

Abstract
Popular bioadhesives, such as fibrin, cyanoacrylate and albumin–glutaraldehyde based materials, have been applied for clinical applications in wound healing, drug delivery, and bone and soft tissue engineering; however, their performances are limited by weak adhesion strength and rapid degradation. We developed a mussel-inspired, nanocomposite–based, biodegradable tissue adhesive by blending poly(lactic-co-glycolic acid) (PLGA) or N-hydroxysuccinimide modified PLGA nanoparticles (PLGA-NHS) with mussel-inspired alginate-dopamine polymer (Alg-Dopa). Adhesive strength measurement of the nanocomposites on porcine skin-muscle constructs revealed that the incorporation of nanoparticles in Alg-Dopa significantly enhanced the tissue adhesive strength compared to the mussel-inspired adhesive alone. The nanocomposite formed by PLGA-NHS nanoparticles showed higher lap shear strength of 33 ± 3 kPa, compared to that of Alg-Dopa hydrogel alone (14 ± 2 kPa). In addition, these nanocomposites were degradable and cytocompatible in vitro, and elicited in vivo minimal inflammatory responses in a rat model, suggesting clinical potential of these nanocomposites as bioadhesives.
Keywords: bioadhesives, nanoparticles, nanocomposites, tissue interfaces, tissue adhesion
Graphical abstract
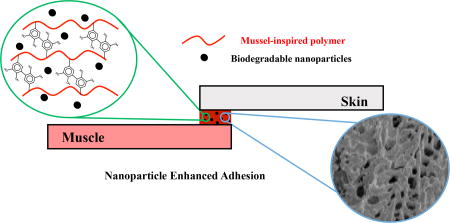
Blending biodegradable nanoparticles into mussel-inspired biodegradable adhesives significantly enhances the adhesive strength between two tissues. The biodegradable nanocomposite adhesive exhibit good cell compatibility in vitro and good tissue compatibility in vivo. It can serve as a safe and strong tissue glue for surgery and tissue repair.
1. Introduction
Tissue adhesives in biomedical research are being developed to replace surgical suturing and stapling of tissues that are more invasive in nature, sometimes require secondary surgeries for removal, cause infections in some cases, and damage to surrounding tissues during the procedure.[1] These adhesives are mainly employed as glues to hold tissues together, as hemostatic agents to control or stop bleeding, or as sealants to close tissue openings or defects.[1, 2] Many of these adhesives are made from either biologically derived or synthetic materials. Biologically derived fibrin glues mimicking the last stage of the physiological coagulation cascade, and synthetic cyanoacrylates are currently the most used tissue adhesives. However, despite having a short curing time and being biodegradable, fibrin glue is not a strong tissue adhesive and has the potential to cause immunogenic responses in the host. The cyanoacrylate based adhesives can cause tissue toxicity although they demonstrate higher adhesive strength and slow degradation behavior over time.[3, 4]
Several approaches have been developed to improve the adhesion strength of bioadhesive in a wet environment. One strategy was to utilize conjugating amino acids onto the backbones of carbohydrate or polyester based polymers to enhance adhesion properties[5]. Other efforts have focused on biomimetic approaches, such as gecko-inspired adhesives[6] or spider silk based adhesives[7] which mimick the nanotopography of gecko limbs or spider silk to adhere to substrates. The gecko or spider silk inspired adhesives, however, only show good adhesion under static and dry conditions.[6, 7] The adhesion strategy employed by some marine creatures such as the blue mussel Mytilus edulis [5, 8–10] is a popular biomimetic approach being currently exploited for synthesizing tissue adhesives.[11–13] [8, 14–16] The strong adhesion ability of the mussels is attributed to a catechol containing amino acid called L-3,4-dihydroxyphenylalanine (L-Dopa) found in the structure of secreted mussel adhesive foot proteins.[9] L-Dopa is responsible for various bonding interactions in the form of bidentate hydrogen bonding, π-π interactions and complex formation with metal and metal oxides and oxidative cross-linking via oxidation to dopa-quinone which have been pointed out as key mediators of mussel adhesion.[17] Mussel-inspired adhesives incorporating either L-Dopa or dopamine onto polymeric backbones have thus being synthesized as tissue adhesives with low to moderate adhesiveness on tissues.[16, 18–26] The major limitations of such mussel-inspired adhesives are that the mechanisms underlying L-Dopa mediated adhesion remain unclear and the chemical nature of the mussels adherence to inorganic and organic substrates has not been fully characterized [17].
Additionally, it has been shown that silica nanoparticles can be used to glue non-adhering gels, with possible applications to glue tissues in vivo.[27] These nanoparticles increased the adhesion of tissues in vivo via their ability to adsorb onto polymer chains and form bridges between two connecting structures. Nanoparticles may also offer excellent potential to function as mediators of adhesion between two surfaces because of their small sizes and high aspect ratios. Thus, we hypothesized that if the nanoparticles and mussel- inspired hydrogels were combined, the nanocomposite might exhibit synergistic effects and achieve a high adhesive strength in a wet environment. Furthermore, because the silica is non-degradable, we selected biodegradable polymer nanoparticles, such as poly(lactide-co-glycolide) (PLGA). Until now, this attractive material design has not been reported.
Specifically, in this work, we explored a combination of hydrogels made of dopamine functionalized alginate and PLGA based nanoparticles to improve tissue-tissue adhesion through catechol reaction with tissues [18–26] and nanoparticle/tissue/hydrogel interaction [27] (Figure 1). In addition, N-hydroxysuccinimide (NHS) was grafted onto the PLGA nanoparticle surface, which can react with the amino groups of proteins in tissues to further enhance the adhesive. The grafted dopamine content of the polymer, and morphology and degradation rates of these nanocomposites were characterized. The adhesive strength of the material was measured using a porcine skin-muscle interface ex vivo model. The in vitro cytocompatibility and in vivo tissue compatibility of the materials were evaluated using human dermal fibroblasts and a rat model, respectively.
Figure 1.

Illustration to glue two tissues using mussel-inspired nanocomposites (MIN). The three interactions between tissues and nanocomposite adhesive might exist to enhance the adhesion. They included that 1) oxidized dopamine molecules in the hydrogel react with amine groups in tissues, 2) the tiny nanoparticles interact with both hydrogel and tissue through physical adsorption, and 3) the NHS groups on the PLGA nanoparticles can covalently react with amine groups in the tissue.
2. Results and Discussion
2.1 Synthesis and characterization of mussel-inspired polymer
The mussel-inspired polymer was synthesized in an aqueous environment by conjugating dopamine with the oxidized alginate through a reductive amination between amine and aldehyde groups. Firstly, the alginate polymer was oxidized using sodium metaperiodate to generate aldehyde groups on its backbone.[28] Secondly, the dopamine was grafted onto the alginate backbone via reductive amination between the aldehyde groups and the amine groups of dopamine (Figure S1A). The FTIR spectrum of sodium alginate (Figure S1B) has specific peaks at 1404 cm−1 and 1596 cm−1corresponding to symmetric and asymmetric COO stretching vibrations, respectively. Other peaks identified for alginate were located at 1023 cm−1 (C-O-C stretching vibration), 946 cm−1 (C–O stretching vibration of uronic acids), 890 cm−1 (C1–H deformation mannuronic acid residues), and 820 cm−1 (mannuronic acid residues).[29] The chemical structure of periodate oxidized alginate was confirmed by aldehyde C=O stretching at 1720 cm−1 in the oxidized alginate spectrum (Figure S1B). The alginate-dopamine polymer structure was verified by C=C aromatic stretching and C-N amine stretching at 1520 cm−1 and 1091 cm−1, respectively (Figure S1B).[30] The dopamine content in mussel foot proteins (Mfps) varies from 10–15 % in Mfp-1, 2–4% in Mfp-2, 25% in Mfp-3, 5% in Mfp-4 and 30% in Mfp-5 whereas Mfp-3 and Mfp-5 are the proteins that contact the surface and help mussels adhere to wet surfaces.[31] Some research groups reported that the dopamine contents incorporated into alginate ranged from 10% to 42%.[30, 32–35] The wide range of dopamine grafting is achieved by altering the molar ratios of alginate to dopamine. The UV-visible absorption spectroscopy at 280 nm of the alginate-dopamine polymer revealed a dopamine content of 26%, which is close to the dopamine content in Mfp-3, and also is within the range of dopamine content reported in other dopamine conjugated alginate polymers.[30, 32–35]
2.2 Nanoparticle characterization
The size and polydispersity of PLGA nanoparticles and N-hydroxysuccinimide (NHS) modified PLGA (PLGA-NHS) nanoparticles were characterized using dynamic light scattering and transmission electron microscopy (Figure S2A–S2B). The PLGA nanoparticles (212 ± 45 nm) were fabricated using a single-emulsion method. The size of PLGA-NHS was 237 ± 106 nm, which is similar to the size of PLGA NPs. The silica nanoparticles (35 nm) were commercially purchased. FTIR was used to confirm the grafting of NHS onto PLGA nanoparticles. The PLGA structure was verified by specific peaks at 2910 cm−1 and 2940 cm−1 (symmetric and asymmetric C-H stretching), 1730 cm−1 (C=O stretching vibration), 1170 cm−1 (C-C-O stretching of ester group) and 1090 cm−1 (O-C-C stretching of ester group) (Figure S2C).[36, 37] For PLGA-NHS nanoparticles (Figure S2C), the specific peaks to identify PLGA-NHS conjugation were located at 1778 cm−1 (symmetric imide C=O stretching in NHS), 1700 cm−1 (asymmetric imide C=O stretching in NHS and ester C=O stretching vibration in PLGA) and 1560 cm−1 (N-O stretching in NHS).
2.3 Mussel-inspired nanocomposite (MIN) formation
The mussel-inspired nanocomposites (MINs) were prepared by blending a range of concentrations from 1 to 40% (w/v) of the mussel-inspired polymer in PBS with PLGA (MIN-PLGA), PLGA-NHS (MIN-PLGA-NHS) or Silica (MIN-Silica) nanoparticles using sodium metaperiodate (PI) as a cross-linker. The nanoparticle concentration was fixed at 12.5% (w/v). The hydrogel from the mussel-inspired polymer (Alg-Dopa) was used as a control. The nanocomposites were dark brown and hydrogel-like, which was the same as the Alg-Dopa. Scanning electron microscopy images of the nanocomposites revealed embedded nanoparticles in MIN-PLGA and MIN-PLGA-NHS groups (Figure 2A). But it is hard to observe the silica nanoparticles due to the small size (30nm). Compared to the morphology of the hydrogel alone without nanoparticles, the nanocomposites appeared more connected and less porous.
Figure 2.

Mussel-inspired nanocomposite (MIN) morphology (A) including SEM images of: (i) Alg-Dopa hydrogel alone, (ii) MIN-PLGA, (iii) MIN-PLGA-NHS, and (iv) MIN-Silica. Left bottom insets showed macroscopic views of the hydrogel and nanocomposites. Right top insets in (ii) and (iii) showed magnified images of the nanocomposites and red arrows pointed to the embedded nanoparticles. (B) In vitro degradation curves of mussel-inspired nanocomposites in PBS at 37°C up to 28 days.
2.4 In vitro degradation of MIN
The periodate oxidation of the alginate leads to a change in conformation of uronate groups in alginate from closed to open chained.[38] This change in conformation makes the oxidized alginate more susceptible to hydrolysis in aqueous solutions thus conferring degradability to it.[28, 38] This degradation of the MINs was characterized by incubation in phosphate buffered saline (PBS, pH 7.2) at 37 °C. The nanocomposites demonstrated a loss of dry weight over time indicating their degradability. The composites degraded to an extent of 53±1% over a period of 4 weeks (Figure 2B).
The degradation rate of the oxidized alginate hydrogels depends on the oxidation degree and the molecular weight of the polymer. Previous studies on the degradation of oxidized alginate hydrogels (molecular weight 270 kDa) in DMEM cell culture media showed 20% loss in dry mass over a period of 4 weeks.[39] Our results exhibited a faster degradation for the alginate-dopamine hydrogel, which may be attributed to the lower molecular weight (12–40 kDa) of alginate used in this work.[39] The incorporation of nanoparticles slightly increased the degradation rate of the nanocomposite. This could be due to the degradable nature of the PLGA nanoparticles. Furthermore, their acidic degradation products, such as lactic acid and glycolic acid, can reduce the surrounding pH and may cause the alginate hydrogel to degrade to a greater extent.[40, 41] However, the nanocomposites containing silica nanoparticles demonstrated similar degradation kinetics with the nanocomposites with PLGA NPs. It might be attributed to the diffusion of some tiny nanoparticles into the PBS when the nanocomposites were immersed in PBS during the period of degradation. High-speed centrifuge might not be able to collect all nanoparticles in the PBS. The possible diffusion, however, might not take place for tissue adhesion in tissues because the diffusion coefficient might be different in solid and/or soft tissues than that of the solution environment.
2.5 Ex vivo adhesion strength of mussel-inspired nanocomposites
The ability of the nanocomposites in gluing tissue interface was evaluated using a setup illustrated in Figure 3A. The lap shear strengths of tissue adhesiveness of the cross-linked polymer at the tissue interface showed a concentration-dependent increasing trend. Previous studies have utilized the mussel-inspired approach to introduce dopamine or L-Dopa into polymers and form adhesive hydrogels, which adhere to tissues with various strengths depending on their dopamine contents.[20, 25, 42, 43] Our mussel-inspired hydrogel at a concentration of 40 %w/v adhered to the pig skin-muscle interface with a lap shear strength of 14 ± 2 kPa, which is comparable to adhesives developed in other studies using the mussel-inspired approach.[18, 20, 25, 42, 43] Furthermore, the adhesive properties of the nanocomposites demonstrated a significant increase in lap shear strengths compared to those without nanoparticles (p<0.05). The highest lap shear strength of 33 ± 3 kPa (Figure 3B) was obtained in the 40% (w/v) Alginate-Dopa with PLGA-NHS nanoparticles. The role of nanoparticles in adhering tissues together was attributed to their ability to adsorb onto surfaces and dissipate energy within the interface which yields resistance to fracture propagation between the two tissue phases.[27] Recently, silica nanoparticles were investigated as a means to adhere liver tissues and gels together where silica nanoparticles could bond two non-adhering surfaces with an adhesive lap shear strength of 0.65±0.25 kPa.[27] Although the nanoparticle alone as a gluing agent provides an attractive method to glue tissues together, the adhesive strength is much lower compared to mussel adhesive hydrogels.[18, 20, 25, 42, 43] The MIN offered improved tissue adhesion performances over nanoparticles alone and mussel inspired hydrogel alone. The adhesive properties of these composites over a 24 hour period increased over time in all groups. It is noted that the nanoparticle-mediated enhancement of adhesion was significant within the first 1 hour of cross-linking and tended to saturate over time. There was no significant difference in additional adhesion strength observed among the mussel-inspired hydrogels and the mussel-inspired nanocomposites at 24 hours. The penetration profile of the nanocomposites was tissue specific with more penetration into the muscle layer compared to the skin layer. It was observed that the inclusion of nanoparticles led to less penetration of the nanocomposites into the muscle tissue compared to that of the hydrogel alone. MIN-PLGA and MIN-PLGA-NHS had lower penetration into the muscle tissue compared to the adhesive alone (Figure 4). Hydrogel materials designed to be used as tissue adhesives require characteristics of both fluidity and cohesive strength to function as an optimal adhesive.[44, 45] The addition of PLGA and PLGA-NHS nanoparticles might increase the cohesiveness of alginate-dopamine hydrogels, which led to the hydrogel solution diffuse less into the tissues. This increase in cohesiveness may also explain the increase in adhesive strengths of PLGA and PLGA-NHS nanocomposites compared to alginate-dopamine hydrogels alone. It was reported that the inclusion of 2% laptonite into poly(acrylic acid) polymer networks resulted in increased cohesion in the hydrogels which correlated to increased adhesion to organic substrates.[46] In addition, for the PLGA-NHS group, the NHS on the nanoparticles can react with the amine groups on the tissue surface, which may limit nanoparticle and hydrogel penetration into the tissues.
Figure 3.

Adhesive properties of nanocomposites. (A) Digital image of an adhered skin-muscle interface (40% w/v, MIN-PLGA-NHS) (B) Tissue adhesion strength of adhesives for porcine skin-muscle interface C) Time dependent adhesion strength of adhesives (Alg-Dopa concentration 40% w/v) on porcine skin-muscle interfaces.
Figure 4.

Penetration depth of nanocomposites into skin-muscle tissue. A) Micrographs of porcine skin-muscle tissue interface showing the extent of penetration of the nanocomposites into tissues. B) Quantification of the mean penetration of the nanocomposites into the skin and muscle tissue using ImageJ analysis.
2.6 In vitro cell compatibility of nanocomposite system
We tested several dilutions (1X, 5X, 10X, 100X) of the cell media that was incubated with the nanocomposites for 24 hours. The nanocomposites with PLGA and PLGA-NHS nanoparticles exhibited good cell compatibility to human dermal fibroblasts. Compared to the positive control Matrigel, the leached products at 1X dilution from the hydrogel alone and all composites showed less cell viability, while the 5X, 10X, 100X dilutions of the hydrogel alone, the MIN-PLGA and MIN-PLGA-NHS exhibited similar cell viability (p>0.05) (Figure 5A). However, the MIN-Silica showed moderate toxicity at all dilutions of the leachable contents (Figure 5A). In Figure 5B, only MIN-Silica exhibited less cell viability than the controls of the Matrigel and cell culture medium alone. No significant toxicity with PLGA and PLGA-NHS nanoparticles on human dermal fibroblasts was observed, whereas silica nanoparticles demonstrated a dose-dependent toxicity towards the cells (Figure S2B). We have previously reported the PLGA nanoparticles showed good cytocompatibility with human dermal fibroblasts, human aortic endothelial cells and human aortic smooth muscle cells up to a concentration of more than 500 µg/ml, which is in agreement with the observed result in this study.[47, 48] The silica nanoparticles exhibit size-dependent cell toxicity towards various human cell lines including dermal fibroblasts.[49–52] The size of silica nanoparticles that induces toxicity usually ranges from 20–60 nm which could explain the toxicity of the 30 nm silica nanoparticles used in this study.[51]
Figure 5.

Cytocompatibility of mussel-inspired nanocomposites (MIN) with human dermal fibroblasts (HDFs). (A) Leachable products cytotoxicity at leachate dilutions of 1×, 5×, 10×, 100×. (B) Adhesive cytocompatibility with human dermal fibroblasts using a Transwell setup.
2.7 In vivo biocompatibility of nanocomposite system
To further evaluate the in vivo biocompatibility of the nanocomposites, the MIN-PLGA-NHS was selected because of its high adhesive strength and good in vitro cytocompatibility. Incisions on the back of rats were successfully sealed by the nanocomposite of MIN-PLGA-NHS with the suture as a control (Figure 6A). At day 7 and day 28, the explanted tissue sections were evaluated for foreign body reactions or immune responses via Hematoxylin and Eosin (H&E) staining and immunohistochemistry analysis. The H&E staining revealed the sutured sections had healed completely both at day 7 and 28 (Figure 6B). The tissue areas filled with the MIN-PLGA-NHS showed cell infiltration and material degradation at day 7, and subsequent clearance of the composite with no residues of the composite visible at day 28. The immunohistochemical analysis revealed the presence of CD11b+ inflammatory cells in the excised tissue sections around the areas sealed with the nanocomposite glue and the control suture at day 7 (Figure 6C). The area sealed with MIN-PLGA-NHS had slightly more inflammatory cells compared to the suture control. This could be because alginate-based materials are generally known to have low immunogenic responses.[53] Alginate has been shown to activate macrophages in a murine model via activation through the NF-κB pathway, leading to the production of interleukins including IL-1β, IL-6, IL-12 and TNF-α, and the promotion of a proinflammatory environment.[54] Some studies have shown that the foreign body reaction towards alginate-based materials varied depending on the content of glucuronic acid, and the higher content (>60%) induced greater foreign body responses.[55–57] The alginate used in this study had less than 50% glucuronate residues, which could achieve the low foreign body response. The H&E stained tissue sections of the area sealed by the MIN nanocomposite showed some evidence of gel fragments at day 7 (Figure 6B). These gel fragments in the micrometer size can possibly activate phagocytes leading to further inflammatory conditions in the tissue.[58] In such cases MΦ macrophages are mainly responsible for sustaining this pro-inflammatory environment.[58] However the presence of inflammatory cells markedly subsided over the course of 28 days in both the suture and the nanocomposite groups, indicating a normal wound healing process. There was no significant difference in the number of CD11b+ cells between the tissue sutured and the one sealed with the MIN-PLGA-NHS (Figure 6C–6D), indicating that the nanocomposite did not generate any lasting inflammatory reaction, Furthermore, the inclusion of PLGA-NHS nanoparticles into the alginate hydrogel did not alter the immunogenicity of alginate hydrogels.
Figure 6.

In vivo biocompatibility of mussel-inspired nanocomposites with PLGA-NHS nanoparticles (MIN-PLGA-NHS) (A) Micrographs of areas sealed by PLGA-NHS nanocomposites and the ones sutured on the back of Sprague-Dawley rats at Day 0 and Day 7 (B) H&E staining of wound areas that were sutured and adhered with MIN-PLGA-NHS at day 7 and day 28. (C) Immunohistochemistry staining of macrophages in tissue sections. (D) Quantification of number of cd11b+ cells per millimeter square area near the tissues adhered with MIN-PLGA-NHS and the suture.
3. Conclusion
The MINs were prepared by incorporating biodegradable nanoparticles into cross-linked mussel-inspired hydrogels. The tissue adhesive properties of the hydrogels were enhanced by blending nanoparticles. The PLGA-NHS nanoparticles performed best to enhance hydrogel adhesion between skin-muscle tissues compared to the silica and PLGA nanoparticles. The adhesive properties of the nanocomposites increased over time within the tissue interface. The degradable adhesive nanocomposites were cytocompatible and exhibited a lower penetration into the pig muscle than that of the hydrogel alone. The in vivo implantation of MIN-PLGA-NHS did not generate significant inflammatory responses. This biodegradable and biocompatible nanocomposite with high adhesive ability may be a suitable candidate for adhering tissue interfaces as a bio-glue in wound closure.
4. Experimental Section
Materials
Sodium alginate (molecular weight (MW)=12–40 kDa), sodium metaperiodate (PI), dopamine hydrochloride, sodium cyanoborohydride, polyvinyl alcohol (PVA, MW=31 kDa), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, (EDC) phosphate buffered saline (PBS), silica nanoparticle (LUDOX TM-50), and 4-morpholineethanesulphonic acid (MES) were purchased from Sigma-Aldrich, Inc, MO. Poly(lactide-co-glycolide) (PLGA, COOH terminated, lactide/glycolide molar ratio =50:50, and MW= 97 kDa) was purchased from Akina PolySciTech, Inc. All chemicals were used as received without further purification.
Synthesis of mussel-inspired polymers
Sodium alginate was oxidized by using periodate aqueous solution.[59] Sodium alginate (1% w/v in deionized water, 90 ml) was reacted with sodium metaperiodate (PI) (10% w/v in deionized water, 10 ml) for a period of 12 hours in the dark. The reaction was then subjected to dialysis (molecular weight cut-off (MWCO) 3.5kDa) against deionized water and subsequently lyophilized to obtain oxidized alginate. The oxidized alginate (0.2 g in deionized water, 14 ml) then reacted with dopamine hydrochloride (0.28 g in deionized water, 6 ml) in the presence of sodium cyanoborohydride (NaBH3CN, 25 mM) for 24 h in a dark setting and under a nitrogen protection. The obtained mussel-inspired polymer was purified via dialysis (MWCO 3.5 kDa) against deionized water at 4°C and subsequently lyophilized.
Fabrication of PLGA nanoparticles and NHS modified PLGA nanoparticles
The PLGA nanoparticles were fabricated using emulsion methods. [21] A PLGA solution (2% w/v in dichloromethane, 5 ml) was added dropwise to a PVA solution (5% w/v in deionized water, 20 ml). This emulsion was sonicated (30 W, 5 minutes) and then stirred overnight at room temperature to allow solvent evaporation and particle formation. The resulting particle suspension was centrifuged (15,000 rpm, 30 min, 10 °C) and the pellet suspension in deionized water was lyophilized to obtain PLGA nanoparticles.
To modify the PLGA particles with NHS, PLGA nanoparticle suspension (0.2 % w/v, 5 ml) in MES buffer (pH 4.75) was mixed with EDC (120 mg, 0.63 mmole) for 30 minutes at room temperature. NHS (180 mg, 1.56 mmole) was then added to the solution and allowed to mix for 2 hours at room temperature to graft NHS onto the surface of PLGA nanoparticles. The NHS modified PLGA nanoparticles (PLGA-NHS) were purified by centrifugation (15,000 rpm, 30 minutes, 10°C), then subsequently washed with deionized water and lyophilized.
Fabrication of mussel-inspired nanocomposites (MIN)
The alginate-dopamine polymer at different concentrations was cross-linked using the cross-linker PI at equimolar concentrations with respect to the dopamine amount in the polymer. Briefly, alginate-dopamine polymer at concentration of 40 % w/v was mixed with 1X PBS (pH 7.2) to prepare a polymer precursor solution. Known amounts of the polymer precursor solution (1, 5, 20 and 40%) were then mixed with a nanoparticle suspension in 1X PBS solution. The nanoparticle concentration was fixed as 12.5% w/v in the composite. The polymer-nanoparticle mixture was then cross-linked using PI for 1 h. Equal moles of PI (0.29, 1.48, 5.95, 11.90 %w/v), with respect to the dopamine content in the alginate-dopamine polymer, were used for cross-linking and to form MINs.
Mussel-inspired polymer characterization
The synthesis of oxidized alginate (Alg-Ox) and mussel-inspired polymer was confirmed by Fourier transform infrared spectroscopy (FTIR). The grafting efficiency of dopamine to Alg-Ox was measured by UV-Visible spectrometry utilizing the absorbance at 280 nm of the aromatic ring in dopamine.[19] The absorbance data from a set of dopamine hydrochloride standards ranging from 0 to 1 mg/mL was used to determine the dopamine concentration in a 1 mg/mL Alg-Dopa solution (n=8). For hydrogel degradation, mussel inspired polymer was mixed in PBS (pH 7.2) at a final concentration of 40%w/v and crosslinked using PI (11.90% w/v) for 1 hour. This cross-linked hydrogel was then immersed in a tube containing 1 ml PBS (n = 6). At predetermined time points each of the tubes were centrifuged (9,000g, 10 min) to collect the hydrogel. This collected hydrogel was lyophilized to achieve the dry hydrogel. The weight of this dry hydrogel (Wt) was recorded and compared with the initial dry weight (W0) to calculate the % weight remaining using Wt/Wo×100%.
Nanoparticle characterization
The hydrodynamic size and polydispersity of the PLGA, PLGA-NHS, and silica nanoparticles were measured via dynamic light scattering technique (DLS) (Brooke Haven, ZETA PALS). Nanoparticle solutions (0.01 % w/v in DI water) were used for this DLS analysis. The grafting of NHS onto PLGA nanoparticles was confirmed via FTIR (Thermo Electron Corporation, NICOLET 6700). The nanoparticles were also observed under high resolution transmission electron microscopy (HR-TEM, Hitachi H-9500).
Nanoparticle cytocompatibility
The cytocompatibility of silica, PLGA and PLGA-NHS nanoparticles was evaluated using human dermal fibroblasts (HDFs, ATCC). HDFs were seeded at a density of 10,000 cells/cm2 into wells of a 24 well plate (n = 8) and incubated in Dulbecco’s Modified Eagle Medium (DMEM, Sigma Aldrich) supplemented with 10% (v/v) fetal bovine serum (FBS, Life Technologies) and 1% (v/v) penicillium-streptomycin (Life Technologies, Inc.) at 37 °C and 5% CO2 for 24 h. Nanoparticles at concentrations of 50, 100, 200, 500, 1000 µg/ml were then incubated with the cells for 24 h. A MTS assay (CellTiter 96® aqueous one solution cell proliferation Assay, Promega, Madison, WI) was performed according to manufacturer’s instructions to assess cell viability. The HDFs exposed to culture media only served as a control.
Mussel-inspired nanocomposite characterization
The cross-sectional morphologies of nanocomposites were examined using scanning electron microscopy. A 40 % w/v solution of the mussel-inspired polymer in PBS (pH 7.2) was combined with a 12.5% w/v solution of the nanoparticles and cross-linked using PI at a final concentration of 11.9%w/v for 1 hour. The cross-linked samples were immersed in liquid nitrogen and cut at cross-section. The frozen cross-sections were freeze-dried and imaged under a scanning electron microscope. For nanocomposite degradation, the mussel-inspired nanocomposites (MIN) containing silica, PLGA and PLGA-NHS nanoparticles were formulated using methods as described above, and the measurement protocol is the same as hydrogel degradation method.
Tissue adhesion strength measurement
The tissue adhesion of the mussel-inspired nanocomposites and the mussel-inspired polymer was tested on porcine skin and muscle samples (n=6) using guidelines from the American Society for Testing and Materials (ASTM) standard F2255-05. Freshly obtained porcine skin and muscle were cut into sections (30mm × 10mm × 3mm), and the mussel-inspired polymer at concentrations ranging from 1 to 40% (w/v) in PBS (pH 7.2) with or without the incorporation of nanoparticle suspensions (12.5% w/v) was allowed to cross-link on an area of 10 mm × 5 mm between the skin-muscle interface for a period of 1 hour at 37°C (Figure S3). To ensure the skin-muscle samples were moist and retained water, the skin-muscle interface was kept in a humid environment in a standard cell culture incubator, maintaining humidity levels greater than 90% during the incubation time. The samples were then subjected to lap shear adhesion tests on an MTS insight workstation (cross-head speed of 10 mm min−1; 500 N load cell; room temperature), and then the lap shear strength was measured. The same parameters of MTS testing were also used to measure the changes in tissue adhesion of the mussel-inspired polymer over a 24 hour time period by measuring the lap shear strengths of the mussel-inspired hydrogel and the nanocomposites at predetermined time points.
Nanocomposite cytocompatibility
The HDFs were used to determine cytotoxic effects of the Alg-Dopa alone and the mussel-inspired nanocomposite systems (MINs). The HDFs were seeded at a density of 10,000 cells/cm2 into the wells of a 24 well plate (n = 8) and incubated in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillium-streptomycin at 37 °C and 5% CO 2 for 24 hours. These seeded cell culture plates were then used to determine the cytotoxicity of the leached products of Alg-Dopa and MINs as well as to study the effects of the incubation of these systems on the cells.
The leached products were formulated using the systems (40% w/v polymer and 12.5% w/v nanoparticles) as described above, UV sterilized and incubated them in a known amount of DMEM cell culture medium at different dilutions (1X, 5X, 10X, 100X) at 37 °C and 5% CO2 for 24 hours. Post incubation, all solutions were neutralized to pH 7.2 and added to the seeded HDFs for a period of 24 hours. MTS assays were performed on each group after the 24-hour time-point following manufacturer’s instructions. The cell culture medium only and Matrigel were used as controls to compare with the viability of HDFs exposed to treated groups. The Matrigel was prepared at a concentration of 1% w/v and applied to the Transwell plates at a volume to surface area ratio of 50 µL/cm2.
To assess the cytotoxicity of incubated hydrogels and nanocomposites, the formulated systems post UV sterilization were also placed in Transwell inserts (Pore size: 0.4 µm, growth area: 0.14 cm2). These inserts were then placed into the cell pre-seeded 24-well plates for 24 h. MTS assays were performed in each group following manufacturer’s instructions. Viability of HDFs in each group was assessed compared to the culture media alone and Matrigel.
Ex vivo tissue penetration depth of nanocomposites
The different groups of MINs and the Alg-Dopa alone were cross-linked within the skin-muscle tissue interfaces for 1 hour. The adhered tissue samples were then embedded in optimal cutting temperature solutions (OCT, Tissue-tek) and frozen at −20 °C for 1 hour. The OCT embedded tissue samples were then cut into slices (10 µm thick). Image analysis was performed on the sections (n = 10) of each group of MINs and the Alg-Dopa using Image J (National Institute of Health, Bethesda, MD, USA). A baseline indicated by the white horizontal lines (Figure 5A) was chosen, and it represented the original contact between the muscle-skin and the glue. The glue penetration depth was measured based on this baseline.
In vivo nanocomposite biocompatibility
All animal experiments were conducted in accordance with the animal welfare and IACUC approved protocols from the University of Texas at Arlington. Sprague-Dawley male rats (Taconic Biosciences), one year old and weighing 300–500 g, were used to test the biocompatibility of the nanocomposite adhesive. The MIN-PLGA-NHS was tested in vivo with suture as a control. Four incisions (1 inch wide × 0.5 inch deep) were made on the back of each rat (n = 4). Two of these incisions were closed by the application of the nanocomposite adhesive, while the other two were closed with silicone treated non-absorbable sutures (4-0 SILK, Davis+Geck). The rats were sacrificed at day 7 and 28, and the implants with surrounding tissues were isolated for histological analysis. The sections were embedded in OCT, and 10 µm thick cryosections were cut for analysis. Hematoxylin and eosin (H&E) staining was performed to evaluate the interaction of the nanocomposite with the host tissue at the site of implantation. Immunohistochemistry analysis using rabbit anti-integrin αM CD11b, H-61 (Santa Cruz Biotechnology, inc Santa Cruz, CA, USA) antibodies against CD11b+ cells were performed on the tissue cryosections at day 7 and 28 to evaluate inflammatory responses in the tissue sealed by the adhesive and the suture control. The cellular infiltration into the tissue areas was quantified by calculating the number of cells per unit area using ImageJ (National Institute of Health, Bethesda, MD, USA). The tissue areas for cell counting were selected randomly around the incision site
Statistical Analysis
All data analysis was executed using one-way and two-way ANOVA (Statview 5.0 software, SAS institute, Cary, NC, USA) to analyze differences between groups with one or more independent variables. p < 0.05 was considered as significant difference. Post hoc analysis was done using Tukey’s honest significant difference (HSD). All data was reported as mean ± standard deviation.
Supplementary Material
Acknowledgments
We acknowledge the support from the startup fund of the University of Texas at Arlington (YH), NSF CAREER grant (#1554835, YH), and NIH grant (#R01HL118498, KN and # R21HD090680, YH).
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Annabi N, Tamayol A, Shin SR, Ghaemmaghami AM, Peppas NA, Khademhosseini A. Nano today. 2014;9:574–589. doi: 10.1016/j.nantod.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehdizadeh M, Yang J. Macromol. Biosci. 2013;13:271–288. doi: 10.1002/mabi.201200332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leggat PA, Smith DR, Kedjarune U. ANZ J. Surg. 2007;77:209–213. doi: 10.1111/j.1445-2197.2007.04020.x. [DOI] [PubMed] [Google Scholar]
- 4.Sierra DH. J. Biomater. Appl. 1993;7:309–352. doi: 10.1177/088532829300700402. [DOI] [PubMed] [Google Scholar]
- 5.Hyon S-H, Nakajima N, Sugai H, Matsumura K. J. Biomed. Mater. Res. Part A. 2014;102:2511–2520. doi: 10.1002/jbm.a.34923. [DOI] [PubMed] [Google Scholar]
- 6.Mahdavi A, Ferreira L, Sundback C, Nichol JW, Chan EP, Carter DJ, Bettinger CJ, Patanavanich S, Chignozha L, Ben-Joseph E, Galakatos A, Pryor H, Pomerantseva I, Masiakos PT, Faquin W, Zumbuehl A, Hong S, Borenstein J, Vacanti J, Langer R, Karp JM. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2307–2312. doi: 10.1073/pnas.0712117105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kluge JA, Rabotyagova O, Leisk GG, Kaplan DL. Trends Biotechnol. 2008;26:244–251. doi: 10.1016/j.tibtech.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Haller CM, Buerzle W, Kivelio A, Perrini M, Brubaker CE, Gubeli RJ, Mallik AS, Weber W, Messersmith PB, Mazza E, Ochsenbein-Koelble N, Zimmermann R, Ehrbar M. Acta Biomater. 2012;8:4365–4370. doi: 10.1016/j.actbio.2012.07.047. [DOI] [PubMed] [Google Scholar]
- 9.Kim BJ, Oh DX, Kim S, Seo JH, Hwang DS, Masic A, Han DK, Cha HJ. Biomacromolecules. 2014;15:1579–1585. doi: 10.1021/bm4017308. [DOI] [PubMed] [Google Scholar]
- 10.Waite JH. Int. J. Adhes. Adhes. 1987;7:9–14. [Google Scholar]
- 11.Zhu W, Yang J, Iqbal J, Peck Y, Fan C, Wang D-A. J. Surg. Res. 2017;215:173–182. doi: 10.1016/j.jss.2017.03.020. [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Wang R, Xu T, Ma X, Yao Z, Chi BO, Xu H. J. Mater. Chem. B. 2017;5:5668–5678. [Google Scholar]
- 13.Jeon EY, Hwang BH, Yang YJ, Kim BJ, Choi B-H, Jung GY, Cha HJ. Biomaterials. 2015;67:11–19. doi: 10.1016/j.biomaterials.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H, Bre LP, Zhao T, Zheng Y, Newland B, Wang W. Biomaterials. 2014;35:711–719. doi: 10.1016/j.biomaterials.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Kord Forooshani P, Lee BP. J. Polym. Sci. A Polym. Chem. 2017;55:9–33. doi: 10.1002/pola.28368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo J, Kim GB, Shan D, Kim JP, Hu J, Wang W, Hamad FG, Qian G, Rizk EB, Yang J. Biomaterials. 2017;112:275–286. doi: 10.1016/j.biomaterials.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee H, Scherer NF, Messersmith PB. Proc. Natl. Acad. Sci. U. S. A. 2006;103:12999–13003. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen B, Pinkas O, Foox M, Zilberman M. Acta Biomater. 2013;9:9004–9011. doi: 10.1016/j.actbio.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Foster LJ, Karsten E. J Vis Exp. 2012 doi: 10.3791/35273527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan C, Fu J, Zhu W, Wang DA. Acta biomaterialia. 2016;33:51–63. doi: 10.1016/j.actbio.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Shin J, Lee JS, Lee C, Park H-J, Yang K, Jin Y, Ryu JH, Hong KS, Moon S-H, Chung H-M, Yang HS, Um SH, Oh J-W, Kim D-I, Lee H, Cho S-W. Adv. Funct. Mater. 2015;25:3814–3824. [Google Scholar]
- 22.Han L, Lu X, Liu K, Wang K, Fang L, Weng L-T, Zhang H, Tang Y, Ren F, Zhao C, Sun G, Liang R, Li Z. ACS Nano. 2017;11:2561–2574. doi: 10.1021/acsnano.6b05318. [DOI] [PubMed] [Google Scholar]
- 23.Fan C, Fu J, Zhu W, Wang D-A. Acta Biomater. 2016;33:51–63. doi: 10.1016/j.actbio.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 24.Han L, Yan L, Wang K, Fang L, Zhang H, Tang Y, Ding Y, Weng L-T, Xu J, Weng J, Liu Y, Ren F, Lu X. NPG Asia Mater. 2017;9:e372. [Google Scholar]
- 25.Ji Y, Ji T, Liang K, Zhu L. J. Mater. Sci. Mater. Med. 2016;27:30. doi: 10.1007/s10856-015-5649-2. [DOI] [PubMed] [Google Scholar]
- 26.Lu D, Wang H, Li Te, Li Y, Dou F, Sun S, Guo H, Liao S, Yang Z, Wei Q, Lei Z. ACS Appl. Mater. Interfaces. 2017;9:16756–16766. doi: 10.1021/acsami.6b16575. [DOI] [PubMed] [Google Scholar]
- 27.Rose S, Prevoteau A, Elziere P, Hourdet D, Marcellan A, Leibler L. Nature. 2014;505:382–385. doi: 10.1038/nature12806. [DOI] [PubMed] [Google Scholar]
- 28.Kristiansen KA, Potthast A, Christensen BE. Carbohydr. Res. 2010;345:1264–1271. doi: 10.1016/j.carres.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Papageorgiou SK, Kouvelos EP, Favvas EP, Sapalidis AA, Romanos GE, Katsaros FK. Carbohydr. Res. 2010;345:469–473. doi: 10.1016/j.carres.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Jiang Z, Shi J, Zhang C, Zhang W, Wu H. Ind. Eng. Chem. Res. 2013;52:14828–14836. [Google Scholar]
- 31.Hwang DS, Zeng H, Lu Q, Israelachvili J, Waite JH. Soft Matter. 2012;8:5640–5648. doi: 10.1039/C2SM25173F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee C, Shin J, Lee JS, Byun E, Ryu JH, Um SH, Kim DI, Lee H, Cho SW. Biomacromolecules. 2013;14:2004–2013. doi: 10.1021/bm400352d. [DOI] [PubMed] [Google Scholar]
- 33.Hou J, Li C, Guan Y, Zhang Y, Zhu XX. Polym. Chem. 2015;6:2204–2213. [Google Scholar]
- 34.Alegre-Requena JV, Haring M, Herrera RP, Diaz Diaz D. New J. Chem. 2016;40:8493–8501. [Google Scholar]
- 35.Kim S, Moon J-M, Choi JS, Cho WK, Kang SM. Adv. Funct. Mater. 2016;26:4099–4105. [Google Scholar]
- 36.Jalali N, Moztarzadeh F, Mozafari M, Asgari S, Motevalian M, Alhosseini SN. Colloids Surf., A. 2011;392:335–342. [Google Scholar]
- 37.Silva ATCR, Cardoso BCO, Silva MESRe, Freitas RFS, Sousa RG. J Biomater Nanobiotechnol. 2015;06:8–19. [Google Scholar]
- 38.Bouhadir KH, Lee KY, Alsberg E, Damm KL, Anderson KW, Mooney DJ. Biotechnol. Prog. 2001;17:945–950. doi: 10.1021/bp010070p. [DOI] [PubMed] [Google Scholar]
- 39.Boontheekul T, Kong HJ, Mooney DJ. Biomaterials. 2005;26:2455–2465. doi: 10.1016/j.biomaterials.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 40.Makadia HK, Siegel SJ. Polymers. 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zolnik BS, Burgess DJ. J. Control. Release. 2007;122:338–344. doi: 10.1016/j.jconrel.2007.05.034. [DOI] [PubMed] [Google Scholar]
- 42.Cencer M, Murley M, Liu Y, Lee BP. Biomacromolecules. 2015;16:404–410. doi: 10.1021/bm5016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim HJ, Hwang BH, Lim S, Choi B-H, Kang SH, Cha HJ. Biomaterials. 2015;72:104–111. doi: 10.1016/j.biomaterials.2015.08.055. [DOI] [PubMed] [Google Scholar]
- 44.Lee BP, Messersmith PB, Israelachvili JN, Waite JH. Annual review of materials research. 2011;41:99–132. doi: 10.1146/annurev-matsci-062910-100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peak CW, Wilker JJ, Schmidt G. Colloid. Polym. Sci. 2013;291:2031–2047. [Google Scholar]
- 46.Shen M, Li L, Sun Y, Xu J, Guo X, Prud’homme RK. Langmuir. 2014;30:1636–1642. doi: 10.1021/la4045623. [DOI] [PubMed] [Google Scholar]
- 47.Menon JU, Kona S, Wadajkar AS, Desai F, Vadla A, Nguyen KT. J. Biomed. Mater. Res. Part A. 2012;100A:1998–2005. doi: 10.1002/jbm.a.34040. [DOI] [PubMed] [Google Scholar]
- 48.Kona S, Dong J-F, Liu Y, Tan J, Nguyen KT. Int. J. Pharm. 2012;423:516–524. doi: 10.1016/j.ijpharm.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Sun L, Jin M, Du Z, Liu X, Guo C, Li Y, Huang P, Sun Z. Toxicol. In Vitro. 2011;25:1343–1352. doi: 10.1016/j.tiv.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Sun L, Li Y, Liu X, Jin M, Zhang L, Du Z, Guo C, Huang P, Sun Z. Toxicol. In Vitro. 2011;25:1619–1629. doi: 10.1016/j.tiv.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 51.Napierska D, Thomassen LCJ, Rabolli V, Lison D, Gonzalez L, Kirsch-Volders M, Martens JA, Hoet PH. Small. 2009;5:846–853. doi: 10.1002/smll.200800461. [DOI] [PubMed] [Google Scholar]
- 52.Park Y-H, Bae HC, Jang Y, Jeong SH, Lee HN, Ryu W-I, Yoo MG, Kim Y-R, Kim M-K, Lee JK, Jeong J, Son SW. MOL CELL TOXICOL. 2013;9:67–74. [Google Scholar]
- 53.Zimmermann U, Klöck G, Federlin K, Hannig K, Kowalski M, Bretzel RG, Horcher A, Entenmann H, Sieber U, Zekorn T. ELECTROPHORESIS. 1992;13:269–274. doi: 10.1002/elps.1150130156. [DOI] [PubMed] [Google Scholar]
- 54.Yang D, Jones KS. J Biomed Mater Res A. 2009;90:411–418. doi: 10.1002/jbm.a.32096. [DOI] [PubMed] [Google Scholar]
- 55.Tam SK, Dusseault J, Bilodeau S, Langlois G, Hallé J-P, Yahia LH. J. Biomed. Mater. Res. Part A. 2011;98A:40–52. doi: 10.1002/jbm.a.33047. [DOI] [PubMed] [Google Scholar]
- 56.Lee KY, Mooney DJ. Prog. Polym. Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thambi T, Phan VHG, Lee DS. Macromol. Rapid Commun. 2016;37:1881–1896. doi: 10.1002/marc.201600371. [DOI] [PubMed] [Google Scholar]
- 58.Jiang WW, Su SH, Eberhart RC, Tang L. J Biomed Mater Res A. 2007;82:492–497. doi: 10.1002/jbm.a.31175. [DOI] [PubMed] [Google Scholar]
- 59.Jeon O, Alt DS, Ahmed SM, Alsberg E. Biomaterials. 2012;33:3503–3514. doi: 10.1016/j.biomaterials.2012.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.