

Abstract
Objective
To provide an overview of clinical and MRI characteristics of the different variants of the leukodystrophy megalencephalic leukoencephalopathy with subcortical cysts (MLC) and identify possible differentiating features.
Methods
We performed an international multi-institutional, cross-sectional observational study of the clinical and MRI characteristics in patients with genetically confirmed MLC. Clinical information was obtained by questionnaires for physicians and retrospective chart review.
Results
We included 204 patients with classic MLC, 187 of whom had recessive mutations in MLC1 (MLC1 variant) and 17 in GLIALCAM (MLC2A variant) and 38 patients with remitting MLC caused by dominant GLIALCAM mutations (MLC2B variant). We observed a relatively wide variability in neurologic disability among patients with classic MLC. No clinical differences could be identified between patients with MLC1 and MLC2A. Patients with MLC2B invariably had a milder phenotype with preservation of motor function, while intellectual disability and autism were relatively frequent. Systematic MRI review revealed no MRI features that distinguish between MLC1 and MLC2A. Radiologic improvement was observed in all patients with MLC2B and also in 2 patients with MLC1. In MRIs obtained in the early disease stage, absence of signal abnormalities of the posterior limb of the internal capsule and cerebellar white matter and presence of only rarefied subcortical white matter instead of true subcortical cysts were suggestive of MLC2B.
Conclusion
Clinical and MRI features did not distinguish between classic MLC with MLC1 or GLIALCAM mutations. Absence of signal abnormalities of the internal capsule and cerebellar white matter are MRI findings that point to the remitting phenotype.
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is an infantile-onset inherited disorder characterized by cerebral white matter edema.1–3 MRI shows diffuse signal abnormalities of the cerebral hemispheric white matter. The swelling of the abnormal cerebral white matter is most prominent in the first few years of life.1 Subcortical cysts are typically located in the anterior temporal region and less consistently elsewhere.4 Two different MLC phenotypes can be distinguished: a classic, deteriorating phenotype, and a remitting phenotype.5 Classic MLC is caused by recessive mutations in the MLC1 gene (MIM 605908) in the majority of cases: this variant is called MLC1 (MIM 604004).6 Classic MLC caused by recessive GLIALCAM mutations (also known as HEPACAM, MIM 613925) is called MLC2A.6 Patients with remitting MLC or MLC2B (MIM 613926) have dominant GLIALCAM mutations.7 Classic MLC starts with increasing macrocephaly in the first year of life; after a few years, patients develop neurologic signs, most commonly ataxia, spasticity, and epilepsy.1 Mortality is thought to be low, although no systematic study on survival has been performed. Remitting MLC initially resembles classic MLC: patients generally present with progressive macrocephaly and may have developmental delay, but neurologic deterioration does not occur and the MRI abnormalities improve or normalize.
Among patients with MLC1, a relatively broad variation in disease severity has been described, also for siblings and unrelated patients with the same mutations.8,9 So far, no clinical differences have been recognized between patients with MLC2A vs MLC1. It is unknown whether MRI features allow distinction among MLC1, MLC2A, and MLC2B before improvement occurs in the latter. MLC is a rare disorder and large studies on the disease course are scarce.8,10,11 Systematic review of MRI characteristics has not been performed. The objective of the current study is to identify potential different and perhaps discriminating clinical and MRI features for different MLC variants, aiming at improved clinical recognition and avoidance of unnecessary genetic testing.
Methods
Study design
We performed an international, cross-sectional observational multicenter study among all patients with genetically proven MLC enrolled in the Amsterdam database of leukoencephalopathies between January 1991 and January 2017. The database contains patients from over the world referred to VU University Medical Center for MRI review and mutational analysis. A prerequisite for genetic testing was review of clinical and MRI data in Amsterdam.1 We performed analysis of the MLC1 gene as previously described,12 if necessary including analysis of MLC1 copy DNA12 and multiplex ligation-dependent probe amplification.13 Analysis of the GLIALCAM gene was also performed as previously described.7 By DNA analysis in the parents, we confirmed that recessive variants were biallelic.
Standard protocol approvals, registrations, and patient consents
We received approval from the ethical standards committee for clinical evaluation, systematic MRI review, and review of the DNA findings and obtained informed consent from the patients/guardians.
Clinical information
We analyzed clinical information available at the start of the study, supplemented by information obtained via a clinical questionnaire for physicians. We focused on measures that could be retrospectively assessed, including head circumference, motor deterioration (especially loss of ambulation), seizures, and death. As formal, standardized assessment of motor and cognitive development, autistic features, behavioral problems, and current cognitive function was not feasible, these items were subjectively assessed by physicians and caretakers or derived from medical records. The patients' status at last examination was assessed by quantitative, validated, and widely used 5-level classification systems for gross motor function (Gross Motor Function Classification System [GMFCS]), manual ability (Manual Ability Classification System [MACS]), and communication function (Communication Function Classification System [CFCS]).14 Scores on these scales range from I (no limitations) to V (severe limitations; table e-1, links.lww.com/WNL/A356). Patients with comorbidities affecting neurologic function were excluded from the study.
MRI scoring
We scored all available MRIs according to a standardized protocol.15 Specifically, cysts were defined as areas with the same signal intensity of CSF on all pulse sequences including fluid-attenuated inversion recovery, while in rarefaction the signal intensity was close but not the same.16
Statistical analysis
We used summary statistics to describe the clinical characteristics. Results were reported by means ± SD for continuous variables that were normally distributed, and median with 25th and 75th percentiles for non-normally distributed data and ranges. We performed time-to-event analysis of the events “start of motor deterioration,” “start of cognitive decline,” “first seizure,” “loss of walking without support,” “loss of walking with or without support,” and “death,” with age as time variable. Individuals in whom the respective event had not occurred at the last follow-up were indicated as censored for the respective analysis. Nonambulatory patients younger than 18 months were not included in the analysis of loss of ambulation. We estimated the median ages at which the events had occurred by plotting Kaplan-Meier curves. Group differences regarding disease variant were analyzed with the log-rank test. We performed linear regression analysis to compare GMFCS, MACS, and CFCS scores in relation to age per disease variant. Statistical analysis was performed using SPSS version 22 (IBM, Armonk, NY) and GraphPad Prism version 6.07 (San Diego, CA).
Results
Patients
A total of 245 patients with MLC from 207 families were diagnosed with MLC by DNA analysis. Three patients were excluded because of comorbidity (Turner syndrome, asphyxia, pituitary adenoma). In the case of limited clinical information due to loss to follow-up or nonresponse, patients were selectively included in the analyses on the basis of availability of information. Throughout the Results section we report the number of patients included in the different analyses in parentheses or in the respective tables. In total, 242 patients with MLC, of whom 187 had MLC1, 17 had MLC2A, and 38 had MLC2B, were included.
Clinical characteristics
Details are presented in table 1. Macrocephaly in the first year of life was the most common first disease sign and had been present in almost all patients in infancy. Macrocephaly persisted in more than half of patients. Secondary normocephaly was particularly common in patients with MLC2B. Initial motor development was reported as mildly delayed in the majority of patients and normal in a smaller number. All patients except 12 patients with MLC1 achieved unsupported walking, at a mean age of 16 months (±8, range 10–72). Initial cognitive development was reported as normal in just over half of patients with MLC1, MLC2A, and MLC2B. Kaplan-Meier curves on start of motor deterioration indicated that both patients with MLC1 and patients with MLC2A showed the first signs of motor deterioration at a median age of 5 years (range 6 months–41 years); there were no significant differences between the curves (p = 0.98; figure 1A). None of the patients with MLC2B had a decline of motor function; in 34% (10/29), some clumsiness remained.
Table 1.
Clinical characteristics at latest phenotyping
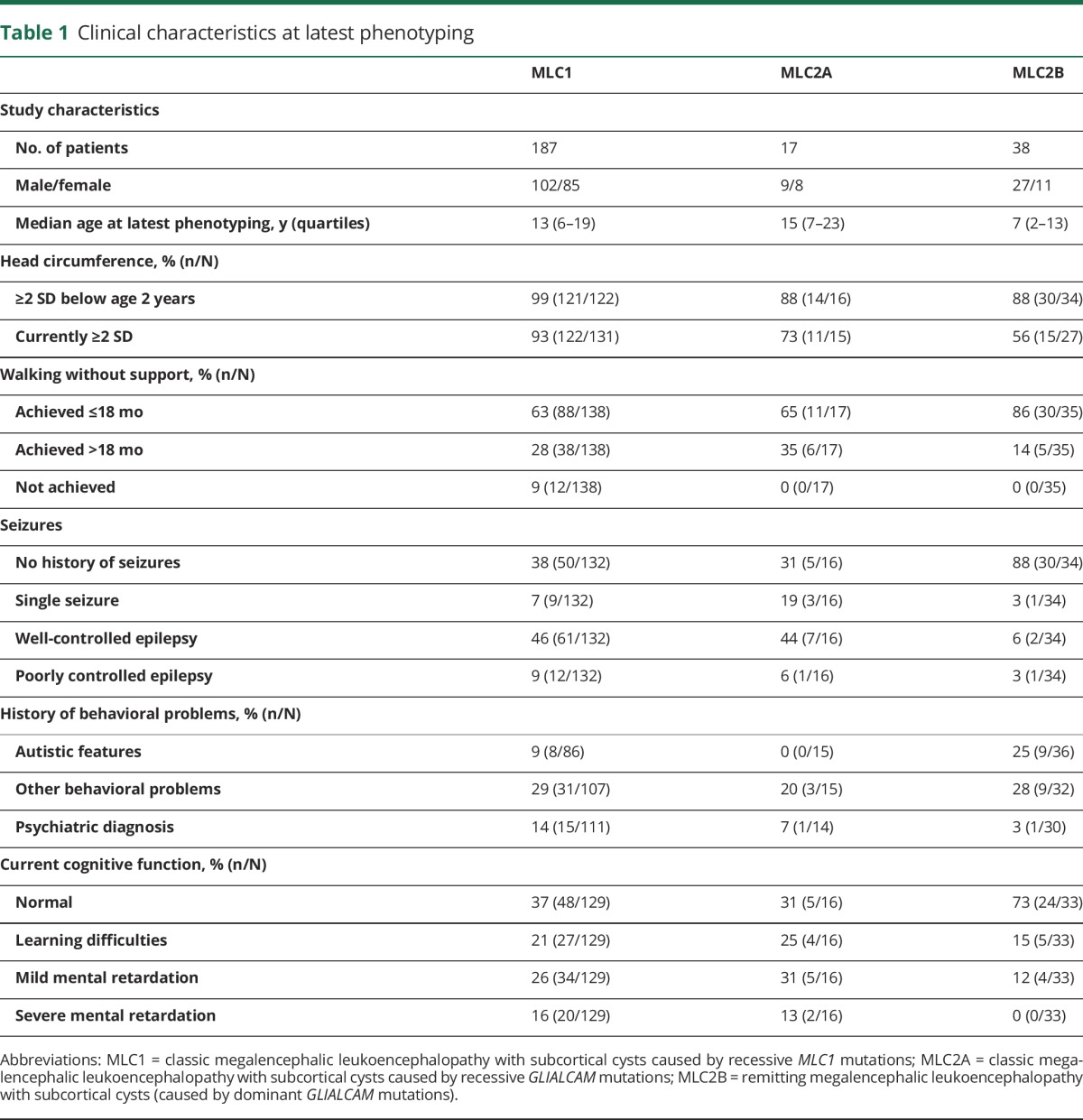
Figure 1. Disease course.

Kaplan-Meier plots on (A) onset of motor deterioration, (B) loss of walking without support, (C) loss of walking with or without support (full wheelchair dependency), (D) onset of seizures, and (E) survival, grouped by disease variant. Censored patients (absence of motor deterioration, still walking without support, still walking with or without support, absence of seizures, or still alive at last follow-up) are indicated by crosses. MLC1 = classic megalencephalic leukoencephalopathy with subcortical cysts caused by recessive MLC1 mutations; MLC2A = classic megalencephalic leukoencephalopathy with subcortical cysts caused by recessive GLIALCAM mutations; MLC2B = remitting megalencephalic leukoencephalopathy with subcortical cysts caused by dominant GLIALCAM mutations.
Loss of walking without support occurred in the majority of patients with MLC1 and MLC2A at ages ranging from 18 months to 43 years; Kaplan-Meier curves showed no significant differences between MLC1 and MLC2A (p = 0.66; figure 1B). Median age at loss of walking without support was estimated to be 15 years in MLC1 and 11 years in MLC2A. Patients with MLC2B all remained ambulatory.
Loss of walking with or without support (full wheelchair dependency) occurred in fewer than half of patients with MLC1 and MLC2A and none of the patients with MLC2B. There were no significant differences between patients with MLC1 and patients with MLC2A (p = 0.58; figure 1C). Patients were most likely to become wheelchair dependent before the age of 15 years; after this age, patients generally remained ambulatory, with or without support.
Behavioral problems were relatively common for all MLC variants; autism was most common among patients with MLC2B. Delayed onset cognitive decline was reported in almost half of patients with MLC1 and MLC2A, and none of the patients with MLC2B. However, stable cognitive impairment was reported in a quarter of patients with MLC2B. The age at start of cognitive decline did not statistically differ between MLC1 and MLC2A (log rank p = 0.10). The patients' cognitive levels at time of the inventory are presented in table 1.
At the time of clinical phenotyping, 109 patients had had at least one seizure, of whom the majority had occasional seizures that were generally well-controlled with medication (table 1). Kaplan-Meier curve analysis indicated that approximately 75% of patients with MLC1 and MLC2A had had one or multiple seizures by age 20 years; the median age at the first seizure was 3 years, mode 2 years (figure 1D). Seizures were less common among patients with MLC2B (n = 4/34). There were no significant differences in onset of seizures between patients with MLC1 and MLC2A (p = 0.64).
Overview of GMFCS, MACS, and CFCS scores in relation to age and disease variant showed that there was a wide variability in clinical severity among patients with MLC1 and MLC2A (figure 2). Linear regression analysis showed no significant differences between the patients with MLC1 and MLC2A regarding GMFCS (p = 0.30), MACS (p = 0.18), and CFCS scores (p = 0.89). Gross motor function and dexterity were generally well-preserved in patients with MLC2B and communication function to a lesser degree.
Figure 2. Function levels.
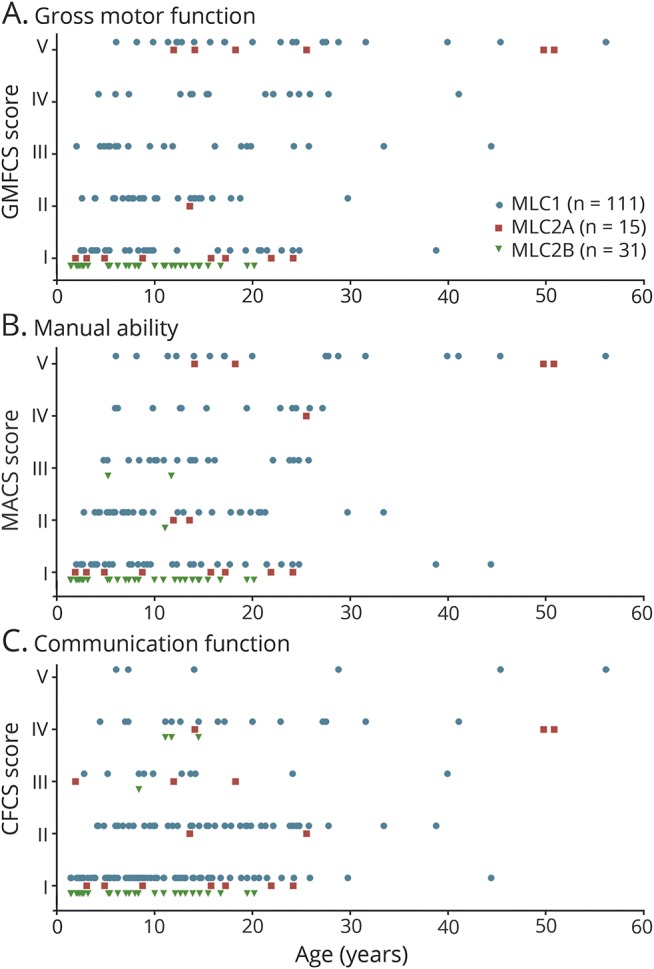
Overview of megalencephalic leukoencephalopathy with subcortical cysts (MLC) patients' scores on (A) the Gross Motor Function Classification System (GMFCS), (B) Manual Ability Classification System (MACS), and (C) Communication Function Classification System (CFCS) in relation to age, grouped by disease variant: MLC1 (classic MLC caused by recessive MLC1 mutations; blue circles), MLC2A (classic MLC caused by recessive GLIALCAM mutations; red squares) and MLC2B (remitting MLC caused by dominant GLIALCAM mutations; green triangles). Scores range from I (no limitations) to V (severe limitations; table e-1, links.lww.com/WNL/A356).
Several patients with classic MLC had very mild disease: 25% (17/68) of patients with MLC1 and 45% (5/11) with MLC2A who were at least 12 years at latest clinical evaluation were able to walk without support and had normal cognition or only learning problems. For ages 12 years and above, 32% (19/59) of patients with MLC1 and 50% (5/10) with MLC2A had GMFCS, MACS, and CFSC scores of I or II.
Survival was assessed in all 242 patients; 7 were deceased (figure 1E). Five deceased patients had MLC1: 4 male patients died at age 7–22 years and 1 female patient at age 56 years. Causes of death were respiratory insufficiency after neurologic deterioration following head trauma, suspected sudden unexpected death in epilepsy (SUDEP), sepsis as complication of surgical procedure, cachexia, and status epilepticus. One deceased male patient with MLC2A and 1 deceased male patient with MLC2B were suspected of SUDEP at ages 23 and 3 years, respectively.
To assess a possible genotype–phenotype correlation in patients with MLC1 and MLC2A, we compared loss of ambulation, wheelchair dependency, and seizures in groups of patients with 3 or more patients from 2 or more families with the same mutations, who were at least 10 years of age at the clinical inventory. The number of patients with MLC2A was too small for evaluation. Six groups of informative patients with MLC1 were available for comparison; characteristics were very divergent within each group (table e-2, links.lww.com/WNL/A356), arguing against a genotype–phenotype correlation.
MRI characteristics
MRIs were available for 187 patients and for 53 patients 1 or more follow-up scans were available; we evaluated a total of 268 MRIs. Patients had their first MRI at a median age of 2 years (range 4 months–55 years). Follow-up scans were obtained at a median age of 5 years (range 11 months–43 years). The median interval between the first and last MRIs was 4 years (range 3 months–15 years).
Details of the first MRIs are presented in table e-3 (links.lww.com/WNL/A356). Extensive, confluent signal abnormalities of the cerebral white matter were invariably present and swelling was observed in all patients, except for one, in whom the first MRI was obtained at 30 years. All patients had sparing of the optic radiation and most patients had sparing in one or more regions of subcortical white matter (figure 3). Several patients also had some regional white matter sparing, mostly in the occipital region (figure 3). There were no clear differences in abnormalities between patients with MLC1 and MLC2A. They all had an abnormal signal of the posterior limb of the internal capsule, while in 54% of patients with MLC2B, in whom the MRI was obtained before the age of 2 years, the signal of the internal capsule was already normal (figure 3). Widening of the ventricles and enlargement of the subarachnoid space was rare in patients with MLC1 and patients with MLC2A in the early disease stage and more common in MRIs performed in adolescence or adulthood. In patients with MLC2B, some enlargement of the subarachnoid spaces was already observed before the age of 2 years in nearly half of patients.
Figure 3. MRI characteristics.

MRI findings in a patient with megalencephalic leukoencephalopathy with subcortical cysts (MLC) caused by recessive MLC1 mutations (MLC1) at age 3 years (top), a patient with MLC caused by recessive GLIALCAM mutations (MLC2A) at age 6 years (middle), and a patient with MLC caused by dominant GLIALCAM mutations (MLC2B) at age 10 months (bottom). Sagittal T1-weighted images show anterior-temporal and frontoparietal cysts in the patient with MLC1 (A), an anterior-temporal cyst in the patient with MLC2A (D), and some near-cystic rarefaction in the anterior-temporal region in the patient with MLC2B (G). The T2-weighted axial images of the hemispheres show diffuse white matter abnormalities with some swelling (B, E, H). The sagittal images show some sparing of the subcortical white matter in all patients, especially in the occipital region (A, D, G); in the patient with MLC2B, the white matter is relatively preserved in the occipital region (G). The patients with classic MLC have a double-line-shaped abnormal signal of the posterior limb of the internal capsule (B, E), while the signal is normal in the patient with MLC2B (H). The T2-weighted axial images of the cerebellum show a mildly hyperintense signal of the cerebellar white matter in the patients with classic MLC (C, F) and a normal signal in the patient with MLC2B (I).
Subcortical cysts were present in all patients with MLC1 and almost all patients with MLC2A. Forty-three percent of patients with MLC2B did not have true cysts, but only near-cystic rarefaction of subcortical white matter. Cysts or near-cystic rarefaction were almost invariably present in both anterior temporal lobes. Additional cysts were located in the frontal and parietal lobes, but never in the occipital lobe. Near-cystic rarefaction of subcortical white matter could be located in all 4 lobes.
The majority of patients with MLC1 and patients with MLC2A (∼85%) had mild signal abnormalities of the cerebellar white matter, which were less pronounced than the cerebral white matter signal abnormalities (figure 3). Patients with MLC2B never had cerebellar white matter signal abnormalities; a few patients younger than 10 months showed some delay in myelination featured by a hyperintense rim of the subcortical cerebellar white matter (figure e-1, links.lww.com/WNL/A355). The cerebellar white matter was never swollen. Almost all patients with MLC1, MLC2A, and MLC2B had mild signal abnormalities in the brainstem.
Change over time
Details are presented in table 2. Normalization of the signal and reversal of the swelling of the white matter only occurred in patients with MLC2B. In patients with MLC1 and patients with MLC2A, the white matter signal abnormalities persisted or increased, with the exception of 2 patients with MLC1 who showed improvement. The number of cysts generally remained the same or increased among patients with MLC1 and MLC2A; in MLC2B, a decrease of cysts and overall quantity of cystic white matter generally occurred, although in all but one patient some cysts or near-cysts persisted.
Table 2.
MRI characteristics: Change over time
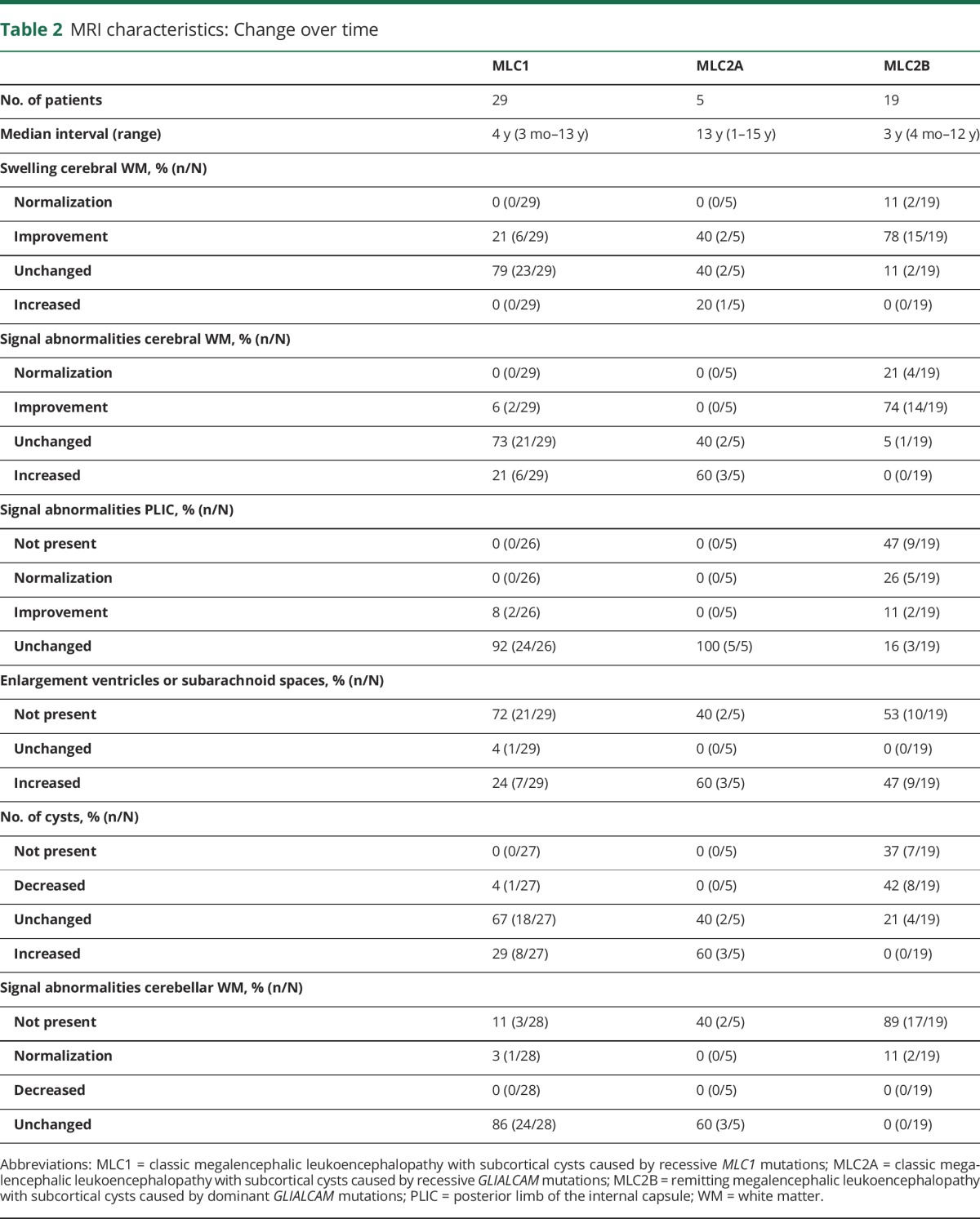
Existing signal abnormalities of the posterior limb of the internal capsule and cerebellar white matter never improved in patients with MLC1 and MLC2A. The few patients with MLC2B who had a rim of slightly abnormal cerebellar white matter on the initial MRI all showed normalization over time.
Discussion
MLC is caused by mutations in MLC1 or GLIALCAM. MLC1 is an astrocyte-specific membrane protein involved in brain ion and water homeostasis.3,17 GlialCAM is a chaperone of MLC1 ensuring its localization in astrocytic endfeet.7,18,19 So, both MLC1 and GLIALCAM mutations affect MLC1 protein function. In addition, GlialCAM is involved in transport of other proteins, such as connexin 43 and the chloride channel ClC2.20,21 Recessive CLCN2 mutations cause a leukoencephalopathy with childhood or adult onset, characterized by mild motor dysfunction and often retinopathy.22 Brain MRI shows restricted diffusion in the posterior limbs of internal capsules, brainstem structures, and cerebellar white matter, suggestive of myelin microvacuolization.22 Pediatric patients have diffuse, mild cerebral white matter signal abnormalities, while these are limited in adult-onset cases. By contrast, in MLC the cerebral white matter abnormalities are profound and diffusion is highly increased, consistent with myelin macrovacuolization and increased extracellular spaces.23 Considering that GlialCAM is supposed to be a chaperone for both MLC1 and ClC2, one might expect GLIALCAM mutations to cause disease features of both MLC1- and CLCN2-associated diseases. The present study, however, confirms that MLC2A is indistinguishable from MLC1 and by no means shares the MRI features of the CLCN2-related disease. The exact roles of MLC1, GlialCAM, and ClC2 in brain ion and water homeostasis and their interaction remain to be elucidated.
The knowledge obtained from our study concerning a relatively large cohort of patients can help physicians in clinical counseling of patients and families. MLC is a fairly mild and slow disorder, with low mortality compared to other leukodystrophies.24 Nevertheless, there is a rather broad variation in clinical severity. Some patients become wheelchair-dependent a few years after onset, while others remain ambulatory during adulthood. An interesting point is that slow motor deterioration often occurs from a few years after presentation onwards, but that patients who are ambulatory with or without support at the age of 15 years most likely remain ambulatory. The study confirms that epilepsy is a common feature in MLC.25 It is puzzling that most patients have well-controlled epilepsy while few patients have refractory epilepsy and ∼25% remain seizure-free. A salient observation is that a considerable proportion of patients with classic MLC exhibit only very mild signs during the disease course; in 2 patients with MLC1, a considerable improvement of the MRI abnormalities occurred. The observed variability in disease severity—also among patients who share the same mutations—may depend on individual differences in compensatory volume regulatory mechanisms. The fact that remarkable improvement of cerebral swelling may occur, as mostly observed in patients with MLC2B but also in a few patients with classic MLC, suggests there is a window of opportunity for therapy.
The multi-institutional and observational nature of this study comes with certain limitations. In the absence of formal testing, measures like cognitive function and presence of autistic features were scored based on subjective assessment by physicians and caretakers. The application of time-to-event analysis and standardized scales such as GMFCS resulted in a robust representation of disease progression, but missing data and interobserver differences may have hampered the evaluation of the clinical course. There are, however, no indications that nonresponse was in any way systematic or that censoring in the time-to-event analyses was informative. The study limitations are at least in part compensated by the study size considering the disease is very rare. The available results give an already informative delineation of the clinical spectrum of MLC. Our ongoing data collection will help include larger numbers of patients with MLC2A and extend the follow-up.
One of the aims of this study was to identify features that can help distinguish different MLC variants in early disease stages. Absence of MRI signal abnormalities of the posterior limb of the internal capsule and cerebellar white matter and presence of only rarefied subcortical white matter instead of actual cysts are features suggestive of MLC2B. This knowledge can be applied to direct genetic testing in patients suspected of the remitting phenotype. No distinguishing features have been identified to discern MLC1 and MLC2A. Considering the much higher prevalence of MLC1 gene defects, in cases of a classic presentation of MLC, this gene should be tested first.
Acknowledgment
The authors thank the patients and families for their cooperation and contribution.
Glossary
- CFCS
Communication Function Classification System
- GMFCS
Gross Motor Function Classification System
- MACS
Manual Ability Classification System
- MLC
megalencephalic leukoencephalopathy with subcortical cysts
- MLC1
classic megalencephalic leukoencephalopathy with subcortical cysts caused by recessive MLC1 mutations
- MLC2A
classic megalencephalic leukoencephalopathy with subcortical cysts caused by recessive GLIALCAM mutations
- MLC2B
remitting megalencephalic leukoencephalopathy with subcortical cysts caused by dominant GLIALCAM mutations
- SUDEP
suspected sudden unexpected death in epilepsy
Footnotes
CME Course: NPub.org/cmelist
Contributor Information
Collaborators: MLC Research Group, Hugo H. Abarca Barriga, Samer Abdelrazeq, Gül Aktan Serdaroğlu, P. Ian Andrews, Richard Appleton, Lucia Argandoña Palacios, Brenda Banwell, Florian Bauder, Gulcin Benbir, Tim Benke, Susan Blaser, Annette Bley, Cristiana Brenner, Knut Brockmann, Rafael Camino, Coriene Catsman-Berrevoets, Yanick Crow, Marguerite Dalton, María de la Luz Arenas-Sordo, Linda de Meirleir, Ana Isabel Dias, Francis J. DiMario, Maria Alice Donati, Nihal Olgac Dundar, Francois Feillet, Maria-Jose Fonseca, Emilio Franzoni, Jeremy Freeman, Katsunori Fujii, Soumya Ghosh, Scott Gold, Solange Gril, Barbara Hallinan, Agnes Herczegfalvi, Jozef Hertecant, Joannie Hui, David Hunt, Parul Jayakar, Bulent Kara, Çiğdem S. Kasapkara, Gulsen Kocaman, David M. Koeller, Wolfgang Köhler, Alfried Kohlschütter, Marja Koivusalo, Urania Kotzaeridou, Roshan Koul, Ingeborg Krägeloh-Mann, Ruzica Kravljanac, Gerhard Kurlemann, Julian Lara Herguedas, Silvia Laurentino, Richard Leventer, Bryan Lynch, Oliver Maier, Sascha Meyer, Olivera Miljanovic, José Paulo Monteiro, Ellen Moran, Teresa Moreno, Jacques Motte, Chris Moyes, Lakshmi Nagarajan, Marie-Cecile Nassogne, Slavica Ostojic, Peter Pietsch, Iliana Porfiri, Sofia Quintas, Maria Belen Ramos, Deborah Renaud, Biserka Rešić, Carolina Rivera Nieto, Jutta Rummel, Robert Rusina, Mustafa A. Salih, Sabine Scholl-Bürgi, Bitten Schönewolf-Greulich, Snehal Shah, Suvasini Sharma, Gabriella Silvestri, Komudi Siriwardena, Victoria Siu, Anne-Bine Skytte, Zeyneb Soysal, Carlos Eduardo Speck Martins, Angela Sun, Burak Tatli, Gareth Thomas, Virpi Toivio, Leyla Tümer, Jean-Claude Turpin, Adeline Vanderver, Helene Verhelst, Ishwar Verma, Rocio Villafuerte-de la Cruz, Roberta Vittorini, Evangeline Wassmer, Claudia Weiß, Janine Whale, Sau Wei Wong, Elif Yilmaz Gulec, and Uluc Yiş
Author contributions
Eline M.C. Hamilton collected and analyzed patient and imaging data and wrote the manuscript. Pinar Tekturk collected and analyzed patient and imaging data. Fia Cialdella collected and analyzed patient and imaging data. Diane F. van Rappard collected and analyzed patient data. Nicole I. Wolf collected and analyzed patient and imaging data. Cengiz Yalcinkaya collected and analyzed patient and imaging data. Ümran Çetinçelik collected and analyzed patient and imaging data. Ahmad Rajaee collected and analyzed patient and imaging data. Ariana Kariminejad collected and analyzed patient and imaging data. Justyna Paprocka collected and analyzed patient and imaging data. Zuhal Yapici collected and analyzed patient and imaging data. Vlatka Mejaški Bošnjak collected and analyzed patient and imaging data. Marjo S. van der Knaap conceptualized and coordinated the project, collected and analyzed patient and imaging data, and wrote the manuscript.
Study funding
This study received financial support from the Optimix Foundation for Scientific Research.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.van der Knaap MS, Barth PG, Stroink H, et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol 1995;37:324–334. [DOI] [PubMed] [Google Scholar]
- 2.Singhal BS, Gursahani RD, Udani VP, Biniwale AA. Megalencephalic leukodystrophy in an Asian Indian ethnic group. Pediatr Neurol 1996;14:291–296. [DOI] [PubMed] [Google Scholar]
- 3.Ridder MC, Boor I, Lodder JC, et al. Megalencephalic leucoencephalopathy with cysts: defect in chloride currents and cell volume regulation. Brain 2011;134:3342–3354. [DOI] [PubMed] [Google Scholar]
- 4.van der Knaap MS, Valk J, Barth PG, Smit LM, van Engelen BG, Tortori Donati P. Leukoencephalopathy with swelling in children and adolescents: MRI patterns and differential diagnosis. Neuroradiology 1995;37:679–686. [DOI] [PubMed] [Google Scholar]
- 5.van der Knaap MS, Lai V, Kohler W, et al. Megalencephalic leukoencephalopathy with cysts without MLC1 defect. Ann Neurol 2010;67:834–837. [DOI] [PubMed] [Google Scholar]
- 6.van der Knaap MS, Boor I, Estevez R. Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol 2012;11:973–985. [DOI] [PubMed] [Google Scholar]
- 7.Lopez-Hernandez T, Ridder MC, Montolio M, et al. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet 2011;88:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singhal BS, Gorospe JR, Naidu S. Megalencephalic leukoencephalopathy with subcortical cysts. J Child Neurol 2003;18:646–652. [DOI] [PubMed] [Google Scholar]
- 9.Patrono C, Di Giacinto G, Eymard-Pierre E, et al. Genetic heterogeneity of megalencephalic leukoencephalopathy and subcortical cysts. Neurology 2003;61:534–537. [DOI] [PubMed] [Google Scholar]
- 10.Kariminejad A, Rajaee A, Ashrafi MR, et al. Eight novel mutations in MLC1 from 18 Iranian patients with megalencephalic leukoencephalopathy with subcortical cysts. Eur J Med Genet 2015;58:71–74. [DOI] [PubMed] [Google Scholar]
- 11.Cao B, Yan H, Guo M, et al. Ten novel mutations in Chinese patients with megalencephalic leukoencephalopathy with subcortical cysts and a long-term follow-up Research. PLoS One 2016;11:e0157258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ilja Boor PK, de Groot K, Mejaski-Bosnjak V, et al. Megalencephalic leukoencephalopathy with subcortical cysts: an update and extended mutation analysis of MLC1. Hum Mutat 2006;27:505–512. [DOI] [PubMed] [Google Scholar]
- 13.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hidecker MJ, Paneth N, Rosenbaum PL, et al. Developing and validating the communication function classification system for individuals with cerebral palsy. Dev Med Child Neurol 2011;53:704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Knaap MS, Breiter SN, Naidu S, Hart AA, Valk J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999;213:121–133. [DOI] [PubMed] [Google Scholar]
- 16.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubey M, Bugiani M, Ridder MC, et al. Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Ann Neurol 2015;77:114–131. [DOI] [PubMed] [Google Scholar]
- 18.Lopez-Hernandez T, Sirisi S, Capdevila-Nortes X, et al. Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum Mol Genet 2011;20:3266–3277. [DOI] [PubMed] [Google Scholar]
- 19.Capdevila-Nortes X, Lopez-Hernandez T, Apaja PM, et al. Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum Mol Genet 2013;22:4405–4416. [DOI] [PubMed] [Google Scholar]
- 20.Jeworutzki E, Lopez-Hernandez T, Capdevila-Nortes X, et al. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron 2012;73:951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu M, Moh MC, Schwarz H. HepaCAM associates with connexin 43 and enhances its localization in cellular junctions. Sci Rep 2016;6:36218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Depienne C, Bugiani M, Dupuits C, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 2013;12:659–668. [DOI] [PubMed] [Google Scholar]
- 23.van der Voorn JP, Pouwels PJ, Hart AA, et al. Childhood white matter disorders: quantitative MR imaging and spectroscopy. Radiology 2006;241:510–517. [DOI] [PubMed] [Google Scholar]
- 24.Brimley CJ, Lopez J, van Haren K, et al. National variation in costs and mortality for leukodystrophy patients in US children's hospitals. Pediatr Neurol 2013;49:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubey M, Brouwers E, Hamilton EM, et al. Seizures and disturbed brain potassium dynamics in the leukodystrophy MLC. Ann Neurol Epub 2018 Feb 21. [DOI] [PMC free article] [PubMed] [Google Scholar]