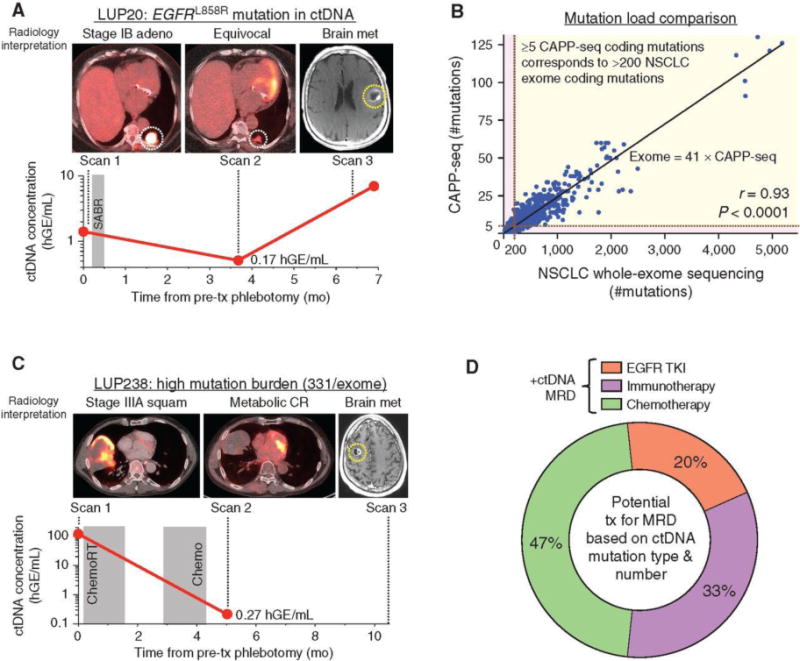
Analysis of ctDNA for assessment of potential treatment options following ctDNA MRD detection. A, Example of patient with stage IB EGFR-mutant lung adenocarcinoma with detectable ctDNA MRD. B, Mutation load comparison between NSCLC whole-exome sequencing and CAPP-seq. NSCLC mutations from 1,178 tumors determined by whole-exome sequencing by TCGA were intersected with the CAPP-seq lung selector to determine number of mutations that would have been called by CAPP-seq. Linear correlation (Pearson r = 0.93) with equation as shown with ≥5 CAPP-seq nonsynonymous mutations corresponding to >200 whole-exome nonsynonymous mutations. C, Example of patient with stage IIIA NSCLC with detectable ctDNA MRD. D, Analysis of treatment strategies that could potentially have been offered to patients with detectable MRD based on mutation type (i.e., presence of EGFR activating mutation) and mutation load (for selection of patients for immunotherapy). mo, months; tx, treatment; adeno, adenocarcinoma; squam, squamous cell carcinoma; hGE, haploid genome equivalents; SABR, stereotactic ablative radiotherapy; chemoRT, chemoradiotherapy; CR, complete response; TKI, tyrosine kinase inhibitor.
Although expression of PD-L1 is the best established predictive biomarker for immune checkpoint inhibitors (19), patients with NSCLC whose tumors harbor >200 nonsynonymous mutations per exome also appear to be enriched for responders (20). Tumor genotyping using smaller gene panels can be used to infer tumor mutation burden (TMB) in tumor biopsies (21), but this approach has not been applied to ctDNA. We therefore derived an equation relating CAPP-seq mutation burden to whole-exome mutation burden using data from TCGA (Fig. 4B). We validated this equation by performing both CAPP-seq and whole-exome sequencing on DNA from 5 NSCLC tumor samples (Supplementary Fig. S10). Using this equation, we identified patients with NSCLC in our cohort whose predicted TMB exceeded 200 variants. One such patient (LUP238) with stage IIIA lung squamous cell carcinoma whose tumor was predicted to harbor 331 exome mutations by CAPP-seq achieved a complete metabolic response by PET/CT to curative-intent first-line concurrent chemoradiotherapy (Fig. 4C). Nevertheless, we detected ctDNA at 0.27 hGE/mL at the MRD landmark, and the patient developed a symptomatic brain metastasis 5 months later that had not been present on pretreatment brain MRI and which was treated with radiosurgery. The patient developed biopsy-proven widespread metastases 4 months later, which were refractory to chemotherapy. Similarly, another patient with stage III disease (LUP241) was predicted to harbor 207 nonsynonymous mutations and developed brain metastases 6 months after treatment that resulted in death and were not present on the pretreatment brain MRI. It is possible that these patients may have benefitted from early initiation of immunotherapy.
Extending this analysis to all patients with detectable ctDNA MRD, we found that 20% could have been potential candidates for early administration of EGFR TKIs, 33% for immune checkpoint inhibitors, and the remaining 47% for chemotherapy (Fig. 4D). Importantly, these analyses are exploratory and hypothesis-generating and will need to be tested in prospective clinical trials before any consideration of routine clinical application.
Discussion
Our findings suggest that ctDNA analysis is a promising approach for MRD detection in patients with localized lung cancers and that it can identify recurrence significantly earlier than routine CT imaging. Within our cohort, all patients with detectable ctDNA during posttreatment surveillance developed progressive disease, whereas all patients whose ctDNA remained undetectable remained disease-free. The sensitivity of our approach for detecting MRD in patients who ultimately recurred was higher than seen in recent ctDNA studies for other cancer types, likely due to a combination of technical and biological differences (7,8).
We found that analysis of ctDNA detected disease recurrence earlier than imaging in 72% of patients with a median lead time of 5.2 months, opening a window of opportunity in which to treat patients while tumor burden and heterogeneity are at their lowest. Given the poor outcomes we observed in patients with detectable posttreatment ctDNA MRD, it is likely that this subgroup could benefit from adjuvant treatment. Previous trials of adjuvant chemotherapy in nonmetastatic NSCLC demonstrated a ∼5% absolute survival benefit at 5 years (22), meaning that ∼20 patients need to be treated per patient who benefits. This relatively large number is in part because a significant subset of patients enrolled in these trials were cured by local therapy and thus received no benefit from the additional treatment. We anticipate that selection of patients for adjuvant therapy based on detection of MRD, rather than based on nodal status or clinical risk factors such as primary tumor size and nuclear grade, will better enrich for patients who need adjuvant treatment while sparing those unlikely to benefit from toxicity.
In our cohort, ctDNA MRD detection was highly prognostic for both node-negative and node-positive patients with lung cancer. We thus envision that detection of MRD will be useful for patients in both groups, with an important caveat that our analysis included only 9 evaluable patients with node-negative disease. For patients with node-negative disease, adjuvant systemic therapy is currently usually not given, because the majority of these patients are cured by surgery and/or radiotherapy and because adjuvant chemotherapy may be detrimental in some of these patients (1,22). Administration of adjuvant chemotherapy in these patients remains controversial and is currently based on clinical risk factors such as tumor size (1). Thus, there is an opportunity for testing the utility of adjuvant systemic treatment based on detection of MRD. For patients with node-positive disease, consolidation systemic therapy is currently part of the standard of care in most patients receiving chemoradiotherapy (1). However, it is likely that a subset of these patients is cured by chemoradiotherapy alone, with multiple randomized trials showing no survival benefit of consolidation chemotherapy (23,24). Thus, personalization of adjuvant/consolidation treatment decisions could potentially be beneficial for both node-positive and node-negative patients with stage I–III NSCLC. Importantly, the utility of chemotherapy treatment based on ctDNA MRD analysis will need to be tested in prospective clinical trials.
Targeted therapeutics are currently not administered first-line in patients with localized NSCLC and instead are reserved for treatment of recurrence. It is tempting to speculate that patients with detectable MRD who are candidates for such targeted agents might benefit from early initiation of treatment. Selection of agents could be based on analysis of pretreatment tissue samples, including mutation testing and PD-L1 expression (19, 20). Our exploratory analysis suggests that assessment of actionable mutations and mutational load in ctDNA could serve as an additional approach for identifying patients who may benefit from early administration of tyrosine kinase or immune checkpoint inhibitors, particularly when diagnostic tissue specimens have been consumed or cannot be obtained. Notably it remains unclear if early targeted intervention based on MRD detection will improve lung cancer patient outcomes, and prospective trials will be required to test this concept.
One attractive feature of ctDNA-based assays is their high specificity compared with existing clinical approaches for determining risk of recurrence. Our approach had a specificity of 96% in healthy controls and 100% in patients with lung cancer who did not develop recurrence. This is similar to specificities observed in recent studies on breast and colorectal cancer ctDNA MRD using other assays (7, 8). Of note, these specificities are significantly higher than those for clinical risk factors that are currently used for informing adjuvant chemotherapy recommendations in stage I NSCLC. For example, the most commonly applied risk factor of primary tumor size ≥ 4 cm has a specificity of only ∼50% for predicting recurrence (25), and therefore future trials basing adjuvant treatment decisions on the presence ctDNA MRD may lead to less overtreatment. That said, applications of ctDNA MRD detection to patients with very low risks of recurrence, such as patients with very small stage IA tumors, could lead to higher false-positive rates. If necessary, specificity of ctDNA-based MRD approaches could likely be further increased by increasing stringency of detection thresholds or repeat assaying.
Many of the patients in our cohort were treated with radiotherapy, which causes pulmonary tissue inflammation and fibrosis that can be difficult to distinguish from residual/recurrent disease on cross-sectional imaging (3). Accordingly, in our cohort the majority of surveillance scans were clinically interpreted as being equivocal, even though nearly half of these scans were subsequently followed by recurrence. Measurement of ctDNA served as an arbiter of equivocal imaging, with high sensitivity and specificity for predicting recurrence. Thus, ctDNA has the potential to supplement analysis of surveillance imaging by aiding the interpretation of equivocal scans.
Despite biological differences between SCLC and NSCLC, we included both histologies in this study. Our rationale was based on the premise that MRD detection and correlation with outcomes was relevant and broadly applicable to diverse lung cancer histologies, including NSCLC and SCLC. Additionally, the employed CAPP-seq panel covered mutations present in both histologic subtypes. Notably, when we considered NSCLC alone, correlation of ctDNA MRD detection and clinical outcomes remained highly significant. Our results suggest that future studies focused specifically on MRD detection in SCLC are warranted.
Using a next-generation sequencing–based approach that involves creation of personalized assays for each patient, the TracerX consortium recently also found that detection of ctDNA during surveillance of patients with early-stage NSCLC precedes imaging-based recurrence (26). Unlike the current study, the TracerX study did not specifically assess the prognostic value of early ctDNA MRD detection (26). Additionally, in the TracerX cohort pretreatment ctDNA was detected in only 19% of lung adenocarcinomas (26), compared with 89% in our study. Reasons for the lower detection rates in the TracerX study compared with our study are likely due at least in part to differences in patient cohorts, such as the higher percentage of stage I patients in the TracerX study and the higher analytic sensitivity of the ctDNA detection method we used (10, 26).
The somatic mutation burden in lung cancers correlates with the duration of tobacco exposure (27), but a minority of such mutations involve genes known to drive lung cancers (12, 13, 27, 28). In a separate study, the TracerX consortium recently reported a comprehensive analysis of mutational heterogeneity using deep multiregion exome sequencing of 327 tumor regions from 100 patients (28). Although the vast majority of all detected mutations were classified as nondrivers, such variants were only slightly less likely to be clonal than driver mutations (57% vs. 64%) and most fit a mutational signature associated with smoking that dominates during early tumorigenesis (28). Consistent with this observation that passenger mutations are often clonal, we found that inclusion of nondriver mutations improved noninvasive disease detection using ctDNA without compromising specificity or predictive value for residual disease after definitive therapy. Therefore, as in tumor specimens, most somatic variants we detected in the plasma of patients with lung cancer were in nondriver genes. However, these nondriver mutations appear to faithfully reflect the clonal burden of disease and are useful for posttreatment MRD detection and surveillance.
Limitations of our study include a relatively long accrual period, which could have introduced unknown selection biases but conversely resulted in a relatively long median follow-up time. Additionally, although all patients were treated with definitive local therapy, the types of treatment were heterogeneous and included mostly patients treated with radiotherapy. Thus, in addition to validation of our findings in similar cohorts, future studies focused on surgically treated patients are warranted. Finally, it is important to note that our approach is unique compared with other ctDNA detection methods because of the dedicated bioinformatic approach for tracking groups of previously identified mutations posttreatment and the high analytic sensitivity. It is unclear whether other techniques for ctDNA detection would yield similar results.
In conclusion, we found that ctDNA is a promising biomarker for early detection of MRD in patients with localized lung cancer and can reliably identify patients at high risk for recurrence. Tracking multiple mutations improves the sensitivity of MRD detection, and both driver and passenger mutations are useful for tracking and monitoring disease. Validation of our findings and prospective clinical trials testing therapeutic strategies based on ctDNA MRD assessment will be required to establish clinical utility.
Methods
Study Design and Patients
The samples analyzed in this article were collected as part of two observational registry studies focused on molecular analysis of thoracic malignancies and other tumors (NCT01385722 and NCT00349830). For our study, we identified a subset of patients from these registries treated between June 2010 and March 2016, to analyze retrospectively with the primary goal of analyzing the association of ctDNA MRD with FFP after definitive therapy of localized lung cancers. Eligible patients included in this study were age >18 years with untreated primary lung cancers, had AJCC v7 stage IB, II, or III disease with WHO NSCLC or SCLC histology and received curative-intent treatment with radiotherapy, chemotherapy, and/or surgery (Supplementary Fig. S1). The study statistical plan used the assumption that 50% of enrolled patients would have detectable posttreatment ctDNA (based on 1-year progression-free survival data from the RTOG 0617 standard-dose arm; ref. 29), such that an accrual of 35 patients would be expected to yield 86% power to detect a difference of 75% versus 25% risk of progression for patients with positive or negative posttreatment ctDNA, with a two-sided alpha of 0.05. A goal of 45 patients was targeted to account for attrition. Blood samples from 5 patients were included in a prior publication (9).
Eligible patients underwent pretreatment imaging by chest CT and whole-body PET-CT and genotyping with CAPP-seq on tumor tissue or plasma using matched germline DNA. All patients with stage II or higher disease underwent pretreatment brain MRI, as did the majority of patients with stage I disease. This was followed by treatment with surgery or radiotherapy (with or without chemotherapy). After first-line therapy, patients were followed every 3 to 6 months with cross-sectional imaging and blood collections (Fig. 1A). For all but 2 patients the second blood sample was collected after completion of all treatments (LUP127 collected during and LUP235 collected before consolidation chemotherapy). The median time between the end of all treatment and the first posttreatment blood sample was 56 days. Healthy adult blood donors (n = 54) were recruited through the Stanford Blood Center (Supplementary Fig. S1). All samples were collected with informed consent and institutional review board approval in accordance with the Declaration of Helsinki. All plasma samples were analyzed by CAPP-seq as previously described (9, 10).
Criteria for ctDNA MRD Detection andPosttreatment Monitoring
ctDNA MRD and serial posttreatment plasma samples were analyzed for presence of mutations identified pretreatment using CAPP-seq on plasma and plasma-depleted whole blood as previously described, with an additional clonal hematopoiesis filter (9, 10). Briefly, ctDNA MRD analysis was performed at a prespecified landmark that occurred within 4 months after treatment completion and typically coincided with the first posttreatment CT scan. For MRD and serial posttreatment time points, the set of mutations identified pretreatment were assessed as a group in the posttreatment blood sample, and a Monte Carlo–based ctDNA detection index was measured to determine significance. Given concern for clonal hematopoiesis, variants with reads in peripheral blood mononuclear cells had to also be called by the CAPP-seq variant caller (9, 10) for detection. At each time point, ctDNA detection status was determined by CAPP-seq using a Monte Carlo–based ctDNA detection index cutoff point of ≤0.05, as previously established (9, 10). If ctDNA detection index was >0.05, ctDNA was classified as not detected at that time point, whereas if it was ≤0.05 it was classified as detected, in accordance with our prior studies (9, 10). The ctDNA-mutant AF at each time point was calculated by averaging the mutant AFs for all mutations used for detection calling. ctDNA concentration was calculated by multiplying the mutant AF by the cell-free DNA concentration determined by Qubit (ThermoFisher Scientific) and using the assumption that each haploid genomic equivalent weighs 3.3 pg.
Landmark Analyses and Definition of MRD
To protect against guarantee-time bias, we used landmark analysis and time-dependent Cox models (16, 30, 31). The “MRD landmark” for ctDNA response was prespecified as the first phlebotomy collection following completion of curative-intent first-line therapy, and occurring no more than 4 months from the end of therapy. MRD was defined as Monte Carlo–based ctDNA detection at the MRD landmark using mutations identified pretreatment (ctDNA index ≤ 0.05). ctDNA detection at the MRD landmark was used to categorize patients as posttreatment MRD positive or negative.
Statistical Analyses
Our primary aim was to test the hypothesis that detection of residual ctDNA at the first blood draw after definitive local therapy is associated with high risk of recurrence. Our secondary aim was to test the hypothesis that patients who ever have ctDNA detected after local therapy have worse outcomes. We considered the following survival endpoints: FFP (event defined as RECIST 1.1–based radiographic, ref. 17; or clinical progression, with nonprogressors censored at last radiographic follow-up), event-free survival (EFS; event defined as posttreatment ctDNA detection or RECIST 1.1–based radiographic progression, ref. 17), DSS (event defined as death from cancer), and OS (event defined as death from any cause). Categorical time-to-event analyses of clinical endpoints including FFP, EFS, DSS, and OS were conducted using the Kaplan–Meier method with log-rank test to estimate P values and the Cox exp(beta) method to estimate hazard ratios. The relationship of ctDNA concentration as a continuous variable with outcome was assessed using Cox proportional hazards regression. The Wald test was used to assess the significance of covariates, and hazard ratios were calculated by the exp(beta) method. Time-dependent Cox regression was performed as previously described (16). See Supplementary Methods for details.
Supplementary Material
Significance.
This study shows that ctDNA analysis can robustly identify posttreatment MRD in patients with localized lung cancer, identifying residual/recurrent disease earlier than standard-of care radiologic imaging, and thus could facilitate personalized adjuvant treatment at early time points when disease burden is lowest.
Acknowledgments
We are grateful to the patients and families involved in this study.
Grant Support
This work was supported with grants from the Radiological Society of North America (A.A. Chaudhuri), the National Science Foundation (J.J. Chabon; DGE-114747), the Department of Defense (M. Diehn), the National Cancer Institute (M. Diehn and A.A. Alizadeh; R01CA188298), the US National Institutes of Health Director’s New Innovator Award Program (M. Diehn; 1-DP2-CA186569), the Virginia and D.K. Ludwig Fund for Cancer Research (M. Diehn and A.A. Alizadeh), a Stanford Cancer Institute-Developmental Cancer Research Award (M. Diehn and A.A. Alizadeh), and the CRK Faculty Scholar Fund (M. Diehn). Funding sources played no role in the writing of this manuscript or the decision to submit it for publication.
A.F. Lovejoy is principal scientist I at Roche Sequencing Solutions. A.M. Newman has ownership interest in CiberMed, Inc., is a co-inventor on patents related to ctDNA detection and other cancer biomarkers, and is a consultant/advisory board member for Roche and Merck. S.K. Padda is a consultant/advisory board member for G1 Therapeutics, AstraZeneca, and Janssen. M.F. Gensheimer reports receiving a commercial research grant from Varian Medical Systems. J.W. Neal reports receiving commercial research grants from Genentech/Roche, Merck, ArQule, Novartis, Boehringer Ingelheim, Nektar, and ARIAD, and is a consultant/advisory board member for Clovis Oncology, Boehringer Ingelheim, ARMO Biosciences, Nektar Therapeutics, ARIAD Pharmaceuticals/Takeda, Eli Lilly & Co., and Physician Resource Management. A.A. Alizadeh has ownership interest in CiberMed, is a co-inventor for patent filings related to cancer biomarkers, and is a consultant/advisory board member for Genentech, Roche, Gilead, and Celgene. M. Diehn reports receiving a commercial research grant from Varian Medical Systems, has ownership interest in CiberMed and in patent filings related to cancer biomarkers including ctDNA, and is a consultant/advisory board member for Roche.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: A.A. Chaudhuri, A.F. Lovejoy, M.F. Gensheimer, J.B. Shrager, J.W. Neal, H.A. Wakelee, A.A. Alizadeh, M. Diehn
Development of methodology: A.A. Chaudhuri, J.J. Chabon, A.F. Lovejoy, H. Stehr, D.M. Kurtz, A.A. Alizadeh, M. Diehn
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A.A. Chaudhuri, J.J. Chabon, A.F. Lovejoy, T.D. Azad, C. Say, J.N. Carter, D.J. Merriott, J.C. Dudley, L. Modlin, S.K. Padda, J.B. Shrager, J.W. Neal, H.A. Wakelee, B.W. Loo Jr, A.A. Alizadeh, M. Diehn
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A.A. Chaudhuri, J.J. Chabon, A.F. Lovejoy, A.M. Newman, H. Stehr, T.D. Azad, M.S. Khodadoust, M.S. Esfahani, F. Scherer, D.M. Kurtz, M.S. Binkley, A.A. Alizadeh, M. Diehn
Writing, review, and/or revision of the manuscript: A.A. Chaudhuri, J.J. Chabon, A.F. Lovejoy, H. Stehr, T.D. Azad, C.L. Liu, F. Scherer, D.M. Kurtz, J.C. Dudley, M.S. Binkley, S.K. Padda, M.F. Gensheimer, R.B. West, J.B. Shrager, J.W. Neal, H.A. Wakelee, B.W. Loo Jr, A.A. Alizadeh, M. Diehn
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): A.A. Chaudhuri, C.L. Liu, L. Zhou, F. Scherer, C. Say, J.N. Carter, D.J. Merriott, L. Modlin, R.B. West, J.B. Shrager, A.A. Alizadeh, M. Diehn
Study supervision: A.A. Alizadeh, M. Diehn
References
- 1.Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, et al. Non-Small Cell Lung Cancer, Version 5.2017, NCCN ClinicalPractice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:504–35. doi: 10.6004/jnccn.2017.0050. [DOI] [PubMed] [Google Scholar]
- 2.Kalemkerian GP, Akerley W, Bogner P, Borghaei H, Chow LQ, Downey RJ, et al. Small cell lung cancer. J Natl Compr Canc Netw. 2013;11:78–98. doi: 10.6004/jnccn.2013.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kocak Z, Evans ES, Zhou SM, Miller KL, Folz RJ, Shafman TD, et al. Challenges in defining radiation pneumonitis in patients with lung cancer. Int J Radiat Oncol Biol Phys. 2005;62:635–8. doi: 10.1016/j.ijrobp.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 4.Huang K, Dahele M, Senan S, Guckenberger M, Rodrigues GB, Ward A, et al. Radiographic changes after lung stereotactic ablative radiotherapy (SABR)–can we distinguish recurrence from fibrosis? A systematic review of the literature. Radiother Oncol. 2012;102:335–42. doi: 10.1016/j.radonc.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 5.Hung JJ, Hsu WH, Hsieh CC, Huang BS, Huang MH, Liu JS, et al. Post-recurrence survival in completely resected stage I non-small cell lung cancer with local recurrence. Thorax. 2009;64:192–6. doi: 10.1136/thx.2007.094912. [DOI] [PubMed] [Google Scholar]
- 6.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumorDNA. J Clin Oncol. 2014;32:579–86. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 8.Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92. doi: 10.1126/scitranslmed.aaf6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–54. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–55. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–6. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giobbie-Hurder A, Gelber RD, Regan MM. Challenges of guaranteetime bias. J Clin Oncol. 2013;31:2963–9. doi: 10.1200/JCO.2013.49.5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 18.Larici AR, del Ciello A, Maggi F, Santoro SI, Meduri B, Valentini V, et al. Lung abnormalities at multimodality imaging after radiation therapy for non-small cell lung cancer. Radiographics. 2011;31:771–89. doi: 10.1148/rg.313105096. [DOI] [PubMed] [Google Scholar]
- 19.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–56. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 20.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campesato LF, Barroso-Sousa R, Jimenez L, Correa BR, Sabbaga J, Hoff PM, et al. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget. 2015;6:34221–7. doi: 10.18632/oncotarget.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pignon JP, Tribodet H, Scagliotti GV, Douillard JY, Shepherd FA, Stephens RJ, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26:3552–9. doi: 10.1200/JCO.2007.13.9030. [DOI] [PubMed] [Google Scholar]
- 23.Ahn JS, Ahn YC, Kim JH, Lee CG, Cho EK, Lee KC, et al. Multinational randomized phase III trial with or without consolidation chemotherapy using docetaxel and cisplatin after concurrent chemoradiation in inoperable stage III non-small-cell lung cancer: KCSG-LU05-04. J Clin Oncol. 2015;33:2660–6. doi: 10.1200/JCO.2014.60.0130. [DOI] [PubMed] [Google Scholar]
- 24.Keller SM, Adak S, Wagner H, Herskovic A, Komaki R, Brooks BJ, et al. A randomized trial of postoperative adjuvant therapy in patients with completely resected stage II or IIIA non-small-cell lung cancer. Eastern Cooperative Oncology Group. N Engl J Med. 2000;343:1217–22. doi: 10.1056/NEJM200010263431703. [DOI] [PubMed] [Google Scholar]
- 25.Butts CA, Ding K, Seymour L, Twumasi-Ankrah P, Graham B, Gandara D, et al. Randomized phase III trial of vinorelbine plus cisplatin compared with observation in completely resected stage IB and II non-small-cell lung cancer: updated survival analysis of JBR-10. J Clin Oncol. 2010;28:29–34. doi: 10.1200/JCO.2009.24.0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–51. doi: 10.1038/nature22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–34. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–21. doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 29.Bradley JD, Paulus R, Komaki R, Masters G, Blumenschein G, Schild S, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015;16:187–99. doi: 10.1016/S1470-2045(14)71207-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response. J Clin Oncol. 1983;1:710–9. doi: 10.1200/JCO.1983.1.11.710. [DOI] [PubMed] [Google Scholar]
- 31.Bouwhuis MG, Suciu S, Collette S, Aamdal S, Kruit WH, Bastholt L, et al. Autoimmune antibodies and recurrence-free interval in melanoma patients treated with adjuvant interferon. J Natl Cancer Inst. 2009;101:869–77. doi: 10.1093/jnci/djp132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.