

ABSTRACT
Human adenoviruses (HAdV) are significant human pathogens. Although only a subset of HAdV serotypes commonly cause gastroenteritis in humans, most HAdV species replicate in the gastrointestinal tract. Knowledge of the complex interaction between HAdVs and the human intestinal epithelium has been limited by the lack of a suitable cell culture system containing relevant cell types. Recently, this need has been met by the stable and prolonged cultivation of primary intestinal epithelial cells as enteroids. Human enteroids have been used to reveal novel and interesting aspects of rotavirus, norovirus, and enterovirus replication, prompting us to explore their suitability for HAdV culture. We found that both prototype strains and clinical isolates of enteric and nonenteric HAdVs productively replicate in human enteroids. HAdV-5p, a respiratory pathogen, and HAdV-41p, an enteric pathogen, are both sensitive to type I and III interferons in human enteroid monolayers but not A549 cells. Interestingly, HAdV-5p, but not HAdV-41p, preferentially infected goblet cells. And, HAdV-5p but not HAdV-41p was potently neutralized by the enteric human alpha-defensin HD5. These studies highlight new facets of HAdV biology that are uniquely revealed by primary intestinal epithelial cell culture.
IMPORTANCE Enteric adenoviruses are a significant cause of childhood gastroenteritis worldwide, yet our understanding of their unique biology is limited. Here we report robust replication of both prototype and clinical isolates of enteric and respiratory human adenoviruses in enteroids, a primary intestinal cell culture system. Recent studies have shown that other fastidious enteric viruses replicate in human enteroids. Therefore, human enteroids may provide a unified platform for culturing enteric viruses, potentially enabling isolation of a greater diversity of viruses from patients. Moreover, both the ability of interferon to restrict respiratory and enteric adenoviruses and a surprising preference of a respiratory serotype for goblet cells demonstrate the power of this culture system to uncover aspects of adenovirus biology that were previously unattainable with standard cell lines.
KEYWORDS: adenovirus, defensins, enteric viruses, enteroid, goblet cell, interferon
INTRODUCTION
Human adenoviruses (HAdV) are a family of DNA viruses that are important human pathogens, causing a wide variety of diseases, including respiratory infections, conjunctivitis, cystitis, and gastroenteritis (1). Much of our understanding of adenovirus replication and host interaction is due to studies of human adenovirus 5 (HAdV-5), a serotype of species C (HAdV-C). However, HAdV-F serotypes (HAdV-40 and -41), which are common causes of childhood gastroenteritis (2), differ significantly from HAdV-C (3, 4). They encode two different fiber proteins (long and short) (5), and their penton base proteins lack the canonical RGD sequence used by all other known HAdV serotypes for binding to integrin coreceptors (6). The importance of these unique features for cell entry and pathogenesis is unresolved, largely due to an inability to grow either prototype strains or clinical isolates of HAdV-F serotypes to high titers in standard transformed cell lines or nonintestinal primary cells (4, 7, 8).
HAdV-F species are naturally tropic for the gastrointestinal (GI) tract (9). More broadly, the GI tract is a site of infection and replication leading to intermittent shedding and persistence of HAdV serotypes that cause disease at other sites (e.g., the respiratory tract) or have no known disease association (2, 10, 11). In this regard, virus watch programs in Seattle and New York during the 1960s documented fecal shedding of respiratory serotypes of HAdV that were known at that time, including HAdV-1, -2, -3, and -5 (10, 11). HAdV-C and -D are also frequently isolated from fecal samples of HIV patients (12), and HAdV-D has been found at high prevalence in diarrheal samples from children (12–14). Thus, in addition to HAdV-A and -F, which have strong causative associations with childhood diarrhea (13), even serotypes more commonly associated with respiratory and ocular infections can be fecal-orally transmitted and in some cases can cause diarrhea in young children (2, 13). More recently, several species of HAdVs have been shown to persist in the lamina propria of the small intestine and colon and, upon immunosuppression, reactivate to infect the intestinal epithelium, likely from the basolateral surface (9). Furthermore, the capacity of HAdV-B and -E serotypes, which are frequent causes of acute respiratory disease in military recruits, to replicate in the intestine was exploited as a vaccine strategy (15). These studies highlight the importance of the GI tract for HAdV replication and transmission.
Until the recent development of human intestinal enteroids (16), stable culture systems containing the diversity of cell types found in the intestinal epithelium and in which human enteric viral replication might be studied in a more physiologic setting did not exist. Human enteroids consist of primary, untransformed cells and recapitulate much of the cellularity of the GI tract (16, 17). The structures arise from adult intestinal stem cells that are cultured in vitro in an extracellular matrix with a complex growth medium. Although they are untransformed, enteroids can be maintained in culture for extended periods of time and cryopreserved to establish a repository (17). The enteroids are differentiated into mature epithelial cell types found in the gut and maintain characteristics unique to the tissue from which they are derived (17, 18).
Human noroviruses, rotaviruses, and enteroviruses have been successfully cultured in human enteroids (19–24), demonstrating the utility of this system for culturing fastidious enteric viruses. Therefore, we sought to determine whether enteroids would support HAdV replication. We found that four species of HAdV replicate in human enteroids and that human enteroids are a suitable system for culturing clinical isolates of enteric and respiratory HAdVs. We show that the prototype strains HAdV-5p and HAdV-41p are sensitive to interferon in primary intestinal epithelial cells but not transformed lung cells. Furthermore, we found that HAdV-41p is resistant to but HAdV-5p is sensitive to human defensin 5 (HD5), an innate host defense peptide expressed in the GI tract. Surprisingly, we uncovered a preference of HAdV-5p but not HAdV-41p for goblet cells (GC). Collectively, these studies demonstrate the utility of using human enteroids to study HAdV tropism and innate immune control of HAdV infection.
RESULTS
Human ileal enteroid culture.
Human enteroid cultures were established from normal human deidentified ileal tissue obtained from surgical resections. Enteroids were propagated in a largely undifferentiated state in medium containing specific growth factors and small molecules; however, to recapitulate the cellular composition of the mature intestinal epithelium, the medium formulation was modified to promote differentiation. Since human small intestinal enteroid culture is not standardized, we characterized differentiation under our culture conditions, which were derived from published protocols (16, 17, 25). We observed consistent upregulation of markers for mature enterocytes (solute carrier family 10 member 2, encoded by SLC10A2) and goblet cells (trefoil factor 3, TFF3) after 5 days in differentiation medium compared to undifferentiated enteroids in multiple independent cultures from two separate donors, HIE5 (Fig. 1A) and HIE3 (Fig. 1B). Upregulation of the enteroendocrine cell marker chromogranin B (CHGB) was more consistent in samples from one donor than the other. Expression of a Paneth cell-specific gene (human defensin 5, DEFA5) was undetectable in all samples on day 1 and detected in only 3 of 6 replicates on day 5 for human ileal enteroid 5 (HIE5) but in all 4 replicates for human ileal enteroid 3 (HIE3) (Fig. 1A and B). It is unclear why there is variability in CHGB and DEFA5 expression within and between cultures of human enteroids.
FIG 1.
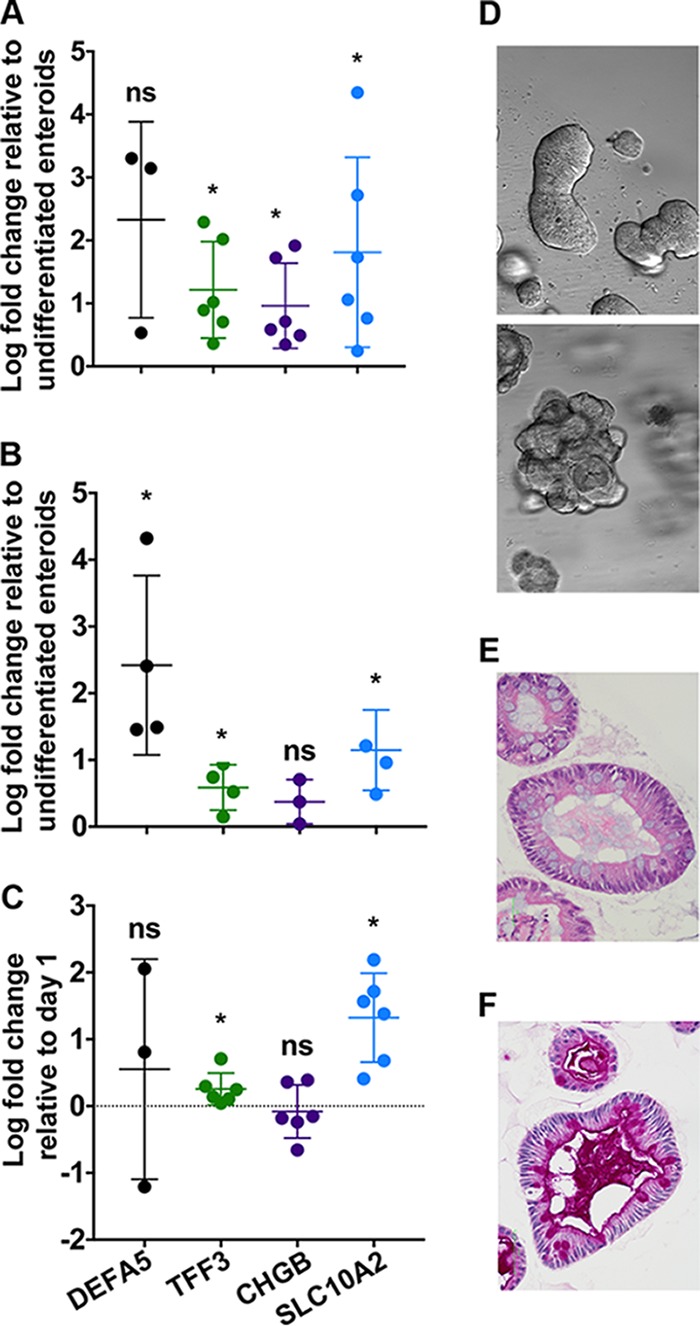
Human intestinal enteroids contain differentiated intestinal epithelial cell types found in the small intestine. Expression of human defensin 5 (DEFA5, Paneth cells), trefoil factor 3 (TFF3, goblet cells), chromogranin B (CHGB, enteroendocrine cells), and a bile acid transporter (SLC10A2, enterocytes) in differentiated HIE5 (A), differentiated HIE3 (B), and differentiated monolayers derived from HIE5 (C). For panels A and B, log fold increase in gene expression was calculated by comparing gene expression on day 5 to undifferentiated enteroids. For panel C, log fold increase in gene expression was calculated by comparing gene expression on day 5 to day 0 postplating. Each dot is an independent biological replicate. Note that although all 4 of the samples in panel B had detectable DEFA5 expression, this was true for only 3 of 6 samples in panel A and 2 of 6 samples in panel C. Individual replicates are plotted with the mean values ± standard deviations (SD) for each gene. (D) Bright-field images of differentiated enteroids representative of morphology with (top) and without (bottom) budding (4× objective). (E and F) Representative images of hematoxylin and eosin-stained (E) and periodic acid-Schiff-stained (F) differentiated human ileal enteroids (40× objective). For panels A to C, data were analyzed using a one-sample t test, *, P < 0.05; ns, not significant.
As has been observed by others (19), the enteroids within a single sample exhibited heterogeneous morphology with approximately 40% of differentiated human enteroids forming budding structures (Fig. 1D, top) reminiscent of the crypt-villus axis of the small intestine, while the other 60% formed small, dense cystic structures without overt budding (Fig. 1D, bottom). A single-cell-thick epithelium with polarized nuclei was apparent by hematoxylin and eosin (H&E) staining in all cases (Fig. 1E). And mature, functional goblet cells were identified as cells with large cytoplasmic vacuoles by H&E (Fig. 1E) or were positive for periodic acid-Schiff (PAS) staining in enteroids with both morphologies (Fig. 1F). PAS-positive debris was also found in the lumen, indicating goblet cell secretion. Notably absent in these cultures were cells with the distinct morphology of Paneth cells, even in samples where expression of the Paneth cell-specific gene DEFA5 was robust.
Infection and replication of prototype HAdV strains in cell lines.
Reports of enteric HAdV-F replication in transformed cells are inconsistent (4, 7, 8, 26, 27). We therefore compared the replications of the prototype strains HAdV-41p (HAdV-F), HAdV-5p (HAdV-C), and HAdV-16p (HAdV-B) in two standard cell lines, A549 cells (lung carcinoma) and 293 (embryonic kidney), to determine whether the culture defect for HAdV-F was at the level of initial infection, replication, or both. Note that the 293 cells used here (293β5) overexpress integrin β5, rendering the cells more adherent. HAdV-5p and HAdV-16p are associated primarily with respiratory disease and use coxsackievirus and adenovirus receptor (CAR) and CD46, respectively, as primary receptors (28, 29). Infected cells were quantified by immunofluorescence for hexon production 24 h postinfection. In 293β5 cells, HAdV-5p was the most infectious, followed by HAdV-16p and HAdV-41p, which were equivalent (Fig. 2A). In A549 cells, HAdV-41p infection was substantially more limited than infection by the respiratory viruses, and HAdV-5p was ∼3-fold more infectious than HAdV-16p (Fig. 2B). We also evaluated HAdV-41p infection on 293 cells with an attenuated interferon response due to the expression of the V protein of simian virus 5 (293-SV5/V). Despite being developed to facilitate the propagation of HAdV-F serotypes (30), 293-SV5/V cells were not more permissive than 293β5 cells (Fig. 2C).
FIG 2.
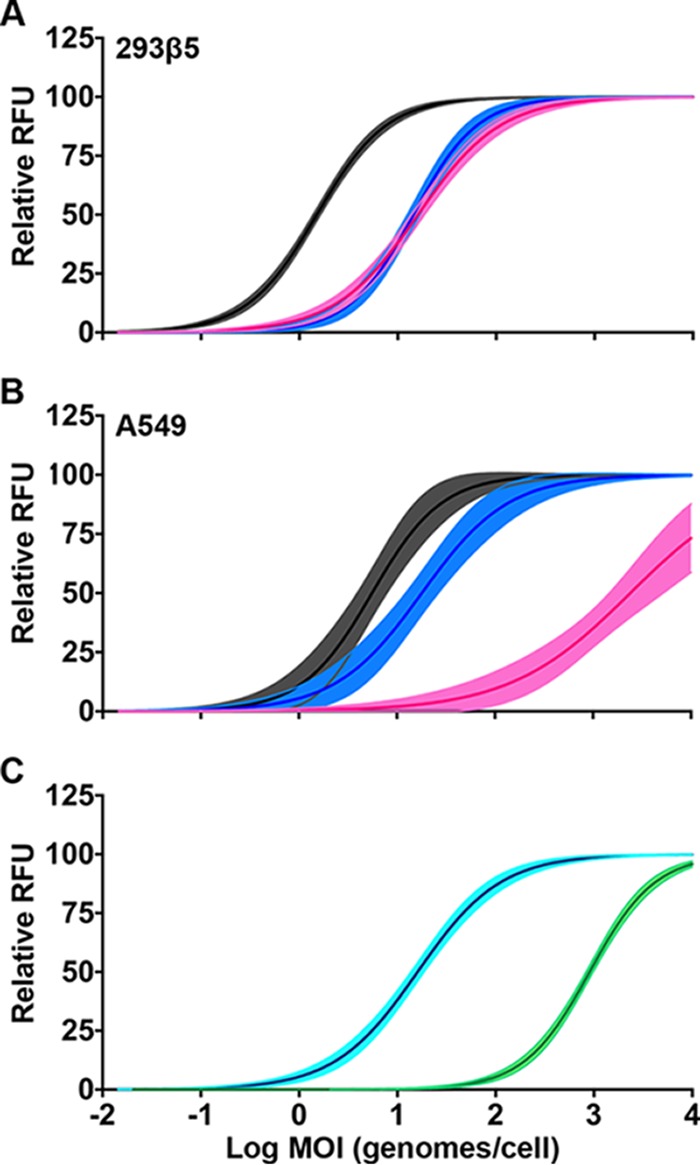
Infection of cell lines. (A and B) 293β5 cells (A) and A549 cells (B) were infected with serial dilutions of HAdV-5p (black), HAdV-16p (blue), and HAdV-41p (pink). (C) 293β5 (cyan) and 293-SV5/V (green) cells were infected with serial dilutions of HAdV-41p. Lines were fitted to the mean values for three biological replicates; 95% confidence intervals of the nonlinear regression are shaded.
We next compared replication of the three HAdV serotypes in the most permissive cell line, 293β5. Parallel cultures of cells were infected and incubated at 4°C for 45 min. The inoculum was then replaced with fresh medium, and samples were shifted to 37°C. The multiplicity of infection (MOI; 5 to 10 genomes/cell) was equivalent among serotypes in a given experiment but varied slightly between experiments. Total viral genomes in the cells and supernatant were quantified every 24 h for 5 days by quantitative real-time PCR (qPCR). We used the same primer pair, which is specific for a region of hexon conserved in all three serotypes, to facilitate direct comparison. All three serotypes amplified rapidly within 24 h. HAdV-5p replication then proceeded at a slower, constant rate through 5 days postinfection (Fig. 3A), while that of HAdV-16p reached a plateau on day 1 (Fig. 3B) and that of HAdV-41p reached a plateau on day 2 (Fig. 3C) postinfection. The fold increase in HAdV-41p replication was higher than for HAdV-16p but lower than for HAdV-5p (Fig. 3D). Taken together, our experiments demonstrate that 293β5 cells are more easily infected than other cell lines and support genome amplification of all HAdV serotypes tested.
FIG 3.
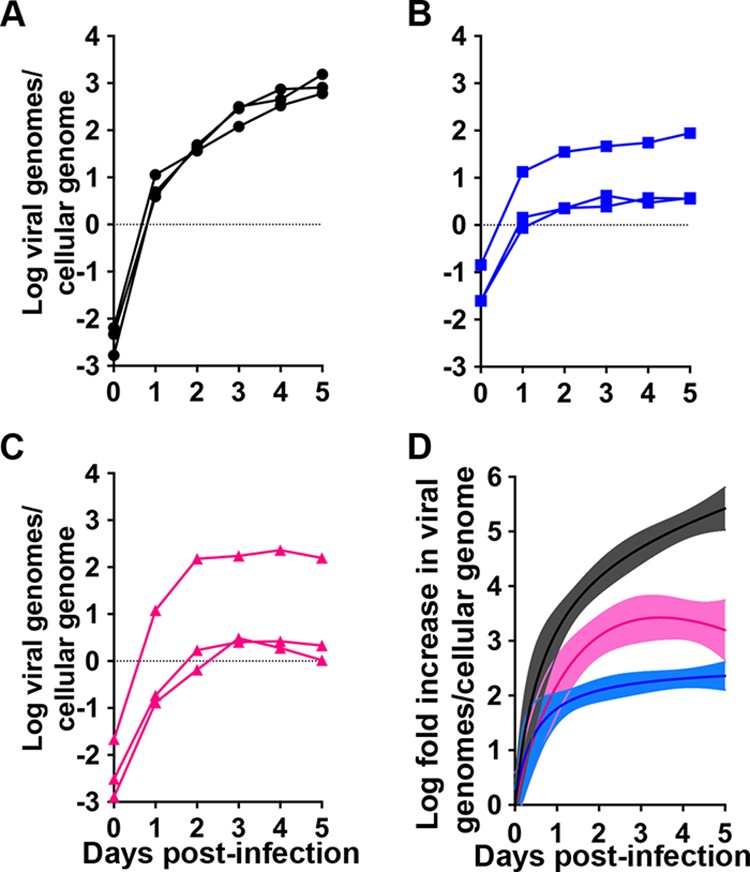
Replication in 293β5 cells. (A to C) HAdV-5p (black) (A), HAdV-16p (blue) (B), and HAdV-41p (pink) (C) replication in 293β5 cells infected at an MOI of 5 to 10 genomes/cell. Data are in viral genomes per cellular genome, and lines connect data points from individual replicates (n = 3). (D) Transformation of the data in panels A to C to fold increases relative to day 0. Coloring is consistent with panels A to C. Lines were fitted to the mean values for the three biological replicates; 95% confidence intervals of the nonlinear regression are shaded.
Replication of prototype HAdV strains in primary ileal enteroids.
Unlike transformed cell lines, human enteroids are composed of primary cells. Because they are more easily cultured, we first infected undifferentiated enteroids with HAdV-5p, HAdV-16p, and HAdV-41p at the same MOI (3,000 genomes/cell). Samples taken 2 h postinfection (day 0) indicated that the numbers of cell-associated genomes for HAdV-5p and HAdV-41p were lower than that for HAdV-16p but equivalent to each other. Similar to their replication kinetics in 293β5 cells, HAdV-5p (Fig. 4A and D) and HAdV-16p (Fig. 4B and D) amplified rapidly within 24 h and then continued to replicate at a slower, constant rate. HAdV-41p replication was characterized by a gradual increase in genomes over 5 days (Fig. 4C and D). The fold increase in viral genomes/cellular genome for HAdV-41p was less than HAdV-5p and -16p, which were similar (Fig. 4D). Therefore, all three HAdVs replicated in undifferentiated enteroids.
FIG 4.
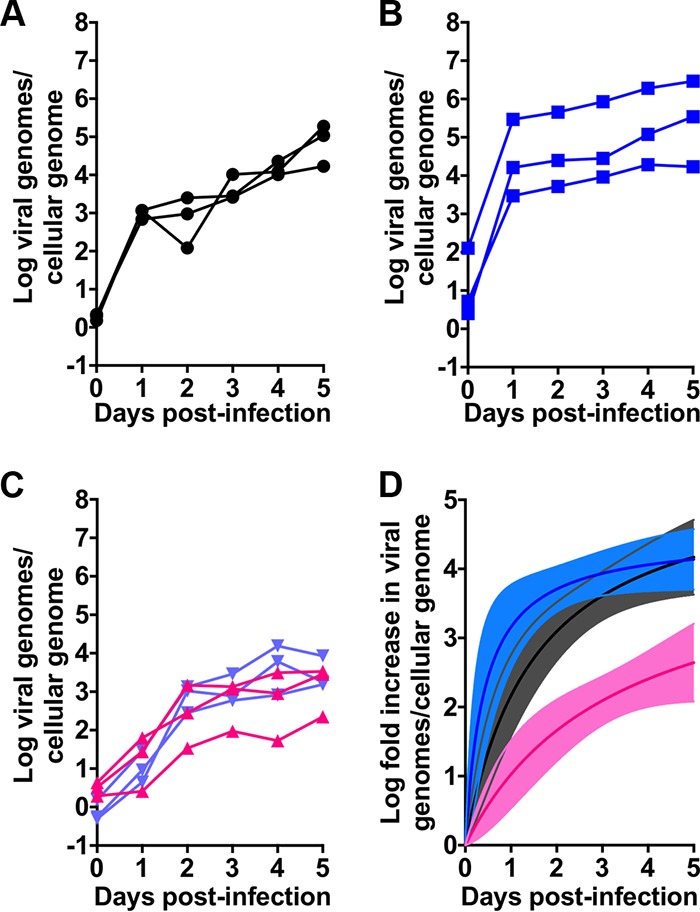
HAdVs replicate in ileal enteroids. (A to C) HAdV-5p (A), HAdV-16p (B), and HAdV-41p (C) replication in human enteroids. Data are viral genomes per cellular genome, and lines connect data points from individual replicates (n = 3). Coloring is consistent with Fig. 3. All data are from undifferentiated enteroids except for the purple lines in panel C, which are from differentiated enteroids. (D) Transformation of HAdV replication in undifferentiated enteroids from panels A to C to fold increase relative to day 0. Coloring is consistent with panels A to C. Lines were fitted to the mean values for the three biological replicates; 95% confidence intervals of the nonlinear regression are shaded.
We next infected differentiated enteroids, which contain mature intestinal epithelial cell types (Fig. 1A and E), with HAdV-41p. Replication in differentiated enteroids was similar to that in undifferentiated enteroids (Fig. 4C). Thus, both differentiated and undifferentiated enteroids express the HAdV-41 receptor and can support replication.
Replication of clinical HAdV isolates in human enteroids.
Many enteric viruses, including HAdV, are difficult to culture from clinical samples. And, prior publications have reported conflicting results of HAdV-F replication and cytopathic effects (CPE) in primary cells (4, 7, 8, 26, 27). Thus, we sought to determine whether human enteroids could support the growth of clinical isolates of HAdV-F. We obtained samples of HAdV-41 from the CDC and from the NYSDOH that were minimally passaged on transformed cells. We used undifferentiated human enteroids for these studies, because they can be cultured indefinitely. All four HAdV-41 isolates amplified over two passages (Fig. 5). We observed a 10-fold to 1,000-fold increase in genomes per cell over a span of 2 to 3 weeks. Cultures were terminated when complete CPE was observed, and our ability to passage the virus demonstrates that infectious particles were produced. We were unable to obtain clinical samples of HAdV-40, but HAdV-40p also replicated. Similarly, we cultured an HAdV-A serotype (HAdV-12p) that was originally isolated from stool (Fig. 5). In contrast to clinical isolates of enteric HAdVs, a clinical isolate of respiratory HAdV-C (HAdV-2, V-2375-18) amplified rapidly in undifferentiated enteroids to high titers within 5 days. We verified the identity of the clinical isolates obtained from external sources by HAdV fiber PCR (31). For all clinical isolates, the virus that amplified in the enteroids matched the serotype detected in the source material (Fig. 5). These experiments show that enteroids are a suitable system for culturing prototype and clinical isolates of HAdVs, including those that are the etiologic agents of respiratory and gastrointestinal disease.
FIG 5.
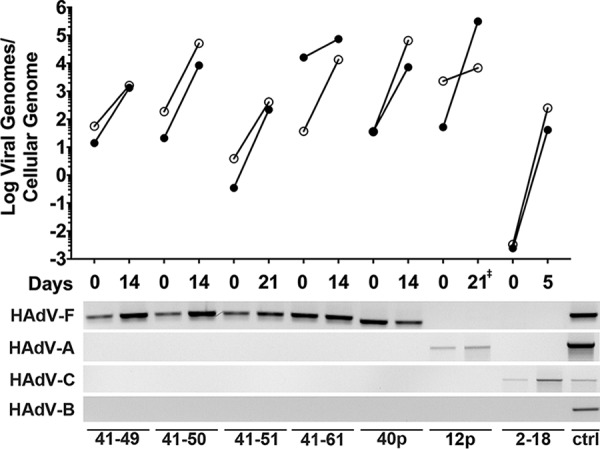
Clinical isolates of HAdVs replicate in undifferentiated ileal enteroids. Undifferentiated human ileal enteroids were infected basolaterally with lysates of each clinical isolate or prototype strain, which are labeled with the serotype and the last two digits of the isolate designation. Data are viral genomes per cellular genome, and lines connect data points from individual passages (top panel). Closed symbols are for viral passage 1 and open symbols for viral passage 2. Time to 80% CPE for each virus was approximately the same for both passages, except for HAdV-12p, for which passage 1 took 21 days while passage 2 took 9 days (‡). Species identification of the inoculum and at the end of passage 2 was determined for each strain by endpoint PCR using species-specific fiber primers (bottom panel). The positive control (ctrl) contained a mixture of HAdV-5p, HAdV-12p, HAdV-16p, HAdV-40p, and HAdV-41p DNA.
IFN attenuates HAdV replication in enteroids.
HAdVs contain multiple mechanisms to prevent interferon (IFN) induction (1). Once induced, replication of respiratory serotypes is sensitive to IFN; however, efficient inhibition is apparent only in primary cells (32). We investigated the impact of IFN pretreatment on HAdV-F replication in primary intestinal epithelial cells. For these experiments, we created monolayers composed of differentiated human enteroid cells. Like the differentiated enteroids, the enteroid monolayers contained cells with upregulated expression of genes associated with differentiation (Fig. 1C). Moreover, enteroid monolayers have been shown to be polarized and express tight-junction proteins (17, 20). Monolayers were pretreated with 1,000 IU/ml IFN-β or 500 ng/ml IFN-λ3 for 24 h and then infected with HAdV-5p for 24 h or HAdV-41p for 48 h. In uninfected cells, the interferon-stimulated genes (ISGs) OAS1 and ISG54 were upregulated at this time point (Fig. 6F). We found that IFN-λ3 and IFN-β inhibited both HAdV-5p replication (105% and 86%, respectively) (Fig. 6A) at 24 h and HAdV-41p replication (110% and 82%, respectively) (Fig. 6B) at 48 h postinfection. There was no induction of ISGs upon infection of these cells in the absence of IFN pretreatment (Fig. 6E). Interestingly, neither IFN-β nor IFN-λ3 pretreatment of A549 cells inhibited HAdV-5p infection at 24 h (Fig. 6C) despite comparable ISG upregulation (Fig. 6F). For HAdV-41p, although a low level of replication was detectable only in the untreated well at 24 h postinfection, both treatments were equivalent to control at 48 h postinfection (Fig. 6D). Thus, like primary airway cells (32), primary intestinal epithelial cells adopt an IFN-induced antiviral state capable of limiting HAdV infection. And, the inability of IFN pretreatment to inhibit HAdV replication in A549 cells is not due to an absolute inability to respond to IFN.
FIG 6.

Interferon pretreatment inhibits replication of HAdV in differentiated human enteroid monolayers but not A549 cells. Human enteroid monolayers and A549 cells were pretreated with 1,000 IU/ml of IFN-β (purple) or 500 ng/ml IFN-λ3 (blue) for 24 h or left untreated (black). (A and B) Enteroid monolayers were infected with HAdV-5p at an MOI of 15 (A) or HAdV-41p at an MOI of 6 (B) to achieve similar levels of infection. (C and D) By the same rationale, A549 cells were infected with HAdV-5p at an MOI of 0.5 (C) or HAdV-41p at an MOI of 80 (D). Data are fold changes in viral genomes per cellular genome relative to day 0, and lines connect the mean values ± SD for each time point (n = 3 for each condition). (E) Enteroid monolayers were infected with HAdV-5p at an MOI of 30 (black) or HAdV-41p at an MOI of 200 (pink) or treated with 1,000 IU/ml of IFN-β (purple) for 24 h. Data are the means ± SD of the fold changes in gene expression relative to untreated, uninfected cells (n = 3 for each condition). (F) Enteroid monolayers and A549 cells were treated with 1,000 IU/ml of IFN-β (purple) or 500 ng/ml IFN-λ3 (blue) for 24 h. Data are the means ± SD of the fold changes in gene expression relative to untreated, uninfected cells (n = 3 for each condition). For panels A to D, significance in comparing untreated to IFN treated at each time point: *, P < 0.05; **, P < 0.01; ns, not significant. For panels E and F, data were analyzed using a one-sample t test: *, P < 0.05; **, P < 0.01, ***, P < 0.001; ns, not significant.
HAdV-41 is resistant to the neutralizing activity of human enteric α-defensin HD5.
Enteric human defensins are secreted by Paneth cells in the small intestine (33). HD5 has been shown to both inhibit and enhance HAdV infection in vitro in a serotype-dependent manner (34, 35). Furthermore, enteric defensins have been shown to play complex roles during mouse AdV infection of mice (36, 37). HAdV-5p infection is potently inhibited and HAdV-41p infection moderately enhanced by HD5 on A549 cells (34). Since these experiments were performed on transformed cells, we next sought to assess whether HD5 exhibited the same activities on infection of primary intestinal epithelial cells, which for HAdV-5p and HAdV-41p can be infected at a lower MOI. Human enteroid monolayers were infected with HAdV-5p and HAdV-41p in the presence or absence of 15 μM HD5. Consistent with previous experiments using A549 cells, HAdV-5p infection of human enteroid monolayers was neutralized (90.1% ± 9.6%) by HD5 (Fig. 7A), while HAdV-41p infection was largely resistant but not enhanced (Fig. 7A).
FIG 7.

HAdV-5p, but not HAdV-41p, is neutralized by HD5 and preferentially infects goblet cells. (A) Human enteroid monolayers were infected at MOIs of 30 and 200 for HAdV-5p and HAdV-41p, respectively, in the presence or absence of 15 μM HD5. Infected cells were enumerated by flow cytometry at 48 h postinfection. Data are from four individual replicates graphed as the percentage of HAdV infection in the presence of HD5 compared to infection in the absence of HD5. Bars are means ± SD. (B) HAdV-5p- and HAdV-41p-infected cells from the samples in panel A were analyzed for the percentage of MUC2+ goblet cells (GC) and MUC2− nongoblet cells (non-GC) that were infected in the absence of HD5. (C) Data from panel A analyzed as the ratio of HAdV-5p infected GC to non-GC as a function of %GCs. (D to G) Representative flow cytometry plots. For panel A, the percentages of cells infected under each condition were compared. For panel B, the percentages of infected GC and non-GC were compared by ratio paired t tests. *, P < 0.05; ns, not significant.
HAdV-5p preferentially infects goblet cells over nongoblet cells.
As part of these experiments, we also examined the tropism of HAdV-5p and HAdV-41p for goblet cells (GC), since recent studies of enterovirus replication in human enteroids revealed preferential infection of enteroendocrine cells by echovirus 11 coupled with a lack of goblet cell infection (23). We costained the infected enteroid monolayers for MUC2, a marker of goblet cells, and hexon. While goblet cells comprised only 0.5 to 1.5% of the cells in the monolayer, 18.9% ± 8.3% of MUC2-positive goblet cells were infected with HAdV-5p compared to 4.4% ± 3.1% of the MUC2-negative cells (Fig. 7B). In contrast, HAdV-41 infected the two populations equally (3.9% ± 1.2% versus 3.1% ± 3.0%). Thus, HAdV-5p but not HAdV-41p preferentially infects goblet cells. And, although the MUC2-positive population is small, this preference was observed over a range of GC population sizes (Fig. 7C). To our knowledge, this is the first reported preference of HAdV for a specific cell type in the GI tract.
DISCUSSION
Human enteroids provide a unique opportunity to investigate host-pathogen interactions in an in vitro system that more accurately represents the cellularity of the GI tract. They are composed of untransformed cells, which have intact signal transduction pathways, and contain a mixture of mature, differentiated intestinal epithelial cell types (16). Both of these features are absent from standard cell lines. Our medium conditions for culturing human enteroids combine elements from several previously published formulations (16, 17, 25). Human enteroids differentiated under our conditions contain upregulated transcriptional markers of enterocytes, enteroendocrine cells, and goblet cells and distinct morphology of a polarized epithelium with functional goblet cells. However, expression of the Paneth cell marker DEFA5 was the most variable. Although we have also tried a variety of different published medium compositions, we have only rarely identified Paneth cell morphology by histology even in samples with robust DEFA5 expression. Because Wnt-dependent expression of DEFA5 is separable from the gene program required for Paneth cell morphology (38–40), it is likely that the culture of human enteroids lacks a key component required for Paneth cell morphology. This has also been noted by others (41). Nonetheless, we have validated suitable conditions for stable culture, prolonged passage, and differentiation of human enteroids.
Our experiments add HAdVs to the set of viruses that can be cultured in human enteroids, which also includes rotavirus, norovirus, and enterovirus (19–21, 24). Thus, enteroids have the potential to be a unified platform for amplifying human enteric viruses from clinical samples. HAdV-41p replicates to a similar extent in differentiated and undifferentiated enteroids; however, differentiated enteroids are less suitable for culturing slowly replicating viruses because of their limited life span (∼10 days) and inability to be passaged. In contrast, undifferentiated enteroids can be passaged for an indefinite period, allowing time for viral amplification. Although human rotaviruses can replicate in both undifferentiated and differentiated human enteroids (19, 24), human rotavirus infection was significantly increased in differentiated enteroids (19). Human norovirus replicates in enteroid monolayers (20), and human echovirus 11 replicates in differentiated human enteroids (21, 23). It is currently unknown whether these viruses can replicate in undifferentiated enteroids. Since rotavirus and norovirus replication efficiency is greatly impacted by the secretor status of the enteroids (19, 20), the differentiation state of the enteroids might affect the secretor phenotype. Furthermore, norovirus replication is enhanced by bile acid in human enteroid monolayers, but not standard cell lines (20). Thus, when isolating enteric viruses from clinical samples, the potential virus species should dictate the differentiation state of the human enteroids. The generally greater replication rate of RNA viruses is also an advantage, although the one respiratory isolate (HAdV-2) that we tested also replicated rapidly. The reason for the slow replication kinetics of HAdV-F is unclear, but it has also been observed in cell lines (7, 8, 42) and may reflect some unique aspect of enteric HAdV biology that remains to be elucidated. Since all of the inocula were lysates and the titer of the HAdV-2 V-2375-18 lysate was 2.5 log lower than the lowest HAdV-41 clinical isolate titer, low MOI or other characteristics of the inoculum are unlikely to explain the discrepancy in replication kinetics. Nonetheless, human enteroids are a remarkable system for supporting replication of human enteric viruses. Moreover, other primary human cell types (embryonic kidney cells, lung fibroblasts, and skin fibroblasts) have been tested for their capacity to support HAdV-F replication (4, 7, 27), but results have been inconsistent. Additionally, most of these primary cells were derived from locations not normally infected by enteric viruses, further complicating their utility in understanding HAdV-F biology. Interestingly, previous attempts to culture HAdV-F in fetal intestinal organ cultures did not result in infectious virus (43). Thus, human enteroids are the only viable system for studying HAdV-F interactions with mature primary intestinal epithelial cells.
As identified previously (4, 7, 8), there were differences in HAdV-41p infection and replication among transformed cell lines. Moreover, different groups have reported different results with the same cell lines, and sublines of the same cells have various abilities to support enteric adenovirus replication (7). Despite considerable investigation, the factors contributing to these differences are not well defined. We found that HAdV-41p can infect 293β5 and A549 lung cells, but infection was inferior to both respiratory serotypes in A549 lung cells but only to HAdV-5p in 293β5 cells. The disparity in HAdV-41p infection of A549 and 293β5 might reflect several different factors. First, the presence of the HAdV-5 E1 region in 293β5 has been implicated (3, 7, 44). And, E1B gene products of HAdV-2, -5, and -12 rescue HAdV-40 replication in a previously nonpermissive cell line (3, 44). Another potential explanation is the presence of a more intact innate immune response in A549 than in 293β5 cells. In our experiments, HAdV-41p appears to be no more sensitive than HAdV-5p to inhibition by IFN (Fig. 6); however, others have noted that HAdV-41 infection is blocked by IFN under conditions permissive for HAdV-2 replication (45). Caveats of these studies include a poorly characterized mixture of IFN-α and IFN-β derived from Namalwa cells (a B lymphoblast cell line) infected with Sendai virus and infection of cells thought to be conjunctival cells but later shown to be HeLa (45). Nonetheless, in one study, prior infection with HAdV-2 rescued HAdV-41 from IFN, suggesting that HAdV-2 potently inhibited the antiviral state (45). Finally, these differences could reflect tissue tropism and receptor usage. HAdV-F serotypes have two fibers, long and short (5), which likely bind to distinct receptors. They also lack an RGD sequence in their penton base proteins (replaced by RGA and GDD in HAdV-40 and -41, respectively), which is present in all other known HAdVs and mediates internalization via integrin coreceptors (6). Although purified HAdV-41 long fiber binds to CAR (46), this may not equate with receptor usage, as has been observed for other HAdVs (47). Thus, the receptors for both enteric HAdV long and short fibers remain unproven or unknown. Given that infection by the CAR-utilizing HAdV-5p is only ∼4-fold better on 293β5 than A549 cells while that of HAdV-41p is improved more substantially (∼150-fold), a simple model whereby the tropism of both viruses is dictated by CAR interactions alone is not supported. Elucidation of the receptors for enteric HAdV fibers and the need, if any, for a coreceptor is more easily performed in a genetically tractable system (e.g., 293 cells) but should be verified in human enteroids. Although human enteroids can be genetically engineered (48), they are not amenable to high-throughput screening techniques. More than receptor usage dictates tropism of HAdVs in vivo and their association with specific diseases. Experiments in more physiologic systems like enteroids provide a platform to examine the role of additional host factors in tropism.
Recently, an unexpected tropism of echovirus 11 for enteroendocrine cells but not goblet cells was revealed in enteroids (23). Surprisingly, we found that HAdV-5p but not HAdV-41p disproportionately infected goblet cells versus nongoblet cells. Nongoblet cells in these cultures include enterocytes, enteroendocrine cells, stem cells, and partially differentiated intermediates (e.g., transit-amplifying cells) based on gene expression but were not parsed further. A previous study of an enteric mouse adenovirus revealed no preference for a particular cell type in the small intestine (49), although to our knowledge this hasn't been examined in humans. Since goblet cells are also found in the airway, it will be interesting to determine the interactions that contribute to this tropism and if it extends to other mucosal surfaces or other respiratory serotypes. These results illustrate the power of the more-physiologic enteroid system to address fundamental aspects of virology that cannot be modeled in transformed cells.
A salient aspect of primary cell systems like human enteroids is the presence of intact signal transduction pathways of innate immunity, such as those leading to IFN production. Recent studies of HAdV-5p infection of primary bronchial epithelial cells (32) led us to investigate the impact of IFN on HAdV-5p and HAdV-41p replication. Like for airway cells, we were unable to detect IFN production in response to HAdV infection by changes in gene expression. This has also been observed in other cell types (45). However, we found that either IFN-β or IFN-λ3 pretreatment was sufficient to inhibit both HAdV-5p and HAdV-41p replication in human enteroid monolayers but not A549 cells, despite the capacity of A549 cells to respond to type I and type III IFN by other measures (50, 51).
Type III IFN (IFN-λs) plays an important role in controlling viral infection of intestinal epithelial cells, whereas type I IFN (IFN-β) is more important for lamina propria cells (52, 53). We observed comparable inhibition of HAdV by IFN-λ3 and IFN-β, while IFN-β inhibited human rotavirus replication in human enteroids to a greater extent than IFN-λ1 and IFN-λ3 (22). This discrepancy in the antiviral effects of IFN-λ could reflect differences in the antagonism of IFN signaling by the two viruses, their respective replication cycles, or the enteroid platforms used: three-dimensional (3D) enteroids versus enteroid monolayer. That concentrations of IFN sufficient to inhibit replication in primary intestinal epithelial cells were insufficient to suppress HAdV replication in transformed cells underscores the importance of using relevant primary cells to study viral infection and host response.
There are many questions in enteric adenovirology that might be addressed in the entroid system. First, the route of respiratory HAdVs seeding the GI tract is unclear. They could apically infect the intestinal epithelium through the fecal-oral route, or transient viremia could seed the intestinal epithelium basolaterally. An apical versus basolateral preference, even if the receptor(s) remain unknown, could be addressed in transwell assays of polarized enteroid monolayers. In a similar vein, an expanded analysis of the tropism of clinical and prototype isolates of HAdVs for specific cell types is warranted. Finally, the cause for the slow replication kinetics of clinical enteric isolates could be pursued and would help in the development of cell culture systems more permissive to the isolation of enteric viruses from patient samples.
MATERIALS AND METHODS
Cells and cell culture.
293β5 (54), 293-SV5/V (30), and A549 cells (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), penicillin, streptomycin, l-glutamine, and nonessential amino acids.
Viruses.
Prototype strains HAdV-5p (strain Adenoid 75), HAdV-16p (strain Ch. 79), and HAdV-41p (strain Tak) were acquired from ATCC. Large-scale cultures of these viruses from A549 (HAdV-5p and HAdV-16p) or 293β5 or 293-SV5/V cells (HAdV-41p) were twice purified by CsCl density gradient ultracentrifugation as described in reference 54. The purified virus was dialyzed against three changes of 150 mM NaCl, 40 mM Tris, 10% glycerol, and 2 mM MgCl2 (pH 8.1), snap-frozen in liquid nitrogen, and stored at −80°C. The purified stocks were quantified by disrupting the capsids in 1% SDS for 4 min at 100°C and then measuring the concentration of viral DNA by Qubit fluorometric quantitation (Thermo Fisher Scientific). All other inocula used in these studies were virus-containing cell lysates. Enteroids were inoculated directly with lysate containing HAdV-12p (strain Huie) from ATCC. HAdV-40p (strain Dugan) lysate from ATCC was passaged once on 293-SV5/V cells prior to use. HAdV-41 clinical isolates (NY/2010/4845, NY/2010/4849, and NY/2010/4851) were originally detected and identified as HAdV in the laboratory of Howard S. Faden, Division of Infectious Disease, Children's Hospital of Buffalo, and then typed and characterized in the laboratory of Kirsten St. George, Laboratory of Viral Diseases, Wadsworth Center, New York State Department of Health (NYSDOH). They were passaged once on A549 cells and once on 239T cells in the St. George lab prior to our use. Clinical isolates of HAdV-2 V-2375-18 and HAdV-41 V-2161 were kindly provided by the Centers for Disease Control and Prevention (CDC). Prior to our use, HAdV-2 V2375-18 had been passaged twice on HepG2 cells and once on A549 cells, and HAdV-41 V-2161 was passaged once on 293 cells. To measure the amount of virus in lysates, total DNA was isolated using the GeneJET Viral DNA and RNA purification kit (Thermo Fisher Scientific), and viral DNA was quantified by qPCR using primers for a conserved region of hexon (Table 1) against a standard curve of HAdV-5p genomes. HAdV-12p was quantified using primers specific for a region of hexon conserved between HAdV-A and HAdV-5 (Table 1) against the same standard curve. qPCR was performed with Sso Advanced Sybr green Supermix or Sso Fast Sybr green Supermix (Bio-Rad) on a Bio-Rad CFX Connect thermocycler.
TABLE 1.
qPCR primers for virus quantitation
| Primer | Sequence |
|---|---|
| HAdV Hexon FWD1 | GCVCTVGCGCTCGACATGACTTTTGAGGTGGA |
| HAdV Hexon REV1 | TCGATGACGCCGCGGTG |
| HAdV-A Hexon FWD1 | GATGCCGCAGTGGTCTTACATG |
| HAdV-A Hexon REV1 | GCCACMGTGGGRTTYCTAAACT |
| Hprt gDNA qPCR FWD2 | GAAAGGGTGTTTATTCCTCATG |
| Hprt gDNA qPCR REV2 | CAGCTGCTGATGTTTGAAATTA |
Infection of cell lines.
HAdV-5p, HAdV-16p, and HAdV-41p were serially diluted on 293β5, 293-SV5/V, and A549 cells in 96-well black wall plates (Perkins-Elmer). Infected cells were quantified 24 h postinfection by staining fixed cells with anti-hexon antibody 8C4 (1:900; Fitzgerald Industries) and an Alexa Fluor 488-conjugated secondary antibody (1:1,000). Total well fluorescence in relative fluorescent units (RFU) was measured with a Typhoon 9400 scanner. Each sample was normalized to the maximal value for HAdV-5p for each individual experiment and plotted relative to log MOI (genomes/cell).
For the time course, parallel samples of 293β5 cells were infected with the same number of genomes of HAdV-5p, HAdV-16p, and HAdV-41p (MOI, 5 to 10 genomes/cell). Viruses were incubated with cells for 45 min at 4°C with rocking. The inoculum was removed and replaced with complete medium. Every day postinfection, supernatant and cells were harvested together. Total DNA was isolated using the GeneJET viral DNA and RNA purification kit. Viral DNA was quantified by qPCR, as described above. Cellular genomes were quantified by qPCR using a standard curve of A549 genomic DNA and primers for HPRT (Table 1). The ratio of viral genomes to cellular genomes was calculated for each sample.
Wnt3a conditioned medium.
L-Wnt3a cells were obtained from ATCC and propagated in DMEM with 10% FBS. Once cells were >95% confluent, medium was changed to 10% FBS, 10 mM HEPES, and 1× Glutamax in advanced DMEM/F12 (ADF), and the cells were incubated for 4 days. The resulting conditioned medium (CM) was harvested and filtered. Each batch of Wnt3a CM was assayed using a Wnt-sensitive reporter system as follows: M50 Super 8× TOPFlash (Addgene 12456) or M51 Super 8× FOPFlash (Addgene 12457) was transfected into 293β5 cells with pRL-TK (Promega). After 24 h, the medium was changed to Wnt3a CM, and the cells were incubated for an additional 24 h. Cells were lysed, and luciferase activity was measured using the dual luciferase reporter assay system (Promega), per the manufacturer's instructions. The ratio of firefly luciferase activity in TOPFlash-transfected cells to that in the FOPFlash-transfected cells, both normalized to Renilla luciferase activity, was calculated. CM was considered usable if this ratio exceeded the value for an archived batch of Wnt3a CM known to support human enteroids.
Rspondin-1 conditioned medium.
293-Rspondin-1 cells were obtained from Calvin Kuo (Stanford University) and maintained in DMEM with 10% FBS. Once the cells reached >95% confluence, the medium was changed to 10% FBS, 10 mM HEPES, and 1× Glutamax in ADF, and the cells were incubated for 4 days. Rspondin-1 CM was harvested and filtered. Each batch of Rspondin-1 CM was assayed using the Wnt-sensitive reporter system described above, except that a mixture of 10% Rspondin-1 CM and 10% Wnt3a CM in ADF with 10% FBS, 10 mM HEPES, and 1× Glutamax was added to transfected cells. CM was considered usable if the activity in this assay exceeded that of an archived batch of Rspondin-1 CM known to support human enteroids.
Noggin conditioned medium.
293-Noggin cells were a gift from Hans Clevers (Hubrecht Institute) and maintained in DMEM with 10% FBS. Once the cells reached >95% confluence, the medium was changed to 10 mM HEPES and 1× Glutamax in ADF, and the cells were incubated for 7 days. Noggin CM was harvested and filtered. Serial dilutions of each batch of Noggin CM were analyzed by immunoblotting with an anti-Noggin antibody (1:1,000; Abcam). CM was considered usable if the concentration of Noggin was comparable to that of a batch of Noggin CM known to support human enteroids.
Human enteroids.
Adult human ileal tissue was acquired through NW BioTrust and NW BioSpecimen from normal, healthy tissue during surgical resections. The University of Washington Institutional Review Board has determined that the deidentified ileal tissue specimens used in this study do not meet the federal regulatory definition of research on human subjects. Ileal tissue was placed in ADF containing 1× antibiotic-antimycotic, 1× penicillin-streptomycin, and 100 μg/ml gentamicin at room temperature for 15 min. Following antibiotic treatment, the tissue was cut into 1-cm square pieces and placed in dissociation solution (8 mM EDTA and 10 mM dithiothreitol [DTT] in 1× Dulbecco's phosphate-buffered saline [DPBS]) for 90 min at 4°C with gentle rocking. Crypts were dissociated from the intestinal tissue by shaking in ADF supplemented with 20 μM Y-27632 (Abcam) and 0.5 μM Jag-1 (Anaspec). Fractions containing crypts were centrifuged at 400 × g at 4°C for 5 min and then plated in Matrigel (growth factor reduced, phenol red free). Enteroids were cultured in complete crypt culture medium (CCCM): 50% Wnt3a CM, 10% Rspondin-1 CM, 10% Noggin CM, 1× N-2 supplement, 1× B-27 supplement, 10 mM HEPES, 1× Glutamax, 1× antibiotic-antimycotic, 1× penicillin-streptomycin, 1 mM N-acetyl-l-cysteine (Sigma), 10 μM Y-27632, 500 nM A-8301 (Tocris), 50 ng/ml epidermal growth factor (EGF), 10 nM gastrin (Sigma), 10 μM SB202190 (Sigma), 10 mM nicotinamide, and 2.5 μM CHIR99021 (P212121, LLC). Jag-1 (0.5 μM) was added only for the initial crypt culture. All medium components are from Thermo Fisher Scientific, except where noted. Enteroids derived from individual patients were sequentially numbered (e.g., HIE3 or HIE5). Except for gene expression analysis, histology, and pilot experiments, all experiments were performed with enteroids (HIE5) from a single donor. For differentiation, human enteroids were sheared with a 23-gauge needle and plated into differentiation medium (CCCM without nicotinamide or CHIR99021 and with SB202190 reduced to 2.5 μM) for 5 days.
Enteroid monolayers.
Human enteroid monolayers were generated as previously described (20). Briefly, 96-well plates were coated with human placental collagen, type IV (10 μg/cm2; Sigma), in water for 1.5 h at 37°C. Undifferentiated human enteroids were removed from Matrigel using cell recovery solution (Thermo Fisher Scientific), dissociated in 0.05% trypsin at 37°C for 5.5 min, quenched with ADF containing 10% FBS, 10 mM HEPES, and 1× Glutamax, and mechanically dissociated with a pipette. Cells were passed through a 40-μm cell strainer, centrifuged at 400 × g for 5 min, resuspended in CCCM without CHIR99021, and plated onto collagen-coated wells (300,000 cells/well). After 24 h, the medium was changed to differentiation medium without Wnt3a CM, and the cultures were incubated for an additional 4 days prior to infection.
Gene expression.
RNA was extracted from enteroids using RNA-Bee (Tel-Test), and cDNA was synthesized (Promega GoScript). Target gene expression was quantified by qPCR using the primers listed in Table 2. The ΔΔCT method (where CT is threshold cycle) was used to determine the fold increase in gene expression of day 5 differentiated enteroids relative to day 1 differentiated enteroids normalized to HPRT expression. For samples in which the target gene was undetectable (e.g., DEFA5), the maximum number of cycles was imputed (46 cycles).
TABLE 2.
qPCR primers for gene expression
| Primer | Sequence | Reference |
|---|---|---|
| DEF5 FWD | AGG AAA TGG ACT CTC TGC TCT TAG | 17 |
| DEFA5 REV | TTG CAC TGC TTT GGT TTC TAT CTA | 17 |
| Trefoil factor 3 FWD | AGC TCT GCT GAG GAG TAC GTG | 17 |
| Trefoil factor 3 REV | ACA GAA AAG CTG AGA TGA ACA GTG | 17 |
| Chromagranin B FWD | CAG CCA ACG CTG CTT CTC | 16 |
| Chromagranin B REV | TGG CAT GGA ATT GAC AGC | 16 |
| SLC10A2 FWD | GGG TTA CTC CCT GGG GTT TC | 17 |
| SLC10A2 REV | CCA TGA CAT TTC TTG TAT GCC ACA | 17 |
| OAS1 FWD | CTGTGTGTGTGTCCAAGGTG | |
| OAS1 REV | AGTGGTGAGAGGACTGAGGA | |
| ISG54 FWD | ACGGTATGCTTGGAACGA | 55 |
| ISG54 REV | AACCCAGAGTGTGGCTGATG | 55 |
| HPRT FWD | TGA CCT TGA TTT ATT TTG CAT ACC | 16 |
| HPRT REV | CGA GCA AGA CGT TCA GTC CT | 16 |
Histology.
Differentiated human enteroids were removed from Matrigel using cell recovery solution, concentrated by centrifugation at 400 × g, and fixed in 4% paraformaldehyde (PFA). Enteroids were then stained with hematoxylin (Gill's formula) to facilitate visualization, resuspended in Histogel (Thermo Scientific), stored overnight in 10% neutral buffered formalin, and embedded in paraffin. They were then sectioned at 4 μm and stained with hematoxylin and eosin or periodic acid-Schiff stain using standard methods.
Image analysis.
Bright-field images were acquired using a Nikon Eclipse Ti inverted microscope fitted with a 4× objective, a charge-coupled-device (CCD) camera, and image acquisition software (μManager; Open Imaging). Images of histology were acquired using a 40× objective.
Enteroid infections.
The number of cells in each well of enteroids was estimated by counting dissociated enteroids from a representative well on a hemocytometer. Matrigel containing enteroids was sheared with a 23-gauge needle, and purified HAdV-5p, HAdV-16p, or HAdV-41p at an MOI of 3,000 genomes/cell in CCCM supplemented with 1 μM Jag-1 was added. The sample was incubated for 45 min at 4°C with agitation. Enteroids were pelleted, the inoculum was removed, and the enteroids were replated in Matrigel. Each Matrigel plug was overlaid with 500 μl of CCCM or differentiation medium. The day 0 sample was harvested 2 h postwarming. One well of infected enteroids was then harvested every day for 5 days. Viral and cellular DNAs were isolated by GeneJET Viral DNA and RNA purification kit and quantified by qPCR as described above.
Infections with clinical isolates and with HAdV-12p and HAdV-40p were performed as described above with slight modifications: the inoculum consisted of lysate rather than purified virus, the enteroids were not sheared prior to infection, and the entire volume of enteroid-virus mixture was mixed with Matrigel and plated. Infected enteroids were passaged on days 7, 14, and 21, if needed, until greater than 80% of the enteroids showed signs of cytopathic effect (CPE), at which point a lysate was made and used to infect new enteroids. Each clinical isolate underwent two viral passages.
For Fig. 6A to D, human enteroid monolayers or A549 cells were treated with 1,000 IU/ml interferon-β (IFN-β) or 500 ng/ml IFN-λ3 (R&D Systems) 24 h prior to infection. Human enteroid monolayers were infected at MOIs of 15 and 6 genomes/cell for HAdV-5p and HAdV-41p, respectively. A549 cells were infected at MOIs of 0.75 and 85 genomes/cell for HAdV-5p and HAdV-41p, respectively. Inocula were chosen to achieve similar levels of infection between serotypes at 2 h postinfection. Viral genomes and cellular genomes were quantified at 2, 24, and 48 h (HAdV-41p only) postinfection as described above. For Fig. 6E, human enteroid monolayers were infected at an MOI of 200 and 30 for HAdV-41p and HAdV-5p, respectively, for 24 h. Expression of OAS1 and ISG54 was quantified at 24 h postinfection relative to uninfected cells, as described above. For Fig. 6F, human enteroid monolayers and A549 cells were treated with 1,000 IU/ml IFN-β or 500 ng/ml IFN-λ3 for 24 h, and expression of OAS1 and ISG54 was quantified at 24 h posttreatment.
For Fig. 7, HAdV-41p and HAdV-5p were incubated in the presence or absence of 15 μM HD5 for 45 min on ice. The virus was then added to human enteroid monolayers for 2 h at 37°C. The medium was then replaced. Human enteroid monolayers were infected at MOIs of 200 and 30 for HAdV-41p and HAdV-5p, respectively. Cells were trypsinized 48 h postinfection, fixed in 2% PFA, and stained with a mixture of 8C4 mouse anti-hexon antibody and rabbit anti-MUC2 (Santa Cruz H-300, 1:50) followed by a mixture of Alexa Fluor 488-conjugated anti-mouse and Alexa Fluor 647-conjugated anti-rabbit (1:200) secondary antibodies. Flow cytometry data were acquired on a Canto II (BD) and analyzed with FlowJo version 9.9.4 (BD) with doublet exclusion and using isotype controls for gating.
Verification of clinical isolates.
Total DNA isolated by the GeneJET viral DNA and RNA purification kit from the lysates of HAdV-12p- and HAdV-40p-infected cells, lysates containing primary isolates from NYSDOH and CDC, and lysates from enteroid cultures at the end of the second passage of each virus were analyzed by PCR to determine HAdV species present in each sample as described previously (31). Note that the PCRs for each HAdV species were performed separately rather than multiplexed, the melting temperatures (Tm) for the HAdV-A, -B, and -C reactions were modified to 56°C, a new forward primer (5′-GGATAVGCDGT-NGTRCTKGGCAT-3′) was designed for the HAdV-B reaction to match HAdV-16, and the extension temperature was lowered to 68°C per the manufacturer's instructions (Roche).
Statistical analysis.
All statistical analysis was performed using Prism 7.0c. For Fig. 1 and 6E and F, the log transformation of the fold increase in gene expression was compared to a theoretical value of zero using a one-sample t test. For Fig. 2, 3, and 4, curves and 95% confidence intervals were fitted by nonlinear regression to the mean data for three biological replicates. For Fig. 6A to D, we compared untreated to IFN-treated samples by one-way analysis of variance (ANOVA) with post hoc Tukey's test. For Fig. 7, we compared HAdV infection in the presence of HD5 to that in the absence of HD5 (as 100% infection) and HAdV infection of goblet cells and nongoblet cells using ratio paired t tests.
ACKNOWLEDGMENTS
We thank Youngmee Sul for her assistance in passaging human enteroids and generation of conditioned media.
J.G.S. was supported by R01 AI104920 from the National Institute of Allergy and Infectious Diseases. M.K.H. was supported by Public Health Service, National Research Service Award T32 GM007270 from the National Institute of General Medical Sciences. NW BioTrust, a core service for patient consenting, and NWBioSpecimen, a core service for procurement and annotation of research biospecimens, are supported by National Cancer Institute grant P30 CA015704 (Gary Gilliland, principal investigator [PI]), Institute of Translational Health Sciences grant UL1 TR000423 (Mary L. Disis, PI), the University of Washington School of Medicine and Department of Pathology, and the Fred Hutchinson Cancer Research Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Wold WSM, Ison MG. 2013. Adenoviruses, p 1732–1767. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 6th ed Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Uhnoo I, Wadell G, Svensson L, Johansson ME. 1984. Importance of enteric adenoviruses 40 and 41 in acute gastroenteritis in infants and young children. J Clin Microbiol 20:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevenson F, Mautner V. 2003. Aspects of the molecular biology of enteric adenoviruses, p 389–406. In Desselberger U, Gray JJ (ed), Viral gastroenteritis. Elsevier Science, Amsterdam, Netherlands. [Google Scholar]
- 4.Tiemessen CT, Kidd AH. 1994. Adenovirus type 40 and 41 growth in vitro: host range diversity reflected by differences in patterns of DNA replication. J Virol 68:1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berk A. 2013. Adenoviridae, p 1704–1731. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 6th ed Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Albinsson B, Kidd AH. 1999. Adenovirus type 41 lacks an RGD alpha(v)-integrin binding motif on the penton base and undergoes delayed uptake in A549 cells. Virus Res 64:125–136. doi: 10.1016/S0168-1702(99)00087-8. [DOI] [PubMed] [Google Scholar]
- 7.de Jong JC. 1983. Candidate adenoviruses 40 and 41: fastidious adenoviruses from human infant stool. J Med Virol 11:215–231. [DOI] [PubMed] [Google Scholar]
- 8.Witt DJ, Bousquet EB. 1988. Comparison of enteric adenovirus infection in various human cell-lines. J Virol Methods 20:295–308. doi: 10.1016/0166-0934(88)90133-4. [DOI] [PubMed] [Google Scholar]
- 9.Kosulin K, Geiger E, Vecsei A, Huber WD, Rauch M, Brenner E, Wrba F, Hammer K, Innerhofer A, Potschger U, Lawitschka A, Matthes-Leodolter S, Fritsch G, Lion T. 2016. Persistence and reactivation of human adenoviruses in the gastrointestinal tract. Clin Microbiol Infect 22:381.e1–381.e8. doi: 10.1016/j.cmi.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 10.Fox JP, Brandt CD, Wassermann FE, Hall CE, Spigland I, Kogon A, Elveback LR. 1969. The virus watch program: a continuing surveillance of viral infections in metropolitan New York families. VI. Observations of adenovirus infections: virus excretion patterns, antibody response, efficiency of surveillance, patterns of infections, and relation to illness. Am J Epidemiol 89:25–50. [DOI] [PubMed] [Google Scholar]
- 11.Fox JP, Hall CE, Cooney MK. 1977. The Seattle virus watch. VII. Observations of adenovirus infections. Am J Epidemiol 105:362–386. [DOI] [PubMed] [Google Scholar]
- 12.De Jong JC. 2003. Epidemiology of enteric adenoviruses 40 and 41 and other adenoviruses in immunocompetent and immunodeficient individuals, p 407–445. In Desselberger U, Gray JJ (ed), Viral gastroenteritis. Elsevier Science, Amsterdam, Netherlands. [Google Scholar]
- 13.Schmitz H, Wigand R, Heinrich W. 1983. Worldwide epidemiology of human adenovirus infections. Am J Epidemiol 117:455–466. doi: 10.1093/oxfordjournals.aje.a113563. [DOI] [PubMed] [Google Scholar]
- 14.Magwalivha M, Wolfaardt M, Kiulia NM, van Zyl WB, Mwenda JM, Taylor MB. 2010. High prevalence of species D human adenoviruses in fecal specimens from Urban Kenyan children with diarrhea. J Med Virol 82:77–84. doi: 10.1002/jmv.21673. [DOI] [PubMed] [Google Scholar]
- 15.Lyons A, Longfield J, Kuschner R, Straight T, Binn L, Seriwatana J, Reitstetter R, Froh IB, Craft D, McNabb K, Russell K, Metzgar D, Liss A, Sun X, Towle A, Sun W. 2008. A double-blind, placebo-controlled study of the safety and immunogenicity of live, oral type 4 and type 7 adenovirus vaccines in adults. Vaccine 26:2890–2898. doi: 10.1016/j.vaccine.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 16.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. 2011. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 17.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, Ciorba MA, Stappenbeck TS. 2015. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64:911–920. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Middendorp S, Schneeberger K, Wiegerinck CL, Mokry M, Akkerman RD, van Wijngaarden S, Clevers H, Nieuwenhuis EE. 2014. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells 32:1083–1091. doi: 10.1002/stem.1655. [DOI] [PubMed] [Google Scholar]
- 19.Saxena K, Blutt SE, Ettayebi K, Zeng XL, Broughman JR, Crawford SE, Karandikar UC, Sastri NP, Conner ME, Opekun AR, Graham DY, Qureshi W, Sherman V, Foulke-Abel J, In J, Kovbasnjuk O, Zachos NC, Donowitz M, Estes MK. 2015. Human intestinal enteroids: a new model to study human rotavirus infection, host restriction, and pathophysiology. J Virol 90:43–56. doi: 10.1128/JVI.01930-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, Kou B, Opekun AR, Burrin D, Graham DY, Ramani S, Atmar RL, Estes MK. 2016. Replication of human noroviruses in stem cell-derived human enteroids. Science 353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drummond CG, Nickerson CA, Coyne CB. 2016. A three-dimensional cell culture model to study enterovirus infection of polarized intestinal epithelial cells. mSphere 1(1):e00030-15. doi: 10.1128/mSphere.00030-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saxena K, Simon LM, Zeng XL, Blutt SE, Crawford SE, Sastri NP, Karandikar UC, Ajami NJ, Zachos NC, Kovbasnjuk O, Donowitz M, Conner ME, Shaw CA, Estes MK. 2017. A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc Natl Acad Sci U S A 114:E570–E579. doi: 10.1073/pnas.1615422114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drummond CG, Bolock AM, Ma C, Luke CJ, Good M, Coyne CB. 2017. Enteroviruses infect human enteroids and induce antiviral signaling in a cell lineage-specific manner. Proc Natl Acad Sci U S A 114:1672–1677. doi: 10.1073/pnas.1617363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B, Ding S, Feng N, Mooney N, Ooi YS, Ren L, Diep J, Kelly MR, Yasukawa LL, Patton JT, Yamazaki H, Shirao T, Jackson PK, Greenberg HB. 2017. Drebrin restricts rotavirus entry by inhibiting dynamin-mediated endocytosis. Proc Natl Acad Sci U S A 114:E3642–E3651. doi: 10.1073/pnas.1619266114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin X, Farin HF, van Es JH, Clevers H, Langer R, Karp JM. 2014. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat Methods 11:106–112. doi: 10.1038/nmeth.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pieniazek D, Pieniazek NJ, Macejak D, Coward J, Rayfield M, Luftig RB. 1990. Differential growth of human enteric adenovirus 41 (TAK) in continuous cell lines. Virology 174:239–249. doi: 10.1016/0042-6822(90)90072-Y. [DOI] [PubMed] [Google Scholar]
- 27.Pieniazek D, Pieniazek NJ, Macejak D, Luftig RB. 1990. Enteric adenovirus 41 (Tak) requires low serum for growth in human primary cells. Virology 178:72–80. doi: 10.1016/0042-6822(90)90380-A. [DOI] [PubMed] [Google Scholar]
- 28.Gaggar A, Shayakhmetov DM, Lieber A. 2003. CD46 is a cellular receptor for group B adenoviruses. Nat Med 9:1408–1412. doi: 10.1038/nm952. [DOI] [PubMed] [Google Scholar]
- 29.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 30.Sherwood V, Burgert HG, Chen YH, Sanghera S, Katafigiotis S, Randall RE, Connerton I, Mellits KH. 2007. Improved growth of enteric adenovirus type 40 in a modified cell line that can no longer respond to interferon stimulation. J Gen Virol 88:71–76. doi: 10.1099/vir.0.82445-0. [DOI] [PubMed] [Google Scholar]
- 31.Xu W, McDonough MC, Erdman DD. 2000. Species-specific identification of human adenoviruses by a multiplex PCR assay. J Clin Microbiol 38:4114–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng Y, Stamminger T, Hearing P. 2016. E2F/Rb family proteins mediate interferon induced repression of adenovirus immediate early transcription to promote persistent viral infection. PLoS Pathog 12:e1005415. doi: 10.1371/journal.ppat.1005415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bevins CL, Salzman NH. 2011. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 34.Smith JG, Silvestry M, Lindert S, Lu W, Nemerow GR, Stewart PL. 2010. Insight into the mechanisms of adenovirus capsid disassembly from studies of defensin neutralization. PLoS Pathog 6:e1000959. doi: 10.1371/journal.ppat.1000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith JG, Nemerow GR. 2008. Mechanism of adenovirus neutralization by Human alpha-defensins. Cell Host Microbe 3:11–19. doi: 10.1016/j.chom.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Gounder AP, Myers ND, Treuting PM, Bromme BA, Wilson SS, Wiens ME, Lu W, Ouellette AJ, Spindler KR, Parks WC, Smith JG. 2016. Defensins potentiate a neutralizing antibody response to enteric viral infection. PLoS Pathog 12:e1005474. doi: 10.1371/journal.ppat.1005474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson SS, Bromme BA, Holly MK, Wiens ME, Gounder AP, Sul Y, Smith JG. 2017. Alpha-defensin-dependent enhancement of enteric viral infection. PLoS Pathog 13:e1006446. doi: 10.1371/journal.ppat.1006446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das S, Yu S, Sakamori R, Vedula P, Feng Q, Flores J, Hoffman A, Fu J, Stypulkowski E, Rodriguez A, Dobrowolski R, Harada A, Hsu W, Bonder EM, Verzi MP, Gao N. 2015. Rab8a vesicles regulate Wnt ligand delivery and Paneth cell maturation at the intestinal stem cell niche. Development 142:2147–2162. doi: 10.1242/dev.121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering M, Pawson T, Clevers H. 2002. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 111:251–263. doi: 10.1016/S0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 40.van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, Thiele A, van den Born M, Begthel H, Brabletz T, Taketo MM, Clevers H. 2005. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol 7:381–386. doi: 10.1038/ncb1240. [DOI] [PubMed] [Google Scholar]
- 41.Petersen N, Reimann F, Bartfeld S, Farin HF, Ringnalda FC, Vries RG, van den Brink S, Clevers H, Gribble FM, de Koning EJ. 2014. Generation of L cells in mouse and human small intestine organoids. Diabetes 63:410–420. doi: 10.2337/db13-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mautner V, Steinthorsdottir V, Bailey A. 1995. Enteric adenoviruses, p 229–282. In Doerfler W, Böhm P (ed), The molecular repertoire of adenoviruses III. Springer, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 43.Tiemessen CT, Ujfalusi M, Kidd AH. 1993. Subgroup F adenovirus growth in foetal intestinal organ cultures. Arch Virol 132:193–200. doi: 10.1007/BF01309853. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto S, Sakakibara N, Kumai H, Nakai M, Sakuma S, Chiba S, Fujinaga K. 1991. Fastidious human adenovirus type 40 can propagate efficiently and produce plaques on a human cell line, A549, derived from lung carcinoma. J Virol 65:2429–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tiemessen CT, Kidd AH. 1992. Sensitivity of subgroup F adenoviruses to interferon. Arch Virol 128:1–13. [DOI] [PubMed] [Google Scholar]
- 46.Roelvink PW, Lizonova A, Lee JGM, Li Y, Bergelson JM, Finberg RW, Brough DE, Kovesdi I, Wickham TJ. 1998. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol 72:7909–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Bergelson JM. 2005. Adenovirus receptors. J Virol 79:12125–12131. doi: 10.1128/JVI.79.19.12125-12131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, Sato T. 2015. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med 21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 49.Takeuchi A, Hashimoto K. 1976. Electron microscope study of experimental enteric adenovirus infection in mice. Infect Immun 13:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei H, Wang S, Chen Q, Chen Y, Chi X, Zhang L, Huang S, Gao GF, Chen JL. 2014. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus infection. PLoS Pathog 10:e1003845. doi: 10.1371/journal.ppat.1003845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stoltz M, Ahlm C, Lundkvist A, Klingstrom J. 2007. Lambda interferon (IFN-lambda) in serum is decreased in hantavirus-infected patients, and in vitro-established infection is insensitive to treatment with all IFNs and inhibits IFN-gamma-induced nitric oxide production. J Virol 81:8685–8691. doi: 10.1128/JVI.00415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, Staeheli P, Hornef MW. 2011. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci U S A 108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, Nice TJ, Virgin HW. 2017. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against norovirus and reovirus. J Virol 91(7):e02079-16. doi: 10.1128/JVI.02079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen EK, Nemerow GR, Smith JG. 2010. Direct evidence from single-cell analysis that human {alpha}-defensins block adenovirus uncoating to neutralize infection. J Virol 84:4041–4049. doi: 10.1128/JVI.02471-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773–E1781. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]