

Abstract
Parkinson’s disease (PD) is a movement disorder with widespread neurodegeneration in the brain. Significant oxidative, reductive, metabolic, and proteotoxic alterations have been observed in PD postmortem brains. The alterations of mitochondrial function resulting in decreased bioenergetic health is important and needs to be further examined to help develop biomarkers for PD severity and prognosis. It is now becoming clear that multiple hits on metabolic and signaling pathways are likely to exacerbate PD pathogenesis. Indeed, data obtained from genetic and genome association studies have implicated interactive contributions of genes controlling protein quality control and metabolism. For example, loss of key proteins that are responsible for clearance of dysfunctional mitochondria through a process called mitophagy has been found to cause PD, and a significant proportion of genes associated with PD encode proteins involved in the autophagy-lysosomal pathway. In this review, we highlight the evidence for the targeting of mitochondria by proteotoxic, redox and metabolic stress and the role autophagic surveillance in maintenance of mitochondrial quality. Furthermore, we summarize the role of α-synuclein, LRRK2, and tau in modulating mitochondrial function and autophagy. Among the stressors that can overwhelm the mitochondrial quality control mechanisms, we will discuss 4-hydroxynonenal (HNE) and nitric oxide. The impact of autophagy is context depend and as such can have both beneficial and detrimental effects. Furthermore, we highlight the potential of targeting mitochondria and autophagic function as an integrated therapeutic strategy and the emerging contribution of the microbiome to PD susceptibility.
Keywords: Bioenergetic health index, 4-hydroxynonenal, thiol, post-translational modification, glucose metabolism, endosome-lysosomes, microglia, α-synuclein, cathepsin D
Graphical Abstract
This review provides highlights on recent observations regarding the multifacet impact of pathological proteins, endogenously produced reactive species, environmental toxins, and metabolism including glucose and fatty acid metabolism, on mitochondria - autophagy function, in the context of Parkinson’s disease. The review also discuss future studies of designing targeted strategies to aid the treatment of Parkinson’s Disease.
Introduction
Parkinson’s disease (PD) was described 200 years ago as a disease of shaking palsy (Obeso et al. 2017). We now know that PD is a condition with both motor and non-motor symptoms including cognitive impairment, due to neurodegeneration of dopaminergic and non-dopaminergic neurons, with prodromal symptoms with gastrointestinal, olfactory, and REM sleep disturbances. Approximately 1 in 100 Americans older than 60 years are affected by this disease and it is estimated that between 2010 and 2030, the number of individuals 65 years or older with PD will increase by 77% (Dorsey et al. 2013, Fritsch et al. 2012). The Parkinson’s Disease Foundation has estimated that the combined annual direct and indirect cost of PD in the United States is $25 billion. Worldwide, it affects 1–3% of the population of 50 years or older (Dehay et al. 2015).
A key pathological feature of PD is the appearance of Lewy bodies and Lewy Neurite in the brain. Protease resistant α-synuclein aggregates are a major component in the Lewy Bodies and Lewy Neurites. A 140 amino acid protein, α-synuclein has been found in the cytoplasm, mitochondria, and the nucleus, although its physiological function is still unclear other than it may be involved in synaptic function (Abeliovich et al. 2000). Pathologically α-synuclein may exert neurotoxicity by disrupting protein trafficking, or interfering with autophagy-lysosomal and mitochondrial function, thus contributing to neurodegeneration and PD pathogenesis and progression (Lee & Trojanowski 2006, Lashuel et al. 2013, Schmid et al. 2013). Cellular damage can also be spread from a donor cell to neighboring cells by inter-cellular transfer of α-synuclein (Luk et al. 2012a, Luk et al. 2012b, Volpicelli-Daley et al. 2011, Luk et al. 2009, Steiner et al. 2011).
There are currently no available pharmacological approaches for preventing or attenuating neuronal loss in PD; consequently, treatment options are limited to symptomatic management. Therefore, a better understanding of the fundamental processes that affect PD development and progression is of paramount importance and has been an area of intense research. Because of the importance of mitochondria and the autophagy-lysosomal activities in neurons and PD, in this review, we highlight some of the recent studies that focused on oxidative, metabolic and proteotoxic stress and discuss how they affect mitochondrial and autophagic function. Finally, we will overview the progress in developing mitochondria and autophagy targeted therapeutic strategies (Fig. 1).
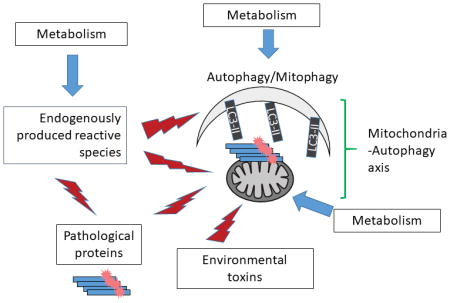
Figure 1. Mitochondrial function and autophagy: integrating redox, metabolic and proteotoxic stress in Parkinson’s disease.
Mitochondria and the autophagy machinery are essential for neuronal function and health. Mitochondria are not only the major source of cellular energy but also a signaling nexus for metabolites and modulating redox status and the bioenergetic health may be used as biomarkers for disease prognosis and response to thereapeutics. Because of the high demand of cellular energy and the high content of fatty acids in neurons, reactive oxygen and nitrogen species can be generated intracellularly, as well as from neighboring glia cells, or even due to exposure to environmental toxins. Metabolic regulation of redox signaling, mitochondrial function and autophagy is of importance in the central nervous system via pathways including but not limited to glycolysis, hexoamine biosynthesis, and fatty acid oxidation. Furthermore, protein homeostasis, as represented by α-synuclein and tau homeostasis is important for Parkinson’s disease. α-synuclein can target to and inhibit mitochondrial function, and itself is subject to HNE modification and nitration. Autophagy is a major pathway for α-synuclein degradation and the clearance of damaged mitochondria, but is also targeted by proteotoxic proteins including α-synuclein, tau and LRRK2. In this review, we highlight the evidence of how mitochondria are targeted by proteotoxic, redox and metabolic stress, while maintained by the autophagic surveillance. These areas present opportunities to better understand Parkinson’s disease pathogenesis and potential for development of effective therapies.
A. The mitochondrial-autophagy axis in Parkinson’s disease
Accumulation of toxic proteins and damaged mitochondria have been noted as key features of PD (Goedert et al. 2017). Damaged mitochondria can be a source and further increase reactive species production (Murphy 2009). Removal of these toxic cellular components is therefore essential for neuronal health, and this is largely dependent on the lysosomal-mediated autophagic function. Both mitochondrial and autophagy-lysosomal dysfunction have been found in postmortem Parkinson’s disease brains (Chu Y 2009, Schapira 2007, Ryan et al. 2015, Swerdlow 2009, Beal 2003, Parker & Swerdlow 1998, Schapira et al. 1990, Bindoff et al. 1989), as well as dopaminergic neurons derived from patients with idiopathic and familial PD (Burbulla et al. 2017, Wang et al. 2017). A recent study also demonstrated downregulation of mRNAs for key autophagy genes in the peripheral blood mononuclear cells of patients with PD (Miki et al. 2017). Here we will review the evidence of mitochondrial and autophagy dysfunction in PD. Understanding of the cause and consequences of this dysfunction is crucial to help find interventions for therapeutics for PD.
1. Mitochondria as targets for environmental toxins that contribute to Parkinson’s disease risk
The mitochondrion is a multifunctional organelle that plays an essential role not only in energy production, but also in thermogenesis, calcium homeostasis, generating and maintaining key cellular metabolites, and redox signaling (Hill et al. 2012). With age, mitochondrial function declines, while mtDNA damage/mutations increase, leading to increased risk for PD (Schapira & Gegg 2011, Reeve et al. 2013). In this section, we will discuss how environmental toxins contribute to proteotoxicity and the formation of Lewy bodies.
Mitochondria contain several hundred redox centers that have the potential to react with xenobiotics generating secondary reactive metabolites or can be secondary targets of ROS (reactive oxygen species) generated within the cell. Since these damaged redox proteins in mitochondria can exhibit a gain of function in which they amplify oxidative damage or disrupt normal redox signaling then it is essential that they are removed. If this does not occur, damaged respiratory proteins and mtDNA can impair the capacity of the organelle quality control mechanisms and thereby amplify the initial insult through a progressively increased metabolic dysfunction and oxidative stress (Hill et al. 2012, Murphy 2009). These patterns of responses have been shown experimentally in the models of PD, which have used xenobiotics that are mitochondrial toxins. For example, 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP), a by-product in the drug abused by a group of young adults in California was found to be responsible for causing parkinsonism, through a mechanism which was later found to be associated with mitochondrial complex I inhibition (Ramsay et al. 1987, Nicklas et al. 1987, Mizuno et al. 1987). Analogous data has been reported for the complex I inhibitor rotenone which is found in plants and used to control fish populations (Clark 1933). There are possibly a range of environmental toxins which increase the susceptibility to PD including pesticides such as paraquat which directly or indirectly target mitochondrial function (Priyadarshi et al. 2001, Priyadarshi et al. 2000, van der et al. 2012, Tanner et al. 2011, Giordano et al. 2012, Giordano et al. 2014b) (Fig. 1).
The targets involved in the environmental xenobiotics are concordant with the biological pathways that are thought to be critical for the development of PD. For example, it has been shown that mitochondrial dysfunction in dopaminergic neurons leads to parkinsonian phenotypes (Ekstrand et al. 2007). Further, the protein α-synuclein associated with PD pathogenesis, targets the mitochondria and decreases mitochondrial function (Devi et al. 2008, Liu et al. 2009, Loeb et al. 2010b, Elkon et al. 2002, Ryan et al. 2015, Martinez & Greenamyre 2012, Swerdlow 2009, Schapira 2007, Beal 2003, Parker & Swerdlow 1998, Schapira et al. 1990, Bindoff et al. 1989). If these mechanisms act together, exposure to a xenobiotic, such as rotenone, can exacerbate an underlying synucleinopathy since the cell will be less capable of meeting the energetic requirements for protection against proteotoxicity. In this paradigm, sub-threshold levels of proteotoxicity or metabolic impairment can synergize to promote disease progression. It is likely that these effects are not solely confined to the central nervous system and can be expressed in other tissues and organs. This raises the possibility that circulating peripheral blood mononuclear cells or platelets can serve as biomarkers of deteriorating mitochondrial quality control (Kramer et al. 2014). This is now amenable to testing in human populations. Cellular Bioenergetics can be determined using the mitochondrial stress test which is based on the sequential addition of well-characterized inhibitors of oxidative phosphorylation to cells while measuring their rate of oxygen consumption and extracellular acidification (Hill et al. 2012). We have proposed that the resulting bioenergetic profile in cells isolated from normal compared to patients can be used to determine a Bioenergetic Health Index (BHI) (Chacko et al. 2014). Cellular bioenergetics health can then be compared to mitochondrial function in a parallel experiment using the same cells, by assessing the activity of protein complexes involved in oxidative phosphorylation including complex I and IV (Salabei et al. 2014). This new concept may help in developing a modifiable biomarker for PD development, prognosis and response to therapy as well as lead to a better understanding of mitochondrial damage and quality control in the disease.
2. Autophagy and mitophagy in neurodegeneration
Since different mechanisms converge to promote proteotoxicity and disrupt metabolic function, enhancing repair pathways would be beneficial irrespective of these diverse mechanisms of cell impairment. The removal of damaged proteins and mitochondria rely on the highly regulated lysosomal-mediated autophagy pathway (Zhang 2013). Autophagy integrates signals ranging from nutrient availability to cellular stress and, as shown recently, oxidative protein damage including oxidized lipids (Levonen et al. 2014, de Duve & Wattiaux 1966, Takeshige et al. 1992, Mizushima et al. 2008, Zhang 2015). Since its discovery in the 1960s and the identification of the genes encoding the autophagic machinery in the 1990s (de Duve & Wattiaux 1966, Takeshige et al. 1992), autophagy has been found to play an important role in health and disease (Mizushima et al. 2008). More than 38 autophagy related proteins and many lysosomal proteins are involved in the process (Zhang 2015).
At a technical level, the extension of the autophagy field to encompass a broad range of pathologies has been enabled by straightforward experimental strategies (Klionsky et al. 2016, Liang et al. 2013, Mitchell et al. 2013). For example, the formation and degradation by the lysosomes of the double membraned vesicles, which contain lipidated microtubule associated protein 1A/1B light chain 3 (LC3-II), has been used for assessment of autophagic activities (Klionsky et al. 2016, Liang et al. 2013, Mitchell et al. 2013). Accumulation of p62 protein in the absence of its transcription activation, and accumulated ubiquitinated proteins have been used as additional markers for autophagy inhibition (Jaber et al. 2012a, Jaber et al. 2012b, Parekh et al. 2013, Parekh et al. 2017). It is now technically possible to monitor autophagy and mitophagy in live cells. For example, RFP-GFP-LC3, MitoKeima and Mito-QC mice have been generated to facilitate autophagy/mitophagy flux analyses based on sensitivity of green fluorescent protein to the acidic environment of the lysosome. Using these approaches translocation of autophagosomes and mitochondria to the lysosomes for degradation can be monitored over a broad range of conditions (Li et al. 2014, Sun et al. 2015, Williams et al. 2017) (Fig. 2).
Figure 2. Assessment of autophagic and mitophagic flux.
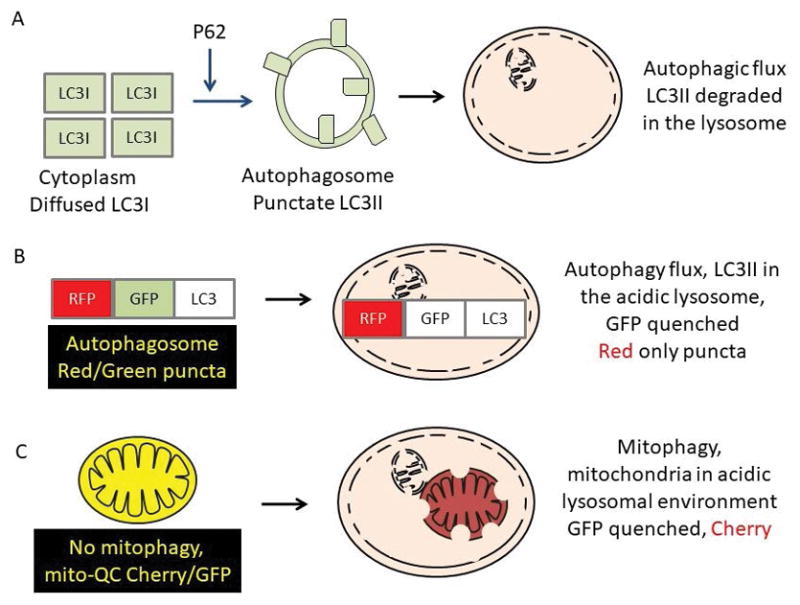
A. The amount of LC3II in the presence and absence of inhibitors that blocks lysosomal degradation of LC3II, and the p62 accumulation in the absence of enhanced p62 transcription activation is used to assess autophagic flux. B. Cells or animals expressing reporter gene RFP-GFP-LC3 can also be examined for autophagic flux based on the amount of LC3 that reaches the lysosomes due to the pH sensitivity of GFP. C. Cells or animals expressing reporter gene mitochondrial matrix targeted Cherry-GFP-LC3 can be examined for mitophagic flux based on the amount of LC3 that reaches the lysosomes due to the pH sensitivity of GFP.
Key findings from these studies supporting the pathological importance of the autophagy pathway have come from a number of different model systems. For example, deficits in key autophagy genes, Atg5 or Atg7, in knockout mice, leads to accumulation of ubiquitinated proteins and neurodegeneration (Hara et al. 2006, Komatsu et al. 2006). Mitophagy plays a key role in mitochondrial quality control (Kim & Lemasters 2011, Lemasters 2005) and proteins that regulate mitophagy such as Parkin, PINK1 and DJ-1, are associated with familial PD (Redmann et al. 2016, Lubbe & Morris 2014, Redmann et al. 2014). Recent studies demonstrate that the most frequently mutated protein for familial PD, LRRK2, also plays a role in regulating autophagy, and its mutation may be synergistic with α-synuclein in impairing autophagic flux (Saha et al. 2015). With more studies and a better understanding of mitophagy regulation (Kim et al. 2016, Williams et al. 2017), there is hope that new effective therapeutic targets may be developed to focus on improving mitochondrial quality control and cellular metabolism.
3. The importance of the lysosome
In the previous sections, we focused on mitochondria as both an initiator and target of proteotoxicity. Of course dysfunctional protein responses can have broad affects in the cell and in this regard as a key player in the autophagy pathway, lysosomes play a central role in integrating autophagic and endosomal activities and in degrading damaged lipids, proteins, and organelles. Fifty hydrolases perform degradative activities in the lysosomes. Nonetheless, many of the hydrolases are essential for lysosomal activities and organismal health. A single enzyme deficiency could produce lysosomal storage diseases (LSD) and neuronal ceroid lipofuscinosis (NCL) (Schneider & Zhang 2010). A significant proportion of the loci associated with PD are involved in or disrupt vesicular trafficking and the autophagy-lysosome pathway, including LRRK2 and GBA (Gan-Or et al. 2015, Diana Chang 2017, Schneider & Zhang 2010, Benitez et al. 2016, Chiasserini et al. 2015, Mata et al. 2015), suggesting a significant impact of lysosomal perturbation in the disease. Among the studies worth noting, a recent study further demonstrated deficient lysosomal enzyme alpha-Galactosidase A in sporadic PD postmortem brains (Nelson et al. 2018). LRRK2 has been shown to impact endolysosomes and autophagy, and may have both autophagy promoting and autophagy inhibitory roles depending on the time/location/cellular context (Manzoni 2017).
Insufficient lysosomal turnover of the pathologic α-synuclein can result in α-synuclein accumulation. This is most evident from studies with the cathepsins that are lysosomal proteases responsible for autophagic degradation. For example, it has been shown that cathepsin D levels are decreased in substantia nigra neurons of PD patients compared to age-matched controls (Chu Y 2009). Furthermore, overexpression of cathepsin D, but not cathepsin B, in C. elegans, decreases α-synuclein toxicity (Qiao et al. 2008). Interestingly, cathepsin B is implicated in genome-wide association studies to be associated with PD (Diana Chang 2017). It has been shown that microglial secreted cathepsin B induces neuronal cell death (Kingham & Pocock 2001) and that cathepsin B enhances aggregate formation and production of reactive oxygen species induced by exogenous α-synuclein fibrils (Tsujimura et al. 2014, Freeman et al. 2013).
B. Proteotoxic signaling regulates mitochondrial function and autophagy
As introduced above the proteotoxic potential of α-synuclein is an important aspect of the molecular mechanisms of cell dysfunction in neurodegenerative disease. The pathologies of PD are known to involve Lewy bodies and Lewy neurites, while the major component of these structures was identified as α-synuclein. Among others, α-synuclein, LRRK2 and PARK7 gene mutations have been found to be responsible for a subset of familial PD (Obeso et al. 2017). α-synuclein is a 140 amino acid protein expressed in neurons, while its accumulation occurs in PD, Alzheimer’s disease, and Dementia with Lewy Bodies brains. Because α-synuclein is associated with pathology, the appearance of phosphorylated or oligomerized forms of α-synuclein in the cerebral spinal fluid are being explored as potential biomarkers with more improved and specific antibodies (Majbour et al. 2016). LRRK2 is a large protein of 2,537 amino acids encompassing both kinase and GTPase domains, its mutations are the most frequently found among familial PD (Obeso et al. 2017). Parkin, PINK1 and PARK7/DJ-1 are recessive PD, whose loss of function contribute to PD pathogenesis and all of these three genes are implicated in regulating mitophagy (Obeso et al. 2017).
1. α-synuclein
Although exact mechanisms are unclear, α-synuclein is normally involved in synaptic function (Abeliovich et al. 2000), and misfolding, mutation, or post-translational modification leading to its accumulation is potentially pathogenic (Lashuel et al. 2013, Goedert et al. 2017). Deficiencies in lysosomal-dependent α-synuclein protein degradation lead to α-synuclein accumulation and neurodegeneration in animal models (Qiao et al. 2008, Cullen et al. 2009). Lysosomal perturbation has been implicated in PD or parkinsonism genetic mutations or polymorphisms, including LRRK2, SNCA, LAMP3, GBA, and ATP13A2 (Gan-Or et al. 2015). Pathological α-synuclein can disrupt protein trafficking, induce neuroinflammation, and interfere with the autophagy-lysosomal pathway and mitochondrial function (Lee & Trojanowski 2006, Lashuel et al. 2013, Schmid et al. 2013, Zhou et al. 2016), toxicity is not only intracellular, but also can be intercellular due to the fact that α-synuclein can spread from a donor cell to neighboring cells and thus propagate cellular damage (Luk et al. 2012a, Luk et al. 2012b, Volpicelli-Daley et al. 2011, Luk et al. 2009, Steiner et al. 2011, Rutherford et al. 2015). Endocytosis, direct penetration, trans-synaptic transmission, membrane receptors, and Parkin regulation of lipid rafts have all been suggested to mediate α-synuclein spreading (Lashuel et al. 2013, Cha et al. 2015). Autophagic failure has been shown to promote the intercellular transfer of α-synuclein (Desplats et al. 2009, Lee et al. 2013, Ejlerskov et al. 2013).
Not surprisingly, α-synuclein interacts with key redox regulated pathways and proteins in the cell. In the cytosol, α-synuclein interacts with Cu, Zn superoxide dismutase and affects its dimerization without affecting its activity (Helferich et al. 2016). α-synuclein targets mitochondria and perturbs mitochondrial function in cell culture and transgenic mouse studies (Elkon et al. 2002, Devi et al. 2008, Shavali et al. 2008, Parihar et al. 2009, Liu et al. 2009, Loeb et al. 2010a, Chinta et al. 2010, Loeb et al. 2010b, Zhu et al. 2011, Imaizumi et al. 2012, Braidy et al. 2013, Perfeito et al. 2014, Chen et al. 2015a, Di Maio et al. 2016). A recent study using a novel protein oxidation detection method also demonstrated α-synuclein radical formation after exposure to the pesticides Maneb and paraquat and caused cytochrome c release to the cytosol (Kumar et al. 2016). In total, one of the key mediators of the proteotoxicity associated with PD has clear mechanistic links to metabolism and redox biology.
2. Other neurotoxic proteins
In addition to α-synuclein, a few other proteins are also either prone to aggregation or connected to autophagy/mitophagy regulation in PD. Mechanisms of their actions may involve their interactions with α-synuclein or each other. The microtubule associated protein tau has been shown to co-localize with α-synuclein in dopaminergic neurons (Wills et al. 2010). The co-localization may play a role in sequestering tau from its normal function as α-synuclein fibrils have been shown to promote tau aggregation, resulting in decreased microtubule polymerization (Oikawa et al. 2016). The ability of LRRK2 to interact with tau has been demonstrated in HEK-293 cells, where LRRK2 overexpression can increase tau protein, its formation of oligomers/aggregates in these cells, and its secretion with the underlying mechanism involving proteasomal but not autophagy impairment (Guerreiro et al. 2016). LRRK2 G2019S mutant induced dendrite degeneration in Drosophila in a tau dependent manner (Lin et al. 2010). Interestingly, as Tau, LRRK2 has also been shown to interact with cytoskeleton proteins, and may play a role in microtubule dynamics, and microtubule-mediated vesicular transport (Blanca Ramirez et al. 2017).
The impact of LRRK2 on autophagy and protein trafficking pathway is complicated, and observations vary depending on the cell type and on whether LRRK2 mutation, silencing, or kinase inhibition was used (Manzoni 2017). Among potential mechanisms of LRRK2 regulation of autophagy, in SH-SY5Y cells and in rat cortical neurons, overexpression of wild type and G2019S mutant LRRK2 induces mitochondrial fragmentation, and this appears to be dependent on Drp1 in SH-SY5Y cells (Wang et al. 2012). In HEK294 cells, it has been shown that LRRK2 overexpression can induce autophagy by a calcium-dependent mechanism mediated by CaMKK-β/AMPK pathway, LRRK2 has been found to phosphorylate NSF at T645 in the ATP binding pocket and thereby increases SNARE disassembly (Belluzzi et al. 2016), which in turn modulates autophagosome-lysosome fusion (Zhang 2015, Yu et al. 2017). Although autophagy can be neuroprotective in clearance of damaged proteins and organelles, excessive autophagy in mouse cortical dendrites promoted by overexpression of LRRK2 mutant protein can be detrimental and this is also associated with calcium imbalance, and may be mediated by a pathological increase in mitochondrial calcium uptake (Verma et al. 2017) (Cherra et al. 2013).
PD recessive genes, Parkin, PINK1 and PARK7/DJ-1, have also been shown to play a role in mitochondrial quality control and development of approaches targeting these proteins as therapeutic strategies will likely be important (Goedert et al. 2017, Obeso et al. 2017, Zhang et al. 2017b, Barodia et al. 2016, Scott et al. 2017). As α-synuclein, PARK7/DJ-1 accumulates in PD brains (Choi et al. 2006), and can be a substrate of chaperone-mediated autophagy (Cuervo et al. 2004, Wang et al. 2016). In a mouse midbrain dopamine neuron progenitor cell line, MPP+ exposure increased DJ-1 carbonyl formation and degradation (Wang et al. 2016). Overexpression of LAMP2A, the lysosomal receptor of chaperone-mediated autophagy, enhances DJ-1 degradation and cell survival in response to MPP+ (Wang et al. 2016). Interestingly, recent studies also demonstrated a role of DJ-1 in microglia in autophagy and α-synuclein uptake and clearance (Nash et al. 2017).
C. Redox stress in the regulation of mitochondrial function and autophagy
1. Redox stress
The redox regulation of cellular environments is now emerging as an important area of research integrating metabolic and redox-dependent signaling. Interestingly, maintaining a stable intracellular redox environment is critical in both preventing proteotoxicity, and removing by autophagy the resulting damage that occurs before the toxic proteins are removed. It is now abundantly clear that both oxidative and reductive stress can arise if at a given cellular location and time the redox tone is not maintained within physiological boundaries. The regulation of these redox specific intracellular domains is controlled by the Keap1/Nrf2 system (Dodson et al. 2015). Interestingly, Nrf2 over activation increases the expression of pro-reductant pathways leading to a reductive stress and proteotoxicity in the heart (Dodson et al. 2015, Badia et al. 2013, Ralser & Benjamin 2008, Rajasekaran et al. 2008, Russell et al. 1999). Whether this also occurs in the brain is not well understood. In the context of the “redox signaling paradigm”, reactive oxygen species are produced by controlled by specific pathways and dysfunction arises not from non-specific oxidation of macromolecules but the loss of domain controlled signaling (Dodson et al. 2013a). In this context although it is well known that mitochondria can produce reactive oxygen species including superoxide and hydrogen peroxide it is less well appreciated that these can be tightly controlled through and play a role in cell signaling (Murphy 2009, Lee et al. 2012a, Dodson et al. 2013a, Quinlan et al. 2013, Wong et al. 2017, Brand 2016). In addition, NADPH oxidase, dual oxidases, monoamine oxidase, cytochrome p450, and xanthine oxidase also generate hydrogen peroxide and superoxide as their normal cellular function (Halliwell & Gutteridge 2015). Excessive reactive oxygen species are dynamically reduced by superoxide dismutases and catalase. Without these enzymes, these reactive species, together with nitric oxide generated by nitric oxide synthases can damage lipid and proteins, and resulting lipid peroxidation product such as HNE can cause further damage to cellular constituents (Dodson et al. 2013a). Oxidative post-translational modification of DNA, lipid, and protein is associated with a number of age related pathologies including PD (Halliwell 2012, Kalyanaraman 2013, Sies 2015). Interestingly, reactive lipid species also have a signaling function so the interface between pathological and physiological oxidative lipid signaling is critical to understand (Levonen et al. 2014, Higdon et al. 2012b). Since redox protein and lipid modifications are essential for normal physiology, the emerging concept is that pathology arises from loss of control of non-physiological redox modifications and the toxic gain of function of oxidized proteins and lipids (Wende et al. 2016b).
2. Oxidative stress and dopaminergic neurodegeneration
In neurons, there are some specific mechanism that can promote oxidative protein modification and result in toxicity. For example, a robust literature has reported an increased in oxidative stress markers in Parkinson’s postmortem brains, including elevated lipid peroxidation-protein adducts and protein carbonyls, oxidized complex I subunits, nitration of α-synuclein, increased thiol-DOPA conjugates, and increased glutathione and Coenzyme Q10 oxidation (Giordano et al. 2014a, Selley 1998, Yoritaka et al. 1996). Oxidation of proteins can contribute to proteotoxicity by blocking the sites needed for proteasomal degradation, or by promoting the formation of insoluble aggregates. An example of a lipid peroxidation-dependent modification is the lipid aldehyde 4-hydroxynonenal (HNE) which forms stable adducts which change protein function by modification of histidine, arginine, lysine and cysteine residues (Higdon et al. 2012a). Once formed, the protein adducts can only be removed by controlled degradation of the protein. Their accumulation can result in a toxic gain of function through promotion of protein aggregation or, in the case of redox proteins, such as those found in the mitochondrion, increased levels of reactive oxygen species. Because of the physiological role of oxidized lipids in signaling, it is thus important to distinguish what constitutes a pathological level of these molecules which may vary according to cell type and age (Higdon et al. 2012c). As will be discussed further below α-synuclein can be modified by HNE and the modification promotes its aggregation and toxicity (Xiang et al. 2015). Aldehyde dehydrogenase 1A1 (ALDH1A1), an enzyme important for metabolizing nonenal as well as dopamine metabolites, is decreased in PD brains, and ALDH1A1 and ALDH2 double knockout mice exhibited increased HNE and 3,4-dihydroxyphenylacetaldehyde (DOPAL) and significant dopaminergic neurodegeneration (Grunblatt & Riederer 2016).
3. Oxidative stress and mitochondria
Since the reactive lipid species or neuronal specific reactive species discussed above can target mitochondria impaired detoxification can lead to the slow accumulation of adducted proteins and a greater demand on the autophagy pathway. In support of this concept experimental evidence has established that the mitochondria are a nexus for both the physiological and pathological effects of reactive lipid species and as such are an important target for potential dysfunction in neurodegenerative disease (Higdon et al. 2012b). Indeed, mitochondrial dysfunction occurs in the brains of PD patients (Ryan et al. 2015, Swerdlow 2009, Schapira 2007, Beal 2003, Parker & Swerdlow 1998, Schapira et al. 1990, Bindoff et al. 1989). Neurotoxins have been shown to target mitochondria in Parkinsonism patients and in animal models (Martinez & Greenamyre 2012, Bove et al. 2005). Specifically, mitochondria are a target for HNE modification and this can influence bioenergetic function in a variety of cell types (Dranka et al. 2011a, Dranka et al. 2011b, Ravi et al. 2015). HNE adducts with mitochondrial respiratory chain subunits have been reported and are associated with enhanced reactive species production and a decrease in the mitochondrial bioenergetic reserve capacity, all of which contribute to a decreased ability of the cell to combat metabolic and proteotoxic stress (Reed et al. 2008, Lee et al. 2012b, Breitzig et al. 2016, Galam et al. 2016, Harris et al. 2015, Fritz et al. 2011, Singh et al. 2015). If these damaged mitochondria, which are capable of generating secondary reactive oxygen species and amplifying the initial oxidative insult, are not removed, the threshold for pathology is lowered (Hill et al. 2012, Lee et al. 2012a, Redmann et al. 2014).
4. Oxidative stress and autophagy
The importance of thresholds for toxicity in neuronal cells depends on age, genetics and other factors and can be assessed in vitro. For example, in primary neurons, HNE inhibits mitochondrial function and although autophagy is initially protective, it can be overwhelmed as HNE concentrations increase in neurons (Dodson et al. 2017, Zhang et al. 2017a). It is now possible to identify the HNE modified proteins using sensitive tagging techniques that can help relate specific targets to changes in metabolism and autophagy (Dodson et al. 2017, Dodson et al. 2013a, Dodson et al. 2015, Dodson et al. 2013b). Protein thiols targeted by HNE and other oxidants have been demonstrated for autophagy-lysosomal pathway proteins including ATG4, LC3, PARK7/DJ-1, and cathepsins that in the cysteine proteinases family (Lee et al. 2012a, Dodson et al. 2015, Dodson et al. 2013a, Wani et al. 2015). ATG4 thiol modification at Cys81 decreases its activity by suppressing LC3 delipidation (Scherz-Shouval et al. 2007). The lipidation mechanism is a possible link to inflammation since a 12/15-lipoxygenase-derived oxidized phospholipid, 12-hydroxyeicosatetraenoic acid-phosphatidylethanolamine, was found to be a preferred substrate compared to native phosphatidylethanolamine for yeast Atg8 lipidation (Morgan et al. 2015). Sulfinic acid formation at Cys106 of PARK7/DJ-1, a protein encoded by a recessive PD gene, is important for its function in maintaining mitochondrial connectivity and in cell protection when exposed to the mitochondrial toxin rotenone (Blackinton et al. 2009). A recent study demonstrated that wildtype but not C106 mutant PARK7/DJ-1 can convert methylglyoxal (pyruvaldehyde)-adducted glutathione to free glutathione and lactate suggesting that it is potentially involved in aldehyde detoxification (Matsuda et al. 2017).
Lysosomal cysteine proteases cathepsin B and L, but not aspartate proteases are inactivated when murine macrophage-like cells are exposed to protein hydroperoxides, consistent with the sensitivity of the active site thiol to oxidative stress (Rahmanto et al. 2010). Nonetheless, cathepsin D oxidative modification has been found in the brains of Down syndrome patients with a slight increase of enzyme activity, which could be viewed as a potential compensatory response to increased plaques and tangles (Di et al. 2016). We propose that autophagy is critical in maintaining cellular redox homeostasis and by removing damaged mitochondria and oxidized proteins it serves a cellular antioxidant function in removing toxic proteins and organelles (Giordano et al. 2014a). α-synuclein contains 1 histidine (H50) and 15 lysine residues that are targets for modification by electrophilic lipid peroxidation products such as HNE. Interestingly, it has been shown that HNE promotes α-synuclein toxicity (Xiang et al. 2013). This result is intriguing as H50Q is responsible for a subset of familial PD cases (Kiely et al. 2015). It turns out that although H50Q α-synuclein is less susceptible to HNE modification than wildtype α-synuclein, it forms more aggregates and cells expressing H50Q are more sensitive to hydrogen peroxide-induced cell death (Xiang et al. 2015).
5. The impact of nitric oxide on mitochondrial function and autophagy in the context of Parkinson’s disease
The previous discussion has focused on either endogenous xenobiotics or the products of lipid or protein oxidation as mediators of toxicity. However, it is important to note that the controlled production of reactive species is an important part of normal physiology. This in itself is potential vulnerability if control of these pathways is disrupted. An important example is nitric oxide (NO) has contrasting roles in neurodegeneration with evidence for both protective and toxic effects. As we have discussed previously, these are best understood in the concentration dependent engagement of distinct cellular targets in the cell (Hill et al. 2010). There are essentially two heme based targets; guanylate cyclase and cytochrome c oxidase. The effects on mitochondrial function are more complex. Nitric oxide can reversibly inhibit mitochondrial complex IV at high concentrations or when oxygen tension is low (Cleeter et al. 1994, Benavides et al. 2013). This can be protective by maintaining physiological oxygen gradients but if NO compromises ATP production then it will become detrimental (Shiva et al. 2005). When nitric oxide reacts with superoxide to generate peroxynitrite, the effects on the modified lipid and proteins can be irreversible and detrimental (Benavides et al. 2013, Radi et al. 1991a, Radi et al. 1991b, Ischiropoulos et al. 1992, Beckman & Crow 1993, Szabo et al. 2007). Interestingly, the mitochondria are thought to be one of the main targets of peroxynitrite in neuronal cells (Estevez et al. 2017). In the central nervous system, nitric oxide is generated in the neurons by nNOS, induced by NMDA and glutamate receptor activation, and in microglia by iNOS induced by inflammatory signals. nNOS variants have been found to be associated with PD in conjunction with organophosphate pesticides (Paul et al. 2016). Nitric oxide produced by either neurons or glia significantly impacts susceptibility of neurons to toxic insults, as evidence by that nNOS or iNOS gene deletion protects mice against MPTP induced dopaminergic neurodegeneration (Przedborski et al. 1996, Liberatore et al. 1999). As iNOS production in microglia can be induced by LPS and TNFα, it has been hypothesized that neuroinflammation may play a role in nitric oxide production and impact PD pathogenesis. The important role of microglia, a major source of nitric oxide, in PD is further demonstrated by the finding that a single nucleotide polymorphism (SNP) in the human leukocyte antigen (HLA-DRA) gene, which encodes HLA-DR antigens expressed in microglia, has been identified as a risk factor for PD (Wissemann et al. 2013, Tansey & Goldberg 2010). It remains to be determined whether microglial expansion, activation or morphological changes are beneficial or detrimental factors in PD pathogenesis.
Although a direct hit of excessive nitric oxide to neurons appear to be detrimental to its mitochondrial function, the role of systemic nitric oxide is dependent on its source and cellular context. Nitric oxide generated from inorganic nitrite has been shown to play a protective role in variety of pathologies including cardiovascular diseases (Weitzberg et al. 2010, Rocha et al. 2016). Nitrite administration in zebrafish protected dopaminergic neurodegeneration induced by 1-methyl-4-phenylpyridinium (MPP+), in rat protected dopaminergic neurodegeneration induced by rotenone or 6-hydroxydopamine, and in dermal fibroblasts from human PD patients with LRRK2 mutations (Chiara Milanese 2017). Since, as mentioned above, nNOS appears to be detrimental in this context then an alternative mechanism of action for nitrite is implied. Indeed, in MPP+ treated SH-SY5Y cells, it appeared that the mechanisms of nitrite protection involve nitrosation of mitochondrial complex I as well as activation of transcription factor Nrf2 (Chiara Milanese 2017).
It has been shown that in a variety of cell types, nitric oxide can inhibit autophagy (Benavides et al. 2013, Sarkar et al. 2011), although how the regulation of autophagy by nitric oxide affects PD pathogenesis is still unclear. Transient autophagosome accumulation is induced by kainic acid, an agonist of glutamate receptors, and delayed increase of p-Akt and p-mTOR follows (Shacka et al. 2007). The regulation of autophagy by kainic acid may be mediated by nitric oxide as glutamate receptor activation has been shown to activate nNOS (Dawson & Dawson 1998). Inhibition of autophagy by nitric oxide during hypoxia-reoxygenation may contribute to bioenergetics impairment and promote cell death in neurons (Benavides et al. 2013).
D. Metabolic signaling regulates mitochondrial function and autophagy
1. Glucose metabolism
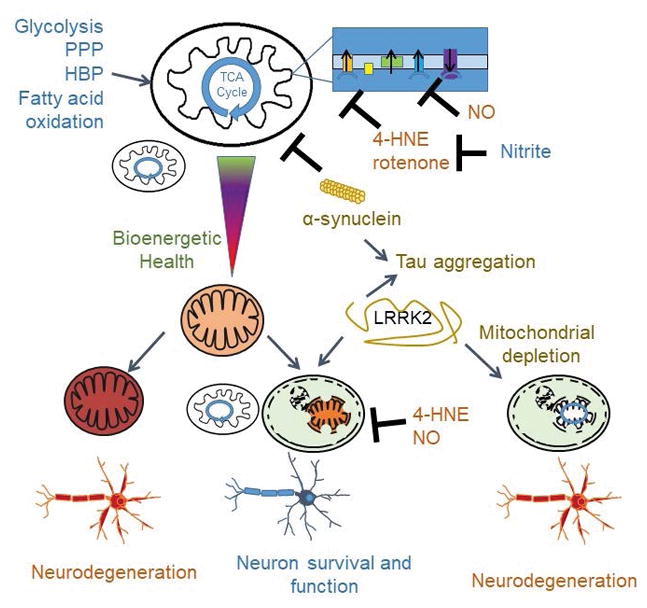
From the previous discussion it is evident that physiological metabolism utilizes a broad range of redox reactions and is essential for normal neuronal function. For this reason, attention has increasingly focused on cellular bioenergetics and metabolism in health and diseases (Holmes et al. 2008). Deficiencies of PD genes, Parkin and PINK1, affect mitochondrial homeostasis or quality control (Narendra et al. 2012). Parkinsonism gene Atg13a2 mutations have been shown to cause lysosomal (Ramirez et al. 2006, Usenovic et al. 2012), mitochondrial (Grunewald et al. 2012), and glycolytic dysfunction (Park et al. 2016). As introduced above, MPTP and pesticides that induce Parkinsonism or increase PD risk target mitochondrial complex I, thereby depressing mitochondrial function (Ramsay et al. 1987, Nicklas et al. 1987, Mizuno et al. 1987, Priyadarshi et al. 2001, Priyadarshi et al. 2000, van der et al. 2012, Tanner et al. 2011, Giordano et al. 2012, Giordano et al. 2014b). In addition to direct targeting of mitochondrial electron transport proteins, additional signaling mechanisms may impact PD pathogenesis, including modification of mitochondrial fission and fusion, and modification of mitochondrial movement within the neuron (Yan et al. 2013). Because complex and chronic diseases such as PD are affected by systemic as well as organ-specific metabolism, changes in metabolic activities may be monitored as biomarkers for progression of the diseases as well as therapeutic responses (Chacko et al. 2014).
While there is some emphasis on glycolysis to provide ATP in the brain, glucose metabolism is also essential to provide NADPH to maintain lipid oxidation products including HNE within the physiological range. This link between metabolism and redox signaling is an important part of normal physiology. In addition, it is known that glucose metabolism can regulate autophagy through several signaling mechanisms in a variety of cell types (Moruno et al. 2012). Key enzymes in the glucose metabolism pathway, hexokinase and GAPDH, have been shown to interact with mTOR or Rheb and decreases mTOR signaling when glucose is low (Eng & Abraham 2011, Roberts & Miyamoto 2015). The integration of glucose metabolism and autophagy has also been demonstrated by a recent study that autophagy proteins, ULK1 and ULK2, can also directly phosphorylate enzymes in the glycolytic and gluconeogenic pathways to enhance glucose uptake and decrease gluconeogenesis in order to maintain cellular ATP production and redox homeostasis (Li et al. 2016). Whether similar regulatory mechanisms play a role in neurons and in the pathogenesis of PD is currently unclear. Assessment of glucose metabolism by 18-F-2-fluoro-deoxyglucose and positron emission tomography has shown a decline of glucose metabolism in aging (Zuendorf et al. 2003). This assessment has been used in Alzheimer’s disease diagnosis (Nordberg et al. 2010). Furthermore, it has been shown that diabetes increases Alzheimer’s disease risk (Holscher 2011). Despite of these findings, the association between glucose metabolism and non-demented PD has not been firmly established (Cereda et al. 2013, Hu et al. 2000).
A significant amount of glucose is metabolized through the hexosamine biosynthetic pathway, which produces UDP-GlcNAc. UDP-GlcNAc can be added to protein Ser/Thr residues and modify protein functions. This modification may be of importance to Alzheimer’s and PD through direct modification of tau or α-synuclein (Yuzwa et al. 2008, Yuzwa et al. 2012, Yuzwa et al. 2014, Wani et al. 2016, Wani et al. 2017), and through regulation of mitochondrial function and autophagy (Hu et al. 2009, Makino et al. 2011, Pekkurnaz et al. 2014, Lozano et al. 2014, Dassanayaka et al. 2015, Trapannone et al. 2016, Wani et al. 2017, Guo et al. 2014, Roberts et al. 2014, Jo et al. 2016). The extent and biological impact of O-GlcNAc modification of proteins in the context of mitochondrial function, autophagy and PD still needs to be further defined. Other metabolites that have been shown to play an important role in regulating mitochondrial and autophagic activities include glutamine and acetyl CoA (Tan et al. 2017, Hjelmeland & Zhang 2016, Marino et al. 2014, Shi & Tu 2015). Although studies have been performed regarding changes of metabolites in toxin and transgenic mice, the involvement of particular metabolites in PD are currently uncertain (Chen et al. 2015b, Worth et al. 2014).
2. Fatty acid oxidation
Although glucose is the preferred fuel for the brain, fatty acid oxidation products are also important substrates under conditions of hypoglycemia (Romano et al. 2017). A higher uptake of hydrogenated and saturated fat in the brain has been associated with development of insulin resistance and Alzheimer’s disease (Holscher 2011), while a ketogenic diet has been shown to be neuroprotective in a variety of conditions (Danial et al. 2013, Romano et al. 2017). The connection between lipid metabolism and autophagy has been further demonstrated by the finding that SREBP1 and SREBF1 can regulate autophagy and mitophagy (Ivatt et al. 2014, Jegga et al. 2011). As SREBF1 is a risk locus for sporadic PD, this finding provides insight into the connection between fatty acid metabolism, mitochondria, and mitophagy in PD pathogenic mechanisms. This important link between metabolism and PD is further supported by the observations that gastrointestinal abnormalities and sleep perturbations are often symptoms preceding motor dysfunction in PD patients.
3. Systemic regulation of metabolism
Naturally, we have largely focused on the local effects of metabolism and protein quality control in the central nervous system. An important and emerging area relates to how the systemic effects of metabolism and redox regulation interact with the pathophysiology of neurodegenerative disease. α-synuclein pathology has been shown to occur early in the enteric nervous system in PD patients (Del Tredici & Braak 2016). A recent study has shown benefit for motor function of a glucagon-like peptide-1 receptor agonist approved for treatment of type 2 diabetes on PD patients in a clinical trial (Athauda et al. 2017). Furthermore, systemic control of metabolism is particularly interesting with respect to the microbiome which has recently been shown to be a potential player in PD (Nair et al. 2018, Hill-Burns et al. 2017). Recent studies identified differences in microbiome in PD patients compared to controls (Scheperjans et al. 2015). In mice overexpressing α-synuclein, both germ-free condition and antibiotic treatment have shown to ameliorate motor deficits, α-synuclein accumulation, microglial activation and cytokine production (Sampson et al. 2016). Colonization with microbiota from PD patients, or oral administration of short-chain fatty acids, which are produced during viral infection by the gut microbiome and are decreased by germ-free condition or antibiotic treatment, exacerbates these phenotypes (Sampson et al. 2016). The interconnection of the gut environment, the metabolites generated, and PD pathogenesis warrants further investigation.
Circadian rhythms integrate metabolism with time-of-day demands for normal physiology, and disruption is associated with increased susceptibility to disease (Wende et al. 2016a, Abbott et al. 2018). Indeed, sleep perturbation is an important clinical feature of PD with as many as 64% PD patients reporting sleep problems. The mechanisms are still emerging but it has been shown that BMAL1, a clock regulatory protein, is decreased in the peripheral blood of PD patients and is no longer rhythmic (Breen et al. 2014). As circadian clock is a strong regulator of metabolism, and recent studies identified key autophagy genes under circadian regulation (Wende et al. 2016a), the clock-metabolism-autophagy connection may play a key role in PD pathogenesis and may be targeted for therapeutic purposes (Musiek & Holtzman 2016, Breen et al. 2014, Wende et al. 2016a).
Therapeutic strategies and future directions
In this review, we have highlighted the integrated nature of metabolism, the response to stress and how the limited capacity of repair systems contributes to the pathophysiology of PD. At the level of the proteotoxicity mechanisms, multiple strategies have been explored to target α-synuclein for the development of new therapies for PD (Olanow & Kordower 2017, Valera & Masliah 2016). Proteasomal and lysosomal pathways can degrade α-synuclein in vivo but these functions decline both with age and with the development of PD (Chu Y 2009). There is an urgent unmet need to investigate novel approaches to enhance the clearance of α-synuclein protein. Autophagy is an attractive therapeutic strategy because it has the potential to not only remove excessive or toxic α-synuclein species, but also degrade other damaged cellular constituents. In this regard, rapamycin and other autophagy activators have been shown to be protective in cell and animal models of neurodegenerative diseases (Hochfeld et al. 2013). However, rapamycin may promote a starvation response that degrades both healthy and unhealthy components of the cells. Furthermore, the potential benefits of inducing autophagy initiation are dependent on optimal lysosomal function, which appears to be inhibited in PD. Thus, it is important to find a strategy to restore the efficiency of clearing damaged cellular proteins and organelles in the lysosomes, without depleting those that are still functional. Studies have identified roles of TFEB as a transcription factor activating the expression of genes encoding lysosomal proteins (Decressac et al. 2013, Settembre et al. 2011). Transcriptional activation of a wide range of autophagy proteins is effective in animal models (Decressac et al. 2013), although it may also lead to excessive autophagy which depletes cellular contents and is then detrimental. Thus, it is important to increase the efficiency of the clearance of damaged cellular proteins and organelles without affecting the undamaged cell constituents.
In this regard, lysosomal cathepsin D is a viable target for enhancement of autophagic clearance. As discussed early, cathepsin D is a major lysosomal aspartate protease, its levels are decreased to <50% in PD substantia nigra neurons with α-synuclein inclusions compared to age-matched controls (Chu Y 2009), and its deficiencies have pathological consequences. Cathepsin D inactivation in humans caused neuronal ceroid lipofuscinosis and early lethality (Siintola et al. 2006, Steinfeld et al. 2006). In humans, sheep and mice, cathepsin D mutations resulted in accumulation of α-synuclein (Qiao et al. 2008, Cullen et al. 2009). The therapeutic potential of overexpression of cathepsin D has been demonstrated in worms and mammalian cells to decrease α-synuclein aggregation and toxicity (Qiao et al. 2008), although its effectiveness in mammalian models in vivo has not been examined. Strategies need to be explored for enhancing cathepsin D activities in vivo by neutraceutical and pharmaceutical agents or by allosteric activators (Ambrosi et al. 2015, McNeill et al. 2014, Parker et al. 2017) or by better delivery strategies that allow BBB penetration (Spencer et al. 2015). In addition to lysosomal enhancement, in light of the special importance of mitochondrial quality control, specific mitophagy enhancement strategies may be effective in disease cases when mitochondrial damage occurs (Hertz et al. 2013, Zhang et al. 2017b).
Since oxidative stress has been associated with PD, various antioxidants have been tested and found to show efficacy in pre-clinical models of PD, although so far largely ineffective in clinical trials (Giordano et al. 2014a). An important and emerging aspect of redox biology is the recognition that the redox state of the cell must be retained at defined set points which vary within different regions of the cell. This finding in itself requires that redox modulatory therapeutics must be targeted to the intracellular compartment that is in a hyperoxidative or reductive state relative to the physiological set point. At the present time, we know little about how the redox status of the intracellular organelles varies in PD and this is an essential piece of information for the development of targeted therapeutics in this area. Nevertheless, testing natural products and anti-inflammatory reagents are still ongoing with majority of studies relying on pre-clinical models (Baranyi et al. 2016, Chen et al. 2015c, Zhou et al. 2016). A more mechanistic approach with an underlying understanding of the different phases of redox biology which occur during the life history of the disease is an urgent need as more potent and druggable targets emerge from the pre-clinical studies.
Because of the importance of mitochondria as an energy generating and signaling organelle and autophagy as the cellular quality control machinery, therapeutics targeting both mitochondria and the autophagy pathway are being actively considered. In this regard, several mito-centric approaches have been used. For example, toxin-resistant complex I subunit, enhancing complex II activities, mitochondrially targeted therapeutics, or stabilizing mitochondrial membrane potential, have all shown some degree of efficacy (Marella et al. 2008, Tieu et al. 2003, Reeves et al. 2007). A recent study also demonstrated that administration of inorganic nitrite ameliorates pathology in animal models of PD by complex I S-nitrosation and activation of antioxidant Nrf2 pathway (Chiara Milanese 2017). Enhancement of mitochondrial biogenesis has been shown to be neuroprotective in human dopaminergic neurons (Makela et al. 2016). In further support for enhancing mitochondrial function as a therapeutic development, it has also been shown that an FDA approved anti-helminth drug nitazoxanide may induce mitochondrial uncoupling and protects against MPP+ toxin-induced parkinsonism in mice (Amireddy et al. 2017). Mitochondrial targeted antioxidants show promises in some but not all clinical scenarios that involve oxidative stress, and thus more effective means to counter cellular damage need to be developed (Logan & Murphy 2017, Jin et al. 2014). Efforts have also been made to directly target potential antioxidant or modulatory molecules (catalase, SS-31 peptide or mitoQ) to the mitochondria (Basisty et al. 2016, Dai et al. 2016, Chaturvedi & Beal 2013, Reily et al. 2013, Dashdorj et al. 2013, Solesio et al. 2013, Gioscia-Ryan et al. 2014, Miquel et al. 2014, Dare et al. 2015, Stucki et al. 2016, Rehman et al. 2016, Logan & Murphy 2017). Effectiveness of these approaches on PD needs to be further investigated. Metabolomics studies have also help identify new targets for cardioprotection (Chouchani et al. 2014, Vinje et al. 2014). Similar strategies may also help develop new target for PD therapeutics. Last but not least, with better understanding of how circadian rhythm plays a role in regulating metabolism, mitochondrial function and autophagy, pharmacological regulation of circadian behavior and metabolic processes is an area which is as yet relatively unexplored (Breen et al. 2014, Solt et al. 2012) (Fig. 3).
Figure 3. Potential targets and therapeutic strategies for treating Parkinson’s disease based on autophagy-lysosomal pathway, bioenergetics and metabolism, as well as redox signaling and oxidative-reductive stress.
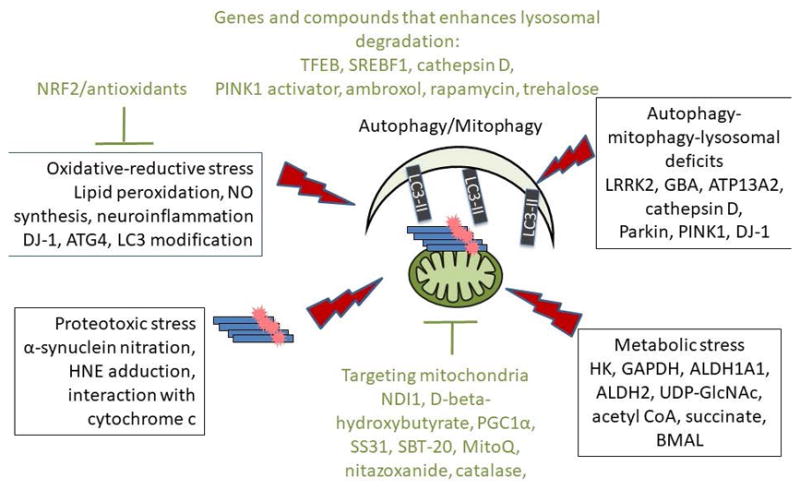
Major efforts have been made in recent years to develop broad spectrum and/or organelle targeted antioxidant strategies, and enhanced autophagy, mitophagy, and lysosomal activities. Highlighted in this figure are some of the key events of metabolic, oxidative-reductive, and proteotoxic stress (in boxes), as well as molecules that have been tested in in vitro and animal models (in green) that target metabolism and autophagy/mitophagy/lysosomal pathways.
Although progress has been made regarding mitochondrial and autophagy function regulated by redox, metabolic and proteotoxic signaling, there is still a great deal we do not know. Some key questions include the following:
In susceptible neurons and glia, are there specific targets for specific redox signals, metabolites, and/or toxic protein species?
Which steps are reversible or can be counteracted to attenuate disease progression?
Can we design targeted and effective antioxidants, efficient and stress resistant mitochondria, and specific lysosomal enhancement strategies to attenuate Parkinson’s disease?
Can targeting the microbiome change the progression or susceptibility to neurodegenerative disease?
Research effort to address these questions will be informative in the next decades.
Acknowledgments
This work was partially supported by UAB Blue Sky program, UAB AMC21 reload multi-investigator grant, Nathan Shock Center P30 G050886, and NIHR01-NS064090.
Abbreviation List
- 3MA
3-methyladenine
- ADH1A1
aldehyde dehydrogenase 1A1
- ADH2
aldehyde dehydrogenase 2
- AMPK
adenosine monophosphate (AMP)-activated protein kinase
- ATG5
autophagy related 5
- ATG7
autophagy related 7
- ATP13A2
ATPase type 13A2
- BECN1
beclin 1, autophagy related
- BHI
bioenergetic health index
- BMAL1
brain and muscle Arnt-like protein 1
- CaMKK
calcium-dependent protein kinase kinase
- CQ
chloroquine
- Drp1
dynamin 1-like
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhdrazone
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GBA
glucosidase, β, acid
- HBP
hexosamine biosynthetic pathway
- HK
hexokinase
- HNE
4-hydroxynonenal
- LRRK2
leucine-rich repeat kinase 2
- LC3
microtubule-associated protein 1 light chain 3 alpha/beta
- MFN1
mitofusin 1
- MFN2
mitofusin 2
- mtDNA
mitochondrial DNA
- mTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NO
nitric oxide
- NRF2
Nuclear factor (erythroid-derived 2)-like 2
- OCR
oxygen consumption rate
- OPA1
mitochondrial dynamin like GTPase
- PD
Parkinson’s Disease
- PINK1
PTEN-induced putative kinase 1
- PGC1α
peroxisome proliferative activated receptor, gamma, coactivator 1 alpha
- PPP
pentose phosphate pathways
- REM
rapid eye movement
- SNARE
Soluble NSF-Attachment protein Receptor
- SOD2
superoxide dismutase 2, mitochondrial
- SQSTM1/p62
sequestosome 1
- SREBF1
sterol regulatory element binding transcription factor 1
- TCA
tricarboxylic acid
- TFAM
transcription factor A, mitochondrial
- TFEB
transcription factor EB
- ULK
unc-51 Like Autophagy Activating Kinase
- VDAC1
voltage-dependent anion channel 1
Footnotes
Conflict of interest: none
References
- Abbott SM, Knutson KL, Zee PC. Health implications of sleep and circadian rhythm research in 2017. Lancet Neurol. 2018;17:17–18. doi: 10.1016/S1474-4422(17)30410-6. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Schmitz Y, Farinas I, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis. 2015;82:235–242. doi: 10.1016/j.nbd.2015.06.008. [DOI] [PubMed] [Google Scholar]
- Amireddy N, Puttapaka SN, Vinnakota RL, Ravuri HG, Thonda S, Kalivendi SV. The Unintended Mitochondrial Uncoupling Effects of the FDA Approved Anti-Helminth Drug Nitazoxanide Mitigates Experimental Parkinsonism in Mice. J Biol Chem. 2017 doi: 10.1074/jbc.M117.791863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–1675. doi: 10.1016/S0140-6736(17)31585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badia MC, Giraldo E, Dasi F, Alonso D, Lainez JM, Lloret A, Vina J. Reductive stress in young healthy individuals at risk of Alzheimer disease. Free Radic Biol Med. 2013;63:274–279. doi: 10.1016/j.freeradbiomed.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Baranyi M, Porceddu PF, Goloncser F, et al. Novel (Hetero)arylalkenyl propargylamine compounds are protective in toxin-induced models of Parkinson’s disease. Mol Neurodegener. 2016;11:6. doi: 10.1186/s13024-015-0067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barodia SK, Creed RB, Goldberg MS. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res Bull. 2016 doi: 10.1016/j.brainresbull.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basisty N, Dai DF, Gagnidze A, et al. Mitochondrial-targeted catalase is good for the old mouse proteome, but not for the young: ‘reverse’ antagonistic pleiotropy? Aging Cell. 2016;15:634–645. doi: 10.1111/acel.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Crow JP. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem Soc Trans. 1993;21:330–334. doi: 10.1042/bst0210330. [DOI] [PubMed] [Google Scholar]
- Belluzzi E, Gonnelli A, Cirnaru MD, et al. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol Neurodegener. 2016;11:1. doi: 10.1186/s13024-015-0066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides GA, Liang Q, Dodson M, Darley-Usmar V, Zhang J. Inhibition of autophagy and glycolysis by nitric oxide during hypoxia-reoxygenation impairs cellular bioenergetics and promotes cell death in primary neurons. Free Radic Biol Med. 2013;65:1215–1228. doi: 10.1016/j.freeradbiomed.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez BA, Davis AA, Jin SC, et al. Resequencing analysis of five Mendelian genes and the top genes from genome-wide association studies in Parkinson’s Disease. Mol Neurodegener. 2016;11:29. doi: 10.1186/s13024-016-0097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindoff LA, Birch-Machin M, Cartlidge NE, Parker WD, Jr, Turnbull DM. Mitochondrial function in Parkinson’s disease. Lancet. 1989;2:49. doi: 10.1016/s0140-6736(89)90291-2. [DOI] [PubMed] [Google Scholar]
- Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS, Cookson MR, Wilson MA. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem. 2009;284:6476–6485. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanca Ramirez M, Lara Ordonez AJ, Fdez E, Hilfiker S. LRRK2: from kinase to GTPase to microtubules and back. Biochem Soc Trans. 2017;45:141–146. doi: 10.1042/BST20160333. [DOI] [PubMed] [Google Scholar]
- Bove J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx. 2005;2:484–494. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Gai WP, Xu YH, et al. Uptake and mitochondrial dysfunction of alpha-synuclein in human astrocytes, cortical neurons and fibroblasts. Transl Neurodegener. 2013;2:20. doi: 10.1186/2047-9158-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Breen DP, Vuono R, Nawarathna U, Fisher K, Shneerson JM, Reddy AB, Barker RA. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014;71:589–595. doi: 10.1001/jamaneurol.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitzig M, Bhimineni C, Lockey R, Kolliputi N. 4-Hydroxy-2-nonenal: a critical target in oxidative stress? Am J Physiol Cell Physiol. 2016;311:C537–C543. doi: 10.1152/ajpcell.00101.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science. 2017;357:1255–1261. doi: 10.1126/science.aam9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereda E, Barichella M, Pedrolli C, Klersy C, Cassani E, Caccialanza R, Pezzoli G. Diabetes and risk of Parkinson’s disease. Mov Disord. 2013;28:257. doi: 10.1002/mds.25211. [DOI] [PubMed] [Google Scholar]
- Cha SH, Choi YR, Heo CH, Kang SJ, Joe EH, Jou I, Kim HM, Park SM. Loss of parkin promotes lipid rafts-dependent endocytosis through accumulating caveolin-1: implications for Parkinson’s disease. Mol Neurodegener. 2015;10:63. doi: 10.1186/s13024-015-0060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko BK, Kramer PA, Ravi S, et al. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (Lond) 2014;127:367–373. doi: 10.1042/CS20140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Beal MF. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol Cell Neurosci. 2013;55:101–114. doi: 10.1016/j.mcn.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Chen L, Xie Z, Turkson S, Zhuang X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci. 2015a;35:890–905. doi: 10.1523/JNEUROSCI.0089-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xie C, Sun L, Ding J, Cai H. Longitudinal Metabolomics Profiling of Parkinson’s Disease-Related alpha-Synuclein A53T Transgenic Mice. PLoS One. 2015b;10:e0136612. doi: 10.1371/journal.pone.0136612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang DQ, Liao Z, et al. Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson’s disease. Mol Neurodegener. 2015c;10:4. doi: 10.1186/1750-1326-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ, 3rd, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol. 2013;182:474–484. doi: 10.1016/j.ajpath.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara Milanese VT, Gabriels Sylvia, Cerri Silvia, Levandis Giovanna, Blandini Fabio, Tresini Maria, Shiva Sruti, Timothy Greenamyre J, Gladwin Mark T, Mastroberardino Pier G. Mitochondrial complex I reversible S-nitrosation improves bioenergetics and is protective in Parkinson’s disease. Antioxidants and Redox Signaling. 2017 doi: 10.1089/ars.2017.6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasserini D, Paciotti S, Eusebi P, et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener. 2015;10:15. doi: 10.1186/s13024-015-0010-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 2010;486:235–239. doi: 10.1016/j.neulet.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Sullards MC, Olzmann JA, et al. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem. 2006;281:10816–10824. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu YDH, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobio Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Clark EP. The Occurrence of Rotenone and Related Compounds in the Roots of Cracca Virginiana. Science. 1933;77:311–312. doi: 10.1126/science.77.1995.311. [DOI] [PubMed] [Google Scholar]
- Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Cullen V, Lindfors M, Ng J, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2:5. doi: 10.1186/1756-6606-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Cheung E, Alleman RJ, Perry JB, Allen ME, Brown DA, Kloner RA. Cardioprotective Effects of Mitochondria-Targeted Peptide SBT-20 in two Different Models of Rat Ischemia/Reperfusion. Cardiovasc Drugs Ther. 2016;30:559–566. doi: 10.1007/s10557-016-6695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Hartman AL, Stafstrom CE, Thio LL. How does the ketogenic diet work? Four potential mechanisms. J Child Neurol. 2013;28:1027–1033. doi: 10.1177/0883073813487598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dare AJ, Logan A, Prime TA, et al. The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J Heart Lung Transplant. 2015;34:1471–1480. doi: 10.1016/j.healun.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashdorj A, Jyothi KR, Lim S, et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013;11:178. doi: 10.1186/1741-7015-11-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassanayaka S, Readnower RD, Salabei JK, et al. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem J. 2015;467:115–126. doi: 10.1042/BJ20141018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog Brain Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- de Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110:E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Bourdenx M, Gorry P, et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14:855–866. doi: 10.1016/S1474-4422(15)00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Tredici K, Braak H. Review: Sporadic Parkinson’s disease: development and distribution of alpha-synuclein pathology. Neuropathol Appl Neurobiol. 2016;42:33–50. doi: 10.1111/nan.12298. [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di DF, Tramutola A, Perluigi M. Cathepsin D as a therapeutic target in Alzheimer’s disease. Expert Opin Ther Targets. 2016;20:1393–1395. doi: 10.1080/14728222.2016.1252334. [DOI] [PubMed] [Google Scholar]
- Di Maio R, Barrett PJ, Hoffman EK, et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med. 2016;8:342ra378. doi: 10.1126/scitranslmed.aaf3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana Chang MAN, Hallgrímsdóttir Ingileif B, Hunkapiller Julie, van der Brug Marcel, Cai Fang, Kerchner Geoffrey A, Ayalon Gai, Bingol Baris, Sheng Morgan, Hinds David, Behrens Timothy W, Singleton Andrew B, Bhangale Tushar R, Graham Robert R International Parkinsons Disease Genomics Consortium, 23andMe Research Team. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nature Genetics. 2017 doi: 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M, Darley-Usmar V, Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med. 2013a;63:207–221. doi: 10.1016/j.freeradbiomed.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M, Liang Q, Johnson MS, Redmann M, Fineberg N, Darley-Usmar VM, Zhang J. Inhibition of glycolysis attenuates 4-hydroxynonenal-dependent autophagy and exacerbates apoptosis in differentiated SH-SY5Y neuroblastoma cells. Autophagy. 2013b;9:1996–2008. doi: 10.4161/auto.26094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M, Redmann M, Rajasekaran NS, Darley-Usmar V, Zhang J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem J. 2015;469:347–355. doi: 10.1042/BJ20150568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M, Wani WY, Redmann M, et al. Regulation of autophagy, mitochondrial dynamics and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy. 2017 doi: 10.1080/15548627.2017.1356948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey ER, George BP, Leff B, Willis AW. The coming crisis: obtaining care for the growing burden of neurodegenerative conditions. Neurology. 2013;80:1989–1996. doi: 10.1212/WNL.0b013e318293e2ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Benavides GA, Diers AR, et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011a;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka BP, Benavides GA, Diers AR, et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radical Biology and Medicine. 2011b;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlerskov P, Rasmussen I, Nielsen TT, Bergstrom AL, Tohyama Y, Jensen PH, Vilhardt F. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. doi: 10.1074/jbc.M112.401174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon H, Don J, Melamed E, Ziv I, Shirvan A, Offen D. Mutant and wild-type alpha-synuclein interact with mitochondrial cytochrome C oxidase. J Mol Neurosci. 2002;18:229–238. doi: 10.1385/JMN:18:3:229. [DOI] [PubMed] [Google Scholar]
- Eng CH, Abraham RT. The autophagy conundrum in cancer: influence of tumorigenic metabolic reprogramming. Oncogene. 2011 doi: 10.1038/onc.2011.220. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Ramdial K, Franco MC. Cellular mechanisms of peroxynitrite-induced neuronal death. In. Brain Res Bull. 2017 doi: 10.1016/j.brainresbull.2017.05.008. [DOI] [PubMed] [Google Scholar]
- Freeman D, Cedillos R, Choyke S, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8:e62143. doi: 10.1371/journal.pone.0062143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch T, Smyth KA, Wallendal MS, Hyde T, Leo G, Geldmacher DS. Parkinson disease: research update and clinical management. South Med J. 2012;105:650–656. doi: 10.1097/SMJ.0b013e318273a60d. [DOI] [PubMed] [Google Scholar]
- Fritz KS, Galligan JJ, Smathers RL, Roede JR, Shearn CT, Reigan P, Petersen DR. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem Res Toxicol. 2011;24:651–662. doi: 10.1021/tx100355a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galam L, Rajan A, Failla A, Soundararajan R, Lockey RF, Kolliputi N. Deletion of P2X7 attenuates hyperoxia-induced acute lung injury via inflammasome suppression. Am J Physiol Lung Cell Mol Physiol. 2016;310:L572–L581. doi: 10.1152/ajplung.00417.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy. 2015;11:1443–1457. doi: 10.1080/15548627.2015.1067364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014a;2:82–90. doi: 10.1016/j.redox.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S, Dodson M, Ravi S, Redmann M, Ouyang X, Darley-Usmar VM, Zhang J. Bioenergetic adaptation in response to autophagy regulators during rotenone exposure. J Neurochem. 2014b;131:625–633. doi: 10.1111/jnc.12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S, Lee J, Darley-Usmar VM, Zhang J. Distinct Effects of Rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on Cellular Bioenergetics and Cell Death. PLoS ONE. 2012;7:e44610. doi: 10.1371/journal.pone.0044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014;592:2549–2561. doi: 10.1113/jphysiol.2013.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG. The Synucleinopathies: Twenty Years On. J Parkinsons Dis. 2017;7:S53–S71. doi: 10.3233/JPD-179005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Riederer P. Aldehyde dehydrogenase (ALDH) in Alzheimer’s and Parkinson’s disease. J Neural Transm (Vienna) 2016;123:83–90. doi: 10.1007/s00702-014-1320-1. [DOI] [PubMed] [Google Scholar]
- Grunewald A, Arns B, Seibler P, Rakovic A, Munchau A, Ramirez A, Sue CM, Klein C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33:1843e1841–1847. doi: 10.1016/j.neurobiolaging.2011.12.035. [DOI] [PubMed] [Google Scholar]
- Guerreiro PS, Gerhardt E, Lopes da Fonseca T, Bahr M, Outeiro TF, Eckermann K. LRRK2 Promotes Tau Accumulation, Aggregation and Release. Mol Neurobiol. 2016;53:3124–3135. doi: 10.1007/s12035-015-9209-z. [DOI] [PubMed] [Google Scholar]
- Guo B, Liang Q, Li L, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014;16:1215–1226. doi: 10.1038/ncb3066. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Free radicals and antioxidants: updating a personal view. Nutr Rev. 2012;70:257–265. doi: 10.1111/j.1753-4887.2012.00476.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. booksgooglecom. 2015. Free Radicals in biology and medicine. [Google Scholar]
- Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Harris PS, Roy SR, Coughlan C, Orlicky DJ, Liang Y, Shearn CT, Roede JR, Fritz KS. Chronic ethanol consumption induces mitochondrial protein acetylation and oxidative stress in the kidney. Redox Biol. 2015;6:33–40. doi: 10.1016/j.redox.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helferich AM, McLean PJ, Weishaupt JH, Danzer KM. Commentary: alpha-synuclein interacts with SOD1 and promotes its oligomerization. J Neurol Neuromedicine. 2016;1:28–30. doi: 10.29245/2572.942x/2016/7.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell. 2013;154:737–747. doi: 10.1016/j.cell.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem J. 2012a;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. The Biochemical journal. 2012b;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon AN, Landar A, Barnes S, Darley-Usmar VM. The Electrophile Responsive Proteome: Integrating Proteomics and Lipidomics with Cellular Function. Antioxidants & redox signaling. 2012c doi: 10.1089/ars.2012.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill-Burns EM, Debelius JW, Morton JT, et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord. 2017;32:739–749. doi: 10.1002/mds.26942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BG, Benavides GA, Lancaster JR, Jr, Ballinger S, Dell’italia L, Zhang J, Darley-Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BG, Dranka BP, Bailey SM, Lancaster JR, Jr, Darley-Usmar VM. What part of NO don’t you understand? Some answers to the cardinal questions in nitric oxide biology. J Biol Chem. 2010;285:19699–19704. doi: 10.1074/jbc.R110.101618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland A, Zhang J. Metabolic, autophagic, and mitophagic activities in cancer initiation and progression. Biomed J. 2016;39:98–106. doi: 10.1016/j.bj.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochfeld WE, Lee S, Rubinsztein DC. Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol Sin. 2013;34:600–604. doi: 10.1038/aps.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E, Wilson ID, Nicholson JK. Metabolic phenotyping in health and disease. Cell. 2008;134:714–717. doi: 10.1016/j.cell.2008.08.026. [DOI] [PubMed] [Google Scholar]
- Holscher C. Diabetes as a risk factor for Alzheimer’s disease: insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem Soc Trans. 2011;39:891–897. doi: 10.1042/BST0390891. [DOI] [PubMed] [Google Scholar]
- Hu MT, Taylor-Robinson SD, Chaudhuri KR, et al. Cortical dysfunction in non-demented Parkinson’s disease patients: a combined (31)P-MRS and (18)FDG-PET study. Brain. 2000;123(Pt 2):340–352. doi: 10.1093/brain/123.2.340. [DOI] [PubMed] [Google Scholar]
- Hu Y, Suarez J, Fricovsky E, et al. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem. 2009;284:547–555. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi Y, Okada Y, Akamatsu W, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain. 2012;5:35. doi: 10.1186/1756-6606-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- Ivatt RM, Sanchez-Martinez A, Godena VK, Brown S, Ziviani E, Whitworth AJ. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc Natl Acad Sci U S A. 2014;111:8494–8499. doi: 10.1073/pnas.1321207111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Chen JS, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012a;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Lin RZ, Zhang J, Zong WX. Mammalian PIK3C3/VPS34: the key to autophagic processing in liver and heart. Autophagy. 2012b;8:707–708. doi: 10.4161/auto.19627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy. 2011;7:477–489. doi: 10.4161/auto.7.5.14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim Biophys Acta. 2014;1842:1282–1294. doi: 10.1016/j.bbadis.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YK, Park NY, Park SJ, et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget. 2016;7:57186–57196. doi: 10.18632/oncotarget.11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol. 2013;1:244–257. doi: 10.1016/j.redox.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely AP, Ling H, Asi YT, et al. Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener. 2015;10:41. doi: 10.1186/s13024-015-0038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Lemasters JJ. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol. 2011;300:C308–C317. doi: 10.1152/ajpcell.00056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Fiesel FC, Belmonte KC, Hudec R, Wang WX, Kim C, Nelson PT, Springer W, Kim J. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1) Mol Neurodegener. 2016;11:55. doi: 10.1186/s13024-016-0121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingham PJ, Pocock JM. Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem. 2001;76:1475–1484. doi: 10.1046/j.1471-4159.2001.00146.x. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kramer PA, Ravi S, Chacko B, Johnson MS, Darley-Usmar VM. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biol. 2014;2:206–210. doi: 10.1016/j.redox.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Ganini D, Mason RP. Role of cytochrome c in alpha-synuclein radical formation: implications of alpha-synuclein in neuronal death in Maneb- and paraquat-induced model of Parkinson’s disease. Mol Neurodegener. 2016;11:70. doi: 10.1186/s13024-016-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med. 2013;45:e22. doi: 10.1038/emm.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012a;441:523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Lee WC, Wong HY, Chai YY, Shi CW, Amino N, Kikuchi S, Huang SH. Lipid peroxidation dysregulation in ischemic stroke: plasma 4-HNE as a potential biomarker? Biochem Biophys Res Commun. 2012b;425:842–847. doi: 10.1016/j.bbrc.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- Levonen AL, Hill BG, Kansanen E, Zhang J, Darley-Usmar VM. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic Biol Med. 2014;71C:196–207. doi: 10.1016/j.freeradbiomed.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang ZV, Hill JA, Lin F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol. 2014;25:305–315. doi: 10.1681/ASN.2013040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TY, Sun Y, Liang Y, et al. ULK1/2 Constitute a Bifurcate Node Controlling Glucose Metabolic Fluxes in Addition to Autophagy. Mol Cell. 2016;62:359–370. doi: 10.1016/j.molcel.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Liang Q, Benavides GA, Vassilopoulos A, Gius D, Darley-Usmar V, Zhang J. Bioenergetic and autophagic control by Sirt3 in response to nutrient deprivation in mouse embryonic fibroblasts. Biochem J. 2013;454:249–257. doi: 10.1042/BJ20130414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tsai PI, Wu RM, Chien CT. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J Neurosci. 2010;30:13138–13149. doi: 10.1523/JNEUROSCI.1737-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang C, Yin J, et al. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett. 2009;454:187–192. doi: 10.1016/j.neulet.2009.02.056. [DOI] [PubMed] [Google Scholar]
- Loeb V, Yakunin E, Saada A, Sharon R. The Transgenic Overexpression of {alpha}-Synuclein and Not Its Related Pathology Associates with Complex I Inhibition. J Biol Chem. 2010a;285:7334–7343. doi: 10.1074/jbc.M109.061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb V, Yakunin E, Saada A, Sharon R. The transgenic overexpression of alpha-synuclein and not its related pathology associates with complex I inhibition. J Biol Chem. 2010b;285:7334–7343. doi: 10.1074/jbc.M109.061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan A, Murphy MP. Using chemical biology to assess and modulate mitochondria: progress and challenges. Interface Focus. 2017;7:20160151. doi: 10.1098/rsfs.2016.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano L, Lara-Lemus R, Zenteno E, varado-Vasquez N. The mitochondrial O-linked N-acetylglucosamine transferase (mOGT) in the diabetic patient could be the initial trigger to develop Alzheimer disease. Exp Gerontol. 2014;58:198–202. doi: 10.1016/j.exger.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Lubbe S, Morris HR. Recent advances in Parkinson’s disease genetics. J Neurol. 2014;261:259–266. doi: 10.1007/s00415-013-7003-2. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012a;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012b;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majbour NK, Vaikath NN, van Dijk KD, et al. Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol Neurodegener. 2016;11:7. doi: 10.1186/s13024-016-0072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela J, Tselykh TV, Kukkonen JP, Eriksson O, Korhonen LT, Lindholm D. Peroxisome proliferator-activated receptor-gamma (PPARgamma) agonist is neuroprotective and stimulates PGC-1alpha expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology. 2016;102:266–275. doi: 10.1016/j.neuropharm.2015.11.020. [DOI] [PubMed] [Google Scholar]
- Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, Dillmann WH. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1296–R1302. doi: 10.1152/ajpregu.00437.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem Soc Trans. 2017;45:155–162. doi: 10.1042/BST20160265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M, Seo BB, Nakamaru-Ogiso E, Greenamyre JT, Matsuno-Yagi A, Yagi T. Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson’s disease. PLoS ONE. 2008;3:e1433. doi: 10.1371/journal.pone.0001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino G, Pietrocola F, Eisenberg T, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal. 2012;16:920–934. doi: 10.1089/ars.2011.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Jang Y, Kim CH, et al. The RAB39B p.G192R mutation causes X-linked dominant Parkinson’s disease. Mol Neurodegener. 2015;10:50. doi: 10.1186/s13024-015-0045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Kimura M, Queliconi BB, et al. Parkinson’s disease-related DJ-1 functions in thiol quality control against aldehyde attack in vitro. Sci Rep. 2017;7:12816. doi: 10.1038/s41598-017-13146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A, Magalhaes J, Shen C, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014;137:1481–1495. doi: 10.1093/brain/awu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Shimoyama S, Kon T, et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol Aging. 2017;63:33–43. doi: 10.1016/j.neurobiolaging.2017.11.006. [DOI] [PubMed] [Google Scholar]
- Miquel E, Cassina A, Martinez-Palma L, et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic Biol Med. 2014;70:204–213. doi: 10.1016/j.freeradbiomed.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Mitchell T, Johnson MS, Ouyang X, et al. Dysfunctional mitochondrial bioenergetics and oxidative stress in Akita+/Ins2-derived beta-cells. Am J Physiol Endocrinol Metab. 2013;305:E585–E599. doi: 10.1152/ajpendo.00093.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Sone N, Saitoh T. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J Neurochem. 1987;48:1787–1793. doi: 10.1111/j.1471-4159.1987.tb05737.x. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AH, Hammond VJ, Sakoh-Nakatogawa M, Ohsumi Y, Thomas CP, Blanchet F, Piguet V, Kiselyov K, O’Donnell VB. A novel role for 12/15-lipoxygenase in regulating autophagy. Redox Biol. 2015;4:40–47. doi: 10.1016/j.redox.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moruno F, Perez-Jimenez E, Knecht E. Regulation of autophagy by glucose in Mammalian cells. Cells. 2012;1:372–395. doi: 10.3390/cells1030372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 2016;354:1004–1008. doi: 10.1126/science.aah4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AT, Ramachandran V, Joghee NM, Antony S, Ramalingam G. Gut Microbiota Dysfunction as Reliable Non-invasive Early Diagnostic Biomarkers in the Pathophysiology of Parkinson’s Disease: A Critical Review. J Neurogastroenterol Motil. 2018;24:30–42. doi: 10.5056/jnm17105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash Y, Schmukler E, Trudler D, Pinkas-Kramarski R, Frenkel D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J Neurochem. 2017;143:584–594. doi: 10.1111/jnc.14222. [DOI] [PubMed] [Google Scholar]
- Nelson MP, Boutin M, Tse TE, Lu H, Haley ED, Ouyang X, Zhang J, Auray-Blais C, Shacka JJ. The lysosomal enzyme alpha-Galactosidase A is deficient in Parkinson’s disease brain in association with the pathologic accumulation of alpha-synuclein. Neurobiol Dis. 2018;110:68–81. doi: 10.1016/j.nbd.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas WJ, Youngster SK, Kindt MV, Heikkila RE. MPTP, MPP+ and mitochondrial function. Life Sci. 1987;40:721–729. doi: 10.1016/0024-3205(87)90299-2. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Rinne JO, Kadir A, Langstrom B. The use of PET in Alzheimer disease. Nat Rev Neurol. 2010;6:78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Stamelou M, Goetz CG, et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32:1264–1310. doi: 10.1002/mds.27115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Nonaka T, Terada M, Tamaoka A, Hisanaga S, Hasegawa M. alpha-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J Biol Chem. 2016;291:15046–15056. doi: 10.1074/jbc.M116.736355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Kordower JH. Targeting alpha-Synuclein as a therapy for Parkinson’s disease: The battle begins. Mov Disord. 2017;32:203–207. doi: 10.1002/mds.26935. [DOI] [PubMed] [Google Scholar]
- Parekh VV, Pabbisetty SK, Wu L, Sebzda E, Martinez J, Zhang J, Van Kaer L. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8alpha+ dendritic cells. Proc Natl Acad Sci U S A. 2017;114:E6371–E6380. doi: 10.1073/pnas.1706504114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh VV, Wu L, Boyd KL, et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J Immunol. 2013;190:5086–5101. doi: 10.4049/jimmunol.1202071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol. 2009 doi: 10.1016/j.biocel.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Park JS, Koentjoro B, Davis RL, Sue CM. Loss of ATP13A2 impairs glycolytic function in Kufor-Rakeb syndrome patient-derived cell models. Parkinsonism Relat Disord. 2016;27:67–73. doi: 10.1016/j.parkreldis.2016.03.018. [DOI] [PubMed] [Google Scholar]
- Parker CG, Galmozzi A, Wang Y, et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell. 2017;168:527–541. doi: 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker WD, Jr, Swerdlow RH. Mitochondrial dysfunction in idiopathic Parkinson disease. Am J Hum Genet. 1998;62:758–762. doi: 10.1086/301812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul KC, Sinsheimer JS, Rhodes SL, Cockburn M, Bronstein J, Ritz B. Organophosphate Pesticide Exposures, Nitric Oxide Synthase Gene Variants, and Gene-Pesticide Interactions in a Case-Control Study of Parkinson’s Disease, California (USA) Environ Health Perspect. 2016;124:570–577. doi: 10.1289/ehp.1408976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014;158:54–68. doi: 10.1016/j.cell.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito R, Lazaro DF, Outeiro TF, Rego AC. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol Cell Neurosci. 2014;62:51–59. doi: 10.1016/j.mcn.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson’s disease: a metaanalysis. Environ Res. 2001;86:122–127. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA, Shrivastava S. A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology. 2000;21:435–440. [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Hamamichi S, Caldwell KA, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991a;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991b;266:4244–4250. [PubMed] [Google Scholar]
- Rahmanto AS, Morgan PE, Hawkins CL, Davies MJ. Cellular effects of peptide and protein hydroperoxides. Free Radic Biol Med. 2010;48:1071–1078. doi: 10.1016/j.freeradbiomed.2010.01.025. [DOI] [PubMed] [Google Scholar]
- Rajasekaran NS, Firpo MA, Milash BA, Weiss RB, Benjamin IJ. Global expression profiling identifies a novel biosignature for protein aggregation R120GCryAB cardiomyopathy in mice. Physiol Genomics. 2008;35:165–172. doi: 10.1152/physiolgenomics.00297.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralser M, Benjamin IJ. Reductive stress on life span extension in C. elegans. BMC Res Notes. 2008;1:19. doi: 10.1186/1756-0500-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Grundemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- Ramsay RR, Kowal AT, Johnson MK, Salach JI, Singer TP. The inhibition site of MPP+, the neurotoxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine is near the Q-binding site of NADH dehydrogenase. Arch Biochem Biophys. 1987;259:645–649. doi: 10.1016/0003-9861(87)90531-5. [DOI] [PubMed] [Google Scholar]
- Ravi S, Johnson MS, Chacko BK, et al. Modification of platelet proteins by 4-hydroxynonenal: Potential Mechanisms for inhibition of aggregation and metabolism. Free Radic Biol Med. 2015 doi: 10.1016/j.freeradbiomed.2015.10.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmann M, Darley-Usmar V, Zhang J. The Role of Autophagy, Mitophagy and Lysosomal Functions in Modulating Bioenergetics and Survival in the Context of Redox and Proteotoxic Damage: Implications for Neurodegenerative Diseases. Aging Dis. 2016;7:150–162. doi: 10.14336/AD.2015.0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmann M, Dodson M, Boyer-Guittaut M, Darley-Usmar V, Zhang J. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol. 2014;53C:127–133. doi: 10.1016/j.biocel.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Reeve A, Meagher M, Lax N, Simcox E, Hepplewhite P, Jaros E, Turnbull D. The impact of pathogenic mitochondrial DNA mutations on substantia nigra neurons. J Neurosci. 2013;33:10790–10801. doi: 10.1523/JNEUROSCI.3525-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science. 2007;316:1345–1348. doi: 10.1126/science.1142984. [DOI] [PubMed] [Google Scholar]
- Rehman H, Liu Q, Krishnasamy Y, et al. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int J Physiol Pathophysiol Pharmacol. 2016;8:14–27. [PMC free article] [PubMed] [Google Scholar]
- Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015;22:248–257. doi: 10.1038/cdd.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell. 2014;53:521–533. doi: 10.1016/j.molcel.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha BS, Lundberg JO, Radi R, Laranjinha J. Role of nitrite, urate and pepsin in the gastroprotective effects of saliva. Redox Biol. 2016;8:407–414. doi: 10.1016/j.redox.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano A, Koczwara JB, Gallelli CA, Vergara D, Micioni Di Bonaventura MV, Gaetani S, Giudetti AM. Fats for thoughts: An update on brain fatty acid metabolism. Int J Biochem Cell Biol. 2017;84:40–45. doi: 10.1016/j.biocel.2016.12.015. [DOI] [PubMed] [Google Scholar]
- Russell RL, Siedlak SL, Raina AK, Bautista JM, Smith MA, Perry G. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch Biochem Biophys. 1999;370:236–239. doi: 10.1006/abbi.1999.1404. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Sacino AN, Brooks M, Ceballos-Diaz C, Ladd TB, Howard JK, Golde TE, Giasson BI. Studies of lipopolysaccharide effects on the induction of alpha-synuclein pathology by exogenous fibrils in transgenic mice. Mol Neurodegener. 2015;10:32. doi: 10.1186/s13024-015-0029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40:200–210. doi: 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Saha S, Ash PE, Gowda V, Liu L, Shirihai O, Wolozin B. Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol Neurodegener. 2015;10:26. doi: 10.1186/s13024-015-0022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson TR, Debelius JW, Thron T, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell. 2016;167:1469–1480. e1412. doi: 10.1016/j.cell.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial dysfunction in Parkinson’s disease. Cell Death Differ. 2007;14:1261–1266. doi: 10.1038/sj.cdd.4402160. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Gegg M. Mitochondrial contribution to Parkinson’s disease pathogenesis. Parkinsons Dis. 2011;2011:159160. doi: 10.4061/2011/159160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheperjans F, Aho V, Pereira PA, et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord. 2015;30:350–358. doi: 10.1002/mds.26069. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid AW, Fauvet B, Moniatte M, Lashuel HA. Alpha-synuclein Post-translational Modifications as Potential Biomarkers for Parkinson Disease and Other Synucleinopathies. Mol Cell Proteomics. 2013;12:3543–3558. doi: 10.1074/mcp.R113.032730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Zhang J. Lysosomal function in macromolecular homeostasis and bioenergetics in Parkinson’s disease. Mol Neurodegener. 2010;5:14. doi: 10.1186/1750-1326-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L, Dawson VL, Dawson TM. Trumping neurodegeneration: Targeting common pathways regulated by autosomal recessive Parkinson’s disease genes. Exp Neurol. 2017;298:191–201. doi: 10.1016/j.expneurol.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selley ML. (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson’s disease. Free Radic Biol Med. 1998;25:169–174. doi: 10.1016/s0891-5849(98)00021-5. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di MC, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414:57–60. doi: 10.1016/j.neulet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shavali S, Brown-Borg HM, Ebadi M, Porter J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett. 2008;439:125–128. doi: 10.1016/j.neulet.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. 2015;33:125–131. doi: 10.1016/j.ceb.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Oh JY, Landar AL, Ulasova E, Venkatraman A, Bailey SM, Darley-Usmar VM. Nitroxia: the pathological consequence of dysfunction in the nitric oxide-cytochrome c oxidase signaling pathway. Free Radic Biol Med. 2005;38:297–306. doi: 10.1016/j.freeradbiomed.2004.10.037. [DOI] [PubMed] [Google Scholar]
- Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynela J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–1445. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- Singh P, Sharma R, McElhanon K, Allen CD, Megyesi JK, Benes H, Singh SP. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic Biol Med. 2015;86:90–101. doi: 10.1016/j.freeradbiomed.2015.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solesio ME, Prime TA, Logan A, Murphy MP, Del Mar Arroyo-Jimenez M, Jordan J, Galindo MF. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim Biophys Acta. 2013;1832:174–182. doi: 10.1016/j.bbadis.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Solt LA, Wang Y, Banerjee S, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–68. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, Valera E, Rockenstein E, Trejo-Morales M, Adame A, Masliah E. A brain-targeted, modified neurosin (kallikrein-6) reduces alpha-synuclein accumulation in a mouse model of multiple system atrophy. Mol Neurodegener. 2015;10:48. doi: 10.1186/s13024-015-0043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JA, Angot E, Brundin P. A deadly spread: cellular mechanisms of alpha-synuclein transfer. Cell Death Differ. 2011;18:1425–1433. doi: 10.1038/cdd.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W, Saftig P, Gartner J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet. 2006;78:988–998. doi: 10.1086/504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucki DM, Ruegsegger C, Steiner S, Radecke J, Murphy MP, Zuber B, Saxena S. Mitochondrial impairments contribute to Spinocerebellar ataxia type 1 progression and can be ameliorated by the mitochondria-targeted antioxidant MitoQ. Free Radic Biol Med. 2016;97:427–440. doi: 10.1016/j.freeradbiomed.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Sun N, Yun J, Liu J, et al. Measuring In Vivo Mitophagy. Mol Cell. 2015;60:685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH. The neurodegenerative mitochondriopathies. J Alzheimers Dis. 2009;17:737–751. doi: 10.3233/JAD-2009-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HWS, Sim AYL, Long YC. Glutamine metabolism regulates autophagy-dependent mTORC1 reactivation during amino acid starvation. Nat Commun. 2017;8:338. doi: 10.1038/s41467-017-00369-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112:892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapannone R, Mariappa D, Ferenbach AT, van Aalten DM. Nucleocytoplasmic human O-GlcNAc transferase is sufficient for O-GlcNAcylation of mitochondrial proteins. Biochem J. 2016;473:1693–1702. doi: 10.1042/BCJ20160092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura A, Taguchi K, Watanabe Y, Tatebe H, Tokuda T, Mizuno T, Tanaka M. Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous alpha-synuclein fibrils. Neurobiol Dis. 2014;73C:244–253. doi: 10.1016/j.nbd.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera E, Masliah E. Therapeutic approaches in Parkinson’s disease and related disorders. J Neurochem. 2016;139(Suppl 1):346–352. doi: 10.1111/jnc.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der MM, Brouwer M, Kromhout H, Nijssen P, Huss A, Vermeulen R. Is pesticide use related to Parkinson disease? Some clues to heterogeneity in study results. Environ Health Perspect. 2012;120:340–347. doi: 10.1289/ehp.1103881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J Neurosci. 2017;37:11151–11165. doi: 10.1523/JNEUROSCI.3791-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinje S, Stroes E, Nieuwdorp M, Hazen SL. The gut microbiome as novel cardio-metabolic target: the time has come! Eur Heart J. 2014;35:883–887. doi: 10.1093/eurheartj/eht467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, Feng D, Gao G, Yang Q. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy. 2016;12:1215–1228. doi: 10.1080/15548627.2016.1179401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Telpoukhovskaia MA, Bahr BA, Chen X, Gan L. Endo-lysosomal dysfunction: a converging mechanism in neurodegenerative diseases. Curr Opin Neurobiol. 2017;48:52–58. doi: 10.1016/j.conb.2017.09.005. [DOI] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Boyer-Guittaut M, Dodson M, Chatham J, Darley-Usmar V, Zhang J. Regulation of autophagy by protein post-translational modification. Lab Invest. 2015;95:14–25. doi: 10.1038/labinvest.2014.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Chatham JC, Darley-Usmar V, McMahon LL, Zhang J. O-GlcNAcylation and neurodegeneration. Brain Res Bull. 2016 doi: 10.1016/j.brainresbull.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Ouyang X, Benavides GA, Redmann M, Cofield S, Shacka JJ, Darley-Usmar V, Chatham JC, Zhang J. O-GlcNAc regulation of autophagy and α-synuclein homeostasis; implications for Parkinson’s disease. Molecular Brain. 2017 doi: 10.1186/s13041-017-0311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzberg E, Hezel M, Lundberg JO. Nitrate-nitrite-nitric oxide pathway: implications for anesthesiology and intensive care. Anesthesiology. 2010;113:1460–1475. doi: 10.1097/ALN.0b013e3181fcf3cc. [DOI] [PubMed] [Google Scholar]
- Wende AR, Young ME, Chatham J, Zhang J, Rajasekaran NS, Darley-Usmar VM. Redox biology and the interface between bioenergetics, autophagy and circadian control of metabolism. Free Radic Biol Med. 2016a;100:94–107. doi: 10.1016/j.freeradbiomed.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Young ME, Chatham J, Zhang J, Rajasekaran NS, Darley-Usmar VM. Redox biology and the interface between bioenergetics, autophagy and circadian control of metabolism. Free Radic Biol Med. 2016b;100:94–107. doi: 10.1016/j.freeradbiomed.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Zhao K, Jin S, Ding WX. New methods for monitoring mitochondrial biogenesis and mitophagy in vitro and in vivo. Exp Biol Med (Maywood) 2017;242:781–787. doi: 10.1177/1535370216688802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills J, Jones J, Haggerty T, Duka V, Joyce JN, Sidhu A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol. 2010;225:210–218. doi: 10.1016/j.expneurol.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissemann WT, Hill-Burns EM, Zabetian CP, et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am J Hum Genet. 2013;93:984–993. doi: 10.1016/j.ajhg.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem. 2017;292:16804–16809. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth AJ, Basu SS, Snyder NW, Mesaros C, Blair IA. Inhibition of neuronal cell mitochondrial complex I with rotenone increases lipid beta-oxidation, supporting acetyl-coenzyme A levels. J Biol Chem. 2014;289:26895–26903. doi: 10.1074/jbc.M114.591354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Menges S, Schlachetzki JC, Meixner H, Hoffmann AC, Schlotzer-Schrehardt U, Becker CM, Winkler J, Klucken J. Posttranslational modification and mutation of histidine 50 trigger alpha synuclein aggregation and toxicity. Mol Neurodegener. 2015;10:8. doi: 10.1186/s13024-015-0004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang W, Schlachetzki JC, Helling S, et al. Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci. 2013;54:71–83. doi: 10.1016/j.mcn.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. 2013;62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2017 doi: 10.1080/15548627.2017.1378838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Cheung AH, Okon M, McIntosh LP, Vocadlo DJ. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J Mol Biol. 2014;426:1736–1752. doi: 10.1016/j.jmb.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- Zhang J. Autophagy and Mitophagy in Cellular Damage Control. Redox Biol. 2013;1:19–23. doi: 10.1016/j.redox.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox Biology. 2015;4:242–259. doi: 10.1016/j.redox.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Eitan E, Mattson MP. Early involvement of lysosome dysfunction in the degeneration of cerebral cortical neurons caused by the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 2017a;140:941–954. doi: 10.1111/jnc.13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nguyen DT, Olzomer EM, Poon GP, Cole NJ, Puvanendran A, Phillips BR, Hesselson D. Rescue of Pink1 Deficiency by Stress-Dependent Activation of Autophagy. Cell Chem Biol. 2017b;24:471–480. e474. doi: 10.1016/j.chembiol.2017.03.005. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lu M, Du RH, Qiao C, Jiang CY, Zhang KZ, Ding JH, Hu G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener. 2016;11:28. doi: 10.1186/s13024-016-0094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Duan C, Lu L, Gao H, Zhao C, Yu S, Ueda K, Chan P, Yang H. alpha-Synuclein overexpression impairs mitochondrial function by associating with adenylate translocator. Int J Biochem Cell Biol. 2011;43:732–741. doi: 10.1016/j.biocel.2011.01.014. [DOI] [PubMed] [Google Scholar]
- Zuendorf G, Kerrouche N, Herholz K, Baron JC. Efficient principal component analysis for multivariate 3D voxel-based mapping of brain functional imaging data sets as applied to FDG-PET and normal aging. Hum Brain Mapp. 2003;18:13–21. doi: 10.1002/hbm.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]