

Abstract
Two widely used anticancer drugs, doxorubicin (DOX) and paclitaxel (PTX), possess distinct physical property and chemotherapy specificity. In order to investigate their interaction mechanism with single-walled carbon nanotube (SWCNT), co-loading on, and releasing from the SWCNT, all atom molecular dynamics (MD) simulations were firstly carried out for different SWCNT systems, followed by the binding free energy calculation with MM-PBSA. The results indicate that the co-loading of DOX and PTX to the pristine SWCNT is exothermic and spontaneous. The DOX molecules predominantly interact with the SWCNT via π–π stacking through the conjugated aromatic rings, while the separated aromatic rings of the PTX also primarily interacts with the SWCNT through π-π stacking, yet supplemented by X-π (X=C-H, N-H and C=O). Moreover, the strongest binding of DOX and PTX with the pristine SWCNT shows similar strength (∆G: −32.0 vs −33.8 kcal/mol). For the chitosan functionalized SWCNT (f-SWCNT) the DOX and PTX molecules still prefer binding to the sidewall of CNT rather than binding with the polymer, and the non-covalent functionalization of the SWCNT with chitosan decreases the binding of DOX and PTX with the sidewall of the f-SWCNT as compared with the system DOX/PTX-SWCNT (∆G: −24.0 and −21.9 kcal/mol). The protonation of chitosan and drug molecules further weakens the interaction between DOX/PTX with the f-SWCNT, and shows a consequent displacement of the drug molecules and triggering the release of the drugs. The variation of binding strength for the three systems (DOX/PTX-SWCNT, DOX/PTX-f-SWCNT, and DOXH+/PTXH+-f-SWCNT) was also discussed in terms of histogram or frequency of the distance from the drugs to the SWCNT. In addition, the encapsulation of two DOX in the f-SWCNT are considerably stronger than the binding of other six drug molecules to the sidewall, indicating the encapsulation of anticancer drugs may also play very important role and should be considered in the drug delivery.
Keywords: Carbon nanotube, doxorubicin, paclitaxel, molecular dynamics simulations
Graphical abstract
Co-loading and release of doxorubicin and paclitaxel using chitosan functionalized single-walled carbon nanotube through pH-controlled drug delivery system
1. Introduction
A great deal of attention has already been paid to the promising carbon nanotube (CNT)-based drug delivery system (DDS) due to CNT’s unique properties such as stability, robustness, high drug carrying capacity, and ability to penetrate cell membranes.1–4 In comparison with other nanomaterials, CNTs appear to be more dynamic and attractive as transporters for the delivery of biomolecules and drugs.5,6 In addition to offering potential drug delivery, CNTs have intrinsic properties that facilitate tracing and chemtherapy. However, pristine CNTs are highly hydrophobic, and the main obstacle in the utilization of CNTs in biological and medicinal chemistry is their poor solubility and dispersability. The toxicity of CNTs also remains an important issue. In order to improve the drug carrying performance and reduce the toxicity of the CNTs, biomolecular functionalization has been found to be an effective option to overcome such defects.7,8
The single-walled carbon nanotubes (SWCNTs) facilitate the interaction of various molecules both along the sidewall and within the hollow cylindrical cavity. Covalent and non-covalent surface modifications with polymers are common practices, but the advantage of noncovalent functionalization is that it would not damage the nanotube structure as it does with covalent functionalization. The noncovalent type of functionalization allows the controlled release of drugs, and has thus been widely used in developing CNT-based nanomedicine, and therapeutic drug release systems with improved bioavailability, increased specificity and sensitivity, and reduced pharmacological toxicity.5,9,10 The chitosan modified SWCNTs show a good solubility and dispersion in aqueous solution.11,12 Chitosan, a polysaccharide biopolymer consisting of β-(1,4)-linked D-glucosamine (GCS or G) units together with some N-acetyl-D-glucosamine (NAG or N) units, can be obtained from the deacetylation of natural chitin that is the main component of crustacean shells, cell walls of fungi, and the exoskeleton of insects. Chitosan has been widely used in medicinal and pharmaceutical applications because of its low toxicity, biodegradability, and biocompatibility.13–15 All these interesting characteristics have led to the development of numerous applications of chitosan and its derivatives in pharmacy, as matrices in compressed tablets, membranes and microparticles for drug delivery.16
Doxorubicin (DOX, Scheme 1) is anthracycline cytostatic antibiotic used in cancer chemotherapy due to its efficacy in fighting a wide range of cancers including breast cancer, ovarian cancer, lung carcinoma, acute leukemia and several sarcomas.17,18 Paclitaxel (PTX) is a potent microtubule-stabilizing agent that triggers cell cycle arrest, and it has often been studied in conjunction with other chemotherapeutic agents to enhance therapeutic effectiveness and to reduce its toxicity.19,20 The combination of DOX and PTX has been used in the treatment of metastatic breast cancer.21 Recently, the recombinant high-density lipoprotein nanoparticle was exploited for the co-delivery of DOX and PTX for efficacious combination of chemotherapy.22 In another study, the co-loaded system Graphene-Curcumin-PTX shows high potency against A549 and MDA cells, compared to the treatments with single drugs.20 It seems that co-delivery of anticancer drugs with desired chemotherapeutic property is becoming very attractive.
Scheme 1.
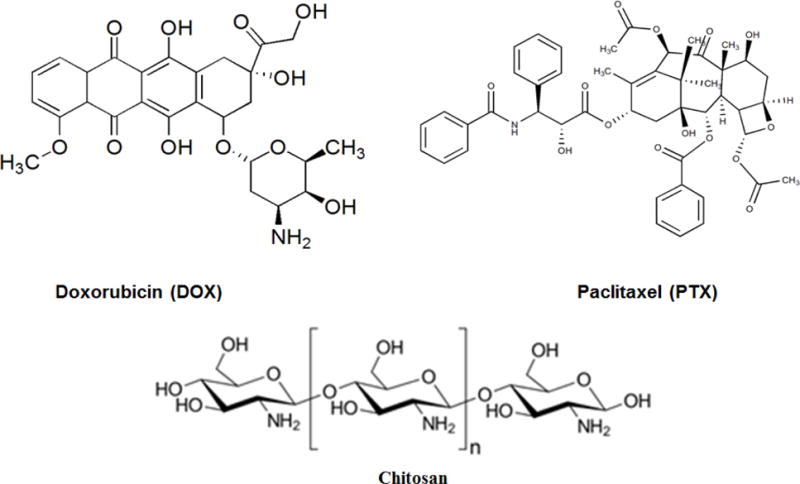
Chemical structure of doxorubicin, paclitaxel, and chitosan
Carbon nanotubes are considered promising drug delivery vehicles for cancer therapy based on their ability to prolong drug circulation time, reduce systemic toxicity, and increase drug accumulation at tumor sites through the enhanced permeation effect.23 Binding of molecules to carbon nanotubes and their release can be controlled by varying the pH.24,25 The increasing importance of co-delivery of the anticancer drugs with different desired chemotherapy, together with the peculiarities of CNT and chitosan motivate us to understand the interaction of two anticancer drugs with CNT systems on a molecular level. In this study we applied molecular dynamics (MD) simulations to investigate the underlying mechanisms of DOX and PTX co-loading using the pristine SWCNT and chitosan functionalized SWCNT (f-SWCNT), and also the release of the protonated DOX (DOXH+) and PTX (PTXH+) using the protonated and functionalized SWCNT (p-f-SWCNT). All atom molecular dynamics (MD) simulations were firstly carried out for different SWCNT systems, followed by the binding free energy calculation with MM-PBSA. Through the current systematic investigation, the new insights into the distinct interaction mechanisms of PTX and DOX with single-walled carbon nanotube (SWCNT), and the effects of noncovalent functionalization and protonation on their binding will be provided, which will eventually help understand co-loading as well as releasing of the two anti-cancer drugs.
2. Materials and Methods
2.1 System Preparation
(12,12) armchair SWCNT terminated with hydrogen atoms with a diameter of approximately 16.5Å and 50.0Å in length was generated using the Nanotube Structure Generator tool TubeGen 3.4.26 The helix chitosan chain consists of 30 units in 3:2 ratio of G:N (G: GCS; N: NAG) was obtained with Material Studio visualizer. The f-SWCNT is generated by initially placing chitosan parallel to SWCNT. In all of the systems DOX and PTX are initially placed approximately 10Å away from the sidewall of SWCNT. The four initial systems are listed as follows. (1) The pristine SWCNT with four DOX and four PTX molecules; (2) The chitosan functionalized SWCNT (f-SWCNT) with four DOX and four PTX molecules; (3) The f-SWCNT with protonated chitosan (p-f-SWCNT) with four DOXH+ and four PTXH+ molecules, where amine groups of the drugs and the polymer chain are protonated; (4) The f-SWCNT with two DOX molecules encapsulated, and two DOX molecules and four PTX molecules adsorbed on the sidewall of SWCNT.
2.2 Molecular Dynamics Simulations
MD simulations were performed using AMBER16 package.27 All complexes (shown in Table 1) were solvated with TIP3P water in an octahedron box keeping buffering distance of 12Å between the complex surface and box boundary. The charges for DOX, PTX, DOXH+, PTXH+, chitosan and protonated chitosan were obtained by fitting electrostatic potential for the optimized structure at HF/3-21G level in Gaussian09 package.28 The drug molecules and SWCNT were parameterized by using the updated general AMBER force field (GAFF), whereas chitosan is parameterized using GLYCAM 06j-1 force field. The TIP3P model for water, and GLYCAM06 for chitosan have also been applied to chitosan functionalized carbon nanotubes carrying DOX.25 The charge neutrality was maintained throughout, and p-f-SWCNT system was neutralized with chloride ions using tLEaP module of AMBER 16.
Table 1.
The different SWCNT systems used in our study.
| System | DOX | PTX | Functionalization | Protonation |
|---|---|---|---|---|
| SWCNT | 4 outside | 4 outside | Pristine | No |
| f-SWCNT | 4 outside | 4 outside | functionalized | No |
| p-f-SWCNT | 4 outside | 4 outside | functionalized | Yes |
| f-SWCNT@2DOX | 2 outside, 2 inside | 4 outside | functionalized | No |
All prepared systems were minimized in two steps. The water molecules and ions were firstly minimized by keeping force restraints over complexes and then followed by minimization of the whole system in the second setup. The first 1000 steps of energy minimization were run with steepest descent method and the remaining 2000 steps with conjugate gradient method. After minimization, each system was gently heated from 0K to 300K in 50ps at constant volume and equilibrated at 300K for another 200ps at constant pressure. Finally, a production run was performed with no restraints imposed for 100ns in an isothermal isobaric (NPT) ensemble, and a time step of 2fs was adopted. The SHAKE algorithm was employed on all atoms so as to constrain the bonds of all hydrogen atoms. The temperature was regulated at 300 K using the Langevin thermostat with a collision frequency of 1.0ps−1. The Berendsen barostat method was used to apply a pressure of 1 atm with isotropic position scaling, with a pressure relaxation time constant of 1.0 ps. The cutoff for van der Waals interaction was set to 10Å during heating and MD simulations. The long-range electrostatic interactions were treated with the particle-mesh Ewald method.29
2.3 Binding Free Energy Calculations
The binding free energy of different SWCNT and anticancer drugs were evaluated using MM/PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) program in AMBER16. In the MM-PBSA strategy the explicit solvent and ions are stripped from the trajectory files to hasten convergence by preventing solvent-solvent interactions. The MM-PBSA following explicit solvent MD was also recently applied to the chitosan functionalized SWCNT for DOX loading.25 For each complex, a total of 1000 snapshots were extracted along MD trajectory from the last 10ns MD simulations. The binding energy (ΔG) in condensed phase can be simply defined by the following equations.30,31
| (1) |
| (2) |
Where ΔG is the binding free energy in solution that consists of the molecular mechanics energy in the gas phase (ΔEMM), the solvation free energy (ΔGsol) and the conformational entropy effect due to the binding (TΔS).
| (3) |
Where ΔEvdw and ΔEele correspond to the van der Waals and electrostatic interactions in gas phase, respectively.
| (4) |
Where ΔGpb and ΔGnp are the polar and non-polar contributions to the solvation free energy, respectively. The ΔGsol was calculated with the PBSA module, where the dielectric constant is set to 1 inside the solute and 80.0 in the solvent. The nonpolar contribution of the solvation free energy was estimated as a function of the solvent accessible surface area (SASA), as follows:
| (5) |
Where, solvent accessible surface area (SASA) was estimated using the MSMS program, with a solvent probe radius of 1.4 Å. The values of empirical constants γ and β were set to 0.000542 kcal mol−1 Å−2 and 0.92 kcal/mol, respectively. The entropy was calculated for the last 1000 snapshots extracted from last 10ns using the Quasi-harmonic entropy approximation method.
2.4 Trajectory Analysis
The distance and radial distribution function (RDF) calculations were performed using the CPPTRAJ.32 In this study, we calculated the RDF of different SWCNT systems to obtain the approaching distance of drug molecules to the SWCNT surface, and we chose the carbon atoms of SWCNT and a few key atoms of drug molecules for RDF calculations.
3. Results and Discussion
3.1 Co-loading of Anti-cancer Drugs (DOX and PTX) on Pristine SWCNT
MD simulations for the co-loading of anti-cancer drugs (DOX and PTX) on the pristine SWCNT were explored. The final MD simulation snapshot of SWCNT co-loaded with DOX and PTX were shown in Figures 1a-b, and the exterior-bound position of DOX and PTX was shown in Figures 1c-d, respectively. In spite of far from the SWCNT at the beginning, the MD results indicate that both DOX and PTX have strong affinity towards pristine SWCNT. During simulation one DOX molecule overlaps on the other DOX through hydrogen bonds, and the other three DOX molecules are quite stable throughout the simulation, which were observed that the planar tricyclic moiety of the DOX attaches to the aromatic surface of the SWCNT via π-π stacking. The DOX molecules show a uniform array along the tube sidewall, as observed from the final snapshots shown in Figure 1. The comparison between PTX and DOX reveals significant differences in their interactions with the SWCNT. Although PTX does not possess the conjugated aromatic rings and three aromatic benzene rings are separated, it is also stably adsorbed through π-π stacking by one benzene ring that is parallel to the sidewall of the SWCNT, whereas another benzene ring is perpendicular and interacts with the SWCNT through CH-π, and third benzene ring doesn’t fall towards the sidewall.
Figure 1.
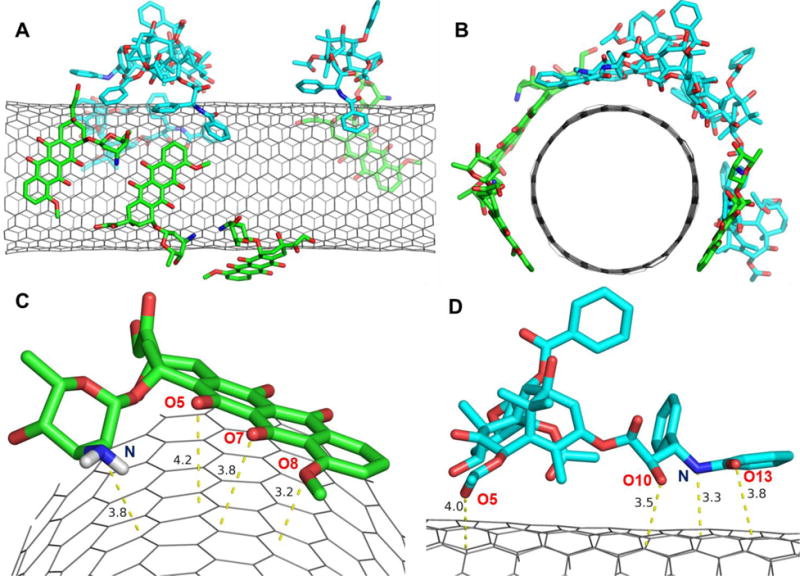
Loading of four DOX and four PTX molecules on pristine SWCNT. A) front view; B) side view; C) DOX; and D) PTX close orientation with respect to the side wall of SWCNT. The dashed lines indicate the nearest distance to the sidewall in Å.
To quantitatively address the different binding of DOX and PTX to the surface of the SWCNT, the binding enthalpies and free energies were tabulated in Table 2. All of the bindings are exothermic (∆H<0), and in spite of entropy loss they still tend to be spontaneous (∆G<0). The decomposition of the binding free energy contributions shows that the contribution of van der Waals is predominant. The binding Gibbs free energies range from −8.2 to −32.0 kcal/mol for four DOX molecules, and −26.0 to −33.8 kcal/mol for PTX molecules. DOX-4 shows the strongest binding with a free energy of −32.0 kcal/mol of four DOX, whereas PTX-4 has the highest binding free energy of −33.8 kcal/mol among four PTX molecules. If the binding modes and free energy of DOX molecules are compared, three DOX molecules parallel to SWCNT for the most part of simulations show similar binding free energy (−28.8-32.0 kcal/mol), whereas the DOX molecule that mainly interacts with other drug through H-bonds shows rather low binding free energy (−8.2 kcal/mol). In order to further understand the binding, the distances were calculated between the carbon atoms of the SWCNT and DOX atoms (O8, O7, O5 and N) (Figure 1c), and those from the carbon atoms of the SWCNT to the PTX atoms (O13, N, O10 and O5) (Figure 1d). The distances of approximately 3.2-3.8Å indicate the π-π stacking that are primarily responsible for the stable loading of both DOX and PTX on the surface of the SWCNT. The π-π stacking can be expected from their vertical distances as compared with the layer distance (3.4Å) for the typical π-π stacking of graphite. In addition, to clearly verify the stacking and other sorts of van der Waals interaction the reduced density gradient (RDG) for the MD snapshots was added as supporting Figure S3. According to the color code green shows vdW interaction and red strong nonbonded overlap.44 The isosurface of sign(λ2)×ρ in Figure S3 provides a clear visualization of the π-π stacking between aromatic moiety (three conjugated rings) of DOX with CNT, the π-π stacking of the aromatic ring of PTX with CNT.
Table 2.
ΔH, −TΔS, ΔG (kcal/mol) for the binding of DOX and PTX molecules with the pristine SWCNT.
| ΔH | −TΔS | ΔG | |
|---|---|---|---|
| DOX-1 | −36.1 | 27.9 | −8.2 |
| DOX-2 | −35.5 | 6.7 | −28.8 |
| DOX-3 | −36.1 | 6.6 | −29.5 |
| DOX-4 | −34.3 | 2.3 | −32.0 |
| PTX-1 | −33.7 | 7.6 | −26.0 |
| PTX-2 | −36.0 | 5.3 | −30.6 |
| PTX-3 | −40.8 | 9.0 | −31.7 |
| PTX-4 | −40.6 | 6.8 | −33.8 |
The similar binding enthalpies of DOX and PTX to pristine SWCNT are also supported by the result from the PM6-DH2 method, a semiempirical quantum mechanical method PM6 corrected by the dispersion as well as hydrogen bonds, implemented in MOPAC2009 package.33 The PM6-DH2 including the implicit solvent model (COSMO) was used to fully optimized the adsorptions of five as well as individual drug molecules on (12,12) SWCNT (Figure S1), bringing about the average binding enthalpy of −31.0 and −29.0 kcal/mol for DOX and PTX, respectively. The binding enthalpy for individual DOX and PTX molecule gets even closer, −32.7 vs −32.6 kcal/mol.
To assess the drug orientation on the sidewall of the SWCNT, the time-average RDF was calculated for the molecules, DOX-4 and PTX-4 that possess the strongest binding to the pristine SWCNT. The RDF plots between the selected atoms of DOX-4 and carbon atoms of the SWCNT are shown in Figure 2A. The most probable distances from the N atom of DOX-4 is shown at ~8.0 Å maximum, oxygen atoms (O5, O7, O8) shown at ~4.0 Å maximum to the SWCNT surface, whereas the center of mass (COM) of DOX-4 is located 5 Å away from the surface of the SWCNT. The RDF plot between the selected atoms of PTX-4 and the SWCNT in Figure 2b shows that the distance from the O10 and N atoms of PTX-4 to the carbon atoms of SWCNT is located at ~6.0 Å maximum, and the COM of PTX-4 is approximately 6 Å away from the surface of SWCNT. The O5 atom shows 6.5 Å maximum to the surface, whereas O13 atom shows around 3.5 and 7.5Å.
Figure 2.
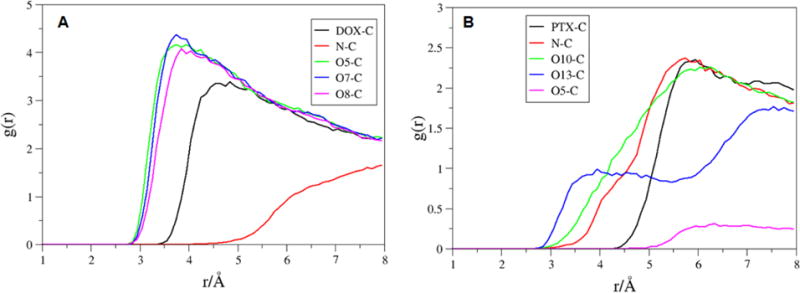
RDFs for selected atoms. A) DOX-4 (DOX center of mass, N, O5, O7 and O8); and B) PTX-4 (PTX center of mass, N, O10, O13 and O5) to the carbon atoms of the pristine SWCNT.
3.2 Functionalization of SWCNT with Chitosan
Noncovalent functionalization of SWCNT offers the possibility of attaching chemical bundles without affecting the electronic network of the tubes, and it is able to also improve dispersibility and biocompatibility of SWCNT, which eventually helps in successful transfer of drugs into the target cells.34 In order to get the chitosan wrapped on the SWCNT surface, the MD simulations were performed by initially placing the chitosan in parallel to the SWCNT (Figure 3, 0ns). The process of polymer wrapping on the surface of SWCNT is shown in Figure 3. The chitosan takes several nano-seconds to form a compact wrapping conformation on the nanotube surface. After 10ns it was observed that polymer stretches as a chain and starts wrapping around SWCNT, and after 20ns the chitosan encircles SWCNT as a wobble. Almost 95% of the chitosan wrapped in around 50ns and it is quite stable till 100ns time scale. The close view of chitosan and the SWCNT was shown in Figure 4. The strong van der Waals interactions between SWCNT and chitosan are the driving force for such complexation. The glucose rings of the chitosan tend to be parallel to the sidewalls of the SWCNT in order to maximize the van der Waals interaction. The hydrophobic acetyl group of NAG approaches to and interacts with the sidewall of the SWCNTs through van der Waals interactions such as C=O…π and C-H…π interactions, spreading the alkyl groups on the sidewalls of the SWCNT. The isosurface of sign(λ2)×ρ in Figure S4 provides a clear visualization of N-H···π as well as C-H···π between the chitosan and CNT.
Figure 3.
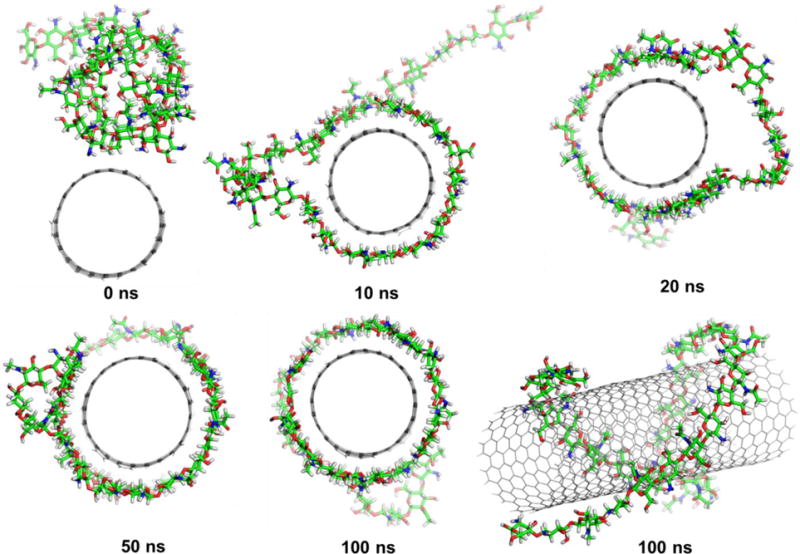
The time evolutions of the functionalization of the SWCNT with chitosan.
Figure 4.
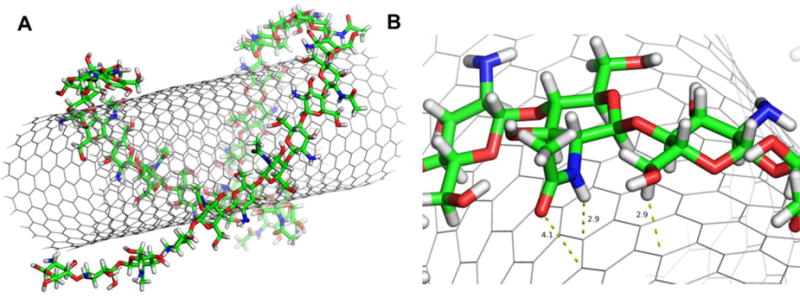
A) Structure of SWCNT and chitosan complex; B) Close view of chitosan interacting with SWCNT.
The root-mean-square deviation (RMSD) of the f-SWCNT relates to their initial minimized complex structure is shown in Figure 5. The RMSD plot reveals that the SWCNT and chitosan form a stable complex after 25 ns simulations. The final stable f-SWCNT conformer will be used to understand co-loading and release of DOX and PTX molecules. Further, the binding free energies of chitosan and the SWCNT is calculated for the last 10ns MD trajectory. The total binding free energy of the complex is −91.1kcal/mol, where the enthalpy including solvation energy (ΔGtotal) is −271.4 kcal/mol and the entropy (-TΔS) is 180.2kcal/mol. The binding free energy for the complex is predominately contributed by van der Waals free energy.
Figure 5.
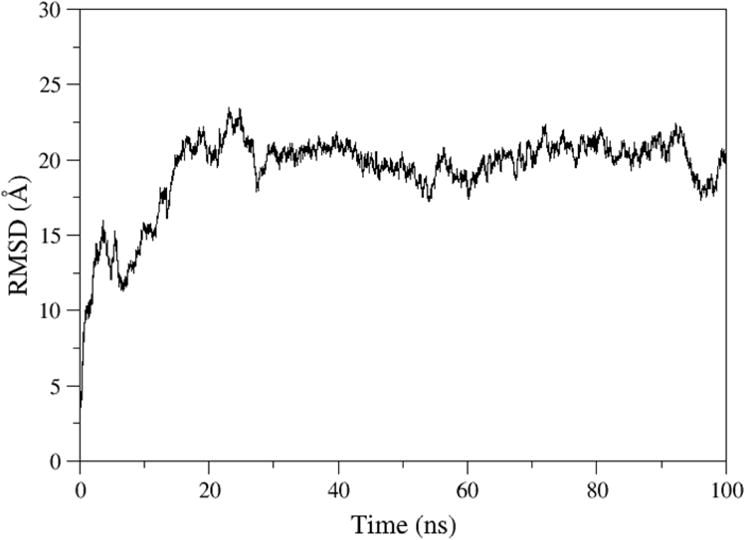
The root-mean-squared deviations (RMSD) of f-SWCNT relative to their initial minimized complex structures as a function of time.
3.3 Loading of DOX and PTX on f-SWCNT
The colloidal stabilization of CNTs is a very important issue for their biomedical applications. Many attempts were made in order to make CNTs dispersible by non-covalent functionalization.35 A thorough understanding on the various factors that govern the non-covalent adsorption and desorption behaviors of DOX and PTX on different types of SWCNT is useful in helping design strategies to control the loading and releasing of molecules from CNTs.36 The PTX loaded on the SWCNT functionalized with phospholipid-branched polyethylene glycol (pl-PEG) shows higher efficacy in suppressing tumor growth than Taxol in murine 4T1 breast cancer model, owing to prolonged blood circulation and 10-fold higher PTX uptake by SWCNT delivery.37,38 It was also reported that the co-delivery of DOX and curcumin with nanoparticle system exhibits higher anti-tumor activity in vitro.39 Starting with the above final stabilized f-SWCNT and placing the DOX and PTX molecules approximately 10Å away from the surface of the f-SWCNT, all atom MD simulations were performed. After a few ns the four DOX and four PTX molecules approaches to the sidewall and are loaded on the f-SWCNT. The final snapshot of fully loaded drug molecules on f-SWCNT was shown in Figure 6. It was observed that the DOX and PTX molecules still prefer binding to the remaining space of the f-SWCNT wrapped by the chitosan rather than the polymer. Similar to the pattern for the pristine SWCNT, the drug molecules are also well adsorbed on the surface of the f-SWCNT, mainly through the π-π stacking between the conjugated aromatic rings of DOX and the sidewalls of the SWCNT for DOX molecules, and the π-π stacking of the two separated aromatic rings of PTX and other sorts of van der Waals interactions for PTX molecules.
Figure 6.
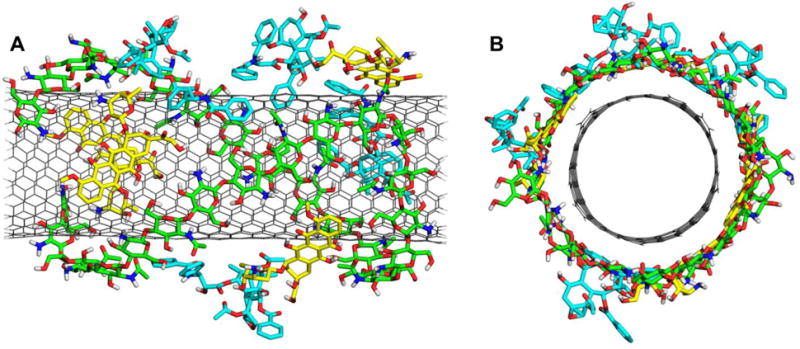
The final stabilized structure of f-SWCNT loaded with four DOX and four PTX molecules. (A) front view; (B) side view.
To quantitatively discuss the difference for the loading on the pristine-SWCNT and f-SWCNT, the binding free energies of drug molecules (four DOX and four PTX) towards f-SWCNT were illustrated in Table 3. When we consider f-SWCNT (both chitosan and SWCNT) as a receptor, in spite of still rather similar exothermic the binding free energies become positive due to significant entropy loss (Table S1). In general, the functionalization of the SWCNT with chitosan weakens the binding of the DOX and PTX drugs to the f-SWCNT to some degree, yet it will improve the dispersion of the DDS. The binding free energies range from −18.5 to −24.0 kcal/mol for the DOX molecules, and −16.4 to −21.9 kcal/mol for the PTX molecules. In the DDS, DOX-2 shows the strongest binding with a free energy of −24.0 kcal/mol among four DOX molecules, whereas PTX-4 shows the highest binding free energy of −21.9 kcal/mol among four PTX molecules. They are weaker than the bindings with the pristine SWCNT by approximately 8.0 and 12.0 kcal/mol, respectively. The DOX molecules show slightly stronger binding to f-SWCNT than PTX does. Rungnim et al. reported that DOX molecules are rather weakly bound to the chitosan f-SWCNT with binding enthalpy of −3~−10 kcal/mol.25 If including the entropy term, their binding Gibbs free energies would tend to be positive.
Table 3.
ΔH, −TΔS, ΔG (kcal/mol) for the binding of DOX and PTX molecules with the SWCNT of the f-SWCNT.
| ΔH | −TΔS | ΔG | |
|---|---|---|---|
| DOX-1 | −34.7 | 16.2 | −18.5 |
| DOX-2 | −36.1 | 12.1 | −24.0 |
| DOX-3 | −35.4 | 15.3 | −20.1 |
| DOX-4 | −34.8 | 13.2 | −21.6 |
| PTX-1 | −39.9 | 20.9 | −19.1 |
| PTX-2 | −33.4 | 17.0 | −16.4 |
| PTX-3 | −41.1 | 20.0 | −21.1 |
| PTX-4 | −34.8 | 12.9 | −21.9 |
The RDF plots for the DOX-2 and PTX-4 (the molecules with the strongest binding) on the f-SWCNT were shown in Figure 7. The COM of DOX-2 is located 4Å away from the surface of the SWCNT, whereas PTX-4 is located 6Å away. The average distance evolutions from the COM of the SWCNT to those of diketo benzene and benzene ring of four DOX and four PTX molecules were calculated. The average distances for the f-SWCNT system (Figure 9) are less than those for pristine SWCNT (Figure 8), and the distance frequencies (Figure S2) provide a similar story that the DOX/PTX- f-SWCNT is more compact than the pristine SWCNT. The results may explain why the entropy loss (-TS) for the f-SWCNT system is generally bigger than that for the pristine SWCNT, although the binding enthalpies are comparable.
Figure 7.
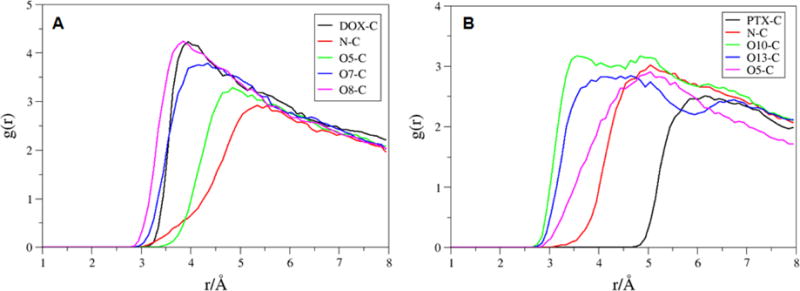
RDFs for selected atoms of the A) DOX-2 (DOX center of mass, N, O5, O7 and O8) and B) PTX-4 (PTX center of mass, N, O10, O13 and O5) to the carbon atoms of the f-SWCNT.
Figure 9.
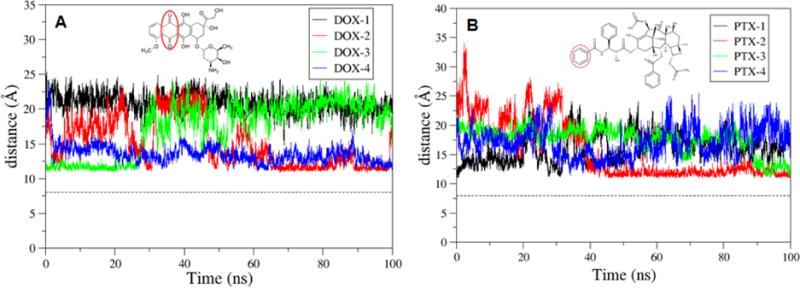
The time evolution of the distance between the COM of the f-SWCNT and that of diketo benzene, and benzene ring of DOX and PTX, respectively.
Figure 8.
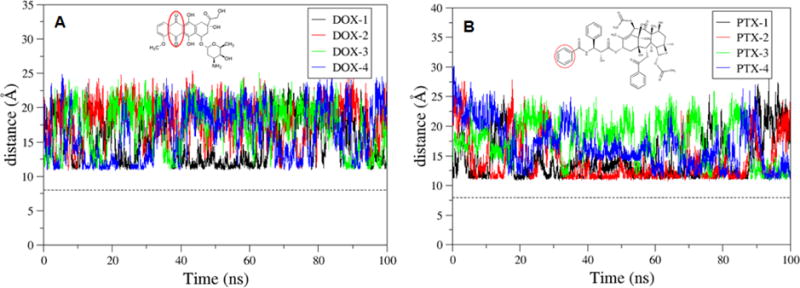
The time evolution of the distance between the center of mass (COM) of the pristine SWCNT and the COM of diketo and benzene ring of DOX and PTX, respectively.
3.4 Releasing of DOX and PTX using p-f-SWCNT
It is well known that in the neighborhood of a cancerous tumor, the local pH is about 5-5.5, while the regular blood has a neutral pH level of 7.4. It was shown that DOX can release more readily in acidic environment after protonation.35,40–42 The acidic media of a tumor microenvironment may induce DOX and PTX release from the DDS. PEG-DOX-Curcumin prodrug nanoparticle was designed for simultaneous delivery of DOX and curcumin as a combined therapy to treat cancer, and they also release more efficiently in acidic environment at pH 5.0 than at pH 7.4.39 This pH sensitive release of the drugs is useful in targeted delivery of DOX and PTX to cancer cells because of acidic tumor microenvironment. In addition, as the internal pH environment of lysosomes is acidic (pH 5.5), the release of DOX and PTX from CNTs may also be triggered automatically. In our study the above final snapshot of f-SWCNT system consisting of SWCNT, chitosan, four DOX and four PTX molecules was used to protonate the systems by adding proton to the relevant amine groups. The effect of protonation on the binding of f-SWCNT and anti-cancer drugs was explored. From the MD simulations it was observed that PTXH+-4 tends to release from the p-f-SWCNT surface, while during the simulation other molecules are still loosely packed on the outer surface of the p-f-SWCNT (Figure 10). Consequently, the binding of PTXH+-4 to p-f-SWCNT (only SWCNT) is reduced drastically to −8.6 kcal/mol as compared with the pristine SWCNT and f-SWCNT systems (shown in Table 4). When p-f-SWCNT (both protonated chitosan and SWCNT) is considered as a receptor, the binding free energies were more positive compared to f-SWNCT system (Table S2). The DOXH+ molecules with p-f-SWCNT also generally show less binding as compared with pristine SWCNT as well as f-SWCNT systems, which again indicates that the molecules are more loosely adhered to the surface in acidic environment. The charge-charge repulsion between the protonated drug and protonated chitosan may be also partially responsible for decreasing the binding free energy. The higher protonation of chitosan could trigger the release of drug molecules. Rungnim et al., also found that high protonation of chitosan weaken the binding of DOX molecules with f-SWCNT, e.g., as half of 30 glucosamines in 50-unit chitosan chain are protonated the binding enthalpies of eight DOX change to ~ −3 to −8 kcal/mol for six DOX and even 0-2 kcal/mol for other two DOX (−3 to −10 kcal/mol for eight DOX at neutral).25
Figure 10.
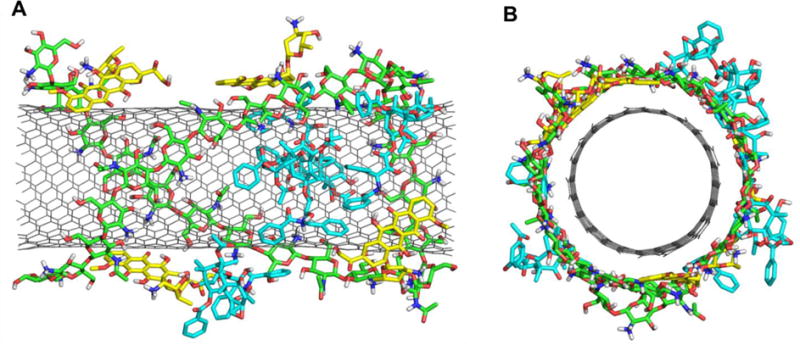
The final stabilized structure of P-f-SWCNT loaded with four DOXH+ and four PTXH+ molecules. (a) front view (b) side view.
Table 4.
ΔH, −TΔS, ΔG (kcal/mol) for the binding of DOX+ and PTX+ molecules with the SWCNT of the p-f-SWCNT.
| ΔH | −TΔS | ΔG | |
|---|---|---|---|
| DOX-1 | −35.0 | 16.3 | −18.6 |
| DOX-2 | −34.1 | 14.7 | −19.5 |
| DOX-3 | −34.6 | 15.3 | −19.2 |
| DOX-4 | −34.5 | 7.6 | −26.9 |
| PTX-1 | −37.6 | 17.3 | −20.3 |
| PTX-2 | −33.5 | 16.6 | −16.9 |
| PTX-3 | −39.5 | 20.2 | −19.3 |
| PTX-4 | −32.5 | 23.9 | −8.6 |
The distances were analyzed between the COM of the SWCNT and diketo benzene of DOXH+, and the benzene ring of PTXH+, respectively (Figure 11). Further, the average distance is calculated between the COM of SWCNT (pristine, functionalized, and protonated) to those of DOX and PTX molecules (Table 5). The distances between the COM of p-f-SWCNT and those of two molecules DOX-2 and PTX-4 are 21.0 and 22.9 Å, respectively. The bigger distance for the molecules shows that the drug molecules are really in the rather released state. The protonated system shows that the average distance between COM of SWCNT and drug molecules are bigger when compared to pristine and f-SWCNT. The RDF plots for DOXH+-2 and PTXH+-4 around the surface carbon atoms of SWCNT were also shown in Figure 12.
Figure 11.

The time evolution of the distance between the center of mass of the p-f-SWCNT and the center of mass of diketo benzene and benzene ring of DOXH+ and PTXH+, respectively.
Table 5.
The average distance (Å) between the center of mass of the SWCNT and those of DOX and PTX molecules
| SWCNT | f-SWCNT | p-f-SWCNT | |
|---|---|---|---|
| DOX-1 | 15.6 | 20.2 | 18.8 |
| DOX-2 | 17.3 | 14.6 | 21.0 |
| DOX-3 | 16.6 | 16.6 | 17.6 |
| DOX-4 | 15.7 | 13.0 | 12.9 |
| PTX-1 | 15.0 | 14.7 | 14.5 |
| PTX-2 | 15.0 | 17.5 | 15.0 |
| PTX-3 | 16.7 | 13.9 | 16.1 |
| PTX-4 | 16.3 | 19.1 | 22.9 |
Figure 12.
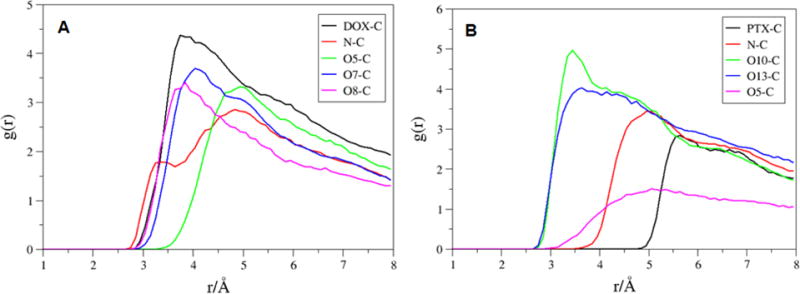
RDFs for selected atoms of the a) DOXH+ (DOX center of mass, N, O5, O7 and O8) and b) PTXH+ (PTX center of mass, N, O10, O13 and O5) to the carbon atoms of the p-f-SWCNT.
3.5 Encapsulation of DOX
In our previous study, we explored the adsorption of DOX on the surface of SWCNTs as well as its encapsulation in SWCNTs, and their dependence on the protonation of the NH2 group of DOX, solvent, and the diameter of armchair (n,n) SWCNTs were systematically investigated using the theoretical methods such as PM6-DH2 and M06-2X in the scheme of ONIOM.43 Compared to loading on the surface, the encapsulation provides a sought after alternative towards restoring the activity of the molecules and preventing any undesirable degradation of its properties. In the present study, two DOX molecules were placed inside the tube, and other two DOX and four PTX molecules are still placed on the surface of f-SWCNT. The binding free energy results were shown in Table 6. The binding free energies of f-SWCNT with two encapsulated DOX molecules DOX-1 and DOX-2 are −52.8 and −43.4 kcal/mol, respectively, which are considerably stronger than its binding to the sidewall. As shown by previous work,43 the encapsulation is even more sensitive to pH than the loading on the sidewall of CNT, getting further weaken in acidic environment. The encapsulation of anticancer drugs may also play very important role in the drug delivery.43 The structure of f-SWCNT with the encapsulated DOX-1 was illustrated in Fig. 13. The RDF plot for the encapsulated DOX-1 is shown in Fig. 14. The distance from the O5,O6,O7, and N atoms to the carbon atoms of SWCNT is shown ~3.5 Å maximum, the center of mass of DOX is also located 5 Å away from the surface of SWCNT. The encapsulated DOX is very stable compared to adsorbed molecules.
Table 6.
ΔH, −TΔS, ΔG (kcal/mol) for the binding with f-SWCNT of two encapsulated DOX (DOX-1 and DOX-2), two DOX and four PTX.
| ΔH | −TΔS | ΔG | |
|---|---|---|---|
| DOX-1 | −63.3 | 10.5 | −52.8 |
| DOX-2 | −53.4 | 9.9 | −43.4 |
| DOX-3 | −35.8 | 18.5 | −17.3 |
| DOX-4 | −35.0 | 15.2 | −19.9 |
| PTX-1 | −35.6 | 18.2 | −17.5 |
| PTX-2 | −20.7 | 22.6 | 1.9 |
| PTX-3 | −32.8 | 20.7 | −12.0 |
| PTX-4 | −40.8 | 23.7 | −17.1 |
Fig. 13.
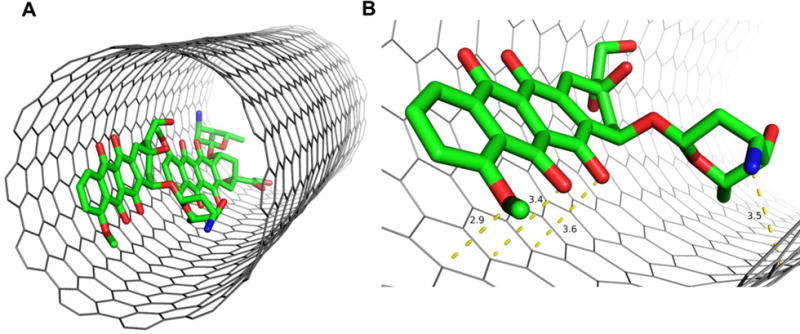
A) Structure of the encapsulated DOX in the f-SWCNT; B) Close view of the encapsulated DOX interacting with f-SWCNT.
Fig. 14.
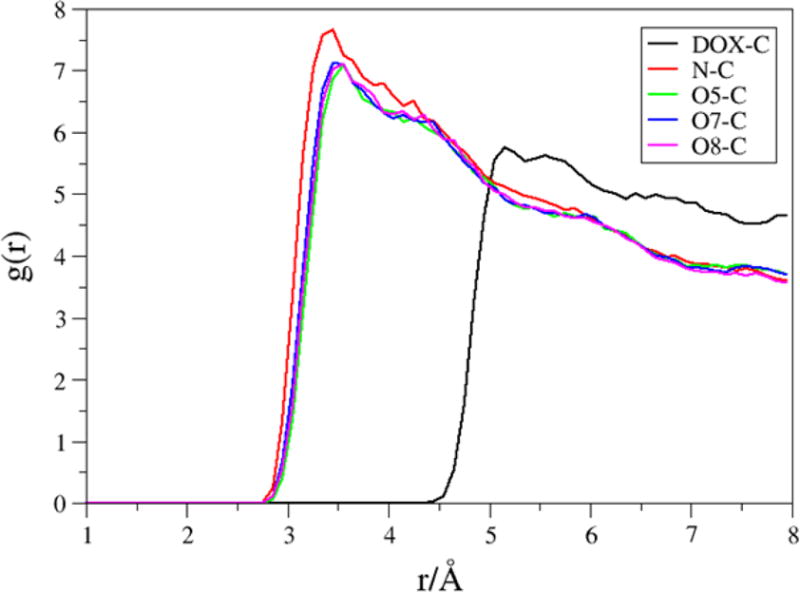
RDFs for selected atoms of the encapsulated DOX-1 (center of mass of DOX, N, O5, O7 and O8) to the carbon atoms of the f-SWCNT.
4. Conclusion
All atom molecular dynamic (MD) simulations was applied to investigate the co-loading and releasing of two anti-cancer drugs (DOX and PTX) with the pristine SWCNT, chitosan functionalized SWCNT (f-SWCNT), and protonated f-SWCNT (p-f-SWCNT) systems. DOX and PTX bind with pristine SWCNT in a similar strength, where the former is primarily driven by the π–π stacking of the conjugated aromatic rings with the sidewall of the SWCNT, and the latter by the π-π stacking of two separated rings supplemented by X-π(X=C-H, N-H and C=O). The co-loading of DOX and PTX to the pristine SWCNT is exothermic and spontaneous. The DOX and PTX molecules still prefer binding to the sidewall of the f-SWCNT wrapped by the chitosan rather than binding with the polymer, and the non-covalent functionalization of the SWCNT (f-SWCNT) with chitosan decreases the binding free energies by a few kcal/mol. However, both the average distance evolutions from the center of mass of the f-SWCNT to those of drug molecules, and the distance frequencies show that the DOX/PTX- f-SWCNT is more compact than the the DOX/PTX-SWCNT, indicating the effective co-loading of DOX and PTX molecules. At acidic pH modeled by the protonation of drugs and chitosan, one PTX shows releasing from the the f-SWCN, and the further decrease in binding was observed together with loosing of other drugs from the f-SWCNT. We can conclude that the f-SWCNT is plausible for the coloading and releasing of DOX and PTX.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Science of the National Institute of Health (SC3GM105576).
References
- 1.Iijima S. Nature. 1991;354:56–58. [Google Scholar]
- 2.Heister E, Neves V, Lamprecht C, Silva SRP, Coley HM, McFadden J. Carbon. 2012;50:622–632. [Google Scholar]
- 3.Bianco A, Kostarelos K, Partidos CD, Prato M. Chem Commun. 2005:571–577. doi: 10.1039/b410943k. [DOI] [PubMed] [Google Scholar]
- 4.Fabbro C, Ali-Boucetta H, Ros TD, Kostarelos K, Bianco A, Prato M. Chem Commun. 2012;48:3911–3926. doi: 10.1039/c2cc17995d. [DOI] [PubMed] [Google Scholar]
- 5.Madani SY, Naderi N, Dissanayake O, Tan A, Seifalian AM. Int J Nanomedicine. 2011;6:2963–2979. doi: 10.2147/IJN.S16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Son KH, Hong JH, Lee JW. Int J Nanomedicine. 2016;11:5163–5185. doi: 10.2147/IJN.S112660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prato M, Kostarelos K, Bianco A. Acc Chem Res. 2008;41:60–68. doi: 10.1021/ar700089b. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Bai Y, Yan B. Drug Discov Today. 2010;15:428–435. doi: 10.1016/j.drudis.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu P. Ind Eng Chem Res. 2013;52:13517–13527. [Google Scholar]
- 10.Ghadi A, Mahjoub S, Tabandeh F, Talebnia F. Caspian J Intern Med. 2014;5:156–161. [PMC free article] [PubMed] [Google Scholar]
- 11.Yan LY, Poon YF, Chan-Park MB, Chen Y, Zhang Q. J Phys Chem C. 2008;112:7579–7587. [Google Scholar]
- 12.Najeeb CK, Lee JH, Kim JH, Kim D. Colloids Surf B Biointerfaces. 2013;102:95–101. doi: 10.1016/j.colsurfb.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Cheung RC, Ng TB, Wong JH, Chan WY. Mar Drugs. 2015;13:5156–5186. doi: 10.3390/md13085156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang XY, Zhang L, Wei XH, Wang Q. Biomaterials. 2013;34:1843–1851. doi: 10.1016/j.biomaterials.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 15.Iamsamai C, Hannongbua S, Ruktanonchai U, Soottitantawat A, Dubas ST. Carbon. 2010;48:25–30. doi: 10.1016/j.cplett.2011.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peniche H, Peniche C. Polym Int. 2011;60:883–889. [Google Scholar]
- 17.Lown JW. Pharmacol Ther. 1993;60:185–214. doi: 10.1016/0163-7258(93)90006-y. [DOI] [PubMed] [Google Scholar]
- 18.Matyszewska D. Surface Innovations. 2014;2:201–210. [Google Scholar]
- 19.Altmann KH, Gertsch J. Nat Prod Rep. 2007;24:327–357. doi: 10.1039/b515619j. [DOI] [PubMed] [Google Scholar]
- 20.Muthoosamy K, Abubakar IB, Bai RG, Loh HS, Manickam S. Sci Rep. 2016;6:32808. doi: 10.1038/srep32808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Fang J, Kim YJ, Wong MK, Wang P. Mol Pharmaceutics. 2014;11:1651–1661. doi: 10.1021/mp5000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rui M, Xin Y, Li R, Ge Y, Feng C, Xu X. Mol Pharmaceutics. 2017;14:107–123. doi: 10.1021/acs.molpharmaceut.6b00732. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Tozer T, Alisaraie L. J Phys Chem C. 2016;120:4061–4070. [Google Scholar]
- 24.Panczyk T, Wolski P, Lajtar L. Langmuir. 2016;32:4719–4728. doi: 10.1021/acs.langmuir.6b00296. [DOI] [PubMed] [Google Scholar]
- 25.Rungnim C, Rungrotmongkol T, Poo-Arporn RP. J Mol Graph Model. 2016;70:70–76. doi: 10.1016/j.jmgm.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 26.Frey J, Doren D. TubeGen 3.4. University of Delaware; Newark, DE: 2011. http://turinnss.udel.edu/research/tubegenonline.html. [Google Scholar]
- 27.Case DA, Betz RM, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, Le Grand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, Kollman PA. AMBER 2016. University of California; San Francisco: 2016. [Google Scholar]
- 28.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09 D. Gaussian Inc; Wallingford CT: 2016. [Google Scholar]
- 29.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 30.Miller BR, 3rd, McGee TD, Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 31.Genheden S, Ryde U. Expert Opin Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roe DR, Cheatham TE., 3rd J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 33.James J, Stewart P. MOPAC2009. Computational Chemistry; Colorado Springs, CO, USA: [Google Scholar]
- 34.Klumpp C, Kostarelos K, Prato M, Bianco A. Biochim Biophys Acta. 2006;1758:404–412. doi: 10.1016/j.bbamem.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 35.Wolski P, Nieszporek K, Panczyk T. Phys Chem Chem Phys. 2017;19:9300–9312. doi: 10.1039/c7cp00702g. [DOI] [PubMed] [Google Scholar]
- 36.Wong BS, Yoong SL, Jagusiak A, Panczyk T, Ho HK, Ang WH, Pastorin G. Adv Drug Deliv Rev. 2013;65:1964–2015. doi: 10.1016/j.addr.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Cancer Res. 2008;68:6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lay CL, Liu HQ, Tan HR, Liu Y. Nanotechnology. 2010;21:065101. doi: 10.1088/0957-4484/21/6/065101. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Yang C, Wang W, Liu J, Liu Q, Huang F, Chu L, Gao H, Li C, Kong D, Liu Q, Liu J. Sci Rep. 2016;6:21225. doi: 10.1038/srep21225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z, Sun X, Nakayama-Ratchford N, Dai H. ACS Nano. 2007;1:50–56. doi: 10.1021/nn700040t. [DOI] [PubMed] [Google Scholar]
- 41.Gu YJ, Cheng J, Jin J, Cheng SH, Wong WT. Int J Nanomedicine. 2011;6:2889–2898. doi: 10.2147/IJN.S25162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahdavi M, Rahmani F, Nouranian S. J Mater Chem B. 2016;4:7441–7451. doi: 10.1039/c6tb00746e. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Xu Z. RSC Adv. 2016;6:314–322. doi: 10.1039/C5RA20866A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson ER, Keinan S, Mori-Sanchez P, Contreras-Garcia J, Cohen AJ, Yang W. J A Chem Soc. 2010;132:6498. doi: 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.