

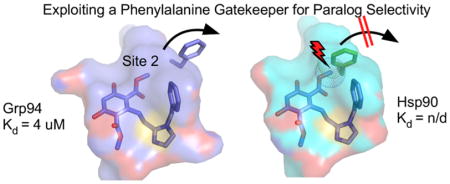
Abstract
Grp94 and Hsp90, the ER and cytoplasmic hsp90 paralogs, share a conserved ATP-binding pocket that has been targeted for therapeutics. Paralog-selective inhibitors may lead to drugs with fewer side effects. Here, we analyzed 1 (BnIm), a benzyl imidazole resorcinylic inhibitor, for its mode of binding. The structures of 1 bound to Hsp90 and Grp94 reveal large conformational changes in Grp94, but not Hsp90 that expose Site 2, a binding pocket adjacent to the central ATP cavity that is ordinarily blocked. The Grp94:1 structure reveals a flipped pose of the resorcinylic scaffold that inserts into the exposed Site 2. We exploited this flipped binding pose to develop a Grp94-selective derivative of 1. Our structural analysis shows that the ability of the ligand to insert its benzyl imidazole substituent into Site 1, a different side pocket off the ATP binding cavity, is the key to exposing Site 2 in Grp94.
Keywords: Hsp90, Grp94, Chaperone, Paralog selectivity, Crystal structure, ITC, Radicicol, Resorcinol
Graphical abstract
Introduction
The hsp90 molecular chaperones play essential roles in the folding and maturation of a diverse set of client proteins that are involved in numerous regulatory and signal transduction pathways 1, 2. Given the centrality of these clients in the control of critical cellular functions, hsp90s have emerged as an attractive target for therapeutic intervention against a wide variety of pathologies including cancer, neurodegenerative diseases, and infection.
Higher eukaryotes contain four hsp90 paralogs. Hsp90α and Hsp90β are localized to the cytoplasm, Grp94 is in the endoplasmic reticulum, and Trap-1 is mitochondrial. For all of the paralogs, chaperone action is associated with conformational changes driven by the binding of ATP to the N-terminal domain of the protein 3. The route to therapeutic intervention has largely targeted this domain with small molecule compounds that act as competitive inhibitors of ATP binding 4.
The majority of hsp90 inhibitors identified to date bind with similar affinities to the individual paralogs. The resulting indiscriminate suppression of multiple hsp90 clients with such compounds, however, has been associated clinically with toxicity and other adverse effects, and with the upregulation of Hsp70 and other compensatory chaperoning pathways 4, 5. Our understanding of the biology of hsp90 chaperones has also been closely tied to the discovery of highly selective inhibitors 1, 6, 7. The available pan-hsp90 inhibitors, however, are of limited use in deconvoluting the biological role of individual hsp90 paralogs. To potentially reduce toxicity by targeting fewer housekeeping clients, avoid up-regulation of compensatory chaperone pathways, and to provide reagents for probing the biology of hsp90 paralogs, inhibitor design efforts have begun to include approaches that selectively target individual hsp90 paralogs 8.
Grp94 has emerged as an attractive target for paralog-selective ligand development. Compared to cytoplasmic Hsp90, Grp94 has a more limited set of clients, consisting of proteins destined for the cell surface or excretion 2. Despite this more limited clientele, however, increased expression of Grp94 in cancers is uniformly associated with poor clinical prognosis 9–12, and studies have identified a critical role for Grp94 in breast cancer, multiple myeloma, and colon and liver cancers. Mechanistically, this reflects the role Grp94 plays in chaperoning strategically important clients in oncogenesis including integrins, Wnt co-receptor, toll-like receptors, Insulin-like growth factors, and the surface receptor for TGFβ, LRRC3213. Through these clients Grp94 controls a diverse set of strategically important oncogenic processes including proliferation, differentiation, apoptosis, angiogenesis, epithelial-mesenchymal transition, stemness and metastasis. In addition to cancer, Grp94 has also been implicated in diseases such as glaucoma via its client protein myocillin14.
The design of Grp94-selective inhibitors represents a significant challenge in light of the high sequence and structural conservation in the ATP binding cavity of the N-terminal domains of the hsp90 paralogs. For the four human paralogs, the 29 amino acids that make up the ATP binding cavity are over 70% identical; the remaining 30% are highly similar. Recent work, however, has led to a deeper understanding of the structure of the hsp90 ATP binding cavity. In particular, the ATP binding cavity comprises a central nucleotide-binding pocket surrounded by a halo of conserved side pockets, termed Sites 1, 2, and 3 8, 15. These pockets form pairwise binding sites for multi-functional ligands, with the central nucleotide-binding pocket accommodating the main scaffold of the ligand while an adjoining side pocket provides the binding site for attached substituents. To date, the paralog-selective binding of a ligand has reflected differences in the ability of individual paralogs to allow access to a side pocket. A key insight to emerge from this work is that intrinsic conformational differences among the paralogs, or conformational changes to the protein driven by ligand binding, govern access to Sites 1, 2 or 3. The ability of one hsp90 paralog and not another to undergo these conformational changes results in selective access to these side pocket sites, and selective ligand binding.
The Grp94-selective inhibitors described to date contain flexible substituents attached to a structural scaffold that sits in the central cavity of the ATP binding pocket. Three main classes of Grp94-selective inhibitors have thus far been identified. First among these was N-ethyl carboximino adenosine (NECA). The 5′ substituent of NECA accessed Site 3 in Grp94, a pocket that is exposed in Grp94 but is partially occluded in Hsp9016. Early binding17 and pulldown18, 19 data suggested that NECA exhibited high selectivity for Grp94, although this conclusion has been questioned recently20. Subsequent development of inhibitors preferentially targeting Site 3 in Grp94 includes a series of compounds based on the resorcinylic scaffold21–25. The substituted resorcinol of these compounds sits in the central ATP binding cavity and the pendant flexible substituent occupies Site 3. The progenitor of this series, Radamide,21, 22 showed a slight preference for Grp94 over Hsp90, but subsequent compounds containing benzyl imidazole23 and phenyl-linked benzyl substituents24, 25 were reported to exhibit sub-micromolar affinity for Grp94 and IC50 binding preferences of 12- to 70- fold for Grp94 over Hsp90 in a fluorescence polarization competition assay. Finally, Purine-based (PU) compounds contain a central purine scaffold connected to an aryl substituent via a sulfur linkage from the 8 position of the purine6, 15. Structural analysis6 showed that the flexibly-attached 8-aryl group occupied Site 2 in Grp94 and Site 1 in Hsp90. The loss of the hydrophobic stabilizing interactions from Site 2 resulted in a binding affinity that was 1–2 orders of magnitude lower for Hsp90 compared to Grp94. The general conclusion from these sets of experiments is that preferential binding to Grp94 occurs when a flexible substituent off the central scaffold can access one of the nearby side pockets. In the process of doing so, the ligand makes favorable interactions with the Grp94 ATP binding pocket that are not recapitulated when it binds to Hsp90. Interestingly, the use of rigidly attached moieties to target a selective side pocket, in place of the flexible substituent, has not been explored. This strategy may improve paralog selectivity by steric exclusion of the rigid moiety if the selective side pocket is blocked. This approach, however, requires an understanding of the ligand and protein determinants that cause a favorable side pocket to be exposed or blocked in each paralog.
In this report, we analyzed the binding of a recently-described inhibitor, BnIm (1) (Figure 1), to Hsp90 and Grp94. 1 is a resorcinylic benzyl imidazole that showed modest Grp94 selectivity in cell based assays (termed Compound 2 in this report) 23 and 12-fold Grp94 IC50 preference over Hsp90 in a displacement binding assay 15. Here, we provide an in-depth evaluation of the Grp94 and Hsp90 binding properties of 1 by determining the co-crystal structures of 1 bound to Hsp90 and Grp94. These structures reveal ligand-driven conformational differences in the two paralogs that expose Site 2 in Grp94, but not in Hsp90. The structures also reveal a novel difference in the binding pose of the resorcinylic scaffold when bound to Grp94. We exploited this previously unknown alternate binding pose, along with the observed conformational changes in Grp94, to develop 2, a compound with rigidly attached resorcinilyic moieties that is sterically restricted from binding to Hsp90 and exhibits high selectivity for Grp94.
Figure 1.

Ligands described in this report.
Results
The resorcinol moiety of 1 exhibits different poses when bound to Grp94N and Hsp90N
Inhibitor 1 (Figure 1) is a resorcinylic benzyl imidazole that has been reported to selectively inhibit Grp94 in vivo26,23 but whose in vitro paralog selectivity properties and mechanism of potential selectivity have not been evaluated. To probe the mechanism of selectivity, we determined the co-crystal structures of 1 bound to the N-terminal domains of human Hsp90α and canine Grp94, at 1.79 Å and 2.75 Å resolution. Electron density maps for both protein and ligand (Figure S1) were readily interpretable. As seen in Figures 2A and C, when bound to Hsp90N, the resorcinol ring of 1 sits in the ATP binding pocket where it is stabilized by hydrogen bonds between the phenolic hydroxyls and Asp93 and two water molecules. A van der Waals contact between the 3-chloro of the resorcinol ring and the side chain of Phe138, along with a hydrogen bond between the ester carbonyl and Thr184, further recapitulate the ATP binding pocket interactions observed previously with radamide (Figure 1) 22 and the diaryl resorcinylic inhibitors 27–30.
Figure 2. 1 (BnIm) binds with different poses to Hsp90 and Grp94.

A) Stereo view of 1 bound to Hsp90. 1 is drawn in yellow, Hsp90 in green. Water molecules are represented as red spheres, and hydrogen bonds are drawn as dashes. B) Stereo view of 1 bound to Grp94. C, D) Schematic representation of 1 contacts in Hsp90 (Cl-in pose) and Grp94 (Cl-out pose). Hydrogen bonds are drawn as dashes and van der Waals contacts are drawn as bold red arcs. Weak non-polar interactions are drawn as fine red arcs. Residues corresponding to Site 2 in Grp94 are indicated by purple boxes.
The crystal structure of Grp94N in complex with 1 was also determined and reveals an unexpected difference in the bound pose of the ligand. As seen in Figures 2B,D, the resorcinol of Grp94-bound 1 is flipped by 180° about the axis of the resorcinol-imidazole linker such that, compared to Hsp90N:1, the positions of the chloro and ester carbonyl substituents are reversed in the ATP binding pocket. Whereas the chloro atom in Hsp90N:1 resides in the back of the ATP binding pocket (“chloro-in”) and makes a van der Waals contact with Phe138, in Grp94 the resorcinylic chloro faces the mouth of the ATP binding pocket (“chloro-out”). This flip leaves the symmetrically opposed ester carbonyl inserted into the back of the pocket of Grp94 where it sits in the mouth of Site 2, the hydrophobic cavity formed by Val211, Ile247, Ala202, Val204, Leu249, Phe100, and Phe199. We confirmed the location of the chloro atom in each complex by anomalous difference Fouriers calculated from chlorine anomalous diffraction data collected from crystals of Grp94N:1 and Hsp90N:1 (Figure S2).
Until now, all resorcinylic chlorines bound to hsp90 family members have been found to adopt the chloro-in pose. The chloro-out pose found in Grp94-bound 1 is the first example of a chloro-substituted resorcinol bound to an Hsp90 family member in this flipped-out conformation.
The benzyl imidazole substituent sits in Site 1 of Grp94 and Hsp90
In both Hsp90 and Grp94, the benzyl imidazole of 1 is inserted into Site 1 and stabilizes the binding of 1 to the ATP binding pocket (Figure 2). In Hsp90, the tight, strongly hydrophobic Site 1 is defined by the side chains of Trp162, Val150, Phe138, Leu107, Met98, and Leu103. The benzyl moiety of 1 is situated ~3.7 Å from the side chain of Phe138 and makes an edge-to-face stacking interaction with this residue. A further contribution to the overall binding energetics comes from van der Waals stacking interactions between the cis face of the imidazole ring of 1 and the side chain of Asn106 from helix 3.
The benzyl imidazole substituent of Grp94-bound 1 adopts a conformation similar to that seen in Hsp90N:1 and also inserts into Site 1 of the ATP binding pocket. Site 1 in Grp94 is larger than the equivalent site in Hsp90 and is defined by Trp223, Val211, Phe199, Met154, and Leu163. Unlike Hsp90N:1, however, the benzyl moiety of 1 is not favorably positioned for an interaction with the side chain of Grp94 Phe199 (equivalent to Hsp90 Phe138). Instead, it makes a more modest van der Waals interaction with the backbone of Asn162. Interestingly, in Grp94N:1, the imidazole is 3.56 Å away from Phe195 benzyl ring and forms a face-on pi-pi stacking interaction with the benzyl side chain of Phe195. This interaction stabilizes the benzyl imidazole in Site 1 and may help compensate for loss of the benzyl-Phe199 interaction.
Extensive Structural changes to Grp94 but not to Hsp90 upon binding of 1
Compared to its unliganded form (PDB code 1YT1), the structure of inhibitor-bound Grp94 shows significant conformational changes in the ATP binding pocket and the lid region. In Grp94N:1, the interior of the ATP binding pocket is altered by the 4.3 Å movement of Phe199 away from its apo-Grp94 position (Figure 3A). This prevents a steric clash between the ester carbonyl of 1 and Phe199 (Figure 3B), and exposes Site 2, which accommodates the resorcinylic ester carbonyl moiety and stabilizes the flipped resorcinol pose (“chloro-out”).
Figure 3.

Conformational changes to the Grp94 lid and pocket upon 1 binding. A) Superposition of Grp94N in the open conformation (PDB code 1QY5) and Grp94N:1. Grp94N:1 is colored purple and red, Grp94-open is colored blue and yellow. The helix 4/5 lid is shown as α-carbons. Disordered residues 173–185 in Grp94N:1 are represented by a dashed line. Distances between selected pairs of α-carbons are indicated. B) Modeling 1 bound to the open conformation of Grp94 reveals potential clashes. Clashing Grp94-open residues are colored magenta and shown with their van der Waals radii drawn as dots. Corresponding Grp94N:1 residues are colored dark blue.
A distinctive characteristic of Grp94N:ligand complexes is the effect that bound ligands have on the conformation of the lid (residues 164–196) and helix 4,5. Apo, NECA, radicicol, and geldanamycin binding to Grp94 yield the “open” lid conformation 16, 22, 31; ATP, ADP, and AMP binding result in the “extended open” conformation of the lid 32; and the selective purine inhibitor PU-H54 yields a “partially closed” conformation of the lid 6. The structure of Grp94N:1 now contributes a fourth lid conformation to this set. As seen in Figure 3A, the distal end of helix 3 (residues 161–164) is unwound and the trajectory of the 165–169 linker that connects helices 3 and 4 is diametrically opposed to that seen in apo-Grp94. This remodeling places Ser169, the start of helix 4, 25.5 Å away from its apo-Grp94 counterpart. The orientation of helix 5 in Grp94N:1 is also changed. In this case, a 90° rotation of helix 5 about a pivot centered on Gly196 results in a helical axis that is perpendicular to its apo-Grp94 counterpart. The result of this movement is that Ala186, the end of helix 5, is displaced by 25.3 Å compared to its counterpart in apo-Grp94. This novel reorientation of helix 5 also dramatically repositions the side chain of Phe195 to a location 13 Å away from its apo Grp94 position. This allows for the strong pi-stacking stabilization between the side chain of Phe195 and the trans face of the imidazole ring of 1 (Figure 2B).
Finally, apo- and 1-bound Grp94 differ in the conformation of helix 3. Modeling the insertion of the benzyl imidazole group into Site 1 of apo-Grp94 shows that the moiety would impinge on the side chains of Asn162 and Leu163. As predicted from the modeling, these side chains move in Grp94N:1 to accommodate the ligand.
The structure of Hsp90 when bound to 1 remains essentially unchanged from the unliganded state (PDB code 3T0H) except for the region of helix 3 between Ile104 to Ala111, which remodels to form a pseudo-continuous helix with helix 4. This previously observed remodeling 33, moves the side chains of Asn106 and Leu107 away from the interior of the Site 1 binding pocket, where they would make a steric clash with the benzyl imidazole substitutent of 1.
The pose of Grp94-bound 1 is incompatible with the binding pocket of Hsp90
The observed poses of the Grp94-bound (1grp) or Hsp90-bound (1hsp) 1 ligand are similar except for the flipped configuration of the resorcinol ring, which results in the end-for-end swapping of the chloro and ester carbonyl moieties within the ATP binding pocket. Superposition of the Grp94N:1grp complex onto Hsp90N:1hsp shows that the resorcinol scaffolds of 1grp and 1hsp, as well as the benzyl groups, align well, indicating that the placement of the ligand scaffold in the ATP binding cavity is the same in both paralogs (Figure 4). From this analysis, however, it is also clear that the flipped 1grp pose is incompatible with the Hsp90 binding pocket due to a steric clash between the methyl ester of 1grp and Phe138 of Hsp90. As noted above, this steric constraint in Grp94N:1 is mitigated by displacing the corresponding Phe199 residue in the Grp94 ATP binding pocket, thus exposing Site 2 and providing space for the ester carbonyl moiety of 1grp. A similar movement of Phe138 in Hsp90 does not occur in Hsp90N:1. This predicted steric clash thus precludes the binding of 1 to Hsp90 in the flipped 1grp configuration, and explains the conformation of 1hsp in Hsp90.
Figure 4.

Superposition of Hsp90N:1 and Grp94N:1. Inhibitor 1 bound to Hsp90 is shown in white, and 1 bound to Grp94 is shown in yellow. Hsp90 side chains are colored green, and those from Grp94 are colored blue. Van der Waals radii of Hsp90 side chains making potential clashes with the 1Grp are shown as dots. F199 from Grp94-open (PDB code 1QY5) is colored cyan.
Characterizing the Binding of 1 to Grp94 and Hsp90 by ITC and FP
We used isothermal titration calorimetry (ITC) to measure the energetic consequences of the two poses of 1 observed upon binding to Grp94 and Hsp90. As seen in Figure 5A,D and Table 2, at 25 °C 1 binds to Grp94 with a Kd of 1.38 μM, and to Hsp90 with a Kd of 0.62 μM. The slightly better binding of 1 to Hsp90 compared to Grp94 may reflect the modestly improved stabilization of the benzyl imidazole substituent in the more tightly packed Site 1 of Hsp90, compared to the looser accommodation of this substituent in Site 1 of Grp94. This phenomenon was recently exploited in the design of benzamadine tetrahydroindolones that exhibited preferential binding to Hsp90 over Grp94 34, 35.
Figure 5.

Affinity and selectivity profiles of compounds. (A-F) ITC of ligand binding to Hsp90 and Grp94. Panels A, C, D, E, F, 400 μM Hsp90N or Grp94N were titrated into 40 μM 1, 2, or 3 at 25 °C in a buffer containing 40 mM Hepes, pH 7.5, 100 mM NaCl, 2% DMSO. For Panel B, 750 μM Hsp90N was titrated into 81 μM 2. Kd values were calculated from the average of 2 replicate experiments, ± SD. G) Graphical summary of the thermodynamic binding parameters derived from ITC titrations. H) Western blot of lysates from MDA-MB-231 cells treated for 24 h with vehicle (D), Geldanamycin (5 nM, G), or indicated amounts of 2. Probe antibodies are listed at the side of each blot.
Table 2.
Thermodynamic parameters of ligand binding
| Ligand | Protein | Temp (K) | N | Kd (μM) | ΔG (kcal/mol) | ΔH (kcal/mol) | TΔS (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 1 | Grp94N | 298 | 0.98 ± 0.02 | 1.38 ± 0.11 | −8.00 ± 0.04 | −11.53 ± 0.26 | −3.53 ± 0.22 |
| 1 | Grp94NMC | 298 | 1.12 ± 0.11 | 1.73 ± 0.21 | −7.87 ± 0.07 | −9.57 ± 1.75 | −1.70 ± 1.82 |
| 1 | Hsp90N | 298 | 1.00 ± 0.09 | 0.62 ± 0.13 | −8.48 ± 0.12 | −6.96 ± 0.13 | 1.52 ± 0.25 |
| 1 | Hsp90NMC | 298 | 1.07 ± 0.01 | 0.81 ± 0.10 | −8.31 ± 0.11 | −6.52 ± 0.12 | 1.80 ± 0.30 |
| 2 | Grp94N | 298 | 0.95 ± 0.0 | 4.20 ± 0.22 | −7.35 ± 0.03 | −11.33 ± 0.19 | −3.98 ± 0.16 |
| 2 | Hsp90N | 298 | - | n/d | - | - | - |
| 3 | Grp94N | 298 | 0.92 ± 0.00 | 0.65 ± 0.02 | −8.44 ± 0.02 | −10.96 ± 0.07 | −2.52 ± 0.09 |
| 3 | Hsp90N | 298 | 0.93 ± 0.01 | 0.32 ± 0.01 | −8.85 ± 0.02 | −8.70 ± 0.04 | 0.16 ± 0.06 |
| Rdc | Grp94N | 298 | 0.93 | 0.0048 | −11.40 | −29.30 | −17.90 |
From the ITC data, the entropic component of the binding energetics of 1 is significantly different for Grp94 compared to Hsp90. For Grp94, the calculated ΔS was −11.8 cal/mol/K. This results in a highly unfavorable entropic contribution to the ΔG of binding (Figure 5G). On the other hand, for Hsp90, the calculated ΔS was +5.1 cal/mol/K, indicating a favorable entropic contribution to the binding. The entropic penalty for 1 binding to Grp94 adds an additional +3.5 kcal/mol to the ΔG of binding, or more than 300-fold to the Kd, at 25 °C. The entropic penalty associated with the binding of 1 to Grp94 correlates with the significant structural rearrangements of the protein observed in the Grp94N:1 crystal structure.
An earlier report compared the binding of 1 to Grp94 and Hsp90 at 4 °C using a fluorescence polarization (FP) competition assay 15. In that study, which we have reproduced here (Table 3), a 12-fold preference in the IC50 for 1 binding to Grp94 over Hsp90 was observed. The differences between the ITC data presented here and the FP data can be reconciled in large part by converting the observed IC50 value to Ki’s, which takes into account significant differences for each paralog in the intrinsic affinity of the fluorescent probe used in the competitive displacement reaction36. When expressed as Kis, the FP data shows good agreement with the ITC data. The remaining differences between the FP and ITC data may reflect differences in the temperature dependence of the Hsp90 and Grp94 binding reactions, as seen from the opposite signs of their respective entropic components (Figure 5G, Table 2). At the lower temperature of the FP measurement compared to the ITC study (4 °C vs. 25 °C), the positive ΔS for Hsp90 would lower the measured affinity, while the negative ΔS for Grp94 would improve the measured affinity. A similar discrepancy between ITC and FP IC50 measurements for Hsp90 has been reported previously37 and is seen for other ligands described later in this report (Table 3) and for other ligand classes (D. Gewirth et al., in preparation).
Table 3.
FP Competition binding data
| Ligand | Protein | Temp (K) | IC50 (μM) | Ki (μM) | Ki/Kd1 |
|---|---|---|---|---|---|
| 1 | Grp94 | 277 | 1.1 | 0.44 | 0.3 |
| 1 | Hsp90 | 277 | 13.2 | 1.8 | 2.9 |
| 2 | Grp94 | 277 | 17.4 | 4.1 | 1.0 |
| 2 | Hsp90 | 277 | >100 | >13 | n/d |
| 3 | Grp94 | 277 | 1.2 | 0.44 | 0.7 |
| 3 | Hsp90 | 277 | 6.2 | 0.84 | 2.6 |
Kd data from Table 2
The effects of buffer composition and pH on the ITC binding of 1 to both Hsp90 and Grp94 were tested and found to have no significant impact on binding constants. ITC titrations with the full-length proteins yielded Kds that were similar to those obtained using the N-terminal domains (Table 2). Together the data suggests that in vitro, at physiologic temperatures, there is no preference by 1 for Grp94 over Hsp90.
Structure-based design and testing of Grp94-specific derivatives of 1
To develop a Grp94-selective derivative of 1, we exploited the modeled incompatibility of the “flipped” pose of inhibitor 1 with the Hsp90 binding pocket. We reasoned that replacing the chloro atom on the resorcinol ring with a bulkier substituent would disfavor binding to Hsp90 due to predicted steric clashes with Phe138. We therefore tested the more bulky bis-methyl ester (2) derivative of 1 (Figure 1) for its ability to bind to Hsp90 and Grp94 by ITC. As seen in Figure 5B,E, ITC titrations show that 2 binds to Grp94 with a Kd of 4.20 μM but generated almost no heat when titrated with Hsp90, indicating very weak or absent binding to this paralog. Compound 2 thus exhibits a strong preference for Grp94. The preference of 2 for Grp94 was confirmed by in vivo testing (Figure 5H), where treatment of MDA-MB-231 cells with 2 suppressed the levels of the Grp94 client Integrin α2 but showed no induction of Hsp70 or Akt degradation, which are both markers of Hsp90 inhibition, even at the highest levels tested (75 μM).
We determined the 2.7 Å co-crystal structure of Grp94N:2. In this structure, the conformation adopted by Grp94 bound with 2, and the overall pose of 2, is identical to that of Grp94N:1, confirming the modeling predictions that led to the design of 2 (Figure 6A). We also determined the 1.9 Å structure of 2 soaked into crystals of apo Hsp90N. The high concentration of the protein in the crystal (23 mM) and the soaked ligand (5 mM) allowed us to visualize very low affinity interactions that are beyond the range of detection of solution binding techniques such as ITC. Nevertheless, the overall quality of the electron density for the ligand was poorer than that observed for other complexes in this study, and correlates with the weaker observed binding of 2 to Hsp90. Compared to the structure of Hsp90N:1, the structure of Hsp90N:2 shows that the resorcinol ring of 2, for which there is strong electron density (Figure S1), is rotated in the ATP binding pocket by ~9° in order to accommodate the bulkier methyl ester group (Figure 6B,D). This rotation of the resorcinol results in an unfavorable positioning of the benzyl imidazole moiety so that it no longer occupies Site 1, but instead makes only weak van der Waals interactions with Asp54, Ala55, and Lys58 of helix 2 (Figure 6B,C). The rotation of the resorcinol ring and the potential inability to access Site 1 may account for the poor binding of 2 to Hsp90.
Figure 6.

Binding of ligands 2 and 3 in Grp94 and Hsp90. A) Superposition of Grp94N:1, Grp94N:2, and Grp94N:3 showing similar poses for the ligand. B) The binding pose of 2 in Hsp90 is distorted. Overview alignment of Hsp90:1 (green) and Hsp90:2 (cyan) crystal structures showing the good superposition of the two protein structures except for the region between residues 103 and 111. The benzyl imidazole groups from 1 (grey) and 2 (magenta) are found in different poses in the two structures. C) Superposition of Grp94N:2 and Hsp90N:2 showing the different poses for the bound ligand and potential clashes between the Grp94 pose and the Hsp90 pocket. D) The resorcinol scaffolds of 1 (gray) and 2 (magenta) make similar hydrogen bonds in the Hsp90 ATP binding pocket but 2 is rotationally displaced by about 9 degrees in order to prevent clashes between the larger methyl ester group and Phe138, which blocks Site 2 in Hsp90. E) Phe199 in Grp94 exposes Site 2 in Grp94N:3 despite having the smaller chloro moieties on the resorcinol ring. The residues of Site 2 are shown as sticks with dot surfaces.
Our modeling also predicts that swapping the ester moiety of 1 with a less bulky substituent such as chlorine would eliminate any preference for the Grp94 binding pocket. We therefore also tested the bis-chloro (3) derivative of 1 and measured the binding to each paralog by ITC. As seen in Figure 5C,E, the less bulky 3 binds to Hsp90 and Grp94 with Kds of 0.32 μM and 0.65 μM, respectively, indicating that this compound is not selective for either paralog, as predicted. Thus, the steric restriction imposed by the bulkier bis-ester moieties is a key determinant that imparts Grp94 selectivity to the resorcinol scaffold.
We determined the co-crystal structure of Grp94N:3. Interestingly, as seen in Figure 6E, despite replacement of the bulky bis-ester moiety with the smaller chloro group, Phe199, the gatekeeper residue for Site 2, is rotated into the position that exposes Site 2. This is the same conformation of Phe199 seen in the Grp94N:1 and Grp94N:2 structures, and suggests that the movement of Phe199 that exposes Site 2 is not driven by the size of the resorcinylic substitutents at the 3 position.
Discussion and Conclusions
In this report, we show that the structures of 1 in complex with the N domains of Hsp90 and Grp94 reveal two binding poses for the chlorinated resorcinylic moiety of the inhibitor: the “chloro-in” pose as seen in Hsp90 and the “chloro-out” orientation adopted in Grp94. The two poses arise when the resorcinol moiety is flipped about the resorcinol-imidazole linker. The chloro-out pose was unexpected, since to date only the chloro-in pose has been observed in all reported examples of resorcinylic ligands bound to hsp90 family members. The chloro-out pose requires rearrangement of the ATP binding pocket of Grp94 to expose Site 2.
Steric considerations and the flexibility of the N domain of each paralog dictate the pose allowed for the binding of 1. The chloro-in pose of Hsp90N:1 positions the bulkier methyl ester substituent into the solvent exposed mouth of the ATP binding pocket, where it is easily accommodated. In contrast, the resorcinylic methyl ester in Grp94 is thrust towards the rear of the pocket where it occupies Site 2 after displacing Phe199. The equivalent residue Phe138 is not displaced in Hsp90 and thereby prevents Hsp90 from binding 1 in the chloro-out pose.
We tested the effect of forcing Hsp90 to bind 1 in the chloro-out (methyl ester-in) pose with 2, which we designed based on replacement of the 3-chloro substituent with a methyl ester. The bis-methyl ester resorcinylic ring is now symmetric around the apendant imidazole group and, when bound to Hsp90, has no choice but to accommodate one of the two esters into the main ATP binding cavity. While the structure of Grp94N:2 showed that the ligand is accommodated in the same manner as 1, the structure of Hsp90N:2 showed significant distortions to the pose of the bound ligand. The structural data correlates with the ITC analysis that showed that 2 exhibited good binding to Grp94 but failed to bind Hsp90. Together with the data that shows that the bis-chloro derivative 3 binds equally well to Grp94 and Hsp90, this supports the conclusion that the distortions to the ligand pose needed to accommodate the bis-methyl ester substituted resorcinol in the Hsp90 pocket are energetically unfavorable.
Exposing Site 2 in Grp94 depends on perturbing Site 1
To date, Grp94-selective ligands have accessed Site 2, a deep, strongly hydrophobic pocket adjacent to the ATP binding cavity. Access to Site 2 in Grp94 is gated by Phe199 (Figure 7). Upon 1 binding to Grp94, the movement of Phe199 allows the ester carbonyl to insert into Site 2. Earlier analyses of the ATP binding cavity of hsp90s did not identify Site 2 as a potential ligand binding site, however, since in apo or ATP-bound Grp94 the mouth of this cavity is blocked by the position of the conserved Phe199 (Phe138 in Hsp90), which is part of the hydrophobic core of the N-terminal domain. Indeed, Site 2 was only recognized as a paralog-specific druggable pocket when its exposure was observed experimentally in the structure of the complex between Grp94 and the selective ligand PU-H54 6.
Figure 7.

Summary of structural and binding data showing Site 1, F195, and F199 act in concert to open Site 2. F199 and F138 block access to Site 2 in Grp94 and Hsp90. Insertion of the benzyl imidazole into Site 1 of Grp94 is stabilized by a repositioned F195, opens F199, and exposes Site 2, which can accommodate the larger resorcinylic methyl ester moiety. F138 does not open in Hsp90, resulting in an unstable binding of the resorcinylic methyl ester and benzyl imidazole in 2.
The residues of Site 2 and the ATP binding pocket are identical or highly homologous among hsp90 family members. This raises the question of what causes Phe199 to move in response to 1 binding to Grp94, and why when 1 binds to Hsp90 the equivalent Hsp90 residue, Phe138, does not move. Modeling of 1 into apo Grp94 shows that the ester carbonyl group of 1 makes steric clashes with Phe199 in the back of the pocket. However, the need to accommodate the incoming ester carbonyl moiety does not satisfactorily explain the movement of Phe199 in Grp94N:1. This is because the resorcinol moiety also has the option of adopting the 1hsp pose that places the 3-chloro at the “in” position and the 5-ester at the “out” position, thus avoiding any potential steric clashes. Indeed, a closely related compound, radamide 21 (Figure 1), contains the same 3-chloro and 5-ester carbonyl substituents on its resorcinol group as 1, yet when radamide binds to Grp94 (PDB code 2GFD) it adopts the Hsp90-like chloro-in, ester-out pose, rather than the chloro-out, ester-in pose seen in Grp94N:1 22 (Figure 7). If the chloro-in pose reflects the blockage of Site 2 by Phe199, this suggests that the movement of Phe199 depends on the identity and placement of the other resorcinylic substituents.
Comparison of radamide and 1 shows that they differ in the substituent located at the distal end of the ethylene linker, with 1 containing a benzyl imidazole at this position and radamide having an amide-linked quinone. The crystal structures of these complexes show that these substituents occupy different side pockets off the ATP binding cavity. In Grp94N:radamide, the amide-linked quinone occupies Site 3. Insertion of substituents into Site 3 is structurally benign and does not trigger rearrangements in the N-terminal domain in any of the three known examples where Site 3 is utilized (Grp94N:NECA, PDB code 1QY5, Grp94N:Radamide, PDB code 2GFD, and Grp94N:KUNG29, PDB code 5IN9).
In Grp94N:1, on the other hand, the benzyl imidazole moiety sits in Site 1, where it is stabilized by a highly favorable stacking interaction between the imidazole ring and the face of Phe195. In contrast to Site 3, placement of substituents into Site 1 of Grp94 trips a structurally destabilizing trigger that is formed by the interaction between Leu163 and Phe199. In apo Grp94, Leu163 and Phe199 make a van der Waals contact that blocks Site 1. Upon insertion of the benzyl imidazole into Site 1, however, Phe199 and Leu163 move in tandem away from the benzyl imidazole, while maintaining their van der Waals interaction. The consequences of maintaining this contact are extensive. Among the structural rearrangements that result are the movement of the Grp94 lid, the repositioning of Phe195, and the exposure of Site 2, all of which were seen in the Grp94N:PU-H54 complex as well6 (PDB code 3O2F).
The structure of Grp94N:3 supports the Site 1 trigger hypothesis. In that structure, even though the smaller bis-chloro resorcinylic substituents do not require access to Site 2, Site 2 was nonetheless exposed. The benzyl imidazole of 3 occupies Site 1 and the structural rearrangements described above are observed. This suggests that insertion of substituents into Site 1 is the key to exposing Site 2 (Figure 7).
The observation that the benzyl imidazole of 1 occupies Site 1 also explains the observation 23 that linkers between the benzyl group and the imidazole group of greater than one carbon exhibit no binding to Grp94 in fluorescence displacement assays, and markedly reduced inhibition of client production in cell based assays. Modeling the longer linker compounds into Grp94N:1 shows that, in any orientation, the benzyl group at the end of the longer linkers would make severe steric clashes with one or more of the residues lining Site 1, including Phe199, Trp223, Asn162, and Leu163, thereby preventing stable binding.
In apo-Hsp90, Site 1 is also blocked by Leu107 and Phe138, the Hsp90 equivalents of Leu163 and Phe199 in Grp94. Moieties that insert into Site 1 must therefore also disrupt this interaction to gain access to the site. In contrast to Grp94, however, the structural response to the insertion of ligands into Site 1 of Hsp90 is not the same as in Grp94. In Grp94N:1, the Phe199-Leu163 van der Waals interaction is preserved, at the cost of large rearrangements in the N-terminal domain, whereas in Hsp90N:1 the Phe138-Leu107 contact is disrupted. This forces Leu107 into a new local environment, losing its apo Hsp90 interactions with Phe138 and Leu103, but gaining new stabilization from insertion into a pocket formed by Phe20, Phe22, Ile26, and Ile104. This response has been observed not only in Hsp90N:1 but in a number of Hsp90N:ligand complexes 33, 38. Importantly, however, in Hsp90N:1 and in all of the other examples of Site 1 usage, the position of Phe138 is unaltered and Site 2 in Hsp90 is not exposed.
Prospects for selective ligand development
Compared to the tightly constrained nature of the Hsp90 ATP binding cavity, the plasticity of its Grp94 counterpart can be exploited for paralog selective inhibitor design. Compound 2, the bis-methylester derivative of 1, shows little detectable binding to Hsp90. This is presumably because stable accommodation of the resorcinol into the central ATP binding cavity requires the insertion of the methyl ester moiety into Site 2, which is blocked in Hsp90, or unfavorable distortion of the 2 binding pose. Examination of the crystal structures of Grp94N:1 and Grp94N:2 shows that the methyl ester moiety only partially fills the deep, hydrophobic pocket of Site 2. This raises the possibility that derivatives of 2 with bulkier substituents on the 1 and 3 positions of the resorcinol ring might show even greater selectivity for Grp94 over Hsp90. Simple modeling suggests that bis-ethylester-, propylester-, butylester-, pentylester-, and methylbenzyl-carbonyl derivatives of 2 could easily be accommodated in Site 2 of Grp94. All of these would be sterically excluded from binding to Hsp90. The binding of compound 2 to Grp94 has a measured Kd of 4.2 μM. It is possible that a tighter fit of the bulkier 1- or 3-moieties of derivatives of 2 into Site 2, coupled with their greater hydrophobicity, might also improve the overall affinity of such compounds for Grp94.
Experimental Procedures
Reagents
Radicicol was purchased from AG Scientific. Compound 1 was synthesized as previously described 23. All inhibitors were dissolved in DMSO and stored at −20 °C.
Chemistry
Synthetic procedures and full characterization of 2 and 3 are provided in the Supporting Information. Thin layer chromatography was used to confirm that compounds were ≥ 95% pure.
Protein Expression and Purification
Canine Grp94NΔ41 (residues 69–337; 41 amino acid from the charged linker consisting of residues 286–328 was replaced by four glycine residues) was expressed in E. coli BL21star (DE3) as a GST fusion and purified as previously described 16. Canine Grp94N is overall 99.2% identical to human Grp94N and differs at just 1 position (S127A) in the N-terminal domain studied here. Human Hsp90 alpha (residues 1–236) was expressed in E. coli strain BL21star (DE3) or Rosetta2 (DE3) as an N-terminal hexa-histidine fusion and purified as described 38. N-terminally His-tagged canine Grp94-NMC (73–754Δ41) missing the charged linker, the pre-N domain, and the KDEL C-terminus was expressed and purified as before 39. Full length N-terminal His-tagged human Hsp90 (1–693) with the charged domain (225–280) replaced by four glycine residues was expressed in E. coli Rosetta2 (DE3). Purification by Nickel-affinity, Q-Sepharose ion exchange, and gel filtration chromatography was similar to that used for full length Grp94. Protein concentrations were determined by absorption at 280 nm using calculated extinction coefficients.
Crystallization
Complexes of Grp94NΔ41 (30 mg/ml) and hHsp90N (20 mg/ml) with inhibitor were formed by mixing the protein with a 3–5 fold molar excess of the concentrated ligand. Crystallizations were done by the hanging drop vapor diffusion method by mixing 2 μL of the protein:ligand complex with 2 μL of reservoir solution. For the Hsp90:1 complex, crystals were obtained at 4 °C over a reservoir solution containing 25% PEG 2000 MME, 0.8 M sodium formate, 0.1 M sodium cacodylate, pH 6.5. Crystals were cryoprotected by rapid passage through a reservoir solution containing 30% PEG 2000 MME followed by flash freezing in liquid nitrogen. The Grp94NΔ41:1 complex was crystallized at 18 °C over a reservoir solution containing 16% PEG 2000 MME, 0.15 M Li2SO4, 0.1 M sodium cacodylate, pH 6.5. Crystals were looped out of the drop, cryoprotected with reservoir solution supplemented with ethylene glycol and flash frozen in liquid nitrogen. Crystals of Grp94NΔ41:2 and Grp94NΔ41:3 complexes were grown over reservoir solutions containing 0.22 M ammonium phosphate and 1.6 M ammonium sulfate, 0.1 M MES, pH 6.0 solutions, respectively, and cryoprotected with ethylene glycol supplemented reservoir solutions. Crystals of apo hHsp90N were grown at 4 °C over a reservoir solution of 17% PEG 2000 MME, 0.2 M MgCl2, 0.1 M sodium cacodylate, pH 6.5. Crystals of hHsp90N:2 were obtained by adding a concentrated solution of 2 in the crystallization reservoir solution supplemented with glycerol to pre-formed crystals of apo hHsp90N at 4 °C. After 24 hours crystals were looped out of the drops and flash frozen in liquid nitrogen.
Data Collection, structure determination, and refinement
X-ray diffraction data were collected on beamline BL9-2 at the Stanford Synchrotron Radiation Lightsource or on beamlines 23ID-B and 17ID-B at the Advanced Photon Source. Data were reduced and scaled using HKL200040. Structures of the hHsp90 co-crystals were determined by molecular replacement using MolRep from the CCP4 suite 6.141 with the hHsp90N-geldanamycin structure (PDB code 1YER) as the search model. Initial phases for the Grp94NΔ41-inhibitor complexes were obtained by molecular replacement using only the core-region of the search model Grp94N:Radicicol (PDB code 1U0Z). Omitted residues were manually rebuilt using Coot and refined using Phenix 42, 43. Inhibitor and solvent positions were identified from difference density. Ligand parameter and topology files were generated using the Dundee PRODRG server 44. Structure validation was performed using KING and MolProbity45, 46. Data collection and refinement statistics are shown in Table 1, and ligand electron density is shown in Figure S1. Molecular graphics were generated using The PyMol Molecular Graphics System Version 1.8 (Schrodinger, LLC).
Table 1.
Data collection and refinement statistics.
| hHsp90α:1 | Grp94N:1 | hHsp90α:2 | Grp94N:2 | Grp94N:3 | |
|---|---|---|---|---|---|
| PDB code | 5VYY | 5WMT | 6CEO | 6BAW | 6C91 |
| Diffraction data | |||||
| Data set | q228d6b | q229c2a | q289a5b | q279a6b | q530b3h |
| Source | APS 23IDB | SSRL 9-2 | APS 23IDB | APS 23IDB | APS 17ID |
| Space group | C 2 2 21 | P 21 21 21 | I222 | P 21 21 21 | P 43 21 2 |
| a,b,c | 67.08, 92.06, 85.26 | 94.41, 94.73, 179.25 | 65.44, 88.93, 99.93 | 93.65, 93.71,175.41 | 95.05, 95.05, 178.59 |
| Resolution (Å) | 33.51-1.79 | 34.54-2.64 | 50 – 1.90 | 50-2.70 | 50-2.90 |
| Last shell (Å) | 1.83-1.80 | 2.64-2.60 | 1.93-1.90 | 2.75-2.70 | 2.95-2.90 |
| Total (Unique) reflections | 90663 (24883) | 238406 (49257) | 72064 (23393) | 245839 (41640) | 362816 (18991) |
| % Complete (last shell) | 99.4 (98.9) | 98.1 (94.5) | 99.3 (99.0) | 96.8 (98.5) | 100.0 (99.9) |
| Average I/σI (last shell) | 30.8 (3.2) | 34.8 (1.6) | 26.5 (2.2) | 9.0 (1.1) | 38.4 (2.1) |
| Redundancy | 3.6 | 4.8 | 3.1 | 5.9 | 19.1 |
| Rsym | 0.066 (0.43) | 0.059 (0.988) | 0.050 (0.422) | 0.187 (1.0) | 0.081 (1.15) |
| Refinement | |||||
| Resolution range (Å) | 33.51-1.79 | 34.54-2.75 | 34.52-1.90 | 41.91-2.70 | 33.61-2.90 |
| Reflections | 24866 | 41977 | 23232 | 41569 | 18920 |
| Total atoms (non H) | 1838 | 6903 | 1824 | 7046 | 3444 |
| Solvent atoms | 165 | 60 | 159 | 176 | 38 |
| RMSD Bond lengths (Å) | 0.010 | 0.002 | 0.007 | 0.002 | 0.009 |
| RMSD Bond angles (°) | 1.085 | 0.514 | 0.844 | 0.461 | 1.035 |
| R value | 0.184 | 0.208 | 0.185 | 0.214 | 0.196 |
| Rfree | 0.218 | 0.236 | 0.211 | 0.257 | 0.241 |
| Structure solution | MR | MR | MR | MR | MR |
| All atom clashscore | 2.42 | 4.57 | 2.45 | 3.12 | 8.01 |
| Ramachand. outliers (%) | 0.0 | 0.12 | 0.0 | 0.24 | 0.71 |
Chlorine anomalous diffraction
Chlorine anomalous diffraction data were collected from crystals of hHsp90N:1 and Grp94NΔ41:1 at beamline 23ID-B at the APS with the X-ray wavelength set to 1.984 Å (6.25 keV). Data were processed using HKL2000, keeping the F+ and F- reflections separate. The location of the chlorine atoms in the bound ligand was visualized using anomalous difference Fouriers using phases derived from the previously refined hHsp90N:1 and Grp94NΔ41:1 complexes.
Isothermal Titration Calorimetry
Proteins were equilibrated into DTT-free assay buffer by repeated concentration and dilution using an ultrafiltration spin filter (Millipore). Concentrated ligand solutions in DMSO, typically 100 mM, were diluted into assay buffer. The standard ITC assay buffer contained 40 mM Hepes pH 7.5, 100 mM NaCl, and 2% DMSO. Titrations were carried out at 25 °C using a VP-ITC calorimeter (Microcal, Inc.) with the protein solution (300–400 μM) loaded into the titration syringe and the inhibitor (30–40 μM) into the cell. The effect of buffer and pH was also determined by repeating the titrations using PBS (pH 7.5), 40 mM MOPS, 100 mM NaCl (pH 6.5), and 40 mM TAPS, 100 mM NaCl (pH 8.5). ITC titrations against radicicol, a well-characterized Hsp90 inhibitor, was used as a benchmark for both instrument and protein quality control.
ITC assays consisted of 29 injections of 10 μL (2 μL for the first injection) each, with 5 minute intervals between injections, and the stirring speed set to 310 rpm. The first injection was discarded in all titrations. Reference power was set at 10 μCal s−1. Data were fitted to a one-site model using Origin 7 Software. Titrations using Grp94 and Hsp90 N-terminal domains were repeated two times and errors reported as standard deviation from the mean.
Fluorescence Polarization Binding Assay
Titrations were incubated for 5 hours at 4 °C in the presence of 10 nM recombinant full length Grp94 or hHsp90α, and 6 nM FITC-Geldanamycin, essentially as described47. Ki values were calculated from the IC50 measurements using the method of Nikolovska-Coleska et al.48, using values for the Kd of the tracer ligand of 1.4 nM for Hsp90 and 6.6 nM for Grp94, as described36.
Western blot
MDA-MB-231 cells were maintained in Advanced DMEM/F12 (Gibco) supplemented with 1% L-glutamine (2 mM), 1% penicillin/Streptomycin and 10% FBS. Cells were grown to confluence in a humidified atmosphere (37° C, 5% CO2), seeded (2000/well, 100 μL) in the appropriate plates, and allowed to attach overnight. Cells were treated with various concentrations of compound 2, Geldanamycin in DMSO (0.1% DMSO final concentration, 5 nM), or vehicle (DMSO) for 24 h. Cells were harvested in cold PBS and lysed in mammalian protein extraction reagent (MPER, Pierce) lysis buffer containing protease and phosphatase inhibitor cocktails (Roche) on ice for 1 h. Protein concentrations were determined using the Pierce BCA protein assay kit. Equal amounts of protein (5 μg) were electrophoresed under reducing conditions (8% polyacrylamide gel), transferred to a polyvinylidene fluoride membrane (PVDF), and immunoblotted with antibodies for Hsp70, Integrin α2, Akt, or Actin. Membranes were incubated with an appropriate horseradish peroxidase-labeled secondary antibody, developed with a chemiluminescent substrate, and visualized.
Supplementary Material
Acknowledgments
D.T.G. is funded by grants from the NIH (R01-CA095130, P01-CA186866) and the Richard and Mae Stone Goode Foundation of Buffalo. B.S.J.B. is supported by NIH EY024232 and V.M.C. is supported by F99212467 from the NCI. X-ray diffraction data was collected at the Advanced Photon Source beamlines 23ID-B, 17-ID and the Stanford Synchrotron Radiation Lab beamline BL9-2.
Abbreviations used
- FITC
Fluorescein isothiocyanate
- FP
fluorescence polarization
- Grp94
Glucose regulated protein 94 kilodaltons
- HER2
Receptor tyrosine-protein kinase erbB-2
- Hsp90
Heat shock protein 90 kilodaltons
- ITC
isothermal titration calorimetry
- LRP6
LDL Receptor Related protein 6
- MME
mono methyl ether
- MOPS
3-morpholinopropane-1-sulfonic acid
- NECA
N-ethylcarboximido adenosine
- TAPS
[trismethylamino] propanesulfonic acid
- Wnt
Wingless-related integration site
Footnotes
PDB ID Codes: Grp94N:1 – 5WMT; Hsp90N:1 – 5VYY; Grp94N:2 – 6BAW; Hsp90N:2 – 6CEO; Grp94N:3 – 6C91. Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions: N.S.Q. and D.T.G. designed experiments, determined all crystal structures, carried out ITC binding analyses, designed derivatives, analyzed the structures, and wrote the manuscript. V.M.C., A.S.D., J.Z., C.K., and B.S.J.B. synthesized compounds 1–3, carried out FP and Western blot analyses, and reviewed the manuscript.
Ancillary Information
Supporting Information: 2 supplemental figures; chemical synthesis and characterization; molecular formula strings.
References
- 1.Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS One. 2011;6:e26044. doi: 10.1371/journal.pone.0026044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hong F, Mohammad Rachidi S, Lundgren D, Han D, Huang X, Zhao H, Kimura Y, Hirano H, Ohara O, Udono H, Meng S, Liu B, Li Z. Mapping the interactome of a major mammalian endoplasmic reticulum heat shock protein 90. PLoS One. 2017;12:e0169260. doi: 10.1371/journal.pone.0169260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prodromou C. Mechanisms of Hsp90 regulation. Biochem J. 2016;473:2439–2452. doi: 10.1042/BCJ20160005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, Gewirth DT, Chiosis G. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9:677–684. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 8.Gewirth DT. Paralog specific Hsp90 inhibitors - a brief history and a bright future. Curr Top Med Chem. 2016;16:2779–2791. doi: 10.2174/1568026616666160413141154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2012 doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dejeans N, Glorieux C, Guenin S, Beck R, Sid B, Rousseau R, Bisig B, Delvenne P, Buc Calderon P, Verrax J. Overexpression of GRP94 in breast cancer cells resistant to oxidative stress promotes high levels of cancer cell proliferation and migration: implications for tumor recurrence. Free Radic Biol Med. 2012;52:993–1002. doi: 10.1016/j.freeradbiomed.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 11.McLaughlin M, Vandenbroeck K. The endoplasmic reticulum protein folding factory and its chaperones: new targets for drug discovery? Br J Pharmacol. 2011;162:328–345. doi: 10.1111/j.1476-5381.2010.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ansa-Addo EA, Thaxton J, Hong F, Wu BX, Zhang Y, Fugle CW, Metelli A, Riesenberg B, Williams K, Gewirth DT, Chiosis G, Liu B, Li Z. Clients and oncogenic roles of molecular chaperone gp96/grp94. Curr Top Med Chem. 2016;16:2765–2778. doi: 10.2174/1568026616666160413141613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suntharalingam A, Abisambra JF, O’Leary JC, 3rd, Koren J, 3rd, Zhang B, Joe MK, Blair LJ, Hill SE, Jinwal UK, Cockman M, Duerfeldt AS, Tomarev S, Blagg BS, Lieberman RL, Dickey CA. Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J Biol Chem. 2012;287:40661–40669. doi: 10.1074/jbc.M112.384800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel HJ, Patel PD, Ochiana SO, Yan P, Sun W, Patel MR, Shah SK, Tramentozzi E, Brooks J, Bolaender A, Shrestha L, Stephani R, Finotti P, Leifer C, Li Z, Gewirth DT, Taldone T, Chiosis G. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem. 2015;58:3922–3943. doi: 10.1021/acs.jmedchem.5b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278:48330–48338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- 17.Rosser MF, Nicchitta CV. Ligand interactions in the adenosine nucleotide-binding domain of the Hsp90 chaperone, GRP94. I. Evidence for allosteric regulation of ligand binding. J Biol Chem. 2000;275:22798–22805. doi: 10.1074/jbc.M001477200. [DOI] [PubMed] [Google Scholar]
- 18.Hutchison KA, Fox IH. Purification and characterization of the adenosine A2-like binding site from human placental membrane. J Biol Chem. 1989;264:19898–19903. [PubMed] [Google Scholar]
- 19.Hutchison KA, Nevins B, Perini F, Fox IH. Soluble and membrane-associated human low-affinity adenosine binding protein (adenotin): properties and homology with mammalian and avian stress proteins. Biochemistry. 1990;29:5138–5144. doi: 10.1021/bi00473a020. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Street TO. 5′-N-ethylcarboxamidoadenosine is not a paralog-specific Hsp90 inhibitor. Protein Sci. 2016;25:2209–2215. doi: 10.1002/pro.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clevenger RC, Blagg BS. Design, synthesis, and evaluation of a radicicol and geldanamycin chimera, radamide. Org Lett. 2004;6:4459–4462. doi: 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 22.Immormino RM, Metzger LEt, Reardon PN, Dollins DE, Blagg BS, Gewirth DT. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. J Mol Biol. 2009;388:1033–1042. doi: 10.1016/j.jmb.2009.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, Blagg BS. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134:9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowley VM, Khandelwal A, Mishra S, Stothert AR, Huard DJ, Zhao J, Muth A, Duerfeldt AS, Kizziah JL, Lieberman RL, Dickey CA, Blagg BS. Development of Glucose Regulated Protein 94-selective inhibitors based on the BnIm and Radamide scaffold. J Med Chem. 2016;59:3471–3488. doi: 10.1021/acs.jmedchem.6b00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley VM, Huard DJE, Lieberman RL, Blagg BSJ. Second generation Grp94-selective inhibitors provide opportunities for the inhibition of metastatic cancer. Chemistry. 2017;23:15775–15782. doi: 10.1002/chem.201703398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stothert AR, Suntharalingam A, Huard DJ, Fontaine SN, Crowley VM, Mishra S, Blagg BS, Lieberman RL, Dickey CA. Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum Mol Genet. 2014;23:6470–6480. doi: 10.1093/hmg/ddu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brough PA, Barril X, Beswick M, Dymock BW, Drysdale MJ, Wright L, Grant K, Massey A, Surgenor A, Workman P. 3-(5-Chloro-2,4-dihydroxyphenyl)-pyrazole-4-carboxamides as inhibitors of the Hsp90 molecular chaperone. Bioorg Med Chem Lett. 2005;15:5197–5201. doi: 10.1016/j.bmcl.2005.08.091. [DOI] [PubMed] [Google Scholar]
- 28.Cheung KM, Matthews TP, James K, Rowlands MG, Boxall KJ, Sharp SY, Maloney A, Roe SM, Prodromou C, Pearl LH, Aherne GW, McDonald E, Workman P. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg Med Chem Lett. 2005;15:3338–3343. doi: 10.1016/j.bmcl.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 29.Dymock BW, Barril X, Brough PA, Cansfield JE, Massey A, McDonald E, Hubbard RE, Surgenor A, Roughley SD, Webb P, Workman P, Wright L, Drysdale MJ. Novel, potent small-molecule inhibitors of the molecular chaperone Hsp90 discovered through structure-based design. J Med Chem. 2005;48:4212–4215. doi: 10.1021/jm050355z. [DOI] [PubMed] [Google Scholar]
- 30.Kreusch A, Han S, Brinker A, Zhou V, Choi HS, He Y, Lesley SA, Caldwell J, Gu XJ. Crystal structures of human HSP90alpha-complexed with dihydroxyphenylpyrazoles. Bioorg Med Chem Lett. 2005;15:1475–1478. doi: 10.1016/j.bmcl.2004.12.087. [DOI] [PubMed] [Google Scholar]
- 31.Dollins DE, Immormino RM, Gewirth DT. Structure of unliganded GRP94: the endoplasmic reticulum Hsp90: Basis for nucleotide-Induced conformational change. J Biol Chem. 2005;280:30438–30447. doi: 10.1074/jbc.M503761200. [DOI] [PubMed] [Google Scholar]
- 32.Immormino RM, Dollins DE, Shaffer PL, Soldano KL, Walker MA, Gewirth DT. Ligand-induced conformational shift in the N-terminal domain of GRP94: an Hsp90 chaperone. J Biol Chem. 2004;279:46162–46171. doi: 10.1074/jbc.M405253200. [DOI] [PubMed] [Google Scholar]
- 33.Wright L, Barril X, Dymock B, Sheridan L, Surgenor A, Beswick M, Drysdale M, Collier A, Massey A, Davies N, Fink A, Fromont C, Aherne W, Boxall K, Sharp S, Workman P, Hubbard RE. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem Biol. 2004;11:775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 34.Ernst JT, Liu M, Zuccola H, Neubert T, Beaumont K, Turnbull A, Kallel A, Vought B, Stamos D. Correlation between chemotype-dependent binding conformations of HSP90alpha/beta and isoform selectivity-implications for the structure-based design of HSP90alpha/beta selective inhibitors for treating neurodegenerative diseases. Bioorg Med Chem Lett. 2014;24:204–208. doi: 10.1016/j.bmcl.2013.11.036. [DOI] [PubMed] [Google Scholar]
- 35.Ernst JT, Neubert T, Liu M, Sperry S, Zuccola H, Turnbull A, Fleck B, Kargo W, Woody L, Chiang P, Tran D, Chen W, Snyder P, Alcacio T, Nezami A, Reynolds J, Alvi K, Goulet L, Stamos D. Identification of novel HSP90alpha/beta isoform selective inhibitors using structure-based drug design. demonstration of potential utility in treating CNS disorders such as Huntington’s disease. J Med Chem. 2014;57:3382–3400. doi: 10.1021/jm500042s. [DOI] [PubMed] [Google Scholar]
- 36.Taldone T, Patel PD, Patel M, Patel HJ, Evans CE, Rodina A, Ochiana S, Shah SK, Uddin M, Gewirth D, Chiosis G. Experimental and structural testing module to analyze paralogue-specificity and affinity in the hsp90 inhibitors series. J Med Chem. 2013;56:6803–6818. doi: 10.1021/jm400619b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 38.Immormino RM, Kang Y, Chiosis G, Gewirth DT. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J Med Chem. 2006;49:4953–4960. doi: 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- 39.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Afonine PV, Grosse-Kunstleve RW, Chen VB, Headd JJ, Moriarty NW, Richardson JS, Richardson DC, Urzhumtsev A, Zwart PH, Adams PD. phenix.model_vs_data: a high-level tool for the calculation of crystallographic model and data statistics. J Appl Crystallogr. 2010;43:669–676. doi: 10.1107/S0021889810015608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 45.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen VB, Davis IW, Richardson DC. KING (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci. 2009;18:2403–2409. doi: 10.1002/pro.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra SJ, Ghosh S, Stothert AR, Dickey CA, Blagg BS. Transformation of the non-selective aminocyclohexanol-based Hsp90 inhibitor into a Grp94-seletive scaffold. ACS Chem Biol. 2017;12:244–253. doi: 10.1021/acschembio.6b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.