

Abstract
We introduce an approach for identifying disease-relevant tissues and cell types by analyzing gene expression data together with genome-wide association study (GWAS) summary statistics. Our approach uses stratified LD score regression to test whether disease heritability is enriched in regions surrounding genes with the highest specific expression in a given tissue. We apply our approach to gene expression data from several sources together with GWAS summary statistics for 48 diseases and traits (average N=169K), detecting significant tissue-specific enrichments (FDR<5%) for 34 traits. In our analysis of multiple tissues, we detect a broad range of enrichments that recapitulate known biology. In our brain-specific and immune-specific analyses, significant enrichments include an enrichment of inhibitory over excitatory neurons for bipolar disorder but excitatory over inhibitory neurons for schizophrenia and body mass index. Our results demonstrate that our polygenic approach is a powerful way to leverage gene expression data for interpreting GWAS signal.
INTRODUCTION
There are many diseases whose causal tissues or cell types are uncertain or unknown. Identifying these tissues and cell types is critical for developing systems to explore gene regulatory mechanisms that contribute to disease. In recent years, researchers have been gaining an increasingly clear picture of which parts of the genome are active in a range of tissues and cell types: for example, which parts of the genome are accessible, which enhancers are active, and which genes are expressed1–3. Combining this type of information with GWAS data offers the potential to identify causal tissues and cell types for disease.
Many different types of data characterizing tissue- and cell-type-specific activity have been analyzed together with GWAS data to identify disease-relevant tissues and cell types, including histone marks4–8, DNase I hypersensitivity (DHS)9–12, eQTLs3,13, and gene expression data14–17. Of these data types, gene expression data (without genotypes or eQTLs) has the advantage of being available in the widest range of tissues and cell types. Previous studies have shown that gene expression data are informative for disease-relevant tissues and cell types, and have led to biological insights about the diseases and traits studied14–17. However, the methods applied in these studies restrict their analyses to subsets of SNPs that pass a significance threshold. To our knowledge, no previous study has modeled genome-wide polygenic signals to identify disease-relevant tissues and cell types systematically from GWAS and gene expression data.
Here, we apply stratified LD score regression7, a method for partitioning heritability from GWAS summary statistics, to sets of specifically expressed genes to identify disease-relevant tissues and cell types across 48 diseases and traits with an average GWAS sample size of 169,331. We first analyze two gene expression data sets3,17,18 containing a wide range of tissues to infer system-level enrichments. We then analyze chromatin data from the Roadmap Epigenomics and ENCODE projects1,2 across the same set of diseases and traits to validate these results. Finally, we analyze gene expression data sets that allow us to achieve higher resolution within a system3,19–21, identifying enriched brain regions, brain cell types, and immune cell types for several brain- and immune-related diseases and traits; we validate several of our immune enrichments using independent chromatin data. Our results underscore that a heritability-based framework applied to gene expression data allows us to achieve high-resolution enrichments, even for very polygenic traits.
RESULTS
Overview of methods
We analyzed the five gene expression data sets listed in Table 1, mapping mouse genes to orthologous human genes when necessary. To assess the enrichment of a focal tissue for a given trait, we follow the procedure described in Figure 1. We begin with a matrix of normalized gene expression values across genes, with samples from multiple tissues including the focal tissue. For each gene, we compute a t-statistic for specific expression in the focal tissue (Online Methods). We rank all genes by their t-statistic, and define the 10% of genes with the highest t-statistic to be the gene set corresponding to the focal tissue; we call this the set of specifically expressed genes, but we note that this includes not only genes that are strictly specifically expressed (i.e. only expressed in the focal tissue), but also genes that are weakly specifically expressed (i.e. higher average expression in the focal tissue). For a few of the data sets analyzed, we modified our approach to constructing the set of specifically expressed genes to better take advantage of the data available (Online Methods). We add 100kb windows on either side of the transcribed region of each gene in the set of specifically expressed genes to construct a genome annotation corresponding to the focal tissue. (The choice of the parameters 10% and 100kb is discussed in the Supplementary Note; our results are robust to these choices (see below).) Finally, we apply stratified LD score regression7 to GWAS summary statistics to evaluate the contribution of the focal genome annotation to trait heritability (Online Methods). We jointly model the annotation corresponding to the focal tissue, a genome annotation corresponding to all genes, and the 52 annotations in the “baseline model”7 (including genic regions, enhancer regions, and conserved regions; see Table S1). A positive regression coefficient for the focal annotation in this regression represents a positive contribution of this annotation to trait heritability, conditional on the other annotations. We report regression coefficients, normalized by mean per-SNP heritability, together with a P-value to test whether the regression coefficient is significantly positive. Stratified LD score regression requires GWAS summary statistics for the trait of interest, together with an LD reference panel (e.g. 1000 Genomes22), and has been shown to produce robust results with properly controlled type I error7. We have released open source software implementing our approach, and have also released all genome annotations derived from the publicly available gene expression data that we analyzed (see URLs). We call our approach LD score regression applied to specifically expressed genes (LDSC-SEG).
Table 1.
List of gene expression data sets used in this study. We analyzed five gene expression data sets: two (GTEx and Franke lab) containing a wide range of tissues and three (Cahoy, PsychENCODE, ImmGen) with more detailed information about a particular tissue.
Figure 1.
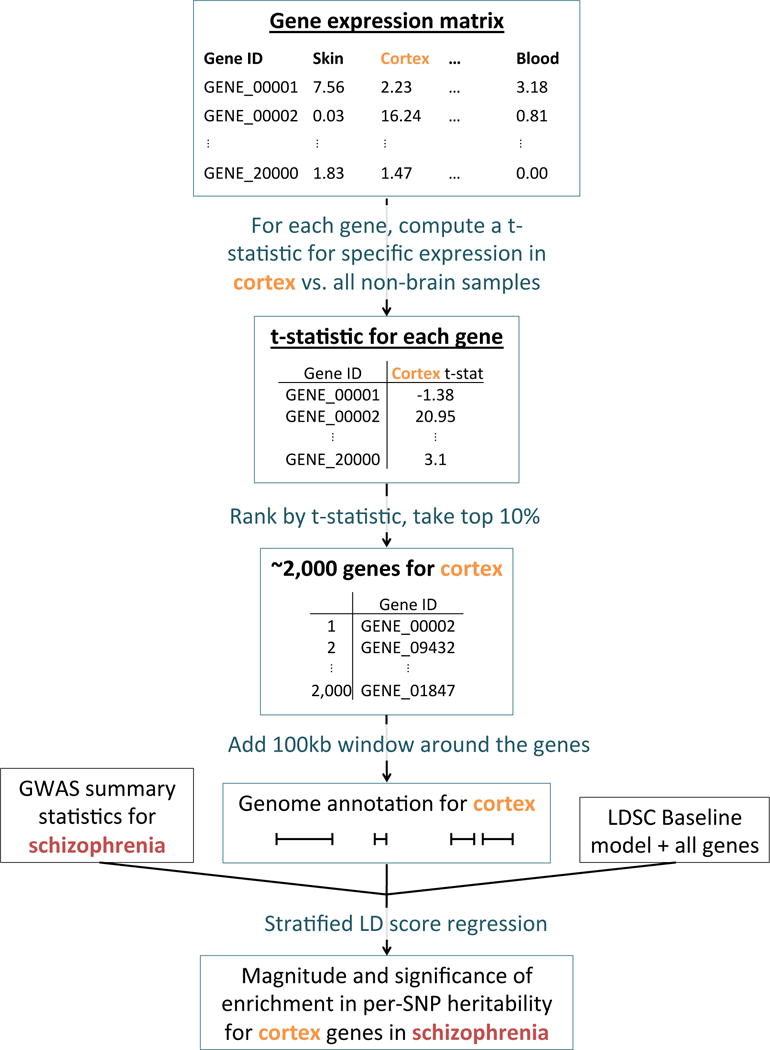
Overview of the approach. For each tissue in our gene expression data set, we compute t-statistics for differential expression for each gene. We then rank genes by t-statistic, take the top 10% of genes, and add a 100kb window to get a genome annotation. We use stratified LD score regression7 to test whether this annotation is significantly enriched for per-SNP heritability, conditional on the baseline model7 and the set of all genes.
Analysis of 48 complex traits across multiple tissues
We first analyzed two gene expression data sets — GTEx and a dataset which we call the Franke lab data set — and we classified the 205 tissues and cell types in these data sets into nine categories for visualization (Tables S2 and S3, Online Methods). We analyzed GWAS summary statistics for 48 diseases and traits from the UK Biobank23 (Online Methods), the Brainstorm Consortium16,24–32, and publicly available sources33–43, with an average sample size of 169,331 (Table S4), applying LDSC-SEG for each of the 205 specifically expressed gene annotations in turn. We excluded the HLA region from all analyses, due to its unusual genetic architecture and pattern of LD.
For 34 of the 48 traits, at least one tissue was significant at FDR<5% (Figure 2, Figure S1 and Tables S5 and S6). Several of our results recapitulate known biology: immunological traits exhibit immune cell-type enrichments, psychiatric traits exhibit strong brain enrichment, LDL and triglycerides exhibit liver-specific enrichments, BMI-adjusted waist-hip ratio exhibits adipose enrichment, type 2 diabetes exhibits enrichment in the pancreas, and height exhibits enrichments in a variety of tissues in a pattern similar to previous analyses of this trait44. In addition, several of our results validate very recent findings from other genetic analyses: in particular, smoking status, years of education, BMI, and age at menarche show robust brain enrichments that recapitulate results from our previous analysis of genetic data together with chromatin data7. Our results were robust to the choice of percent of genes used (10%) and to the size of the window used (100kb) (Figure S2). We assessed correlations in enrichment patterns for pairs of traits (Online Methods), and found large and significant correlations among many brain-related phenotypes, among many immune-related phenotypes, and among a third set of phenotypes including height and blood pressure that tended to have enrichments in the musculosketal/connective, cardiovascular, and other categories (Figure S3). The most significant annotation for each of these 34 traits spanned 11%-23% (mean 16%) of the genome and explained 21%-62% (mean 36%) of SNP-heritability, with enrichments varying from 1.4× to 4.7× (mean 2.3×) (Table S5).
Figure 2.
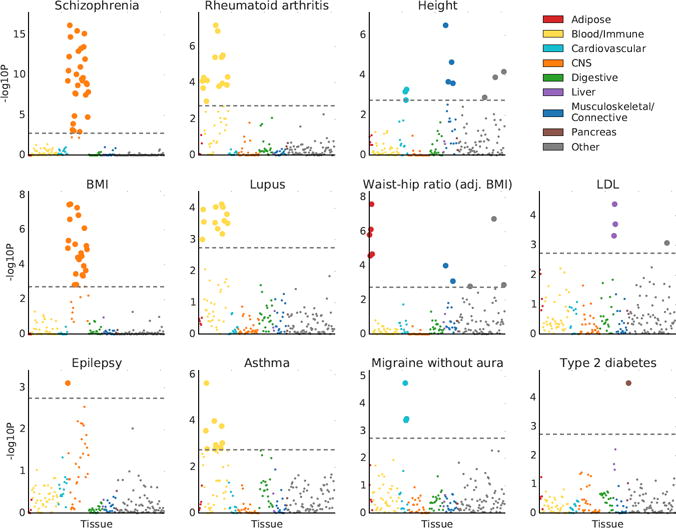
Results of the multiple-tissue analysis for selected traits. Results for the remaining traits are displayed in Figure S1. Each point represents a tissue/cell type from either the GTEx data set or the Franke lab data set. Large points pass the FDR<5% cutoff, –log10(P)=2.75. GWAS data is described in Table S4, gene expression data is described in the Online Methods and Tables S2-3, and the statistical method is described in the Overview of Methods and the Online Methods. Numerical results are reported in Table S6.
Because related tissues have highly overlapping gene sets and we fit each tissue without adjusting for the other tissues, related tissues often appear enriched as a group. In this analysis and the analysis in the next section, both focused on identifying system-level enrichments, these correlated results do not limit interpretability. In later sections, we focus on differentiating among related tissues/cell types within a system. We note also that the correlation structure among annotations can lead to a distribution of P values that is highly non-uniform (Online Methods).
Validation using independent chromatin data
We analyzed the same 48 diseases and traits using stratified LD score regression7 in conjunction with chromatin data from the Roadmap Epigenomics and ENCODE projects1,2 (see URLs) instead of gene expression data, with three goals: (1) to validate the results from our analysis of gene expression data using a different type of data from an independent source, (2) to identify new enrichments using chromatin data that we did not observe using gene expression data, and (3) to compare enrichments from the two types of data. The ENCODE data we used was from a subproject called EN-TEx, which includes epigenetic data on a set of tissues that match a subset of the tissues from the GTEx project but are from different donors. In total, we analyzed 489 tissue-specific chromatin-based annotations from peaks for six epigenetic marks (Online Methods).
We considered two types of validation for the results of the multiple-tissue analysis of gene expression described above: validation at the system level and validation at the tissue/cell-type level. For validation at the system level, we classified the top tissue or cell type for each trait with a significant enrichment into one of nine systems (Online Methods), and we considered an enrichment to be validated if a tissue or cell type from the same system passed FDR < 5% for the same phenotype in the chromatin analysis. For validation at the tissue/cell-type level, we only analyzed the 27 tissues present in both GTEx and EN-TEx, and we considered an enrichment of a tissue in GTEx to be validated if any mark in the same tissue in EN-TEx passed FDR < 5% for the same phenotype. The top enrichment from our multi-tissue analysis of gene expression was validated at the system level for 33 out of 34 phenotypes (Figure 3a, Table S5), and the top enrichment of a tissue or cell type shared between GTEx and EN-TEx was validated at the tissue/cell-type level for 13 out of 20 phenotypes, rising to 16 with a more lenient definition (Table S5, Online Methods). In many instances, the analysis of chromatin data detected more enrichments, larger enrichments, and/or enrichments at higher significance levels than the analysis of gene expression data, though this was not always the case (Figures S4-S5, Table S7, Online Methods). The enrichment correlations in this analysis showed a similar pattern to the gene expression analysis above (Figure S6).
Figure 3.
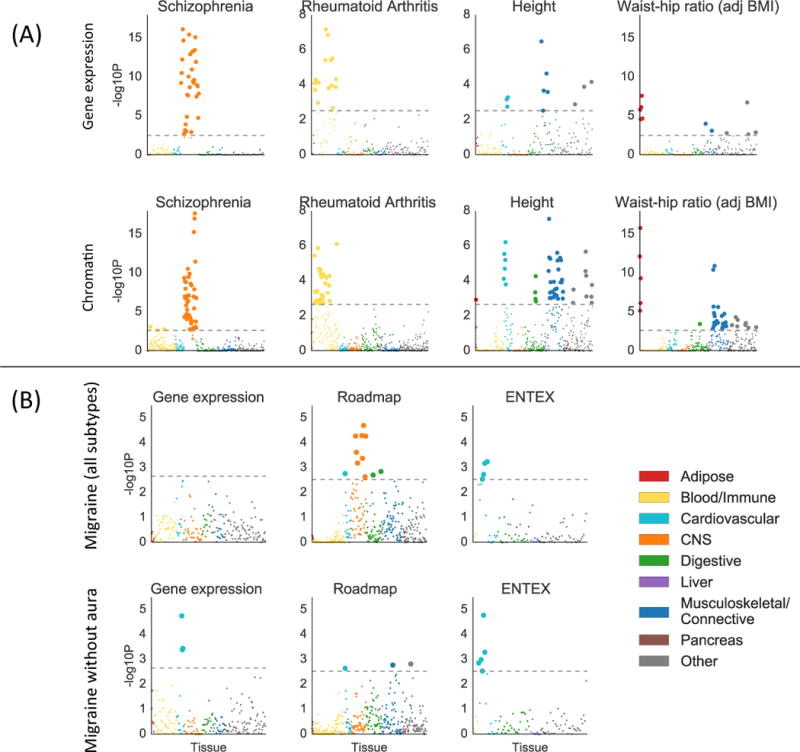
Validation of gene expression results with chromatin data. (A) Examples of validation using chromatin data (bottom) of results from gene expression data (top), for selected traits. Results using chromatin data for all traits are displayed in Figure S5, with numerical results in Table S7. For the chromatin results, each point represents a track of peaks for H3K4me3, H3K4me1, H3K9ac, H3K27ac, H3K36me3, or DHS in a single tissue/cell type. (B) Results using gene expression data (including GTEx), Roadmap, and EN-TEx, for migraine (all subtypes) and migraine without aura. For both subfigures, large points pass the FDR<5% cutoff, –log10(P)=2.85 (chromatin) or –log10(P)=2.75 (gene expression). GWAS data is described in Table S4; gene expression data and chromatin data are described in the Online Methods, Tables S2-3, and Table S7; and the statistical method is described in the Overview of Methods and the Online Methods.
There is a long-standing scientific debate as to whether migraine has a primarily neurological or vascular basis45. We analyzed GWAS summary statistics for migraine with aura, migraine without aura, and migraine (all subtypes)16. The migraine (all subtypes) data set contained the data sets for migraine with aura and for migraine without aura, as well as a large number of additional subjects whose subtype was unknown. We found cardiovascular enrichments for migraine without aura with gene expression data, and for migrane without aura and migraine (all subtypes) with EN-TEx data, consistent with previous work16 (Figure 3b). Our analysis of Roadmap data, however, yielded qualitatively different results: the strongest enrichment for migraine (all subtypes) was a neurological enrichment. The top two annotations were neurospheres and fetal brain, neither of which was present in the gene expression data we analyzed nor in EN-TEx. The correlation in enrichments between migraine (all subtypes) and migraine without aura in the gene expression analysis was estimated to be 0.48 (s.e. 0.15), and in the chromatin data was estimated to be 0.60 (s.e. 0.13). Our results are consistent with the hypothesis that migraine without aura does indeed have a vascular component, and that another subtype of migraine may have a neurological basis which is sufficiently cell-type specific that the relevant cell types are not represented in either the GTEx or Franke lab data sets. These results highlight the importance of having as many tissues and cell types as possible represented in a multiple-tissue analysis.
A major advantage of gene expression data is that it is available at finer tissue/cell-type resolution within several systems. In the within-system analyses that follow, we investigate these finer patterns of tissue/cell-type specificity.
Analysis of 12 brain-related traits using fine-scale brain expression data
We identified 12 traits with CNS enrichment at FDR<5% in our gene expression and/or chromatin analyses (Online Methods). We first investigated whether some brain regions are enriched over other brain regions for these traits using gene expression data from GTEx (Figure S7, Online Methods). The results are displayed in Figure 4a and Table S8a. We identified significant enrichments in the cortex relative to other brain regions at FDR<5% for bipolar disorder, schizophrenia, depressive symptoms, and BMI, and in the striatum for migraine. These enrichments are consistent with our understanding of the biology of these traits46–49, but to our knowledge have not previously been reported in any integrative analysis using genetic data. We also identified enrichments in cerebellum for bipolar disorder, years of education, and BMI. However, we caution that differential gene expression in samples from different brain regions can reflect the cell type composition of these brain regions as well as their function. In particular, the cerebellum is known to have a very high concentration of neurons50, and thus cerebellar enrichments could indicate either that the cerebellum is a region that is important in disease etiology, or that neurons are an important cell type. While many pairs of phenotypes had high estimated enrichment correlations in this analysis, migraine tended to have low enrichment correlations with other phenotypes (Figure S8); for example, the estimated enrichment correlation between migraine and schizophrenia was 0.06 (s.e.=0.30) while the estimated enrichment correlation between bipolar disorder and schizophrenia was 0.96 (s.e.=0.05).
Figure 4.
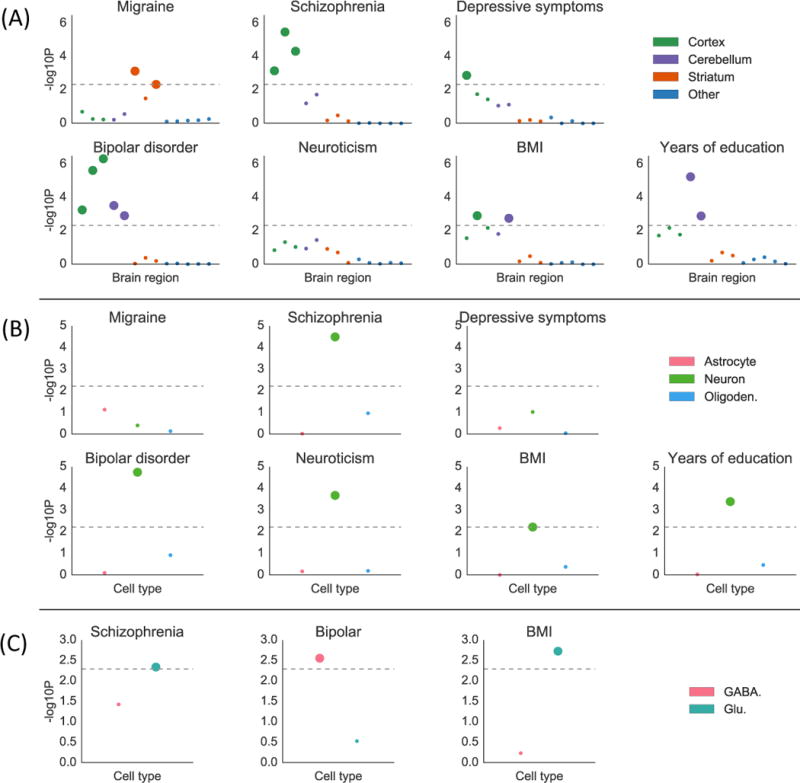
Results of the brain analysis for selected traits. Numerical results for all traits are reported in Table S8. (A) Results from within-brain analysis of 13 brain regions in GTEx, classified into four groups, for seven of 12 brain-related traits. Large points passed the FDR<5% cutoff, –log10(P)=2.34. (B) Results from the data of Cahoy et al. on three brain cell types for seven of 12 brain-related traits. Large points passed the FDR<5% cutoff, –log10(P)=2.22. (C) Results from PyschENCODE data on two neuronal subtypes for three of five neuron-related traits. Large points passed the Bonferroni significance threshold in this analysis, –log10(P)=2.06. GWAS data is described in Table S4, gene expression data is described in the Online Methods and Table S8, and the statistical method is described in the Overview of Methods and the Online Methods.
To address the question of the relative importance of brain cell types, as opposed to brain regions, we analyzed the same set of traits using a publicly available data set of specifically expressed genes identified from different brain cell types purified from mouse forebrain19 (Online Methods). The results of this analysis are displayed in Figure 4b and Table S8b. We identified neuronal enrichments at FDR<5% for five traits: bipolar disorder, schizophrenia, years of education, BMI, and neuroticism. The other cell types did not exhibit significant enrichment for any of the 12 brain-related traits. The enrichment of neurons for all three of the traits with enrichment in cerebellum in the brain-region analysis supports the hypothesis that analyses of brain regions may be confounded by cell-type composition.
To more precisely characterize the neuronal enrichments, we analyzed the five traits with neuronal enrichment at FDR<5% using t-statistics computed by the PsychENCODE consortium20 on differential expression in glutamatergic (excitatory) vs. GABAergic (inhibitory) neurons (Online Methods). The results are displayed in Figure 4c and Table S8c; we used Bonferroni correction in this analysis, as we were testing only 5×2=10 hypotheses. For bipolar disorder, genes that are specifically expressed in GABAergic neurons exhibited heritability enrichment, while genes specific to glutamatergic neurons did not. This result supports the theory that pathology in GABAergic neurons can contribute causally to risk for bipolar disorder51,52. For BMI and schizophrenia, on the other hand, we found significant enrichment in glutamatergic neurons but not in GABAergic neurons.
We were unable to validate the results of these analyses using independent chromatin data. For the two analyses of brain cell types, this was because we were not aware of any available data sets with analogous chromatin data. For the analysis of brain regions, this was because the chromatin annotations that we analyzed were highly correlated across different brain regions and thus some phenotypes showed enrichment in nearly every brain region; we did not consider these non-specific enrichments to be a meaningful validation of our region-specific results using gene expression data.
Analysis of 25 immune-related traits using immune cell expression data
We identified 25 traits with immune enrichment at FDR<5% in our gene expression and/or chromatin analyses (Online Methods). We investigated cell-type-specific enrichments for these traits using gene expression data from the ImmGen project21, which contains microarray data on in 292 immune cell types from mice (Online Methods). This data set contains data for many immune cell types that are not available in the multiple-tissue analysis, and because we compute t-statistics within the data set—i.e., each immune cell vs. other immune cells—the gene sets are less overlapping than those of immune cell types in the multiple-tissue analysis.
We identified enrichments at FDR<5% for 16 traits. Results are displayed in Figure 5, Figure S9 and Tables S9 and S10, and reveal highly trait-specific patterns of enrichment. For primary biliary cirrhosis, the largest and most significant enrichment was in B cells, consistent with literature on the importance of B cells for this trait54. Alzheimer’s disease exhibits enrichment in myeloid cells, as seen previously from genetics55. Asthma and eczema both exhibited enrichment in T and NKT cells; several subclasses of T cells have been shown to be important in asthma,57 and a previous study using chromatin data found an enrichment in T cells for asthma but not in other immune cell types6. Rheumatoid arthritis, Crohn’s disease, inflammatory bowel disease, and multiple sclerosis all exhibited enrichments in a variety of cell types, consistent with complex etiologies for these diseases that involve many different immune cell types58–60. Schizophrenia and bipolar disorder both exhibited an enrichment in T cells. Patients with bipolar disorder have been shown to have a reduction in certain types of T cells, but have equal levels of B cells, NK cells, and monocytes compared to controls61. T cell levels have been shown to vary between schizophrenia cases and controls, but existing literature is not consistent in its description of the direction of effect62. Note that our analysis excludes the HLA region; a previous analysis of the HLA region for schizophrenia implicated the complement system through its role in synaptic pruning, a signal that is distinct from the signal we observe here63. Finally, we identified an enrichment in stromal cells for both diastolic and systolic blood pressure. For each of these two traits, we identified enrichments in the musculoskeletal/connective category in the multiple-tissue analysis that were stronger than the immune enrichments in that analysis, and thus we hypothesize that the enrichment in stromal cells is not providing better resolution on the immune enrichment but instead reflects the more general importance of connective tissue. In enrichment correlation analyses, schizophrenia and bipolar disorder clustered with immunological diseases, while metabolic traits, neurological diseases, and other psychiatric diseases did not (Figure S10).
Figure 5.
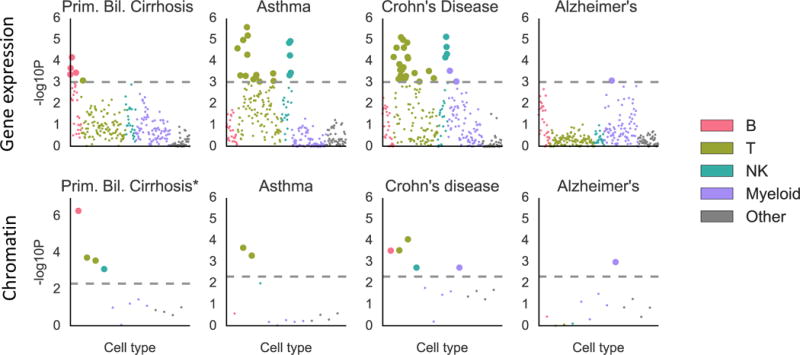
Results of the analysis of ImmGen gene expression data (top) and hematopoiesis ATAC-seq data (bottom) for selected traits. Results for the remaining traits are displayed in Figure S9. Large points passed the FDR<5% cutoff, –log10(P)=3.03 (Gene expression) or –log10(P)=2.32 (Chromatin). Numerical results are reported in Table S10. GWAS data is described in Table S4, gene expression and chromatin data is described in the Online Methods and Table S10, and the statistical method is described in the Overview of Methods and the Online Methods.
To validate these results, we analyzed ATAC-seq (chromatin) data from 13 cell types spanning the hematopoietic hierarchy in humans64. We validated 10 out of 14 top results (Table S9, Online Methods). The only immunological disease whose result was not validated was lupus; the top result for lupus in the ImmGen analysis was a myeloid cell type, while the largest and most significant enrichment in the hematopoiesis data set was a B cell enrichment, consistent with other genetic studies of this trait14.
DISCUSSION
We have shown that applying stratified LD score regression to sets of specifically expressed genes identifies disease-relevant tissues and cell types. Our approach, LDSC-SEG, allows us to take advantage of the large amount of gene expression data available—including fine-grained data for which we do not currently have a comparable chromatin counterpart—to ask questions ranging in resolution from whether a trait is brain-related to whether excitatory or inhibitory neurons are more important for disease etiology. Our results improve our understanding of the phenotypes studied here, highlight the power of GWAS as a source of biological insight, and may also be useful for choosing the relevant tissue or cell type for in-vitro experiments to further elucidate molecular mechanisms underlying genome-wide significant loci identified in genome-wide association studies.
There are several key differences between LDSC-SEG, which relies on gene expression data without genotypes or eQTLs, and approaches that require eQTL data3,13 (Online Methods, Figure S11, Supplementary Note). Our polygenic approach also differs from other gene expression-based approaches such as SNPsea14,15 and DEPICT17, which restrict their analyses to subsets of SNPs that pass a significance threshold (Supplementary Note, Figures S12-S16, Tables S11-S15).
We cannot conclusively say whether gene expression or chromatin data is preferable when both types of data are available in the same tissues and cell types (Online Methods, Figure S4, Figure S17, Table S10, Table S16). Instead, we conclude that the question of which type of data is preferable may depend on complex factors such as which chromatin marks were analyzed, the sample size with which the specifically expressed genes are called, and the overall quality of the data set. When gene expression and chromatin data are available on the same set of tissues or cell types, it may be possible to combine these types of data to improve power, for example by restricting an annotation to tissue-specific chromatin marks near specifically expressed genes, or by combining the P-values from separate analyses of the two types of data. We defer a thorough exploration of this set of possibilities to future work.
Our work is based on the assumption that a tissue or cell type is important for a particular disease if and only if SNPs near genes with high specific expression in that tissue/cell type are enriched for heritability. This assumption leads to several limitations of our approach. First, when analyzing gene expression data from different tissues, cell type composition can confound the analysis, as we demonstrated in our comparison of brain regions; this makes enrichments of organs such as the esophagus or uterus hard to interpret. Second, tissues/cell types with similar gene expression profiles to a causal tissue/cell type will be identified as relevant to disease, just as SNPs in LD with a causal SNP will be identified as associated to disease in a GWAS; thus, significant tissues/cell types should be cautiously interpreted as the “best proxy” for the truly causal tissue/cell type, which may be unobserved. Third, our focus on nearby SNPs prevents us from leveraging signal from regulatory SNPs that act at longer distances. Our approach is also fundamentally limited by the availability of gene expression data and cannot rule out the importance of a given cell type; for example, if the tissue/cell type that is most relevant for a disease occurs in a stage of development or under a stimulus that has not been assayed, then we may not identify enrichments in that tissue/cell type. We would also like to highlight that for most of these phenotypes there is likely not just one causal tissue/cell type, but many.
Our use of a heritability-based approach has advantages but also leads to some limitations. First, our approach will not detect strong but highly localized signals. Second, power increases only modestly with sample size at very large sample sizes (Supplementary Note). Also, because our approach uses stratified LD score regression, it cannot be applied to custom array data, it requires a sequenced reference panel that matches the population studied in the GWAS, and can be affected by model misspecification7. Recent augmentations to the baseline model65 have been shown to help ameliorate model misspecification, but we leave further investigation of this in the context of cell-type-specific analyses to future work.
Another limitation of our method is that its results may be difficult to validate. We undertook a type of validation using independent chromatin data, when there was comparable chromatin data available. However, this type of validation involves a number of challenges. First, we often do not have chromatin data in the same tissues and cell types as the gene expression data. Second, it is not clear that we should always expect results to replicate; for example, it is biologically plausible that SNPs near specifically expressed genes in the relevant tissue are enriched, while SNPs in H3K36me3 peaks called in the tissue are not. Third, our gene expression annotations represent relative activity—we select genes with higher expression in the focal tissue compared to other tissues—while the chromatin annotations that we use here represent absolute activity (although relative chromatin annotations are also possible6,66). Despite these limitations, replicating an enrichment for a particular system, tissue, or cell type using independent chromatin data can provide a strong validation for gene expression results.
Our power to identify disease-relevant tissues and cell types will improve as large GWAS sample sizes become available for more phenotypes, and as gene expression data is generated in new tissues and cell types. This will help advance our understanding of disease biology and lay the groundwork for future experiments exploring specific variants and mechanisms.
ONLINE METHODS
Computing t-statistics
When computing the t-statistic of each gene for a focal tissue, we excluded all samples from similar tissues category (described for each data set below). For example, when computing the t-statistic of specific expression for each gene in cortex using GTEx data, we compared expression in cortex samples to expression in all other samples, excluding other brain regions. We chose to exclude other brain regions because we wanted to include genes that are more highly expressed in brain tissues than in non-brain tissues, even if they are not specific to cortex within the brain. This procedure results in a higher correlation among the t-statistics for the different brain regions; in a separate analysis, we compute within-brain t-statistics to disentangle this signal.
Thus, for a focal tissue (e.g., cortex) in a larger tissue category (e.g., brain), we computed the t-statistic for gene g as follows. We first constructed a design matrix X where each row corresponds to a sample either in cortex or outside of the brain. The first column of X has a 1 for every cortex sample and a -1 for every non-brain sample. The remaining columns are an intercept and covariates (see below). The outcome Y in our model is expression. We fit this model via ordinary least squares, and compute a t-statistic for the first explanatory variable in the standard way:
where MSE is the mean squared error of the fitted model; i.e.,
where N is the number of rows in X. This gives us a t-statistic for each gene for the focal tissue. We then select the top 10% of genes, add a 100kb window around their transcribed regions, and apply stratified LD score regression to the resulting genome annotations as described below.
For visualization purposes and discussion of results, it is often useful to color tissues or cell types according to a categorization; the categorization for visualization is not always the same as the categorization for computing t-statistics. We give the categorization for visualization in the supplementary tables listed in the respective figure captions.
Modifications of our approach
For some analyses, we modified our approach to constructing sets of specifically expressed genes to better take advantage of the data available.
Franke lab data set. The values in the publicly available matrix are not a quantification of expression intensity, but rather a quantification of differential expression relative to other tissues in this data set17,18. Thus, it was not appropriate to compute t-statistics in this data set. We used the original values in place of our t-statistics, then proceeded as described in Figure 1.
Cahoy data set. The data set of Cahoy et al. had available sets of specifically expressed genes for the three cell types that each had between 1,700 and 2,100 genes. We took these to be the gene sets for the three cell types, then proceeded as in the standard approach, adding a 100kb window and applying stratified LD score regression.
PsychENCODE data set. The PsychENCODE data set had available t-statistics for GABAergic neurons vs. Glutamatergic neurons. We used these t-statistics, rather than computing our own.
For the other data sets we analyzed (GTEx, GTEx brain regions, ImmGen), we used the approach described in Figure 1. We view it as an advantage of our method that it can be flexibly adapted to many different types of data.
Application of stratified LD score regression
Stratified LD score regression7 is a method for partitioning heritability. Given (potentially overlapping) genomic annotations one of which is the category of all SNPs, we model the causal effect of SNP j on phenotype Y as drawn from a distribution with mean 0 and variance
| (1) |
(If the genomic annotations are real-valued rather than subsets of SNPs, we can replace with any other function of the SNP indices65.) We then model the phenotype Y as depending linearly on genotype: , where X is a vector of SNP values for an individual, and each SNP has been standardized to mean 0 and variance 1 in the population. Because each SNP is standardized, and because has mean zero, we can call the per-SNP heritability of SNP i. (Note that here, because we model as random, our definition of heritability is different from definitions of heritability in which is fixed, and so we are estimating a fundamentally different quantity than some other methods67.)
Under this model, the expected marginal chi-square association statistic for SNP i reflects the causal contributions not only of SNP i but of SNPs in LD with SNP i. Specifically,
where N is the GWAS sample size, a is a constant that reflects population structure and other sources of confounding,68 and is the LD score of SNP i to category , defined as , where is the squared correlation between SNPs i and j in the population. To estimate the , we first estimate from a reference panel, and we then perform weighted regression on , using a jackknife over blocks of SNPs to estimate standard errors.
The regression coefficient quantifies the importance of annotation , correcting for all other annotations in the model; will equal zero if is not enriched, will be negative if belonging to decreases per-SNP heritability accounting for all other annotations included, and will be positive if belonging to increases per-SNP heritability, accounting for all other factors. Thus, as in our previous cell-type-specific anlaysis7, we compute P-values that test whether is positive. When reporting quantitative results, we normalize the coefficient by our estimate of the mean per-SNP heritability to make it comparable across phenotypes. The normalized coefficient can be interpreted as the proportion by which the per-SNP heritability of an average SNP would increase if were added to it. In addition it is possible to estimate the total heritability, defined as , as well as the heritability in category , defined as , by plugging estimates of into Equation (1), and to compare the proportion of heritability, , to the proportion of SNPs, , where M is the total number of SNPs7.
We analyzed autosomes only and excluded the HLA from all analyses. In each analysis, we jointly fit the following annotations:
The annotation created for our focal tissue by adding 100kb windows around the top 10% of genes ranked by t-statistic.
An identical annotation created for all genes included in the gene expression data set being analyzed.
The baseline model with 52 functional categories, described previously7 and listed in Table S1.
GTEx data set
We downloaded the RNA-seq read counts from GTEx v6p (see URLs), removed genes for which fewer than 4 samples had at least one read count per million, removed samples for which fewer than 100 genes had at least one read count per million, and applied TPM normalization69. We analyzed 53 tissues with an average of 161 samples per tissue. We used the “SMTSD” variable (“Tissue Type, more specific detail of tissue type”) to define our tissues and the “SMTS” variable (“Tissue Type, area from which the tissue sample was taken”) to define the tissue categories for t-statistic computation (Table S2). We used age and sex as covariates for our t-statistics.
Franke lab data set
The Franke lab data set is an aggregation of publicly available microarray gene expression data sets comprising 37,427 samples in human, mouse, and rat17,18. We downloaded the publicly available gene expression data from the DEPICT website (see URLs). The available gene expression values already quantify relative expression for a tissue/cell-type rather than absolute expression for a single sample17,18, and so we used these values in place of our t-statistics. We determined that several pairs of tissues had values that were correlated at r2>0.99, including several that had r2=1. We pruned our data so that no two tissues had r2>0.99. Most of the closely correlated pairs were also biologically closely related so that the interpretation did not depend on which tissue we chose to keep (e.g., plasma and plasma cells, joint and joint capsule). For pairs of tissues where one tissue was more specific than the second, we kept the more specific pair (e.g., nose vs. nasal mucosa, quadriceps muscle vs. skeletal muscle). There were two clusters of highly correlated tissues for which we decided to remove the entire cluster, not keeping any of the tissues, because these clusters had very strong but biologically implausible correlations. The first such cluster was made up of eyelids, conjunctiva, anterior eye segment, tarsal bones, foot bones, and bones of the lower extremity. The second such cluster was made up of connective tissue, bone and bones, skeleton, and bone marrow. After pruning, this data set contained 152 tissues, listed in Table S3.
UK Biobank data
We analyzed data from the full N=500K UK Biobank release23 for 13 traits (P.R. Loh et al., unpublished data). The summary statistics were generated using BOLT-LMM v2.3, an unpublished extension of BOLT-LMM70.
Enrichment correlation
For a pair of phenotypes and a set of tissues/cell types, we defined the enrichment correlation to be the correlation between the regression coefficients corresponding to each tissue/cell type. We estimated the enrichment correlation by correlating the estimates of the regression coefficients, and we quantified uncertainty via block jackknife over 200 sets of consecutive SNPs. We note that when the number of tissues/cell types included is small, the true underlying enrichment correlation may be large even though there is no relationship between the two phenotypes, so we only estimate enrichment correlations when there are at least 10 tissues or cell types.
Distribution of P-values
The correlation structure among annotations can lead to a distribution of P values that is highly non-uniform with many P-values close to 0 or 1 (Figure 2). This is caused by our one-sided test for enrichment, testing whether the regression coefficient—which represents the change in per-SNP heritability due to a given annotation, beyond what is explained by the set of all genes as well as the baseline model—is positive. The P-values near 0 occur due to correlated annotations with true signal, and the P-values near 1 occur due to annotations without true signal that, conditional on the baseline model, are negatively correlated to annotations with true signal as a consequence of our construction of sets of specifically expressed genes; these annotations thus have negative regression coefficients.
Chromatin-based annotations
We downloaded narrow peaks from the Roadmap Epigenomics consortium for DNase hypersensitivity and five activating histone marks: H3K27ac, H3K4me3, H3K4me1, H3K9ac, and H3K36me3 (see URLs). Each of these six features was present in a subset of the 88 primary cell types/tissues, for a total of 397 cell-type-/tissue-specific annotations. We also analyzed peaks called using Homer from EN-TEx, a subgroup of the ENCODE project, for four activating histone marks: H3K27ac, H3K4m3, H3K4me1, and H3K36me3. Each of these four features was present in a subset of 27 tissues that were also included in the GTEx data set, for a total of 93 cell-type-/tissue-specific annotations. For each of these two datasets, of each of the annotations, we tested for enrichment by adding the annotation to the baseline model (see Table S1), together with the union of cell-type-specific annotations within each mark and the average of cell-type-specific annotations within each mark. A positive regression coefficient for a tissue-/cell-type-specific annotation represents a positive contribution of the annotation to per-SNP heritability, conditional on the other annotations. We again computed a P-value to test whether the regression coefficient was positive.
Our analysis of chromatin in this work differs from our previous analysis of chromatin data7 in three ways. First, we use a larger range of marks and tissues/cell types: every track available from the Roadmap Epigenomics website (see URLs) for any of six activating marks, H3K27ac, H3K4me1, H3K4me3, H3K9ac, H3K36me3, and DHS, in any of the 88 primary tissues and cell types available, in addition to recent EN-TEx data. Second, for our analysis of Roadmap data, we used narrow peaks from Roadmap for all of the marks. Previously, we analyzed H3K27ac data from one source6 and H3K4me1, H3K4me3, and H3K9ac data from another source5,12; now that there is a single standard source with uniformly processed data for all Roadmap data, we have switched to using this data. Finally, we controlled more strictly for confounders by including the average across cell types of the cell-type-specific annotations for a given mark as an annotation in the model, so that annotations that tend to fall in areas that are more active overall are not falsely interpreted as cell-type-specific signal.
Classification of tissues/cell types for system-level validation of the results of the multiple-tissue analysis of gene expression
We used the classification for visualization used in Figure 2, classifying the top tissue or cell type for each trait with a significant enrichment into one of the eight systems (excluding “Other”) in the Figure 2 legend. There were three phenotypes whose top tissue fell in the “Other” category; two of these we classified into a new “Reproductive” category. The last one, serous membrane, did not have any comparable tissues in our chromatin data and so we instead attempted to replicate the second most significant result for that phenotype.
Multiple-tissue validation results
The top enrichment from our multi-tissue analysis of gene expression was validated at the system level for 33 out of 34 phenotypes, and at the tissue level for 13 out of 20 (Results). If we allowed an enrichment of any artery sample in GTEx to be validated by an enrichment of any artery sample in EN-TEx (instead of requiring strict matching of aorta, tibial artery, and coronary artery), the number of validations rose from 13 to 16. Of the four remaining results that were not validated, three were an enrichment in lung for an immunological disease; for all three diseases, the top enrichment in the analysis of gene expression (not restricting to tissues shared between GTEx and EN-TEx) was an immune category from the Franke lab dataset, and the top enrichment in the analysis of chromatin data was an immune category in the Roadmap dataset. We hypothesize that the lung samples analyzed in GTEx may have contained substantial amounts of blood and thus exhibited a gene expression signature reflecting immune activity; this is supported by a GO enrichment analysis of the lung gene set, in which the top three results were related to antigen presentation, immune response, and cytokine-mediated signaling, respectively.
Heritability enrichments of chromatin-based annotations
Aggregating all results of the Roadmap and EN-TEx chromatin analyses, at least one tissue was significant at FDR<5% for 44 of the 48 traits (Figure S5 and Tables S5 and S7). Averaging across the most significant annotation for each of these 44 traits, the tissue-specific chromatin annotation spanned 3.3% of the genome and explained 43% of the SNP-heritability (Table S5). The sizes of the annotation ranged from 0.8% to 7.8%, and the estimates of enrichment varied from 3.5× to 33×, representing much more variability than for the top annotations in the multiple-tissue gene expression analysis. Because the annotations were much smaller, the estimates of proportion of heritability tended to be much noisier.
Phenotypes with CNS enrichment
The following 12 traits had CNS enrichment at FDR<5% in either the multiple tissue analysis of gene expression or in the analysis of chromatin data above: schizophrenia, bipolar disorder, Tourette syndrome, epilepsy, generalized epilepsy, ADHD, migraine, depressive symptoms, BMI, smoking status, years of education, and neuroticism. The nervous system has been implicated, either with genetic evidence or non-genetic evidence, for each of these traits7,34,24,32,45,71–73.
Analysis of 13 brain regions using data from GTEx
While the multiple-tissue analysis included annotations for many different brain regions, the gene sets for the different brain regions were often highly overlapping so that for many traits, many brain regions were identified as enriched. For example, nearly every brain region in either the GTEx or Franke lab data was found to be enriched at FDR<5% in schizophrenia (Figure 2). To differentiate among brain regions, we restricted ourselves to gene expression data only from samples from the brain in the GTEx data. We computed t-statistics within the brain-only data set; e.g. we computed t-statistics for cortex vs. other brain regions instead of cortex vs. other tissues in GTEx, and we used these new t-statistics to construct and test gene sets as in the multiple-tissue analysis. In this analysis, we set each tissue to be its own category for computation of t-statistics, and we used age and sex as covariates. Individual-level data was not available for the Franke lab data set, and thus we could not compute within-brain t-statistics for this data set.
An alternative approach would be to undertake a joint analysis of the original 13 annotations from the multiple-tissue analysis. However, joint analysis of 13 highly correlated annotations is likely to be underpowered, while re-computing t-statistics within the brain allows us to construct new annotations with lower correlations (Figure S7), increasing our power. Moreover, differential expression within the brain may allow us to isolate signals from cell types or processes that are unique to a single brain region, separately from the cell types or processes that are unique to the brain but shared among brain regions. Thus, we use differential expression within the brain, rather than joint analysis of the original annotations, to differentiate among brain regions.
Data on three brain cell types from Cahoy et al.19
The authors of Cahoy et al.19 purified neurons, astrocytes, and oligodendrocytes from mouse forebrain, and made lists of specifically expressed genes available for each of these three cell types, which we downloaded (see URLs). To obtain a list of all genes, we also downloaded a list of all genes that passed quality control in their analysis (Table S3b of Cahoy et al.). We mapped from mouse to human genes using orthologs from ENSEMBL (see URLs).
Data on two neuron types from PsychENCODE20
PsychENCODE20 generated RNAseq data from the nuclei of GABAergic and Glutamatergic neurons from the dorsolateral prefrontal cortex of four neurotypical human donors, and computed t-statistics using limma74. We used these t-statistics.
Phenotypes with immune enrichment
Twenty-five traits had immune enrichment at FDR<5% in either the multiple tissue analysis of gene expression or in the analysis of chromatin data. This includes many immunological disorders: celiac disease, Crohn’s disease, inflammatory bowel disease, lupus, primary biliary cirrhosis, rheumatoid arthritis, type 1 diabetes, ulcerative colitis, asthma, eczema, and multiple sclerosis. It also includes Alzheimer’s and Parkinson’s diseases, which are neurodegenerative diseases with an immune component previously identified from genetics75,76, as well as several brain-related traits—ADHD, anorexia nervosa, bipolar disorder, schizophrenia, Tourette syndrome, and neuroticism—and HDL, LDL, triglycerides, diastolic and systolic blood pressure, hypertension, and BMI. Several of the brain-related traits have been previously suggested to have an immune component32,77,78; HDL, LDL, and triglycerides have been linked to immune activation79–82; immune cells are causally involved in blood pressure and hypertension83; and obesity, in addition to contributing to inflammation84, can also be induced in mice through alterations of the immune system85.
Data on 292 immune cell types from ImmGen
We downloaded publicly available microarray gene expression data on 292 immune cell types from the ImmGen Consortium (see URLs). We used both Phase 1 (GSE15907) and Phase 2 (GSE37448) data. The data on GEO were on an exponential scale, so we log transformed the data and mapped to human genes using ENSEMBL orthologs. We defined tissue categories for t-statistic computation using the classification on the main page of immgen.org of cell types into categories: B cells, gamma delta T cells, alpha beta T cells, innate lymphocytes, myeloid cells, stromal cells, and stem cells (Table S10). The classification at immgen.org also has a “T cell activation” category that we collapsed into the alpha beta T cell category because it had data on alpha beta T cells at different stages of activation. We did not include any covariates.
Validation of immune results
To validate the results of the ImmGen analysis, we analyzed ATAC-seq peaks from 13 cell types spanning the hematopoietic hierarchy in humans64. The 13 cell types did not allow us to validate at very high resolution; instead, we classified all cell types from ImmGen and from the hematopoiesis data set using the classification for visualization of Figure 5 into five categories: B cells, T cells, NK cells, myeloid cells, and other cells. There were no stromal cells in the hematopoiesis data set and it was not possible to validate the enrichments for diastolic and systolic blood pressure; this left us with 14 phenotypes with an enrichment at FDR<5% in the ImmGen analysis where the top result fell into one of the first four categories (excluding “Other”). We considered one of these 14 results to be validated if any cell type in the same category from the hematopoiesis data set passed FDR<5%. The four phenotypes whose top results did not replicate were Lupus, schizophrenia, bipolar disorder, and neuroticism.
Differences between LDSC-SEG and eQTL-based approaches
Our approach differs in several key ways from approaches that require eQTL data3,13. First, our approach can be applied to expression data sets such as the Franke lab data set, the Cahoy data set, the PsychENCODE data set, and the ImmGen data set that do not have genotypes or eQTLs available (Table 1). Second, methods based on eQTLs require gene expression sample sizes that are large enough to detect eQTLs. In an analysis of data from the GTEx project, we determined that we could identify strong enrichments such as brain enrichment for schizophrenia with just one brain sample, though subtler enrichments had decreasing levels of significance as the gene expression data were down-sampled (Figure S11, Supplementary Note). Results from our analysis of ImmGen data, which has 2.8 samples per cell type on average, confirm that LDSC-SEG can identify significant enrichments even when the gene expression data has a small number of samples per tissue/cell type, in contrast to eQTL-based methods. Finally, we note that a recent study86 tested 30 phenotypes for tissue-specific enrichment in 44 tissues from GTEx using the TWAS approach87 but concluded that their results “did not suggest tissue-specific enrichment at the current sample sizes.” We share their hypothesis that this is because eQTLs are often shared across tissues even when overall expression levels are very different.
Comparison of gene expression and chromatin for cell-type specific analysis
Our estimated enrichments were higher for the chromatin-based annotations than for the gene expression-based annotations, but the gene expression-based annotations are larger and have less LD to the rest of the genome. Some chromatin marks tend to be more cell type-specific than overall gene expression, but our specifically expressed gene sets have low correlation across tissues (Figure S17). There were two instances in which we had gene expression and chromatin data on the same set of tissues/cell types, and we compared the P-values in our analyses of these data sets. First, we compared our results from GTEx (gene expression) and EN-TEx (chromatin) for the tissues shared between these two data sets in the multiple-tissue analysis, and we found that the two data sets had comparable distributions of P-values (Figure S4). On the other hand, the hematopoietic data set that we analyzed64 had matched ATAC-seq and RNA-seq data, and while our analysis of the ATAC-seq peaks lead to significant enrichments for many traits (Figure 5, Table S10), the RNA-seq data set yielded only a single enrichment for a single trait (Table S16).
Data availability
We have released all genome annotations derived from the publicly available gene expression data that we analyzed at http://data.broadinstitute.org/ alkesgroup/LDSCORE/. This includes all annotations used in Figures 2–5 with the exception of the annotations derived from the PsychENCODE data in Figure 4c, for which we did not have permission to release annotations.
Code availability
Open source software implementing our approach is available at http://www.github.com/bulik/ldsc.
Supplementary Material
Acknowledgments
We are thankful to R Herbst, E Hodis, F Hormozdiari, M Kanai, T Pers, S Riesenfeld, J Ulirsch, and A Veres for helpful comments. This research has been conducted using the UK Biobank Resource (Application Number: 16549). This research was funded by NIH grants R01 MH107649, R01 MH109978, U01 CA194393, and U01 HG009379. HKF was supported by the Fannie and John Hertz Foundation and by Eric and Wendy Schmidt. The data on neuron types were generated as part of the PsychENCODE Consortium, supported by: U01MH103339, U01MH103365, U01MH103392, U01MH103340, U01MH103346, R01MH105472, R01MH094714, R01MH105898, R21MH102791, R21MH105881, R21MH103877, and P50MH106934 awarded to: Schahram Akbarian (Icahn School of Medicine at Mount Sinai), Gregory Crawford (Duke), Stella Dracheva (Icahn School of Medicine at Mount Sinai), Peggy Farnham (USC), Mark Gerstein (Yale), Daniel Geschwind (UCLA), Thomas M. Hyde (LIBD), Andrew Jaffe (LIBD), James A. Knowles (USC), Chunyu Liu (UIC), Dalila Pinto (Icahn School of Medicine at Mount Sinai), Nenad Sestan (Yale), Pamela Sklar (Icahn School of Medicine at Mount Sinai), Matthew State (UCSF), Patrick Sullivan (UNC), Flora Vaccarino (Yale), Sherman Weissman (Yale), Kevin White (UChicago) and Peter Zandi (JHU).
Footnotes
AUTHOR CONTRIBUTIONS
H.K.F. and A.L.P. designed the study. H.K.F., Y.A.R., K.S., and S.P. analyzed data. H.K.F. and A.L.P. wrote the manuscript with assistance from Y.A.R., V.A., K.S., A.G., A.B., S.G., P.R.L., C.L., N.S., G.G., A.S., E.M., S.P., J.R.B.P., J.D.B., B.E.B., S.R., S.M., and B.M.N.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
- LDSC software, including LDSC-SEG: https://github.com/bulik/ldsc.
- Gene sets and LD scores from this paper: https://data.broadinstitute.org/alkesgroup/LDSCORE/.
- GTEx: http://www.gtexportal.org)
- Cahoy et al. data: http://jneurosci.org/content/suppl/2008/01/03/28.1.264.DC1, see Tables S4-S6.
- ImmGen, https://www.immgen.org/
- Roadmap Epigenomics: http://www.roadmapepigenomics.org.
- GERA data set (database of Genotypes and Phenotypes (dbGaP), phs000674.v1.p1): http://www-ncbi-nlm-nih-gov.libproxy.mit.edu/projects/gap/cgi-bin/study.cgi?study_id=phs000674.v1.p1.
- makegenes.sh: https://github.com/freeseek/gwaspipeline
References
- 1.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kundaje A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trynka G, et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farh KKH, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2014;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Kellis M. Joint Bayesian inference of risk variants and tissue-specific epigenomic enrichments across multiple complex human diseases. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maurano MT, et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickrell JK. Joint Analysis of Functional Genomic Data and Genome-wide Association Studies of 18 Human Traits. Am J Hum Genet. 2014;94:559–573. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kichaev G, et al. Integrating Functional Data to Prioritize Causal Variants in Statistical Fine-Mapping Studies. PLoS Genet. 2014;10:e1004722. doi: 10.1371/journal.pgen.1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gusev A, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet. 2014;95:535–552. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ongen H, et al. Estimating the causal tissues for complex traits and diseases. bioRxiv. 2016 doi: 10.1038/ng.3981. [DOI] [PubMed] [Google Scholar]
- 14.Hu X, et al. Integrating Autoimmune Risk Loci with Gene-Expression Data Identifies Specific Pathogenic Immune Cell Subsets. Am J Hum Genet. 2011;89:496–506. doi: 10.1016/j.ajhg.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slowikowski K, Hu X, Raychaudhuri S. SNPsea: an algorithm to identify cell types, tissues and pathways affected by risk loci. Bioinformatics. 2014;30:2496–2497. doi: 10.1093/bioinformatics/btu326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gormley P, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet. 2016;48:856–866. doi: 10.1038/ng.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pers TH, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015;6:5890. doi: 10.1038/ncomms6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fehrmann RSN, et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat Genet. 2015;47:115–125. doi: 10.1038/ng.3173. [DOI] [PubMed] [Google Scholar]
- 19.Cahoy JD, et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akbarian S, et al. The PsychENCODE project. Nat Neurosci. 2015;18:1707–1712. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heng TSP, Painter MW, Immunological Genome Project Consortium The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 22.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudlow C, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anttila V, et al. Analysis of shared heritability in common disorders of the brain. bioRxiv. 2016 doi: 10.1126/science.aap8757. 048991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambert JC, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cross-Disorder Group of the Psychiatric Genomics Consortium. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 2014;13:893–903. doi: 10.1016/S1474-4422(14)70171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo D, et al. Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet. 2014;94:511–521. doi: 10.1016/j.ajhg.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traylor M, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol. 2012;11:951–962. doi: 10.1016/S1474-4422(12)70234-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patsopoulos NA, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol. 2011;70:897–912. doi: 10.1002/ana.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nalls MA, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ripke S, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okbay A, et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature. 2016;533:539–542. doi: 10.1038/nature17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okbay A, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat Genet. 2016;48:624–633. doi: 10.1038/ng.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manning AK, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okada Y, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2013;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jostins L, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bradfield JP, et al. A Genome-Wide Meta-Analysis of Six Type 1 Diabetes Cohorts Identifies Multiple Associated Loci. PLOS Genet. 2011;7:e1002293. doi: 10.1371/journal.pgen.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dubois PCA, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bentham J, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47:1457–1464. doi: 10.1038/ng.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cordell HJ, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun. 2015;6:8019. doi: 10.1038/ncomms9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wood AR, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tfelt-Hansen PC, Koehler PJ. One hundred years of migraine research: major clinical and scientific observations from 1910 to 2010. Headache. 2011;51:752–778. doi: 10.1111/j.1526-4610.2011.01892.x. [DOI] [PubMed] [Google Scholar]
- 46.Hanford LC, Nazarov A, Hall GB, Sassi RB. Cortical thickness in bipolar disorder: a systematic review. Bipolar Disord. 2016;18:4–18. doi: 10.1111/bdi.12362. [DOI] [PubMed] [Google Scholar]
- 47.Callicott JH, et al. Physiological Dysfunction of the Dorsolateral Prefrontal Cortex in Schizophrenia Revisited. Cereb Cortex. 2000;10:1078–1092. doi: 10.1093/cercor/10.11.1078. [DOI] [PubMed] [Google Scholar]
- 48.Medic N, et al. Increased body mass index is associated with specific regional alterations in brain structure. Int J Obes. 2016;40:1177–1182. doi: 10.1038/ijo.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maleki N, et al. Migraine attacks the Basal Ganglia. Mol Pain. 2011;7:71. doi: 10.1186/1744-8069-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herculano-Houzel S, Lent R. Isotropic Fractionator: A Simple, Rapid Method for the Quantification of Total Cell and Neuron Numbers in the Brain. J Neurosci. 2005;25:2518–2521. doi: 10.1523/JNEUROSCI.4526-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakai T, et al. Changes in density of calcium-binding-protein-immunoreactive GABAergic neurons in prefrontal cortex in schizophrenia and bipolar disorder. Neuropathology. 2008;28:143–150. doi: 10.1111/j.1440-1789.2007.00867.x. [DOI] [PubMed] [Google Scholar]
- 52.Benes FM, Berretta S. GABAergic Interneurons: Implications for Understanding Schizophrenia and Bipolar Disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 53.Dhirapong A, et al. B cell depletion therapy exacerbates murine primary biliary cirrhosis. Hepatol Baltim Md. 2011;53:527–535. doi: 10.1002/hep.24044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, et al. Ongoing activation of autoantigen-specific B cells in primary biliary cirrhosis. Hepatol Baltim Md. 2014;60:1708–1716. doi: 10.1002/hep.27313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raj T, et al. Polarization of the Effects of Autoimmune and Neurodegenerative Risk Alleles in Leukocytes. Science. 2014;344:519–523. doi: 10.1126/science.1249547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang K, et al. A common haplotype lowers PU1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20:1052–1061. doi: 10.1038/nn.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just TH2 cells. Nat Rev Immunol. 2010;10 doi: 10.1038/nri2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Müller-Ladner U, Pap T, Gay RE, Neidhart M, Gay S. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1:102–110. doi: 10.1038/ncprheum0047. [DOI] [PubMed] [Google Scholar]
- 59.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 60.Sospedra M, Martin R. Immunology of Multiple Sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 61.Barbosa IG, Machado-Vieira R, Soares JC, Teixeira AL. The immunology of bipolar disorder. Neuroimmunomodulation. 2014;21:117–122. doi: 10.1159/000356539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steiner J, et al. Acute schizophrenia is accompanied by reduced T cell and increased B cell immunity. Eur Arch Psychiatry Clin Neurosci. 2010;260:509–518. doi: 10.1007/s00406-010-0098-x. [DOI] [PubMed] [Google Scholar]
- 63.Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corces MR, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gazal S, et al. Linkage disequilibrium dependent architecture of human complex traits reveals action of negative selection. bioRxiv. 2017 doi: 10.1101/082024. 082024. Nature Genetics, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi H, Kichaev G, Pasaniuc B. Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data. Am J Hum Genet. 2016;99:139–153. doi: 10.1016/j.ajhg.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci Theor Den Biowissenschaften. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 70.Loh PR, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47:284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Backenroth D, et al. Tissue-specific functional effect prediction of genetic variation and applications to complex trait genetics. bioRxiv. 2016 [Google Scholar]
- 72.Wilens TE, Biederman J, Spencer TJ. Attention Deficit/Hyperactivity Disorder Across the Lifespan. Annu Rev Med. 2002;53:113–131. doi: 10.1146/annurev.med.53.082901.103945. [DOI] [PubMed] [Google Scholar]
- 73.Davis LK, et al. Partitioning the Heritability of Tourette Syndrome and Obsessive Compulsive Disorder Reveals Differences in Genetic Architecture. PLOS Genet. 2013;9:e1003864. doi: 10.1371/journal.pgen.1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gjoneska E, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature. 2015;518:365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gagliano SA, et al. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s. bioRxiv. 2016 doi: 10.1101/059519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rege S, Hodgkinson SJ. Immune dysregulation and autoimmunity in bipolar disorder: Synthesis of the evidence and its clinical application. Aust N Z J Psychiatry. 2013;47:1136–1151. doi: 10.1177/0004867413499077. [DOI] [PubMed] [Google Scholar]
- 78.Elamin I, Edwards MJ, Martino D. Immune dysfunction in Tourette syndrome. Behav Neurol. 2013;27:23–32. doi: 10.3233/BEN-120295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jin W, Millar JS, Broedl U, Glick JM, Rader DJ. Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J Clin Invest. 2003;111:357–362. doi: 10.1172/JCI16146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Broedl UC, et al. Endothelial lipase promotes the catabolism of ApoB-containing lipoproteins. Circ Res. 2004;94:1554–1561. doi: 10.1161/01.RES.0000130657.00222.39. [DOI] [PubMed] [Google Scholar]
- 81.Feingold KR, Grunfeld C. The role of HDL in innate immunity. J Lipid Res. 2011;52:1–3. doi: 10.1194/jlr.E012138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lo JC, et al. Lymphotoxin beta receptor-dependent control of lipid homeostasis. Science. 2007;316:285–288. doi: 10.1126/science.1137221. [DOI] [PubMed] [Google Scholar]
- 83.Harrison DG. The Immune System in Hypertension. Trans Am Clin Climatol Assoc. 2014;125:130–140. [PMC free article] [PubMed] [Google Scholar]
- 84.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 85.Zlotnikov-Klionsky Y, et al. Perforin-Positive Dendritic Cells Exhibit an Immuno-regulatory Role in Metabolic Syndrome and Autoimmunity. Immunity. 2015;43:776–787. doi: 10.1016/j.immuni.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 86.Mancuso N, et al. Integrating Gene Expression with Summary Association Statistics to Identify Genes Associated with 30 Complex Traits. Am J Hum Genet. 2017;100:473–487. doi: 10.1016/j.ajhg.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gusev A, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have released all genome annotations derived from the publicly available gene expression data that we analyzed at http://data.broadinstitute.org/ alkesgroup/LDSCORE/. This includes all annotations used in Figures 2–5 with the exception of the annotations derived from the PsychENCODE data in Figure 4c, for which we did not have permission to release annotations.