

Abstract
There has been a very limited number of high-throughput screening campaigns carried out with Leishmania drug targets. In part, this is due to the small number of suitable target genes that have been shown by genetic or chemical methods to be essential for the parasite. In this perspective, we discuss the state of genetic target validation in the field of Leishmania research and review the 200 Leishmania genes and 36 Trypanosoma cruzi genes for which gene deletion attempts have been made since the first published case in 1990. We define a quality score for the different genetic deletion techniques that can be used to identify potential drug targets. We also discuss how the advances in genome-scale gene disruption techniques have been used to assist target-based and phenotypic-based drug development in other parasitic protozoa and why Leishmania has lacked a similar approach so far. The prospects for this scale of work are considered in the context of the application of CRISPR/Cas9 gene editing as a useful tool in Leishmania.
Keywords: Leishmania, gene knockouts, null, CRISPR/Cas9, target validation, pathogen, Trypanosoma cruzi, drug discovery
Leishmaniasis is a disease that mainly affects those burdened by extreme poverty, across large swaths of the tropics and subtropics. It consists of a spectrum of human and animal diseases caused by at least 20 species of parasites in the genus Leishmania. The symptoms range from self-limiting ulcers to the destruction of mucocutaneous surfaces to fatal disease caused by visceral leishmaniasis; dermal sequelae can also occur following the cure of primary visceral disease. There are up to 1.2 million cases of cutaneous leishmaniasis and up to 0.4 million cases of visceral leishmaniasis each year, resulting in up to 30 000 deaths.1 Collectively, 350 million people live at risk of catching leishmaniasis due to the wide geographic range of the vector insects, phlebotomine sand flies. There are medicines available to treat these diseases, but they have notable deficiencies due to emerging resistance, poor safety profiles, and the long duration of treatment, so more effective treatments with improved safety and dosing profiles are desirable. The complex picture of a spectrum of diseases, in several organs, caused by different parasite species with differing biology means that several new medicines may be required to satisfy quite stringent target product profiles (TPPs) for visceral and cutaneous leishmaniasis.2,3 New chemical entities will probably require different chemical features for achieving the desired bioavailability in skin versus viscera coupled with the challenge of drugging an intracellular parasite that resides in a phagolysosomal compartment. There is growing appreciation that Leishmania infections can also form quiescent, persistent forms.4 Targeting these stages is desirable, though it may also prove challenging. There are a number of ongoing drug discovery programs that aim to address this need, typically working within defined phenotypic or target-based screening strategies to best achieve success.2,3,5−8 Phenotypic or whole organism screening selects for compounds that cause a loss of parasite fitness, whereas the target-based approach involves screening against a selected target of interest thought to be essential to parasite survival. We will assess these approaches and the role that genetic target validation can play within them.
Target Validation in Drug Discovery Programs
Drug target validation for antiparasitic compounds consists of acquiring the evidence that defines whether a target molecule (usually a protein/enzyme) is selectively inhibited by a chemical entity leading to the death of the parasite as well as the linkage of the target molecule to an essential parasite process. The evidence used to validate targets can arise from genetic manipulations of a pathogen or from the interrogation of the organism with specific chemical probes. The strength of validation will depend on how much evidence is accrued, and well-validated targets will be supported by both genetic and chemical evidence.5,9 For noninfectious diseases, it is estimated that selecting genetically supported targets doubles the success rate in later stages of clinical development,10 highlighting the importance of genetic validation. The best validation of all comes when the molecular target for a compound is known and the compound is available on the market for clinical use, the one example for trypanosomatids being the treatment of human African trypanosomiasis with Eflornithine (difluoromethylornithine, DFMO), a compound that inhibits ornithine decarboxylase.11 It is notable that for most antileishmanial drugs (miltefosine, paromomycin, antimonials, and pentamidine) there are no target proteins identified despite extensive research, although amphotericin B is reported to specifically target ergosterol-containing membranes.12,13
Phenotypic Screening
For Leishmania, several hit compounds have been identified by phenotypic-based drug discovery, which involves screening druglike molecules against Leishmania axenic amastigotes or amastigotes in macrophages to identify compounds that kill the parasites. This approach has the advantage in that it can identify bioactive compounds that have appropriate cell permeability characteristics to kill the parasite within the parasitophorous vacuole. One disadvantage is that the screening is very stringent; consequently, few bioactive compounds are identified.5 Another disadvantage is that one does not immediately know the molecule being targeted by the bioactive compounds. If a large (>1 million compounds) screen is performed, then it may conceivably identify diverse target-class inhibitors whereas sublibraries can be used to increase the chances of hits within a certain target class, for example, by using a protein kinase inhibitor-focused library.14 The optimization of hit compounds by medicinal chemistry involves a level of randomness in which it may not be immediately apparent which functional groups of the molecule are the important pharmacophores. Structural activity analyses are performed in whole cell assays, and it is not possible to know if increases in activity against parasites are concurrent with activity against a specific target. Therefore, a campaign of target deconvolution is often initiated to identify the molecular target of a hit compound. This can involve a combination of forward genetic screens such as the generation of resistant parasites and the identification of mutant alleles in the target gene by whole genome sequencing;15 affinity purification of the target using a compound conjugated to beads;16 and the use of overexpression libraries17,18 or metabolomics to identify blocks in biochemical pathways.19 It is also important to have an understanding of an experimental compound’s mode of action as this can enable an assessment of the likelihood of resistance mechanisms evolving in the parasite. Recent successful examples of the phenotypic screening of large compound libraries or focused subset chemical libraries against Leishmania parasites include the identification of the “Leish-Box” of inhibitors by GSK,20 the identification of a selective proteasome inhibitor (GNF6702) by Novartis,15 and the identification of natural product inhibitors.21
Target-Based Drug Discovery
The alternative strategy of target-based drug discovery is used extensively by the pharmaceutical industry and has been applied to Leishmania; this consists of the selection of a protein target and the development of a biochemical or biophysical assay that can be used to identify inhibitors of that target, followed by a high-throughput screening (HTS) campaign. Hit compounds from such screens are validated and then chemically optimized into lead compounds that have improved properties in terms of potency, selectivity, and bioavailability. This is a resource-intensive and costly process and needs to be built on a solid understanding of why the selected target is an appropriate choice, a process defined as target assessment. Drug targets are evaluated on the basis of gene essentiality in addition to other criteria, for example, the presence of close homologues in the parasite genome that may allow for an easier evolution of resistance through functional redundancy. Alternatively, single-point mutations in a gene may lead to drug resistance with little associated cost to parasite fitness. These examples emphasize the importance of a good understanding of parasite genetics for drug target prioritization. Other parameters for target assessment include the potential druggability of the protein, the availability of in vitro assays, the availability of structural information, and the presence of structurally similar proteins in the host proteome that may present a potential risk for host toxicity. The assessment of a target against each of these criteria can be scored, generating a “traffic lights” rating: red ratings will typically stop a target from progression, but those scoring green should progress more easily through development.22 The level of rigor in building a strong evidence base to support target-focused approaches means that many targets are shown to be invalid. For these reasons as well as due to the limitation of financial and material resources, there have been notably few HTS campaigns against Leishmania proteins, namely, CRK3 (cyclin-dependent kinase),23 NMT (N-myristoyltransferase),24 and PTR1 (pteridine reductase).25 Casein kinase (LmCK1.2) has also been the subject of an HTS campaign14 after being chemically validated as a target.26,27
Drug Repurposing and Piggybacking
Alternatives to developing or identifying new chemical entities to target Leishmania exist in the form of drug repurposing, a strategy where a compound that has been developed to treat one disease is used to treat another, different disease. The benefit of drug repurposing is that the treatment can be used in patients relatively quickly and with lower cost compared to de novo drug discovery. Notable examples for leishmaniasis include the use of miltefosine, amphotericin B, and pentamidine, which were all designed or approved for other indications.6 Piggybacking is the concept of identifying existing pharmaceutical material, investment, and knowledge around a given target or inhibitor class and applying it to infectious disease targets, but this usually stops short of direct drug repurposing.
Genetic Target Validation in Leishmania
Leishmania has some biological features that have made it somewhat difficult to genetically manipulate; it is highly prone to aneuploidy, meaning that genetic modification of essential genes can be challenging or unstable.28−30 RNA interference does not function in most Leishmania species.31 In those species where RNAi does function, it has not been exploited to the same extent as Trypanosoma brucei, primarily due to the lack of an inducible system to investigate the function of essential genes. Leishmania parasites are easily cultured and genetically manipulated as promastigote stages, those found in the insect vector, but these are not the relevant stage for human disease. It is therefore very important to confirm the essentiality of a potential target using the amastigote stages of the parasite lifecycle. Some species of Leishmania, such as L. mexicana, are readily converted to axenic amastigotes in vitro32 and this can facilitate the study of gene function in the pathogenic stages, but phenotypic analysis is perhaps best carried out using macrophages to perform intracellular amastigote growth and survival assays and/or mouse infections. Amastigotes can be genetically manipulated, but this is not a common practice.33 Typically, an in vivo model is used to demonstrate a fitness or virulence defect of a genetically engineered mutant.
Leishmania possess a very efficient homologous recombination pathway for DNA repair that has been exploited as a basis for generating gene deletions, integrating epitope tags, or expressing transgenes.34 (See Table 1 for a definition of terms.) As the technology to manipulate Leishmania has improved over time, so has the ability to achieve more confidence in a gene’s essentiality. On the basis of the literature, we used a scoring system for the strength of validation resulting from gene deletion studies, on a 1–5-star scale (Table 1 and Figure 1), and we discuss these below, with examples. We propose that the higher scoring methods provide better-quality evidence of a gene’s essentiality but acknowledge that they are also associated with increased time and effort to generate the mutants. Despite this, we hope to use this guide to inform good practices for Leishmania genetic manipulation.
Table 1. Definitions of Key Terms.
| term | definition | synonym |
|---|---|---|
| allele | This is a variant form of a gene. For diploid chromosomes, there are two alleles in the nuclear DNA (if identical, these are homozygous; if different, these are heterozygous). Additional alleles or copies of a gene can be complemented on an episome or integrated into the nuclear DNA in wild-type, mutant, or floxed forms. | |
| gene deletion by double homologous replacement | An allele of a gene is replaced by a drug resistance marker, resulting in deletion of the coding DNA sequence of the allele. This is performed twice, in sequential steps, with two different drug resistance markers to result in the replacement of two alleles of a gene, yielding a null mutant if the gene is not essential. | knockout (verb) |
| null mutant | This is a parasite strain that lacks both alleles of a gene of interest after a deletion strategy. | knockout (noun) |
| facilitated null mutant | Both chromosomal alleles can be deleted, but this requires prior genetic (episomal allele) or nutritional supplementation. | episomal rescue, conditional null mutant |
| unforced plasmid shuffle | Drug selection for a plasmid episome that complements a chromosomal null mutant is removed, and the loss or retention of this plasmid is analyzed. | plasmid cure |
| forced plasmid shuffle | The complementing plasmid episome contains both a positive and negative selectable marker, which allows a selection pressure to be imposed to favor parasites that lose the episome. | |
| inducible gene deletion | A parasite line is engineered to contain an allele that can be removed by the addition of a trigger, for example, the rapamycin-induced DiCre recombinase deletion of a floxed allele. | |
| floxed | Flanked by the loxP (locus of X-overP1) site, this term is used to describe alleles of genes that can be inducibly deleted by a Cre or DiCre (inducible dimerizable Cre) recombinase system. | |
| complementation | This involves the restoration of null mutants by the addition of an extra allele of the gene of interest (episomally or genomically) or by the addition of a nutritional supplement in the case of a gene that encodes a metabolic enzyme. | add back |
| episome | A circular DNA molecule that can be replicated independently of the nuclear or kinetoplast DNA. | plasmid |
Figure 1.

Overview of techniques that can be used for genetic target validation in Leishmania. (a) Gene deletion by homologous replacement. Drug resistance markers are targeted to the gene of interest by long homology flanks (0.5–1 kb) in sequential transfections by electroporation. This process can now be facilitated using CRISPR/Cas9 and short homology flanked cassettes in a single transfection. Deletions targeting essential genes will result in cell death and failure to isolate null mutants or in ploidy changes that allow the cell to retain alleles of the wild-type locus as well as drug resistance markers. (b) Facilitated null mutant with unforced plasmid shuffle. An episome expressing the gene of interest is first transfected into the cell line or a nutritional supplement is provided, allowing it to survive the subsequent deletion of the chromosomal alleles of the gene of interest. The drug selection pressure for the episome can be removed and retention of the plasmid can be determined if the gene is not essential; then it will be possible to isolate parasites that lack the episome. (c) Forced plasmid shuffle. As in B, an episome expressing the gene of interest is transfected into the parasite to allow the deletion of the chromosomal alleles of the target gene. The episome also encodes a negative-selectable marker, herpes simplex virus thymidine kinase. Selection with ganciclovir favors the survival of parasites that lack the episome, so if a gene is nonessential, the episome will rapidly be lost from the population but will be retained for an essential gene despite the associated costs. The addition of a second episome containing mutant versions of the gene of interest allows for exploration of the roles of specific domains and residues in the encoded protein for correct gene function by assessing which plasmid of the two is preferentially retained. (d) DiCre inducible gene deletion. One allele of the target gene is replaced by a drug-selectable cassette containing a “floxed” allele, and in a second transfection stage, the remaining allele is replaced by a second drug resistance marker. The addition of rapamycin induces DiCre dimerization and excision of the floxed allele, and the phenotypes that emerge in the induced null mutants can then be analyzed. As in C, complementation allows for the assessment of null mutant specificity and functional assessment of defined domains or residues in the protein. In all panels, the number of stars indicate the quality of the genetic evidence for gene essentiality, with one star being the weakest and five stars being the strongest.
Table 2. Scoring System to Assess the Quality of Evidence for Target Essentiality by Gene Deletion Techniques.
|
Leishmania |
Trypanosoma cruzi |
|||||
|---|---|---|---|---|---|---|
| quality score | definition | issues | number | genes | number | genes |
| * | failure of attempt to perform gene deletion by double homologous replacement | changes in ploidy indicative of essentiality; may be a technical failure | 30 | TR, A2-A2REL, SODB1, LACK, PK, CRN12, TOR1, TOR2, SPASE I, MYO21, HSLU1, LHR1, HUS1, HEMAC, ENDOG, ASNA, ARP, RAD51-6, AIRK, LMIT1, RAD50, TYRRS, ABC3, RAB5A, RAB5B, TWF, SIR2RP2, SODA, ACECS, CPN10 | 15 | TC52, DHOD, GALE, DHFR-TS, ECH1, SUB2, GPI8, GPI12, CRT, IP3R, RPA2, GALF, CYP51, STI1 |
| ** | facilitated null mutant; gene of interest is complemented with an episome or nutritional supplement, allowing genomic alleles to be deleted | shows the gene locus can be targeted for deletion | 20 | TUB, CRK1, NMT, SIR2, RPC2, TOPS, TXN1, GSH1, GLO1, SGT, DHS34, HSLV, NTR, TRYS, CYP51EIF4E,RAD51-3, RPIB, RAD9, LYSRS-1 | 1 | NMT |
| *** | unforced plasmid shuffle; plasmid retained in the absence of antibiotic selection | indirect evidence for a gene being essential; best carried out in vivo | 12 | PRT1, ODC, DHFR-TS, SPSDS, ADOMETDC, ARG, DHCH1, STI1, UMPS, CPS, UPRT | ||
| **** | forced plasmid shuffle or DiCre inducible deletion | death of parasite after induction used as evidence of essentiality | 2 | H2A.Z, H2B.V, MPK4 | ||
| ***** | as above but with analysis in amastigote stages and/or the mouse model | application to amastigotes provides best evidence for essentiality in vivo | 1 | CRK3 | ||
Approaches to the Genetic Manipulation of Leishmania
Deletion by Double Homologous Replacement and Facilitated Null Mutants
In 1990, it was demonstrated that genes could be deleted from Leishmania promastigotes by homologous replacement with linear dsDNA.35 The authors demonstrated proof of concept, which involved deleting a single allele of DHFR-TS in wild-type L. major. This was then repeated in a strain that was already identified as heterozygous for DHFR-TS, which permitted the deletion of the remaining allele using a homology flanked neomycin resistance cassette. The generation of a DHFR-TS null mutant led to thymidine auxotrophy and could only be achieved in the presence of thymidine nutritional supplementation; removal of thymidine supplementation stalled cell growth, demonstrating that DHFR-TS was essential for parasite survival. The complementation of the null mutant with an episome expressing DHFR-TS restored wild-type growth levels, indicating that the function of this gene could be reconstituted genetically as well as chemically. This set the stage for other researchers to begin performing reverse genetic analyses in Leishmania. The development of new resistance markers allowed for gene deletions of two alleles of a gene,36 given that Leishmania was considered at the time to have a diploid genome that allowed for the generation of null mutants from wild-type backgrounds. In the following years, a number of papers demonstrated that upon deletion of both alleles of a suspected essential gene the system would fail. Either no transfectants would survive the drug selections, parasites would survive but had duplicated their genome to retain the gene of interest as well as the drug resistance markers,36 or there were extra copies of the gene remaining on supernumerary chromosomes or ectopic elements (Figure 1A). At the time, this was considered evidence for gene essentiality, but with the inability to perform any further, this is classified as the weakest form of evidence (one star) as the failure to generate a null mutant may result from a simple technical failure. For example, as the technology for performing electroporation became more sophisticated, transfection efficiencies increased, allowing some previously intractable genes to be revealed as nonessential. The Metacaspase gene, MCA, initially could not be deleted from L. major,37 and overexpression caused a growth defect, which was interpreted to mean that the gene played an essential role in cellular proliferation. However, MCA null mutants could be generated in both L. major(38) and L. mexicana(39) using a newer high-efficiency electroporation system that causes minimal cellular damage. Generating this deletion allowed for the characterization of the role of MCA in amastigotes and mice where the null mutant exhibited an increase in virulence.39
Several studies have performed deletions that resulted in parasites auxotrophic for a given metabolite and required chemical supplementation to allow null mutants to be recovered35,40 (Table S1). The advantage of a chemical complementation is that it allows for rapid withdrawal and the resulting phenotype was investigated. Where nutritional supplementation could not be used to facilitate the deletion of essential genes, researchers began to introduce an episomal copy of the gene of interest into the parasite prior to the deletion of the two chromosomal alleles41 (Figure 1B). This approach increases the confidence in the validation as it demonstrates that the failure to delete both alleles of the gene of interest is not a technical failure or due to the locus being refractory to genetic manipulation (Figure 2B, two stars).
Figure 2.
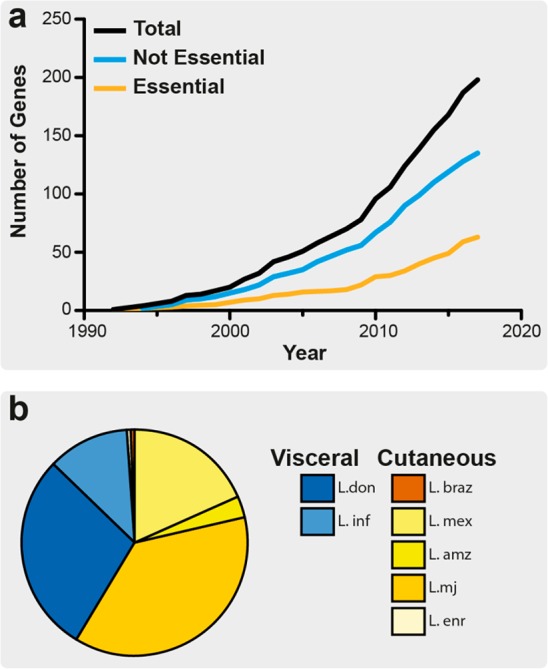
Overview of the number of Leishmania genes with published attempts at the creation of a null mutant. (a) Line graph depicting the cumulative number of genes for which attempts have been made to generate null mutants, for human infective Leishmania species. Only the first attempt at a gene deletion for each individual gene was recorded. Data from this study were ordered by year, and cumulative values of publications per year were derived, where the total number of attempted gene deletions is shown as well as the number of essential genes identified. (b) Pie chart showing the proportion of unique gene deletion attempts by species of Leishmania. Cutaneous species are shaded in yellows, and visceral species, in blues.
An episome expressing a gene in a null mutant background will be retained if it confers a selective advantage for growth, even if the gene is dispensable for parasite viability. In a facilitated null mutant of a nonessential gene, the episome can be lost in the absence of drug selection, and a null mutant is generated. For essential genes, even after prolonged culturing of the facilitated null mutant in a medium that lacks drug selection for the episome or by passaging the parasites through mice, the episome will be retained. This is considered indirect evidence that the gene is essential (Figure 1B, three stars). However, most episomes cannot be forced from the parasite, so this still does not provide direct evidence for gene essentiality or allow the phenotype analysis of loss-of-function mutants.
Facilitated Null with Negative Selection/Forced Plasmid Shuffle
As an improvement in the ability to select for the loss of the episome in a facilitated null mutant, negative selection or forced plasmid shuffle can be achieved using negative selectable marker herpes simplex thymidine kinase (TK). Ganciclovir is used to impose a fitness cost on parasites that retain the plasmid, selecting for parasites that lose the episome. If it is complementing an essential gene, then the parasites must retain the plasmid to remain viable, so any loss will prove fatal. The inclusion of a green fluorescent protein (GFP) reporter gene helps to distinguish cells containing the episome, which is useful for fluorescence-activated cell sorting (FACS) of different populations of cells (Figure 1C).
This strategy was first applied to Leishmania major to investigate the role of 5,10-methylene tetrahydrofolate dehydrogenase (DHCH) in 10-formyl tetrahydrofolate (10-CHO-THF) metabolism42 following the failure of classic gene deletion and nutritional supplementation strategies. DHCH1 was expressed from an episome (also containing TK and GFP), allowing a facilitated null mutant to be generated. The episome was selected against using ganciclovir, and parasites were then flow sorted into GFP bright or dim populations and cloned into 96-well plates. The dim clones, lacking the DHCH1 episome, were not viable and failed to grow. This system has also been used to examine the essentiality of MAP kinase 4 (MPK4) in L. major promastigotes in a manner allowing more detailed functional genetic analysis.43 A facilitated null mutant was achieved by episomal complementation, followed by forced plasmid shuffle to assess gene essentiality. After ascertaining that MPK4 was essential for promastigote viability, the authors complemented with a panel of secondary episomes (lacking TK) containing functionally mutated versions of MPK4. This allowed for exploration of the importance of protein motifs and residues for MPK4 ATP binding, activity, and activation by upstream MKKs. This study highlighted the power of plasmid shuffle in that an MPK4 mutant with altered ATP binding properties, predicted to have reduced protein kinase activity, could replace wild-type MPK4, resulting in resistance to acidic stress. The results from this study support previous facilitated null and unforced plasmid shuffle data suggesting that MPK4 from L. mexicana(44) is essential to the mammalian infection cycle. Profiling of null mutant phenotypes in the amastigote stage is critical to the validation of drug targets, so the compatibility of the plasmid shuffle with this is welcome. However, a limitation of the plasmid shuffle approach is that null mutants of essential genes are never generated in the absence of ganciclovir (which acts as a cytostatic but not a cytocidal drug), so direct analysis of the null mutant phenotypes is not possible. In reference to the suitability of MPK4 as a drug target, it has so far been impossible to identify a biochemical assay for the screening of small-molecule inhibitors, mainly due to its lack of protein kinase activity against generic substrates.43 Currently, this lack of an enzymatic activity assay renders MPK4 a genetically well-validated but poorly tractable target for drug discovery programs.
DiCre Inducible Deletion
In order to directly measure the loss of gene function, a population of cells would ideally be subjected to an inducible gene deletion approach, allowing synchronous, direct analysis of emerging phenotypes in real time. A technique that has recently been applied to Leishmania to directly study gene function is the DiCre inducible gene deletion strategy45 (Figure 1D). This was first used in Leishmania mexicana to inducibly delete the cyclin-dependent protein kinase, CRK3, a gene that had previous indirect evidence for essentiality in Leishmania.46 Prior to the study by Duncan et al., there was a significant body of indirect genetic evidence for CRK3 essentiality, such as changes in ploidy associated with attempts to delete both alleles and that it was possible to delete both chromosomal alleles only in the presence of an episome expressing CRK3, with the episome being retained in the absence of drug selection.46 Other chemical evidence existed for CRK3 to be a potential drug target as known inhibitors of CDKs were shown to induce a G2/M cell cycle block and halt parasite growth.46−48
This DiCre-mediated deletion of CRK3 involved replacing a genomic allele of the gene with a version flanked by loxP sites (floxed), the second copy replaced by a cassette encoding two subunits of the Cre recombinase, fused to rapamycin binding domains45 (Figure 1D). Once this strain was generated, the dimerization of Cre subunits was initiated by the addition of rapamycin, forming a functional Cre recombinase that excised the remaining floxed copy of the gene of interest, resulting in a null mutant (Figure 1D). The advantage of this system is that phenotypes emerging from the gene deletion can be studied over several days, and the mechanism of cell death or dysregulation can be elucidated. The disadvantage is that RNA and protein remain in the cell after DiCre-induced deletion of the gene and the phenotype may take several rounds of cell division before becoming apparent.
By performing the DiCre analysis, Duncan et al. were able to delete the CRK3flx gene from promastigote cells and observe precisely the phenotype resulting in CRK3 null mutant–cell cycle (G2/M) arrest, the initiation of G1 phase in the absence of cytokinesis, and impaired cytokinesis leading to an increase in both multinucleated cells and “zoids” (cells lacking nuclear DNA content) and associated cell death. Cell lines were complemented with both wild-type CRK3 and CRK3T178E (a mutation in the activation loop) genes to demonstrate that the phenotype was specific to CRK3 deletion, that the presence of threonine 178 in the activation loop of the protein kinase domain was important for proper function, and that CRK3T178E could not complement the loss of wild-type CRK3. Not only did this study show the importance of CRK3 in promastigote stages, but the gene was deleted in stationary phase cultures (metacyclic-enriched), allowing the parasites to be inoculated into mice and the progress of the infection to be monitored by noninvasive, bioluminescent imaging using a strain engineered to express red-shifted luciferase. CRK3 was shown to be important for the establishment and maintenance of infection by a reduction in bioluminescence from mice infected with the CRK3-deficient cells. This was ranked as a five-star validation (1, Figure 1D). Despite the now extremely good genetic validation of L. mexicana CRK3 as an essential gene, a previous HTS campaign (against LmxCRK3/CYC6) failed to identify compounds that had bioactivity against the parasite in macrophages.49 The compounds identified failed to inhibit parasite growth in macrophages despite having good physiochemical properties and having different scaffolds, suggesting that these compounds failed because of some unknown aspect of CRK3 biology in Leishmania or unknown bioavailability within the parasitophorous vacuole, thus CRK3/CYC6 has not been fully chemically validated.
A more definitive assessment of essentiality would be to induce gene deletions in amastigotes during animal infections. This experiment is not possible with the DiCre system due to the lack of bioavailability of rapamycin/rapalogues and the toxicity of rapamycin to amastigotes. Such a model would enable researchers to observe that an experimental infection was not compromised during the establishment phase, but upon inducible deletion of an essential gene the infection would fail and be resolved by the host immune system.
Overview of Existing Gene Deletion Attempts
To assess the current state of target validation in Leishmania, a literature search was performed to identify all instances where these genetic techniques have been used to attempt to generate null mutants in human pathogenic Leishmania (Figure 2A, Table S1). We focused on identifying the first instance where researchers had attempted to generate a null mutant for a given gene, but we do not provide a comprehensive review of every genetic study on the same gene in multiple species of Leishmania. The strategy used to attempt the genetic deletion was identified, whether this was successful or not, along with a broad categorization of the phenotype, i.e. essential for promastigotes, conditionally essential, or nonessential. If a complementation was required, it was defined if this was genetic or a nutritional supplementation. Two hundred genes were identified, where attempts have been made to make null mutants in promastigotes of various Leishmania species; 65 were classified as essential, and the remaining 135 were classified as nonessential. The rate of genetic deletion studies being published appeared to accelerate after 2008, possibly due to the publication of the L. major genome in 200550 and the L. braziliensis and L. infantum genomes in 2007,51 but has remained fairly constant between 2008 and 2017. Our literature search identified a range of different species of Leishmania under investigation, including those responsible for both cutaneous and visceral diseases (60 vs 40%, respectively), with L. major (37%) being the most commonly studied, followed by L. donovani (29%) (Figure 2B).
A gene ontology (GO) analysis was performed on the 200 genes using TriTrypDB52 and REVIGO53 (Figure 3, Table S1). GO was analyzed by clustering to describe the biological processes that are performed by the 200 gene products. Clear clusters emerge for processes involved in nutrient transport and stress response, and broader clusters emerge for metabolic processes including nucleotide salvage and synthesis. There are many instances of GO terms associated with oxidation–reduction processes and protein modification such as protein folding, proteolysis, and protein phosphorylation, demonstrating the strength of research on metabolism, peptidases, and protein kinases. Important single gene processes were also identified, such as N-terminal protein myristoylation.
Figure 3.

REVIGO analysis of GO terms associated with targeted Leishmania genes. L. major orthologues for all gene IDs in Table S1 were used to recover the associated GO terms for biological processes. The number of occurrences of each GO term was used to weight a REVIGO analysis, depicted as larger circles and hotter colors. The more frequently occurring biological processes are annotated in the figure, as are key (but less frequent) GO terms such as N-terminal protein myristoylation.
Because of the high variability in the assay protocols used for phenotypic analyses in each study, it is challenging to allocate each gene deletion mutant to a defined phenotypic category, other than essential or nonessential to promastigotes. For example, gene deletions that confer altered virulence may be essential in a given context such as mammalian infection, but the classification of these phenotypes can be quite subtle or dependent on the species, infectious dose, or mouse strain used and is outside the scope of this Perspective. In this regard, there is a clear need for a comparative analysis of Leishmania null mutants, where the same genetic approaches and phenotype analysis can be carried out on a large scale, which is discussed in the next section.
By way of comparison and to extend the analysis, the literature was also reviewed to identify Trypanosoma cruzi genes for which the creation of null mutants had been attempted (Table S2). The genetic tools available for Leishmania are broadly applicable to Trypanosoma cruzi in that this organism also has a functional homologous recombination system and can support the expression of genes from episomes.54,55 As for Leishmania, we focused on identifying the first study to report the deletion attempt for any given gene. On the basis of our five star scoring system, studies on T. cruzi did not reach a high-quality level of drug target validation. Since 1993, only 36 genes had deletion attempts reported in the literature, and 16 were considered to be essential in epimastigotes (Figure 4, Table S2). Only one study, of NMT,56 used a variant of the facilitated null approach to validate the target gene; the deletion of both chromosomal alleles was not possible until a “rescue” allele was integrated into the ribosomal locus of the parasite. This demonstrated that the generation of chromosomal allelic deletions was possible, but only after genetic complementation; therefore, the gene is likely to be essential. However, because the rescue allele was integrated into the genomic DNA there was no possibility to test that it could be lost to create a true facilitated null cell line. Recently, DiCre57 systems were applied to T. cruzi, opening the possibility for better target validation.
Figure 4.

Number of reverse genetic manipulations of Leishmania in comparison to other model parasitic protozoans. (A) Comparison of reverse genetic screening in Leishmania spp. (Table S1), Trypanosoma cruzi (Table S2), Trypanosoma brucei,59Toxoplasma gondii,60 and Plasmodium berghei.67 Pie chart segments depict the proportion of genes in each organism that have been targeted using reverse genetics, with the overall area of the pie depicting the relative sizes of the genomes.
Prospects for Large-Scale Genetic Manipulations of Leishmania: Lessons from Other Protozoa
When compared to other, more genetically tractable protozoan parasites, the situation in Leishmania looks limited, mainly due to the low throughput of the prevailing techniques used to generate Leishmania gene deletions. Through a brief description of tools and screens that have been used in other kinetoplastids and apicomplexans, despite the unique biology and technical challenges each organism and method present, desirable features for future genetic screens in Leishmania can be identified. To date, the genetic manipulation of Leishmania has been on a gene-by-gene basis, yet several other parasitic protozoa such as Trypanosomabrucei and Toxoplasma gondii have been amenable to genome-wide screens58−60 (Figure 4). Combining these massively paralleled genetic depletion resources with various selection pressures can yield sophisticated experiments that are able to interrogate, identify, and validate drug targets and elucidate resistance mechanisms.
Most relevant to Leishmania target validation is the use of RNAi screening in T. brucei, which evolved from gene-by-gene projects to genome-wide RNAi screens58,59 and medium-throughput gene family screens.61,62 These massively paralleled RIT-seq (RNA interference target sequencing) experiments have led to the identification of genes essential for life cycle progression, drug resistance, and the response to stress.63,64 Of particular relevance is the way that this technology was used to identify organelles, proteins, and metabolic pathways responsible for the mode of action of trypanocidal drugs65,66 as this may be used to shed light on pan-kinetoplastid biology and drug targets.
Other model pathogens such as Plasmodium berghei have also been subject to high-throughput gene deletion screens. Although less amenable to genetic manipulation, a herculean effort has allowed half of the P. berhei genome to be assessed for essentiality67,68 (Figure 4). The PlasmoGem project used recombineering to build libraries of long-homology flanked deletion vectors that can be transfected into parasites prior to infecting mice.67,68 Pooling these libraries allowed for more than half of the parasite’s genes to be assessed for their role in parasite fitness by sequencing DNA barcodes that were included in the null mutants. This analysis suggested a wealth of drug targets as ∼60% of the P. berghei genes were considered essential. This compares to ∼30% in T. brucei, and on the basis of our literature search, we expect Leishmania to be similar to 32.6% of the Leishmania genes investigated so far that are essential.
Increased throughput and efficiency for the genetic manipulation of parasitic protozoa has occurred by the adoption of CRISPR/Cas9-mediated gene disruption and editing, with the first genome-wide screen in a parasite performed in Toxoplasma gondii.60T. gondii has a propensity for repairing double-stranded DNA breaks by error-prone, nonhomologous end joining (NHEJ), allowing for the inactivation of genes by frame-shifting insertions or deletions. Next-generation sequencing was used to quantify the remaining sgRNAs in the population and thus calculate a score for how important a given gene is for parasite fitness. When combined with the comparative genomic assessment of essential T. gondii genes to conserved genes in other apicomplexans, the authors identified potential pan-apicomplexan targets, some of which were then investigated and confirmed as essential in Plasmodium falciparum. The recent application of CRISPR/Cas9 to Leishmania may enable such genome-wide screening, but this requires further development.69−73 This is due to Leishmania lacking a functional NHEJ pathway, although it can repair a double-strand break using microhomology-mediated end joining (MMEJ). Despite this, the medium-throughput analysis of gene families in Leishmania is now a reality using the highly efficient L. mexicana T7/Cas9 system. This method uses PCR amplicons for both sgRNA and repair templates with no cloning steps, both alleles can be targeted in a single transfection, and drug selection in pools has a high efficiency.73 These factors permit easy and fast deprioritization of nonessential genes from a family of potential targets.
Combined with the ability to generate facilitated null mutants, CRISPR/Cas9 approaches can begin to provide indirect evidence for a gene’s essentiality. Indeed, we expect that soon the number of CRISPR/Cas9 genetic deletion attempts in Leishmania will rapidly exceed the total number of gene deletions generated between 1990 and 2017, possibly even genome-wide. Because the other genetic techniques, such as DiCre, are more labor-intensive, it would be beneficial to focus them on prioritized genes that cannot be deleted by CRISPR/Cas9; therefore, CRISPR/Cas9 should facilitate faster and more detailed studies on genes that have interesting or essential functions. CRISPR/Cas9 systems have also been developed for T. cruzi, further expanding the toolbox for genetic manipulation.72,74,75
While constructing the spreadsheet of attempted gene deletions, we were struck by the range of different phenotypic analyses that were performed, which restricted placing genes into neat categories based on the type or strength of phenotype observed. One of the benefits of cell library screens or massively paralleled approaches is the ability to simultaneously analyze all of the mutants in the same assay, which allows for an easier comparison of phenotypes. The integration of such data into EuPathDB would make an exceptionally useful resource.52 Target selection for Leishmania can be guided by mining the data sets available in resources such as TriTrypDB, so we encourage fellow researchers to contribute to this community resource with both historical and current data.
Conclusions
The discovery of new drugs for intracellular pathogens is a complex process that usually has a low chance of success. In order to give it the best chance, the evidence needed to support the process must be of a high standard. The optimization of existing genetic techniques, the development of new techniques, and the application of best practices is critical for identifying and validating novel druggable targets. A balanced approach between target-based drug development and phenotypic screening, supported by genetic tools for molecular target validation, is desirable. High-quality genetic tools now available for conducting research on potential drug targets in Leishmania are optimal, and the new technologies available will facilitate and improve the number of genetically well-validated targets suitable for entry into HTS campaigns.
Acknowledgments
Research in the J.C.M. laboratory is supported by a Wellcome Trust Investigator Award (200807/Z/16/Z). The research leading to these results has, in part, received funding form the Research Council United Kingdom Grand Challenges Research Funder under grant agreement ‘A Global Network for Neglected Tropical Diseases’ grant number MR/P027989/1. We apologize for any published gene deletions that have been inadvertently omitted during our search.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.7b00244.
Lists of genes for Leishmania species, where gene deletions have been attempted. This table contains a tab for essential genes and a tab for nonessential genes. (XLSX)
Lists of genes for Trypanosoma cruzi, where gene deletions have been attempted. This table contains a tab for essential genes and a tab for nonessential genes. (XLSX)
Descriptions of Tables S1 and S2 (PDF)
Author Contributions
N.G.J., C.M.C.C.-P., and J.C.M. designed and developed the work. N.G.J., C.M.C.C.-P., A.P.C.A.L., and J.C.M. carried out data analysis. N.G.J., C.M.C.C.-P., and J.C.M. wrote the article.
The authors declare no competing financial interest.
Supplementary Material
References
- Alvar J.; Vélez I. D.; Bern C.; Herrero M.; Desjeux P.; Cano J.; Jannin J.; Boer den. M. (2012) Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7 (5), e35671. 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DNDi . https://www.dndi.org/diseases-projects/leishmaniasis/, accessed November 02, 2017.
- WHO . http://www.who.int/leishmaniasis/en/, accessed November 02, 2017.
- Mandell M. A.; Beverley S. M. (2017) Continual renewal and replication of persistent Leishmania major parasites in concomitantly immune hosts. Proc. Natl. Acad. Sci. U. S. A. 114 (5), E801–E810. 10.1073/pnas.1619265114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field M. C.; Horn D.; Fairlamb A. H.; Ferguson M. A. J.; Gray D. W.; Read K. D.; De Rycker M.; Torrie L. S.; Wyatt P. G.; Wyllie S.; et al. (2017) Anti-trypanosomatid drug discovery: an ongoing challenge and a continuing need. Nat. Rev. Microbiol. 15 (4), 217–231. 10.1038/nrmicro.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle A. S.; Khare S.; Kumar A. B.; Supek F.; Buchynskyy A.; Mathison C. J. N.; Chennamaneni N. K.; Pendem N.; Buckner F. S.; Gelb M. H.; et al. (2014) Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem. Rev. 114 (22), 11305–11347. 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulfiqar B.; Shelper T. B.; Avery V. M. (2017) Leishmaniasis drug discovery: recent progress and challenges in assay development. Drug Discovery Today 22 (10), 1516–1531. 10.1016/j.drudis.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Rajasekaran R.; Chen Y.-P. P. (2015) Potential therapeutic targets and the role of technology in developing novel antileishmanial drugs. Drug Discovery Today 20 (8), 958–968. 10.1016/j.drudis.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Gilbert I. H. (2013) Drug discovery for neglected diseases: molecular target-based and phenotypic approaches. J. Med. Chem. 56 (20), 7719–7726. 10.1021/jm400362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M. R.; Tipney H.; Painter J. L.; Shen J.; Nicoletti P.; Shen Y.; Floratos A.; Sham P. C.; Li M. J.; Wang J.; et al. (2015) The support of human genetic evidence for approved drug indications. Nat. Genet. 47 (8), 856–860. 10.1038/ng.3314. [DOI] [PubMed] [Google Scholar]
- Priotto G.; Pinoges L.; Fursa I. B.; Burke B.; Nicolay N.; Grillet G.; Hewison C.; Balasegaram M. (2008) Safety and effectiveness of first line eflornithine for Trypanosoma brucei gambiense sleeping sickness in Sudan: cohort study. BMJ: British Medical Journal 336 (7646), 705–708. 10.1136/bmj.39485.592674.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A. K.; Mukherjee T.; Bhaduri A. (1986) Mechanism of action of amphotericin B on Leishmania donovani promastigotes. Mol. Biochem. Parasitol. 19 (3), 195–200. 10.1016/0166-6851(86)90001-0. [DOI] [PubMed] [Google Scholar]
- Mbongo N.; Loiseau P. M.; Billion M. A.; Robert-Gero M. (1998) Mechanism of amphotericin B resistance in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 42 (2), 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieu E.; Prina E.; Leclercq O.; Oumata N.; Gaboriaud-Kolar N.; Vougogiannopoulou K.; Aulner N.; Defontaine A.; No J. H.; Ruchaud S.; et al. (2016) From Drug Screening to Target Deconvolution: a Target-Based Drug Discovery Pipeline Using Leishmania Casein Kinase 1 Isoform 2 To Identify Compounds with Antileishmanial Activity. Antimicrob. Agents Chemother. 60 (5), 2822–2833. 10.1128/AAC.00021-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S.; Nagle A. S.; Biggart A.; Lai Y. H.; Liang F.; Davis L. C.; Barnes S. W.; Mathison C. J. N.; Myburgh E.; Gao M.-Y.; et al. (2016) Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537 (7619), 229–233. 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino M.; Choy J. W.; Gushwa N. N.; Oses-Prieto J. A.; Koupparis K.; Burlingame A. L.; Renslo A. R.; McKerrow J. H.; Taunton J. (2013) Hypothemycin, a fungal natural product, identifies therapeutic targets in Trypanosoma brucei. eLife 2, e00712. 10.7554/eLife.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begolo D.; Erben E.; Clayton C. (2014) Drug target identification using a trypanosome overexpression library. Antimicrob. Agents Chemother. 58 (10), 6260–6264. 10.1128/AAC.03338-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazanion É.; Fernández-Prada C.; Papadopoulou B.; Leprohon P.; Ouellette M. (2016) Cos-Seq for high-throughput identification of drug target and resistance mechanisms in the protozoan parasite Leishmania. Proc. Natl. Acad. Sci. U. S. A. 113 (21), E3012–E3021. 10.1073/pnas.1520693113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I. M.; Barrett M. P. (2015) Metabolomic-based strategies for anti-parasite drug discovery. J. Biomol. Screening 20 (1), 44–55. 10.1177/1087057114551519. [DOI] [PubMed] [Google Scholar]
- Peña I.; Pilar Manzano M.; Cantizani J.; Kessler A.; Alonso-Padilla J.; Bardera A. I.; Alvarez E.; Colmenarejo G.; Cotillo I.; Roquero I.; et al. (2015) New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource. Sci. Rep. 5, 8771. 10.1038/srep08771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulfiqar B.; Jones A. J.; Sykes M. L.; Shelper T. B.; Davis R. A.; Avery V. M. (2017) Screening a Natural Product-Based Library against Kinetoplastid Parasites. Molecules 22 (10), 1715. 10.3390/molecules22101715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson J. A.; Wyatt P. G.; Gilbert I. H.; Fairlamb A. H. (2007) Target assessment for antiparasitic drug discovery. Trends Parasitol. 23 (12), 589–595. 10.1016/j.pt.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R. G.; Thomson G.; Malone K.; Nowicki M. W.; Brown E.; Blake D. G.; Turner N. J.; Walkinshaw M. D.; Grant K. M.; Mottram J. C. (2011) High throughput screens yield small molecule inhibitors of Leishmania CRK3:CYC6 cyclin-dependent kinase. PLoS Neglected Trop. Dis. 5 (4), e1033. 10.1371/journal.pntd.0001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell A. S.; Mills J. E.; Williams G. P.; Brannigan J. A.; Wilkinson A. J.; Parkinson T.; Leatherbarrow R. J.; Tate E. W.; Holder A. A.; Smith D. F. (2012) Selective inhibitors of protozoan protein N-myristoyltransferases as starting points for tropical disease medicinal chemistry programs. PLoS Neglected Trop. Dis. 6 (4), e1625. 10.1371/journal.pntd.0001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzuti A.; Paglietti G.; Hunter W. N.; Gamarro F.; Piras S.; Loriga M.; Allecca S.; Corona P.; McLuskey K.; Tulloch L.; et al. (2008) Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. U. S. A. 105 (5), 1448–1453. 10.1073/pnas.0704384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachidi N.; Taly J. F.; Durieu E.; Leclercq O.; Aulner N.; Prina E.; Pescher P.; Notredame C.; Meijer L.; Späth G. F. (2014) Pharmacological assessment defines Leishmania donovani casein kinase 1 as a drug target and reveals important functions in parasite viability and intracellular infection. Antimicrob. Agents Chemother. 58 (3), 1501–1515. 10.1128/AAC.02022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocco J. J.; Donald R.; Zhong T.; Lee A.; Tang Y. S.; Hendrickson R. C.; Liberator P.; Nare B. (2006) Inhibitors of casein kinase 1 block the growth of Leishmania major promastigotes in vitro. Int. J. Parasitol. 36 (12), 1249–1259. 10.1016/j.ijpara.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Sterkers Y.; Lachaud L.; Crobu L.; Bastien P.; Pagès M. (2011) FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major. Cell. Microbiol. 13 (2), 274–283. 10.1111/j.1462-5822.2010.01534.x. [DOI] [PubMed] [Google Scholar]
- Dumetz F.; Imamura H.; Sanders M.; Seblova V.; Myskova J.; Pescher P.; Vanaerschot M.; Meehan C. J.; Cuypers B.; De Muylder G.; et al. (2017) Modulation of Aneuploidy in Leishmania donovani during Adaptation to Different In Vitro and In Vivo Environments and Its Impact on Gene Expression. mBio 8 (3), e00599-17. 10.1128/mBio.00599-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M. B.; Hilley J. D.; Dickens N. J.; Wilkes J.; Bates P. A.; Depledge D. P.; Harris D.; Her Y.; Herzyk P.; Imamura H.; et al. (2011) Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 21 (12), 2129–2142. 10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye L. F.; Owens K.; Shi H.; Murta S. M. F.; Vieira A. C.; Turco S. J.; Tschudi C.; Ullu E.; Beverley S. M. (2010) Retention and Loss of RNA Interference Pathways in Trypanosomatid Protozoans. PLoS Pathog. 6 (10), e1001161. 10.1371/journal.ppat.1001161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates P. A. (1994) Complete developmental cycle of Leishmania mexicana in axenic culture. Parasitology 108 (Pt 1), 1–9. 10.1017/S0031182000078458. [DOI] [PubMed] [Google Scholar]
- Sereno D.; Roy G.; Lemesre J. L.; Papadopoulou B.; Ouellette M. (2001) DNA Transformation of Leishmania infantum Axenic Amastigotes and Their Use in Drug Screening. Antimicrob. Agents Chemother. 45 (4), 1168–1173. 10.1128/AAC.45.4.1168-1173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan S. M.; Jones N. G.; Mottram J. C. (2017) Molecular & Biochemical Parasitology Recent advances in Leishmania reverse genetics: Manipulating a manipulative parasite. Mol. Biochem. Parasitol. 216 (June), 30–38. 10.1016/j.molbiopara.2017.06.005. [DOI] [PubMed] [Google Scholar]
- Cruz A.; Beverley S. M. (1990) Gene replacement in parasitic protozoa. Nature 348 (6297), 171–173. 10.1038/348171a0. [DOI] [PubMed] [Google Scholar]
- Cruz A.; Coburn C. M.; Beverley S. M. (1991) Double targeted gene replacement for creating null mutants. Proc. Natl. Acad. Sci. U. S. A. 88 (16), 7170–7174. 10.1073/pnas.88.16.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambit A.; Fasel N.; Coombs G. H.; Mottram J. C. (2008) An essential role for the Leishmania major metacaspase in cell cycle progression. Cell Death Differ. 15 (1), 113–122. 10.1038/sj.cdd.4402232. [DOI] [PubMed] [Google Scholar]
- Casanova M.; Gonzalez I. J.; Sprissler C.; Zalila H.; Dacher M.; Basmaciyan L.; Späth G. F.; Azas N.; Fasel N. (2015) Implication of different domains of the Leishmania major metacaspase in cell death and autophagy. Cell Death Dis. 6 (10), e1933. 10.1038/cddis.2015.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanys-Muñoz E.; Brown E.; Coombs G. H.; Mottram J. C. (2012) Leishmania mexicana metacaspase is a negative regulator of amastigote proliferation in mammalian cells. Cell Death Dis. 3 (9), e385. 10.1038/cddis.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Roberts S. C.; Jardim A.; Carter N. S.; Shih S.; Ariyanayagam M.; Fairlamb A. H.; Ullman B. (1999) Ornithine decarboxylase gene deletion mutants of Leishmania donovani. J. Biol. Chem. 274 (6), 3781–3788. 10.1074/jbc.274.6.3781. [DOI] [PubMed] [Google Scholar]
- Vergnes B.; Sereno D.; Tavares J.; Cordeiro-da-Silva A.; Vanhille L.; Madjidian-Sereno N.; Depoix D.; Monte-Alegre A.; Ouaissi A. (2005) Targeted disruption of cytosolic SIR2 deacetylase discloses its essential role in Leishmania survival and proliferation. Gene 363, 85–96. 10.1016/j.gene.2005.06.047. [DOI] [PubMed] [Google Scholar]
- Murta S. M. F.; Vickers T. J.; Scott D. A.; Beverley S. M. (2009) Methylene tetrahydrofolate dehydrogenase/cyclohydrolase and the synthesis of 10-CHO-THF are essential in Leishmania major. Mol. Microbiol. 71 (6), 1386–1401. 10.1111/j.1365-2958.2009.06610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher M.; Morales M. A.; Pescher P.; Leclercq O.; Rachidi N.; Prina E.; Cayla M.; Descoteaux A.; Späth G. F. (2014) Probing druggability and biological function of essential proteins in Leishmania combining facilitated null mutant and plasmid shuffle analyses. Mol. Microbiol. 93 (1), 146–166. 10.1111/mmi.12648. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Melzer I. M.; Kruse M.; Sander-Juelch C.; Wiese M. (2005) LmxMPK4, a mitogen-activated protein (MAP) kinase homologue essential for promastigotes and amastigotes of Leishmania mexicana. Kinetoplastid Biol. Dis. 4, 6. 10.1186/1475-9292-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan S. M.; Myburgh E.; Philipon C.; Brown E.; Meissner M.; Brewer J.; Mottram J. C. (2016) Conditional gene deletion with DiCre demonstrates an essential role for CRK3 in Leishmania mexicana cell cycle regulation. Mol. Microbiol. 100 (6), 931–944. 10.1111/mmi.13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan P.; Fergusson D.; Grant K. M.; Mottram J. C. (2001) The CRK3 protein kinase is essential for cell cycle progression of Leishmania mexicana. Mol. Biochem. Parasitol. 113 (2), 189–198. 10.1016/S0166-6851(01)00220-1. [DOI] [PubMed] [Google Scholar]
- Grant K. M.; Dunion M. H.; Yardley V.; Skaltsounis A.-L.; Marko D.; Eisenbrand G.; Croft S. L.; Meijer L.; Mottram J. C. (2004) Inhibitors of Leishmania mexicana CRK3 cyclin-dependent kinase: chemical library screen and antileishmanial activity. Antimicrob. Agents Chemother. 48 (8), 3033–3042. 10.1128/AAC.48.8.3033-3042.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant K. M.; Hassan P.; Anderson J. S.; Mottram J. C. (1998) The crk3 gene of Leishmania mexicana encodes a stage-regulated cdc2-related histone H1 kinase that associates with p12. J. Biol. Chem. 273 (17), 10153–10159. 10.1074/jbc.273.17.10153. [DOI] [PubMed] [Google Scholar]
- Walker R. G.; Thomson G.; Malone K.; Nowicki M. W.; Brown E.; Blake D. G.; Turner N. J.; Walkinshaw M. D.; Grant K. M.; Mottram J. C. (2011) High throughput screens yield small molecule inhibitors of Leishmania CRK3:CYC6 cyclin-dependent kinase. PLoS Neglected Trop. Dis. 5 (4), e1033. 10.1371/journal.pntd.0001033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens A. C.; Peacock C. S.; Worthey E. A.; Murphy L.; Aggarwal G.; Berriman M.; Sisk E.; Rajandream M. A.; Adlem E.; Aert R.; et al. (2005) The genome of the kinetoplastid parasite, Leishmania major. Science (Washington, DC, U. S.) 309 (5733), 436–442. 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock C. S.; Seeger K.; Harris D.; Murphy L.; Ruiz J. C.; Quail M. A.; Peters N.; Adlem E.; Tivey A.; Aslett M.; et al. (2007) Nat. Genet. 39 (7), 839–847. 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrecoechea C.; Barreto A.; Basenko E. Y.; Brestelli J.; Brunk B. P.; Cade S.; Crouch K.; Doherty R.; Falke D.; Fischer S.; et al. (2017) EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Res. 45 (D1), D581–D591. 10.1093/nar/gkw1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F.; Bošnjak M.; Škunca N.; Šmuc T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6 (7), e21800. 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. M.; Ward H. M.; Miles M. A.; Kendall G. (1992) A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Res. 20 (15), 3963–3969. 10.1093/nar/20.15.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burle-Caldas G. de A.; Grazielle-Silva V.; Laibida L. A.; DaRocha W. D.; Teixeira S. M. R. (2015) Expanding the tool box for genetic manipulation of Trypanosoma cruzi. Mol. Biochem. Parasitol. 203 (1–2), 25–33. 10.1016/j.molbiopara.2015.10.004. [DOI] [PubMed] [Google Scholar]
- Roberts A. J.; Torrie L. S.; Wyllie S.; Fairlamb A. H. (2014) Biochemical and genetic characterization of Trypanosoma cruzi N-myristoyltransferase. Biochem. J. 459 (2), 323–332. 10.1042/BJ20131033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangussu-Marcolino M. M.; Cunha A. P.; Avila A. R.; Herman J.-P.; DaRocha W. D. (2014) Conditional removal of selectable markers in Trypanosoma cruzi using a site-specific recombination tool: proof of concept. Mol. Biochem. Parasitol. 198 (2), 71–74. 10.1016/j.molbiopara.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Schumann Burkard G.; Jutzi P.; Roditi I. (2011) Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol. Biochem. Parasitol. 175 (1), 91–94. 10.1016/j.molbiopara.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Alsford S.; Turner D. J.; Obado S. O.; Sanchez-Flores A.; Glover L.; Berriman M.; Hertz-Fowler C.; Horn D. (2011) High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21 (6), 915–924. 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik S. M.; Huet D.; Ganesan S. M.; Huynh M.-H.; Wang T.; Nasamu A. S.; Thiru P.; Saeij J. P. J.; Carruthers V. B.; Niles J. C.; et al. (2016) A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166 (6), 1423–1435.e12. 10.1016/j.cell.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N. G.; Thomas E. B.; Brown E.; Dickens N. J.; Hammarton T. C.; Mottram J. C. (2014) Regulators of Trypanosoma brucei Cell Cycle Progression and Differentiation Identified Using a Kinome-Wide RNAi Screen. PLoS Pathog. 10 (1), e1003886. 10.1371/journal.ppat.1003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Cortes F.; Serafim T. D.; Wilkes J. M.; Jones N. G.; Ritchie R.; Mcculloch R.; Mottram J. C. (2017) RNAi screening identifies Trypanosoma brucei stress response protein kinases required for survival in the mouse. Sci. Rep. 7, 6156. 10.1038/s41598-017-06501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stortz J. A.; Serafim T. D.; Alsford S.; Wilkes J.; Fernandez-Cortes F.; Hamilton G.; Briggs E.; Lemgruber L.; Horn D.; Mottram J. C.; et al. (2017) Genome-wide and protein kinase-focused RNAi screens reveal conserved and novel damage response pathways in Trypanosoma brucei. PLoS Pathog. 13 (7), e1006477. 10.1371/journal.ppat.1006477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony B. M.; MacGregor P.; Ivens A.; Rojas F.; Cowton A.; Young J.; Horn D.; Matthews K. (2014) Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature 505 (7485), 681–685. 10.1038/nature12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsford S.; Eckert S.; Baker N.; Glover L.; Sanchez-Flores A.; Leung K. F.; Turner D. J.; Field M. C.; Berriman M.; Horn D. (2012) High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 482 (7384), 232–236. 10.1038/nature10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N.; Hamilton G.; Wilkes J. M.; Hutchinson S.; Barrett M. P.; Horn D. (2015) Vacuolar ATPase depletion affects mitochondrial ATPase function, kinetoplast dependency, and drug sensitivity in trypanosomes. Proc. Natl. Acad. Sci. U. S. A. 112 (29), 9112–9117. 10.1073/pnas.1505411112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell E.; Gomes A. R.; Sanderson T.; Anar B.; Girling G.; Herd C.; Metcalf T.; Modrzynska K.; Schwach F.; Martin R. E.; et al. (2017) Functional Profiling of a Plasmodium Genome Reveals an Abundance of Essential Genes. Cell 170 (2), 260. 10.1016/j.cell.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes A. R.; Bushell E.; Schwach F.; Girling G.; Anar B.; Quail M. A.; Herd C.; Pfander C.; Modrzynska K.; Rayner J. C.; et al. (2015) A genome-scale vector resource enables high-throughput reverse genetic screening in a malaria parasite. Cell Host Microbe 17 (3), 404–413. 10.1016/j.chom.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. W.; Matlashewski G. (2015) CRISPR-Cas9-mediated genome editing in Leishmania donovani. mBio 6 (4), e00861-15. 10.1128/mBio.00861-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollelis L.; Ghorbal M.; Macpherson C. R.; Martins R. M.; Kuk N.; Crobu L.; Bastien P.; Scherf A.; Lopez-Rubio J. J.; Sterkers Y. (2015) First efficient CRISPR-Cas9-mediated genome editing in Leishmania parasites. Cell. Microbiol. 17 (10), 1405–1412. 10.1111/cmi.12456. [DOI] [PubMed] [Google Scholar]
- Zhang W.-W.; Matlashewski G. (2017) Optimized CRISPR-Cas9 Genome Editing for Leishmania and Its Use To Target a Multigene Family, Induce Chromosomal Translocation, and Study DNA Break Repair Mechanisms. mSphere 2, e00340-16. 10.1128/mSphere.00340-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares Medeiros L. C.; South L.; Peng D.; Bustamante J. M.; Wang W.; Bunkofske M.; Perumal N.; Sanchez-Valdez F.; Tarleton R. L. (2017) Rapid, Selection-Free, High-Efficiency Genome Editing in Protozoan Parasites Using CRISPR-Cas9 Ribonucleoproteins. mBio 8 (6), e01788-17. 10.1128/mBio.01788-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beneke T., Madden R., Makin L., Valli J., Sunter J., and Gluenz E.. A CRISPR Cas9 high-throughput genome editing toolkit for kinetoplastids. R. Soc. Open Sci. 2017, May 10.1098/rsos.170095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D.; Kurup S. P.; Yao P. Y.; Minning T. A.; Tarleton R. L. (2015) CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio 6 (1), e02097-14. 10.1128/mBio.02097-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander N.; Li Z.-H.; Niyogi S.; Docampo R. (2015) CRISPR/Cas9-Induced Disruption of Paraflagellar Rod Protein 1 and 2 Genes in Trypanosoma cruzi Reveals Their Role in Flagellar Attachment. mBio 6 (4), e01012-15. 10.1128/mBio.01012-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.