

Abstract
In pursuit of fast bioorthogonal reactions, reactive moieties have been increasingly employed for selective labeling of biomolecules in living systems, posing a challenge in attaining reactivity without sacrificing selectivity. To address this challenge, here we report a bioinspired strategy in which molecular shape controls the selectivity of a transient, highly reactive nitrile imine dipole. By tuning the shape of structural pendants attached to the ortho position of the N-aryl ring of diaryltetrazoles—precursors of nitrile imines, we discovered a sterically shielded nitrile imine that favors the 1,3-dipolar cycloaddition over the competing nucleophilic addition. The photo-generated nitrile imine exhibits an extraordinarily long half-life of 102 sec in aqueous medium, owing to its unique molecular shape that hinders the approach of a nucleophile as shown by DFT calculations. The utility of this sterically shielded nitrile imine in rapid (~1 min) bioorthogonal labeling of glucagon receptor in live mammalian cells was demonstrated.
Graphical abstract
Introduction
Harnessing chemical reactivity for covalent modification of biomolecules in their native environment is currently attracting a lot of interest.1 The success of this ‘in vivo’ chemistry approach depends critically on robust bioorthogonal ligation reactions that proceed between a pair of reactants at low concentrations without cross-reactivity with the endogenous functional groups.2 Because of their concerted reaction mechanism, fast reaction kinetics, and operational simplicity, cycloaddition-based bioorthogonal ligation reactions have received the most attention. These include the azide-alkyne cycloaddition,3 the nitrone-alkyne cycloaddition,4 the inverse electron demand Diels-Alder reaction5 and the tetrazole-alkene cycloaddition.6 Analogous to the optimization of small molecule ligands with higher affinity and selectivity toward their macromolecular targets, the optimization of bioorthogonal ligation reactions, particularly their reaction rate and chemo-selectivity, i.e., bioorthogonality, is of paramount importance for their use in biological systems.
In optimizing bioorthogonal reactions, two general strategies have been developed: one uses a metal catalyst to accelerate the ligation reaction, e. g., copper-catalyzed azide-alkyne cycloaddition (CuAAC)3; the other activates substrates directly through the use of strain. Because of the well-known copper toxicity,7 recent efforts by many groups have largely focused on the design of strained cycloalkenes and cycloalkynes such as trans-cyclooctene,8 norbornene,9 cyclopropene,10 cyclooctyne,11 thiacycloheptyne,12 and bicyclo[6.1.0]nonyne (BCN).13 For example, a cyclopropane-fused trans-cyclooctene was designed by Fox and co-workers that adopts the high-energy ‘half-chair’ conformation and gives a second-order rate constant, k2, of 22,000 M−1 s−1 in the inverse electron demand Diels-Alder reaction with 3,6-di(2-pyridyl)-s-tetrazine in methanol.14 A major challenge for substrate activation strategy is that it is generally difficult to achieve high reactivity while maintain high stability and selectivity in cellular systems because these factors counteract each other in most circumstances.
We recently reported the design of strained cycloalkenes such as 3,3-disubstituted cyclopropene15 and spiro[2.3]hex-1-ene16 that show drastic rate acceleration in the photoinduced tetrazole-alkene cycloaddition reaction with k2 values approaching 10,000 M−1 s−1. Compared to other fast bioorthogonal reactions such as tetrazine ligation, the tetrazole-alkene cycloaddition requires photoactivation, which is a very attractive attribute to biological applications where the spatiotemporal control is of paramount importance. Because the photo-generated nitrile imine dipoles also exhibit electrophilic character, they are known to undergo nucleophilic thiol and water additions in the absence of a suitable dipolarophile.17,18 To harness the nitrile imine reactivity for bioorthogonal reactions in living systems, it is critical that we can selectively tune up the cycloaddition reactivity while suppress the competing nucleophilic additions. Here, we report a bioinspired strategy whereby an exquisite pendant group was identified that dramatically stabilizes the nitrile imine in aqueous medium, increases its reactivity in the desired cycloaddition reaction, suppresses the undesired nucleophilic additions, and allows robust bioorthogonal labeling of GCGR—a member of class B G protein-coupled receptors—with a small organic fluorophore in live mammalian cells.
Results and discussion
Synthesis and evaluation of sterically shielded diaryltetrazoles
To control the reaction selectivity of the in situ generated nitrile imine, we were inspired by how the bacterial enzymes UbiD/UbiX catalyze the decarboxylation step in bacterial ubiquinone biosynthesis pathway, a rare example of biocatalytic 1,3-dipolar cycloaddition involving azomethine ylide in nature.19 Crystal structures of A. niger Fdc1UbiX (Fdc1 is a fungal homolog of UbiD) revealed that the enzyme utilize a unusual isoprene ring-fused flavin mononucleotide (prFMN) cofactor for this transformation.20 In addition to providing the necessary azomethine ylide, the extra isoprene ring may prevent the competing hydride and carbanion addition to the isoalloazine ring21 through steric effect. Inspired by Nature’s elaborate design, we envisioned that the reactivity of the nitrile imine dipole can be directed through adjacent structural pendants away from the nucleophilic addition and toward the desired 1,3-dipole cycloaddition (Scheme 1).
Scheme 1.

Strategy of employing structural pendants to stabilize the in situ generated nitrile imine and direct the reaction toward 1,3-dipolar cycloaddition over the competing nucleophilic addition.
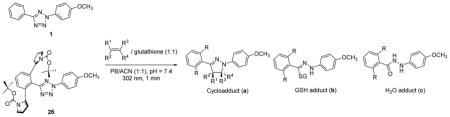
A series of ortho-substituted diphenyltetrazoles were synthesized (Scheme S1-S8 in SI) and their reactivities were assessed in an HPLC-based competition assay using equal amount of excess acrylamide and glutathione (GSH) in mixed acetonitrile/phosphate buffer (Scheme 2 and Table S1). In principle, the in situ photo-generated nitrile imines can produce four different adducts: (i) pyrazoline a from 1,3-dipolar cycloaddition, (ii) hydrazone b from nucleophilic GSH addition, (iii) hydrazide c from water quenching and subsequent tautomerisation, and (iv) adduct d from intramolecular nucleophilic addition (Table 1 scheme). Four series of tetrazoles (Scheme 2) were evaluated in this assay: (i) mono-aliphatic substituted tetrazoles 2–10; (ii) mono-methoxymethyl-substituted tetrazoles 11–14; (iii) dimethyl, dimethoxy, and dimethoxymethyl-substituted tetrazoles 15–17; and (iv) monoaryl and diaryl-substituted tetrazoles 18–29. The di(o-2′-N-Boc-pyrrole)-substituted tetrazole 26 showed the highest cycloaddition selectivity with a:b:c:d ratio of 92:2:6:0 among all the tetrazole analogs (Table S1). Both the N-substituent and the proximity to the nitrile imine center appear to be important as lack of either feature resulted in erosion of this high selectivity (compare tetrazole 26 to 27–29 in Table S1).
Scheme 2.

Potential products from the substituted tetrazoles under the competitive conditions. Four types of substituted tetrazoles were evaluated in this competition assay; see Table S1 in SI for structural details. PB = phosphate buffer; ACN = acetonitrile.
Table 1.
Product distribution and reaction kinetic of tetrazole 26 vs. 1 in 1,3-dipolar cycloaddition reaction with the various alkene dipolarophilesa
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Dipolarophile | Tetrazole 1 | Tetrazole 26 | ||||||
| Product distribution (%) | k2 | Product distribution (%) | k2 | ||||||
| a | b | c | M−1 s−1 | a | b | c | M−1 s−1 | ||
| 1 |
|
47 | 51 | 2 | 2,000 ± 300 | 92 | 2 | 6 | 860 ± 110 |
| 2 |
|
95 | 5 | 0 | 4,700 ± 500 | 99 | 0 | 1 | 2,400 ± 300 |
| 3 |
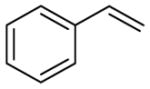
|
5 | 91 | 4 | NA b | 96 | 2 | 2 | 1,000 ±100 |
| 4 |
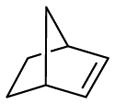
|
20 | 75 | 5 | 2,200 ± 700 | 89 | 3 | 8 | 1,000 ± 250 |
| 5 |
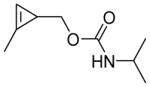
|
0 | 93 | 7 | NA b | 85 | 6 | 9 | 520 ± 140 |
| 6 |
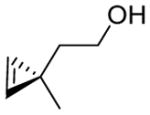
|
4 | 90 | 6 | 2,800 ± 300 | 86 | 2 | 12 | 1,400 ± 130 |
| 7 |
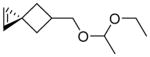
|
62 | 35 | 3 | 3,100 ± 600 | 96 | 0 | 4 | 2,800 ± 200 |
| 8 |
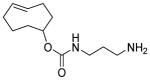
|
60 | 37 | 3 | 3,800 ± 700 | 97 | 0 | 3 | 2,300 ± 300 |
For determination of product distribution, a solution of 25 μM tetrazole and 500 μM each of alkene dipolarophile and glutathione (reduced form; GSH) in 500 μL phosphate buffer/acetonitrile (1:1), pH 7.4, was photoirradiated with a handheld 302-nm UV lamp at room temperature for 1 min. The product distribution was analyzed by reverse-phase HPLC with absorbance set at 254 nm. For kinetic measurement, a mixture of 10 μM tetrazole and 100 μM alkene dipolarophile (50 μM for dimethyl fumarate, Sph, and TCO) in 1 mL phosphate buffer/acetonitrile (1:1), pH 7.4, in a quartz cuvette was photoirradiated with a handheld 302-nm UV lamp at room temperature for a period of 30–40 s, and the reactions were monitored by a fluorimeter. The measurements were repeated three times to derive the mean and standard deviation.
NA = not available because pyrazoline fluorescence was not detected under kinetics conditions.
Reactivity comparison of tetrazole 26 versus tetrazole 1
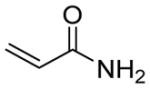
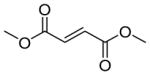
With the highly selective tetrazole 26 in hand, we assessed its reactivity toward a range of alkene dipolarophiles (Table 1). Compared to tetrazole 1, the sterically shielded tetrazole 26 displayed outstanding selectivity (92–99%) toward the conjugated alkenes including acrylamide, dimethyl fumarate and styrene, as well as the highly strained cycloalkenes including spiro[2.3]hex-1-ene (Sph) and trans-cyclooctene (TCO). For these reactive alkenes, GSH adducts were either undetectable or negligible. A reversal of selectivity was observed for less reactive alkenes including norbornene, 1,3- and 3,3-disubstituted cyclopropenes (entries 4–6 in Table 1). The selectivity for photoinduced cycloaddition with tetrazole 26 is robust as the reaction with Sph proceeded in the presence of 10 mM glutathione, 10 mM glutamic acid, or a mixture of nucleophilic amino acids (Figs. S1–S3). Kinetic measurements revealed that the second-order rate constants are lower for tetrazole 26 than those of tetrazole 1 by about two-fold (Table 1 and Figs. S4–S11), suggesting that the increased selectivity for cycloaddition with tetrazole 26 is due to the greater inhibition of GSH addition. For styrene and 1,3-cyclopropene, the cycloadducts were either negligible or non-detectable with tetrazole 1, but became dominant with tetrazole 26 when nucleophilic GSH addition is inhibited (entries 3 and 5 in Table 1). Interestingly, the amount of water adducts remained low and essentially unchanged for all alkene dipolarophiles, indicating that the steric shielding does not suppress the water-quenching process as effectively as the GSH addition presumably due to the small size of water.
Effect of steric shielding on transition state energies
To understand how the steric shield influences the reaction selectivity of the in situ generated nitrile imine, we computed the transition states of the 1,3-dipolar cycloaddition and the nucleophilic addition reactions involving the sterically shielded nitrile imine derived from tetrazole 26 and the unhindered nitrile imine from 1 using density functional theory (Figure 1). In these calculations, 5-methyl-spiro[2.3]hex-1-ene and methyl thiolate were used as the alkene dipolarophile and a model nucleophile, respectively. The 1,3-dipolar cycloadditions of 5-methyl-spiro[2.3]hex-1-ene to the unhindered nitrile imine (TS1) and to the sterically shielded nitrile imine (TS2) occur in a concerted manner. The N-Boc-pyrrole substitution led to increases of activation free energy (ΔG‡) and activation enthalpy (ΔH‡) of the cycloaddition, by 2.2 kcal/mol and 0.4 kcal/mol, respectively. On the other hand, the reactivity difference between the nucleophilic addition of methyl thiolate22 to the unhindered nitrile imine (TS3) and to the sterically hindered nitrile imine (TS4) is much greater (ΔΔG‡ = 5.2 kcal/mol; ΔΔH‡ = 2.8 kcal/mol). In TS4, the N-Boc-pyrrole group perpendicular to the C-phenyl ring effectively creates a steric shield surrounding the electrophilic side of nitrile imine, and therefore blocks the approach of the methyl thiolate nucleophile. As a result, the activation energies for the nucleophilic addition reactions increase from 12.2 kcal/mol for TS3 to 17.4 kcal/mol for TS4. This staggering 5.2 kcal/mol increase in ΔG‡ corresponds to >6000-fold reduction in reaction rate, which is consistent with the low reactivity to form the GSH adduct in reactions with tetrazole 26. The greater sensitivity to steric shielding effect in the nucleophilic addition compared with cycloaddition is attributed to the preferred orientation of the nucleophile, which places the methyl thiolate in close proximity to the C-phenyl ring on the nitrile imine (see TS3 and Fig. S12). In TS4, the bulky N-Boc-pyrrole substitution forces the methyl thiolate to adopt the higher energy orientation. Together, these computational studies indicate that the steric shield formed by the two N-Boc-pyrrole groups in tetrazole 26 causes a dramatic slowdown of the competing thiolate addition, thereby shifting the selectivity toward the 1,3-dipolar cycloaddition (Scheme 1).
Figure 1. Computational studies of the steric shielding effect of the ortho N-Boc-pyrrole substituents.

Computed transition states and activation energies involving nitrile imine-1 and -26 for (a) 1,3-dipolar cycloaddition with 5-methyl-spiro[2.3]hex-1-ene, and (b) nucleophilic addition with methyl thiolate, respectively. DFT calculations were performed at the ωB97x-D/6-311++G(d,p)/SMD(Water) level of theory using geometries optimized by B3LYP-D3/6-31+G(d)/SMD(Water). R = N-Boc. The distances shown are in Å, and energies are in kcal/mol.
Structures of tetrazoles in solid state and in solution
To corroborate computational results, we solved the crystal structures of the sterically shielded tetrazole 26 and its analog 27 (Tables S3 and S4). Figure 2b shows that the pyrrole rings in 26 are nearly perpendicular to the C-phenyl ring with the t-butyl group at one N-Boc-pyrrole extending to the other pyrrole ring. The closest distance between the H1 of t-butyl and H4 of the other pyrrole is 3.1 Å (Figure 2b). This type of unusual side contact generates a continuous bowl-shaped shield on the C-aryl side of the tetrazole, similar to what we observed in TS2. In contrast, when the Boc groups are removed as in tetrazole 27, the pyrrole rings are coplanar with the C-phenyl ring, forcing the tetrazole ring to twist out of the co-planarity with the C-phenyl ring (Figure 2d). The collapse of the three-dimensional bowl-shaped shield likely causes the destabilization of nitrile imine-27, leading to predominant water quenching (Table S1). To probe whether the steric shield exists in solution, we performed a 2D NMR experiment with tetrazole 26 in CDCl3. The proton signals of tetrazole 26 were assigned based on the 1H-1H COSY spectrum (Fig. S13). The spatial proximity between two pyrrole rings was assessed by examining the NOE effect. Strong NOE cross-peaks were detected between H1 of one pyrrole, and H4 of the other pyrrole, and between H1 and H5 of the C-phenyl ring in the ROESY spectrum (Figure 2e), indicating the steric shield remains intact in solution.
Figure 2. Structural characterization of tetrazoles 26 and 27.

(a) Chemical structure of tetrazole 26 with the proton numbering marked on the structure. (b) X-ray crystal structure of tetrazole 26 with depth cue showing co-planarity of the diphenyltetrazole (left) and perpendicular arrangement of N-Boc-pyrrole relative to the diphenyltetrazole (right). CW, clockwise. (c) Chemical structure of tetrazole 27. (d) Crystal structure of 27 with depth cue showing co-planarity between the C-phenyl and pyrrole rings, and perpendicular twisting of the tetrazole and the C-phenyl ring. The structures are viewed from the same angles as in tetrazole 26. (e) Partial 1H-1H ROESY spectrum of tetrazole 26 in CDCl3 showing prominent cross-peaks between tert-butyl protons and proton-4, -5 and -7 as marked on the chemical structure.
Stability of nitrile imine-26 in aqueous medium
Because the in situ generated nitrile imines also undergo water-quenching, we compared the water-quenching rate of nitrile imine-26 to that of nitrile imine-1 by subjecting the corresponding tetrazole to a 15-sec photoirradiation at 302 nm and quantifying the residual nitrile imines post-photoirradiation by intercepting them with excess dimethyl fumarate. To our surprise, despite complete photoinduced tetrazole ring rupture we could not detect residual nitrile imine-1 under these conditions (Fig. S14b, c), suggesting that the half-life of nitrile imine-1 is less than 7.5 sec. In contrast, the half-life of nitrile imine-26 was determined to be 102 sec (Fig. S14d, e), reminiscent of the sterically crowded 2,5-ditritylnitrilimine reported in the literature.23 To probe structural basis of the extraordinarily long half-life of nitrile imine-26, we also examined the nitrile imine derived from Moc-pyrrole-tetrazole 28 and found its half-life to be 21 sec (Fig. S14f, g), 5-times shorter than that of nitrile imine-26, indicating that the Boc groups are crucial to the formation of an effective steric shield. The reduced protection from the less bulky Moc groups may also explain the modest selectivity for the cycloaddition with tetrazole 28 (Table S1).
In situ generation of QBP conformation sensor
Because the photoinduced cycloaddition reaction produces an environmentally sensitive pyrazoline fluorophore24–26 whose emission intensity increases when placed in less polar solvents (Fig. S15), we investigated whether tetrazole 26 is capable of generating a fluorescent sensor in situ for studying protein conformational dynamics. To this end, we selected glutamine binding protein (QBP) because its active site closes after binding to glutamine with reduced polarity near the binding cleft.27–29 Indeed, environmentally sensitive probes have been installed at Asn160 position to sense this conformational transition by fluorescence.30 Accordingly, we expressed the QBP mutants encoding SphK at position-160 and reacted this mutant with a water-soluble tetrazole 30 (Figure 3a). In-gel fluorescence analysis revealed the pyrazoline band for QBP-N160SphK but not for the control mutant QBP-N160BocK, indicating that the cycloaddition-mediated pyrazoline formation is highly specific (Fig. S16). Upon titration with glutamine, the QBP-pyrazoline fluorescence showed concentration-dependent increases with a maxima reached when the glutamine concentration was 10 mM (Figure 3b), indicating reduced polarity of the environment surrounding the pyrazoline fluorophore following the closure of the QBP active site. As a control, the small-molecule pyrazoline did not show the glutamine-dependent fluorescence change (Fig. S15c). Fitting the data to a one-site specific binding model gave a dissociation constant, Kd, of 3.3 mM for glutamine (Figure 3c), significantly higher than the reported Kd values for other fluorophore-modified QBP-N160C mutants (Kd = 0.3 ~ 6.6 μM),28 presumably due to perturbation by the larger pyrazoline. Nevertheless, the result demonstrates that the sterically shielded nitrile imine is capable of generating a fluorescent sensor in situ for monitoring protein conformational change.
Figure 3. Application of the sterically shielded nitrile imine to generate in situ a fluorescent sensor for probing ligand-induced QBP conformational change.

(a) Scheme for in situ synthesis of a pyrazoline-based sensor from QBP-N160SphK mutant and a water-soluble tetrazole 30. The glutamine-free QBP (PDB code: 1GGG) adopts an open conformation27 while the glutamine-bound QBP (PDB code: 1WDN) adopts a closed conformation29. (b) Fluorescence spectra of QBP-pyr-30 upon titration of glutamine (final concentration = 0.1 mM, 1 mM, 5 mM, 10 mM or 15 mM in 4:1 DPBS/acetonitrile). λex = 405 nm; QBP concentration was set at 5 μM. (c) Plot of the change of fluorescence intensity at 480 nm vs. glutamine concentration. The data were fitted to one-site specific binding model with the following equation: ΔFobs = ΔFmaxS/(Kd + S).
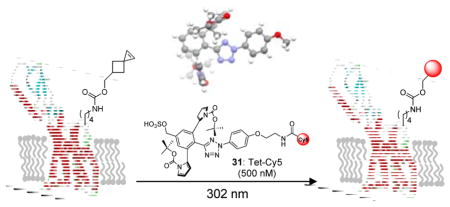
Fluorescent labeling of GCGR in live mammalian cells
To probe whether the sterically shielded nitrile imine reacts selectively with an alkene-encoded protein in cellular environment, we synthesized a Cy5-conjugated tetrazole 31 and verified its selective reactivity in fluorescent labeling of the Sph-encoded sfGFP in bacterial cell lysates (Fig. S17). We then investigated the efficiency of tetrazole 31 in fluorescent labeling of a glucagon receptor (GCGR) mutant encoding SphK at position-372 in mammalian cells. We chose GCGR because: (1) it is a member of class B G protein-coupled receptor family and a validated drug target for diabetes;31,32 (2) the study of GCGR activation is crucial to the design of GCGR antagonists;33 and (3) bioorthogonal fluorescent labeling of GCGR via tetrazine ligation was reported recently.34 Accordingly, HEK 293T cells expressing GCGR-H372SphK-GFP was treated with 500 nM tetrazole 31 followed by a brief exposure to 302-nm photoirradiation (Figure 4a). Strong Cy5 fluorescence was detected at the plasma membrane by confocal microscopy as early as 30 sec with maximum intensity reached at 1 min (Figure 4b). The Cy5 fluorescence co-localized with that of GFP, indicating that the tetrazole 31 mediated GCGR labeling is highly specific. For comparison, HEK 293T cells expressing GCGR-H372SphK-GFP were similarly labelled with a Cy5-conjugated tetrazine,34 though a higher tetrazine concentration (1 μM) and longer incubation time (30 min) were needed to reach maximum fluorescence (Fig. S18). Based on fluorescence-activated cell sorting, 89% of GCGR-H372SphK-GFP-expressing cells were labelled by tetrazole 31 after 1 min (Figure 4c–e; Fig. S19). The labeling specificity is particularly striking as cells without expression of GCGR-GFP show negligible Cy5 signals (Figure 4b, 4e). Together, the confocal and FACS analysis results confirmed that the sterically shielded tetrazole offers a new class of reagents for fast (~1 min) and highly specific bioorthogonal labeling of membrane proteins with a small organic fluorophore in live mammalian cells.
Figure 4. Application of the stabilized nitrile imine to bioorthogonal labeling of GCGR in live cells.

(a) Scheme for bioorthogonal labeling of a GCGR-GFP mutant encoding SphK at position-372 with a Cy5-funtionalised water-soluble tetrazole 31. The C-terminal GFP is not shown for clarity. (b) Confocal micrographs of HEK 293T cells expressing GCGR-H372SphK-GFP after photoirradiation with a handheld 302-nm UV lamp for a period of 0.5, 1, or 2 min in DMEM medium containing 500 nM tetrazole-Cy5. Scale bar = 20 μm. Flow cytometry analysis of HEK 293T cells expressing (c) GCGR-H372SphK, (d) GCGR-H372SphK-GFP before labeling, and (e) GCGR-H372SphK-GFP after labeling with tetrazole 31. Cells were treated with 500 nM of tetrazole 31 and photoirradiated for 1 min. A total of 10,000 cells were analyzed in each measurement. X-axis, GFP channel; y-axis, Cy5 channel; both axes are in log scale.
In summary, we have adopted a bioinspired strategy in the design of sterically shielded tetrazoles for bioorthogonal photoinduced cycloaddition with a suitable alkene dipolarophile. The photo-generated nitrile imine from the sterically shielded tetrazole 26 exhibited dramatically enhanced selectivity for the cycloaddition over the nucleophilic addition. The DFT calculations and structural studies showed that the N-Boc-pyrrole groups adjacent to the in situ generated nitrile imine provide a steric shield to prevent the thiolate addition by raising the corresponding activation energy. Indeed, the photoactivation–interception study of the in situ generated, sterically shielded nitrile imine revealed a remarkably long half-life of 102 sec, compared to less than 7.5 sec for the unshielded parent nitrile imine. Because the tetrazole-alkene cycloaddition produces an environmentally sensitive pyrazoline fluorophore, the sterically shielded tetrazole 26 was used to successfully generate in situ a fluorescent sensor for monitoring ligand-induced conformational transition of QBP. Due to its fast kinetics and extraordinary selectivity, the sterically shielded nitrile imine was also utilized to fluorescently label an alkene-encoded GCGR in live mammalian cells at a concentration as low as 500 nM within 1 min, which is significantly faster than the optimized tetrazine reagent. While the present work focuses on the N-aryl ring of the 2,5-diaryltetrazoles for enhanced selectivity, we expect the half-life, reactivity, and selectivity of this class of sterically shielded nitrile imines can be further tuned through optimization of the C-aryl rings26,35 for their expanded use in chemical biology. In addition, the use of molecular shape to control reaction selectivity could provide a general strategy for optimization of other cycloaddition-based bioorthogonal reactions.
Experimental Section
Expression and purification of SphK or BocK-encoded QBP
BL21(DE3) competent cells were transformed with the pETtrio-QBP160TAG and pEVOL-TCOKRS (for SphK) or pEVOL-PylT-PylRS (for BocK) plasmids. The transformants were recovered in 1 mL SOC medium at 37 °C for 1 h before plating onto LB agar plate containing 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. A single colony was picked and allowed to grow in 5 mL LB medium supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol at 37 °C overnight. This starter culture was used to inoculate 25 mL LB medium supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Cells were allowed to grow at 37 °C with shaking at 280 rpm until OD600 reached 0.6~0.8. Protein expression was then initiated by adding 1 mM IPTG, 0.2% arabinose, and 1 mM SphK or BocK. After 6 hours of incubation, cells were harvested by centrifugation, resuspended in binding buffer (300 mM NaCl, 50 mM Na2HPO4, 10 mM imidazole, pH 8.0), and lysed by sonication. The lysate was clarified by centrifugation. Afterwards, 150 μL Ni-NTA beads (Thermo Scientific) was added to the supernatant and the mixture was rocked at room temperature for 2 h. Beads were collected by centrifugation and washed with washing buffer (300 mM NaCl, 50 mM Na2HPO4, 50 mM imidazole, pH 8.0). The bound proteins were eluted with elution buffer (300 mM NaCl, 50 mM Na2HPO4, 250 mM imidazole, pH 8.0). A buffer exchange was then carried out with DPBS using a 10 kD MWCO spin column (Thermo Scientific). The protein mass was determined by deconvoluting the charge ladder in the LC-MS analysis. The protein concentration was determined by the BCA assay.
Measurement of ligand-dependent QBP-pyrazoline fluorescence change
A concentrated solution of QBP-N160SphK and tetrazole 30 (1 mM dissolved in DMSO) were added to 50 μL of DPBS/ACN (4:1) to obtain a final protein concentration of 10 μM and tetrazole concentration of 20 μM. The mixture was irradiated with a handheld 302 nm UV light for 1 min. An equal volume of L-glutamine solution in DPBS/ACN (4:1) (concentration = 200 μM, 2 mM, 10 mM, 20 mM, or 30 mM) was added to the mixture, and the fluorescence spectra were measured using Horiba FluoroMax-4 spectrofluorometer at 25 °C with the excitation wavelength set at 405 nm.
HEK 293T cell culture and transfection
HEK 293T cells were maintained in a growth medium containing Dulbecco’s modified eagle medium (DMEM, Life Technologies) supplemented with 10% (v/v) fetal bovine serum (FBS, Life Technologies) and 10 μg/mL gentamicin (Life Technologies). Transfection was performed at 70–80% confluency using 3:1 reagent/DNA ratio of Lipofectamine 2000 (Life Technologies) with 2.5 μg total DNA or polyethylenimine (PEI, Polysciences Inc.) with 3 μg total DNA per 35-mm dish. For imaging experiments, cells were maintained in 35-mm glass bottom culture dishes using FluoroBrite™ DMEM medium (Life Technologies) supplemented with 10% FBS, 4 mM L-glutamine, and 25 mM HEPES.
Bioorthogonal labeling of SphK-encoded GCGR-GFP and confocal microscopy
HEK 293T cells were co-transfected with 4:1 ratio of pCMV-MmPylRS-U6-tRNA and pCMV-GCGR-H372TAG-GFP plasmids. A solution of 100 mM SphK in DMSO was diluted in growth medium to obtain a final concentration of 1 mM SphK and the resulting medium was filtered through 0.2 μm polyethersulfone membrane. The medium was added to the cells followed by the transfection mixture. The cells were incubated for additional 24–48 h, and then washed with the growth medium prior to labeling. A fresh solution of tetrazole 31 and DpTz-Cy5 was prepared from 1 mM stock solution in DMSO to a final concentration of 500 nM and 1 μM, respectively, in 1 mL growth medium. For the photoclick chemistry mediated labeling, HEK 293T cells expressing GCGR-H372SphK-GFP were treated with 500 nM tetrazole 31 in 1 mL DMEM medium containing 10% FBS, and the culture was exposed to 302 nm UV light for a brief period (30 sec, 1 min, or 2 min). The tetrazole-containing medium was removed and the cells were washed with growth medium before incubation in 1 mL growth medium at 37 °C and 5% CO2 for additional 30 min. For the tetrazine ligation mediated labeling, HEK 293T cells expressing GCGR-H372SphK-GFP were treated with 1 μM DpTz-Cy5 in 1 mL DMEM medium containing 10% FBS, and the culture was incubated at 37 °C and 5% CO2 for various time (5 min, 10 min, 30 min, or 60 min). The DpTz-Cy5 containing medium was removed, and the cells were washed with growth medium before incubation at 37 °C and 5% CO2 for additional 30 min. The medium was then switched to FluoroBrite™ DMEM before laser scanning confocal microscopy. The confocal images were acquired using a Zeiss LSM 710 equipped with Plan-Apochromat 20×/0.8 M27 or 40×/1.3 Oil DIC M27 objective with ex. 488/em. 493–598 nm for GFP channel and ex. 640/em. 645–759 nm for Cy5 channel.
Flow cytometry analysis of fluorescent labelled HEK 293T cells
HEK 293T cells expressing GCGR-H372SphK, GCGR-H372SphK-GFP, or Cy5-labeled GCGR-H372SphK-GFP (~106 cells each) were detached from the culture dish using trypsin and washed successively with PBS (2×) and FACS buffer (DPBS, 1% BSA, 0.03% NaN3; 2×) at room temperature. The cells were resuspended in FACS buffer, and 10,000 cells were recorded in each fluorescence activated cell sorting.
Computational Details
All calculations were carried out with the Gaussian 09 program package. Geometry optimizations were performed with the B3LYP functional with Grimme’s DFT-D3 dispersion correction, the 6-31+G(d) basis set, and the SMD solvation model with water as the solvent. Frequency analysis was conducted at the same level of theory to verify the stationary points to be minima or saddle points and to obtain zero-point energy (ZPE) and thermal energy corrections at 298.15 K. Single-point energy calculations on the B3LYP-D3-optimized geometries were performed with the ωB97XD functional, using 6-311++G(d,p) basis set and the SMD solvation model with water as the solvent. Computed structures are illustrated using CYLView.
Supplementary Material
Acknowledgments
We gratefully acknowledge the NIH (R01GM085092) for generous financial support. We thank Prof. Wenshe Liu at Texas A&M University for providing the pETtrio-1-AcKRS-pylT(TAA)-QBP141TAA plasmid, Alan Siegel at SUNY Buffalo North Campus Imaging Facility for assistance with confocal microscopy, Alan Connor at SUNY Buffalo for assistance with 2D NMR analysis, and Alan Friedman at SUNY Buffalo School of Engineering and Applied Sciences Instrumentation Center for assistance with high-resolution mass spectrometry.
Footnotes
Supplemental figures, tables and methods, synthetic schemes, characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Boutureira O, Bernardes GJ. Chem Rev. 2015;115:2174–2195. doi: 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- 2.Lang K, Chin JW. Chem Rev. 2014;114:4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 3.Meldal M, Tornoe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 4.MacKenzie DA, Sherratt AR, Chigrinova M, Cheung LL, Pezacki JP. Curr Opin Chem Biol. 2014;21:81–88. doi: 10.1016/j.cbpa.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 5.Oliveira BL, Guo Z, Bernardes GJL. Chem Soc Rev. 2017;46:4895–4950. doi: 10.1039/c7cs00184c. [DOI] [PubMed] [Google Scholar]
- 6.Ramil CP, Lin Q. Curr Opin Chem Biol. 2014;21:89–95. doi: 10.1016/j.cbpa.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennedy DC, McKay CS, Legault MCB, Danielson DC, Blake JA, Pegoraro AF, Stolow A, Mester Z, Pezacki JP. J Am Chem Soc. 2011;133:17993–18001. doi: 10.1021/ja2083027. [DOI] [PubMed] [Google Scholar]
- 8.Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Nat Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J Am Chem Soc. 2012;134:18638–18643. doi: 10.1021/ja3060436. [DOI] [PubMed] [Google Scholar]
- 11.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 12.de Almeida G, Sletten EM, Nakamura H, Palaniappan KK, Bertozzi CR. Angew Chem Int Ed. 2012;51:2443–2447. doi: 10.1002/anie.201106325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dommerholt J, Schmidt S, Temming R, Hendriks LJ, Rutjes FP, van Hest JC, Lefeber DJ, Friedl P, van Delft FL. Angew Chem Int Ed. 2010;49:9422–9425. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor MT, Blackman ML, Dmitrenko O, Fox JM. J Am Chem Soc. 2011;133:9646–9649. doi: 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Z, Pan Y, Wang Z, Wang J, Lin Q. Angew Chem Int Ed. 2012;51:10600–10604. doi: 10.1002/anie.201205352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Z, Lin Q. J Am Chem Soc. 2014;136:4153–4156. doi: 10.1021/ja5012542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahra JA, Abu Thaher BA, El-Abadelah MM, Boese R. Org Biomol Chem. 2003;1:822–825. doi: 10.1039/b211458p. [DOI] [PubMed] [Google Scholar]
- 18.Herner A, Marjanovic J, Lewandowski TM, Marin V, Patterson M, Miesbauer L, Ready D, Williams J, Vasudevan A, Lin Q. J Am Chem Soc. 2016;138:14609–14615. doi: 10.1021/jacs.6b06645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Payne KA, White MD, Fisher K, Khara B, Bailey SS, Parker D, Rattray NJ, Trivedi DK, Goodacre R, Beveridge R, Barran P, Rigby SE, Scrutton NS, Hay S, Leys D. Nature. 2015;522:497–501. doi: 10.1038/nature14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh CT, Wencewicz TA. Nat Prod Rep. 2013;30:175–200. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baunach M, Hertweck C. Angew Chem Int Ed. 2015;54:12550–12552. doi: 10.1002/anie.201507120. [DOI] [PubMed] [Google Scholar]
- 22.The activation free energies and enthalpies of TS3 and TS4 were calculated with respect to the nitrile imine and the methyl thiolate. Since the deprotonated methyl thiolate anion represents a more reactive nucleophile than glutathione, the computed activation free energies of TS3 and TS4 are both expected to be lower than those of the thiolate additions with glutathione. However, the use of methyl thiolate should have a smaller impact on the relative reactivity (ΔΔG‡) of the two nitrile imines in thiolate addition reactions.
- 23.Réau R, Veneziani G, Dahan F, Bertrand G. Angew Chem Int Ed. 1992;31:439–440. [Google Scholar]
- 24.Fahrni CJ, Yang L, VanDerveer DG. J Am Chem Soc. 2003;125:3799–3812. doi: 10.1021/ja028266o. [DOI] [PubMed] [Google Scholar]
- 25.Yu Z, Ohulchanskyy TY, An P, Prasad PN, Lin Q. J Am Chem Soc. 2013;135:16766–16769. doi: 10.1021/ja407867a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.An P, Yu Z, Lin Q. Chem Commun. 2013;49:9920–9922. doi: 10.1039/c3cc45752d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsiao CD, Sun YJ, Rose J, Wang BC. J Mol Biol. 1996;262:225–242. doi: 10.1006/jmbi.1996.0509. [DOI] [PubMed] [Google Scholar]
- 28.Wada A, Mie M, Aizawa M, Lahoud P, Cass AE, Kobatake E. J Am Chem Soc. 2003;125:16228–16234. doi: 10.1021/ja036459l. [DOI] [PubMed] [Google Scholar]
- 29.Sun YJ, Rose J, Wang BC, Hsiao CD. J Mol Biol. 1998;278:219–229. doi: 10.1006/jmbi.1998.1675. [DOI] [PubMed] [Google Scholar]
- 30.Lee HS, Guo J, Lemke EA, Dimla RD, Schultz PG. J Am Chem Soc. 2009;131:12921–12923. doi: 10.1021/ja904896s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wootten D, Miller LJ, Koole C, Christopoulos A, Sexton PM. Chem Rev. 2017;117:111–138. doi: 10.1021/acs.chemrev.6b00049. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Qiao A, Yang D, Yang L, Dai A, de Graaf C, Reedtz-Runge S, Dharmarajan V, Zhang H, Han GW, Grant TD, Sierra RG, Weierstall U, Nelson G, Liu W, Wu Y, Ma L, Cai X, Lin G, Wu X, Geng Z, Dong Y, Song G, Griffin PR, Lau J, Cherezov V, Yang H, Hanson MA, Stevens RC, Zhao Q, Jiang H, Wang MW, Wu B. Nature. 2017;546:259–264. doi: 10.1038/nature22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian H, Fürstenberg A, Huber T. Chem Rev. 2017;117:186–245. doi: 10.1021/acs.chemrev.6b00084. [DOI] [PubMed] [Google Scholar]
- 34.Ramil CP, Dong M, An P, Lewandowski TM, Yu Z, Miller LJ, Lin Q. J Am Chem Soc. 2017;139:13376–13386. doi: 10.1021/jacs.7b05674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Song W, Hu WJ, Lin Q. Angew Chem Int Ed. 2009;48:5330–5333. doi: 10.1002/anie.200901220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.