

Abstract
Many catabolic stimuli, including interleukin-1 (IL-1) in combination with oncostatin M (OSM), promote cartilage breakdown via the induction of collagen-degrading collagenases such as matrix metalloproteinase 1 (MMP1) and MMP13 in human articular chondrocytes. Indeed, joint diseases with an inflammatory component are characterised by excessive extracellular matrix (ECM) catabolism. Importantly, protein kinase C (PKC) signalling has a primary role in cytokine-induced MMP1/13 expression, and is known to regulate cellular functions associated with pathologies involving ECM remodelling. At present, substrates downstream of PKC remain undefined. Herein, we show that both IL-1- and OSM-induced phosphorylation of protein kinase D (PKD) in human chondrocytes is strongly associated with signalling via the atypical PKCι isoform. Consequently, inhibiting PKD activation with a pan-PKD inhibitor significantly reduced the expression of MMP1/13. Specific gene silencing of the PKD isoforms revealed that only PKD3 (PRKD3) depletion mirrored the observed MMP repression, indicative of the pharmacological inhibitor specifically affecting only this isoform. PRKD3 silencing was also shown to reduce serine phosphorylation of signal transducer and activator of transcription 3 (STAT3) as well as phosphorylation of all three mitogen-activated protein kinase groups. This altered signalling following PRKD3 silencing led to a significant reduction in the expression of the activator protein-1 (AP-1) genes FOS and JUN, critical for the induction of many MMPs including MMP1/13. Furthermore, the AP-1 factor activating transcription factor 3 (ATF3) was also reduced concomitant with the observed reduction in MMP13 expression. Taken together, we highlight an important role for PKD3 in the pro-inflammatory signalling that promotes cartilage destruction.
Introduction
The progression of joint diseases such as rheumatoid arthritis (RA) and osteoarthritis (OA) is associated with inflammation, whereby inflammatory mediators released by infiltrating immune cells as well as resident joint cells induce altered gene expression that promotes extracellular matrix (ECM) degradation [1]. Cytokines such as interleukin (IL-)1 and tumour necrosis factor α (TNFα) are key mediators in the inflammatory responses in destructive joint disease [2,3], and we have previously demonstrated enhanced cartilage ECM catabolism by these mediators when combined with IL-6-family cytokines such as oncostatin M (OSM) in vitro and in vivo [4–8]. This co-operative exacerbation in catabolic potential is likely to occur in many inflammatory milieu, such as the arthritides, to promote cartilage ECM catabolism.
Normal articular cartilage is maintained by a single resident cell type, the chondrocyte, which preserve homeostasis by regulating the expression of ECM components and catabolic factors such as the matrix metalloproteinases (MMPs). Collectively, the MMPs can degrade all ECM macromolecules, and during inflammatory joint diseases stimulated chondrocytes secrete elevated levels of MMPs which, once activated, mediate proteolysis of tendon, bone and cartilage [9,10]. MMP-1 and MMP-13 are the collagenolytic MMPs most strongly associated with cartilage collagenolysis, a key proteolytic event in joint diseases since it is essentially irreversible [11]. Indeed, MMP-13 is the most potent collagenase with respect to type II collagen [12], the major structural collagen in articular cartilage. Potent pro-inflammatory stimuli such as IL-1+OSM activate a complex array of signal transduction pathways, shown to be common to a wide variety of pro-inflammatory stimuli [13], which together markedly enhance the induction of MMP1/13 in human chondrocytes [7,13–15]. The cFos/cJun activator protein-1 (AP-1) transcription factor complex is critical for MMP gene expression (see [16] and references therein), although we recently reported a role for the AP-1-binding factor activating transcription factor 3 (ATF3) in selectively mediating MMP13 expression which was nevertheless AP-1-dependent [13]. Furthermore, we have also previously demonstrated the importance of signal transducer and activator of transcription (STAT)3, phosphatidylinositol-3’ kinase (PI3K/Akt) and protein kinase C (PKC) signalling pathways [17–19]. The atypical PKC isoform PKCι has been shown to modulate MMP1/13 induction following IL-1+OSM stimulation [19], but the identity of the downstream signalling components it affects remains unknown, as do potential points of cross-talk.
Protein kinase D (PKD) comprises a family of serine/threonine protein kinases that belong to the Ca2+/calmodulin-dependent kinase superfamily. Three isoforms exist in humans: PKD1, PKD2 and PKD3 (reviewed in [20]). Diverse cellular functions, including cell survival/proliferation and apoptosis, plasma membrane-directed transport, metastasis and inflammation have been reported [20–24] for PKD isoforms. With respect to cartilage remodelling, PKD is reported to regulate both the expression and activity of several MMPs. Studies have shown that PKD1 down-regulates the expression and activity of multiple MMPs in breast cancer cells lines, particularly the gelatinase MMP-9 [22]. Conversely, gene silencing of PKD2 and PKD3 leads to a decrease in the activity of MMP-9 in prostate cancer, suggesting MMP gene regulation by distinct PKD family members is cell-type specific. PKD3 has been reported to regulate nuclear localization and activity of histone deacetylases 5 and 7 (HDAC5/7) [25]. OA cartilage exhibits elevated HDAC7 levels, whilst HDAC7 depletion in SW1353 human chondrosarcoma cells strongly suppressed IL-1-dependent induction of MMP13 [26]. PKD3 can be activated independently of PKC via phosphorylation of tyrosine residues within the pleckstrin homology domain following oxidative stress, which is a feature of OA [27]. Moreover, PKD isoforms have also been shown to be a downstream target of G-protein-coupled receptors, via increases in phospholipase C activity and auto-phosphorylation [28]. Such receptors play roles in innate and adaptive immunity, and have been implicated in the pathology associated with arthritis [29–31].
PKD3 is ubiquitously expressed in adult human tissues [32] and has been shown to be expressed during skeletogenesis in mice, notably in the cartilage primordia of bones [33], which are areas of active ECM turnover (via MMP-13) as cartilage is remodelled prior to mineralization. Interestingly, phenotypic analysis of a mutant mouse strain harbouring a gene-trap deletion of the PKD3 gene (prkd3) revealed a mild skeletal abnormality including decreased mean trabecular bone volume and thickness [34], suggestive of reduced remodelling in cartilage primordia. In this context, downstream PKD3 signalling may also be involved in cytokine-induced MMP13 expression in human chondrocytes. Herein, we show that PKD3 is indeed involved in pro-inflammatory signalling via modulating the expression of the AP-1 genes FOS and JUN, and enhancing MMP expression. PKD3 activity also stimulates the induction of ATF3 which is specifically involved in cytokine-induced MMP13 expression in human chondrocytes.
Materials and methods
Materials
All chemicals were obtained from Sigma Chemical Co (Poole, UK) unless otherwise stated and of the highest purity available. All cytokines were recombinant human. IL-1α was a generous gift from Dr Keith Ray (GlaxoSmithKline, Stevenage, UK). OSM was prepared in-house using expression vectors kindly provided by Prof. J. Heath (University of Birmingham, UK) and methods described [35]. Gö6983 was from Merck Chemicals (Nottingham, UK). PKD inhibitor kb NB 142–70 was from Tocris Bioscience (Bristol, UK). The STAT3 inhibitor S31-201 was from Selleckchem (TX, USA). Kinase inhibitors and small interfering (siRNA) reagents were screened for toxicity using the Toxilight assay of adenylate kinase release (Lonza, Wokingham, UK), and always used at concentrations that did not affect cell viability over the assay period.
Chondrocytes
Human chondrocytes were obtained by the enzymatic digestion of macroscopically normal articular cartilage from OA patients undergoing joint replacement surgery as described [36]. All subjects gave informed consent and the study was approved by the Newcastle and North Tyneside Joint Ethics Committee (REC 14/NE/1212). Chondrocytes were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 IU penicillin, 100 μg/ml streptomycin, 40 U/ml nystatin.
Cell fractionation and immunoblotting
Chondrocyte lysates were prepared as described previously (18) and stimulated with IL-1 (0.2 ng/ml) ± OSM (10 ng/ml) for up to 60 min. In some experiments, chondrocytes were subjected to subcellular fractionation using NE-PER Nuclear and Cytoplasmic Protein Extraction Kit or Subcellular Protein Fractionation Kit (both from ThermoFisher Scientific, Loughborough, UK). GAPDH was used as a loading control for whole cell lysates whilst MEK2 and lamin A/C were used for cytoplasmic and nuclear extracts, respectively. Lysates or fractions were resolved by SDS-PAGE, transferred to PVDF membranes and subsequently probed using the following antibodies: PKCι (#2998), PKCζ (#9372), PKD*S916 (#2051), PKD*S744/748 (#2054: recognises PKD*S731/735 in PKD3), PKD3 (#5655), ERK1/2*T202/Y204 (#9101), JNK (#9252), JNK*T183/Y185 (#9251), p38*T180/Y182 (#9211), Akt*S473 (#4060), cFos (#4384), STAT1*Y701 (#9171), STAT1*S727 (#9177), STAT3*Y705 (#9131), STAT3*S727 (#9134) and lamin A/C (#2032) were from Cell Signaling Technology (Danvers, MA); cJun (sc1694) and ATF3 (sc188) were from Santa Cruz Biotechnology (Santa Cruz, CA); glyceraldehyde 3’-phosphate dehydrogenase (GAPDH; MAB374) was from Millipore (MA, USA); PKD1 (L905) was from Bio-world (Dublin, USA); MEK2 (#04–377) and PKD2 (#1969–1) were from Epitomics (Insight Biotech, Wembley, UK). The specificity of all antibodies was confirmed using chondrocyte lysates (see full-length blots in S1 Fig), whilst blots were cropped for clarity of comparison.
Gene silencing
PRKCZ and PRKCI siRNAs were Dharmacon ON-TARGETplus™ (ThermoFisher Scientific), comprising 4 specific siRNA complexes [16], whilst validated MISSION™ siRNA reagents (Sigma) were used to silence: PRKD2, 5’-CGAUACAUCACGCAUGAGA-3’; PRKD3, 5’-CAUAAACGCUGUGCAUCAA-3’; JNK1, 5’-GUUCUUAUGAAAUGUGUUATT-3’; JNK2, 5’-CUGUAACUGUUGAGAUGUATT-3’. Primary human chondrocytes were prepared and cultured as above, and transfected as described previously (18). After transfection, cells were serum-starved for 24 h prior to stimulation as indicated. Depletion of gene-specific mRNA levels was calculated by comparison of expression levels with cells transfected with 100 nM siCONTROL (siCon: non-targeting siRNA #2, Cat. 001210–02; Dharmacon).
GIPZ lentiviral vector containing non-targeting control shRNA (shCon) or PRKD1 shRNA (shPRKD1: V2LHS-170466) were purchased from OpenBiosystems (Thermo Fisher Scientific). Packaging and envelope plasmids pCMV-dR8.91 and pMD2.G were a kind gift from Prof. Nick Reynolds (Newcastle University). The HEK293T producer cell line (SBI, California, USA), maintained in DMEM plus 10% FBS, was employed for packaging. On the day of transfection, medium was changed to DMEM containing 10% heat-inactivated FBS. Packaging, envelope and shRNA plasmids were co-transfected using JetPEI transfection reagent (Polyplus, Illkirch, France), according to the manufacturer’s instructions. Supernatant was harvested 72 h post-transfection and filtered (0.45 μm). Viral particles were then concentrated 50-fold using a Lenti-X concentrator (Takara Bio Europe/ Clontech, Saint-Germain-en-Laye, France), and viral titre calculated using the Lenti-X qRT-PCR Titration Kit (Takara). Multiplicity of infection (MOI) was then calculated via serial dilution. Primary chondrocytes were prepared and cultured as above, and exposed to lentiviral infection at a MOI of 30 for 72 h. Transduction was assessed using confocal microscopy for green fluorescent protein (GFP). After transduction, cells were serum-starved for 24 h then stimulated as indicated.
Real-time PCR of relative mRNA levels
Primary human chondrocytes were stimulated with IL-1 (0.05 ng/ml) ± OSM (10 ng/ml) for 1 h to measure FOS, JUN and ATF3, or 24 h for MMP mRNAs. RNA was stabilised in cell lysates in a 96-well format, cDNA synthesised and real-time PCR assays conducted using conditions described previously [18]. Primer and probe sequences are as previously detailed [13,19], or MMP8: For, 5’- CACTCCCTCAAGATGACATCGA-3’ and Rev, 5’-ACGGAGTGTGGTGATAGCATCA-3’, Probe, 5’-CAAGCAACCCTATCCAA CCTACTGGACCAA-3’; MMP14: For, 5’-AGGCCGACATCATGATCTTCTTT-3’ and Rev, 5’-AAGTGGGTGTCTCCTCCAATGTT-3’, Probe, 5’-CCATGGCGACAGCACGC CCTT-3’. Some Taqman assays used Universal Probe Library probes (Roche Applied Sciences) as directed: PRKD1: For, 5’- TGTATTACCCTCTTTCAGAATGACA-3’ and Rev, 5’-CCAGAGACAAAATTTCAGATAAAGG-3’, Probe: 38; PRKD2: For, 5’-AGATGCTC TTCGGCCTAGTG-3’ and Rev, 5’-AGCGCTTGTGGTAGTTCAGC-3’, Probe: 46; PRKD3: For, 5’-TGATTTAAAGCCAGAAAATGTGC-3’ and Rev, 5’-CGTGCAAATCC AAAGTCACA-3’, Probe: 21.
Statistical analyses
Statistical differences between sample groups were assessed using one-way analysis of variance (ANOVA) with a post-hoc Bonferroni’s multiple comparison test or Student’s 2-tailed unpaired t-test (1 sample t-test for immunoblot quantification), where ***p<0.001, **p<0.01, *p<0.05. For clarity, only selected comparisons are presented in some Figures.
Results
Inhibition of PKD phosphorylation curtails IL-1+OSM induction of MMP1 and MMP13 in human articular chondrocytes
We previously demonstrated roles for PKCι and PKCζ in IL-1+OSM-mediated expression of the key collagenases MMP1 and MMP13 in human chondrocytes [19]. Moreover, we have also shown both IL-1 and OSM induce PKD phosphorylation such that we sought to confirm that PKD was indeed a downstream substrate of PKC signalling. Cytokine-induced PKD phosphorylation was assessed in the presence of the Gӧ6983 (20 μM), a PKC inhibitor that does not inhibit PKD, which completely abolished both IL-1- and OSM-stimulated PKD phosphorylation (Figs 1A and S2). Assessment of the effect of gene silencing of PRKCI and PRKCZ (coding for PKCι and PKCζ, respectively; S3A Fig) indicated that, of the atypical PKC isoforms, only PKCι played a role in this phosphorylation (Fig 1B and 1C).
Fig 1. Effect of PKD inhibition on collagenase expression in human chondrocytes.

Primary human articular chondrocytes were stimulated with IL-1 (0.2 ng/ml) alone, OSM (10 ng/ml) alone or IL-1+OSM as detailed in the Methods for 20 min (A-C). Cells were either (A) pre-treated with Gӧ6983 (20 μM; PKCi) for 1 h or (B and C) transfected with siRNA for non-targeting siCon (white bars), PRKCZ (grey bars) or PRKCI (black bars) (all 100 nM) for 48 h prior to stimulation. Cell lysates were prepared as described in Methods and subjected to SDS-PAGE and immunoblotting (using the antibodies indicated). Relative densitometry of some blots was performed, scans combined and plotted graphically (C), where ††, p≤0.01 versus siCon-treated IL-1+OSM. (D) Human chondrocytes were pre-treated with NB142-70 (5 μM; PKDi), or a DMSO vehicle control for 1 h prior to stimulation with IL-1 (0.05 ng/ml) + OSM (10 ng/ml) for 24 h. Real-time RT-PCR was performed on isolated RNA to determine relative levels of MMP1 (black bars) and MMP13 (hatched bars), normalised to 18S rRNA housekeeping gene and presented as a percentage of the IL-1+OSM induction (n = 6; mean ± S.E.), where ***, p≤0.001 versus the relevant DMSO-treated vehicle control. Data are representative of at least 3 separate chondrocyte populations.
Since combined PKC/PKD inhibition (using Gӧ6976) suppressed MMP expression and reduced cytokine-induced collagen release from cartilage [16], we confirmed that a specific PKD inhibitor (NB142-70; PKDi) also significantly reduced IL-1+OSM-induced expression of MMP1 and MMP13 expression (Fig 1D). Together, these findings implicate a role for PKD in cytokine-induced MMP expression in human chondrocytes.
PKD3 depletion correlates with reduced MMP1 and MMP13 expression in human chondrocytes
To determine which of the three individual PKD isoforms accounted for the PKDi-mediated reduction in MMP expression, selective ‘knockdown’ of the PKD isoforms (S3B Fig) indicated that only PRKD3 silencing led to a significant decrease in the expression of both MMP1 and MMP13 (Fig 2). Expression of these MMPs was markedly enhanced with PRKD1 silencing but unaltered following PRKD2 depletion (S4A and S4B Figs, respectively). Since these data strongly implicated PKD3 as the only ‘pro-inflammatory’ moiety of PKD signalling for cytokine-induced MMP expression, specificity assessment of PRKD3 silencing for the other collagenolytic MMP species indicated that MMP8, but not MMP14, was also significantly suppressed (S5 Fig).
Fig 2. Effect of PKD isoform silencing on MMP1 and MMP13 expression in human chondrocytes.
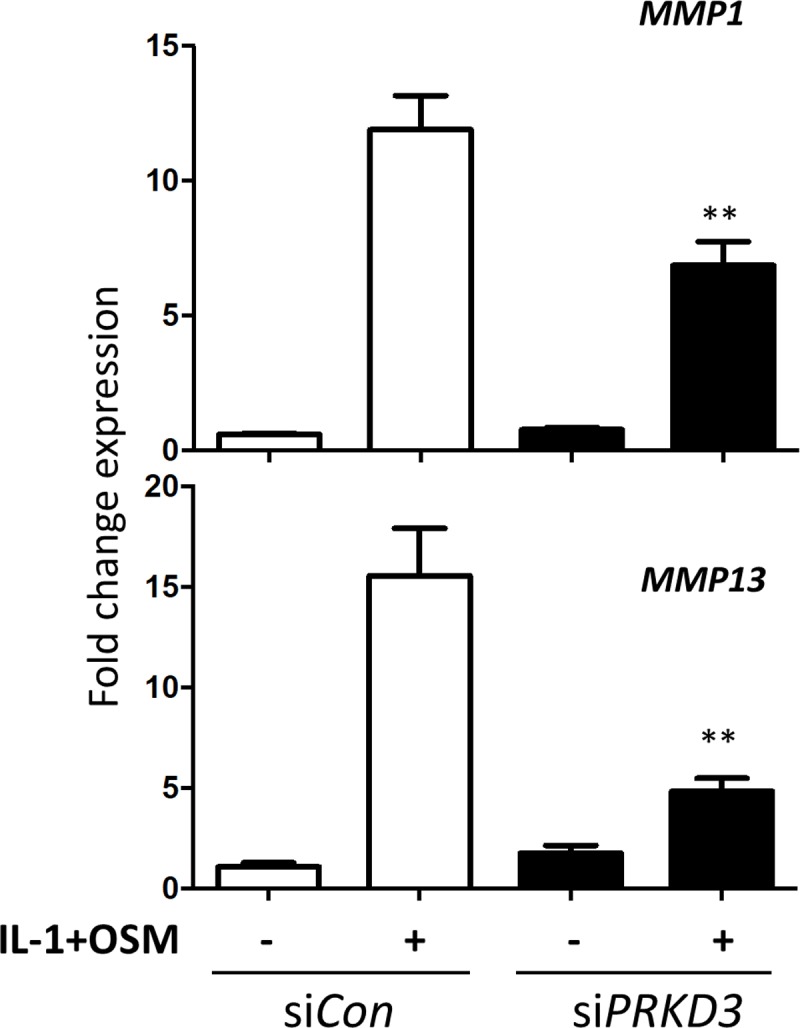
Following transfection with siRNA, either non-targeting siCon (white bars) or one specific for PRKD3 (black bars) (both 100 nM), primary human articular chondrocytes were stimulated with IL-1 (0.05 ng/ml) + OSM (10 ng/ml) for 24 h. Cells were lysed and real-time RT-PCR performed for MMP1 (upper panel) and MMP13 (lower panel), 72 h after the start of transfection as described in the Materials and Methods. Data (mean ± S.E.) are representative of at least three separate chondrocyte populations (each assayed in hextuplicate), presented as relative expression levels normalised to 18S rRNA housekeeping gene, where **, p≤0.01 versus siCon transfection.
PKD3 depletion alters STAT signalling in IL-1+OSM-stimulated human chondrocytes
Analyses of the signalling pathways activated in human chondrocytes following PRKD3 silencing with IL-1+OSM stimulation revealed reductions in phosphorylation of all three MAPK groups but no apparent effect on the phosphorylation of Akt*S473 (Fig 3A and 3B). As dual phosphorylation of STATs occurs for full activity [37,38], we also observed reductions in Ser727 phosphorylation of STAT1 and STAT3 (Fig 3A and 3B), important for transcriptional activity [37], whilst a more detailed analysis using subcellular fractionation indicated that although cytoplasmic (and indeed total) levels of Tyr705 phosphorylation of STAT3 appeared to be relatively unaffected, reduced nuclear levels were observed (Fig 4A) following PRKD3 depletion. In contrast, total and nuclear STAT1*Y701 was unaffected by PRKD3 silencing (Fig 4A).
Fig 3. Effect of PRKD3 silencing on STAT and MAPK phosphorylation in human chondrocytes.

(A) Following transfection with siRNA specific to PRKD3 or non-targeting siCon (100 nM), primary human articular chondrocytes were stimulated with IL-1+OSM as detailed in the Methods for the indicated times. Whole cell lysates were prepared and subjected to SDS-PAGE and immunoblotting using the antibodies indicated. (B) Scans from multiple blots with siCon (white bars) or siPRKD3 (black bars) were combined and the relative densitometry values presented for the individual antibodies used, where **, p≤0.01 vs the relevant siCon time point. The data are representative of at least three separate chondrocyte populations.
Fig 4. The role of STATs in PKD3-mediated MMP expression in human chondrocytes.
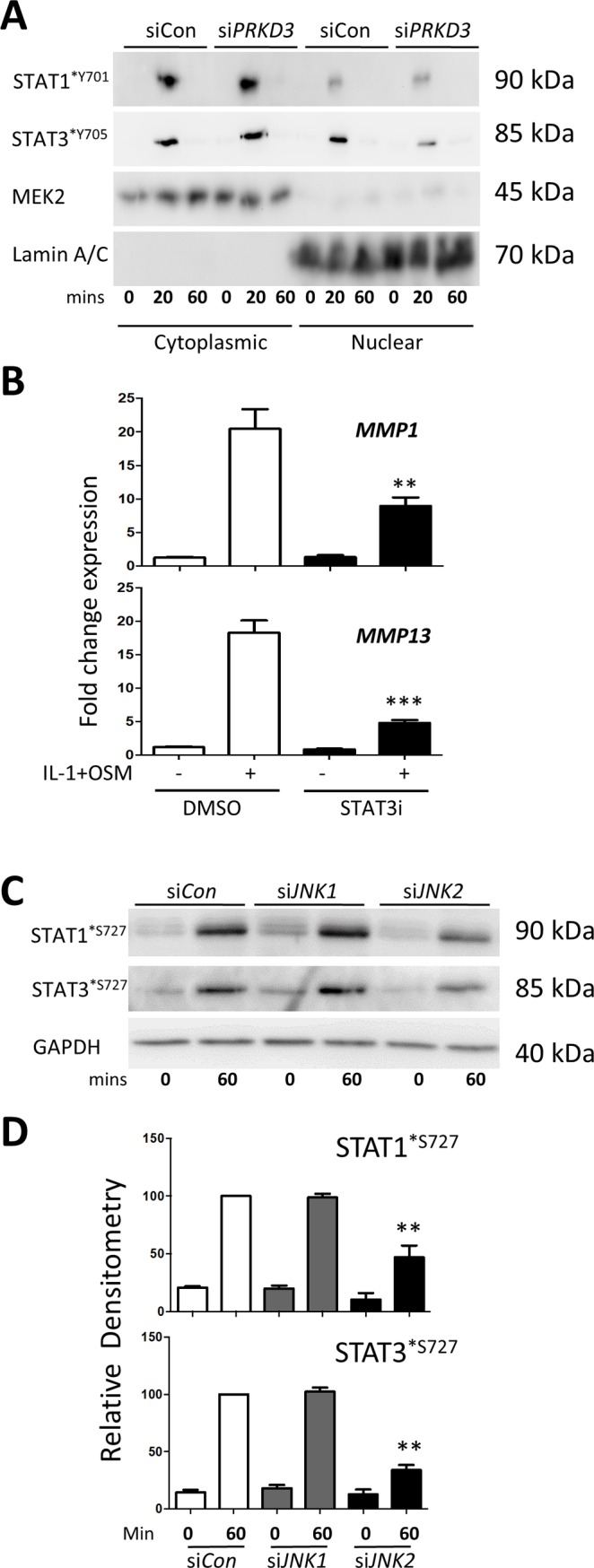
(A) Following transfection with siRNA specific to PRKD3 or non-targeting siCon (100 nM), primary human articular chondrocytes were stimulated with IL-1+OSM as detailed in the Methods for the indicated times. Subcellular fractions were prepared and subjected to SDS-PAGE and immunoblotting using the antibodies indicated. (B) Chondrocytes were pre-treated for 1 h with S31-201 (STAT3i; black bars) or DMSO vehicle control (white bars), and then stimulated with IL-1+OSM for 24 h. Lysates were subjected to real-time RT-PCR (n = 6; mean ± S.E.) for MMP1 and MMP13 as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where ***, p≤0.001, *, p≤0.05 vs the relevant DMSO treatment. (C) Chondrocytes were transfected with siCon (white bars), siJNK1 (grey bars) or siJNK2 (black bars) (all 100 nM) for 48 h prior to stimulation with IL-1+OSM for the indicated durations. Cell lysates were immunoblotted with the antibodies indicated. Scans from multiple blots were combined and the relative density values presented for the individual antibodies used (D), where **, p≤0.01 vs the relevant siCon time point. All data are representative of at least three separate chondrocyte populations.
To further confirm a role for STAT3 in IL-1+OSM-mediated MMP expression [17,19] the STAT3 inhibitor S31-201 significantly reduced both MMP1 and MMP13 expression in human chondrocytes (Fig 4B). Since PRKD3 silencing curtailed IL-1+OSM-mediated JNK activation, and in light of the previously reported PKD-dependent activation of JNK [39] and our previous report indicating no dependency for ERK [19], we determined whether the observed effects on STAT phosphorylation could be mediated by JNK moieties. Gene silencing of JNK1 and JNK2 revealed that JNK2 appeared to have a role in the Ser727 phosphorylation of both STAT1 and STAT3 following IL-1+OSM stimulation (Fig 4C and 4D).
PKD3 indirectly regulates AP-1 factors in IL-1+OSM-mediated MMP13 expression
Previous data have shown a role for STAT3 in the induction of the AP-1 factor cFos [17], and we show here that the effect of PRKD3 silencing on STAT3 phosphorylation is concomitant with a reduction in the expression of both FOS and JUN (Fig 5A) as well as cFos protein and phosphorylated cJun (Fig 5B and 5C). We have recently reported the involvement of ATF3 in cytokine-induced MMP13 expression [13], and assessment of ATF3 expression following PRKD3 silencing confirmed that it was indeed significantly reduced (Fig 6A). This was further reflected in markedly reduced nuclear ATF3 protein in cytokine-stimulated human chondrocytes following PRKD3 gene silencing (Fig 6B and 6C).
Fig 5. Effect of PRKD3 gene silencing on the expression of AP-1 factors in human chondrocytes.

Following transfection with siRNA specific to PRKD3 (black bars) or non-targeting siCon (white bars) (both 100 nM), primary human articular chondrocytes were stimulated with IL-1+OSM as detailed in the Methods for 1 h unless stated otherwise. (A) Cell lysates were subjected to real-time RT-PCR (n = 6; mean ± S.E.) for FOS (upper panel) or JUN (lower panel) as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where **, p≤0.01 versus siCon. (B and C) Nuclear fractions were prepared and subjected to SDS-PAGE and immunoblotting using the antibodies indicated. Scans from multiple blots were combined and the relative density values presented for the individual antibodies used (C), where **, p≤0.01 vs the relevant siCon time point. All data are representative of at least three separate chondrocyte populations.
Fig 6. Effect of PRKD3 gene silencing on ATF3 expression in human chondrocytes.

Following transfection with siRNA specific to PRKD3 (black bars) or non-targeting siCon (white bars) (both 100 nM), primary human articular chondrocytes were stimulated with IL-1+OSM as detailed in the Methods for 75 min unless stated otherwise. (A) Cell lysates were subjected to real-time RT-PCR (n = 6; mean ± S.E.) for ATF3 as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where ***, p≤0.001 versus siCon. (B) Nuclear fractions were prepared and subjected to SDS-PAGE and immunoblotting using the antibodies indicated. Scans from multiple blots were combined and the relative density values presented (C), where **, p≤0.01 vs siCon. All data are representative of at least two separate chondrocyte populations.
Discussion
The signal transduction associated with potent pro-inflammatory stimuli is complex although it is emerging that many of the activated pathways are common to a multitude of inflammatory mediators [13]. The interactions and cross-talk serve to markedly enhance MMP induction in human chondrocytes which ultimately drive ECM cataoblism [6,7,13,14,18,19]. We have used the catabolic stimulus of IL-1+OSM as a model inflammatory stimulus to investigate the signalling that underpins the expression of collagenolytic MMPs, in particular MMP13 due to its high capacity to degrade the major structural component of cartilage, type II collagen [12]. Our own data and those of others [13,17,40] have shown a strong dependence on AP-1 complexes of cFos/cJun in order for MMP gene expression to occur, and this has been linked to AP-1 consensus motifs in the proximal promoters of both MMP1 and MMP13 (see [16]). Phosphorylation of PKC isoforms in human chondrocytes occurs following stimulation with IL-1 or OSM, whilst their silencing has been shown to reduce both MMP1 and MMP13 expression as well as the induction of FOS [19]. These stimuli also promote phosphorylation of the downstream PKC substrate, PKD [19]. Herein we extend these observations through the identification of PKCι as the atypical PKC isoform that mediates downstream PKD phosphorylation. Moreover, pharmacological inhibition of PKD species significantly reduced IL-1+OSM-induced MMP1 and MMP13 expression. However, the PKD inhibitor kb NB142-70 does not discriminate between PKD isoforms, and indeed many early studies on PKD reported findings relating to PKD as a single ‘entity’ rather than considering the three individual protein kinase isoforms. In this context, only silencing of PKD3 (PRKD3) mimicked the inhibition of PKDi on MMP1/13 expression thus highlighting its role in pro-inflammatory signalling in chondrocytes. Moreover, these data also indicate that stimulation of two specific cytokine receptor pathways (IL-1 and OSM, respectively) converges on PKD3 activation via PKCι (Fig 7).
Fig 7. Proposed mechanism for the involvement of PKD3 in cytokine-stimulated MMP13 induction in human chondrocytes.

Stimulation by IL-1 and OSM mediates the phosphorylation and activation of PKCι, whilst OSM stimulation leads to activation of the JAK/STAT pathway including Tyr705 phosphorylation of STAT3. PKCι phosphorylates/activates ERK as well as PKD3 at Ser731/735, leading to PKD3 Ser916 autophosphorylation. In turn, this leads to the phosphorylation of JNK which induces Ser phosphorylation of STAT3, priming it for transcription, as well as stabilising and phosphorylating cJun (PKD3 may also directly phosphorylate cJun). Both STAT3 and ERK (via Elk1 activation) lead to the induction of FOS, and when combined with phospho-cJUN drives the expression of various transcriptional regulators which regulate the resulting MMP induction including ATF3-dependent MMP13 expression. The scheme is based on the current study and input from Refs. (13,17–19,22,39–46).
Earlier findings indicated that STAT3 is a potent mediator of cytokine-induced MMP expression in chondrocytes [17,19]. Evidence is provided here too indicate that PRKD3 knockdown also modulates the activation of STATs 1 and 3 via reducing Ser727 phosphorylation, a requirement for maximal transcriptional activation of target genes [37]. Notably, STAT1/3 tyrosine phosphorylation was unaffected and, since nuclear import of STAT3 is independent of Tyr705 phosphorylation [38], the reduced nuclear STAT3 was presumably due to a PKD3-dependent alteration in the importin-α/importin-β1-Ran pathway [38]. Furthermore, the catalytic activity of PKD3 has been shown to regulate its own nuclear import through auto-phosphorylation and/or interaction with other proteins [41], such that PRKD3 depletion prior to stimulation could markedly restrict subsequent nuclear trafficking to limit STAT3-dependent gene transcription. Indeed, PRKD3 silencing led to a reduction in the expression of both cFOS and cJUN at both the mRNA and protein levels, as previously shown to be critical for MMP1 and MMP13 expression [19]; the induction of FOS has already been attributed to STAT3 activation [19]. Moreover, phosphorylation of cJun by JNK (Ser63/93) as well as by PKD isotypes (Ser58) has been reported [42], thus generating the transcriptionally active AP-1 (cFos/cJun) complexes associated with MMP1/13 transcription [16]. The bone abnormalities in a gene-trap prkd3 deletion mouse [34] also support a key role for PKD3 in the regulation of MMP1/13 expression in chondrocytes and cartilage.
Of the other collagenolytic MMPs, MMP14 does not possess a promoter-proximal AP-1 element [43,44] and was unaffected following PRKD3 silencing as we would predict. Bioinformatics has identified a potential proximal AP-1 for MMP8 [44] and, in line with our findings of reduced AP-1 expression, this MMP was also supressed. However, although many non-canonical AP-1 sequences have been shown to be functional, other reports do not support the presence of a proximal AP-1 element for MMP8 [43,45] which would indicate other regulatory mechanisms being impacted by PKD3 activity following cytokine stimulation.
We provide further evidence for a critical role for cFos in the cytokine-mediated upregulation of MMP1 and MMP13 since the effect of PRKD3 silencing on FOS expression was specific. Importantly, silencing of either PRKD1 or PRKD2 had no modulatory effects on MMP1/13, FOS or JUN (S4 and S6 Figs).
Despite a demonstrable role for the immediate early gene FOS and AP-1(cFos/cJun)-dependent induction of MMP1 and MMP13 (reviewed in [46]), we have also reported an absence of cFos binding to the MMP13 promoter indicative of an indirect role for such cFos/cJun complexes [13]. Indeed, cytokine-induced MMP13 expression involved the bZIP transcriptional regulator ATF3, the expression of which was AP-1(cFos/cJun)-dependent [13]. The obvious corollary therefore was that ATF3 expression would be supressed following PRKD3 silencing thus reducing its ability to transcriptionally regulate MMP13. Our findings herein confirm this mechanism and further highlight that although AP-1 (cFos/cJun) complexes are critical for cytokine-induced expression of genes such as MMP13, their role is indeed indirect: this is also most likely the case for MMP1 [13] although no such regulatory factors have yet been reported. Furthermore, earlier findings indicate that the magnitude of FOS expression following pro-inflammatory stimulation appears to directly influence the subsequent levels of MMP expression [17] which requires the expression of AP-1-dependent transcriptional regulators [13]. This is probably the case for other examples of complex pro-inflammatory stimuli that markedly enhance MMP expression (eg. [5,6,8,14,17,18]. For cytokine-induced MMP13 we further confirm the involvement of ATF3, the expression of which is critically reliant upon phosphorylation of PKD3.
Conclusion
Our data indicate that PKD3 functions downstream of PKCι to affect the cytokine-mediated induction of MMP1 and MMP13 in human chondrocytes. Thus, PKD3 may represent a novel therapeutic target for consideration in the better management of inflammatory joint diseases, as well as other pathologies associated with aberrant collagenase expression and ECM catabolism.
Supporting information
The specificity of each antibody used in the study was confirmed using whole cell lysates, except for MEK2 and lamin A/C (cytoplasmic and nuclear extracts, respectively), prepared as described in the Methods from primary human articular chondrocytes either unstimulated or stimulated with IL-1 (0.2 ng/ml) ± OSM (10 ng/ml). Following SDS-PAGE, proteins were transferred to PVDF membranes and probed with the indicated antibodies. Full-length blots are presented to highlight the specific immuno-reactivity of each antibody (the arrow indicates the expected molecular mass).
(PDF)
Primary human articular chondrocytes were stimulated with IL-1 (0.2 ng/ml) alone, OSM (10 ng/ml) alone or IL-1+OSM as detailed in the Methods for 20 min. Cells were pre-treated with Gӧ6983 (20 μM; PKCi) or a DMSO vehicle control for 1 h prior to stimulation, and then lysed and immunoblotted with the indicated antibodies.
(PDF)
Specificity of shRNA and siRNAs was assessed in primary human articular chondrocytes using immunoblotting (using the antibodies indicated), whilst fluorescent microscopy was also used to assess lentiviral transduction. Following transfection with siRNA specific to PRKCI, PRKCZ, PRKD2, PRKD3, JNK1, JNK2 or non-targeting siCon (100 nM) (A-C) or lentiviral shRNA (MOI = 30) specific to PRKD1 or shCon (B), as described in the Methods, cells were lysed and immunoblotted with the indicated antibodies. Fluorescent microscopy was used to assess lentiviral transduction (B). All data are representative of at least three separate chondrocyte populations.
(PDF)
Following transduction with lentiviral shRNA (MOI = 30) specific to PRKD1 or shCon (A), or transfection with siRNA specific for PRKD2 or a non-targeting siCon (100 nM) (B), primary human articular chondrocytes were stimulated with IL-1+OSM as described in the Methods for 24 h. Cell lysates were then subjected to real-time PCR (n = 6; mean ± S.E.) for MMP1 or MMP13 (upper and lower panels, respectively) as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where ***, p≤0.001, **, p≤0.01 versus the relevant Control (ns = not significant). All data are representative of at least three separate chondrocyte populations.
(PDF)
Following transfection with siRNA specific for PRKD3 or a non-targeting siCon (100 nM), primary human articular chondrocytes were stimulated with IL-1+OSM as described in the Methods for 24 h. Cell lysates were then subjected to real-time PCR (n = 6; mean ± S.E.) for MMP8 and MMP14 (upper and lower panels, respectively) as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where **, p≤0.01, versus siCon (ns = not significant). All data are representative of at least three separate chondrocyte populations.
(PDF)
Following transduction with lentiviral shRNA (MOI = 30) specific to PRKD1 or shCon (A), or transfection with siRNA specific for PRKD2 or a non-targeting siCon (100 nM) (B), primary human articular chondrocytes were stimulated with IL-1+OSM as described in the Methods for 1 h. Cell lysates were then subjected to real-time PCR (n = 6; mean ± S.E.) for FOS or JUN (upper and lower panels, respectively) as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where ns = not significant versus the relevant Control. All data are representative of at least three separate chondrocyte populations.
(PDF)
Acknowledgments
We thank all the collaborators mentioned for their kind gifts, and the surgeons of the Newcastle Hospitals Trust for the generous supply of human tissue.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
ADR and GJL received funding from Arthritis Research UK (grant 19322; http://www.arthritisresearchuk.org/), and the NIHR Newcastle Biomedical Research Centre. Clinical and translational research was supported by the Northumberland, Tyne and Wear Comprehensive Local Research Network. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Sakkas LI, Platsoucas CD. The role of T cells in the pathogenesis of osteoarthritis. Arthritis Rheum. 2007;56(2):409–24. doi: 10.1002/art.22369 [DOI] [PubMed] [Google Scholar]
- 2.Goldring MB, Goldring SR. Osteoarthritis. Journal of cellular physiology. 2007;213(3):626–34. doi: 10.1002/jcp.21258 [DOI] [PubMed] [Google Scholar]
- 3.Koenders MI, Joosten LA, van den Berg WB. Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Annals of the rheumatic diseases. 2006;65 Suppl 3:iii29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hui W, Cawston T, Rowan AD. Transforming growth factor beta 1 and insulin-like growth factor 1 block collagen degradation induced by oncostatin M in combination with tumour necrosis factor alpha from bovine cartilage. Annals of the rheumatic diseases. 2003;62(2):172–4. doi: 10.1136/ard.62.2.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hui W, Rowan AD, Richards CD, Cawston TE. Oncostatin M in combination with tumor necrosis factor alpha induces cartilage damage and matrix metalloproteinase expression in vitro and in vivo. Arthritis Rheum. 2003;48(12):3404–18. doi: 10.1002/art.11333 [DOI] [PubMed] [Google Scholar]
- 6.Koshy PJ, Henderson N, Logan C, Life PF, Cawston TE, Rowan AD. Interleukin 17 induces cartilage collagen breakdown: novel synergistic effects in combination with proinflammatory cytokines. Annals of the rheumatic diseases. 2002;61(8):704–13. doi: 10.1136/ard.61.8.704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowan AD, Hui W, Cawston TE, Richards CD. Adenoviral gene transfer of interleukin-1 in combination with oncostatin M induces significant joint damage in a murine model. The American journal of pathology. 2003;162(6):1975–84. doi: 10.1016/S0002-9440(10)64330-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rowan AD, Koshy PJ, Shingleton WD, Degnan BA, Heath JK, Vernallis AB, et al. Synergistic effects of glycoprotein 130 binding cytokines in combination with interleukin-1 on cartilage collagen breakdown. Arthritis Rheum. 2001;44(7):1620–32. doi: 10.1002/1529-0131(200107)44:7<1620::AID-ART285>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- 9.Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529–43. [DOI] [PubMed] [Google Scholar]
- 10.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nature reviews Molecular cell biology. 2007;8(3):221–33. doi: 10.1038/nrm2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jubb RW, Fell HB. The breakdown of collagen by chondrocytes. J Pathol. 1980;130(3):159–67. doi: 10.1002/path.1711300304 [DOI] [PubMed] [Google Scholar]
- 12.Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271(3):1544–50. [DOI] [PubMed] [Google Scholar]
- 13.Chan CM, Macdonald CD, Litherland GJ, Wilkinson DJ, Skelton A, Europe-Finner GN, et al. Cytokine-induced MMP13 Expression in Human Chondrocytes Is Dependent on Activating Transcription Factor 3 (ATF3) Regulation. J Biol Chem. 2017;292(5):1625–36. doi: 10.1074/jbc.M116.756601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barksby HE, Hui W, Wappler I, Peters HH, Milner JM, Richards CD, et al. Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: implications for cartilage destruction and repair. Arthritis Rheum. 2006;54(2):540–50. doi: 10.1002/art.21574 [DOI] [PubMed] [Google Scholar]
- 15.Koshy PJ, Lundy CJ, Rowan AD, Porter S, Edwards DR, Hogan A, et al. The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: a time-course study using real-time quantitative reverse transcription-polymerase chain reaction. Arthritis Rheum. 2002;46(4):961–7. [DOI] [PubMed] [Google Scholar]
- 16.Rowan AD, Young DA. Collagenase gene regulation by pro-inflammatory cytokines in cartilage. Front Biosci. 2007;12:536–50. [DOI] [PubMed] [Google Scholar]
- 17.Catterall JB, Carrere S, Koshy PJ, Degnan BA, Shingleton WD, Brinckerhoff CE, et al. Synergistic induction of matrix metalloproteinase 1 by interleukin-1alpha and oncostatin M in human chondrocytes involves signal transducer and activator of transcription and activator protein 1 transcription factors via a novel mechanism. Arthritis Rheum. 2001;44(10):2296–310. [DOI] [PubMed] [Google Scholar]
- 18.Litherland GJ, Dixon C, Lakey RL, Robson T, Jones D, Young DA, et al. Synergistic collagenase expression and cartilage collagenolysis are phosphatidylinositol 3-kinase/Akt signaling-dependent. J Biol Chem. 2008;283(21):14221–9. doi: 10.1074/jbc.M710136200 [DOI] [PubMed] [Google Scholar]
- 19.Litherland GJ, Elias MS, Hui W, Macdonald CD, Catterall JB, Barter MJ, et al. Protein kinase C isoforms zeta and iota mediate collagenase expression and cartilage destruction via STAT3- and ERK-dependent c-fos induction. J Biol Chem. 2010;285(29):22414–25. doi: 10.1074/jbc.M110.120121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci. 2006;27(6):317–23. doi: 10.1016/j.tips.2006.04.003 [DOI] [PubMed] [Google Scholar]
- 21.McEneaney V, Dooley R, Harvey BJ, Thomas W. Protein kinase D stabilizes aldosterone-induced ERK1/2 MAP kinase activation in M1 renal cortical collecting duct cells to promote cell proliferation. J Steroid Biochem Mol Biol. 2010;118(1–2):18–28. doi: 10.1016/j.jsbmb.2009.09.014 [DOI] [PubMed] [Google Scholar]
- 22.Eiseler T, Doppler H, Yan IK, Goodison S, Storz P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009;11(1):R13 doi: 10.1186/bcr2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim YI, Park JE, Brand DD, Fitzpatrick EA, Yi AK. Protein kinase D1 is essential for the proinflammatory response induced by hypersensitivity pneumonitis-causing thermophilic actinomycetes Saccharopolyspora rectivirgula. J Immunol. 2010;184(6):3145–56. doi: 10.4049/jimmunol.0903718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JE, Kim YI, Yi AK. Protein kinase D1 is essential for MyD88-dependent TLR signaling pathway. J Immunol. 2009;182(10):6316–27. doi: 10.4049/jimmunol.0804239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, et al. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol. 2006;26(4):1569–77. doi: 10.1128/MCB.26.4.1569-1577.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higashiyama R, Miyaki S, Yamashita S, Yoshitaka T, Lindman G, Ito Y, et al. Correlation between MMP-13 and HDAC7 expression in human knee osteoarthritis. Mod Rheumatol. 2010;20(1):11–7. doi: 10.1007/s10165-009-0224-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavriilidis C, Miwa S, von Zglinicki T, Taylor RW, Young DA. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013;65(2):378–87. doi: 10.1002/art.37782 [DOI] [PubMed] [Google Scholar]
- 28.Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, et al. Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98(1):59–68. doi: 10.1016/S0092-8674(00)80606-6 [DOI] [PubMed] [Google Scholar]
- 29.Milner JM, Patel A, Davidson RK, Swingler TE, Desilets A, Young DA, et al. Matriptase is a novel initiator of cartilage matrix degradation in osteoarthritis. Arthritis Rheum. 2010;62(7):1955–66. doi: 10.1002/art.27476 [DOI] [PubMed] [Google Scholar]
- 30.Jones SW, Brockbank SM, Mobbs ML, Le Good NJ, Soma-Haddrick S, Heuze AJ, et al. The orphan G-protein coupled receptor RDC1: evidence for a role in chondrocyte hypertrophy and articular cartilage matrix turnover. Osteoarthritis Cartilage. 2006;14(6):597–608. doi: 10.1016/j.joca.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 31.Huesa C, Ortiz AC, Dunning L, McGavin L, Bennett L, McIntosh K, et al. Proteinase-activated receptor 2 modulates OA-related pain, cartilage and bone pathology. Ann Rheum Dis. 2016;75(11):1989–97. doi: 10.1136/annrheumdis-2015-208268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi A, Seki N, Hattori A, Kozuma S, Saito T. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta. 1999;1450(1):99–106. [DOI] [PubMed] [Google Scholar]
- 33.Ellwanger K, Pfizenmaier K, Lutz S, Hausser A. Expression patterns of protein kinase D 3 during mouse development. BMC Dev Biol. 2008;8:47 doi: 10.1186/1471-213X-8-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogers EM, Hsiung F, Rodrigues AB, Moses K. Slingshot cofilin phosphatase localization is regulated by receptor tyrosine kinases and regulates cytoskeletal structure in the developing Drosophila eye. Mech Dev. 2005;122(11):1194–205. doi: 10.1016/j.mod.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 35.Staunton D, Hudson KR, Heath JK. The interactions of the cytokine-binding homology region and immunoglobulin-like domains of gp130 with oncostatin M: implications for receptor complex formation. Protein Eng. 1998;11(11):1093–102. [DOI] [PubMed] [Google Scholar]
- 36.Cleaver CS, Rowan AD, Cawston TE. Interleukin 13 blocks the release of collagen from bovine nasal cartilage treated with proinflammatory cytokines. Annals of the rheumatic diseases. 2001;60(2):150–7. doi: 10.1136/ard.60.2.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wen Z, Zhong Z, Darnell JE Jr., Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82(2):241–50. [DOI] [PubMed] [Google Scholar]
- 38.Cimica V, Chen HC, Iyer JK, Reich NC. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PLoS One. 2011;6(5):e20188 doi: 10.1371/journal.pone.0020188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, Zheng S, Storz P, Min W. Protein kinase D specifically mediates apoptosis signal-regulating kinase 1-JNK signaling induced by H2O2 but not tumor necrosis factor. J Biol Chem. 2005;280(19):19036–44. doi: 10.1074/jbc.M414674200 [DOI] [PubMed] [Google Scholar]
- 40.Benbow U, Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol. 1997;15(8–9):519–26. [DOI] [PubMed] [Google Scholar]
- 41.Rey O, Papazyan R, Waldron RT, Young SH, Lippincott-Schwartz J, Jacamo R, et al. The nuclear import of protein kinase D3 requires its catalytic activity. J Biol Chem. 2006;281(8):5149–57. doi: 10.1074/jbc.M508014200 [DOI] [PubMed] [Google Scholar]
- 42.Waldron RT, Whitelegge JP, Faull KF, Rozengurt E. Identification of a novel phosphorylation site in c-jun directly targeted in vitro by protein kinase D. Biochem Biophys Res Commun. 2007;356(2):361–7. doi: 10.1016/j.bbrc.2007.02.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. Journal of cellular physiology. 2007;211(1):19–26. doi: 10.1002/jcp.20948 [DOI] [PubMed] [Google Scholar]
- 44.Clark IM, Swingler TE, Sampieri CL, Edwards DR. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol. 2008;40(6–7):1362–78. doi: 10.1016/j.biocel.2007.12.006 [DOI] [PubMed] [Google Scholar]
- 45.Jeong YJ, Shin JM, Bae YS, Cho HJ, Park KK, Choe JY, et al. Melittin has a chondroprotective effect by inhibiting MMP-1 and MMP-8 expressions via blocking NF-kappaB and AP-1 signaling pathway in chondrocytes. Int Immunopharmacol. 2015;25(2):400–5. doi: 10.1016/j.intimp.2015.02.021 [DOI] [PubMed] [Google Scholar]
- 46.Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4(3):157–64. doi: 10.1186/ar401 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The specificity of each antibody used in the study was confirmed using whole cell lysates, except for MEK2 and lamin A/C (cytoplasmic and nuclear extracts, respectively), prepared as described in the Methods from primary human articular chondrocytes either unstimulated or stimulated with IL-1 (0.2 ng/ml) ± OSM (10 ng/ml). Following SDS-PAGE, proteins were transferred to PVDF membranes and probed with the indicated antibodies. Full-length blots are presented to highlight the specific immuno-reactivity of each antibody (the arrow indicates the expected molecular mass).
(PDF)
Primary human articular chondrocytes were stimulated with IL-1 (0.2 ng/ml) alone, OSM (10 ng/ml) alone or IL-1+OSM as detailed in the Methods for 20 min. Cells were pre-treated with Gӧ6983 (20 μM; PKCi) or a DMSO vehicle control for 1 h prior to stimulation, and then lysed and immunoblotted with the indicated antibodies.
(PDF)
Specificity of shRNA and siRNAs was assessed in primary human articular chondrocytes using immunoblotting (using the antibodies indicated), whilst fluorescent microscopy was also used to assess lentiviral transduction. Following transfection with siRNA specific to PRKCI, PRKCZ, PRKD2, PRKD3, JNK1, JNK2 or non-targeting siCon (100 nM) (A-C) or lentiviral shRNA (MOI = 30) specific to PRKD1 or shCon (B), as described in the Methods, cells were lysed and immunoblotted with the indicated antibodies. Fluorescent microscopy was used to assess lentiviral transduction (B). All data are representative of at least three separate chondrocyte populations.
(PDF)
Following transduction with lentiviral shRNA (MOI = 30) specific to PRKD1 or shCon (A), or transfection with siRNA specific for PRKD2 or a non-targeting siCon (100 nM) (B), primary human articular chondrocytes were stimulated with IL-1+OSM as described in the Methods for 24 h. Cell lysates were then subjected to real-time PCR (n = 6; mean ± S.E.) for MMP1 or MMP13 (upper and lower panels, respectively) as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where ***, p≤0.001, **, p≤0.01 versus the relevant Control (ns = not significant). All data are representative of at least three separate chondrocyte populations.
(PDF)
Following transfection with siRNA specific for PRKD3 or a non-targeting siCon (100 nM), primary human articular chondrocytes were stimulated with IL-1+OSM as described in the Methods for 24 h. Cell lysates were then subjected to real-time PCR (n = 6; mean ± S.E.) for MMP8 and MMP14 (upper and lower panels, respectively) as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where **, p≤0.01, versus siCon (ns = not significant). All data are representative of at least three separate chondrocyte populations.
(PDF)
Following transduction with lentiviral shRNA (MOI = 30) specific to PRKD1 or shCon (A), or transfection with siRNA specific for PRKD2 or a non-targeting siCon (100 nM) (B), primary human articular chondrocytes were stimulated with IL-1+OSM as described in the Methods for 1 h. Cell lysates were then subjected to real-time PCR (n = 6; mean ± S.E.) for FOS or JUN (upper and lower panels, respectively) as described in Methods. Data are presented as relative expression levels normalised to 18S rRNA housekeeping gene, where ns = not significant versus the relevant Control. All data are representative of at least three separate chondrocyte populations.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.